 Lærke J. B. Brandt
Lærke J. B. Brandt Mike B. Barnkob
Mike B. Barnkob Yale S. Michaels
Yale S. Michaels Julia Heiselberg
Julia Heiselberg Torben Barington
Torben Barington- 1Department of Clinical Immunology, Odense University Hospital, University of Southern Denmark, Odense, Denmark
- 2School of Biomedical Engineering, University of British Columbia, Vancouver, BC, Canada
Chimeric antigen receptor (CAR) T cells have emerged as a promising treatment for patients with advanced B-cell cancers. However, widespread application of the therapy is currently limited by potentially life-threatening toxicities due to a lack of control of the highly potent transfused cells. Researchers have therefore developed several regulatory mechanisms in order to control CAR T cells in vivo. Clinical adoption of these control systems will depend on several factors, including the need for temporal and spatial control, the immunogenicity of the requisite components as well as whether the system allows reversible control or induces permanent elimination. Here we describe currently available and emerging control methods and review their function, advantages, and limitations.
Introduction
Chimeric antigen receptor (CAR) T cells have emerged as a promising treatment for patients with advanced B-cell cancers (1–3) but more effective control of the therapy is needed to combat associated toxicity and to expand CAR therapy toward other cancer types. CAR T cells are a personalized immunotherapy, in which allogeneic or autologous T cells are genetically modified to express a synthetic construct, combining an extracellular binding domain, often an antibody-derived single chain variable fragment (scFv), with activating signaling domains from the T-cell-receptor complex, such as CD3ζ, CD28, and 4-1BB. Recognition of cell-surface proteins through the extracellular domain allows CAR T cells to target cancer cells for cytotoxic killing (4).
As a living drug, CAR T cells bear the potential for rapid and massive activation and proliferation, which contributes to their therapeutic efficacy but simultaneously underlies the side effects associated with CAR T-cell therapy. The most well-known toxicity is called cytokine release syndrome (CRS) which is a systemic inflammatory response characterized by fever, hypotension and hypoxia (5–7). CRS is triggered by the activation of CAR T cells and their subsequent production of pro-inflammatory cytokines including IFNγ, IL-6 and IL-2 (8). This is thought to result in additional activation of bystander immune and non-immune cells which further produce cytokines, including IL-10, IL-6, and IL-1 (9). The severity of CRS is associated with tumor burden, and ranges from a mild fever to life-threatening organ failure (10, 11). Neurologic toxicity is another serious adverse event which can occur alongside CRS (12). Although the pathomechanism is unknown, it is believed to be the result of cerebral endothelial dysfunction (13). Finally, since few antigens are truly tumor specific, toxicities can arise if CAR T cells target healthy cells expressing the recognized antigen i.e., on-target, off-tumor activity. Unfortunately, this has led to severe and fatal outcomes, especially when targeting antigens in solid tumors, hampering CAR T-cell application in these patients (14–17).
Current clinically approved CAR designs do not enable control over CAR T cells following infusion, and so management of toxicities depends on immuno-suppression using systemic corticosteroids as well as an IL-6 receptor antibody, tocilizumab. Unfortunately, the use of immunosuppressive drugs severely limits the time span CAR T cells are functional (11). Given the severity of the toxicities, as well as the manufacturing costs, there is a clinical need to regulate CAR T-cell numbers and activity once deployed in patients. In this mini review, we describe existing and emerging approaches to regulation and control of CAR T cells, and discuss each method's advantages and disadvantages.
Passive Control
Passive control methods provide straightforward opportunities to limit CAR T-cell mediated cytotoxicity, but offer no downstream control over engrafted cells following transfusion (Figure 1, left panel).
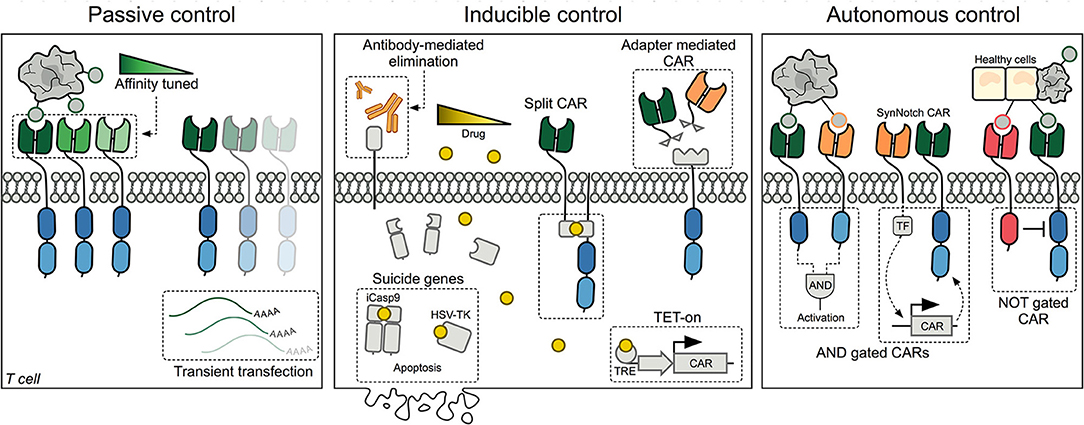
Figure 1. Schematic representation of the three major methods designed for controlling CAR T cells today. Left panel: Passive control methods include affinity tuned CARs and transient transfection of T cells. Middle panel: Inducible control includes methods to eliminate CAR T cells using antibodies or inducible suicide systems. Additionally, different drugs have been utilized to either control CAR expression at the transcriptional level or assembling of a split-CAR, where the extra- and intracellular domains have been separated. Another approach has been to decouple the binding domain from the intracellular signaling domain, such that binding adapters can be supplied and titrated. Right panel: Autonomous CAR T cells are self-regulated and can decide whether to initiate or withhold cytotoxic killing of target cells based on surface proteins expressed by healthy and cancerous cells. CAR, Chimeric Antigen Receptor; TRE, Tetracycline Response Element; TF, Transcription Factor; SynNotch, Synthetic Notch receptor.
Transient Transfection
A simple but effective way of regulating CAR T cells consists of transiently transfecting T cells with CAR-encoding mRNA (18–23). Due to the lack of genomic integration, CAR expression is limited by the degradation of the CAR-encoding mRNA and dilution following each T-cell division (18). The result is a steady decrease in CAR-expressing T-cell numbers, unless new cells are infused. Repeated infusions are however associated with a higher risk of an anaphylactic reaction due to the CAR T cells (24). While the inherently limited persistence of these CAR T cells might compromise continued anti-leukemic effect (25), it also limits long-term hematologic toxicities and off-target effects.
Affinity Tuning
Lowering the binding domain's affinity toward the targeted antigen aims to prevent on-target, off-tumor toxicities from arising in the first place (26, 27). While affinity-tuned CARs retain the ability to bind to cancer cells with a high antigen expression, healthy tissues with lower expression are spared (28). The use of low-affinity CARs is therefore especially interesting when targeting antigens known to be expressed on healthy tissue in low amounts, e.g., HER2 or EGFR (26, 27). This, however, might also lead to cancer cell escape variants with low antigen expression (29). Additionally, both promoter usage and transduction level of T cells might result in heterogeneous expression of the CAR protein, making it hard to ensure consistent behavior among individual CAR T cells as their avidity toward the antigen can vary. One promising strategy to overcome heterogeneous CAR expression is to instead integrate the CAR construct into the endogenous TCR alpha chain (TRAC) locus using the CRISPR/Cas9 system (30).
Inducible Control
Recognizing that CAR T-cell toxicities arise rapidly, researchers have developed several exogenous methods to quickly regulate the activity of the CAR T cells or to eliminate them completely. These methods rely on co-administration of a drug, thereby making their use dependent on pharmacokinetics, tissue availability, and potential adverse effects of the chosen drug (Figure 1, middle panel).
Suicide Genes
Depletion of CAR T cells can be achieved by designing CAR constructs that also express a suicide gene, such as inducible Caspase 9 (iCasp9) (31–39), herpes simplex virus tyrosine kinase (HSV-TK) (40, 41) or human thymidylate kinase (TMPK) (42). In cells expressing iCasp9 and TMPK, elimination is achieved through activation of the caspase 3 apoptotic pathway when a small molecule is administered. The iCasp9 system has successfully been validated in patients receiving haploidentical stem-cell transplants (HSCT) in which iCasp9-expressing T cells were rapidly removed at onset of graft-versus-host disease (GvHD) (31). Likewise, administration of ganciclovir to T cells co-expressing HSV-TK causes formation of a toxic metabolite but cell death may take up to several days as it depends on cell proliferation (40, 41). The use of HSV-TK is severely limited by the high immunogenicity of the virally-derived protein (43). Furthermore, the HSV-TK suicide system is complicated by the fact that ganciclovir is used as a first-line treatment against cytomegalovirus (CMV) infection, a virus which is often reactivated in HSCT and other immunocompromised patients (44). As TMPK and iCasp9 are of human origin, the risk of immunogenicity is low. Indeed long-term engraftment up to several years in patients infused with iCasp9 expressing cells has been reported (45).
Elimination Markers
Co-expression of a cell-surface elimination marker, not normally present on T cells, allows for antibody-mediated degradation and control of the CAR T cells (22, 46–52). By utilizing clinically approved antibodies, e.g., rituximab targeting CD20 (48–50) or cetuximab targeting EGFR (51, 52), complement- or antibody-dependent cytotoxicity (CDC/ADCC) can be achieved toward the CAR T cells (51). Choosing a marker that is co-expressed on cancer cells, allows this method to create additional tumor killing, with the caveat that further collateral toxicity might arise. Cell-surface markers also allow for positive selection of transduced T cells in the manufacturing process, and subsequent monitoring of CAR T-cell levels in vivo. However, the efficacy of the strategy can be compromised by the fact that CDC/ADCC capacity is limited in patients treated with chemotherapy prior to CAR T infusion (53). In addition, antibodies can have limited biodistribution and tissue penetration, especially in poorly vascularized tumors (54). In order to address these problems, researchers have instead created anti-idiotype CARs recognizing murine CD19-specific CARs (55) or incorporated a short peptide epitope, called an E-tag, into the extracellular domain of the CAR, and created anti-E-tag CARs which could then be used to eliminate the anti-tumor CARs (56).
Since the use of suicide genes and elimination markers result in irreversible depletion of this complex treatment, researchers have developed a number of reversible methods to control CAR T cells as well.
Systemic T-Cell Inhibition
Current methods for controlling CAR T cells include systemic immunosuppressive agents, e.g., corticosteroids (57). The lymphocytotoxic anti-CD52 antibody alemtuzumab, has also been proposed as a method of depleting CD4- and CD123-specific CAR T cells (47, 58), as targeting these proteins might cause hematological aplasia and toxicity. More recently, it was shown that CAR signaling could be inhibited using the tyrosine kinase inhibitor dasatinib. Dasatinib inhibits phosphorylation of lymphocyte-specific protein tyrosine kinase (LCK), a critical component in the T-cell signaling pathway. Preclinical studies suggest that treatment with dasatinib can reversibly inhibit CAR T-cell proliferation and cytokine production without negatively affecting viability (59, 60). Although dasatinib cannot adequately inhibit already activated CAR T cells, limiting its usage against acutely arising CRS or neurotoxicity, the drug was shown to be superior to dexamethasone in inhibiting further activation in a preclinical study (59). Finally, while corticosteroids and alemtuzumab cause widespread inhibition or complete elimination of both CAR T cells and healthy lymphocytes, dasatinib has the advantage of acting as a faster on/off switch, due to its short half-life of 4 h (61).
Adapter Mediated CARs
Aiming to specifically control CAR T-cell activity toward the antigen, several models of adapter-mediated CARs, also known as universal CARs, have been developed (62–71). A shared feature is their method of tumor recognition, which is achieved by linking an adaptor, a molecule recognized by the CAR, to an antibody or ligand that recognizes the tumor antigen. While current clinically approved CARs are designed to be constitutively active, adapter-dependent CAR T cells can only recognize and kill when the adapter is administered, allowing for titratable and reversible control of the CAR T cells. A major advantage of this approach is the ability to target different antigens without the need to re-engineer and re-transfuse T cells. Adapters have also been designed to redirect anti-CD19-specific CARs to another target, using CD19-fusion proteins, suggesting that adapter proteins might be used with current clinically approved CAR T cells (72). However, large differences in adapter kinetics and subsequent effects on CAR T cells have been reported, probably reflecting differences in models used, affinity differences between the adapter and both target and CAR T cells, and biodistribution of the adapter molecules.
Split-CARs
Instead of directly regulating CAR binding to antigen, pharmacological inducers can also be used to control the activity of CAR T cells themselves, by splitting the CAR's extracellular antigen-binding domain from its intracellular signaling domains (73, 74). Assembling of the fully functional CAR is therefore dependent on administration of a dimerizing drug, limiting the CAR activity by the half-life of the drug. Consequently, CAR T-cell activation requires two inputs: the tumor antigen and the dimerizing drug. The split-CAR can thus also be considered an AND gate CAR (see below). The split-CAR allows temporal and reversible control over the number of functional CARs, but the design does not prevent on-target, off-tumor toxicities as no spatial control is achieved due to a lack of control over the distribution of the drug.
Protease Inhibitors
Strategic incorporation of the autocleaving hepatitis-C-derived NS3 protease in the CAR construct has also been suggested as a way to control CAR T-cell activity. Juillerat et al. incorporated the NS3 protease between the CAR and a degradation moiety, thereby tagging the CAR construct for degradation when a NS3 protease inhibitor was administered (75). This construct showed reversible as well as tunable control over CAR T-cell cytotoxicity in vitro. As the NS3 protease is virally derived, it holds immunogenic potential which could potentially limit CAR T-cell persistence.
TET-On Regulation
It has also been suggested to regulate CARs at the transcriptional level. Several groups have shown that a drug-inducible CAR system can be generated by controlling CAR transcription using the TET-on system, allowing reversible control of CAR T cells (76–78). CAR mRNA is thus only produced in the presence of doxycycline, although some background CAR expression was observed (76). As the TET-on system is derived from both bacteria and virus, it holds significant immunogenic potential with the risk of host mediated elimination of the CAR T cells. Another drawback is the lack of rapid control, should life-threatening side effects occur, due to the control occurring on a transcriptional level. However, for highly proliferative CAR T cells that are constantly diluting the CAR protein amongst daughter cells, transcriptional regulation may be sufficient to limit the quantity of functional CAR complexes.
Logic Gates and Autonomous Control
As CAR T-cell therapy is applied against solid tumors, the distinction between healthy and malignant tissue becomes increasingly important (79). The use of so-called boolean logic gates and tumor selectivity mechanisms is envisioned to generate autonomous CARs with a higher target specificity, capable of better distinguishing tumor cells from healthy cells (Figure 1, right panel).
AND Gates
One approach to enable better decision-making in CARs is the incorporation of logic AND gates, such that a combination of antigens are required for activation. Often this dual CAR design consists of two extracellular domains, with specificities toward different antigens, each coupled to separate components of the intracellular stimulatory apparatus, e.g., CD3ζ and CD28 or 4-1BB (80–83). Such approaches have been tested in preclinical prostate and breast cancer models and might allow for targeting of proteins that are also present in healthy tissue (81, 82). A worry, however, is that even partial signaling through one receptor may generate sufficient T-cell activity to cause off-target damage (81, 83). Another approach has therefore been to use one receptor exclusively as a priming signal, with no activating signaling capacity itself. This was achieved using so-called SynNotch receptors, which were coupled to orthogonal transcription factors that were released upon binding. This “priming” leads to expression of a fully functional CAR targeting another cancer-associated antigen, ensuring localized activity of the CAR T cell (84, 85). Importantly, in mouse models the SynNotch CARs did not migrate, but retained their function only in dual positive tumors, indicating that this approach ensured good spatial control of the CAR (84, 85).
NOT Gates
Better discrimination between malignant and healthy cells can also be achieved by designing an inhibitory CAR (iCAR). The iCAR contains a binding domain specific for an antigen expressed on healthy cells fused to the signaling domains of CTLA-4 or PD-1, such that recognition of the healthy antigen leads to an inhibitory signaling cascade that overrides activating signals by dephosphorylating the receptor complex (86). The iCAR should restrict CAR T-cell activity to tumor tissue lacking the healthy antigen, limiting on-target, off tumor activity. Such a system might allow for CAR T cells previously shown to cause lethal off-tumor activity, such as ERBB2-specific designs, to be re-introduced.
While the AND gate and iCARs restrict CAR T-cells' activity spatially, they cannot control the intensity of the CAR T-cell activity nor control them in a temporal manner.
Tumor Localizing Mechanisms: Hypoxia Sensitivity and Masked CARs
In order to gain better temporal control and limit CAR T-cell activity to the tumor microenvironment (TME), CAR expression can be controlled by incorporating a hypoxia inducible factor (HIF) in the CAR construct (87). The CAR-HIF construct is continuously targeted for degradation when the CAR T cell is present in a normoxic environment, i.e., most healthy tissues, ensuring that CAR expression only occurs under hypoxic conditions, as seen within parts of the TME. Because degradation of the CAR is regulated at the protein level, control is thought to occur quickly, which is favorable to avoid CAR activity outside of the tumor. This method has only been tested in vitro, and may fail to eradicate tumor cells residing in normoxic tissues, e.g., the peripheral parts of the tumor. Moreover, these CAR T cells may show off-target effects in healthy, hypoxic tissues like the bone marrow.
Another method to constrict CAR activity to the TME is by designing a masked CAR, as proposed by Han et al. (88). Here the CARs antigen-binding site is hidden by a masking peptide with a linker sensitive to proteolytic cleavage. Tumor associated proteases present in the TME can then cleave the linker, removing the masking peptide and allowing CAR T cells to target antigen presenting cells. However, a possibility remains that endogenous proteases cleave the masking peptide, paving the way for on-target, off-tumor toxicities.
CAR T Cells Producing IL-1 Receptor Antagonist (IL-1Ra)
One of the central cytokines involved in CRS is IL-1. Giavridis et al. recently designed a CAR T cell constitutively producing IL-1 receptor antagonist, protecting mice against CRS associated mortality without affecting the anti-tumor efficacy (9). One major advantage of using IL-1 receptor antagonist is its ability to cross the blood-brain barrier, thereby potentially reducing CAR T-cell-related neurotoxicity (89).
While the above mentioned logic gates and hypoxia sensitive CARs seek to enhance tumor specificity, they do not allow the clinicians control over the CAR T cells and as such offer no solution should life-threatening toxicities arise.
Discussion
The recent years' success of CAR T cells in the clinic has revealed the serious and potentially lethal side effects associated with the potent treatment, including off-tumor, on-target effects, systemic inflammatory conditions such as CRS and acute neurotoxicity (90, 91). More recently it has become clear that cardiovascular and gastrointestinal events can also occur post-CAR-T (92, 93). As the range of specificities and tumor-types targeted using CAR T cells increase, new side-effects will likely come to light. It is therefore important to consider how to best regulate and control engineered T cells.
Because CAR-related toxicities often arise acutely, control mechanisms should ideally grant the clinician swift control over CAR T-cell activity. A direct comparison of the on/off kinetics for each method is made difficult by differences in study design, but regulation on the protein level as well as the use of suicide genes or elimination markers are expected to act faster than regulation at the transcriptional level (Table 1). Permanent elimination of CAR T cells however abrogates the long term anti-leukemic effect and many methods therefore aim at reversible control, allowing the clinician to turn off the CAR T cells when toxicities occur. In the future, an appropriately designed recombinase-mediated switch would allow CAR activity to be stably switched into an OFF state with one small molecule, and subsequently flipped back into an ON state using a second molecule (94). This would give clinicians the power to halt CAR T activity without permanently destroying a costly and life-saving therapeutic product, while avoiding the need to constantly administer the suppressive molecule to maintain an OFF state. Ideally, small molecules that have already gained regulatory approval and show minimal side effects can be co-opted to rapidly and reversibly modulate CAR T-cell activity. The choice of drug however must also be guided by the tumor-type targeted, as endothelial barriers, such as the blood-brain-barrier, and poor vascularization can prevent proper biodistribution and concentration in the effected organs.
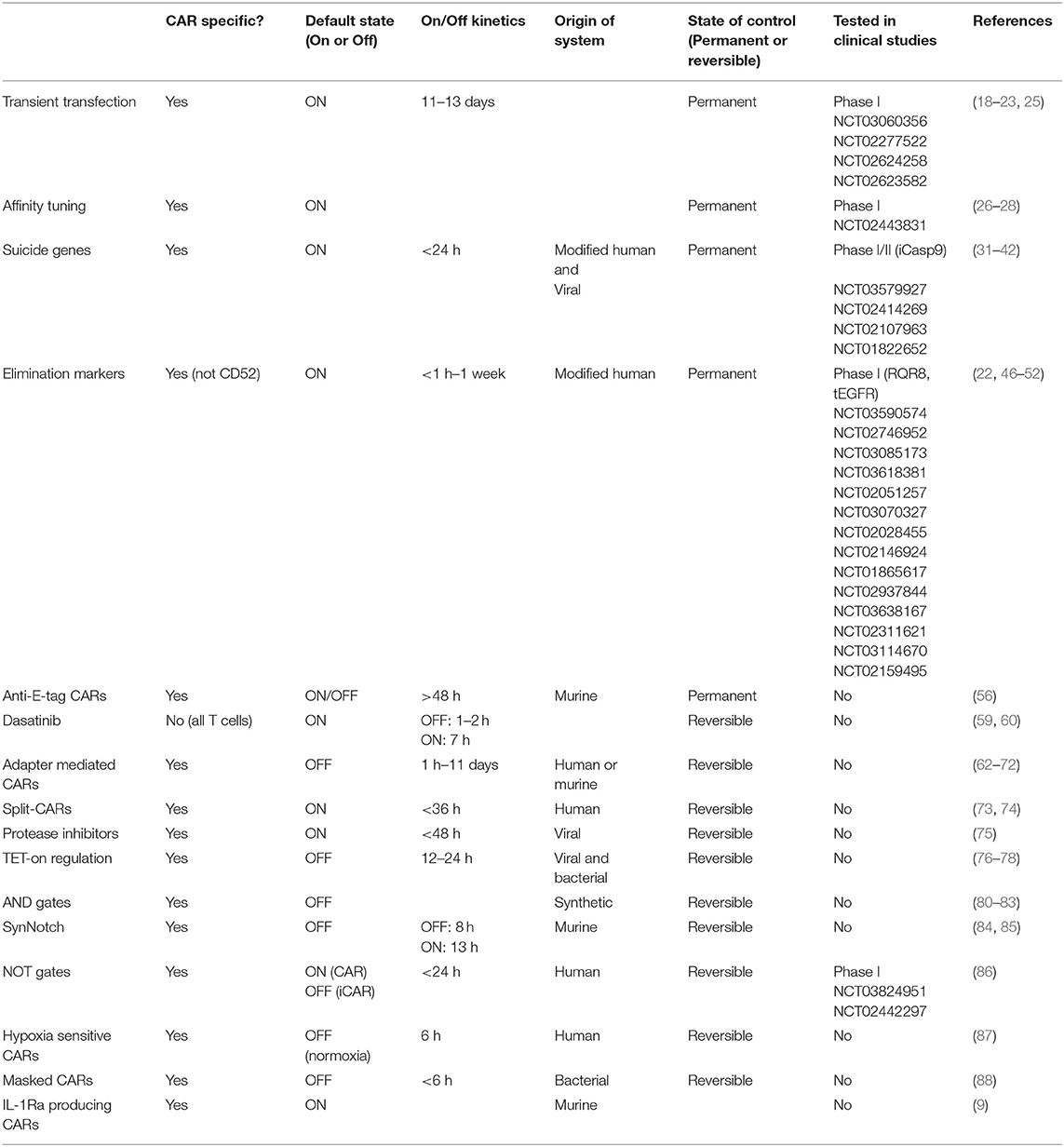
Table 1. Current approaches to regulation and control of CAR T cells.
Another approach is to avoid unwanted immune responses from arising at all. This is the rationale behind many of the logic-gated CAR designs developed, including iCARs and tumor-localized CARs (84, 87). Passive and sustained fine-tuning of CAR expression levels could also be achieved by targeted genomic integration (30) or by using degrons (95) or synthetic miRNA regulation (96). Not only does this have the potential to mitigate dangerous cytokine release, reducing CAR expression has also been shown to combat T-cell exhaustion (30).
The adaptation of CARs to recognize and respond to soluble ligands, such as secreted cytokines, were recently reported by Cheng et al. and creates exciting new possibilities in CAR engineering (97). CARs targeting immunosuppressive soluble ligands, such as TGF-beta, could possibly contribute in overcoming the hostile TME, which has proven a major obstacle especially in solid tumors. Likewise, a possible combination of iCARs or SynNotch receptors with the ability to sense inflammatory cytokines could be used to achieve autonomous dynamic feedback control of CAR activity (98). Consequently, this could lead to the creation of CAR T cells capable of responding to heightened levels of inflammatory cytokines, preventing accompanying toxicities. Simultaneously leveraging inducible and autonomous CAR T-cell control methods could substantially improve the safety of CAR therapy. This might be especially useful when targeting solid tumors, where targeted antigens can often be found in other healthy tissues. While autonomous CAR control designs can restrict cytotoxic activity to the time, location and target cells of interest, also including an inducible kill switch will provide an additional fail-safe in the event the engrafted T cells behave unexpectedly or undergo oncogenic transformation.
Early clinical success and challenges have led to an explosion in new technologies for inducibly, autonomously and passively controlling CAR T cell function, providing the community with a growing menu of solutions for safe and effective anti-cancer therapy. Ultimately the desired regulation of CAR T cells will depend on the location, aggressiveness and targetability of the tumor.
Author Contributions
LB, MB, and YM wrote the manuscript. JH and TB reviewed and edited the manuscript.
Conflict of Interest
YM and MB have filed a patent relating to a technology presented in this manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank Marcela V. Maus, Harvard Medical School, for helpful comments on figure and table.
References
1. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
2. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
3. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. (2013) 5:177ra38. doi: 10.1126/scitranslmed.3005930
4. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
5. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. (2016) 6:664–79. doi: 10.1158/2159-8290.CD-16-0040
6. Turtle CJ, Hanafi L-A, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. (2016) 8:355ra116. doi: 10.1126/scitranslmed.aaf8621
7. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
8. Frey N, Porter D. Cytokine release syndrome with chimeric antigen receptor T cell therapy. J Am Soc Blood Marrow Transplant. (2019) 25:e123–7. doi: 10.1016/j.bbmt.2018.12.756
9. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. (2018) 24:731–8. doi: 10.1038/s41591-018-0041-7
10. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
11. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. (2014) 6:224ra25. doi: 10.1126/scitranslmed.3008226
12. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management. Blood Rev. (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
13. Gust J, Hay KA, Hanafi L-A, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood–brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. (2017) 7:1404–19. doi: 10.1158/2159-8290.CD-17-0698
14. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther J Am Soc Gene Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
15. Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther J Am Soc Gene Ther. (2013) 21:904–12. doi: 10.1038/mt.2013.17
16. Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol Off J Am Soc Clin Oncol. (2006) 24:e20–2. doi: 10.1200/JCO.2006.05.9964
17. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother CII. (2017) 66:1425–36. doi: 10.1007/s00262-017-2034-7
18. Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, et al. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther J Am Soc Gene Ther. (2006) 13:151–9. doi: 10.1016/j.ymthe.2005.07.688
19. Foster JB, Choudhari N, Perazzelli J, Storm J, Hofmann TJ, Jain P, et al. Purification of mRNA encoding chimeric antigen receptor is critical for generation of a robust T-cell response. Hum Gene Ther. (2019) 30:168–78. doi: 10.1089/hum.2018.145
20. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. (2014) 2:112–20. doi: 10.1158/2326-6066.CIR-13-0170
21. Beatty GL, O'Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. (2018) 155:29–32. doi: 10.1053/j.gastro.2018.03.029
22. Tasian SK, Kenderian SS, Shen F, Ruella M, Shestova O, Kozlowski M, et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood. (2017) 129:2395–407. doi: 10.1182/blood-2016-08-736041
23. Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther. (2013) 24:717–27. doi: 10.1089/hum.2013.075
24. Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. (2013) 1:26–31. doi: 10.1158/2326-6066.CIR-13-0006
25. Panjwani MK, Smith JB, Schutsky K, Gnanandarajah J, O'Connor CM, Powell DJ, et al. Feasibility and safety of RNA-transfected CD20-specific chimeric antigen receptor T cells in dogs with spontaneous B cell lymphoma. Mol Ther J Am Soc Gene Ther. (2016) 24:1602–14. doi: 10.1038/mt.2016.146
26. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. (2015) 75:3505–18. doi: 10.1158/0008-5472.CAN-15-0139
27. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-Tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. (2015) 75:3596–607. doi: 10.1158/0008-5472.CAN-15-0159
28. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. (2019) 25:1408–14. doi: 10.1038/s41591-019-0549-5
29. Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC, et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther J Am Soc Gene Ther. (2014) 22:623–33. doi: 10.1038/mt.2013.262
30. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. (2017) 543:113–7. doi: 10.1038/nature21405
31. Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. (2011) 365:1673–83. doi: 10.1056/NEJMoa1106152
32. Straathof KC. An inducible caspase 9 safety switch for T-cell therapy. Blood. (2005) 105:4247–54. doi: 10.1182/blood-2004-11-4564
33. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. (2010) 24:1160–70. doi: 10.1038/leu.2010.75
34. Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, et al. Inducible Caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol Ther J Am Soc Gene Ther. (2017) 25:580–92. doi: 10.1016/j.ymthe.2017.01.011
35. Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS ONE. (2013) 8:e82742. doi: 10.1371/journal.pone.0082742
36. Duong MT, Collinson-Pautz MR, Morschl E, Lu A, Szymanski SP, Zhang M, et al. Two-dimensional regulation of CAR-T cell therapy with orthogonal switches. Mol Ther Oncolytics. (2019) 12:124–37. doi: 10.1016/j.omto.2018.12.009
37. Stavrou M, Philip B, Traynor-White C, Davis CG, Onuoha S, Cordoba S, et al. A rapamycin-activated caspase 9-based suicide gene. Mol Ther J Am Soc Gene Ther. (2018) 26:1266–76. doi: 10.1016/j.ymthe.2018.03.001
38. Minagawa K, Jamil MO, Al-Obaidi M, Pereboeva L, Salzman D, Erba HP, et al. In vitro pre-clinical validation of suicide gene modified anti-CD33 redirected chimeric antigen receptor T-cells for acute myeloid leukemia. PLoS ONE. (2016) 11:e0166891. doi: 10.1371/journal.pone.0166891
39. Quintarelli C, Vera JF, Savoldo B, Giordano Attianese GMP, Pule M, Foster AE, et al. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood. (2007) 110:2793–802. doi: 10.1182/blood-2007-02-072843
40. Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. (1997) 276:1719–24. doi: 10.1126/science.276.5319.1719
41. Tiberghien P, Ferrand C, Lioure B, Milpied N, Angonin R, Deconinck E, et al. Administration of herpes simplex-thymidine kinase-expressing donor T cells with a T-cell-depleted allogeneic marrow graft. Blood. (2001) 97:63–72. doi: 10.1182/blood.V97.1.63
42. Sato T, Neschadim A, Konrad M, Fowler DH, Lavie A, Medin JA. Engineered human tmpk/AZT as a novel enzyme/prodrug axis for suicide gene therapy. Mol Ther J Am Soc Gene Ther. (2007) 15:962–70. doi: 10.1038/mt.sj.6300122
43. Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. (2006) 107:2294–302. doi: 10.1182/blood-2005-08-3503
44. Emery V, Zuckerman M, Jackson G, Aitken C, Osman H, Pagliuca A, et al. Management of cytomegalovirus infection in haemopoietic stem cell transplantation. Br J Haematol. (2013) 162:25–39. doi: 10.1111/bjh.12363
45. Zhou X, Di Stasi A, Tey S-K, Krance RA, Martinez C, Leung KS, et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. (2014) 123:3895–905. doi: 10.1182/blood-2014-01-551671
46. Wu X, Shi B, Zhang J, Shi Z, Di S, Fan M, et al. A Fusion receptor as a safety switch, detection, and purification biomarker for adoptive transferred T cells. Mol Ther J Am Soc Gene Ther. (2017) 25:2270–9. doi: 10.1016/j.ymthe.2017.06.026
47. Ma G, Shen J, Pinz K, Wada M, Park J, Kim S, et al. Targeting T cell malignancies using CD4CAR T-cells and implementing a natural safety switch. Stem Cell Rev Rep. (2019) 15:443–7. doi: 10.1007/s12015-019-09876-5
48. Vogler I, Newrzela S, Hartmann S, Schneider N, von Laer D, Koehl U, et al. An improved bicistronic CD20/tCD34 vector for efficient purification and in vivo depletion of gene-modified T cells for adoptive immunotherapy. Mol Ther J Am Soc Gene Ther. (2010) 18:1330–8. doi: 10.1038/mt.2010.83
49. Griffioen M, van Egmond EHM, Kester MGD, Willemze R, Falkenburg JHF, Heemskerk MHM. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica. (2009) 94:1316–20. doi: 10.3324/haematol.2008.001677
50. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. (2014) 124:1277–87. doi: 10.1182/blood-2014-01-545020
51. Paszkiewicz PJ, Fräßle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest. (2016) 126:4262–72. doi: 10.1172/JCI84813
52. Wang X, Chang W-C, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. (2011) 118:1255–63. doi: 10.1182/blood-2011-02-337360
53. Ericson SG, Guyre CA, Benoit NE, Meehan KR, Mills LE, Fanger MW. Anti-body-dependent cellular cytotoxicity (ADCC) function of peripheral blood polymorphonuclear neutrophils (PMN) after autologous bone marrow transplantation (ABMT). Bone Marrow Transplant. (1995) 16:787–91.
54. Tabrizi M, Bornstein GG, Suria H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J. (2010) 12:33–43. doi: 10.1208/s12248-009-9157-5
55. Ruella M, Barrett DM, Shestova O, Perazzelli J, Posey AD, Hong SJ, et al. A cellular antidote to specifically deplete anti-CD19 chimeric antigen receptor positive cells. Blood. (2020) 135: 505–9. doi: 10.1182/blood.2019001859
56. Koristka S, Ziller-Walter P, Bergmann R, Arndt C, Feldmann A, Kegler A, et al. Anti-CAR-engineered T cells for epitope-based elimination of autologous CAR T cells. Cancer Immunol Immunother CII. (2019) 68:1401–15. doi: 10.1007/s00262-019-02376-y
57. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
58. Tasian SK, Kenderian SS, Shen F, Li Y, Ruella M, Fix WC, et al. Efficient termination of CD123-redirected chimeric antigen receptor T cells for acute myeloid leukemia to mitigate toxicity. Blood. (2015) 126:565. doi: 10.1182/blood.V126.23.565.565
59. Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med. (2019) 11:eaau5907. doi: 10.1126/scitranslmed.aau5907
60. Weber EW, Lynn RC, Sotillo E, Lattin J, Xu P, Mackall CL. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. (2019) 3:711–7. doi: 10.1182/bloodadvances.2018028720
61. Christopher LJ, Cui D, Wu C, Luo R, Manning JA, Bonacorsi SJ, et al. Metabolism and disposition of dasatinib after oral administration to humans. Drug Metab Dispos Biol Fate Chem. (2008) 36:1357–64. doi: 10.1124/dmd.107.018267
62. Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. (2012) 72:1844–52. doi: 10.1158/0008-5472.CAN-11-3890
63. Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, et al. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. (2014) 74:93–103. doi: 10.1158/0008-5472.CAN-13-1365
64. Ma JSY, Kim JY, Kazane SA, Choi S, Yun HY, Kim MS, et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci. (2016) 113:E450–8. doi: 10.1073/pnas.1524193113
65. Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci. (2016) 113:E459–68. doi: 10.1073/pnas.1524155113
66. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. (2018) 173:1426–38.e11. doi: 10.1016/j.cell.2018.03.038
67. Cao Y, Rodgers DT, Du J, Ahmad I, Hampton EN, Ma JSY, et al. Design of switchable chimeric antigen receptor T cells targeting breast cancer. Angew Chem Int Ed. (2016) 55:7520–4. doi: 10.1002/anie.201601902
68. Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, et al. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. (2016) 6:e458. doi: 10.1038/bcj.2016.61
69. Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, et al. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res Off J Am Assoc Cancer Res. (2012) 18:6436–45. doi: 10.1158/1078-0432.CCR-12-1449
70. Kim MS, Ma JSY, Yun H, Cao Y, Kim JY, Chi V, et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J Am Chem Soc. (2015) 137:2832–5. doi: 10.1021/jacs.5b00106
71. Lee YG, Chu H, Lu Y, Leamon CP, Srinivasarao M, Putt KS, et al. Regulation of CAR T cell-mediated cytokine release syndrome-like toxicity using low molecular weight adapters. Nat Commun. (2019) 10:2681. doi: 10.1038/s41467-019-10565-7
72. Klesmith JR, Su L, Wu L, Schrack IA, Dufort FJ, Birt A, et al. Retargeting CD19 chimeric antigen receptor T cells via engineered CD19-fusion proteins. Mol Pharm. (2019) 16:3544–58. doi: 10.1021/acs.molpharmaceut.9b00418
73. Wu C-Y, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. (2015) 350:aab4077. doi: 10.1126/science.aab4077
74. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible activation of MyD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov. (2017) 7:1306–19. doi: 10.1158/2159-8290.CD-17-0263
75. Juillerat A, Tkach D, Busser BW, Temburni S, Valton J, Duclert A, et al. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol. (2019) 19:44. doi: 10.1186/s12896-019-0537-3
76. Sakemura R, Terakura S, Watanabe K, Julamanee J, Takagi E, Miyao K, et al. A Tet-On inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol Res. (2016) 4:658–68. doi: 10.1158/2326-6066.CIR-16-0043
77. Drent E, Poels R, Mulders MJ, van de Donk NWCJ, Themeli M, Lokhorst HM, et al. Feasibility of controlling CD38-CAR T cell activity with a Tet-on inducible CAR design. PLoS ONE. (2018) 13:e0197349. doi: 10.1371/journal.pone.0197349
78. Gu X, He D, Li C, Wang H, Yang G. Development of inducible CD19-CAR T cells with a Tet-on system for controlled activity and enhanced clinical safety. Int J Mol Sci. (2018) 19:3455. doi: 10.3390/ijms19113455
79. Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. (2019) 10:128. doi: 10.3389/fimmu.2019.00128
80. Duong CP, Westwood JA, Berry LJ, Darcy PK, Kershaw MH. Enhancing the specificity of T-cell cultures for adoptive immunotherapy of cancer. Immunotherapy. (2011) 3:33–48. doi: 10.2217/imt.10.81
81. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. (2013) 31:71–5. doi: 10.1038/nbt.2459
82. Wilkie S, van Schalkwyk MCI, Hobbs S, Davies DM, van der Stegen SJC, Pereira ACP, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. (2012) 32:1059–70. doi: 10.1007/s10875-012-9689-9
83. Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. (2013) 1:43–53. doi: 10.1158/2326-6066.CIR-13-0008
84. Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell. (2016) 164:770–9. doi: 10.1016/j.cell.2016.01.011
85. Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell. (2019) 35:489-503.e8. doi: 10.1016/j.ccell.2019.02.003
86. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. (2013) 5:215ra172. doi: 10.1126/scitranslmed.3006597
87. Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. (2017) 7:39833. doi: 10.1038/srep39833
88. Han X, Bryson PD, Zhao Y, Cinay GE, Li S, Guo Y, et al. Masked chimeric antigen receptor for tumor-specific activation. Mol Ther J Am Soc Gene Ther. (2017) 25:274–84. doi: 10.1016/j.ymthe.2016.10.011
89. Gutierrez E, Banks W, Kastin A. Blood-borne interleukin-1 receptor antagonist crosses the blood-brain barrier. J Neuroimmunol. (1994) 55:153–60. doi: 10.1016/0165-5728(94)90005-1
90. Cordeiro A, Bezerra ED, Hirayama AV, Hill JA, Wu QV, Voutsinas J, et al. Late events after treatment with CD19-targeted chimeric antigen receptor modified T cells. J Am Soc Blood Marrow Transplant. (2020) 26:26–33. doi: 10.1016/j.bbmt.2019.08.003
91. Gust J, Finney OC, Li D, Brakke HM, Hicks RM, Futrell RB, et al. Glial injury in neurotoxicity after pediatric CD19-directed chimeric antigen receptor T cell therapy. Ann Neurol. (2019) 86:42–54. doi: 10.1002/ana.25502
92. Alvi RM, Frigault MJ, Fradley MG, Jain MD, Mahmood SS, Awadalla M, et al. Cardiovascular events among adults treated with chimeric antigen receptor T-cells (CAR-T). J Am Coll Cardiol. (2019) 74:3099–108. doi: 10.1016/j.jacc.2019.10.038
93. Abu-Sbeih H, Tang T, Ali FS, Luo W, Neelapu SS, Westin JR, et al. Gastrointestinal adverse events observed after chimeric antigen receptor T-cell therapy. Am J Clin Oncol. (2019) 42:789–96. doi: 10.1097/COC.0000000000000596
94. Robles-Oteiza C, Taylor S, Yates T, Cicchini M, Lauderback B, Cashman CR, et al. Recombinase-based conditional and reversible gene regulation via XTR alleles. Nat Commun. (2015) 6:8783. doi: 10.1038/ncomms9783
95. Chassin H, Müller M, Tigges M, Scheller L, Lang M, Fussenegger M. A modular degron library for synthetic circuits in mammalian cells. Nat Commun. (2019) 10:2013. doi: 10.1038/s41467-019-09974-5
96. Michaels YS, Barnkob MB, Barbosa H, Baeumler TA, Thompson MK, Andre V, et al. Precise tuning of gene expression levels in mammalian cells. Nat Commun. (2019) 10:818. doi: 10.1038/s41467-019-08777-y
97. Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. (2018) 14:317–24. doi: 10.1038/nchembio.2565
Keywords: chimeric antigen receptor, cancer, immunotherapy, T cell, synthetic, regulation, cell therapy
Citation: Brandt LJB, Barnkob MB, Michaels YS, Heiselberg J and Barington T (2020) Emerging Approaches for Regulation and Control of CAR T Cells: A Mini Review. Front. Immunol. 11:326. doi: 10.3389/fimmu.2020.00326
Received: 21 November 2019; Accepted: 10 February 2020;
Published: 26 February 2020.
Edited by:
Axel Schambach, Hannover Medical School, GermanyReviewed by:
Jan Joseph Melenhorst, University of Pennsylvania, United StatesFernando Aranda, University of Navarra, Spain
Copyright © 2020 Brandt, Barnkob, Michaels, Heiselberg and Barington. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Torben Barington, torben.barington@rsyd.dk
†These authors have contributed equally to this work