 Chiara Minotti1,2*†
Chiara Minotti1,2*† Ludovico Graziani1,2†
Ludovico Graziani1,2† Ester Sallicandro3
Ester Sallicandro3 Maria Cristina Digilio1Roberto Falasca3Viola Alesi3
Maria Cristina Digilio1Roberto Falasca3Viola Alesi3 Giuseppe Novelli2,4
Giuseppe Novelli2,4 Maria Lisa Dentici1
Maria Lisa Dentici1 Sara Loddo3
Sara Loddo3 Antonio Novelli3
Antonio Novelli3- 1Medical Genetics Unit, Translational Pediatrics and Clinical Genetics Research Area, Bambino Gesù Children Hospital, Istituto di Ricovero e Cura a Carattere Scientifico, Rome, Italy
- 2Medical Genetics Section, Depepartment of Biomedicine and Prevention, Tor Vergata University of Rome, Rome, Italy
- 3Translational Cytogenomics Research Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 4Medical Genetics Lab, Tor Vergata Hospital, Rome, Italy
Interstitial deletions involving 6q chromosomal region are rare. Less than 30 patients have been described to date, and fewer have been characterized by high-resolution techniques, such as chromosomal microarray. Deletions involving 6q21q22.1 region are associated with an extremely wide and heterogeneous clinical spectrum, thus genotype–phenotype correlation based on the size of the rearranged region and on the involved genes is complex, even among individuals with overlapping deletions. Here we describe the phenotypic and molecular characterization of a new 6q interstitial deletion in a girl with developmental delay, intellectual disability, cerebellar vermis hypoplasia, facial peculiar characteristics, ataxia and ocular abnormalities. Microarray analysis of the proposita revealed a 7.9 Mb interstitial de novo deletion at 6q21q22.1 chromosomal region, which spanned from nucleotides 108,337,770 to 116,279,453 (GRCh38/hg38). The present case, alongside with a systematic review of the literature, provides further evidence that could aid to the definition of the Smallest Region of Overlap and of the genomic traits that are associated with particular phenotypes, focusing on neurological findings and especially on cerebellar anomalies.
Introduction
Deletions of the long arm of chromosome 6 are rare, and less than 30 patients with interstitial deletions involving the 6q21q22.1 region have been described to date (Schwartz et al., 1984; Young et al., 1985; Pandya et al., 1995; Correa-Cerro et al., 1996; Evers et al., 1996; Hopkin et al., 1997; Tsukahara et al., 1997; Duran-Gonzalez et al., 2007; Zherebtsov et al., 2007; Rosenfeld et al., 2012; Toschi et al., 2012; Hudson et al., 2014; Szafranski et al., 2015; Tassano et al., 2015; Milani et al., 2016; Shukla et al., 2016; Donahue and Rohena, 2017; Machida et al., 2022).
Hopkin et al. (1997) first classified deletions of the long arm of chromosome 6 into three groups, based on conventional cytogenetics, with different and recurrent phenotypes: group A, or proximal deletions (6q11q16), group B, or medial deletions (6q15q25), and group C, or terminal deletions (6q25qter) (Hopkin et al., 1997).
Intellectual disability, developmental delay, hypotonia and postnatal growth retardation appear to be common and non-specific features among patients with 6q deletions (Tassano et al., 2015; Hopkin et al., 1997). Medial deletions (6q15q25) are associated with additional recurrent clinical features including intrauterine growth restriction (IUGR), abnormal respiration, hypertelorism, ear anomalies and upper limb malformation (Hopkin et al., 1997; Donahue and Rohena, 2017). Nonetheless, no univocal genotype-phenotype correlation has been determined so far, even comparing overlapping 6q deletions.
Standard cytogenetic techniques were performed in the first reports (Schwartz et al., 1984; Young et al., 1985; Pandya et al., 1995; Correa-Cerro et al., 1996; Evers et al., 1996; Tsukahara et al., 1997; Duran-Gonzalez et al., 2007; Hopkin et al., 1997). More recent studies are based on higher resolution techniques, such as chromosomal microarray analysis (CMA) (Zherebtsov et al., 2007; Rosenfeld et al., 2012; Toschi et al., 2012; Hudson et al., 2014; Szafranski et al., 2015; Tassano et al., 2015; Milani et al., 2016; Shukla et al., 2016; Donahue and Rohena, 2017; Machida et al., 2022), which has allowed a better characterization of the deleted region and of the genes involved, as new and different clinical features emerged in patients described over the years.
Here, we report a girl with a 6q21q22.1 de novo deletion, detected by CMA, and we focus on her neurological findings detected by brain MRI, such as cerebellar vermis hypoplasia (CBVH). We also provide a review of the literature of the reported cases with overlapping rearrangements.
Case report
The patient is a 2-year-old girl, addressed to our Genetics Unit due to developmental delay. She is the only child of healthy non-consanguineous parents with unremarkable family history.
She was born at 41 weeks of gestational age by cesarean section due to fetal distress, after an uneventful pregnancy. Standard chromosome analysis performed prenatally was normal (46, XX). Birth weight, length and occipitofrontal circumference (OFC) were 3.000 gr (−0.92 SDS), 49 cm (−0.13 SDS), and 35 cm (0.85 SDS) respectively. The Apgar score was 9 at 1 min and 10 at 5 min.
Gross motor development delay was reported: she was able to sit with no support at 8–9 months and she could walk independently at 20 months. She presented a speech delay, as she could pronounce her first words at about 2 years of age and was unable to complete intelligible sentences at the time of examination.
At physical examination (2 years old), her weight was 16 kg (1.23 SDS), her height was 99 cm (1.88 SDS) and her OFC was 49.3 cm (0.82 SDS). Phenotypic features included hypertelorism, downslanted palpebral fissures, epicanthal folds, prominent nasal bridge, low-set small ears with thick helix, bilateral pes valgus, and mild generalized hypotonia.
Ophthalmologic evaluation documented oculomotor apraxia and right convergent strabismus. Neurological evaluation demonstrated general clumsiness and ataxia manifested by balance deficit and a wide-based gait, which her parents referred to be congenital.
Brain MRI (Magnetic Resonance Imaging) at 2 years old revealed CBVH, cerebellar volume reduction and a minimal asymmetry of cerebral peduncles (Figure 1).

FIGURE 1. Brain Magnetic Resonance imaging (MRI) analyses of the patient. (A) Mid sagittal T1-weighted image and (B) Coronal T2-weighted image demonstrating cerebellar vermis hypoplasia (B). (C) Axial T1-weighted and (D) T2-weighted images showing mild asymmetry of cerebral peduncles.
According to Griffiths Mental Development Scales (GMDS) (Griffiths, 1970), our patient presented with a General Quotient (GQ) score of 79 and a developmental age of 18 months versus a chronological age of 23 months. Her major deficits consisted in poor oculo-manual coordination and attention defects.
Informed consent was obtained from the parents of the proposita, and CMA was performed. The analysis revealed a 7.9 Mb interstitial deletion at 6q21q22.1 chromosomal region, which spanned from nucleotides 108,337,770 to 116,279,453 (GRCh38). FISH (Fluorescence In Situ Hybridization) on metaphase chromosome preparation from cultured lymphocytes of patient and her parents confirmed a highly likely de novo deletion.
Materials and methods
All data were obtained in agreement with Bambino Gesù Children Hospital ethical standards. CMA was performed using Infinium CytoSNP-850K BeadChip (Illumina, San Diego, CA), according to the manufacturer’s protocol. Array scanning data were generated on the Illumina NextSeq 550 system and the results were analyzed by the BluefuseMulti 4.4 software. Confirmation and segregation tests were performed by FISH on metaphase chromosome preparations of the patient and her parents, using different locus-specific BAC probes [RP11-469I15 (6q21) e RP11-179F7 (6q22.1)]. BACs clones were selected from a genomic library (32 K library; BACPAC Resources, Oakland, CA).
We searched the DECIPHER database and scientific reports on PubMed to identify individuals with overlapping chromosomal imbalances characterized through postnatal CMA, specifically involving the 6q21q22.1 chromosomal region.
Discussion
Interstitial deletions of the 6q region are related with an extremely wide phenotypic spectrum. Intellectual disability, developmental delay, facial peculiar characteristics, hypotonia and postnatal growth retardation are frequent but non-specific features of the condition, and thus present in most chromosome imbalances (Zherebtsov et al., 2007; Hopkin et al., 1997).
We report a new interstitial 6q deletion in a girl with developmental delay, hypotonia, ataxia, facial peculiar characteristics, CBVH and skeletal, ophthalmological, and neurological anomalies.
Although numerous 6q21q22.1 deletion cases have been described, only the most recent ones have been characterized with molecular cytogenetics resolution. In most patients, CMA techniques have demonstrated that chromosomal rearrangements show non-recurrent breakpoints and only partial overlap (Rosenfeld et al., 2012; Szafranski et al., 2015; Milani et al., 2016). The following mechanisms could be hypothesized for the non-recurrent breakpoints observed: Non-Allelic Homologous Recombination (NAHR), Non-Homologous End Joining (NHEJ) or Fork Stalling and Template Switching (FoSTeS) (Gu et al., 2008; Stankiewicz and Lupski, 2002; Zhang et al., 2009). Notwithstanding these limitations, previous authors suggested some genotype-phenotype correlations for 6q interstitial deletion.
Rosenfeld et al. (2012) described a cohort of 12 individuals with variable deletions within the 6q15q22.33 region and compared their clinical features. Heterogeneous clinical expression was reported, even among individuals with overlapping deletions. Some facial peculiar characteristics were variably shared among different cases, with hypertelorism, microcephaly and broad/flat nasal bridge being the most common (Rosenfeld et al., 2012).
Toschi et al. (2012) and then Hudson et al. (2014) described the association between 6q21q22.3 microdeletions and Acrocardiofacial syndrome (ACFS) (Toschi et al., 2012; Hudson et al., 2014). Developmental anomalies of muscular tissue (as in Poland syndrome) have also been reported (Tassano et al., 2015). Milani et al. (2016) hypothesized a critical region for ACFS or at least congenital heart disease (CHD), spanning from nucleotides 107,754,749 to 110,769,883 (GRCh38). They also pointed out a possible role of three genes: SNX3 (MIM*605930), SESN1 (MIM*606103) and FOXO3 (MIM*602681) (Milani et al., 2016). Shukla et al. (2016) reported on a patient who shared similar deletion breakpoints with those described by Toschi et al. and Hudson et al., but lacked any feature suggestive of ACFS (Shukla et al., 2016). We as well describe a patient who does not display any phenotypic feature suggestive of ACFS. This may underline the extreme phenotypic variability in this microdeletion syndrome and also could raise doubts on whether ACFS belongs to the group of microdeletion syndromes (Shukla et al., 2016).
Some authors speculated that interstitial deletion in the 6q21q22.1 region could be a risk factor for structural neurological anomalies, mainly involving the corpus callosum (CC) and the lateral and the third ventricles (Rosenfeld et al., 2012; Toschi et al., 2012; Shukla et al., 2016; Donahue and Rohena, 2017). Furthermore, Szafranski et al. (2015) suggested that the 6q22 region contains important contributors to the onset of childhood epilepsy (Szafranski et al., 2015).
Our patient’s rearrangement contains 90 genes, 13 of which are classified, to current knowledge, as OMIM (Online Mendelian Inheritance in Man) disease-associated (Figure 2): ARMC2 (MIM*618424) armadillo repeat-containing protein 2; CCN6 (MIM*603400) cellular communication network factor 6; CD164 (MIM*603356) CD164 antigen; CDC40 (MIM*605585) cell division cycle 40; CDK19 (MIM*614720) cyclin-dependent kinase 19; COL10A1 (MIM*120110) collagen, type X, alpha-1; DSE (MIM*605942) dermatan sulfate epimerase; FIG4 (MIM*609390) FIG4 phosphoinositide 5-phosphatase; LAMA4 (MIM*600133) laminin, alpha-4; TRAF3IP2 (MIM*607043) TRAF3-interacting protein 2; TSPYL1 (MIM*604714) TSPY-like 1; WASF1 (MIM*605035) WASP protein family, member 1; ZBTB24 (MIM*614064) zinc-finger and BTB domain-containing protein 24.
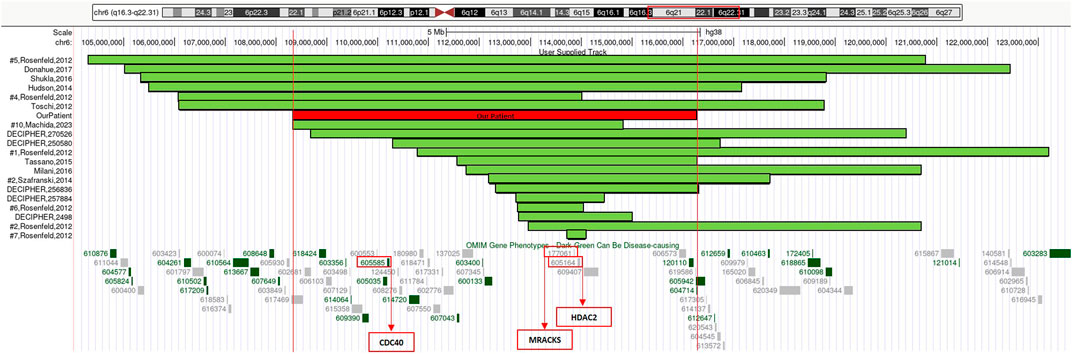
FIGURE 2. 6q21q22.1 deletions aligned according to the proximal breakpoint, reference articles are reported on the left, our patient is outlined in red. At the bottom of the figure is an overview of the region 6q21q22.1 and its OMIM gene phenotypes content according to the UCSC Genome Browser [GRCh38/hg38 assembly]. In red boxes we highlighted three genes considered to be relevant in neurological developmental disorders and in cerebellar development.
We considered the shared 6q21q22.1 chromosomal region to compare our patient’s phenotype with the other cases characterized by CMA in scientific literature and within genomic databases (e.g., DECIPHER) (Table 1) (Figure 2).
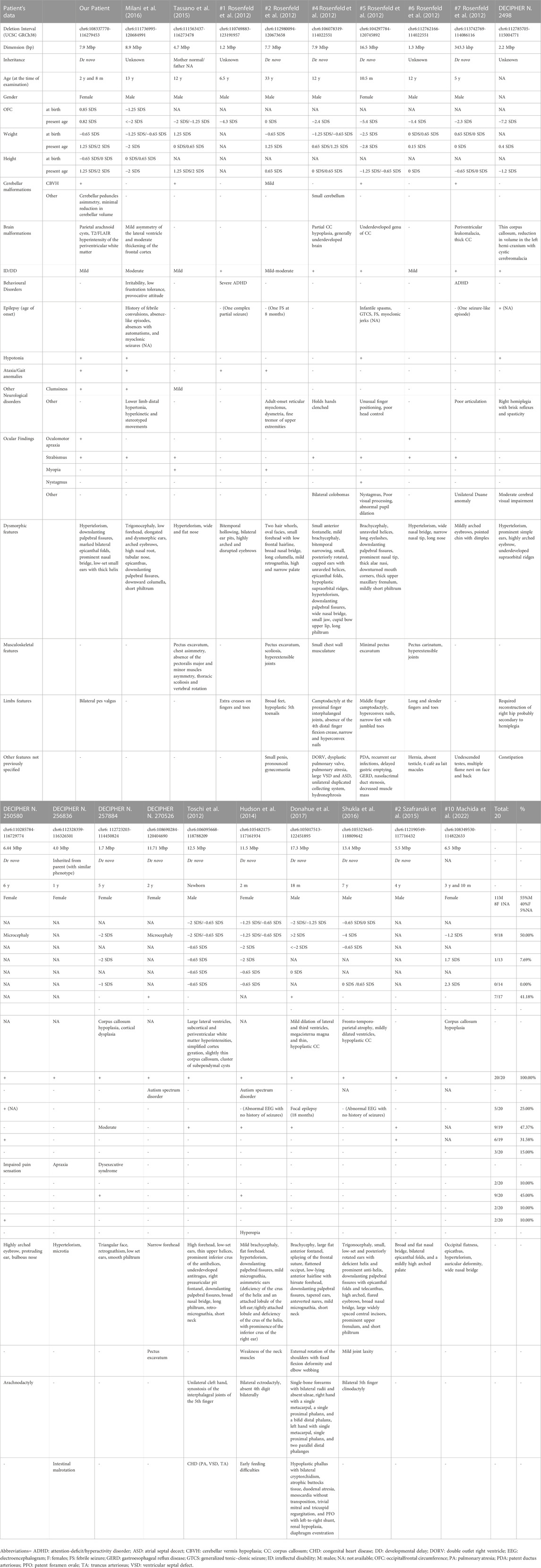
TABLE 1. Comparison of the reported case with individuals with overlapping chromosomal deletions involving the 6q21q22.1 chromosomal region, characterized through postnatal CMA.
The only adult patient described to date, patient #2 reported by Rosenfeld et al. (2012), has presented neurological manifestations over time, such as adult-onset reticular myoclonus and fine tremor of upper extremities (Rosenfeld et al., 2012). However, current literature is limited and further evidence is needed to better clarify any possible association with progressive neurological disease.
Epilepsy is often described in association with 6q interstitial deletions (Cutillo et al., 2023). Nonetheless, our patient never presented seizures, nor a pathological EEG was recorded. Reviewing the scientific literature, Szafranski et al. (2015) narrowed down a possible critical region for epilepsy to a 250 kb segment and suggested NUS1, EST AI858607 and SLC35F1 as candidate genes (Szafranski et al., 2015). This region is not involved in our patient’s rearrangement: this may partially justify the absence of epilepsy in our proposita.
Brain MRI documented the absence of major prosencephalic anomalies in our proposita, and the presence of CBVH with reduced cerebellar volume. These features have been previously reported in other unrelated patients with interstitial 6q deletions who underwent radiological examination (Rosenfeld et al., 2012; Tassano et al., 2015; Donahue and Rohena, 2017). Movement disorders manifested by balance deficit and a wide-based gait in our proposita are well reported in 6q deletion cases with or without CBVH. Inasmuch as to date the development of neuromuscular abnormalities underlying the coordination deficits has not been associated with the pleiotropic effect of a single causative gene: the presence of an oligogenic effect arising within the commonly deleted region has been hypothesized. A defective expression of GOPC (Golgi associated PDZ and coiled-coil motif containing; MIM*606845) (cytogenetic location: 6q22.1), which is involved in autophagy and transduction pathways in cerebellar Purkinje cells (Yue et al., 2002), was speculated to play a role in the development of ataxia and abnormal movements, even though it was not included in the deleted region of several patients presenting with CBVH (Rosenfeld et al., 2012). Interestingly, oculomotor apraxia was documented in our patient, and has been rarely reported in previous 6q interstitial deletion case (Rosenfeld et al., 2012).
Hayashi et al. (2017) studied a cohort of 41 patients through Next-Generation Sequencing and CMA techniques to identify candidate genomic aberrations to pontine and cerebellar hypoplasia.
Haploinsufficiency of both HDAC2 (histone deacetylase 2) (MIM*605164) and MARCKS (myristoylated alanine-rich C kinase substrate) (MIM*177061) genes, which are contained in 6q21q22.1 chromosomal deletion, was speculated to be relevant in neurological developmental disorders (Hayashi et al., 2017).
HDAC2 gene is zinc finger transcription factor (Inouye and Seto, 1994) and plays a role in adult neurogenesis. It is required for full differentiation and survival of adult generated neurons, and it is dispensable during development (Jawerka et al., 2010). In addition, HDAC2 expression could be involved in cell proliferation and differentiation in cell type- and developmental stage-specific patterns of expression in the developing cerebellum (Yoo et al., 2013).
MARCKS gene is expressed in the brain and spinal cord during embryological development. Stumpo et al. (1995) demonstrated a vital role for MARCKS in the normal processes of neurulation, hemisphere fusion, forebrain commissure formation, and formation of cortical and retinal laminations through gene knock-out studies (Stumpo et al., 1995).
Moreover, biallelic partial loss-of-function mutations of CDC40 gene, which is included in 6q21q22.1 region and encodes a core spliceosomal component, were proven to interfere with RNA splicing and neuronal survival. This was recently associated with pontocerebellar hypoplasia and partial agenesis of the CC with microcephaly in humans and mice. In addition, knock-out of the CDC40 gene is lethal in utero in animal models (Chai et al., 2021).
Haploinsufficiency of multiple genes can be speculated to be involved in brain development and, as such, in the neurological anomalies documented in patients with 6q21q22.1 deletion. Nonetheless, further research and the development of tools and databases focusing on this specific rearrangement are needed to advance our understanding of this genetic mechanism.
Conclusion
Interstitial deletions of the long arm of chromosome 6 are associated with an extremely variable phenotype. Disease expression also depends on, but is not limited to, the size and the location of the rearrangement. The review of current literature along with this new report can provide further insights on 6q21q22.1 chromosomal deletions and can help to restrict the Smallest Region of Overlap (SRO) associated with peculiar phenotypes.
Even though there is little evidence yet, we speculate that interstitial 6q deletion might be a risk factor for CBVH and/or cerebellar anomalies. These features should be considered by the clinitian who suspects the involvement of 6q21q22.1 rearrangements as the cause of their patient’s phenotype. We recommend neurological follow-up for patients with 6q interstitial deletions, as neurological symptoms may become evident over time. Further investigation on this aspect is needed in a larger group of genetically confirmed 6q interstitial deletion patients, as cerebellar hypoplasia can run asymptomatically.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies involving humans because the submitted report is derived from a hospital case of a patient with developmental delay, which was sent to our institution by the attending physician. Therefore, Ethical Committee approval was unnecessary, since no supplementary analysis was performed on the patient, except for the diagnostic genetic test for developmental delay. We obtained written consent from the patient beforehand, as required by our regulations. The internal Ethical Committee grants approval for entire research projects and not for reports based on single cases. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by-product of routine care or industry. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
CM: Conceptualization, Data curation, Investigation, Writing–original draft. LG: Conceptualization, Data curation, Investigation, Writing–original draft. ES: Formal Analysis, Methodology, Software, Writing–original draft. MCD: Conceptualization, Methodology, Supervision, Writing–review and editing. RF: Formal Analysis, Methodology, Resources, Writing–review and editing. VA: Investigation, Software, Validation, Writing–review and editing. GN: Formal Analysis, Supervision, Validation, Writing–review and editing. MLD: Conceptualization, Data curation, Formal Analysis, Writing–review and editing. SL: Writing–review and editing, Conceptualization, Data curation, Supervision. AN: Conceptualization, Data curation, Supervision, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by ricerca 5x1000_2023 (to MCD) and by European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability ERN-ITHACA (EU Framework Partnership Agreement ID: 3HP-HP-FPA ERN-01-2016/739516). This work was supported also by the Italian Ministry of Health with “Current Research funds”.
Acknowledgments
This analysis makes use of data collected by the DECIPHER community. A complete list of scientific centers who contributed to the generation of the data is available from: Y29udGFjdEBkZWNpcGhlcmdlbm9taWNzLm9yZw==.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Chai, G., Webb, A., Li, C., Antaki, D., Lee, S., Breuss, M. W., et al. (2021). Mutations in spliceosomal genes PPIL1 and PRP17 cause neurodegenerative pontocerebellar hypoplasia with microcephaly. Neuron 109 (2), 241–256.e9. doi:10.1016/j.neuron.2020.10.035
Correa-Cerro, L., Garcíaz-Cruz, D., Díaz-Castaños, L., Figuera, L. E., and Sanchez-Corona, J. (1996). Interstitial deletion 6q16.2q22.2 in a child with ectrodactyly. Ann. Genet. 39 (2), 105–109.
Cutillo, G., Bonacchi, R., Cecchetti, G., Bellini, A., Vabanesi, M., Zambon, A., et al. (2023). Interstitial 6q deletion in a patient presenting with drug-resistant epilepsy and Prader-Willi like phenotype: an electroclinical description with literature review. Seizure Eur. J. Epilepsy 109, 45–49. doi:10.1016/j.seizure.2023.05.011
Donahue, M. L., and Rohena, L. O. (2017). Rare presentation of 6q16.3 microdeletion syndrome with severe upper limb reduction defects and duodenal atresia. Clin. Case Rep. 5 (6), 905–914. doi:10.1002/ccr3.916
Duran-Gonzalez, J., Gutierrez-Angulo, M., Garcia-Cruz, D., Ayala, M. de la L., Padilla, M., and Davalos, I. P. (2007). A de novo interstitial 6q deletion in a boy with a split hand malformation. J. Appl. Genet. 48 (4), 405–407. doi:10.1007/BF03195240
Evers, L. J., Schrander-Stumpel, C. T., Engelen, J. J., Hoorntje, T. M., Pulles-Heintzberger, C. F., et al. (1996). Deletion of the long arm of chromosome 6: two new patients and literature review. Clin. Genet. 50 (3), 138–144. doi:10.1111/j.1399-0004.1996.tb02368.x
Griffiths, R. (1970). The abilities of babies: a study in mental measurement. London: University of London Press.
Gu, W., Zhang, F., and Lupski, J. R. (2008). Mechanisms for human genomic rearrangements. Pathogenetics 1 (1), 4. doi:10.1186/1755-8417-1-4
Hayashi, S., Uehara, D. T., Tanimoto, K., Mizuno, S., Chinen, Y., Fukumura, S., et al. (2017). Comprehensive investigation of CASK mutations and other genetic etiologies in 41 patients with intellectual disability and microcephaly with pontine and cerebellar hypoplasia (MICPCH). PloS One 12 (8), e0181791. doi:10.1371/journal.pone.0181791
Hopkin, R. J., Schorry, E., Bofinger, M., Milatovich, A., Stern, H. J., Jayne, C., et al. (1997). New insights into the phenotypes of 6q deletions. Am. J. Med. Genet. 70 (4), 377–386. doi:10.1002/(SICI)1096-8628(19970627)70:4<377::AID-AJMG9>3.0.CO;2-Q
Hudson, C., Schwanke, C., Johnson, J. P., Elias, A. F., Phillips, S., Schwalbe, T., et al. (2014). Confirmation of 6q21-6q22.1 deletion in acro-cardio-facial syndrome and further delineation of this contiguous gene deletion syndrome. Am. J. Med. Genet. A 164A (8), 2109–2113. doi:10.1002/ajmg.a.36548
Inouye, C. J., and Seto, E. (1994). Relief of YY1-induced transcriptional repression by protein-protein interaction with the nucleolar phosphoprotein B23. J. Biol. Chem. 269 (9), 6506–6510. doi:10.1016/s0021-9258(17)37400-8
Jawerka, M., Colak, D., Dimou, L., Spiller, C., Lagger, S., Montgomery, R. L., et al. (2010). The specific role of histone deacetylase 2 in adult neurogenesis. Neuron Glia Biol. 6 (2), 93–107. doi:10.1017/S1740925X10000049
Machida, O., Shimojima, K. Y., Shiihara, T., Akamine, S., Kira, R., Hasegawa, Y., et al. (2022). Interstitial deletions in the proximal regions of 6q: 12 original cases and a literature review. Intractable Rare Dis. Res. 11 (3), 143–148. doi:10.5582/irdr.2022.01065
Milani, D., Cagnoli, G. A., Baccarin, M., Alfei, E., Guerneri, S., and Esposito, S. (2016). Insights into 6q21-q22: refinement of the critical region for acro-cardio-facial syndrome. Congenit. Anom. 56 (4), 187–189. doi:10.1111/cga.12164
Pandya, A., Braverman, N., Pyeritz, R. E., Ying, K. L., Kline, A. D., and Falk, R. E. (1995). Interstitial deletion of the long arm of chromosome 6 associated with unusual limb anomalies: report of two new patients and review of the literature. Am. J. Med. Genet. 59 (1), 38–43. doi:10.1002/ajmg.1320590109
Rosenfeld, J. A., Amrom, D., Andermann, E., Andermann, F., Veilleux, M., Curry, C., et al. (2012). Genotype-phenotype correlation in interstitial 6q deletions: a report of 12 new cases. Neurogenetics 13 (1), 31–47. doi:10.1007/s10048-011-0306-5
Schwartz, M. F., Kaffe, S., Wallace, S., and Desnick, R. J. (1984). Interstitial deletion of the long arm of chromosome 6 [del(6) (q16q22)]: case report and review of the literature. Clin. Genet. 26 (6), 574–578. doi:10.1111/j.1399-0004.1984.tb01106.x
Shukla, A., Hebbar, M., Harms, F. L., Kadavigere, R., Girisha, K. M., and Kutsche, K. (2016). Phenotypic variability in patients with interstitial 6q21-q22 microdeletion and Acro-Cardio-Facial syndrome. Am. J. Med. Genet. A 170 (11), 2998–3003. doi:10.1002/ajmg.a.37759
Stankiewicz, P., and Lupski, J. R. (2002). Genome architecture, rearrangements and genomic disorders. Trends Genet. 18 (2), 74–82. doi:10.1016/s0168-9525(02)02592-1
Stumpo, D. J., Bock, C. B., Tuttle, J. S., and Blackshear, P. J. (1995). MARCKS deficiency in mice leads to abnormal brain development and perinatal death. Proc. Natl. Acad. Sci. U. S. A. 92 (4), 944–948. doi:10.1073/pnas.92.4.944
Szafranski, P., Von Allmen, G. K., Graham, B. H., Wilfong, A. A., Kang, S. H., Ferreira, J. A., et al. (2015). 6q22.1 microdeletion and susceptibility to pediatric epilepsy. Eur. J. Hum. Genet. 23 (2), 173–179. doi:10.1038/ejhg.2014.75 Epub 2014 May 14.
Tassano, E., Mirabelli-Badenier, M., Veneselli, E., Puliti, A., Lerone, M., Vaccari, C. M., et al. (2015). Clinical and molecular characterization of a patient with interstitial 6q21q22.1 deletion. Mol. Cytogenet 8 (31), 31. doi:10.1186/s13039-015-0134-7
Toschi, B., Valetto, A., Bertini, V., Congregati, C., Cantinotti, M., Assanta, N., et al. (2012). Acro-cardio-facial syndrome: a microdeletion syndrome? Am. J. Med. Genet. A 158A (8), 1994–1999. doi:10.1002/ajmg.a.35444
Tsukahara, M., Yoneda, J., Azuma, R., Nakashima, K., Kito, N., Ouchi, K., et al. (1997). Interstitial deletion of 6q21-q23 associated with split hand. Am. J. Med. Genet. 69 (3), 268–270. doi:10.1002/(sici)1096-8628(19970331)69:3<268::aid-ajmg10>3.0.co;2-p
Yoo, J. Y. J., Larouche, M., and Goldowitz, D. (2013). The expression of HDAC1 and HDAC2 during cerebellar cortical development. Cerebellum Lond Engl. 12 (4), 534–546. doi:10.1007/s12311-013-0459-x
Young, R. S., Fidone, G. S., Reider-Garcia, P. A., Hansen, K. L., McCombs, J. L., and Moore, C. M. (1985). Deletions of the long arm of chromosome 6: two new cases and review of the literature. Am. J. Med. Genet. 20 (1), 21–29. doi:10.1002/ajmg.1320200105
Yue, Z., Horton, A., Bravin, M., DeJager, P. L., Selimi, F., and Heintz, N. (2002). A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron 35 (5), 921–933. doi:10.1016/s0896-6273(02)00861-9
Zhang, F., Khajavi, M., Connolly, A. M., Towne, C. F., Batish, S. D., and Lupski, J. R. (2009). The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 41 (7), 849–853. doi:10.1038/ng.399
Keywords: 6q21q22.1, interstitial deletion, cerebellar vermis hypoplasia, chromosomal microarray analysis, developmental delay
Citation: Minotti C, Graziani L, Sallicandro E, Digilio MC, Falasca R, Alesi V, Novelli G, Dentici ML, Loddo S and Novelli A (2024) Case report: A new de novo 6q21q22.1 interstitial deletion case in a girl with cerebellar vermis hypoplasia and developmental delay and literature review. Front. Genet. 14:1315291. doi: 10.3389/fgene.2023.1315291
Received: 10 October 2023; Accepted: 04 December 2023;
Published: 06 February 2024.
Edited by:
Paulo Ricardo Gazzola Zen, Federal University of Health Sciences of Porto Alegre, BrazilReviewed by:
Silvia Russo, Italian Auxological Institute (IRCCS), ItalyFrancesca Luisa Sciacca, IRCCS Carlo Besta Neurological Institute Foundation, Italy
Louise Pinto, Hospital Infantil Joana de Gusmão, Brazil
Mona Kamal Mekkawy, National Research Centre, Egypt
Copyright © 2024 Minotti, Graziani, Sallicandro, Digilio, Falasca, Alesi, Novelli, Dentici, Loddo and Novelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Minotti, Y2hpYXJhLm1pbm90dGkwQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work