 Julia Busselaar
Julia Busselaar Sun Tian
Sun Tian Hans van Eenennaam3
Hans van Eenennaam3 Jannie Borst
Jannie Borst- 1Department of Immunology and Oncode Institute, Leiden University Medical Center, Leiden, Netherlands
- 2Aduro Biotech Europe BV, Oss, Netherlands
- 3AIMM Therapeutics BV, Amsterdam, Netherlands
Persistent antigen exposure in chronic infection and cancer has been proposed to lead to cytotoxic T lymphocyte (CTL) “exhaustion”, i.e., loss of effector function and disease control. Recent work identifies a population of poorly differentiated TCF-1+PD-1+ CD8+ T cells as precursors of the terminally exhausted CTL pool. These “predysfunctional” CTLs are suggested to respond to PD-1 targeted therapy by giving rise to a pool of functional CTLs. Supported by gene expression analyses, we present a model in which lack of CD4+ T cell help during CD8+ T cell priming results in the formation of predysfunctional CTLs. Our model implies that predysfunctional CTLs are formed during priming and that the remedy for CTL dysfunction is to provide “help” signals for generation of optimal CTL effectors. We substantiate that this may be achieved by engaging CD4+ T cells in new CD8+ T cell priming, or by combined PD-1 blocking and CD27 agonism with available immunotherapeutic antibodies.
Introduction
In chronic infection and cancer, CD8+ T cells upregulate coinhibitory receptors and display impaired proliferative and cytotoxic capacities, a phenomenon described as “T cell exhaustion”. T cell exhaustion is considered a crucial factor in limiting clinical responses to immunotherapy, but this T cell state is not well understood. Some experts do not envision functions for exhausted T cells, while others surmise a role in host protection (1). Recent data illuminate how exhausted CD8+ T cells are formed. The original model proposed that exhausted CD8+ T cells develop from effector T cells as a result of chronic stimulation via their T cell antigen receptor (TCR) (2). However, new transcriptomic analyses, that include TCR-based lineage tracing, argue that exhausted CD8+ T cells are not derived from functional effector cells. Rather, CD8+ T cells can attain a “predysfunctional” state early after infection or tumorigenesis that may progress into a terminally exhausted state. It is considered that predysfunctional cells may also be “reinvigorated” to become CTL effectors. Blockade of the PD-1/PD-L1 coinhibitory axis may lead to such reinvigoration. Knowledge about the exact molecular and cellular mechanisms underlying CD8+ T cell predysfunction, exhaustion and reinvigoration are clinically relevant in chronic infection and cancer, and likely also in auto-immune and inflammatory diseases.
Here, we first discuss the recent literature on CD8+ T cell predysfunction and exhaustion in a key mouse model of chronic virus infection. This work has recently led to the concept that predysfunction and exhaustion represent aspects of a CD8+ T cell differentiation pathway, distinct from effector and memory differentiation. By connecting studies on infection and cancer, we integrate supporting arguments for this concept. We synthesize these recent insights into a model of progressive fate commitment of primed CD8+ T cells. Supported by gene expression analyses, we introduce the novel perspective that the predysfunctional differentiation state results from CD8+ T cell priming in the absence of CD4+ T cell help. This viewpoint implies that reinvigoration of predysfunctional CD8+ T cells may be achieved by addition of “help” signals. We rationalize that PD-1 targeted checkpoint blockade may lead to delivery of help signals and may be supported by engagement of specific T cell costimulatory receptors.
Methods
No Help CD8+ T Cell Gene Expression Signature
RNAseq fastq files of samples of helped CD8+ T cells (n = 3) and samples of non-helped CD8+ T cells (n = 3) were retrieved from GEO database (GSE89665) (3). FASTQ files were aligned to the mouse genome mm10 (GRCm38.77) using HISAT2 v2.1.0 (4), and number of reads was assigned to genes by using featureCounts v1.6.1 (5). Reads mapped to genes were normalized and differentially expressed gene analysis between non-helped CD8+ T cells and helped CD8+ T cell was performed using edgeR package in R Bioconductor (6). The false discovery rate (FDR) < 0.01 was used as the criteria to select statistically differentially expressed gene lists. In total, a list of 1,331 genes were found differentially expressed between non-helped condition and helped conditions (FDR < 0.01), which represents the No Help signature.
Calculation of No Help Score in Published CD8 T Cell Expression Signatures
RNAseq fastq files were retrieved from GEO database (GSE99531, GSE122713) (7, 8). FASTQ files were aligned to the mouse genome mm10 using HISAT2 v2.1.0 (4), and number of reads was assigned to genes by using featureCounts v1.6.1 (5). Genes with all zero counts were removed. The raw counts were normalized by count per million (CPM) methods (6). For each sample, a “No Help score” was determined by the nearest centroid method on the 1331 genes from the No Help signature. In short, the No Help score was calculated as the difference of Pearson correlations in normalized read counts between a given population and No Help or Help vaccination settings. A higher No Help score indicates greater transcriptional similarity to helpless CD8+ T cells.
Gene Set Enrichment Analysis
RNAseq files of helped or non-helped CD8 T cells, aligned to the mouse genome mm10, were imported into Qlucore Omics Explorer. Genes with less than 5 reads in at least one of the samples were discarded. Mapping quality threshold was set to 10. TNM normalization method was applied. Gene Set Enrichment Analysis was performed using published gene sets of the top 200 up- and downregulated genes from Tcf7-GFP+ versus Tcf7-GFP− P14 cells in chronic LCMV infection (9) or B16-gp33 tumor model (10).
Statistical Analysis
Data was analyzed with GraphPad Prism software using unpaired two-tailed Student’s t-test, or repeated measures one-way ANOVA with Tukey’s multiple comparison test. A P value < 0.05 was considered statistically significant; *P < 0.05, **P < 0.01, and ***P < 0.001.
Illustrations
Illustrations in Figures 1–4 were created with BioRender.
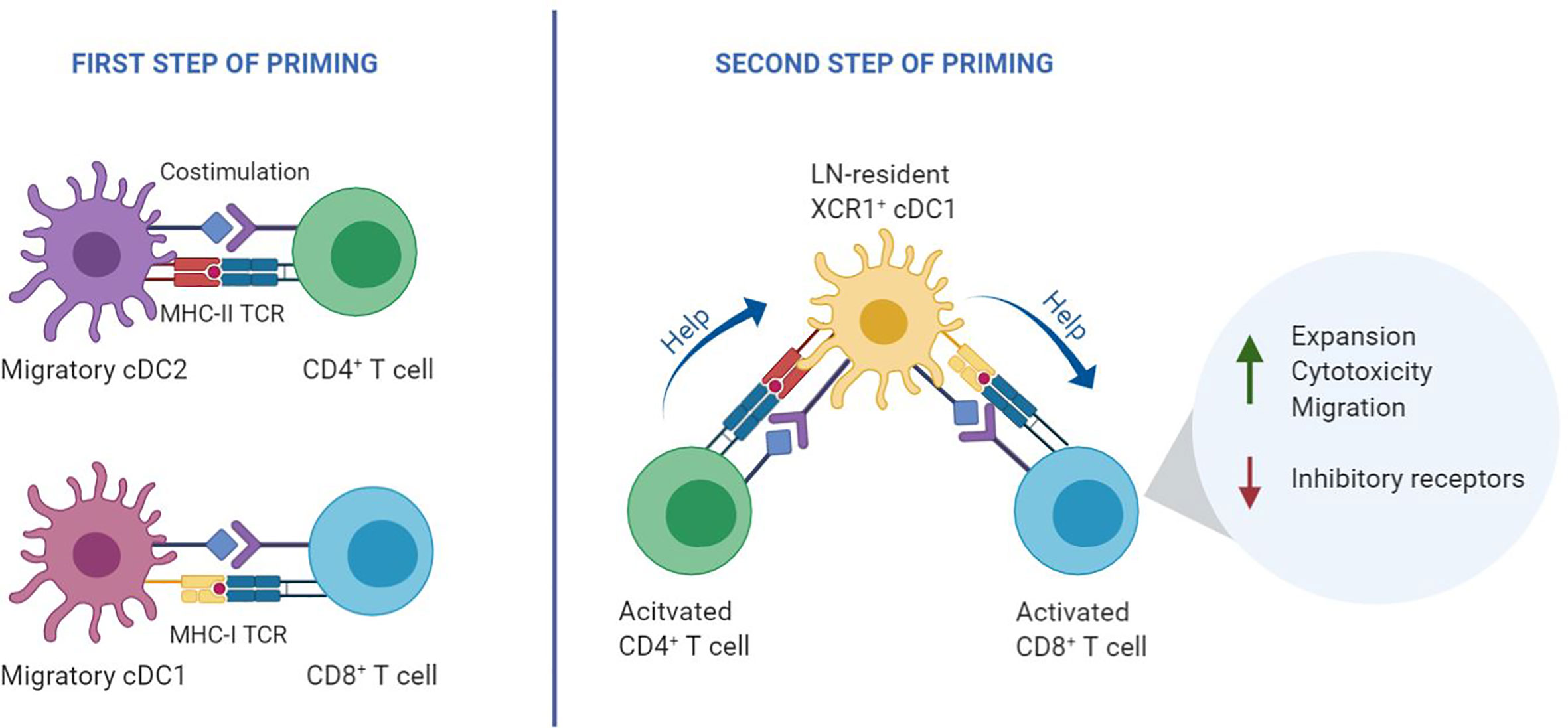
Figure 1 Two-step priming model. During the first step of T cell priming (left), CD8+ T cells and CD4+ T cells are initially activated independently by different DC subtypes that present antigen on MHC class I and class II, respectively. In the second step of priming (right), recently activated CD4+ and CD8+ T cells interact with the same lymph node-resident cDC1 co-expressing MHC class I and MHC class II epitopes. Helped CD8+ T cells undergo optimal priming by signaling via various costimulatory and cytokine signals that emerge from the helped cDC1, resulting in an optimal CTL effector program (11).
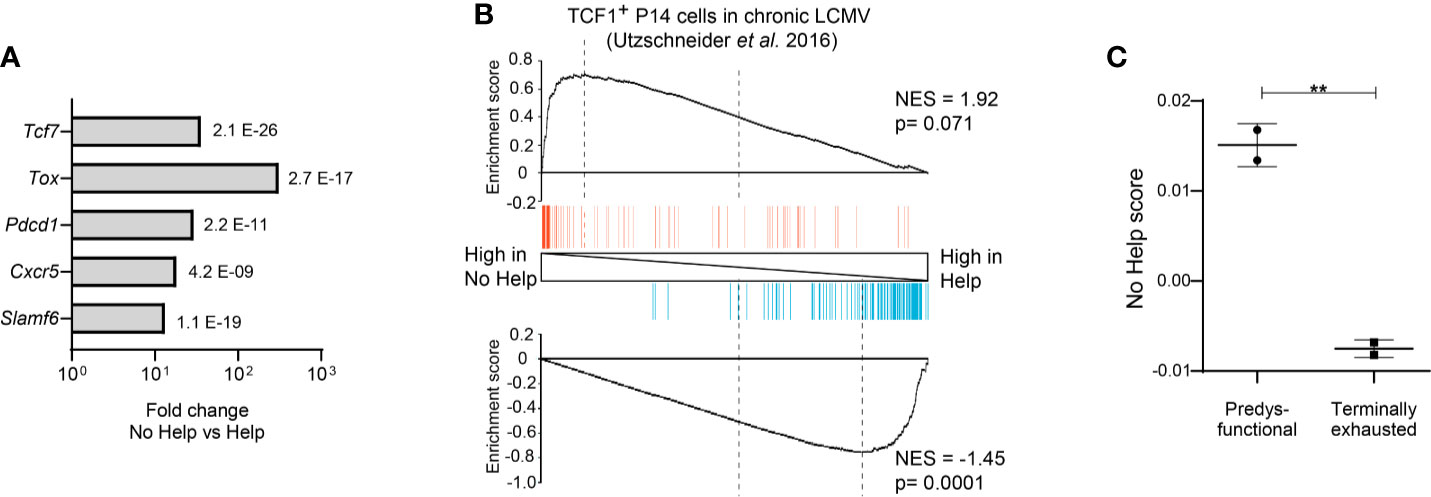
Figure 2 Predysfunctional TCF-1+ CD8+ T cells in a chronic LCMV infection model display a gene expression signature characteristic of helpless antigen-specific CD8+ T cells in a vaccination model. The transcriptional “No Help” signature was determined by differential gene expression (False discovery rate (FDR) < 0.01) of antigen-specific CTLs raised in No Help versus Help vaccination settings (GEO database GSE89665) (3). (A) Differential expression of selected genes characteristic of predysfunctional TCF-1+CD8+ T cells (Table 1) in No Help versus Help settings. FDR is depicted per gene. (B) GSEA of the top 200 upregulated (red)- or downregulated (blue) genes from TCF1+ versus TCF1− virus-specific CD8+ T cells in chronic LCMV infection (9) within the gene expression profiles of CD8+ T cells from the No Help versus Help vaccination settings. NES, normalized enrichment score. (C) No Help score in predysfunctional TCF-1+TIM3− and terminally exhausted TCF-1−TIM3+ CD8+ T cells from a setting of chronic LCMV infection (GEO database GSE122713) (7). The No Help score was calculated as the difference of correlations in gene expression between a given population and No Help or Help vaccination settings. A higher No-Help score indicates greater transcriptional similarity to helpless CD8+ T cells. **P < 0.01 by Student’s t-test.
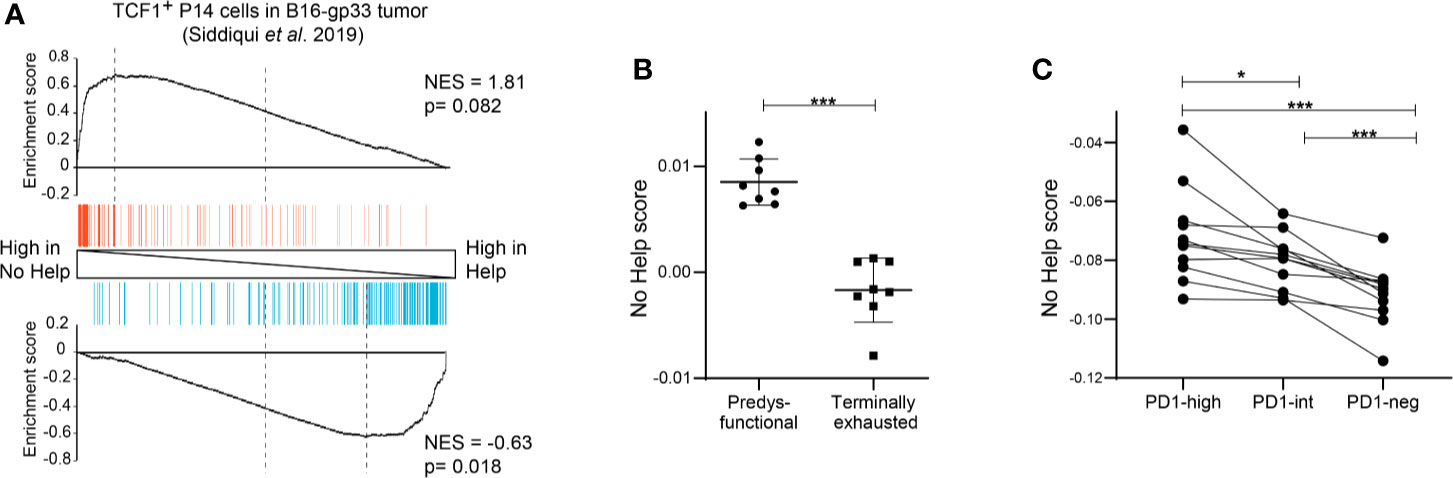
Figure 3 Predysfunctional TCF-1+ CD8+ T cells in human cancer display a gene expression signature characteristic of helpless antigen-specific CD8+ T cells in a mouse vaccination model. (A) GSEA showing enrichment of the top 200 upregulated (red) or downregulated (blue) genes in gp33-specific TCF-1+ CD8+ T cells in a murine B16-gp33 tumor model (10) within the gene expression profiles of vaccine antigen-specific CD8+ T cells in No Help versus Help settings (3). (B) No Help scores, defined in our vaccination model, determined in the transcriptomes of predysfunctional TCF-1+TIM3− and terminally exhausted TCF-1−TIM3+ tumor antigen-specific CD8+ T cells from a murine B16-OVA tumor model (GEO database GSE122713) (7). (C) No Help score defined as in (B), determined in the transcriptome of patient-matched PD-1-high, PD-1-intermediate, and PD-1-negative CD8+ TILs in human melanoma (GEO database GSE99531) (8). *P < 0.05, ***P < 0.001 by Student’s t-test (B) or one-way ANOVA (C).
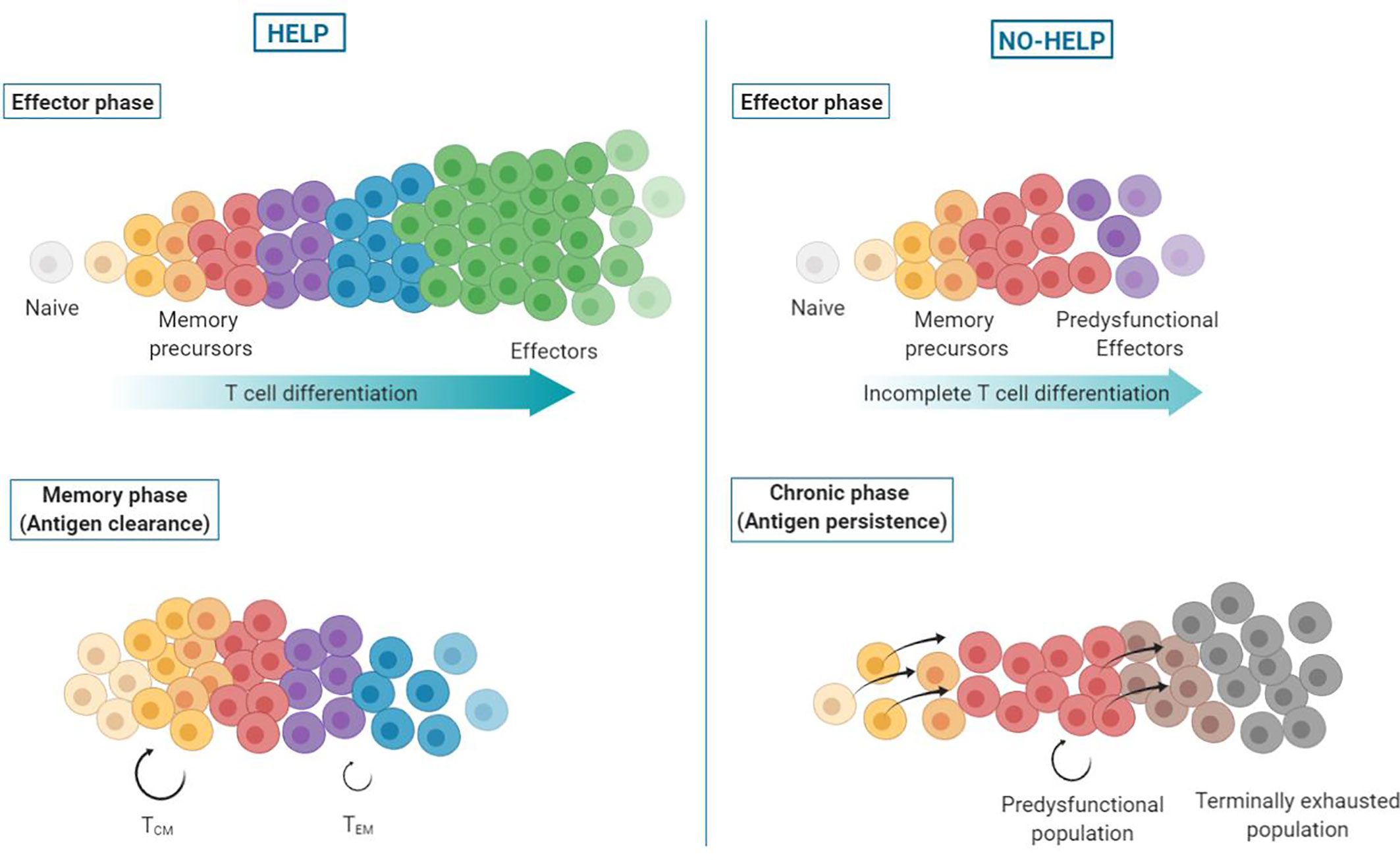
Figure 4 Helpless dysfunction model. Upon priming of CD8+ T cells, a differentiation spectrum is formed, ranging from uncommitted memory precursors to terminally differentiated effector cells. In presence of CD4+ T cell help signals (left), the antigen-specific CD8+ T cell population attains higher differentiation states, with the majority of cells becoming terminally differentiated, short-lived effector CTLs. These helped CTLs clear the antigen source and die. When antigen wanes, memory precursor cells persist and form helped central (TCM) and effector memory (TEM) CD8+ T cells. In absence of help signals (right), antigen-specific CD8+ T cells undergo incomplete effector differentiation and terminally differentiated effector CTLs are lacking. Instead, predysfunctional effector CTLs are formed that are less committed (“memory-like”), i.e., have not fully unfolded their effector program and express coinhibitory receptors. In addition, formation of effector memory CD8+ T cells is impaired. As a result, antigen persists and continuous TCR stimulation of memory precursor cells drives their differentiation into predysfunctional CTLs that self-maintain or differentiate into terminally exhausted cells.
Help Delivery During CD8+ T Cell Priming
Priming of CD4+ and CD8+ T cells relies on three key signals: TCR engagement by peptide/MHC complexes, costimulation by CD28 and members of the TNF receptor family, as well as specific cytokine signaling. Dendritic cells (DCs) can supply these signals, provided that the DC is of the appropriate subset and adequately activated, by pathogen- or danger-derived signals or by CD4+ T cells. In secondary lymphoid organs, CD4+ and CD8+ T cells engage in successive antigen-specific interactions with different DC subtypes. Migratory DCs deliver the antigen from the site of infection, while lymph node-resident DCs pick up the antigen locally. CD4+ and CD8+ T cells are initially activated independent from each other, in different regions of the lymph node by migratory conventional (c)DC1 and cDC2 subsets (12–14). After this first step of priming, a second step of priming takes place on lymph node-resident cDC1s. In this interaction, CD4+ T cell help is delivered that is essential for optimal differentiation of CD8+ T cells into CTL effector and memory cells (11) (Figure 1). CD4+ and CD8+ T cells that have undergone the first step of priming produce specific chemokines that attract lymph node-resident cDC1 (12, 13, 15). In case the cDC1 co-presents recognizable MHC class II- and MHC class I- restricted antigens, it can relay help signals from the CD4+ T cell to the CD8+ T cell. Plasmacytoid (p)DCs likely promote this scenario by the production of type I interferon (IFN), which optimizes maturation and antigen crosspresentation by cDC1s (16).
Upon cognate contact with the CD4+ T cell, the lymph node-resident cDC1 gains expression of various cytokines and co-stimulatory ligands that in concert optimize the CD8+ T cell response (11). Interaction between CD40 ligand on the CD4+ T cell and CD40 on the cDC1 amplifies production of IL-12 and IL-15 by the DC, which improves clonal expansion and effector differentiation of CD8+ T cells (17, 18). Furthermore, CD40 signaling in DCs upregulates CD80/CD86 and CD70, which relay costimulatory signals via CD28 and CD27, respectively (19–21). In both CD4+ and CD8+ T cells, CD28 costimulation amplifies the TCR signal and drives cell division (22), while CD27 costimulation promotes cell survival and effector differentiation (3, 23–25). CD27 costimulation of CD8+ T cells is a key effector pathway of CD4+ T cell help. It promotes CTL differentiation and survival, likely directly, but also by increasing expression of the IL-2 receptor alpha chain, IL-2 and the IL-12 receptor, leading to autocrine IL-2 signaling and responsiveness to DC-derived IL-12 (3, 26–28). IL-21 production by CD4+ T cells also promotes CTL effector differentiation (29).
By transcriptomic analyses in mice, we have discovered how help signals impact effector and memory gene expression programs of CD8+ T cells (3, 30). At the effector stage, “helped” versus “helpless” CTLs differentially expressed about 1,000 transcripts, encoding proteins enabling critical CTL functions, such as cytotoxicity and migratory abilities. From functional studies in a tumor model, we concluded that CD4+ T cell help confers upon CTLs the exact properties desired for effective anti-tumor immunity, as defined by Chen and Mellman in “The cancer immunity cycle” (31). Conversely, helpless CTLs proved to have a dysfunctional phenotype characterized by low cytotoxic capacity and high expression of PD-1 and other co-inhibitory receptors (3), classifying them as “exhausted”, according to the original definition. Other authors defined by micro-array similar gene expression features in helpless CTLs, which proved to resemble exhausted CTLs, as defined in a mouse model of chronic LCMV infection (32). In conclusion, there appears to be a connection between helpless priming of CD8+ T cells and acquisition of the exhausted state. This connection will be clarified in this Hypothesis and Theory article.
Antigen-Specific CD8+ T Cell Fates in Chronic Infection
Exhaustion
Exhaustion of antigen-specific CD8+ T cells was first described in mouse models of chronic infection with LCMV (33). Exhausted virus-specific CD8+ T cells were defined by a diminished ability to display effector functions such as IFNγ production, and high expression of coinhibitory receptors such as PD-1. It was proposed that virus-specific effector CD8+ T cells gradually turn into exhausted cells upon chronic engagement of the TCR by persistent viral antigen. Observations that TCR-regulated transcription factors contribute to exhaustion led to this idea (34–36). In agreement with TCR signaling driving exhaustion, the exhausted virus-specific CD8+ T cell fraction was found to increase in time upon viral persistence (37). However, virus-specific CD8+ T cells can already show impaired effector functions from the beginning of a chronic infection, suggesting causes other than chronic antigen exposure (37). Adoptive transfer experiments demonstrated that exhausted CD8+ T cells in chronic LCMV infections derive from the same progenitors as memory cells and not from terminally differentiated (KLRG1hi) effector T cells (38). This finding suggested that exhausted CD8+ T cells in chronic infection do not follow a normal effector differentiation path (39).
Predysfunction
Despite the persistence of viral antigen, not all virus-specific CD8+ T cells in chronic infection acquire a terminally exhausted phenotype. A subset of virus-specific CD8+ T cells in chronic LCMV infection was found to proliferate and give rise to terminally exhausted cells (40). Other authors defined in the same model a small “memory-like” subpopulation within the virus-specific CD8+ T cell pool that retained proliferative capacities and could re-expand upon secondary infection in an antigen-free host (41). Later, this proliferative population was found to express the transcription factor TCF-1 (9, 42) and the chemokine receptor CXCR5 (43, 44). These studies report that TCF-1+ CXCR5+ CD8+ T population is self-sustaining and constantly replenishes the exhausted CD8+ T cell pool. This population is described by different nomenclature (Table 1), but throughout this article, we will use the term “predysfunctional”. The predysfunctional population is established early in chronic infection with LCMV strain clone 13, before the peak of the T cell response, but is not seen in acute infection with LCMV strain Armstrong (51). TCF-1 is also expressed in memory T cells in acute infection, but predysfunctional TCF1+ T cells in chronic infection can be identified by co-expression of CXCR5, Slamf6 and PD-1 (29, 44). TCF-1 signaling represses effector differentiation and is thereby essential for generation and maintenance of predysfunctional T cells (42, 57, 59).
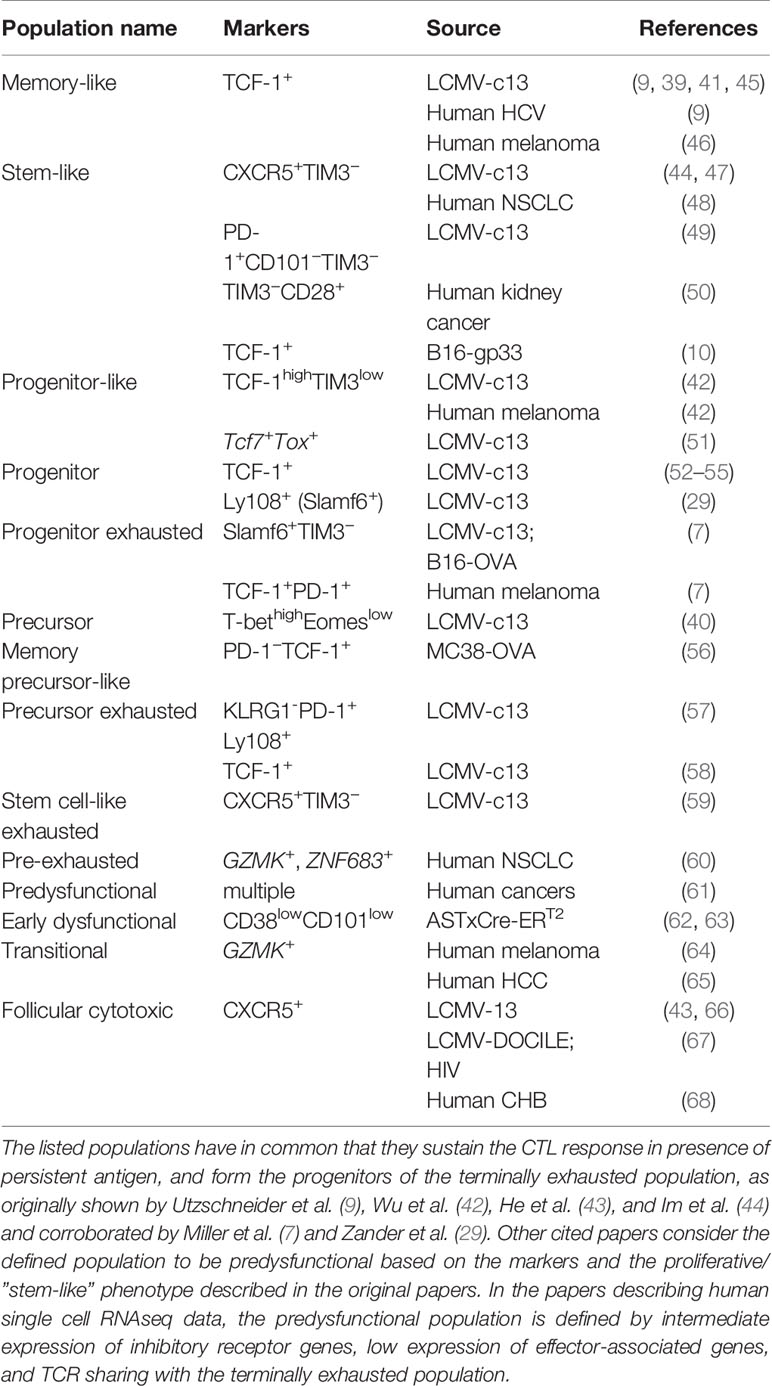
Table 1 Definitions of predysfunctional CD8+ T cell populations in chronic infection and cancer.
From Predysfunction to Exhaustion
Antigenic stimulation of predysfunctional TCF-1+CXCR5+ CD8+ T cells can drive their differentiation into TCF-1− CXCR5− “terminally exhausted” cells (40, 49, 69). During this differentiation process, predysfunctional cells transiently acquire a more effector-like gene signature (49, 57, 70). Terminally exhausted CD8+ T cells are short-lived and display higher expression of coinhibitory receptors than TCF-1+ predysfunctional cells (9, 42–44). Conversion from a predysfunctional to a terminally exhausted state is associated with epigenetic and transcriptional changes involving genes encoding coinhibitory receptors, effector molecules and effector-associated transcription factors (7, 47, 70). The transcription factor TOX plays a critical role in epigenetic imprinting of dysfunction in the TCF-1+ subset and induces fate commitment to a terminally exhausted phenotype (51, 52, 71–73). Both the establishment of the predysfunctional population and the TOX-driven commitment to exhaustion are part of a differentiation path that is separate from effector differentiation, occurring in early stages of chronic LCMV infection (51, 57, 71). Together, these findings provide strong support for the notion that terminally exhausted T cells found in chronic infections are derived from a population of predysfunctional cells, instead of from functional effectors. Similar processes likely take place in human, where virus-specific predysfunctional and terminally exhausted CD8+ T cell populations have been identified in patients with chronic hepatitis C virus (HCV) infection (9). Also, CXCR5+ CD8+ T cells were found in patients infected with human immunodeficiency virus (HIV) or chronic hepatitis B virus (HBV) (67, 68).
Reinvigoration
Importantly, PD-1 blockade unleashes the expansion potential of predysfunctional, but not terminally exhausted virus-specific CD8+ T cells (9, 43, 44). Predysfunctional TCF-1+ CD8+ T cells express PD-1 that supports the maintenance of this population early during chronic LCMV infection (57). Chronic virus infections (LCMV clone 13, HIV) induce chromatin accessibility and permanent demethylation of the Pdcd1 locus (encoding PD-1), causing exhausted CD8+ T cells to stably express PD-1 at high levels (74, 75). Terminally exhausted CD8+ T cells express higher levels of PD-1 and other coinhibitory receptors than predysfunctional cells (9, 42, 43). In the terminally exhausted population, efficacy and durability of virus-specific CD8+ T cell reinvigoration by PD-1 blockade proved to be limited by the epigenetic landscape, including chromatin accessibility and de novo DNA methylation (76, 77). Taken together, these results argue that the predysfunctional virus-specific CD8+ T cell population in chronic infection is reinvigorated by PD-1 blockade. Predysfunctional cells respond to PD-(L)1 blockade by undergoing proliferation, as well as differentiation toward a terminally exhausted phenotype (7). During this differentiation, cells pass through an intermediate or “transitory” state, characterized by a transcriptional signature that resembles that of effector CTLs (49, 70). While these effector-like CD8+ T cells that are reinvigorated by PD-1 blockade are able to produce cytokines and contribute to virus control, they retain expression of inhibitory receptors and eventually convert to a terminally exhausted state upon persistent antigen exposure (49).
Proposition: Helpless Priming Generates Predysfunctional CD8+ T Cells
Establishing a chronic infection in mouse models is often aided by depleting CD4+ T cells (33, 37, 44, 77, 78), suggesting a link between the absence of CD4+ T cell help and infections persisting chronically. Decreased antigen presentation and decreased costimulatory signaling by DCs during priming promote the formation of TCF-1+ cells, suggesting that this population may be generated as a result of suboptimal priming (45). Importantly, CD4+ T cell depletion in chronic LCMV infection impaired the generation of terminally differentiated effector CD8+ T cells, but not of predysfunctional TCF-1+ CD8+ T cells (53). This finding indicates that the predysfunctional TCF-1+ CD8+ T cell population is formed independently of CD4+ T cell help. We propose that this population is formed as a result of helpless priming and provide supporting evidence in this article.
As a model to study CD4+ T cell help for the CTL response, our group made use of a therapeutic DNA vaccination scheme in mice. We used a comparative setting with two vaccines that encode an immunodominant MHC-I restricted peptide from the human papillomavirus (HPV) E7 protein to prime CD8+ T cells, either with or without HPV-unrelated immunodominant MHC-II restricted peptides to induce CD4+ T cell help (79). Genome-wide mRNA deep sequencing of HPV-E7-specific CD8+ T cells at the effector stage of the CTL response yielded “Help” and “No Help” signatures (3). Helpless CTLs expressed many genes characteristic of the predysfunctional CD8+ T cell subset at a higher level than helped CTLs, including Tcf7 (encoding TCF-1), Tox, Pdcd1, Cxcr5, and Slamf6 (Figure 2A) (3). We therefore hypothesized that predysfunctional CD8+ T cells found in chronic LCMV infection are cells that have not experienced CD4+ T cell help during priming. To test this, we determined how predysfunctional CD8+ T cells defined in literature and helpless CD8+ T cells defined in our study are related at the gene expression level, by Gene Set Enrichment Analysis (GSEA). A published gene expression signature characteristic for the predysfunctional TCF-1+ CD8+ T cell population in chronic LCMV infection (9) in mice thus proved to be enriched in the No Help gene expression signature of antigen-specific CD8+ T cells from our vaccination study (Figure 2B). Additionally, using another published dataset from chronic LCMV infection (7), we determined a “No Help score” as a measure of correlation with our No Help gene expression signature. This analysis demonstrated that predysfunctional TCF-1+ CD8+ T cells display a higher No Help score than TCF-1− terminally exhausted cells, indicating that predysfunctional CD8+ T cells are transcriptionally more similar to helpless CD8+ T cells (Figure 2C).
CD8+ T Cell Dysfunction In Cancer
The Parallel
In cancer, tumor antigen-specific CD8+ T cells may be chronically stimulated within the tumor micro-environment (TME), which theoretically can lead to exhaustion, as it does in mouse models of chronic virus infection. However, in the LCMV models, infection is systemic and analysis is generally focused on CD8+ T cells from the spleen. This milieu is distinct from the TME in partially undefined aspects. In both environments, specific conditions are created by interplay between infected cells or growing tumor cells, immune cells and non-immune cells. Intratumoral CD8+ T cells are known to be exposed to various suppressive immune cell types, inhibitory molecules, hypoxia, metabolites and nutrient deprivation (2).
Mouse Models
Using a mouse model of tamoxifen-inducible liver cancer, it was shown that tumor antigen-specific CD8+ T cells taken from the TME early during tumorigenesis could be reinvigorated by PD-1 blockade or recall in an antigen-free host. Late in tumor development, however, these cells could no longer be rendered functional. It was found that tumor-specific CD8+ T cells in the TME over time acquire a fixed dysfunctional phenotype (62). Follow-up research in this model showed that tumor-specific CD8+ T cells in the TME first attain a reversible dysfunctional state and next enter a epigenetically fixed dysfunctional state (63). These data are in agreement with a transition from predysfunction to exhaustion.
In a murine melanoma model, single-cell transcriptomics revealed that among CD8+ tumor infiltrating lymphocytes (TILs), TCF-1+ predysfunctional and TCF-1− terminally exhausted cell subsets can be discerned that are analogous to those defined in chronic LCMV infection. Adoptive transfer experiments demonstrated that TCF-1+ CD8+ T cells can persist long-term inside a tumor and give rise to terminally exhausted cells (7). Like in chronic infection, transcriptional and epigenetic changes underlying this conversion depended on the transcription factor TOX (72, 73).
Human Cancer
Also in human cancer, there is increasing evidence for the existence of predysfunctional and terminally exhausted CD8+ T cell populations. In non-small cell lung cancer (NSCLC), CXCR5 expression was selectively found on CD8+ TILs and not on CD8+ T cells from healthy tissue or blood (48). In kidney cancer, TCF-1+TIM3−CD28+ predysfunctional TILs were found to reside in niches that are rich in antigen-presenting cells, while PD-1+TIM3+ terminally exhausted cells were distributed throughout the tumor tissue. Transcriptional and epigenetic profiles of these human TIL subsets proved to be similar to those described in the mouse. Importantly, TCR repertoire overlap between the two populations indicated that TCF-1+ predysfunctional TILs are indeed the progenitors of terminally exhausted TILs (50). TCR repertoire overlap between a terminally exhausted TIL population, characterized by high expression of coinhibitory receptor genes, and a predysfunctional TIL population, characterized by expression of GZMK, was also found in human melanoma (64), NSCLC (60), colorectal cancer (CRC) (80) and hepatocellular carcinoma (HCC) (65). These findings are consistent with a model where also in human cancer, exhausted TILs derive from a predysfunctional population. However, a strict division of the human TIL pool into predysfunctional or terminally exhausted may be an oversimplification. Rather, CTL dysfunction in human TILs covers a spectrum of differentiation states, ranging from predysfunctional to terminally exhausted (61).
The question remains whether the active CTLs that display effector functions in human tumors are generated from a separate CD8+ T cell pool, or are connected to the (pre)dysfunctional pool. In CRC, HCC and NSCLC studies, TCR sharing was found between GZMK+ predysfunctional TILs and CX3CR1+ effector populations from blood and normal tissue (60, 65, 80). These results support a model in which the predysfunctional population forms a branchpoint from which differentiation trajectories of effector versus exhausted CD8+ T cells emanate, possibly reflecting CD8+ T cells after the first step of priming that subsequently receive CD4+ T cell help, versus CD8+ T cells that do not. However, it was not determined in those studies whether the T cells that shared TCRs were tumor-specific. In melanoma, intratumoral GZMH+ effector CTLs did not share TCRs with the predysfunctional or exhausted CD8+ TIL population, indicating that they formed a separate lineage (64). Interestingly, in this study, tumor reactivity was enriched in the dysfunctional but not in the cytotoxic TIL population, suggesting that the cytotoxic population consists of bystander cells that do not recognize the tumor, as was demonstrated before (81, 82). These data argue that in melanoma, persistent tumor antigen recognition drives the conversion of helpless tumor-specific TILs from the predysfunctional to the terminally exhausted state, while the tumor may also harbor helped bystander cells with an effector phenotype (61). Whether tumor-specific dysfunctional TILs can differentiate within the TME into competent effector CTLs remains to be investigated.
Reinvigoration
In mouse models of melanoma, the TCF-1+ predysfunctional CD8+ TILs proved to be the responders to PD-1 blockade and necessary for tumor control (7, 10, 56). In melanoma patients, an increased fraction of TCF-1+ predysfunctional CD8+ TILs is a positive predictor for response to PD-(L)1 targeted therapy (7, 46). In a murine liver cancer model, CD101 and CD38 marked predysfunctional versus terminally exhausted TILs. These markers were heterogeneously expressed by PD-1high TILs from melanoma and NSCLC patients, suggesting that the human PD-1high TIL population consists of a mixture of predysfunctional and terminally exhausted cells (63).
Helplessness and Predysfunction in Cancer
CD4+ T cell help is less likely to be delivered in cancer than in infection for the following reasons: Tumor cells generally do not express PAMPs and may only exude DAMPs under specific circumstances. Therefore, they are less likely to activate migratory DCs than infected cells. Furthermore, in the suppressive TME, migratory cDC2s, which are essential for the priming of CD4+ T cells (83), are reportedly suppressed by Tregs, resulting in suboptimal priming of CD4+ helper T cells in the tumor-draining lymph node (84). Also, DC-activating signals such as type I IFN that promote crosspresentation functions of the lymph node-resident cDC1 (16), are often lacking. In the blood of melanoma patients, tumor reactivity of CTLs was found to be enriched in the PD-1+ population (85). These data led us to hypothesize that helpless priming may contribute to the dysfunctional phenotype of CD8+ T cells in cancer.
To test this hypothesis, we performed bioinformatic analyses using our previously defined No Help versus Help signatures of mouse CD8+ T cells and datasets from mouse and human cancer. GSEA showed that gene sets characteristic of predysfunctional TCF-1+ CD8+ TILs from a gp33 antigen bearing B16 melanoma mouse model (10) were enriched in the No Help gene expression signature (Figure 3A). In an ovalbumin (OVA) antigen-bearing B16 melanoma model from a different research group (7), TCF-1+ CD8+ TILs displayed a higher No Help score than TCF-1− CD8+ TILs (Figure 3B). These results indicate that also in mouse cancer models, dysfunctional TCF-1+ CD8+ TILs display a gene expression profile that resembles that of helpless cells. In NSCLC patients, the presence of PD-1high TILs was a positive predictor of response to PD-1 blockade therapy. Importantly, PD-1high TIL displayed higher intrinsic tumor reactivity compared to TIL populations with intermediate or no PD-1 expression from the same tumor (8). We used the published gene expression profiles from these matched TIL subsets to calculate their No Help score. Among these patients’ TIL populations, the transcriptome of PD-1high TILs was most similar to that of helpless vaccine antigen-specific CD8+ T cells (Figure 3C). These data from human cancer support our hypothesis that dysfunctional tumor-reactive CD8+ T cells are cells that have lacked help during priming.
Helpless Dysfunction Model
We present a novel model posing that virus-specific or tumor-specific, predysfunctional TCF-1+ CD8+ T cells in chronic infection or cancer result from priming in the absence of CD4+ T cell help. CD4+ T cell help delivered during priming optimizes effector differentiation of antigen-specific CD8+ T cells (3, 53). Additionally, CD4+ T cell help promotes effector memory CD8+ T cell (TEM) generation, and renders these TEM cells more effector-like on a per-cell basis (30). These results are in line with a previously proposed progressive differentiation model for primed CD8+ T cells (86), adding that CD4+ T cell help shifts differentiation of primed CD8+ T cells toward a more effector-like state (Figure 4).
By optimizing CTL function, CD4+ T cell help contributes to antigen clearance, which is necessary for proper memory formation (87, 88). CD4+ T cell help also promotes the long-term maintenance of TCM cells and is necessary for open configuration of gene loci encoding CTL effector molecules in memory CD8+ T cells (30, 89, 90). The epigenetic imprinting induced by help signals during priming allows memory cells to rapidly exert effector functions upon reactivation in a CD4+ T helper cell-independent manner (30, 91).
In the absence of CD4+ T cell help, effector differentiation of CD8+ T cells is incomplete, resulting in predysfunctional CTLs that have limited cytotoxic and migratory potential and express coinhibitory receptors (3, 32), which prohibits antigen clearance. The chronic stimulation of memory precursor cells impairs the formation of a memory pool and instead drives their differentiation into predysfunctional CTLs, as seen in chronic infection and cancer (39, 54). These predysfunctional TCF-1+ cells have self-maintaining properties and form the progenitors of the terminally exhausted TCF-1− CD8+ T cell pool (58). Exhausted CD8+ T cells differ in their epigenetic and transcriptional states from predysfunctional CD8+ T cells. They have a further developed effector differentiation program, but are fixed in their dysfunctional state (55).
Overcoming CTL Dysfunction By Help Signals
Based on our model, we propose that in chronic infection and cancer, CTL dysfunction can be overcome by help signals. In that scenario, help signals would enable the CTLs to progress further toward a terminal effector differentiation state. Adoptive transfer of CD4+ T cells has been shown to increase proliferation of pre-existing TCF-1+ CD8+ T cells in chronic LCMV infection (53). Also, adoptive transfer of IL-21-producing CD4+ T cells into tumor-bearing mice induced generation of a CX3CR1+ effector CD8+ T cell pool, leading to improved tumor control (29). Using help signals to alleviate CTL dysfunction is not yet incorporated into clinical protocols. In the clinic, PD-1 blockade is used as method to “reinvigorate” dysfunctional CTLs.
We here propose that PD-1 blockade recapitulates aspects of CD4+ T cell help and acts on the predysfunctional/helpless CD8+ T cell population. As reviewed in the preceding sections, in chronic LCMV infection and cancer, PD-1 blockade induced proliferation of predysfunctional TCF-1+ CD8+ T cells. The question is whether PD-1 blockade is sufficient to overcome lack of help and—by association—to convert predysfunctional CTLs into fully functional effectors. In chronic LCMV infection, established through transient CD4+ T cell depletion, PD-L1 blockade promoted differentiation of predysfunctional CD8+ T cells into transitional cells that displayed a more effector-like phenotype and contributed to virus control. However, eventually these cells became terminally exhausted (49). Blockade of the PD-L1/PD-1 axis in a helpless setting increases the magnitude of the antigen-specific CD8+ T cell response, but in contrast to CD4+ T cell help, it did not rescue the formation of the effector population that conferred protection against chronic infection and cancer (29). These results suggest that predysfunctional/helpless cells cannot be rescued by PD-1 blockade alone.
The prevailing view is that PD-1 blockade relieves pre-existing dysfunctional CTLs from suppression in the TME. However, accumulating data argue that PD-1 blockade can also facilitate de novo CTL priming. Firstly, PD-(L)1 targeted immunotherapy can be effective while PD-L1 is not expressed in the tumor (92). Secondly, PD-1 signaling impedes TCR as well as CD28 signaling, indicating that it can also impact on costimulation at the T cell/DC interface (93). In agreement with this, tumor regression upon PD-1 blockade in mouse colon carcinoma depended on CD28 co-stimulation (94). Thirdly, the response to PD-1 blockade in mouse colon carcinoma was found to depend on influx of newly activated CD8+ T cells from tumor draining lymph nodes (95).
Recent data from human cancer also argue that PD-1 blockade promotes CD8+ T cell priming: In basal cell carcinoma, new CD8+ T cell clones entered the upon tumor PD-1 blockade (96). TCR repertoire analysis argued that these clones pre-existed in blood and entered the tumor after treatment (97). PD-1 is expressed rapidly after stimulation of naive CD8+ T cells, and inhibits effector differentiation during priming (98). We found that in the CD4+ T cell help-dependent second step of priming, CD8+ T cells downregulate PD-1, whereas helpless cells maintain PD-1 expression (3). This supports a model in which PD-1 serves as a checkpoint in the two-step T cell priming process.
We have shown in the mouse vaccination model, that the effects of CD4+ T cell help on the CTL response could be mimicked by combined PD-1-blockade and CD27 agonism (99). We and others have shown that delivery of CD4+ T cell help is highly dependent on CD70-CD27 signaling and CD27 agonism installs a large part of the Help gene signature into CD8+ T cells during priming (3, 20, 24, 25). The combined effect of PD-1 blockade and CD27 agonism likely recapitulates combined CD28 and CD27 costimulation that are known to complement each other in generation of the CTL effector pool (23). The collective data make a strong case for combining CD27 agonism with PD-(L)1 blockade in cancer immunotherapy.
Concluding Remarks
We here present our hypothesis that CD8+ T cell priming in the absence of CD4+ T cell help leads to CD8+ T cell dysfunction. We pose that exhausted antigen-specific CD8+ T cells observed in infection and cancer derive not from previously active CTLs, but from helpless CD8+ T cells that emerge from the priming process in a dysfunctional state. We pose that provision of CD4+ T cell help, or the key signals that recapitulate help for CD8+ T cells will be crucial for the development of effective immunotherapeutic strategies in chronic infection and cancer. In immunotherapy, reverting exhausted cells back to a functional phenotype is considered an important challenge (1). Alternatively, we argue that in patients with immunogenic cancer types, de novo priming of helped CD8+ T cells will be beneficial for tumor control. For this purpose, potential approaches are antigen-agnostic PD-1/CD27 targeting or antigen-informed therapeutic vaccination. Such vaccines should contain MHC-I and MHC-II epitopes to activate both CD8+ and CD4+ T cells. Other strategies include specific targeting of antigens and activation signals to XCR1+ cDC1s. In these approaches, evaluation of the transcriptional help signature in tumor-specific CD8+ T cells is a potential diagnostic tool.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/.
Author Contributions
JuB performed data analysis and wrote the paper. ST performed data analysis. JaB contributed to writing the paper. HE critically reviewed the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by grant 11079 from the Dutch Cancer Society.
Conflict of Interest
HE and ST were employees of Aduro Biotech. HE has stocks and/or stock options in Aduro Biotech, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Paul Vink and Lars Guelen (Aduro Biotech) for their support of this project.
References
1. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining ‘T cell exhaustion.’. Nat Rev Immunol (2019) 19:665–74. doi: 10.1038/s41577-019-0221-9
2. Xia A, Zhang Y, Xu J, Yin T, Lu X-J. T cell dysfunction in cancer immunity and immunotherapy. Front Immunol (2019) 10:1719. doi: 10.3389/fimmu.2019.01719
3. Ahrends T, Spanjaard A, Pilzecker B, Bąbała N, Bovens A, Xiao Y, et al. CD4+T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity (2017) 47:848–61. doi: 10.1016/j.immuni.2017.10.009
4. Kim D, Langmead B, Salzberg SL. HISAT: A fast spliced aligner with low memory requirements. Nat Methods (2015) 12:357–60. doi: 10.1038/nmeth.3317
5. Liao Y, Smyth GK, Shi W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics (2014) 30:923–30. doi: 10.1093/bioinformatics/btt656
6. Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (2010) 26:139–40. doi: 10.1093/bioinformatics/btp616
7. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
8. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med (2018) 24:994–1004. doi: 10.1038/s41591-018-0057-z
9. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T Cell Factor 1-expressing memory-like CD8+T cells sustain the immune response to chronic viral infections. Immunity (2016) 45:415–27. doi: 10.1016/j.immuni.2016.07.021
10. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity (2019) 50:195–211. doi: 10.1016/j.immuni.2018.12.021
11. Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol (2018) 18:635–47. doi: 10.1038/s41577-018-0044-0
12. Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, et al. Robust anti-viral immunity requires multiple distinct T cell-dendritic cell interactions. Cell (2015) 162:1322–37. doi: 10.1016/j.cell.2015.08.004
13. Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, Mueller SN. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+and CD8+T cell activation to localized viral infection. Immunity (2015) 43:554–65. doi: 10.1016/j.immuni.2015.07.020
14. Kitano M, Yamazaki C, Takumi A, Ikeno T, Hemmi H, Takahashi N, et al. Imaging of the cross-presenting dendritic cell subsets in the skin-draining lymph node. Proc Natl Acad Sci USA (2016) 113:1044–9. doi: 10.1073/pnas.1513607113
15. Castellino F, Germain RN. Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annu Rev Immunol (2006) 24:519–40. doi: 10.1146/annurev.immunol.23.021704.115825
16. Brewitz A, Eickhoff S, Dähling S, Quast T, Bedoui S, Kroczek RA, et al. CD8+T cells orchestrate pDC-XCR1+dendritic cell spatial and functional cooperativity to optimize priming. Immunity (2017) 46:205–19. doi: 10.1016/j.immuni.2017.01.003
17. Greyer M, Whitney PG, Stock AT, Davey GM, Tebartz C, Bachem A, et al. T Cell Help Amplifies Innate Signals in CD8+DCs for Optimal CD8+T Cell Priming. Cell Rep (2016) 14:586–97. doi: 10.1016/j.celrep.2015.12.058
18. Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, et al. Gene Regulation and Chromatin Remodeling by IL-12 and Type I IFN in Programming for CD8 T Cell Effector Function and Memory. J Immunol (2009) 183:1695–704. doi: 10.4049/jimmunol.0900592
19. Prilliman KR, Lemmens EE, Palioungas G, Wolfe TG, Allison JP, Sharpe AH, et al. Cutting Edge: A Crucial Role for B7-CD28 in Transmitting T Help from APC to CTL. J Immunol (2002) 169:4094–7. doi: 10.4049/jimmunol.169.8.4094
20. Taraban VY, Rowley TF, Al-Shamkhani A. Cutting Edge: A critical role for CD70 in CD8 T cell priming by CD40-licensed APCs. J Immunol (2004) 173:6542–6. doi: 10.4049/jimmunol.173.11.6542
21. Sanchez PJ, McWilliams JA, Haluszczak C, Yagita H, Kedl RM. Combined TLR/CD40 Stimulation Mediates Potent Cellular Immunity by Regulating Dendritic Cell Expression of CD70 In Vivo. J Immunol (2007) 178:1564–72. doi: 10.4049/jimmunol.178.3.1564
22. Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol (2003) 3:939–51. doi: 10.1038/nri1248
23. Hendriks J, Xiao Y, Borst J. CD27 Promotes Survival of Activated T Cells and Complements CD28 in Generation and Establishment of the Effector T Cell Pool. J Exp Med (2003) 198:1369–80. doi: 10.1084/jem.20030916
24. Bullock TNJ, Yagita H. Induction of CD70 on dendritic cells through CD40 or TLR stimulation contributes to the development of CD8 + T cell responses in the absence of CD4 + T cells. J Immunol (2005) 174:710–7. doi: 10.4049/jimmunol.174.2.710
25. Feau S, Garcia Z, Arens R, Yagita H, Borst J. The CD4 + T-cell help signal is transmitted from APC to CD8 + T- cells via CD27–CD70 interactions. Nat Commun (2012) 3:948. doi: 10.1038/ncomms1948
26. Obar JJ, Molloy MJ, Jellison ER, Stoklasek TA, Zhang W, Usherwood EJ, et al. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc Natl Acad Sci (2010) 107:193–8. doi: 10.1073/pnas.0909945107
27. Wilson EB, Livingstone AM. Cutting Edge: CD4 + T Cell-Derived IL-2 Is Essential for Help-Dependent Primary CD8 + T Cell Responses. J Immunol (2008) 181:7445–8. doi: 10.4049/jimmunol.181.11.7445
28. Peperzak V, Xiao Y, Veraar EAM, Borst J. CD27 sustains survival of CTLs in virus-infected nonlymphoid tissue in mice by inducing autocrine IL-2 production. J Clin Invest (2010) 120:168–78. doi: 10.1172/JCI40178
29. Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, et al. CD4+ T cell help is required for the formation of a cytolytic CD8+ T cell subset that protects against chronic infection and cancer. Immunity (2019) 53:1–15. doi: 10.1016/j.immuni.2019.10.009
30. Ahrends T, Busselaar J, Severson TM, Bąbała N, De Vries E, Bovens A, et al. CD4+ T cell help creates memory CD8+ T cells with innate and help-independent recall capacities. Nat Commun (2019) 10:5531. doi: 10.1038/s41467-019-13438-1
31. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
32. Provine NM, Larocca RA, Aid M, Penaloza-MacMaster P, Badamchi-Zadeh A, Borducchi EN, et al. Immediate dysfunction of vaccine-elicited CD8 + T cells primed in the absence of CD4 + T cells. J Immunol (2016) 197:1809–22. doi: 10.4049/jimmunol.1600591
33. Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJD, Suresh M, Altman JD, et al. Viral Immune Evasion Due to Persistence of Activated T Cells Without Effector Function. J Exp Med (1998) 188:2205–13. doi: 10.1084/jem.188.12.2205
34. Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity (2015) 42:265–78. doi: 10.1016/j.immuni.2015.01.006
35. Chen J, López-Moyado IF, Seo H, Lio C-WJ, Hempleman LJ, Sekiya T, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature (2019) 567:530–4. doi: 10.1038/s41586-019-0985-x
36. Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, et al. Transcription factor IRF4 promotes CD8+ T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity (2017) 47:1129–41. doi: 10.1016/j.immuni.2017.11.021
37. Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity (2007) 27:670–84. doi: 10.1016/j.immuni.2007.09.006
38. Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol (2012) 86:8161–70. doi: 10.1128/jvi.00889-12
39. Speiser DE, Utzschneider DT, Oberle SG, Münz C, Romero P, Zehn D. T cell differentiation in chronic infection and cancer: Functional adaptation or exhaustion? Nat Rev Immunol (2014) 14:768–74. doi: 10.1038/nri3740
40. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and Terminal Subsets of CD8+ T Cells Cooperate to Contain Chronic Viral Infection. Science (2012) 338:1220–5. doi: 10.1126/science.1229620
41. Utzschneider DT, Legat A, Fuertes Marraco SA, Carrié L, Luescher I, Speiser DE, et al. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol (2013) 14:603–10. doi: 10.1038/ni.2606
42. Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol (2016) 1:eaai8593. doi: 10.1126/sciimmunol.aai8593
43. He R, Hou S, Liu C, Zhang A, Bai Q, Han M, et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature (2016) 537:412–6. doi: 10.1038/nature19317
44. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature (2016) 537:417–21. doi: 10.1038/nature19330
45. Snell LM, MacLeod BL, Law JC, Osokine I, Elsaesser HJ, Hezaveh K, et al. CD8+ T Cell Priming in Established Chronic Viral Infection Preferentially Directs Differentiation of Memory-like Cells for Sustained Immunity. Immunity (2018) 49:678–94. doi: 10.1016/j.immuni.2018.08.002
46. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell (2018) 175:998–1013. doi: 10.1016/j.cell.2018.10.038
47. Jadhav RR, Im SJ, Hu B, Hashimoto M, Li P, Lin J-X, et al. Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc Natl Acad Sci USA (2019) 116:14113–8. doi: 10.1073/pnas.1903520116
48. Brummelman J, Mazza EMC, Alvisi G, Colombo FS, Grilli A, Mikulak J, et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors. J Exp Med (2018) 215:2520–35. doi: 10.1084/jem.20180684
49. Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, et al. Proliferating transitory T cells with an effector-like transcriptional signature emerge from PD-1+ stem-like CD8+ T cells during chronic infection. Immunity (2019) 51:1–16. doi: 10.1016/j.immuni.2019.11.002
50. Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature (2019) 576:465–70. doi: 10.1038/s41586-019-1836-5
51. Yao C, Sun H-W, Lacey NE, Ji Y, Moseman EA, Shih H-Y, et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat Immunol (2019) 20:890–901. doi: 10.1038/s41590-019-0403-4
52. Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature (2019) 571:265–9. doi: 10.1038/s41586-019-1326-9
53. Kanev K, Wu M, Drews A, Roelli P, Wurmser C, von Hösslin M, et al. Proliferation-competent Tcf1+ CD8 T cells in dysfunctional populations are CD4 T cell help independent. Proc Natl Acad Sci USA (2019) 116:20070–6. doi: 10.1073/pnas.1902701116
54. Philip M, Schietinger A. Heterogeneity and fate choice: T cell exhaustion in cancer and chronic infections. Curr Opin Immunol (2019) 58:98–103. doi: 10.1016/j.coi.2019.04.014
55. Mann TH, Kaech SM. Tick-TOX, it’s time for T cell exhaustion. Nat Immunol (2019) 20:1090–9. doi: 10.1038/s41590-019-0478-y
56. Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1 – CD8 + tumor-infiltrating T cells. Immunity (2019) 50:181–94. doi: 10.1016/j.immuni.2018.11.014
57. Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity (2019) 51:840–55. doi: 10.1016/j.immuni.2019.09.013
58. Kallies A, Zehn D, Utzschneider DT. Precursor exhausted T cells: key to successful immunotherapy? Nat Rev Immunol (2020) 20:128–36. doi: 10.1038/s41577-019-0223-7
59. Shan Q, Hu S, Chen X, Danahy DB, Badovinac VP, Zang C, et al. Ectopic Tcf1 expression instills a stem-like program in exhausted CD8+ T cells to enhance viral and tumor immunity. Cell Mol Immunol (2020). doi: 10.1038/s41423-020-0436-5
60. Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med (2018) 24:978–85. doi: 10.1038/s41591-018-0045-3
61. van der Leun AM, Thommen DS, Schumacher TN. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer (2020) 20:218–32. doi: 10.1038/s41568-019-0235-4
62. Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity (2016) 45:389–401. doi: 10.1016/j.immuni.2016.07.011
63. Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature (2017) 545:452–6. doi: 10.1038/nature22367
64. Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell (2019) 176:775–89. doi: 10.1016/j.cell.2018.11.043
65. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell (2017) 169:1342–56. doi: 10.1016/j.cell.2017.05.035
66. Yu D, Ye L. A portrait of CXCR5+ follicular cytotoxic CD8+ T cells. Trends Immunol (2018) 39:965–79. doi: 10.1016/j.it.2018.10.002
67. Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol (2016) 17:1187–96. doi: 10.1038/ni.3543
68. Jiang H, Li L, Han J, Sun Z, Rong Y, Jin Y. CXCR5+CD8+T cells indirectly offer B cell help and are inversely correlated with viral load in chronic hepatitis B infection. DNA Cell Biol (2017) 36:321–7. doi: 10.1089/dna.2016.3571
69. Utzschneider DT, Alfei F, Roelli P, Barras D, Chennupati V, Darbre S, et al. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J Exp Med (2016) 213:1819–34. doi: 10.1084/jem.20150598
70. Beltra J-C, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental relationships of four exhausted CD8+ T Cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity (2020) 52:825–41. doi: 10.1016/j.immuni.2020.04.014
71. Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature (2019) 571:211–8. doi: 10.1038/s41586-019-1325-x
72. Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature (2019) 571:270–4. doi: 10.1038/s41586-019-1324-y
73. Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc Natl Acad Sci USA (2019) 116:12410–5. doi: 10.1073/pnas.1905675116
74. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity (2011) 35:400–12. doi: 10.1016/j.immuni.2011.06.015
75. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science (2016) 354:1165–9. doi: 10.1126/science.aae0491
76. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science (2016) 354:1160–5. doi: 10.1126/science.aaf2807
77. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell (2017) 170:142–57. doi: 10.1016/j.cell.2017.06.007
78. Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol (1994) 68:8056–63. doi: 10.1128/JVI.68.12.8056-8063.1994
79. Oosterhuis K, Aleyd E, Vrijland K, Schumacher TN, Haanen JB. Rational design of DNA vaccines for the induction of human papillomavirus type 16 E6-and E7-specific cytotoxic T-cell responses. Hum Gene Ther (2012) 23:3001–1312. doi: 10.1089/hum.2012.101
80. Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature (2018) 564:268–72. doi: 10.1038/s41586-018-0694-x
81. Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med (2019) 25:89–94. doi: 10.1038/s41591-018-0266-5
82. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature (2018) 557:575–9. doi: 10.1038/s41586-018-0130-2
83. Gerner M, Casey KA, Kastenmuller W, Germain RN. Dendritic cell and antigen dispersal landscapes regulate T cell immunity. J Exp Med (2017) 214:3105–22. doi: 10.1084/jem.20170335
84. Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell (2019) 177:556–71. doi: 10.1016/j.cell.2019.02.005
85. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med (2016) 22:433–8. doi: 10.1038/nm.4051
86. Restifo NP, Gattinoni L. Lineage relationship of effector and memory T cells. Curr Opin Immunol (2013) 25:556–63. doi: 10.1016/j.coi.2013.09.003
87. Kim J, Jeong Ryu S, Oh K, Ju J-M, Yeong Jeon J, Nam G, et al. Memory programming in CD8+ T-cell differentiation is intrinsic and is not determined by CD4 help. Nat Commun (2015) 6:7994. doi: 10.1038/ncomms8994
88. Li Y, Shen C, Zhu B, Shi F, Eisen HN, Chen J. Persistent antigen and prolonged AKT–mTORC1 activation underlie memory CD8 T cell impairment in the absence of CD4 T cells. J Immunol (2015) 195:1591–8. doi: 10.4049/jimmunol.1500451
89. Northrop JK, Wells AD, Shen H. Cutting Edge: Chromatin remodeling as a molecular basis for the enhanced functionality of memory CD8 T cells. J Immunol (2008) 181:865–8. doi: 10.4049/jimmunol.181.2.865
90. Northrop JK, Thomas RM, Wells AD, Shen H. Epigenetic Remodeling of the IL-2 and IFN -γ Loci in Memory CD8 T Cells Is Influenced by CD4 T Cells. J Immunol (2006) 177:1062–9. doi: 10.4049/jimmunol.177.2.1062
91. Cullen JG, McQuilten HA, Quinn KM, Olshansky M, Russ BE, Morey A, et al. CD4+ T help promotes influenza virus-specific CD8+ T cell memory by limiting metabolic dysfunction. Proc Natl Acad Sci USA (2019) 116:4481–8. doi: 10.1073/pnas.1808849116
92. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature (2017) 541:321–30. doi: 10.1038/nature21349
93. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355:1428–33. doi: 10.1126/science.aaf1292
94. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1–targeted therapies is CD28-dependent. Science (2017) 355:1423–7. doi: 10.1126/science.aaf0683
95. Fransen MF, Schoonderwoerd M, Knopf P, Camps MG, Hawinkels LJ, Kneilling M, et al. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight (2018) 3:e124507. doi: 10.1172/jci.insight.124507
96. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med (2019) 25:1251–9. doi: 10.1038/s41591-019-0522-3
97. Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen Y-JJ, Chitre AS, et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature (2020) 579:274–8. doi: 10.1038/s41586-020-2056-8
98. Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, et al. Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci (2018) 115:4749–54. doi: 10.1073/pnas.1718217115
Keywords: CD8+ T cell, CD4+ T cell, exhaustion, dysfunction, cancer, infection
Citation: Busselaar J, Tian S, van Eenennaam H and Borst J (2020) Helpless Priming Sends CD8+ T Cells on the Road to Exhaustion. Front. Immunol. 11:592569. doi: 10.3389/fimmu.2020.592569
Received: 07 August 2020; Accepted: 21 September 2020;
Published: 06 October 2020.
Edited by:
Lewis Z. Shi, University of Alabama at Birmingham, United StatesReviewed by:
Weiyi Peng, University of Houston, United StatesAllan Zajac, University of Alabama at Birmingham, United States
Copyright © 2020 Busselaar, Tian, van Eenennaam and Borst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jannie Borst, ai5nLmJvcnN0QGx1bWMubmw=
†Present address: Sun Tian, Carbon Logic Biotech (HK) Ltd, Hong Kong, China