 Shruti Chaturvedi
Shruti Chaturvedi Robert A. Brodsky1
Robert A. Brodsky1 Keith R. McCrae
Keith R. McCrae- 1Division of Hematology, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 2Hematologic Oncology and Blood Disorders, Taussig Cancer Institute, Cleveland, OH, United States
- 3Department of Cellular and Molecular Medicine, Cleveland Clinic, Cleveland, OH, United States
The antiphospholipid syndrome (APS) is characterized by thrombosis and pregnancy morbidity in the presence of antiphospholipid antibodies (aPL). Complement is a system of enzymes and regulatory proteins of the innate immune system that plays a key role in the inflammatory response to pathogenic stimuli. The complement and coagulation pathways are closely linked, and expanding data indicate that complement may be activated in patients with aPL and function as a cofactor in the pathogenesis of aPL-associated clinical events. Complement activation by aPL generates C5a, which induces neutrophil tissue factor-dependent procoagulant activity. Beta-2-glycoprotein I, the primary antigen for pathogenic aPL, has complement regulatory effects in vitro. Moreover, aPL induce fetal loss in wild-type mice but not in mice deficient in specific complement components (C3, C5). Antiphospholipid antibodies also induce thrombosis in wild type mice and this effect is attenuated in C3 or C6 deficient mice, or in the presence of a C5 inhibitor. Increased levels of complement activation products have been demonstrated in sera of patients with aPL, though the association with clinical events remains unclear. Eculizumab, a terminal complement inhibitor, has successfully been used to treat catastrophic APS and prevent APS-related thrombotic microangiopathy in the setting of renal transplant. However, the mechanisms of complement activation in APS, its role in the pathogenesis of aPL related complications in humans, and the potential of complement inhibition as a therapeutic target in APS require further study.
Introduction
Antiphospholipid syndrome (APS) is a systemic autoimmune disorder characterized by thrombosis affecting the venous or arterial vascular systems, and/or obstetrical morbidity along with the persistent presence of antiphospholipid antibodies (aPL), including lupus anticoagulant, anticardiolipin antibody and anti-beta-2-glycoprotein-I (β2GPI) (1). Rather than binding to anionic phospholipids such as cardiolipin as was previously believed, aPL are directed against phospholipid binding proteins bound to an appropriate biological surface such as a cellular membrane (2). Anti-β2GPI antibodies are the primary pathogenic antibody in APS (3–5), although aPL directed against other antigens such as prothrombin and phosphatidylserine have also been described (3, 6). The mechanisms by which aPL induce thrombosis and pregnancy loss are not fully understood. Though β2GPI is the primary antigen in APS, its inhibition does not directly have thrombotic effects as evidenced by the lack of a thrombotic phenotype in β2GPI deficient individuals (7, 8). Multiple pathogenic mechanisms have been proposed including inhibition of the natural anticoagulant and fibrinolytic systems (9–12), activation of vascular cells including endothelial cells (13), platelets (14), and monocytes (15), procoagulant effects of extracellular vesicles (16), and disruption of the anticoagulant annexin A5 shield on cellular surfaces (17). Over the past two decades, complement activation has emerged as an attractive target for mechanistic and therapeutic investigations based on studies demonstrating evidence of complement activation in patients with APS, and murine models that indicate a critical role of complement in aPL-mediated thrombosis (18–21) and obstetric (22–24) complications (25). It remains unclear whether these are distinct mechanisms that reflect antibody heterogeneity or are linked to an as yet undefined central mechanism. Recurrent thrombosis and fetal loss are common despite standard treatment. The variability of treatment effectiveness in APS suggests that subsets of patients might benefit from treatments beyond anticoagulation.
The complement system, consisting of over 50 plasma proteins involved in host defense, is organized into three pathways; the immune complex mediated classical pathway, the lectin pathway, and the alternative pathway. These pathways converge at the level of complement component C3 and the terminal complement pathway that leads to generation of C5a, a potent pro-inflammatory molecule, and C5b-9 (the membrane attack complex) (Figure 1). Anti- β2GPI antibodies are associated with complement activation, and the complement and coagulation pathways are closely linked (26, 27); C5a induces neutrophil tissue factor-dependent procoagulant activity (26) and may inhibit fibrinolysis though increased activation of thrombin activated fibrinolysis inhibitor (TAFI). C3a and C5a also activate endothelial cells, inducing the expression of adhesion molecules and procoagulant activity (28–30), as well as platelets (31). Complement activation also causes placental inflammation and injury (32), a hallmark of fetal loss in APS (23). This review summarizes the current evidence supporting the role of complement in aPL associated clinical events, the interplay between complement and thrombosis in APS, therapeutic perspectives on complement targeted agents in APS, and areas of future research.
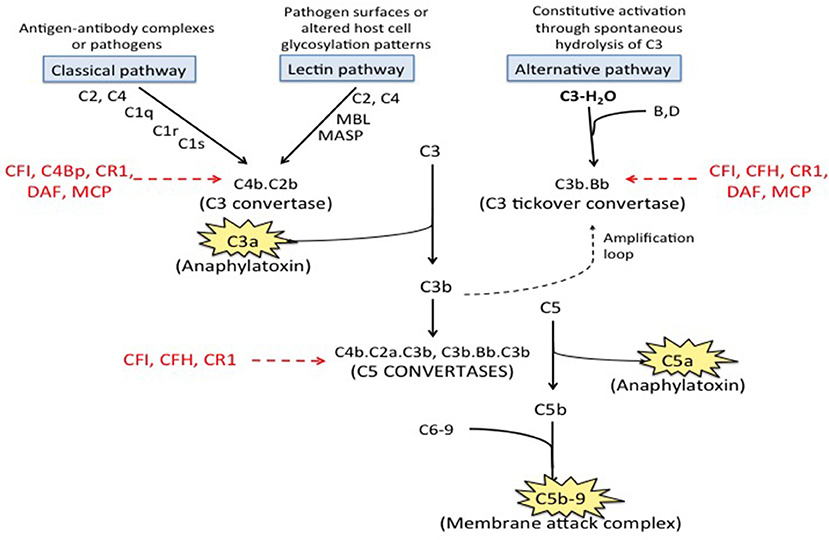
Figure 1. Complement pathways. There are three well-recognized pathways of complement activation; (1) the classical pathway, (2) the lectin pathway, and (3) the alternative pathway. The classical and lectin pathways are activated when specific triggers are recognized by host pattern-recognition receptors while the alternative pathway is constitutively active. Activation of all there pathways ultimately leads to generation of a C3 convertase (C4b.C2a for the classical and lectin pathways and C3b·B for the alternative pathway), which cleave C3 to generate C3a and C3b. C3a is an anaphylatoxin. C3b is quickly inactivated when it lands on a healthy host cell but triggers a rapid amplification loop when it binds to a pathogen or altered host cell. C3b also complexes with the C3 convertases to form the C5 convertases (C4b·C2a·C3b and C3b·Bb·C3b) that cleave C5 into C5a (an anaphylatoxin) and C5b. C5b combines with C6-9 to form C5b-9, also called the membrane attack complex (MAC). Regulatory factors including decay accelerating factor (DAF, CD55), CD59, factor H (CFH), factor I (CFI), membrane cofactor protein (MCP) and C3b/C4b receptor 1 (CR1) act at various stages of the cascade to control complement activation.
Complement in Obstetric APS
Animal Models of Obstetric APS
The earliest and most compelling evidence for complement involvement in APS comes from murine models of aPL-induced pregnancy loss in which Branch et al. showed that passive transfer of IgG fractions from patients with aPL led to fetal loss (33). On histopathologic examination, IgG localized to the decidua, which showed prominent necrosis. A subsequent series of experiments confirmed this finding and elucidated the role of complement in aPL induced fetal loss. In vivo experiments have shown that β2GPI localizes to the decidua (34), and that aPL binding to β2GPI inhibits trophoblastic proliferation, syncytia formation and invasion into maternal decidua, which are required for successful placentation (35).
Holers et al. showed that intraperitoneal injection of IgG from patients with APS into pregnant mice led to fetal resorption in 40% of pregnancies and a 35% fetal weight reduction compared with control mice (36). Inhibition of the complement cascade with the C3 convertase inhibitor complement receptor 1–related gene/protein y (Crry)-Ig prevented aPL mediated fetal resorption. C3 deficient mice (C3−/−) were also resistant to aPL mediated fetal loss (36). Girardi et al. later demonstrated that C5 deficiency or treatment of mice with anti-C5a monoclonal antibody protects against aPL induced pregnancy loss and growth retardation (22). Placentae from the aPL IgG treated mice showed human IgG deposition in the decidua, which demonstrated focal necrosis and apoptosis with neutrophil infiltrates (36). Neutrophils recruited by C5a expressed tissue factor that potentiated neutrophil activation and the respiratory burst leading to trophoblastic injury and fetal loss (24, 32). The absence of aPL-induced growth retardation and fetal resorption in mice deficient in C4 or C5 suggests that the classical pathway is involved in initiating these effects. However, factor B is necessary for aPL mediated fetal loss and its inhibition ameliorates these effects supporting a role of the alternative pathway in amplifying complement activation (37). Taken together, these studies suggest that C3 and C5 activation is central to aPL-mediated fetal loss in this model, with neutrophils and tissue factor playing pro-inflammatory roles. Girardi et al. have also suggested that the protective effect of heparin in APS pregnancies may reflect its inhibitory effects on complement (23).
Complement Activation in Human Studies of Obstetric APS
Studies in humans support the role of complement in aPL mediated pregnancy complications. Hypocomplementemia, suggesting complement activation, has been observed in patients with SLE and APS (38), as well as those with primary APS and obstetric complications (39–41); however others have not found an association with hypocomplementemia and pregnancy complications in APS (42). In the PROMISSE study, which included nearly 500 pregnant women with lupus and/or aPL, adverse pregnancy outcomes were associated with increased serum levels of complement products Bb and C5b-9 early in pregnancy (43). In addition to elevated levels of complement activation products in serum, C4d was deposited at the feto-maternal interface in the placentae of women with SLE or APS, and correlated with fetal loss, decidual vasculopathy, increased syncytial knots and villous infarcts (44, 45). Interestingly, C5b-9 deposition in the trophoblast was not increased compared with control placentae, leading the authors to suggest that C5b-9 may not play a central role in aPL mediated placental injury, which is more likely to be caused by C3a and C5a mediated inflammation (45). Overall, these findings support a role for complement in aPL mediated pregnancy complications; however, the exact mechanisms of complement activation remain to be determined.
Complement in Vascular APS
Animal Models of Thrombotic APS
Animal models of thrombotic APS support a role for complement in aPL mediated thrombosis. Most early models of aPL induced thrombosis included passive transfer of aPL along with direct vessel injury by pinching (19, 46) or other means to induce thrombosis, which was reduced in mice with deficiencies of complement proteins C3, C5, or C6 (19), or in the presence of an inhibitory antibody against C5 (18). However, mechanical or chemical endothelial injury to initiate thrombosis that is propagated in the presence of aPL differs from the usual events in APS, in which a localized vascular insult is usually absent. Fischetti et al. used rats primed with lipopolysaccharide, which does not cause thrombosis by itself (20). Administration of aPL IgG to LPS primed mice led to thrombosis while administration of control IgG did not. Intravascular microscopy showed thrombosis in mesenteric vessels, and immunofluorescence staining confirmed co-localization of IgG and C3 in the vessel wall (20). Thrombosis was markedly attenuated in C6 deficient (C6−/−) rats or animal treated with a C5 inhibitor (20). In another set of experiments, a recombinant single-chain fragment variable recognizing domain 1 of β2GPI induced thrombosis in wild type male Wistar rats primed with lipopolysaccharide and pregnancy loss in female mice, but these effects were blocked in C6 deficient rats or C5 depleted mice (21). A CH2 deleted version of this antibody still recognized β2GPI but failed to fix complement and did not induce thrombosis or pregnancy loss. In addition to demonstrating the critical role of complement in aPL induced thrombosis, these experiments show that unlike effects of anti-β2GPI on the placenta, the procoagulant effects of aPL require a priming factor or “second hit” provided by an inflammatory stimulus such as lipopolysaccharide (34). In these murine models of thrombotic APS, C9 is deposited on the vascular endothelium indicating the presence of the membrane attack complex (20, 21). The membrane attack complex triggers the extrinsic pathway of coagulation by inducing tissue factor expression on the endothelial surface (47).
Complement Activation in Patients With APS
A role for complement activation in patients with thrombotic APS was first suggested nearly 25 years ago by the demonstration of higher serum levels of C5b-9 in patients with aPL and stroke compared with non-APS related stroke (48). Others have reported hypocomplementemia (39) and higher levels of complement fragment Bb and C3a (40, 49) in patients with APS; however, the association with APS-related thrombotic events or serologic characteristics is inconsistent (39, 49). More recently, deposits of C1q, C4, C3, and C5b-9 were noted to co-localize with β2GPI and IgG in the affected artery wall of a patient with primary APS and arterial thrombosis who also had increased plasma levels of C5a and C5b-9 (50).
A minority (1%) of patients with aPL develop catastrophic APS (CAPS), manifesting as small vessel thrombosis in three or more organs within the span of a week in the absence of small vessel inflammation on histopathologic examination (51); CAPS is fatal in over 40% of cases (52). Increased serum C5b-9 has been detected in CAPS with clinical improvement after treatment with eculizumab correlating with a reduction in serum C5b-9 and increase in C3 and C4 (53). While animal studies and anecdotal success of complement inhibitory therapy support a role for complement in aPL induced thrombosis (Table 1) (50, 54, 61), there is a paucity of controlled clinical research studies, and the mechanisms of complement activation in APS and its correlation with vascular events remains incompletely understood.
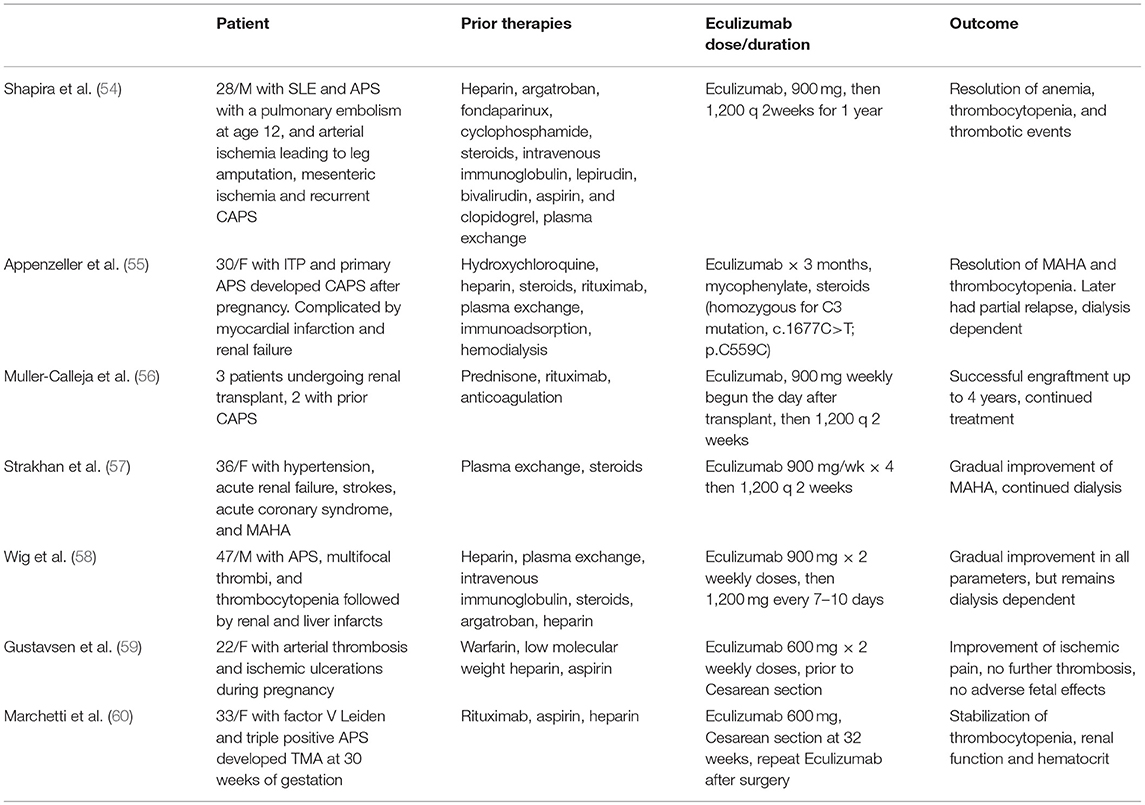
Table 1. Reports of Eculizumab therapy for patients with catastrophic APS or severe APS.
Mechanisms of Complement Activation in APS
The aPL profile can predict risk of thrombosis and pregnancy morbidity. For example, LA was associated with a higher risk of thrombosis (OR 3.6, 95% CI 1.2–10.9) than anti-β2GPI (OR 2.4, 95%CI 1.3–4.2) and anti-prothrombin (anti-PT) antibodies (OR 1.4, 95% CI 1.0–2.1) in the Leiden thrombophilia study (62). Retrospective and prospective studies have not shown a consistent association between thrombosis and aCL (3, 63). However, the differential ability of different aPL to activate complement has not been studied extensively. Since β2GPI is the primary antigen in APS, the anti-β2GPI antibody has been proposed as the more clinically significant and predictive aPL (62, 64, 65). Available studies highlight the role of β2GPI as a complement regulator (66), though others suggest a role of anti-C1q antibodies (67).
The mechanisms of complement activation in APS are not fully understood. Some have suggested that immune complexes in APS bind to C1q and activate the classical complement pathway (68). However, aCL and anti-β2GPI are frequently of the IgG2 subclass (69, 70), which has relatively weak ability to activate complement compared with IgG1. Anti-C1q antibodies have been detected in patients with SLE, in whom they correlate with clinical manifestations particularly lupus nephritis (71, 72). In a mouse model, an anti-C1q monoclonal antibody enhanced complement activation by the classical pathway and caused renal injury (67). Oku et al. (73) have reported that antibodies against C1q were more prevalent in patients with primary APS (36%) than controls with other non-SLE autoimmune disorders, and concluded that these antibodies contribute to complement activation, since titers of anti-C1q correlated with levels of C4a. However, anti-C1q antibodies did not correlate with thrombosis or pregnancy loss although a small subset of patients with recurrent thrombosis had a higher rate of anti-C1q antibodies (73). Finally, there is essentially no data on activation of complement by IgM anti-β2GPI antibodies.
Gropp et al. have reported a complement regulatory role of β2GPI, which inhibits complement activation by enhancing C3/C3b degradation (66). It has been proposed that when bound to a surface, β2GPI undergoes a conformational change from a circular form to an elongated form that can bind C3; in turn, C3 undergoes a conformational change to expose binding sites that make it susceptible to degradation by complement factor H (CFH) and factor I (66). In addition to its inhibitory effects on complement activation, CFH also has structural similarity to β2GPI and appears to share its property of inhibiting of contact pathway activation triggered by anionic phospholipids (74). Some APS patients, particularly those with recurrent thrombotic events, have autoantibodies against CFH suggesting a role of these antibodies in the predisposition to thrombosis (75, 76). A recent study reported low levels of CFH in patients with primary APS who also had low C3 suggesting complement activation (77).
Complement and Vascular Thrombosis
Though the mechanisms of complement activation in APS are unclear, several mechanisms by which activated complement may contribute to thrombotic events have been suggested. Activation of complement leads to cleavage of C5, generating C5a and C5b (leading to membrane attack complex formation). Ritis et al. demonstrated that aPL-induced complement activation may lead to neutrophil expression of TF mediated through the C5a receptor, leading to expression of procoagulant activity (Figure 2) (26). C5a also induces TF expression on monocytes and endothelial cells (78, 79). In addition, deposition of C5b-9 on the endothelial surface leads to secretion of high molecular weight multimers of von Willebrand factor (80), expression of P selectin (81), and plasma membrane vesiculation that exposes a catalytic surface for the prothrombinase complex (82). Interestingly, a recent experiment by Müller-Calleja et al. indicated that a cofactor (β2GPI) -independent aPL induced exposure of procoagulant phosphatidylserine and activated TF on monocytes and induced thrombosis, and that C3 but not C5 was required (56). Complement activation can also contribute to depressed fibrinolysis, a recognized thrombogenic mechanism in APS (83–85). Events occurring during thrombosis and fibrinolysis can also activate complement leading to the generation of C5a and a functional membrane attack complex (86, 87), further amplifying activation of coagulation and thrombosis.
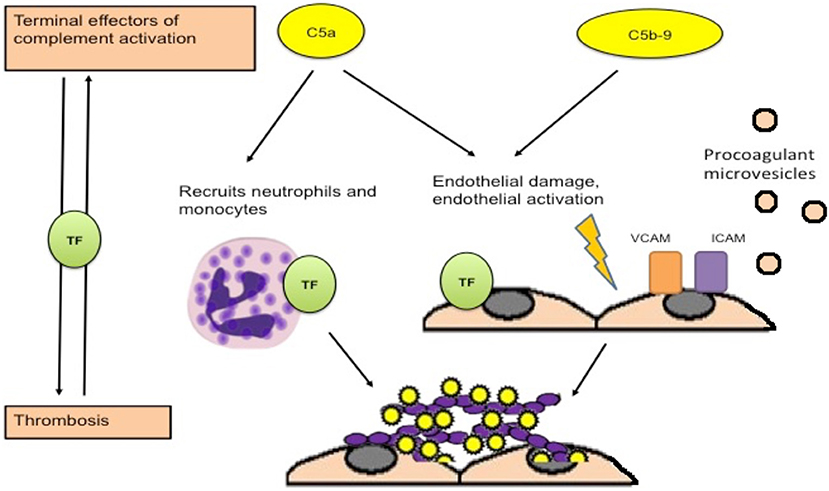
Figure 2. Procoagulant effects of complement activation. Activation of complement leads to generation of C5a and C5b, which combines with other terminal complement components to form the membrane attack complex. C5a is an anaphylatoxin that recruits neutrophils and leads to expression of tissue factor on neutrophils, monocytes and endothelial cells, which is associated with procoagulant activity. Deposition of the membrane attack complex on the endothelium leads to endothelial injury and procoagulant changes including expression of adhesion molecules, secretion of von Willebrand factor, and release of procoagulant microvesicles [adapted from Ritis et al. (26)].
Therapeutic Implications
Though long-term anticoagulation with vitamin K antagonists and a combination of aspirin with low molecular weight heparin are the mainstay of therapy for thrombotic and obstetric APS, respectively, some patients develop recurrent aPL-related clinical events despite “adequate” therapies, indicating a need for other treatments (52). The expanding data supporting a role of complement in aPL associated complications makes complement inhibition an attractive clinical target in APS. Moreover, complement targeted therapeutics are an active area of investigation, and the terminal complement inhibitor, eculizumab, is already widely used for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Although no complement inhibitors are approved for use in APS, eculizumab has been used successfully in patients with CAPS (54, 57, 61), to prevent recurrent CAPS in patients undergoing renal transplantation (88), and to prevent re-thrombosis in a patient with APS and recurrent arterial thrombosis (50). Recent reports of its successful use in CAPS (Table 1) are particularly encouraging since mortality in CAPS is as high as 40% with current treatment modalities (89). Pregnancy is considered a high-risk period for patients with aPL, with a high rate of fetal loss and pregnancy complications, and may serve as the “second hit” that leads to vascular complications. Preeclampsia and HELLP syndrome are also more common in women with aPL (55, 60), and may be complement mediated (90–92). Eculizumab crosses the placenta only minimally and does not affect the fetus (93). Hence, complement blockade may be an effective therapeutic modality for severe aPL related complications, including CAPS, during pregnancy (59, 94, 95).
Despite these encouraging data, there is still insufficient data to support the routine use of anti-complement therapy in APS, particularly patients without CAPS, and further mechanistic studies and randomized clinical trials are required. Ideally, complement-related biomarkers would be able to identify patients who are more likely to be refractory to standard therapy, and those who would benefit from complement inhibition as an adjunct to anticoagulation and antiplatelet therapy. However, standard measures of circulating complement cleavage products have not yet been shown to correlate with or predict the development of thrombosis.
Conclusions
Recent experimental data indicate that complement activation plays a critical role in the pathogenesis of thrombosis and pregnancy complications in APS (25). However, the mechanisms by which aPL activate complement are not fully understood. Complement inhibition may provide a useful adjunctive therapy for patients with APS refractory to standard therapies, which is supported by reports of successful use of complement inhibition in patients with CAPS (54, 61). Mechanistic and clinical studies are needed to evaluate the efficacy of complement inhibition in APS and to develop biomarkers that can identify patients who might benefit from complement inhibition.
Author Contributions
SC wrote the primary manuscript with the assistance and editing by KM and RB.
Funding
This work was supported by HL123098 from the US National Heart Lung and Blood Institute (to KM).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. (2006) 4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x
2. Bevers EM, Galli M, Barbui T, Comfurius P, Zwaal RF. Lupus anticoagulant IgG's (LA) are not directed to phospholipids only, but to a complex of lipid-bound human prothrombin. Thromb Haemost. (1991) 66:629–32. doi: 10.1055/s-0038-1646476
3. Galli M, Borrelli G, Jacobsen EM, Marfisi RM, Finazzi G, Marchioli R, et al. Clinical significance of different antiphospholipid antibodies in the WAPS (warfarin in the antiphospholipid syndrome) study. Blood. (2007) 110:1178–83. doi: 10.1182/blood-2007-01-066043
4. McNeil HP, Simpson RJ, Chesterman CN, Krilis SA. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: beta 2-glycoprotein I (apolipoprotein H). Proc Natl Acad Sci USA. (1990) 87:4120–4. doi: 10.1073/pnas.87.11.4120
5. Arad A, Proulle V, Furie RA, Furie BC, Furie B. Beta(2)-Glycoprotein-1 autoantibodies from patients with antiphospholipid syndrome are sufficient to potentiate arterial thrombus formation in a mouse model. Blood. (2011) 117:3453–9. doi: 10.1182/blood-2010-08-300715
6. Bertolaccini ML, Sciascia S, Murru V, Garcia-Fernandez C, Sanna G, Khamashta MA. Prevalence of antibodies to prothrombin in solid phase (aPT) and to phosphatidylserine-prothrombin complex (aPS/PT) in patients with and without lupus anticoagulant. Thromb Haemost. (2013) 109:207–13. doi: 10.1160/TH12-07-0527
7. Bancsi LF, van der Linden IK, Bertina RM. Beta 2-glycoprotein I deficiency and the risk of thrombosis. Thromb Haemost. (1992) 67:649–53. doi: 10.1055/s-0038-1648517
8. Hoeg JM, Segal P, Gregg RE, Chang YS, Lindgren FT, Adamson GL, et al. Characterization of plasma lipids and lipoproteins in patients with beta 2-glycoprotein I (apolipoprotein H) deficiency. Atherosclerosis. (1985) 55:25–34. doi: 10.1016/0021-9150(85)90163-7
9. Marciniak E, Romond EH. Impaired catalytic function of activated protein C: a new in vitro manifestation of lupus anticoagulant. Blood. (1989) 74:2426–32.
10. Urbanus RT, de Laat B. Antiphospholipid antibodies and the protein C pathway. Lupus. (2010) 19:394–9. doi: 10.1177/0961203309360841
11. Liestøl S, Sandset PM, Jacobsen EM, Mowinckel MC, Wisløff F. Decreased anticoagulant response to tissue factor pathway inhibitor type 1 in plasmas from patients with lupus anticoagulants. Br J Haematol. (2007) 136:131–7. doi: 10.1111/j.1365-2141.2006.06385.x
12. Bu C, Gao L, Xie W, Zhang J, He Y, Cai G, et al. Beta2-glycoprotein i is a cofactor for tissue plasminogen activator-mediated plasminogen activation. Arthritis Rheum. (2009) 60:559–68. doi: 10.1002/art.24262
13. Zhang J, McCrae KR. Annexin A2 mediates endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood. (2005) 105:1964–9. doi: 10.1182/blood-2004-05-1708
14. Shi T, Giannakopoulos B, Yan X, Yu P, Berndt MC, Andrews RK, et al. Anti-beta2-glycoprotein I antibodies in complex with beta2-glycoprotein I can activate platelets in a dysregulated manner via glycoprotein Ib-IX-V. Arthritis Rheum. (2006) 54:2558–67. doi: 10.1002/art.21968
15. Sorice M, Longo A, Capozzi A, Garofalo T, Misasi R, Alessandri C, et al. Anti-beta2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis Rheum. (2007) 56:2687–97. doi: 10.1002/art.22802
16. Chaturvedi S, Alluri R, McCrae KR. Extracellular vesicles in the antiphospholipid syndrome. Semin Thromb Hemost. (2018) 44:493–504. doi: 10.1055/s-0037-1599081
17. Rand JH, Wu XX, Guller S, Gil J, Guha A, Scher J, et al. Reduction of annexin-V (placental anticoagulant protein-I) on placental villi of women with antiphospholipid antibodies and recurrent spontaneous abortion. Am J Obstet Gynecol. (1994) 171:1566–72. doi: 10.1016/0002-9378(94)90403-0
18. Pierangeli SS, Girardi G, Vega-Ostertag M, Liu X, Espinola RG, Salmon J. Requirement of activation of complement C3 and C5 for antiphospholipid antibody-mediated thrombophilia. Arthritis Rheum. (2005) 52:2120–24. doi: 10.1002/art.21157
19. Carrera-Marín A, Romay-Penabad Z, Papalardo E, Reyes-Maldonado E, García-Latorre E, Vargas G, et al. C6 knock-out mice are protected from thrombophilia mediated by antiphospholipid antibodies. Lupus. (2012) 21:1497–505. doi: 10.1177/0961203312458839
20. Fischetti F, Durigutto P, Pellis V, Debeus A, Macor P, Bulla R, et al. Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood. (2005) 106:2340–6. doi: 10.1182/blood-2005-03-1319
21. Agostinis C, Durigutto P, Sblattero D, Borghi MO, Grossi C, Guida F, et al. A non-complement-fixing antibody to beta2 glycoprotein I as a novel therapy for antiphospholipid syndrome. Blood. (2014) 123:3478–87. doi: 10.1182/blood-2013-11-537704
22. Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest. (2003) 112:1644–54. doi: 10.1172/JCI200318817
23. Girardi G, Redecha P, Salmon JE. Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat Med. (2004) 10:1222–6. doi: 10.1038/nm1121
24. Redecha P, Tilley R, Tencati M, Salmon JE, Kirchhofer D, Mackman N, et al. Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood. (2007) 110:2423–31. doi: 10.1182/blood-2007-01-070631
25. Tedesco F, Borghi MO, Gerosa M, Chighizola CB, Macor P, Lonati PA, et al. Pathogenic role of complement in antiphospholipid syndrome and therapeutic implications. Front Immunol. (2018) 9:1388. doi: 10.3389/fimmu.2018.01388
26. Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. (2006) 177:4794–802. doi: 10.4049/jimmunol.177.7.4794
27. Foley JH, Conway EM. Cross talk pathways between coagulation and inflammation. Circ Res. (2016) 118:1392–408. doi: 10.1161/CIRCRESAHA.116.306853
28. Facciabene A, De Sanctis F, Pierini S, Reis ES, Balint K, Facciponte J, et al. Local endothelial complement activation reverses endothelial quiescence, enabling t-cell homing, and tumor control during t-cell immunotherapy. Oncoimmunology. (2017) 6:e1326442. doi: 10.1080/2162402X.2017.1326442
29. Riedl M, Noone DG, Khan MA, Pluthero FG, Kahr WHA, Palaniyar N, et al. Complement activation induces neutrophil adhesion and neutrophil-platelet aggregate formation on vascular endothelial cells. Kidney Int Rep. (2017) 2:66–75. doi: 10.1016/j.ekir.2016.08.015
30. Noone DG, Riedl M, Pluthero FG, Bowman ML, Liszewski MK, Lu L, et al. Von Willebrand factor regulates complement on endothelial cells. Kidney Int. (2016) 90:123–34. doi: 10.1016/j.kint.2016.03.023
31. Subramaniam S, Jurk K, Hobohm L, Jäckel S, Saffarzadeh M, Schwierczek K, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood. (2017) 129:2291–302. doi: 10.1182/blood-2016-11-749879
32. Redecha P, Franzke CW, Ruf W, Mackman N, Girardi G. Neutrophil activation by the tissue factor/Factor VIIa/PAR2 axis mediates fetal death in a mouse model of antiphospholipid syndrome. J Clin Invest. (2008) 118:3453–61. doi: 10.1172/JCI36089
33. Branch DW, Dudley DJ, Mitchell MD, Creighton KA, Abbott TM, Hammond EH, et al. Immunoglobulin G fractions from patients with antiphospholipid antibodies cause fetal death in BALB/c mice: a model for autoimmune fetal loss. Am J Obstet Gynecol. (1990) 163(1 Pt. 1):210–6. doi: 10.1016/S0002-9378(11)90700-5
34. Agostinis C, Biffi S, Garrovo C, Durigutto P, Lorenzon A, Bek A, et al. In vivo distribution of beta2 glycoprotein I under various pathophysiologic conditions. Blood. (2011) 118:4231–8. doi: 10.1182/blood-2011-01-333617
35. Di Simone N, Meroni PL, de Papa N, Raschi E, Caliandro D, De Carolis CS, et al. Antiphospholipid antibodies affect trophoblast gonadotropin secretion and invasiveness by binding directly and through adhered beta2-glycoprotein I. Arthritis Rheum. (2000) 43:140–50. doi: 10.1002/1529-0131(200001)43:1<140::AID-ANR18>3.0.CO;2-P
36. Holers VM, Girardi G, Mo L, Guthridge JM, Molina H, Pierangeli SS, et al. Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J Exp Med. (2002) 195:211–20. doi: 10.1084/jem.200116116
37. Thurman JM, Kraus DM, Girardi G, Hourcade D, Kang HJ, Royer PA, et al. A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody-induced pregnancy loss in mice. Mol Immunol. (2005) 42:87–97. doi: 10.1016/j.molimm.2004.07.043
38. Hazeltine M, Rauch J, Danoff D, Esdaile JM, Tannenbaum H. Antiphospholipid antibodies in systemic lupus erythematosus: evidence of an association with positive Coombs' and hypocomplementemia. J Rheumatol. (1988) 15:80–6.
39. Oku K, Atsumi T, Bohgaki M, Amengual O, Kataoka H, Horita T, et al. Complement activation in patients with primary antiphospholipid syndrome. Ann Rheum Dis. (2009) 68:1030–5. doi: 10.1136/ard.2008.090670
40. Breen KA, Seed P, Parmar K, Moore GW, Stuart-Smith SE, Hunt BJ. Complement activation in patients with isolated antiphospholipid antibodies or primary antiphospholipid syndrome. Thromb Haemost. (2012) 107:423–9. doi: 10.1160/TH11-08-0554
41. De Carolis S, Botta A, Santucci S, Salvi S, Moresi S, Di Pasquo E, et al. Complementemia and obstetric outcome in pregnancy with antiphospholipid syndrome. Lupus. (2012) 21:776–8. doi: 10.1177/0961203312444172
42. Reggia R, Ziglioli T, Andreoli L, Bellisai F, Iuliano A, Gerosa M, et al. Primary anti-phospholipid syndrome: any role for serum complement levels in predicting pregnancy complications? Rheumatology. (2012) 51:2186–90. doi: 10.1093/rheumatology/kes225
43. Kim MY, Guerra MM, Kaplowitz E, Laskin CA, Petri M, Branch DW, et al. Complement activation predicts adverse pregnancy outcome in patients with systemic lupus erythematosus and/or antiphospholipid antibodies. Ann Rheum Dis. (2018) 77:549–55. doi: 10.1136/annrheumdis-2017-212224
44. Cohen D, Buurma A, Goemaere NN, Girardi G, le Cessie S, Scherjon S, et al. Classical complement activation as a footprint for murine and human antiphospholipid antibody-induced fetal loss. J Pathol. (2011) 225:502–11. doi: 10.1002/path.2893
45. Shamonki JM, Salmon JE, Hyjek E, Baergen RN. Excessive complement activation is associated with placental injury in patients with antiphospholipid antibodies. Am J Obstet Gynecol. (2007) 196:167.e161–5. doi: 10.1016/j.ajog.2006.10.879
46. Pierangeli SS, Vega-Ostertag M, Liu X, Girardi G, Pierangeli SS, Vega-Ostertag M, et al. Complement activation: a novel pathogenic mechanism in the antiphospholipid syndrome. Ann N Y Acad Sci. (2005) 1051:413–20. doi: 10.1196/annals.1361.083
47. Saadi S, Holzknecht RA, Patte CP, Stern DM, Platt JL. Complement-mediated regulation of tissue factor activity in endothelium. J Exp Med. (1995) 182:1807–14. doi: 10.1084/jem.182.6.1807
48. Davis WD, Brey RL. Antiphospholipid antibodies and complement activation in patients with cerebral ischemia. Clin Exp Rheumatol. (1992) 10:455–60.
49. Devreese KM, Hoylaerts MF. Is there an association between complement activation and antiphospholipid antibody-related thrombosis? Thromb Haemost. (2010) 104:1279–81. doi: 10.1160/TH10-06-0410
50. Meroni PL, Macor P, Durigutto P, De Maso L, Gerosa M, Ferraresso M, et al. Complement activation in antiphospholipid syndrome and its inhibition to prevent rethrombosis after arterial surgery. Blood. (2016) 127:365–7. doi: 10.1182/blood-2015-09-672139
51. Asherson RA, Cervera R, de Groot PG, Erkan D, Boffa MC, Piette JC, et al. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. (2003) 12:530–4. doi: 10.1191/0961203303lu394oa
52. Cervera R, Serrano R, Pons-Estel GJ, Ceberio-Hualde L, Shoenfeld Y, de Ramón E, et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis. (2015) 74:1011–8. doi: 10.1136/annrheumdis-2013-204838
53. Barratt-Due A, Fløisand Y, Orrem HL, Kvam AK, Holme PA, Bergseth G, et al. Complement activation is a crucial pathogenic factor in catastrophic antiphospholipid syndrome. Rheumatology. (2016) 55:1337–9. doi: 10.1093/rheumatology/kew040
54. Shapira I, Andrade D, Allen SL, Salmon JE. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. (2012) 64:2719–23. doi: 10.1002/art.34440
55. Appenzeller S, Souza FH, Wagner Silva de Souza A, Shoenfeld Y, de Carvalho JF. HELLP syndrome and its relationship with antiphospholipid syndrome and antiphospholipid antibodies. Semin Arthritis Rheum. (2011) 41:517–23. doi: 10.1016/j.semarthrit.2011.05.007
56. Muller-Calleja N, Ritter S, Hollerbach A, Falter T, Lackner KJ, Ruf W. Complement C5 but not C3 is expendable for tissue factor activation by cofactor-independent antiphospholipid antibodies. Blood Adv. (2018) 2:979–86. doi: 10.1182/bloodadvances.2018017095
57. Strakhan M, Hurtado-Sbordoni M, Galeas N, Bakirhan K, Alexis K, Elrafei T. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. (2014) 2014:704371. doi: 10.1155/2014/704371
58. Wig S, Chan M, Thachil J, Bruce I, Barnes T. A case of relapsing and refractory catastrophic anti-phospholipid syndrome successfully managed with eculizumab, a complement 5 inhibitor. Rheumatology. (2016) 55:382–4. doi: 10.1093/rheumatology/kev371
59. Gustavsen A, Skattum L, Bergseth G, Lorentzen B, Floisand Y, Bosnes V, et al. Effect on mother and child of eculizumab given before caesarean section in a patient with severe antiphospholipid syndrome: a case report. Medicine. (2017) 96:e6338. doi: 10.1097/MD.0000000000006338
60. Marchetti T, de Moerloose P, Gris JC. Antiphospholipid antibodies and the risk of severe and non-severe pre-eclampsia: the NOHA case-control study. J Thromb Haemost. (2016) 14:675–84. doi: 10.1111/jth.13257
61. Zikos TA, Sokolove J, Ahuja N, Berube C. Eculizumab induces sustained remission in a patient with refractory primary catastrophic antiphospholipid syndrome. J Clin Rheumatol. (2015) 21:311–3. doi: 10.1097/RHU.0000000000000290
62. de Groot PG, Lutters B, Derksen RH, Lisman T, Meijers JC, Rosendaal FR. Lupus anticoagulants and the risk of a first episode of deep venous thrombosis. J Thromb Haemost. (2005) 3:1993–7. doi: 10.1111/j.1538-7836.2005.01485.x
63. Urbanus RT, Siegerink B, Roest M, Rosendaal FR, de Groot PG, Algra A. Antiphospholipid antibodies and risk of myocardial infarction and ischaemic stroke in young women in the RATIO study: a case-control study. Lancet Neurol. (2009) 8:998–1005. doi: 10.1016/S1474-4422(09)70239-X
64. Urbanus RT, de Groot PG. Antiphospholipid antibodies–we are not quite there yet. Blood Rev. (2011) 25:97–106. doi: 10.1016/j.blre.2010.12.001
65. Devreese K, Hoylaerts MF. Challenges in the diagnosis of the antiphospholipid syndrome. Clin Chem. (2010) 56:930–40. doi: 10.1373/clinchem.2009.133678
66. Gropp K, Weber N, Reuter M, Micklisch S, Kopka I, Hallström T, et al. Beta(2)-glycoprotein I, the major target in antiphospholipid syndrome, is a special human complement regulator. Blood. (2011) 118:2774–83. doi: 10.1182/blood-2011-02-339564
67. Trouw LA, Groeneveld TW, Seelen MA, Duijs JM, Bajema IM, Prins FA, et al. Anti-C1q autoantibodies deposit in glomeruli but are only pathogenic in combination with glomerular C1q-containing immune complexes. J Clin Invest. (2004) 114:679–88. doi: 10.1172/JCI200421075
68. Arfors L, Lefvert AK. Enrichment of antibodies against phospholipids in circulating immune complexes (CIC) in the anti-phospholipid syndrome (APLS). Clin Exp Immunol. (1997) 108:47–51. doi: 10.1046/j.1365-2249.1997.d01-973.x
69. Sammaritano LR, Ng S, Sobel R, Lo SK, Simantov R, Furie R, et al. Anticardiolipin IgG subclasses: association of IgG2 with arterial and/or venous thrombosis. Arthritis Rheum. (1997) 40:1998–2006. doi: 10.1002/art.1780401112
70. Samarkos M, Davies KA, Gordon C, Walport MJ, Loizou S. IgG subclass distribution of antibodies against beta(2)-GP1 and cardiolipin in patients with systemic lupus erythematosus and primary antiphospholipid syndrome, and their clinical associations. Rheumatology. (2001) 40:1026–32. doi: 10.1093/rheumatology/40.9.1026
71. Akhter E, Burlingame RW, Seaman AL, Magder L, Petri M. Anti-C1q antibodies have higher correlation with flares of lupus nephritis than other serum markers. Lupus. (2011) 20:1267–74. doi: 10.1177/0961203311411597
72. Chen Z, Wang GS, Wang GH, Li XP. Anti-C1q antibody is a valuable biological marker for prediction of renal pathological characteristics in lupus nephritis. Clin Rheumatol. (2012) 31:1323–9. doi: 10.1007/s10067-012-2017-1
73. Oku K, Amengual O, Hisada R, Ohmura K, Nakagawa I, Watanabe T, et al. Autoantibodies against a complement component 1 q subcomponent contribute to complement activation and recurrent thrombosis/pregnancy morbidity in anti-phospholipid syndrome. Rheumatology. (2016) 55:1403–11. doi: 10.1093/rheumatology/kew196
74. Ferluga J, Kishore U, Sim RB. A potential anti-coagulant role of complement factor H. Mol Immunol. (2014) 59:188–93. doi: 10.1016/j.molimm.2014.02.012
75. Foltyn Zadura A, Memon AA, Stojanovich L, Perricone C, Conti F, Valesini G, et al. Factor H autoantibodies in patients with antiphospholipid syndrome and thrombosis. J Rheumatol. (2015) 42:1786–93. doi: 10.3899/jrheum.150185
76. Foltyn Zadura A, Zipfel PF, Bokarewa MI, Sturfelt G, Jönsen A, Nilsson SC, et al. Factor H autoantibodies and deletion of Complement Factor H-Related protein-1 in rheumatic diseases in comparison to atypical hemolytic uremic syndrome. Arthritis Res Ther. (2012) 14:R185. doi: 10.1186/ar4016
77. Nakamura H, Oku K, Ogata Y, Ohmura K, Yoshida Y, Kitano E, et al. Alternative pathway activation due to low level of complement factor H in primary antiphospholipid syndrome. Thromb Res. (2018) 164:63–8. doi: 10.1016/j.thromres.2018.02.142
78. Ikeda K, Nagasawa K, Horiuchi T, Tsuru T, Nishizaka H, Niho Y. C5a induces tissue factor activity on endothelial cells. Thromb Haemost. (1997) 77:394–8. doi: 10.1055/s-0038-1655974
79. Wolberg AS, Roubey RA. Mechanisms of autoantibody-induced monocyte tissue factor expression. Thromb Res. (2004) 114:391–6. doi: 10.1016/j.thromres.2004.06.012
80. Hattori R, Hamilton KK, McEver RP, Sims PJ. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J Biol Chem. (1989) 264:9053–60.
81. Foreman KE, Vaporciyan AA, Bonish BK, Jones ML, Johnson KJ, Glovsky MM, et al. C5a-induced expression of P-selectin in endothelial cells. J Clin Invest. (1994) 94:1147–55. doi: 10.1172/JCI117430
82. Hamilton KK, Hattori R, Esmon CT, Sims PJ. Complement proteins C5b-9 induce vesiculation of the endothelial plasma membrane and expose catalytic surface for assembly of the prothrombinase enzyme complex. J Biol Chem. (1990) 265:3809–14.
83. Vikerfors A, Svenungsson E, Ågren A, Mobarrez F, Bremme K, Holmström M, et al. Studies of fibrin formation and fibrinolytic function in patients with the antiphospholipid syndrome. Thromb Res. (2014) 133:936–44. doi: 10.1016/j.thromres.2014.02.023
84. Krone KA, Allen KL, McCrae KR. Impaired fibrinolysis in the antiphospholipid syndrome. Curr Rheumatol Rep. (2010) 12:53–7. doi: 10.1007/s11926-009-0075-4
85. Grosso G, Vikerfors A, Woodhams B, Adam M, Bremme K, Holmström M, et al. Thrombin activatable fibrinolysis inhibitor (TAFI) - A possible link between coagulation and complement activation in the antiphospholipid syndrome (APS). Thromb Res. (2017) 158:168–73. doi: 10.1016/j.thromres.2017.06.028
86. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. (2006) 12:682–7. doi: 10.1038/nm1419
87. Foley JH, Walton BL, Aleman MM, O'Byrne AM, Lei V, Harrasser M, et al. Complement activation in arterial and venous thrombosis is mediated by plasmin. EBioMed. (2016) 5:175–82. doi: 10.1016/j.ebiom.2016.02.011
88. Lonze BE, Zachary AA, Magro CM, Desai NM, Orandi BJ, Dagher NN, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. (2014) 14:459–65. doi: 10.1111/ajt.12540
89. Cervera R, Bucciarelli S, Plasín MA, Gómez-Puerta JA, Plaza J, Pons-Estel G, et al. Catastrophic antiphospholipid syndrome (CAPS): descriptive analysis of a series of 280 patients from the “CAPS Registry”. J Autoimmun. (2009) 32:240–5. doi: 10.1016/j.jaut.2009.02.008
90. Burwick RM, Feinberg BB. Eculizumab for the treatment of preeclampsia/HELLP syndrome. Placenta. (2013) 34:201–3. doi: 10.1016/j.placenta.2012.11.014
91. Burwick RM, Fichorova RN, Dawood HY, Yamamoto HS, Feinberg BB. Urinary excretion of C5b-9 in severe preeclampsia: tipping the balance of complement activation in pregnancy. Hypertension. (2013) 62:1040–5. doi: 10.1161/HYPERTENSIONAHA.113.01420
92. Vaught AJ, Braunstein EM, Jasem J, Yuan X, Makhlin I, Eloundou S, et al. Germline mutations in the alternative pathway of complement predispose to HELLP syndrome. JCI Insight. (2018) 3:99128. doi: 10.1172/jci.insight.99128
93. Hallstensen RF, Bergseth G, Foss S, Jæger S, Gedde-Dahl T, Holt J, et al. Eculizumab treatment during pregnancy does not affect the complement system activity of the newborn. Immunobiology. (2015) 220:452–9. doi: 10.1016/j.imbio.2014.11.003
94. Kronbichler A, Frank R, Kirschfink M, Szilágyi Á, Csuka D, Prohászka Z, et al. Efficacy of eculizumab in apatient with immunoadsorption-dependent catastrophic antiphospholipid syndrome: a case report. Medicine. (2014) 93:e143. doi: 10.1097/MD.0000000000000143
Keywords: antiphosholipid antibodies, complement, thrombosis, endothelial, beta2 - glycoprotein I
Citation: Chaturvedi S, Brodsky RA and McCrae KR (2019) Complement in the Pathophysiology of the Antiphospholipid Syndrome. Front. Immunol. 10:449. doi: 10.3389/fimmu.2019.00449
Received: 25 November 2018; Accepted: 19 February 2019;
Published: 14 March 2019.
Edited by:
Ricard Cervera, Hospital Clínic de Barcelona, SpainCopyright © 2019 Chaturvedi, Brodsky and McCrae. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keith R. McCrae, mccraek@ccf.org