 Alecia Seliga1Michael Hweemoon Lee2Nicole C. Fernandes1Viviana Zuluaga-Ramirez1Marta Didukh1
Alecia Seliga1Michael Hweemoon Lee2Nicole C. Fernandes1Viviana Zuluaga-Ramirez1Marta Didukh1 Yuri Persidsky1
Yuri Persidsky1 Raghava Potula1
Raghava Potula1 Stefania Gallucci2
Stefania Gallucci2 Uma Sriram1*
Uma Sriram1*
- 1Department of Pathology and Laboratory Medicine, Temple University, Philadelphia, PA, United States
- 2Laboratory of Dendritic Cell Biology, Department of Microbiology and Immunology, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
The Kallikrein–Kinin System (KKS), comprised of kallikreins (klks), bradykinins (BKs) angiotensin-converting enzyme (ACE), and many other molecules, regulates a number of physiological processes, including inflammation, coagulation, angiogenesis, and control of blood pressure. In this report, we show that KKS regulates Type I IFN responses, thought to be important in lupus pathogenesis. We used CpG (TLR9 ligand), R848 (TLR7 ligand), or recombinant IFN-α to induce interferon-stimulated genes (ISGs) and proteins, and observed that this response was markedly diminished by BKs, klk1 (tissue kallikrein), or captopril (an ACE inhibitor). BKs significantly decreased the ISGs induced by TLRs in vitro and in vivo (in normal and lupus-prone mice), and in human PBMCs, especially the induction of Irf7 gene (p < 0.05), the master regulator of Type I IFNs. ISGs induced by IFN-α were also suppressed by the KKS. MHC Class I upregulation, a classic response to Type I IFNs, was reduced by BKs in murine dendritic cells (DCs). BKs decreased phosphorylation of STAT2 molecules that mediate IFN signaling. Among the secreted pro-inflammatory cytokines/chemokines analyzed (IL-6, IL12p70, and CXCL10), the strongest suppressive effect was on CXCL10, a highly Type I IFN-dependent cytokine, upon CpG stimulation, both in normal and lupus-prone DCs. klks that break down into BKs, also suppressed CpG-induced ISGs in murine DCs. Captopril, a drug that inhibits ACE and increases BK, suppressed ISGs, both in mouse DCs and human PBMCs. The effects of BK were reversed with indomethacin (compound that inhibits production of PGE2), suggesting that BK suppression of IFN responses may be mediated via prostaglandins. These results highlight a novel regulatory mechanism in which members of the KKS control the Type I IFN response and suggest a role for modulators of IFNs in the pathogenesis of lupus and interferonopathies.
Introduction
The kallikrein–kinin system (KKS) is a metabolic cascade that releases vasoactive kinins (bradykinin-related peptides) upon triggering and has many pharmacological activities; KKS is involved in inflammation and cooperates with important pathways like the renin–angiotensin, coagulation, and complement (1–3). Bradykinin (BK) is the end product of the KKS that is formed from plasma or tissue kallikreins (klks) (4) and acts via their receptors (B1 or B2) (5) to induce responses via nitric oxide and/or prostaglandin production (3). The angiotensin-converting enzyme (ACE), which is part of the renin-angiotensin system (6), not only releases angiotensin II but also inactivates BK (6–8). Inhibitors of ACE have been used successfully in a variety of clinical conditions related to heart or kidney functions (9). The beneficial effects of administration of ACE inhibitors (ACEis) have been attributed to enhancing the actions of BK via its receptor (1, 7).
There is increasing evidence that the KKS is involved in systemic lupus erythematosus (SLE) (10–12). Giving exogenous klks has been shown to ameliorate lupus in mice, and lupus-prone mice have been found to produce less klks than wild-type mice (12). SLE is a systemic autoimmune disease of complex etiology in which immune complex deposition and complement activation lead to inflammation and tissue damage (13). Type I IFN signature is the hallmark of the disease, and efforts have been underway to attenuate the IFN responses in lupus (14–18). Current therapeutic candidates under serious testing include agents that block Type I IFN directly (16) or IFN generated via TLR ligands as TLR7/9 (19, 20). No link has so far been made between Type I IFNs and the KKS.
Molecules of the KKS have been shown to ameliorate end-stage kidney disease in mouse models (klk and BK) (11, 21) and in lupus patients (ACEis) (10) So far, the use of BKs and ACEis have been focused mainly on the renal- and cardio-protective effects, especially treatment upon onset of glomerulonephritis (GN) (22). In this study, we asked the question if exogenous KKS decreases the IFN responses induced by either TLR (TLR7 and TLR9) or direct IFN-α stimulation in normal and lupus-prone mice, as well as in human PBMCs. We show that BKs and other molecules of the KKS system (klks and ACEi) can suppress an ongoing Type I IFN response by decreasing the interferon-stimulated gene (ISG) expression, especially IRF7 the master regulator of Type I IFNs, both in mouse and human cells. Our results strongly suggest a novel pathway of Type I IFN regulation by the KKS.
Materials and Methods
Mice
Female C57BL/6 (B6) mice and B6.NZM Sle1/Sle2/Sle3 (Sle) mice were bought from The Jackson Laboratory and used between 7 and 12 weeks of age. We have shown before that Sle mice are pre-disease at this age and do not make any detectable autoantibodies (23). We used only female mice because of the higher prevalence of Sle in women than men (9:1 ratio, women to men) (24). Animals were maintained in the animal facility in accordance to the guidelines of the Institutional Animal Care and Use Committees of Temple University, which is an American Association for the Accreditation of Laboratory Animal Care-accredited facility.
Reagents
The following reagents were used to stimulate cells: Recombinant human IFN-α (1,000 U/ml, PBL Assay Science, Piscataway, NJ, USA), mouse IFN-α (1,000 U/ml, PBL Assay Science), CpG-B 1826 (10 µg/ml), CpG-A-2336 (5 µg/ml) (IDT Biotechnologies, Coralville, IA, USA), CpG-A-1585 (1 µg/ml) (Invivogen, San Diego, CA, USA), resiquimod (1 µg/ml) (R848; Invivogen) (23, 25–27), bradykinin peptide (10 µM), Lys-des-Arg(9)-bradykinin, which is a kinin breakdown product and a selective bradykinin B1 receptor agonist (10 µM), Arg–Pro–Hyp–Gly–Phe–Ser–Pro–Phe–Arg B2 receptor agonist (10 µM), B1 receptor antagonist ([des-Arg10-HOE140]- DH-1 μg/ml), B2 receptor antagonist (HOE140–H–10 μM) (all bradykinin agonists and antagonists were purchased from Sigma-Aldrich, St. Louis, MO, USA) (28), recombinant human klk1 (1 µg/ml) (Creative Biomart, Shirley, NY, USA), captopril (20 μM)(Sigma) (29), and indomethacin (indo) (1 µg/ml) (Sigma) (30).
Murine Bone Marrow Dendritic Cell (DC) Culture
We used bone-marrow-derived dendritic cells (BMDCs) as our murine cell model. BMDCs were generated as previously described (27). Briefly, bone marrow precursors were flushed from femurs and tibias of mice and the cells were seeded at 1 million cells/well in complete IMDM (Mediatech, Manassas, VA, USA) (10% FBS, penicillin/streptomycin, gentamicin, and 2-ME) (Life Technologies, Grand Island, NY, USA) enriched with 3.3 ng/ml GMCSF (BD Biosciences, San Jose, CA, USA) in 24-well plates. One milliliter of medium was added on day 2, and half was replaced on day 5 and subsequently each day until the culture was used (day 6/7). Some experiments were also performed using bone marrow precursors that were differentiated into DCs in medium containing Flt3L (23, 27). Briefly, bone marrow cavities were flushed, and cells were plated at 1 million cells/well in a 24-well plate in complete RPMI medium containing 10% FBS, l-glutamine, penicillin/streptomycin, 2-mercaptoethanol, and 10% supernatant from Flt3L cell line as described in Ref. (27). Cells were stimulated and harvested on day 8 or 9. Resting DC cultures were stimulated and BMDCs were harvested after 7 h of stimulation for RNA analysis and after 24 h for FACS analysis of surface activation markers. Culture supernatants were collected at the same time-points, for cytokine analysis by ELISA.
Isolation of Human PBMCs
PBMCs were isolated from leukotrap filters (31) from normal human donors (from Red Cross, Philadelphia, PA, USA). The samples received from American Red Cross, used in this study, were de-identified samples and no information about the donors is available to the investigators. Briefly, leukotrap filters were flushed with PBS containing 5 mM EDTA and spun at 400 g at room temperature (RT) without brakes for 10 min. The clear supernatant was removed, and the blood was layered onto Ficoll–Hypaque (GE Healthcare Lifesciences, Pittsburg, PA, USA) and spun at RT for 40 min at 400 g without brakes. The buffy interface was isolated, washed in medium, counted and plated at 2.5 million cells/ml in 24-well plates in complete RPMI medium containing 10% FBS, penicillin/streptomycin and 10 mM HEPES.
In Vivo Injections
B6 or Sle mice were injected intraperitoneally with CpG-A-1585 (10 μg/mouse) overnight to induce IFN responses. BK (100 µM) was injected via retro-orbital route and after 2 h mice were euthanized, and spleens were harvested for gene expression analysis.
Magnetic Bead Sorting of Splenic DCs and Ex Vivo Stimulation
Dendritic cells from B6 mice were sorted using MACS beads (Miltenyi Biotech, Auburn, CA, USA), stimulated and harvested for gene expression analysis. Briefly, spleens were incubated in medium containing 8 mg/ml collagenase (Worthington Biochemical Corporation, Lakewood, NJ, USA) and 1,000 U/ml DNase (Sigma) for 45 min, and single-cell suspensions were prepared by passing through a 100-µm mesh filter (BD Biosciences). Cells were blocked with Fc blocker (anti-CD16/32—clone 2.4G2 clone) (BD Biosciences) for 15 min and incubated with CD11c beads and isolated as per manufacturer’ protocol. Cells were plated at 1 million per well in a 24-well plate, stimulated with CpG (2 h) and BKs (2 h) and harvested for RNA and analyzed by real-time RT-PCR for ISG expression.
Flow Cytometry
Bone-marrow-derived dendritic cells were harvested from the wells, washed in cold PBS, incubated with Fc blocker (2.4G2 monoclonal antibody, BD Biosciences) for 10 min, and then stained for 30 min on ice with allophycocyanin-conjugated hamster anti-mouse CD11c, FITC conjugated mouse anti-mouse H2Kb (MHC Class I) (all antibodies purchased from BD Biosciences). Cells were fixed in 1% formaldehyde (eBioscience, San Diego, CA, USA) and analyzed on a FACS Canto II cytometer (BD Biosciences). FlowJo software (FlowJO LLC, Ashland, OR, USA) was used for data analysis.
Gene Expression Analysis
Gene expression in BMDCs was analyzed by real-time RT-PCR using TaqMan probes. Briefly, RNA was extracted using Zymo quick kit (Zymo Research, Irvine, CA, USA) following the manufacturer’s protocols. Complementary DNA was synthesized using the cDNA archive kit (Life Technologies, Carlsbad, CA, USA). TaqMan primers and probes for both mouse and human genes (Irf7—Mm00516788_m1, IRF7—Hs01014809_g1; Isg15—Mm01705338_s1, ISG15-Hs01921425_s1, Ifnβ—Mm00439546_s1, and Cxcl10—Mm00445235, CXCL10-Hs00171042_m1) were purchased from Applied Biosystems (Foster City, CA, USA). Cyclophilin (Mouse—Mm02342430_g1) was used as the reference gene for normalization for mouse cells and GAPDH (Human—4310884E) was used for human cells. The primer sequences are available from the Applied Biosystems website. The cycle threshold (Ct) method of relative quantification of gene expression was used (ddCt), and the normalized Ct values (against cyclophilin or GAPDH for mouse and human cells, respectively) were calibrated against the control (untreated cells) in each experiment. Percent fold change was calculated by normalizing the values to the CpG, R848, or IFN-α treatments alone in each experiment. Mean and SEM were obtained from at least three independent experiments from independent cultures from different mice or different human donors, and are presented in the graphs.
Western Blot Analysis
Western blotting was performed as described previously (25) using 25–50 µg of total proteins from DC cell lysates. In brief, protein samples were denatured by boiling for 10 min and loaded onto 4–20% Bis–Tris gels (Invitrogen, Carlsbad, CA, USA). After electrophoresis, proteins were transferred to nitrocellulose membranes. Membranes were blocked for 1 h with blocking buffer (2% non-fat milk in PBS), then incubated overnight at 4°C with the primary antibodies diluted in blocking buffer with 0.1% Tween 20. Rabbit polyclonal anti-STAT1 and STAT2, phospho-STAT1 (Tyr701) and phospho-STAT2 (Tyr689) (Millipore) were used as primary antibodies. Anti-actin Ab (Santa-Cruz Biotech) was used as a loading control. After incubating with primary antibodies, the membranes were washed with PBS containing 0.1% Tween 20 (PBST) three times. Membranes were then incubated for 1 h with IR Dye 800 goat anti-rabbit and IR Dye 680 goat anti-mouse (LI-COR Biosciences, Lincoln, NE, USA) diluted in blocking buffer plus 0.1% Tween 20. The blots were then washed three times with PBST and rinsed with PBS. Proteins were visualized by scanning the membrane on an Odyssey Infrared Imaging System (LI-COR Biosciences) in both 700 and 800 nm channels. ImageJ software (Image processing program developed at the National Institutes of Health) was used to calculate the band intensities.
ELISA
We used ELISA kits (sandwich elisa) to measure the levels of mouse cytokines IL-6, IL12p70 (BD Biosciences, San Jose, CA, USA), and CXCL10 (R&D Systems, Minneapolis, MN, USA) in the supernatants of BMDC cultures stimulated for 24 h with TLR ligands, or medium alone, as per the manufacturer’s protocol.
Statistical Analysis
Mean and SEM were calculated from data from three to seven independent experiments performed with independent bone marrow cultures or spleens obtained from individual mouse in each experiment or from at least three to five human donors performed independently. Prism software (GraphPad, San Diego, CA, USA) was used for statistical analysis: Two-tailed paired t-test for comparison between two groups and one-way ANOVA with Tukey’s post hoc test for multiple groups were used for statistic, as appropriate. p-values less than 0.05 were considered significant (marked in the figures as *p < 0.05, **p < 0.01, ***p < 0.001).
Results
BKs Suppress ISGs in Normal and Lupus-Prone Murine DCs In Vitro and in Human PBMCs
Several studies have shown increased production of IFN-α in lupus, both in spontaneous lupus-prone mouse models (23, 32) and in patients (16, 33). We, and others, have also shown that stimulation via TLR7 and TLR9 is important in the induction of Type I IFNs in lupus (14, 19, 23). We, therefore, used ligands against TLR7 (R848) and TLR9 (CpG-A and -B) and also directly stimulated cells with recombinant IFN-α to test if the ISGs’ expression was modulated by BKs in normal and lupus-prone mouse DCs. We tested both a short-time (3 h) as well as long-time (20 h) stimulation with the TLRs and then added BKs without washing out the initial stimuli and analyzed the responses after 4 h. Using bone-marrow-derived DCs grown in GMCSF (GM-cDCs), a murine model of pro-inflammatory DCs, we tested three types of BKs, B1 and B2 receptor agonists and bradykinin peptide and we combined the results and presented in Figure 1. BKs significantly decreased the induction of Irf7 upon short-time stimulation (total of 7 h) with CpG, R848, or IFN-α in B6 GM-cDCs (Figure 1A). The fold changes (mean ± SE) of Irf7 from untreated control (that was normalized to 1) in GM-cDCs stimulated with TLR ligands without BK vs. with BK treatment were: CpG-A—14.9 ± 3.2 vs. 8.7 ± 5.2; CpG-B—53.4 ± 18.9 vs. 24.4 ± 8.7; R848—13.3 ± 2.4 vs. 6.0 ± 1.6; IFN-α—82.6 ± 48.8 vs. 37 ± 21.6. The ubiquitin-like Isg15 gene is one of the predominant genes induced by Type I IFNs (IFNα/β) (34) that we have previously shown to be induced by CpG and R848 in B6 BMDCs (26). We found that BK significantly suppressed the gene expression of Isg15 induced by CpG-B and R848 in GM-cDCs (Figure 1B). The fold changes (mean ± SE) of Isg15 from untreated control (that was normalized to 1) in GM-cDCs stimulated with TLR ligands without BK vs. with BK treatment were: CpG-A—36.8 ± 6 vs. 26.1 ± 2.9; CpG-B—53.1 ± 23.4 vs. 30.5 ± 10.1; R848—26.8 ± 3.3 vs. 13.0 ± 4.9; IFN-α—179.9 ± 124.8 vs. 93.4 ± 50.1. To determine further whether BK can modulate a sustained IFN response, we stimulated B6 GM-cDCs with CpG-A for 20 h and then treated with BK for 4 h and analyzed Irf7 (mean ± SE—no BK vs. BK—173.8 ± 47.3 vs. 89.5 ± 26.8) and ISG15 (no BK vs. BK—108.3 ± 28.8 vs. 55.7 ± 18.4) gene expression. BK significantly suppressed the ongoing production of both Irf7 and Isg15 genes in B6 GM-cDCs (Figure 1C). These results suggest that BK can also strongly suppress the autocrine positive feedback loop that Type I IFNs generate on ISG production upon a sustained response (35). We observed that Isg15 suppression reached statistical significance after 20 h stimulation with CpG-A (Figure 1C) that was not observed at the shorter time (3 h) (Figure 1B).
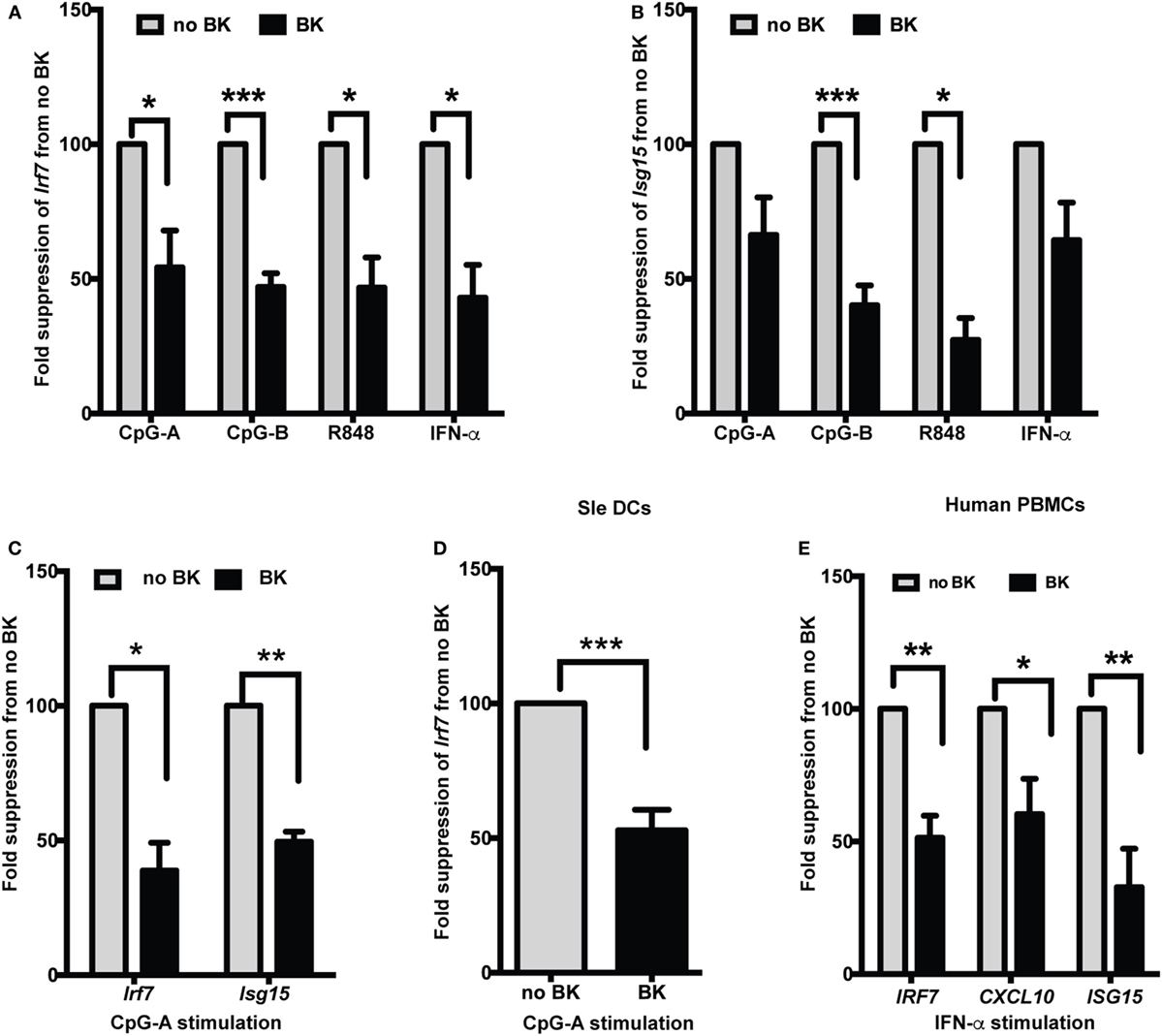
Figure 1. Bradykinins (BKs) suppress interferon-stimulated genes (ISGs) in mouse and human cells. (A–C) Murine bone-marrow-derived dendritic cells (BMDCs) grown in GMCSF containing medium were treated or not with CpG-A, CpG-B, R848, or recombinant IFN-α for 3 h, treated with BK for 4 h and then analyzed expression of Irf7 (A) and Isg15 (B) by real-time qPCR. (C) GMCSF dendritic cells (DCs) were stimulated overnight with CpG-A and with BK for a further 4 h and Irf7 and Isg15 responses were analyzed by qPCR. (D) Sle DCs grown in Flt3L containing medium were stimulated with CpG-A followed by BK and Irf7 gene expression was analyzed. (E) Human PBMCs were stimulated with IFN-α for 3 h followed by BK for a further 3 h and then IRF7, CXCL10, and ISG15 gene expression was analyzed. Percent fold change in expression was calculated by normalizing with no BK treatment for each stimulation in each experiment. Bar graphs represent the average (mean + SEM) of least three independent experiments, performed with independent BMDC culture from each mouse or PBMCs from each donor; *p < 0.05, **p < 0.01, ***p < 0.001.
We tested the effects of BK in the DCs from Sle mice and found that a sustained Irf7 gene expression was strongly suppressed by BK (mean ± SE—no BK vs. BK—35.9 ± 9.7 vs. 17.3 ± 5.0) (Figure 1D). This suggests that BK can also suppress the IFN response ongoing in Sle DCs, which we have previously shown to be amplified compared to the IFN response of B6 DCs upon CpG stimulation (23).
We further wanted to determine if this phenomenon could be reproduced in human cells. We isolated PBMCs from normal human donors and stimulated them with recombinant IFN-α for 3 h and then we added BKs for another 3 h and analyzed the gene expression of IRF7, CXCL10, and ISG15. The upregulation of all three genes were strongly suppressed by BK (mean ± SE—no BK vs. BK—IRF7—21.5 ± 6.4 vs. 10.2 ± 1.9; CXCL10—748 ± 290.7 vs. 332.8 ± 78.2; ISG15—239.8 ± 48.1 vs. 54 ± 19.8) (Figure 1E). None of the BKs tested induced any IFN response by themselves in the mouse or human cells (data not shown). The concentrations of BKs used in these experiments also did not affect the DC percentage or cause any toxicity to the cells in culture (data not shown). By performing titrations, we found that BKs showed the strongest inhibition of the IFN responses at 10 µM.
BKs Inhibit Type I IFN-Induced MHC Class I Upregulation, and CXCL10 Production in Murine DCs In Vitro
We have shown before that MHC Class I was strongly upregulated by Type I IFNs (25) and by TLR ligands via autocrine production of Type I IFNs (27). We have also shown that chemokine CXCL10 is very strongly upregulated in Sle DCs upon CpG stimulation (23) and that CXCL10 is a highly IFN-dependent cytokine (27). In this study, we tested if BKs can suppress the induction of these molecules. Treatment with IFN-α induced an increase in the expression of MHC Class I [untreated vs. IFN-α-median fluorescence intensity (MFI)—1178 ± 84 vs. 1460 ± 21] at 24 h in B6 GM-cDCs, as analyzed by flow cytometry. BK inhibited this MHC Class I upregulation (Figure 2A), with a small but consistent decrease (differences in MFI reached p = 0.05). We also measured the production of CXCL10 in supernatants from mouse DC cultures generated in Flt3-L-conditioned medium [which are considered a model of sentinel DCs and a mixed population of cDCs and pDCs (36)], by ELISA after 24 h stimulation with CpG-A. BK strongly suppressed the production of CXCL10 upon CpG stimulation in both in B6 and Sle DCs (Figure 2B; mean ± SE—CXCL10 picograms/milliliter—no BK vs. BK—B6—4,383 ± 80 vs. 2,689 + 1; Sle—2,322 ± 1,354 vs. 1,331 ± 736). We also tested the BK effects on CXCL10 induced by R848, but interestingly BK did not suppress this response (data not shown). These results suggest that BK inhibit ISG responses both at RNA transcription and protein translation levels.
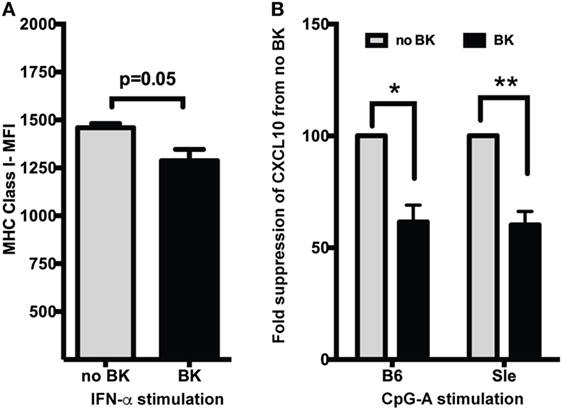
Figure 2. Bradykinins (BKs) suppress MHC Class I upregulation and CXCL10 production in murine dendritic cells (DCs). (A) Mouse bone-marrow-derived dendritic cells (BMDCs) were treated or not with recombinant IFN-α for 3 h and then stimulated with BK for 21 h. Cells were harvested and analyzed for MHC Class I expression by flow cytometry. Cells were also stained with CD11c and gated on CD11c DC populations. Data in the bar graphs represent averages (mean + SE) of median fluorescence intensity (MFI) of the markers analyzed from at least three independent cultures. (B) Supernantants from B6 and Sle BMDCs treated with CpG-A followed by BK for a total of 24 h were analyzed for CXCL10 production by ELISA. Percent change in CXCL10 production was calculated from no BK treatment for each experiment. Bar graphs are mean + SE of three independent experiments analyzed in duplicate in each experiment. *p < 0.05; **p < 0.01.
BKs Strongly Suppress Pro-inflammatory Cytokine Production in Murine DCs
We next analyzed the effects of BK on pro-inflammatory cytokines IL-6 and IL12p70, in the supernatants of B6 and Sle BMDCs cultured in Flt3L medium (Figure 3A). We observed that BK significantly inhibited the production of IL-6 in B6 (p < 0.01) and Sle DCs (p < 0.05) upon CpG-A stimulation (Figure 3A; Figure S1A in Supplementary Material). There was a significant reduction of IL-6 upon R848 stimulation in the B6 DCs but not in the Sle DCs (Figure 3A; Figure S1B in Supplementary Material). Interestingly, IL12p70 was not downregulated by BK upon CpG-A stimulation in the B6 DCs, but was strongly suppressed in the Sle DCs (Figure 3B; Figure S1C in Supplementary Material). However, IL12p70 induced by R848 was strongly suppressed by BK both in the B6 and Sle BMDCs (B6, p < 0.01; Sle, p < 0.05) (Figure 3B; Figure S1D in Supplementary Material). We also tested the effect of BK in the GM-cDCs and found that CpG-B induced IL-6 and IL12p70 were significantly reduced by BK (Figures 3C,D; Figure S2 in Supplementary Material), while the same cytokines were unaffected by BK upon R848 stimulation (Figures 3C,D; Figure S2 in Supplementary Material). Untreated B6 or Sle DCs did not make detectable cytokines. In total, the results indicate that BK can suppress the upregulation of ISGs in both GMCSF or Flt3L -DC cultures (Figures 1A–D), but the effects on the cytokines upregulated by specific stimuli (CpG vs. R848) were very different in the different cultures, suggesting that there may be some post-translational effects that vary in the mixed Flt3L cultures vs. the GMCSF cDC cultures. Moreover, we observed a more consistent suppressive effect of BK on the cytokines induced by CpG (A and B) but not with R848, suggesting different susceptibility to BK of the distinct signaling pathways downstream of TLR9 and TLR7 (Figures 3C,D and 2B).
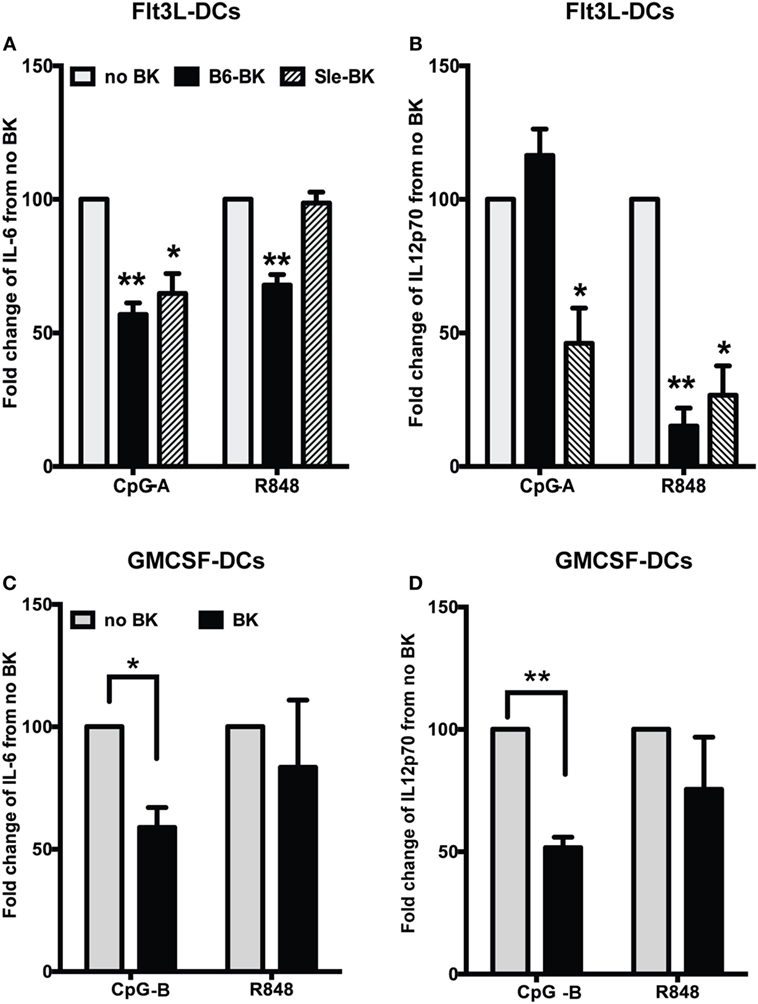
Figure 3. Bradykinins (BKs) suppress pro-inflammatory cytokine production in murine bone-marrow-derived dendritic cells (BMDCs). (A,B) Flt3L grown BMDCs from B6 or Sle mice were stimulated with CpG-A or R848 overnight and then stimulated with BK. Cell supernatants were collected at 24 h of total stimulation to analyze the cytokines, IL-6 (A) and IL12p70 (B) by ELISA. (C,D) GMCSF-grown BMDCs from B6 mice were stimulated with CpG-B or R848 overnight and then stimulated with BK and cell supernatants were collected at 24 h of total stimulation to analyze the cytokines, IL-6 (C) and IL12p70 (D) by ELISA. Percent change in cytokine production was calculated from no BK treatment for each experiment. Bar graphs are mean + SE of at three independent experiments (*p < 0.05; **p < 0.01).
Effects of BKs on the Phosphorylation of Signal Transducers and Activator of Transcription (STATs) upon IFN-α Stimulation in Murine DCs
Classical Type I IFN signaling takes place via IFNAR1 and IFNAR2 receptors that activate the Janus kinase–STAT pathway, leading to transcription of ISGs (37). To determine the effects of BK on the signaling pathway downstream of IFNARs, we chose to analyze the effects on the phosphorylation of STAT1 and STAT2 molecules. We found that IFN- α-induced phosphorylation of STAT2 was suppressed by BK, while there was moderate effect on STAT1, as analyzed by western blotting. BK alone did not induce any phosphorylation of STAT1 or STAT2 (Figure 4). These results indicate that BK might suppress the activation of key molecules that mediate the IFN signaling. Since STAT1 is downstream of the receptors of other cytokines like IFN-γ, while STAT2 molecule is specifically downstream of Type I IFN signaling (37), our results suggest that BK may specifically regulate Type I IFNs.
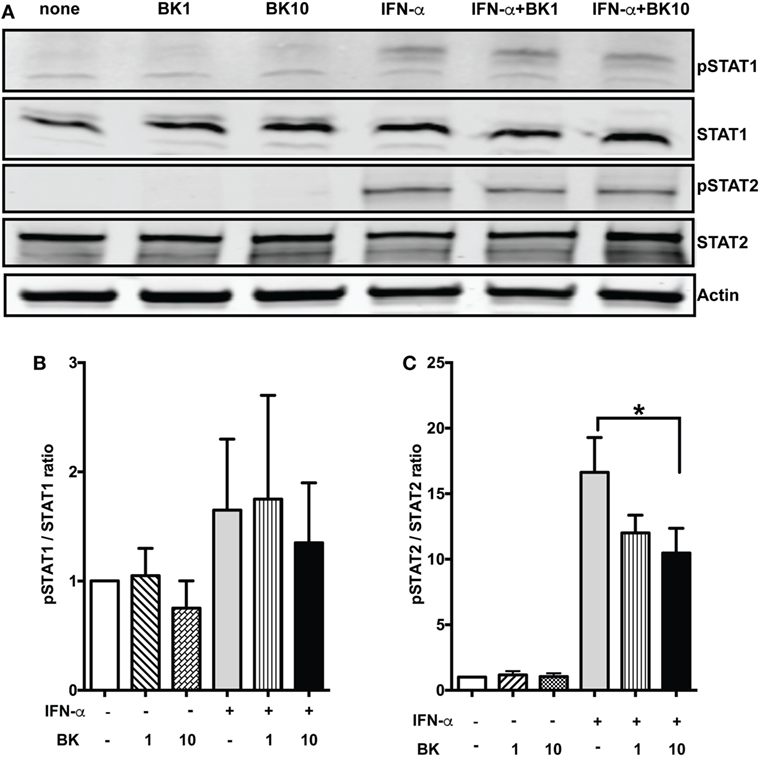
Figure 4. Bradykinins (BKs) suppress STAT2 phosphorylation. (A) Phosphorylation (p) and expression of STAT1 and STAT2 in bone-marrow-derived dendritic cells (BMDCs) stimulated with IFN-α for 15 min followed by BK for 30 min was analyzed by western blot. Actin was used as loading control. (B,C) Bars represent the ratios of intensities of each pSTAT and corresponding signal transducer and activator of transcription band, normalized to the respective loading controls. A representative blot of three independent experiments conducted with three independent BMDC cultures is shown. The differences in pSTAT2/STAT2 reached statistical significance; IFN-α—no BK vs. IFN-α-BK 10—*p < 0.05.
BKs Suppress ISGs in Normal and Lupus-Prone Murine Splenocytes In Vivo and DCs of Normal Mouse Spleen Ex Vivo
To determine the effects of BK in vivo, we analyzed the effects of CpG-induced IFN responses both in B6 and Sle mice. We injected CpG-A intraperitoneally in mice and, after overnight stimulation, we administered BK retro-orbitally. BK is a short-lived peptide (38) and, therefore, the in vivo stimulation was performed very quickly and 2 h after BK injection we collected the spleen and isolated RNA from total splenocytes to analyze the expression of the IFN-inducible genes by real-time qPCR. We found a strong induction of Irf7 and Cxcl10 upon CpG stimulation both in the B6 and Sle splenocytes, that was significantly downregulated upon BK administration (p < 0.05) (Figures 5A,B). We also observed that the constitutive expression Irf7 and Cxcl10 expression was also slightly downregulated in the Sle splenocytes (Figure 5B). The constitutive expression of ISGs in the Sle splenocytes was greater than twofold as compared to the B6 splenocytes (data not shown). To further determine the effects of BK on in vivo differentiated DCs, CD11c positive DCs from spleens of B6 mice were sorted and cultured ex vivo. We stimulated the splenic DCs with CpG for 2 h and then with BK for 3 h. We found that BK strongly suppressed the induction of Ifn-β (p < 0.05; Figure 5C) indicating that BK is a negative regulator of the IFN response in immune cells in general, and in cDCs in particular, in vitro and in vivo.
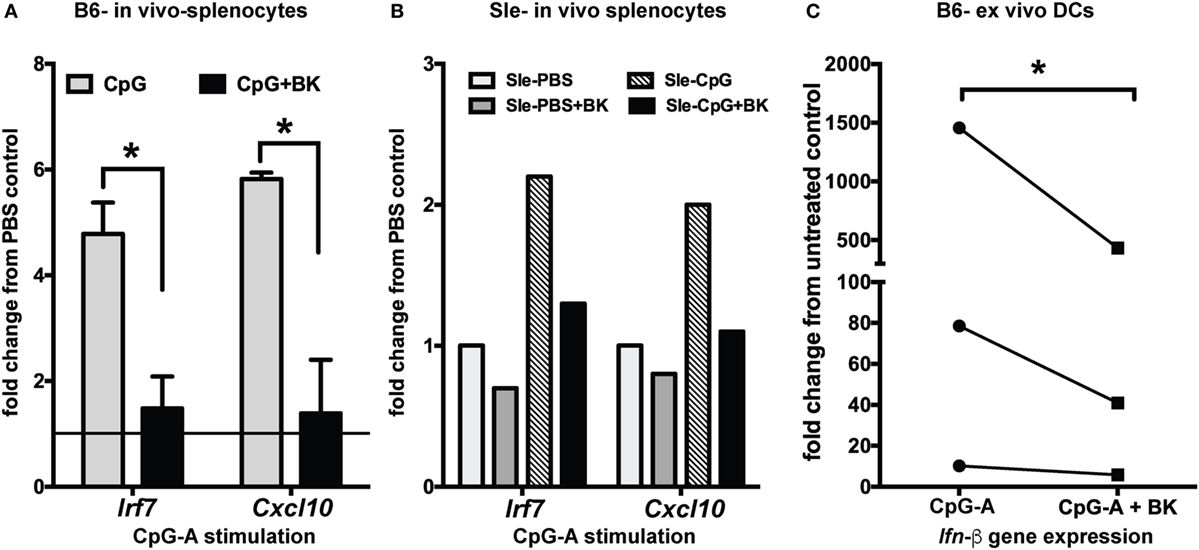
Figure 5. Bradykinins (BKs) suppress interferon-stimulated genes in splenocytes in vivo and in ex vivo mouse dendritic cells (DCs). (A) B6 and Sle mice were injected with CpG-A intraperitoneally for overnight stimulation, BK was administered retro-orbitally for 2 h and expression of the IFN-inducible genes was analyzed from total splenocytes by real-time qPCR. All of the conditions were calibrated against the untreated control in each experiment. Graphs are averages of three independent B6 mice sets (A) and one representative Sle set (B). (C) DCs from B6 mouse spleen were sorted by Miltenyi magnetic beads, plated in 24-well plates, stimulated with CpG-A (2 h) followed by BK (3 h) and analyzed for Ifn-β expression. Line graphs are fold change in gene expression normalized to untreated control in each experiment; *p < 0.05.
klks and Captopril (ACEi) Suppress IFN-Induced ISGs in Murine DCs and Human PBMCs
Earlier reports have shown that antibody-induced GN in lupus-prone mice is ameliorated by exogenous administration of klk1, one of the molecules of the KKS (21). In order to determine if klks can affect Type I IFN responses in vitro, we induced an IFN response with CpG-A and then added recombinant klk1 (1 µg/ml) and analyzed Irf7 gene expression. Klk1 significantly suppressed CpG-A-induced Irf7 gene expression in mouse DCs (p < 0.001) (Figure 6A). To further test if klk1 acted via BK signaling to bring about this suppression, we blocked both B2 and B1 receptors with their inhibitors HOE140 (H) (10 µM) and des-Arg10-HOE140 (DH) (1 µg/ml), respectively. Klk1 could suppress Irf7 gene expression even after blocking the BK receptors (p < 0.001), suggesting that klk may use an alternate and distinct pathway, specific of klks, possibly via protease-activated receptors (PARs) to bring about this suppression. To further test other important candidates of the KKS system, we focused on ACE, because captopril, an ACE inhibitor is a widely prescribed drug for lupus patients with kidney disease (10, 39) and we anticipated captopril to have other immunoregulatory effects (40) other than regulating blood pressure. We measured the effects of captopril (20 µM) on ISGs in mouse DCs and found that it can efficiently suppress Irf7 gene expression (p < 0.05) (Figure 6A). Blocking of the BK receptors and then giving captopril reverted the suppressive effect, suggesting that captopril may primarily affect the availability of BK to signal via BK receptors on the cDCs to bring about downregulation of IFN-induced responses. Blocking the B1 and B2 receptors with the respective antagonists at the indicated concentrations did block the effects of further stimulation with BK, as analyzed by Irf7 gene expression (data not shown). A strong suppression of IRF7 (p < 0.01) and CXCL10 (p < 0.001), gene expression was also observed in human PBMCs stimulated with IFN-α and then treated with captopril (20 µM) (Figure 6B).
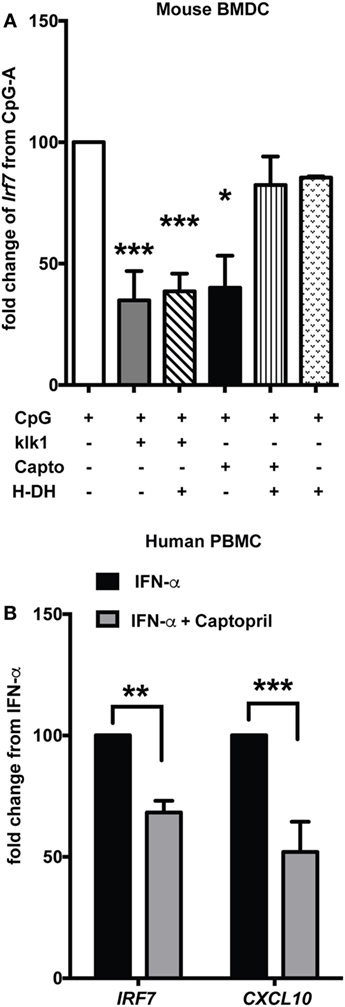
Figure 6. klk1 and captopril also suppress interferon-stimulated genes. (A) Murine bone-marrow-derived dendritic cells (BMDCs) were stimulated with CpG-A for 2.5 h treated with antagonists for 30 min to block both B1 (DH—1 µg/ml) and B2 (H—10 µM) receptors and then stimulated with klk1 (1 µg/ml) or captopril (20 µM) for a further 4 h. Irf7 gene expression was analyzed by qPCR. Percent fold change was calculated by normalizing to CpG-A stimulation alone. (B) Human PBMCs were first treated with IFN-α for 3 h, 20 µM captopril was added and harvested after further 3 h and IRF7 and CXCL10 gene expression was analyzed by qPCR. Percent fold change was calculated by normalizing with IFN-α stimulation alone. Panels (A,B) represent average (mean + SEM) of at least three independent experiments, performed with independent BMDC cultures from each mouse or PBMCs from each donor; *p < 0.05, **p < 0.01, ***p < 0.001.
BKs Suppress IFN Responses via PGE2
Production of prostaglandins via BK receptor signaling has been documented in several studies (41, 42). A recent study indicates that prostaglandins, especially PGE2 can suppress IFN responses in pDCs (43). To determine if the BK suppression of IFN responses is mediated via PGE2, we used indo to inhibit PGE2 production and analyzed ISG expression (Figure 7). The BK-induced suppression of ISGs, both with direct IFN stimulation (Figure 7A) or upon TLR (CpG-B) stimulation (Figure 7B), was significantly reverted with indo treatment, suggesting that BKs, may bring about suppression of IFN responses, at least in part, via prostaglandins.
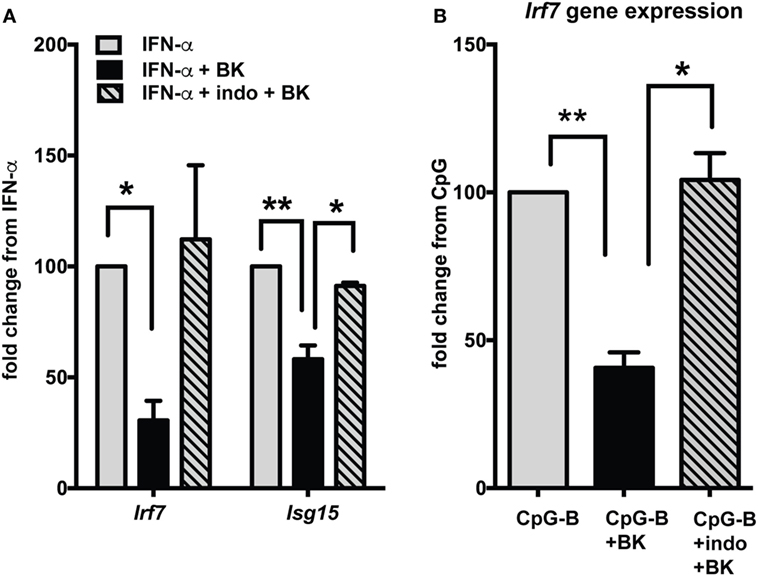
Figure 7. Bradykinin (BK) suppression of interferon-stimulated genes (ISGs) is mediated via PGE2. Murine bone-marrow-derived dendritic cells were stimulated with IFN-α (A) or CpG-B (B) for 2.5 h then 1 µg/ml indomethacin (indo) (COX1 and -2 inhibitor) was added and incubated for 30 min followed by stimulation with BK for a further 4 h. Cells were harvested and ISGs were analyzed by qPCR. Percent fold change was calculated by normalizing with IFN-α stimulation alone (A) or CpG-B stimulation alone (B); *p < 0.05; **p < 0.01.
Discussion
Studies in the past decade have discovered the role of the KKS in regulating immune responses (5, 44) in several models of autoimmunity (45) including lupus (12, 46) and cancer (47). Type I IFNs have been investigated both for their pathogenic role in diseases such as lupus (48, 49) and for its therapeutic role as in MS (50) or in some forms of cancer and chronic viral infections (51). Besides the classic pathways of Type I IFN production via the TLRs, many cytosolic nucleic acid sensors have also been shown to induce IFN production (52) adding to the complexity of the regulatory mechanisms of the IFN system (53). Although the role of KKS has been studied in some immunological pathways (44, 54–57), the effect of this system in regulating IFN responses has never been addressed.
In order to determine the effects of KKS in modulating Type I IFN responses, we tested the BKs which are central in the KKS system (1, 21, 58), tissue klk1 which has been shown to ameliorate GN in antibody-mediated kidney disease (12) and captopril, a widely prescribed ACEi (10, 59). We clearly demonstrated that all these molecules suppressed Type I IFN responses induced by the classical TLR pathway (using TLR7 and -9 agonists) or directly by recombinant IFN-α. BKs suppressed important ISGs that we have previously shown to be induced upon TLR7 and -9 stimulation in lupus dendritic cells (DCs) (23), especially the IRF7, a key regulator of Type I IFN responses (60). BKs efficiently suppressed both short and sustained IFN responses in DCs of normal and lupus-prone Sle mice grown in vitro. We have previously reported induction of MHC Class I as an IFN-specific response (25). The observation that BKs can suppress the upregulation of MHC Class I may have important implications for the innate IFNs response during viral infections (61). To study the immunomodulatory effects of BK, we used two in vitro models of DCs, the GM-cDCs and the FLt3L-DCs and found similar results, indicating that BK can regulate different subsets of DCs. Moreover, the induction of IFN responses in splenocytes in vivo, as well as in mature splenic DCs isolated and stimulated ex vivo, were also strongly suppressed by BK. Altogether, these results suggest that BKs are important negative regulators of the Type I IFN response in immune cells. The BK suppressive effect of IFN responses is further validated by our demonstration of this phenomenon in human PBMCs. The results suggest that different cell types in human blood show decreased responses upon BK stimulation, similar to mouse DCs studied in vitro.
Bradykinins also suppressed the IFN-induced phosphorylation of STAT2 molecules, a key step for further signaling, translocation to the nucleus and induction of IFN responsive genes (37, 62). However, suppression of STAT1 phosphorylation was only marginal. Since STAT1 is downstream of many cytokine receptors such as IFN-γ, while STAT2 is specifically downstream of Type I IFN receptor (62), our results suggest that BK may specifically regulate Type I IFNs. Taken together, these results strongly indicate that signaling via BK has an important role in regulating IFN-induced responses.
We have shown before that CXCL10 is a strongly IFN-dependent cytokine (27). Serum CXCL10 has been shown to be increased in patients with various autoimmune diseases including rheumatoid arthritis, systemic lupus, and Sjogren’s syndrome (63). Urine CXCL10 has been significantly correlated with renal activity in lupus kidney disease (64), and CXCL10 has also been shown to be very high in the cerebrospinal fluid of patients with central nervous system lupus (65). In this study, we observed a strong suppressive effect of BK on CXCL10 both at gene (Figure 1E) and protein levels (Figure 2B), suggesting that drugs that increase BK, such as ACEis (Figure 6B), may downregulate this cytokine in lupus disease and thereby bring about beneficial effects.
Bradykinins have been shown to be pro-inflammatory and induce IL12 production in DCs (28) and IL-6 production in airway smooth muscle cells (66). The BKs that we tested in our DC cultures did not induce production of IL12p70, IL-6, or CXCL10 (data not shown). The IL12p70 production reported by Aliberti et al. (28) was observed in splenic DCs and their responses could be different from in vitro cultured BMDCs. Nevertheless, we found that BK inhibited the production of TLR-induced proinflammatroy cytokines, a result consistent with the inhibition of the IFN responses in DCs shown in this study.
We also found that tissue klk1 decreased the IFN response. klks are serine proteases that cleave low molecular-weight kininogen into BKs that exert their biological functions via triggering the kinin receptors, B1R and B2R (1). Liu et al. have indicated that klks are protective disease-associated genes in immune complex–induced nephritis and lupus nephritis in mice and in SLE patients (21). Our results are consistent with these findings and suggest that some of the protective effects of klks in lupus could be due to dampening IFN responses. Recent studies suggest that klk1 may also activate protease-activated receptors (PARs) in inflammatory diseases (67). PARs are a subfamily of G protein-coupled receptors that participate in cell signaling via cleavage or activation by klk1. Our results suggest that the regulation of IFN responses by klk is independent from the known kk regulation of BK and their receptors; other pathways such as PARs may be involved in IFN regulation and warrants further investigation.
Angiotensin-converting enzyme inhibitors (ACEi), which increase kinin availability in vivo (2), have been found to be beneficial in ameliorating kidney disease in mouse models (39, 68) as well as lupus patients (10, 22). Captopril is one of the most popularly prescribed drugs to control blood pressure (69, 70). ACEis have also been shown to be beneficial in other autoimmune diseases, such as rheumatoid arthritis (71), and prevention of complications in insulin-dependent diabetes mellitus (72). The suppressive effects of captopril on the IFN-induced ISGs in PBMC makes it even more important to further investigate the pathways that lead to the beneficial effects of this drug, not only in lupus but in other diseases as well. The rescue from the suppression of ISGs by captopril by blocking of the kinin receptors clearly indicates that captopril suppresses IFN response by increasing the BKs. It will be interesting to study the effects of other ACEis in regulating IFN responses.
To determine the molecular mechanism the mediate the regulation of the interferon response by BK, we considered some key molecules, including PGE2 (37). PGE2 is a major lipid mediator that is released at the sites of inflammatory and it has been shown to possess pro-inflammatory (73) and anti-inflammatory (74) properties depending on the cell type, the stimulation and the experimental models of inflammatory/autoimmune diseases used. In particular, PGE2 regulated inflammation in several cell types: PGE2 has been shown to be an important inhibitory modulator of airway inflammation (75); PGE2 reduced IL12 pro-inflammatory cytokine production in DCs (76) and also inhibited IL-27 production (76, 77). The role of PGE2 in IFN regulation has been reported recently (39). Production of PGE2 by monocytes has been shown to be one of the mechanisms by which IgG immunoglobulin attenuates IFN production in pDCs (39). Our results of the rescue of the inhibitory function of BK on the IFN response by indo, that inhibits the production of PGE2, suggest that this novel pathway of Type I IFN regulation by the KKS presented in this study is mediated via production of PGE2.
Type I IFN signaling refuels an autocrine positive feedback loop and amplifies its response (78). The system is controlled at several negative check-points such as the SOCS molecules (suppressors of cytokine signaling) (79). The dysregulation in these pathways may increase the production of IFN and cause more amplified responses in lupus (79). Our results indicate that BKs can suppress the ongoing IFN response efficiently, a pathway that is very promising to attenuate the IFN responses in this disease. The results that molecules of the KKS and molecules that induce an increase in BKs, such as the FDA approved ACEis, can suppress an ongoing IFN response suggest that they can be therapeutic in a clinical situation of chronic immune response, as in lupus disease that is characterized by a consistent IFN signature. This phenomenon warrants further investigation to design better therapeutic strategies for IFN-mediated autoimmune diseases.
Ethics Statement
Animals were maintained in the animal facility in accordance to the guidelines of the Institutional Animal Care and Use Committees of Temple University, which is an American Association for the Accreditation of Laboratory Animal Care-accredited facility. According to Temple University IRB, human samples that are procured in an already de-identified state is not considered “human subjects research,” The samples received from American Red Cross, used in this study were de-identified samples and no information about the donors is available to the investigators.
Author Contributions
US conceived the idea, designed experiments, and wrote the manuscript. AS, ML, NF, VR, MD, and US performed experiments and analyzed the data. All authors contributed to interpreting the data and writing of the manuscript. All authors have reviewed and approved the manuscript, and agreed with its submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Drs. Philip L. Cohen and Slava Rom for critically reading the manuscript. The authors thank Nancy Reichenbach for editorial assistance and Brona Ranieri for technical assistance. This work was fully supported by NIH-NIAMS grant R03-AR065157 to US and in part, from NIH grants R01-DA031064 to RP, R01-AI076423 to SG, and R01-MH65151 to YP.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2018.00156/full#supplementary-material.
References
1. Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev (1992) 44(1):1–80.
2. Moreau ME, Garbacki N, Molinaro G, Brown NJ, Marceau F, Adam A. The kallikrein-kinin system: current and future pharmacological targets. J Pharmacol Sci (2005) 99(1):6–38. doi:10.1254/jphs.SRJ05001X
3. Kakoki M, Smithies O. The kallikrein-kinin system in health and in diseases of the kidney. Kidney Int (2009) 75(10):1019–30. doi:10.1038/ki.2008.647
4. Lundwall A, Brattsand M. Kallikrein-related peptidases. Cell Mol Life Sci (2008) 65(13):2019–38. doi:10.1007/s00018-008-8024-3
5. Marceau F, Regoli D. Bradykinin receptor ligands: therapeutic perspectives. Nat Rev Drug Discov (2004) 3(10):845–52. doi:10.1038/nrd1522
6. Te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH. Hypertension: renin-angiotensin-aldosterone system alterations. Circ Res (2015) 116(6):960–75. doi:10.1161/CIRCRESAHA.116.303587
7. Linz W, Wiemer G, Gohlke P, Unger T, Scholkens BA. Contribution of kinins to the cardiovascular actions of angiotensin-converting enzyme inhibitors. Pharmacol Rev (1995) 47(1):25–49.
8. Yang HY, Erdos EG, Levin Y. Characterization of a dipeptide hydrolase (kininase II: angiotensin I converting enzyme). J Pharmacol Exp Ther (1971) 177(1):291–300.
9. Heart Outcomes Prevention Evaluation Study Investigators, Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med (2000) 342(3):145–53. doi:10.1056/NEJM200001203420301
10. Duran-Barragan S, McGwin G Jr, Vila LM, Reveille JD, Alarcon GS. Angiotensin-converting enzyme inhibitors delay the occurrence of renal involvement and are associated with a decreased risk of disease activity in patients with systemic lupus erythematosus – results from LUMINA (LIX): a multiethnic US cohort. Rheumatology (Oxford) (2008) 47(7):1093–6. doi:10.1093/rheumatology/ken208
11. Herlitz H, Svalander C, Tarkowski A, Westberg G. Effect of captopril on murine systemic lupus erythematosus disease. J Hypertens Suppl (1988) 6(4):S684–6. doi:10.1097/00004872-198812040-00215
12. Li QZ, Zhou J, Yang R, Yan M, Ye Q, Liu K, et al. The lupus-susceptibility gene kallikrein downmodulates antibody-mediated glomerulonephritis. Genes Immun (2009) 10(5):503–8. doi:10.1038/gene.2009.7
13. Craft JE. Dissecting the immune cell mayhem that drives lupus pathogenesis. Sci Transl Med (2011) 3(73):73s9. doi:10.1126/scitranslmed.3002138
14. Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol (2007) 37(12):3582–6. doi:10.1002/eji.200737815
15. Bertsias GK, Salmon JE, Boumpas DT. Therapeutic opportunities in systemic lupus erythematosus: state of the art and prospects for the new decade. Ann Rheum Dis (2010) 69(9):1603–11. doi:10.1136/ard.2010.135186
16. Crow MK. Interferon-alpha: a therapeutic target in systemic lupus erythematosus. Rheum Dis Clin North Am (2010) 36(1):173–86, x. doi:10.1016/j.rdc.2009.12.008
17. Kalunian KC, Merrill JT, Maciuca R, McBride JM, Townsend MJ, Wei X, et al. A phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-alpha) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis (2016) 75(1):196–202. doi:10.1136/annrheumdis-2014-206090
18. Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, et al. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis (2016) 75(11):1909–16. doi:10.1136/annrheumdis-2015-208562
19. Lenert PS. Targeting toll-like receptor signaling in plasmacytoid dendritic cells and autoreactive B cells as a therapy for lupus. Arthritis Res Ther (2006) 8(1):203. doi:10.1186/ar1888
20. Barrat FJ, Coffman RL. Development of TLR inhibitors for the treatment of autoimmune diseases. Immunol Rev (2008) 223:271–83. doi:10.1111/j.1600-065X.2008.00630.x
21. Liu K, Li QZ, Delgado-Vega AM, Abelson AK, Sanchez E, Kelly JA, et al. Kallikrein genes are associated with lupus and glomerular basement membrane-specific antibody-induced nephritis in mice and humans. J Clin Invest (2009) 119(4):911–23. doi:10.1172/JCI36728
22. Mok CC. Understanding lupus nephritis: diagnosis, management, and treatment options. Int J Womens Health (2012) 4:213–22. doi:10.2147/IJWH.S28034
23. Sriram U, Varghese L, Bennett HL, Jog NR, Shivers DK, Ning Y, et al. Myeloid dendritic cells from B6.NZM Sle1/Sle2/Sle3 lupus-prone mice express an IFN signature that precedes disease onset. J Immunol (2012) 189(1):80–91. doi:10.4049/jimmunol.1101686
24. Weckerle CE, Niewold TB. The unexplained female predominance of systemic lupus erythematosus: clues from genetic and cytokine studies. Clin Rev Allergy Immunol (2011) 40(1):42–9. doi:10.1007/s12016-009-8192-4
25. Sriram U, Biswas C, Behrens EM, Dinnall JA, Shivers DK, Monestier M, et al. IL-4 suppresses dendritic cell response to type I interferons. J Immunol (2007) 179(10):6446–55. doi:10.4049/jimmunol.179.10.6446
26. Sriram U, Xu J, Chain RW, Varghese L, Chakhtoura M, Bennett HL, et al. IL-4 suppresses the responses to TLR7 and TLR9 stimulation and increases the permissiveness to retroviral infection of murine conventional dendritic cells. PLoS One (2014) 9(1):e87668. doi:10.1371/journal.pone.0087668
27. Xu J, Lee MH, Chakhtoura M, Green BL, Kotredes KP, Chain RW, et al. STAT2 is required for TLR-induced murine dendritic cell activation and cross-presentation. J Immunol (2016) 197(1):326–36. doi:10.4049/jimmunol.1500152
28. Aliberti J, Viola JP, Vieira-de-Abreu A, Bozza PT, Sher A, Scharfstein J. Cutting edge: bradykinin induces IL-12 production by dendritic cells: a danger signal that drives Th1 polarization. J Immunol (2003) 170(11):5349–53. doi:10.4049/jimmunol.170.11.5349
29. Odaka C, Mizuochi T. Angiotensin-converting enzyme inhibitor captopril prevents activation-induced apoptosis by interfering with T cell activation signals. Clin Exp Immunol (2000) 121(3):515–22. doi:10.1046/j.1365-2249.2000.01323.x
30. Kocieda VP, Adhikary S, Emig F, Yen JH, Toscano MG, Ganea D. Prostaglandin E2-induced IL-23p19 subunit is regulated by cAMP-responsive element-binding protein and C/AATT enhancer-binding protein beta in bone marrow-derived dendritic cells. J Biol Chem (2012) 287(44):36922–35. doi:10.1074/jbc.M112.402958
31. Dietz AB, Bulur PA, Emery RL, Winters JL, Epps DE, Zubair AC, et al. A novel source of viable peripheral blood mononuclear cells from leukoreduction system chambers. Transfusion (2006) 46(12):2083–9. doi:10.1111/j.1537-2995.2006.01033.x
32. Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity (2006) 25(3):383–92. doi:10.1016/j.immuni.2006.08.010
33. Niewold TB, Clark DN, Salloum R, Poole BD. Interferon alpha in systemic lupus erythematosus. J Biomed Biotechnol (2010) 2010:948364. doi:10.1155/2010/948364
34. Morales DJ, Lenschow DJ. The antiviral activities of ISG15. J Mol Biol (2013) 425(24):4995–5008. doi:10.1016/j.jmb.2013.09.041
35. Trinchieri G. Type I interferon: friend or foe? J Exp Med (2010) 207(10):2053–63. doi:10.1084/jem.20101664
36. Brasel K, De Smedt T, Smith JL, Maliszewski CR. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood (2000) 96(9):3029–39.
37. Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol (2005) 23:307–36. doi:10.1146/annurev.immunol.23.021704.115843
38. Murphey LJ, Hachey DL, Oates JA, Morrow JD, Brown NJ. Metabolism of bradykinin in vivo in humans: identification of BK1-5 as a stable plasma peptide metabolite. J Pharmacol Exp Ther (2000) 294(1):263–9.
39. Shimazu H, Kinoshita K, Hino S, Yano T, Kishimoto K, Nagare Y, et al. Effect of combining ACE inhibitor and statin in lupus-prone mice. Clin Immunol (2010) 136(2):188–96. doi:10.1016/j.clim.2010.03.008
40. Li HQ, Zhang Q, Chen L, Yin CS, Chen P, Tang J, et al. Captopril inhibits maturation of dendritic cells and maintains their tolerogenic property in atherosclerotic rats. Int Immunopharmacol (2015) 28(1):715–23. doi:10.1016/j.intimp.2015.05.052
41. Brechter AB, Lerner UH. Bradykinin potentiates cytokine-induced prostaglandin biosynthesis in osteoblasts by enhanced expression of cyclooxygenase 2, resulting in increased RANKL expression. Arthritis Rheum (2007) 56(3):910–23. doi:10.1002/art.22445
42. Catalioto RM, Valenti C, Maggi CA, Giuliani S. Enhanced Ca(2+) response and stimulation of prostaglandin release by the bradykinin B2 receptor in human retinal pigment epithelial cells primed with proinflammatory cytokines. Biochem Pharmacol (2015) 97(2):189–202. doi:10.1016/j.bcp.2015.07.034
43. Wiedeman AE, Santer DM, Yan W, Miescher S, Kasermann F, Elkon KB. Contrasting mechanisms of interferon-alpha inhibition by intravenous immunoglobulin after induction by immune complexes versus toll-like receptor agonists. Arthritis Rheum (2013) 65(10):2713–23. doi:10.1002/art.38082
44. Chao J, Chao L. Kallikrein-kinin in stroke, cardiovascular and renal disease. Exp Physiol (2005) 90(3):291–8. doi:10.1113/expphysiol.2004.028464
45. Hebb AL, Bhan V, Wishart AD, Moore CS, Robertson GS. Human kallikrein 6 cerebrospinal levels are elevated in multiple sclerosis. Curr Drug Discov Technol (2010) 7(2):137–40. doi:10.2174/157016310793180611
46. Ponticelli C, Meroni PL. Kallikreins and lupus nephritis. J Clin Invest (2009) 119(4):768–71. doi:10.1172/JCI38786
47. Avgeris M, Mavridis K, Scorilas A. Kallikrein-related peptidase genes as promising biomarkers for prognosis and monitoring of human malignancies. Biol Chem (2010) 391(5):505–11. doi:10.1515/BC.2010.056
48. Ronnblom L, Eloranta ML, Alm GV. The type I interferon system in systemic lupus erythematosus. Arthritis Rheum (2006) 54(2):408–20. doi:10.1002/art.21571
49. Hagberg N, Ronnblom L. Systemic lupus erythematosus – a disease with a dysregulated type I interferon system. Scand J Immunol (2015) 82(3):199–207. doi:10.1111/sji.12330
50. La Mantia L, Di Pietrantonj C, Rovaris M, Rigon G, Frau S, Berardo F, et al. Interferons-beta versus glatiramer acetate for relapsing-remitting multiple sclerosis. Cochrane Database Syst Rev (2016) 11:CD009333. doi:10.1002/14651858.CD009333.pub3
51. Konerman MA, Lok AS. Interferon treatment for hepatitis B. Clin Liver Dis (2016) 20(4):645–65. doi:10.1016/j.cld.2016.06.002
52. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol (2014) 32:461–88. doi:10.1146/annurev-immunol-032713-120156
53. Chen K, Liu J, Cao X. Regulation of type I interferon signaling in immunity and inflammation: a comprehensive review. J Autoimmun (2017) 83:1–11. doi:10.1016/j.jaut.2017.03.008
54. Avgeris M, Mavridis K, Scorilas A. Kallikrein-related peptidases in prostate, breast, and ovarian cancers: from pathobiology to clinical relevance. Biol Chem (2012) 393(5):301–17. doi:10.1515/hsz-2011-0260
55. Blaber SI, Ciric B, Christophi GP, Bernett MJ, Blaber M, Rodriguez M, et al. Targeting kallikrein 6 proteolysis attenuates CNS inflammatory disease. FASEB J (2004) 18(7):920–2. doi:10.1096/fj.03-1212fje
56. Kayashima Y, Smithies O, Kakoki M. The kallikrein-kinin system and oxidative stress. Curr Opin Nephrol Hypertens (2012) 21(1):92–6. doi:10.1097/MNH.0b013e32834d54b1
57. Scharfstein J, Andrade D, Svensjo E, Oliveira AC, Nascimento CR. The kallikrein-kinin system in experimental Chagas disease: a paradigm to investigate the impact of inflammatory edema on GPCR-mediated pathways of host cell invasion by Trypanosoma cruzi. Front Immunol (2012) 3:396. doi:10.3389/fimmu.2012.00396
58. Bertram CM, Baltic S, Misso NL, Bhoola KD, Foster PS, Thompson PJ, et al. Expression of kinin B1 and B2 receptors in immature, monocyte-derived dendritic cells and bradykinin-mediated increase in intracellular Ca2+ and cell migration. J Leukoc Biol (2007) 81(6):1445–54. doi:10.1189/jlb.0106055
59. Albuquerque D, Nihei J, Cardillo F, Singh R. The ACE inhibitors enalapril and captopril modulate cytokine responses in Balb/c and C57Bl/6 normal mice and increase CD4(+)CD103(+)CD25(negative) splenic T cell numbers. Cell Immunol (2010) 260(2):92–7. doi:10.1016/j.cellimm.2009.09.006
60. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434(7034):772–7. doi:10.1038/nature03464
61. Vogel K, Thomann S, Vogel B, Schuster P, Schmidt B. Both plasmacytoid dendritic cells and monocytes stimulate natural killer cells early during human herpes simplex virus type 1 infections. Immunology (2014) 143(4):588–600. doi:10.1111/imm.12337
62. Schindler C, Plumlee C. Inteferons pen the JAK-STAT pathway. Semin Cell Dev Biol (2008) 19(4):311–8. doi:10.1016/j.semcdb.2008.08.010
63. Lee EY, Lee ZH, Song YW. CXCL10 and autoimmune diseases. Autoimmun Rev (2009) 8(5):379–83. doi:10.1016/j.autrev.2008.12.002
64. Marie MA, Abu Khalil RE, Habib HM. Urinary CXCL10: a marker of nephritis in lupus patients. Reumatismo (2013) 65(6):292–7. doi:10.4081/reumatismo.2013.719
65. Okamoto H, Katsumata Y, Nishimura K, Kamatani N. Interferon-inducible protein 10/CXCL10 is increased in the cerebrospinal fluid of patients with central nervous system lupus. Arthritis Rheum (2004) 50(11):3731–2. doi:10.1002/art.20598
66. Huang CD, Tliba O, Panettieri RA Jr, Amrani Y. Bradykinin induces interleukin-6 production in human airway smooth muscle cells: modulation by Th2 cytokines and dexamethasone. Am J Respir Cell Mol Biol (2003) 28(3):330–8. doi:10.1165/rcmb.2002-0040OC
67. Chao J, Shen B, Gao L, Xia CF, Bledsoe G, Chao L. Tissue kallikrein in cardiovascular, cerebrovascular and renal diseases and skin wound healing. Biol Chem (2010) 391(4):345–55. doi:10.1515/BC.2010.042
68. De Albuquerque DA, Saxena V, Adams DE, Boivin GP, Brunner HI, Witte DP, et al. An ACE inhibitor reduces Th2 cytokines and TGF-beta1 and TGF-beta2 isoforms in murine lupus nephritis. Kidney Int (2004) 65(3):846–59. doi:10.1111/j.1523-1755.2004.00462.x
69. Cleland JG, Dargie HJ, Hodsman GP, Ball SG, Robertson JI, Morton JJ, et al. Captopril in heart failure. A double blind controlled trial. Br Heart J (1984) 52(5):530–5. doi:10.1136/hrt.52.5.530
70. Seneviratne B, Moore GA, West PD. Effect of captopril on functional mitral regurgitation in dilated heart failure: a randomised double blind placebo controlled trial. Br Heart J (1994) 72(1):63–8. doi:10.1136/hrt.72.1.63
71. Martin MF, Surrall KE, McKenna F, Dixon JS, Bird HA, Wright V. Captopril: a new treatment for rheumatoid arthritis? Lancet (1984) 1(8390):1325–8. doi:10.1016/S0140-6736(84)91821-X
72. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med (1993) 329(20):1456–62. doi:10.1056/NEJM199311113292004
73. Hoxha M. A systematic review on the role of eicosanoid pathways in rheumatoid arthritis. Adv Med Sci (2017) 63(1):22–9. doi:10.1016/j.advms.2017.06.004
74. Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol (2012) 188(1):21–8. doi:10.4049/jimmunol.1101029
75. Barry T, Delamere F, Holland E, Pavord I, Knox A. Production of PGE2 by bovine cultured airway smooth muscle cells: regulation by cAMP. J Appl Physiol (1985) (1995) 78(2):623–8. doi:10.1152/jappl.1995.78.2.623
76. Kalinski P, Hilkens CM, Snijders A, Snijdewint FG, Kapsenberg ML. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J Immunol (1997) 159(1):28–35.
77. Hooper KM, Yen JH, Kong W, Rahbari KM, Kuo PC, Gamero AM, et al. Prostaglandin E2 inhibition of IL-27 production in murine dendritic cells: a novel mechanism that involves IRF1. J Immunol (2017) 198(4):1521–30. doi:10.4049/jimmunol.1601073
78. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14(1):36–49. doi:10.1038/nri3581
Keywords: kallikrein–kinin system, bradykinins, ACE inhibitors, dendritic cells, PBMC, TLR, Type I IFNs, lupus
Citation: Seliga A, Lee MH, Fernandes NC, Zuluaga-Ramirez V, Didukh M, Persidsky Y, Potula R, Gallucci S and Sriram U (2018) Kallikrein–Kinin System Suppresses Type I Interferon Responses: A Novel Pathway of Interferon Regulation. Front. Immunol. 9:156. doi: 10.3389/fimmu.2018.00156
Received: 16 November 2017; Accepted: 17 January 2018;
Published: 02 February 2018
Edited by:
Annalisa Del Prete, University of Brescia, ItalyReviewed by:
Marco De Andrea, University of Turin, ItalyLuciana D’Apice, Consiglio Nazionale Delle Ricerche (CNR), Italy
Copyright: © 2018 Seliga, Lee, Fernandes, Zuluaga-Ramirez, Didukh, Persidsky, Potula, Gallucci and Sriram. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Uma Sriram, c3JpcmFtQHRlbXBsZS5lZHU=