 Luigi Tornillo
Luigi Tornillo- Institute of Pathology, University of Basel, Basel, Switzerland
Gastrointestinal stromal tumors (GISTs) are the most frequent mesenchymal tumors of the gastrointestinal tract. The discovery that these tumors, formerly thought of smooth muscle origin, are indeed better characterized by specific activating mutation in genes coding for the receptor tyrosine kinases (RTKs) CKIT and PDGFRA and that these mutations are strongly predictive for the response to targeted therapy with RTK inhibitors has made GISTs the typical example of the integration of basic molecular knowledge in the daily clinical activity. The information on the mutational status of these tumors is essential to predict (and subsequently to plan) the therapy. As resistant cases are frequently wild type, other possible oncogenic events, defining other “entities,” have been discovered (e.g., succinil dehydrogenase mutation/dysregulation, insuline growth factor expression, and mutations in the RAS-RAF-MAPK pathway). The classification of disease must nowadays rely on the integration of the clinico-morphological characteristics with the molecular data.
Introduction
Gastrointestinal stromal tumors (GISTs) are the most frequent mesenchymal tumors of the gastrointestinal tract, with an incidence between 15 and 20 new cases/10,00,000/year (1, 2). The actual incidence may be, however, higher, as incidental GISTs are relatively frequent (3–6). The most frequent localization is the stomach, followed by the small intestine, the colon–rectum, and the esophagus (7). The existence of true extragastrointestinal GISTs is controversial. Although the concept that mesenchymal tumors of the gastrointestinal tract with leiomyomatous morphology are “bizarre” or “blastomatous” had been established almost 70 years ago by Stout (8, 9), the actual histogenesis of these tumors was defined about 30 years ago (in the pre-immunohistochemistry era) by Mazur and Clark (10), who proposed the non-committal term of “Gastrointestinal stromal tumors.” With the development of immunohistochemistry, the concept could be better specified. Most cases (70%) were positive for CD34, and the expression of smooth muscle markers was seen in less than 50%. CD34 is normally expressed in endothelia, in hematopoietic stem cells, perivascular fibroblasts, and stromal fibroblasts in various localization, thus underlining the “stromal,” but uncommittal nature of these tumors (11). Moreover, CD34 is expressed in a proportion of interstitial cells of Cajal (ICC), the pacemaker cells of the gastrointestinal tract (12). In 1998, two groups (13, 14) showed independently that more than 80% of these enigmatic tumors harbor constitutively activating mutations of the ckit gene that encode an important receptor tyrosine kinase (RTK) type III. CKIT (also known as CD117) immunohistochemical expression was contemporarily reported in more than 95% of GIST cases, thus becoming an important tool for the diagnosis (15, 16) (Figure 1). The immunophenotype is shared by the ICC (17), so that Kindblom et al. proposed in their paper the term “GIPACT,” GastroIntestinal PAcemaker Cells Tumor. This term was, however, not retained. Although sometimes questioned, the theory of origin from ICC is nowadays generally accepted. In 2003, it was shown that a substantial fraction of CKIT-wild-type GISTs’ harbor-activating mutations in pdfgra (platelet-derived growth factor alpha) gene, coding for another important RTK type III (18). This confirms that the oncogenesis of GISTs is probably related to early activation of RTKs. Interestingly, the immunohistochemical positivity for CD117 is independent from the mutational status of RTK genes (19). Another almost pathognomonic IHC marker is DOG-1, corresponding to the potassium transporter ANO1 (Figure 1D). The importance of the RTK mutational status is also underlined by the fact that CKIT and PDGFRA are very good target for the targeted therapy with the RTK inhibitor imatinib mesylate (Gleevec®, Novartis Pharma AG, Basel, Switzerland). Imatinib is now the first-line option in the medical treatment of GISTs (1, 20). Imatinib is approved in US and in Europe for adjuvant therapy (21) and may be used also in a neoadjuvant setting to reduce the tumor mass (1). Imatinib is very effective on sensitive GISTs as it was shown in the communication of Joensuu published in “The New England Journal of Medicine” in 2003 (22).
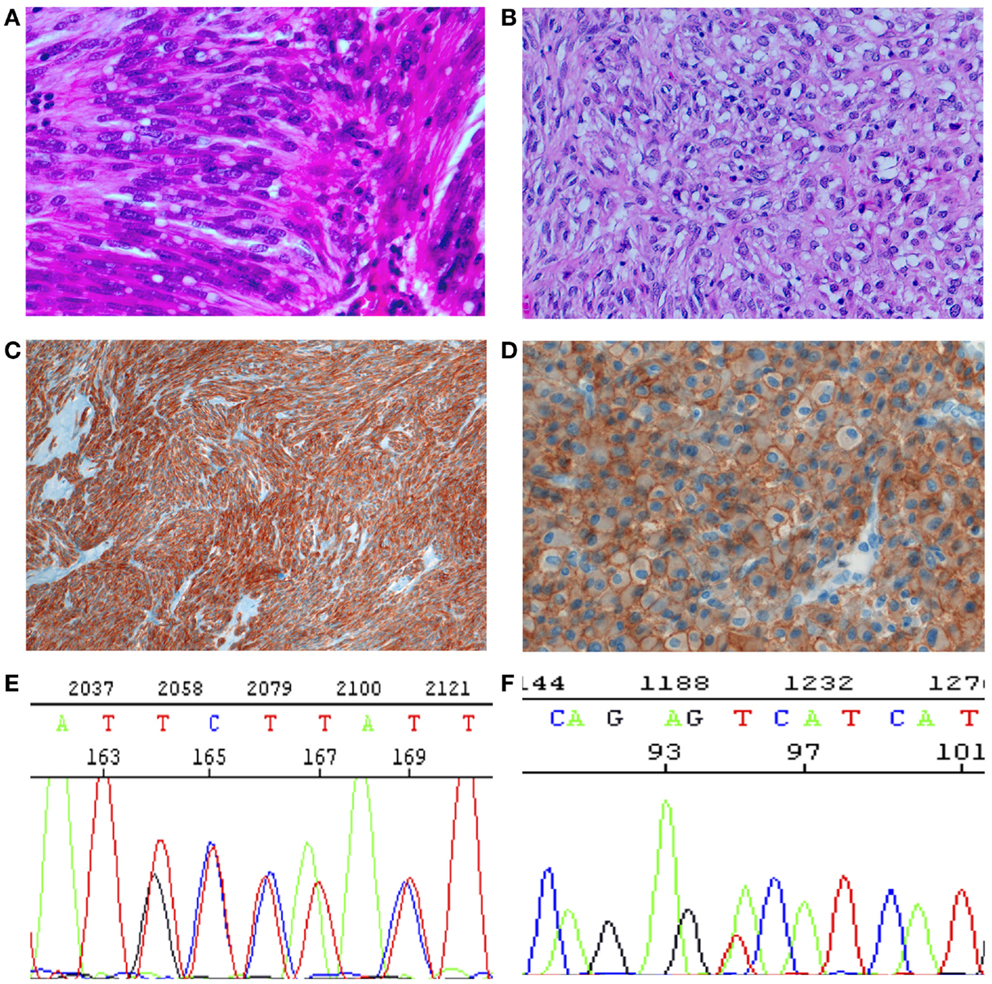
Figure 1. Histology, immunohistochemistry, and molecular genetics of GISTs. (A,B) Histology. (A) Spindle cell tumor, with paranuclear vacuoles (so-called “leiomyoblastoma”). HE 40×. (B) Epitheloid tumor. Large, clear cytoplasm with central nucleus. HE 40×. (C) Spindle cells diffusely positive for CKIT (CD117). IHC 10×. (D) Epitheloid cells strongly and diffusely positive for DOG1, with evident membrane enhancement. IHC 40×. (E) Sanger sequencing with a duplication of GCC TAT in positions 502–503 (p.A502-Y503 dup). Mutation associated with sensitivity to imatinib. (F). Sanger sequencing with a substitution (A–C) in position 842, (p.D842V), imatinib resistant.
The therapy with imatinib has become the paradigm of targeted therapy in solid tumors. In spite of this great success, primary and secondary resistance to targeted therapy remains a problem to solve. Beneath a minority of GISTs that simply do not respond to the therapy with imatinib (primary resistance), due probably to their genetic constitution (see below), half of the patients develop disease progression after 2 years of treatment with imatinib (23). The main predictor of the response to therapy is represented by the mutation in the RTK genes (7, 21, 24). The genetic alterations in the RTK genes are important early events in the oncogenesis of GISTs, and define the response to targeted therapy.
Recently, mutations in BRAF and KRAS, both belonging to the RAS-RAF-MAPK pathway (25, 26), and hyperexpression of the transcription factor ETV1 (27) have been described.
The understanding of GISTs biology has stimulated the development of RTK inhibitors. Nowadays, there are at least three molecules that can be used against KIT and/or PDGFRA. The evolution of the idea of GISTs represents also how our paradigm of classification of disease is changing. From a “morphologic/etiologic” classification, we are going to more dynamic and flexible criteria, where the “old” clinicopathologic parameters are integrated with/substituted by the molecular alterations. This process is advancing also in other fields of oncology, as shown e.g., by the fourth edition of the WHO/IARC “blue books” (http://www.iarc.fr/en/research-groups/sec4/).
This review will, therefore, focus on the biology and molecular pathology of GISTs, and on the evolution of their classification.
Molecular Alterations in GISTs
RTK III
CKIT and PDGFRA are RTK III, together with PDGFRB, macrophage colony-stimulating-factor receptor (CSFR1), FLT1, Flk/KDR, and Fl cytokine receptor (FLT3) (28, 29). RTK III have five Ig-like extracellular domains, one transmembrane domain, one intracellular juxtamembrane regulatory domain, and two intracellular tyrosine kinase domain with autophosphorylating capacity (29) (Figure 2). CKIT and PDGFRA are located on the same chromosomal region (4q12) and are very similar, both in the sequence and in the structure (29). The ligands (stem cell factor, SCF for CKIT and platelet-derived growth factor, PDGF for PDGFRA) cause homodymerization of the receptor. Subsequently, the TK domains autophosphorylate and activate, triggering the metabolic pathways of RAS-RAF-MAPK, PI3K-AKT, and signal transducer and activator of transcription 3 (STAT3) (30–34).
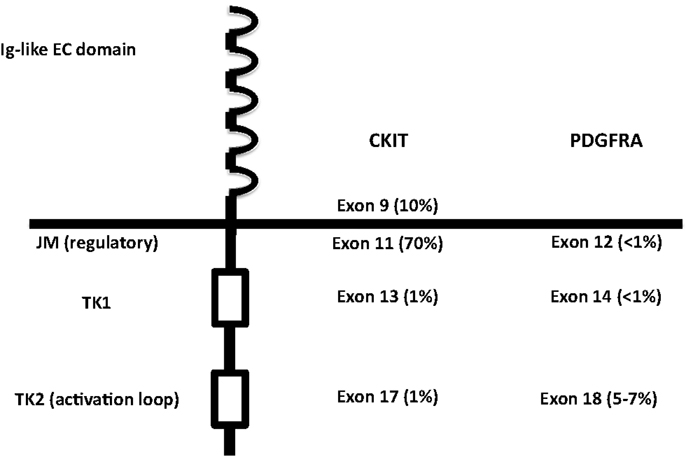
Figure 2. Structure of RTK III, with localization of the activating mutations in KIT and PDGFRA. EC, extracellular; JM, juxtamembrane; TK, tyrosine kinase.
CKIT
CKIT is crucial in the development of different cell types, in particular melanocytes, hematopoietic progenitor cells, mast cells, primordial germ cells, and, last but not least ICC, the probable cells of origin of GISTs (17, 21, 35). KIT-activating mutations are important for the genesis and development of several human tumor types: seminoma/dysgerminoma, mastocytosis, acute myeloid leukemia, melanoma, and GIST (36–39). CKIT can be, therefore, considered as an important oncogenetic factor in various different tissues.
Germline KIT mutations are very rare and they are associated with familial GISTs (21), while activating mutations are generally somatic and cause homodymerization of KIT and subsequent tyrosine kinase activation without SCF binding. Activating mutations are most frequently (60–70% of cases) found in exon 11 of CKIT gene (7), corresponding to the juxtamembrane intracellular regulatory domain of the protein (Figure 2). Because of exon 11 mutations, the protein changes its structure from the inactive into the active state (40). There are many different mutation types (in-frame deletions, insertions, substitutions) in various combinations (24, 29, 41). The kind of the alteration is clearly associated with clinicopathologic characteristics, such as prognosis and localization. For instance, deletions, of codons 557–558, are associated with shorter overall and disease-free survival (42–45), whereas tandem internal duplications (Figure 1E) are associated with a relatively indolent course (46, 47).
Exon 9 (corresponding to the extracellular domain of the KIT molecule) mutations are present in almost 10% of GIST cases (7, 48) and are almost only duplications of six nucleotides, corresponding to the A502_Y503 residues of the protein (49). The mutations probably induce conformational changes similar to SCF binding, leading to autoactivation. Exon 9-mutated GISTs are more often localized in small intestine or colon and have poorer prognosis (47–53). They have also different gene expression signatures (50).
Mutations in tyrosine kinase domain (exon 17) and ATP-binding site (exon 13) are rare and generally not primary (7, 34, 42, 51–60). They are generally secondary mutations arising during the targeted treatment with receptor tyrosine kinase inhibitors (RTKI), and induce secondary resistance (58–60) (see below). Most KIT exon 13 mutations are single substitutions leading to K642E in the aminoacidic sequence (52).
Recently, deletion of codon 419 in exon 8 of KIT (corresponding to the extracellular domain) have been described in a very small subset of GIST (≈0.2% of all cases) (61). This mutation was found also in systemic mastocytosis. Interestingly, it seems that this mutation is associated with sensitivity to RTKI.
Activating mutations of KIT trigger pathways, such as MAPK, PI3K-AKT, and STAT3 (30, 33, 62–66). The MAPK pathway is functionally related to several transcription factors (e.g., MYC, ELK, and CREB) and finally positively controls the cell cycle (67). The PI3K pathway, through phosphorylation of AKT and PDK1, acts against the apoptosis and indirectly stimulates the cell cycle (68). Phosphorylation of STAT3 triggers its transport in the nucleus and behaviors as a transcription factor, with a positive effect on proliferation and a negative effect on apoptosis (69).
Activation of RTK is surely pivotal in the oncogenetic pathway of GISTs, and represents an early event, but is not the exclusive factor for the acquisition of the transformed phenotype. For instance, a very interesting and elegant study has been published (27), suggesting that ETV1 (ETS translocation variant 1) may interact with KIT in the oncogenesis. The clinical importance of ETV1, however, is controversial (70, 71).
Proteasome degradation regulates physiological levels of KIT. Mutated KIT is degraded more slowly than wild-type KIT, probably because of physical interaction with heat-shock protein 90 (72, 73). HSP90 inhibitors are indeed effective in experimental models of GISTs (74).
PDGFRA
PDGFRA has similar sequence and function as KIT. It is mutated in GISTs and AML and translocated on FILP1 in hematologic malignancies (21). Activating mutations in KIT and PDGFRA are mutually exclusive in GISTs (18, 31). Mutations in PDGFRA are in exons 12, 14, and 18, corresponding to the juxtamembrane regulatory domain and the tyrosine kinase domain of the protein, respectively (exons 11, 13, and 17 of KIT). PDGFRA-mutated GISTs are 10–12% of all cases (53, 56, 57, 75–79). They have epitheloid morphology and are generally gastric tumors, showing an indolent course (80–82). They also have different gene expression signatures (32, 83). On the other hand, the immunohistochemical phenotype (positivity for CD117, DOG-1, and PKC-Theta), the chromosomal alteration (deletion of 14q and 22q), and the biochemical properties (activated pathways and stabilization by HSP90) of PDGFRA-mutated GISTs overlap with CKIT-mutated tumors (84–87). The most frequent mutations in PDGFRA involve the aspartic acid in position 842 (Figure 1F) and are generally not sensitive to imatinib (18, 79, 88–95). Insertions and duplications are very rare, while deletions and deletions/insertions between codon 840 and 849 are more frequent. In exon 12, the most frequent mutation is a substitution T - > A, leading to a Val561Asp (19). In-frame deletions are also relatively frequent in exon 12, clustering in codons 559–72. Mutations in exon 14 are exceptional.
RAS-RAF-MAPK Pathway
In all, 10–15% of GISTs are wild type both for KIT and PDGFRA. This genotype is not related to the immunophenotype, namely with the positivity to KIT (CD117) and DOG1 stainings. Several “wt”-GISTs are strongly positive for CKIT (CD117), and the involvement of RTK has been proved functionally (phosphorylation and subsequent activation) (30). These so-called “wild-type” GISTs are a very heterogeneous group, showing different, probably oncogenic mutations. BRAF V600E has been described in 13% of “wild-type” GISTs (25, 58, 96). At the beginning, it was thought that the BRAF mutations were characteristic of “wild-type” GISTs. In a recent study, Miranda et al. (26) have suggested that BRAF is mutated in ca., 2% of GISTs carrying KIT or PDGFRA mutation, thus suggesting a further mechanism of primary resistance to imatinib treatment (see below). In the same study, 5% of GISTs carrying mutations in KIT or PDGFRA showed mutation in codon 2 of KRAS (G12A or G13D). Also, mutations in HRAS and NRAS have been found, but they are very rare. The presence and, above all, the meaning of mutations in KRAS were questioned by a recent study on 450 GISTs sequenced with Sanger’s method (97).
Neurofibromatosis 1
Neurofibromatosis 1 (NF1) (von Recklinghausen disease) is an autosomal dominant inherited disease, characterized by multiple neurofibromas, café-au-lait spots, and other mesenchymal tumors. NF1 is caused by inactivating mutations in the gene NF1, localized on chromosome 17, coding for neurofibromin (98). Patients with NF1 have an increased risk to develop multiple GISTs, with a spindle cell morphology and with predominant intestinal location (99). The tumors have rarely, “uncommon” mutations in RTK, but CKIT and PDGFRA most often are not mutated (100–104). Neurofibromin is analog to guanosine triphosphatase (GTPase) activating proteins (GAPs), which control the level and activity of RAS. Loss of NF1 leads to high levels of active RAS. Hyperactivation of the MAPK pathway is a consequence of somatic inactivation of wild-type NF1 allele in GISTs (105). From the cytogenetic point of view, they share deletion of 14q and 22q with classical GISTs.
Succinate Dehydrogenase Complex
Succinate dehydrogenase complex (SDH) is a heterotetramer composed by four subunits (SDHA-D), localized in the inner mitochondrial membrane. Subunit A oxidizes succinate to fumarate in the Krebs’ cycle. Subunit B participates in the electron transport chain for the oxidation of ubiquinone to ubiquinol, and subunits C and D (SDHC and SDHD) are membrane-anchoring subunits (106). SDH deficiency characterizes subsets of different tumors (e.g., GISTs, paragangliomas, renal cell carcinomas, and pituitary adenomas) (107, 108). SDH-deficient GISTs (identified by immunohistochemical negativity for SDHB) are the largest subgroup of “wt-GIST” (109, 110). They are always found in the stomach, are epitheloid, and often multiple and resistant to imatinib. Moreover, in contrast to “classical” GISTs, they metastasize to lymph nodes, and show activation of insuline growth factor receptor (IGFR). Their prognosis is not determined only by size, site, and mitotic index (110–116). They have an indolent course and even with liver metastases, the patients live long. SDH-deficient GISTs are the majority of pediatric GISTs in the stomach (110) and are part of two syndromes: the Carney triad (association of paranganglioma, pulmonary chondroma, and gastric GIST) and the Carney-Stratakis syndrome (association of GISTs and paragangliomas) (113, 117). The genetic events involved in the tumorigenesis of these tumors are not yet completely clarified (118, 119) and, in half of them, no mutation has been identified, although the IHC staining for SDH is negative (120). Defects of SDHx (independent from the involved subunit) induce accumulation of succinate, which inhibits degradation of HIF, subsequent increase of HIF levels, and its translocation in the nucleus, where HIF triggers the transcription of VEGF and IGF1R (121, 122), with aberrant proliferation and tumorigenic responses. A review of more than 1000 GISTs has shown that IGF1R expression is preferentially expressed in gastric SDH-deficient GISTs, but never in intestinal GISTs (123). The role of the axis IGF-IGF1R in maintaining the proliferative activity in GISTs has also been confirmed, both experimentally and on a series of SDH-deficient GISTs (124, 125).
Chromosomal Alterations
Chromosomal losses are much more frequent than chromosomal gains in GISTs. Losses of 14q, 22q, and 1p are the most frequent losses (126–128) and may subclassify GISTs in subsets with specific characteristics (129). Other losses are −9p, −11p, −17p, −10q, −13q, and −15q. Combining array CGH and transcriptome analysis Ylpaa et al. (130) have shown that the accumulation of cytogenetic changes (chromosomal losses) parallels the evolution of the tumor and could be seen as “genetic staging.” Specific genes (e.g., OXA1L on 14q and AKAP13 on 15q) are differentially expressed in the different “genetic stages.”
Cytogenetic gains are relatively rare and localized, but are associated with malignancy. In particular, gains on the loci for CCND1 and MDM2 gains have been shown to be associated with malignancy (131). On the other hand, genes involved in cell cycle control are dysregulated in high-risk tumors. Deletion or epigenetic inactivation of the gene CDKN2A, encoding for p16 and p14, two regulators of the cell cycle, is associated with malignant behavior (132–134). p16 downregulation may cause through Rb phosphorylation E2F1-dependent transcription of genes essential for late G1/S phase transition (135).
Targeted Therapy
The prognosis of high-risk/advanced GISTs has been very poor until 2000. Surgery was the exclusive therapy, and the median survival was less than 18 months (136). The introduction of imatinib mesylate in the therapy changed dramatically this situation. Imatinib was originally developed for chronic myeloid leukemia (CML) (29). It has a very strong inhibitory activity against KIT, fixing it in its inactive conformation. After the first communication of the dramatic success of the therapy in a case of advanced metastatic GIST in a 50-year-old woman (22), it was rapidly introduced in the therapy of metastatic non-resectable tumors and is now approved also in the adjuvant setting, with a clear-cut improving of the median survival (5 years). The most important predictive factor of the response to targeted therapy is the mutational status of the RTK genes (19, 21, 97). The best response rate is achieved with mutations in exon 11 of CKIT gene that are generally associated with a high response rate (≈80%), whereas mutations in exon 9 are associated with a response rate of ≈45% (91, 137) and deserve a higher doses of RTKI (Table 1). Mutations in exon 13 and exon 17 of CKIT are generally not responsive, such as mutations in codon 842 of PDFGRA and wt-GISTs (see below).
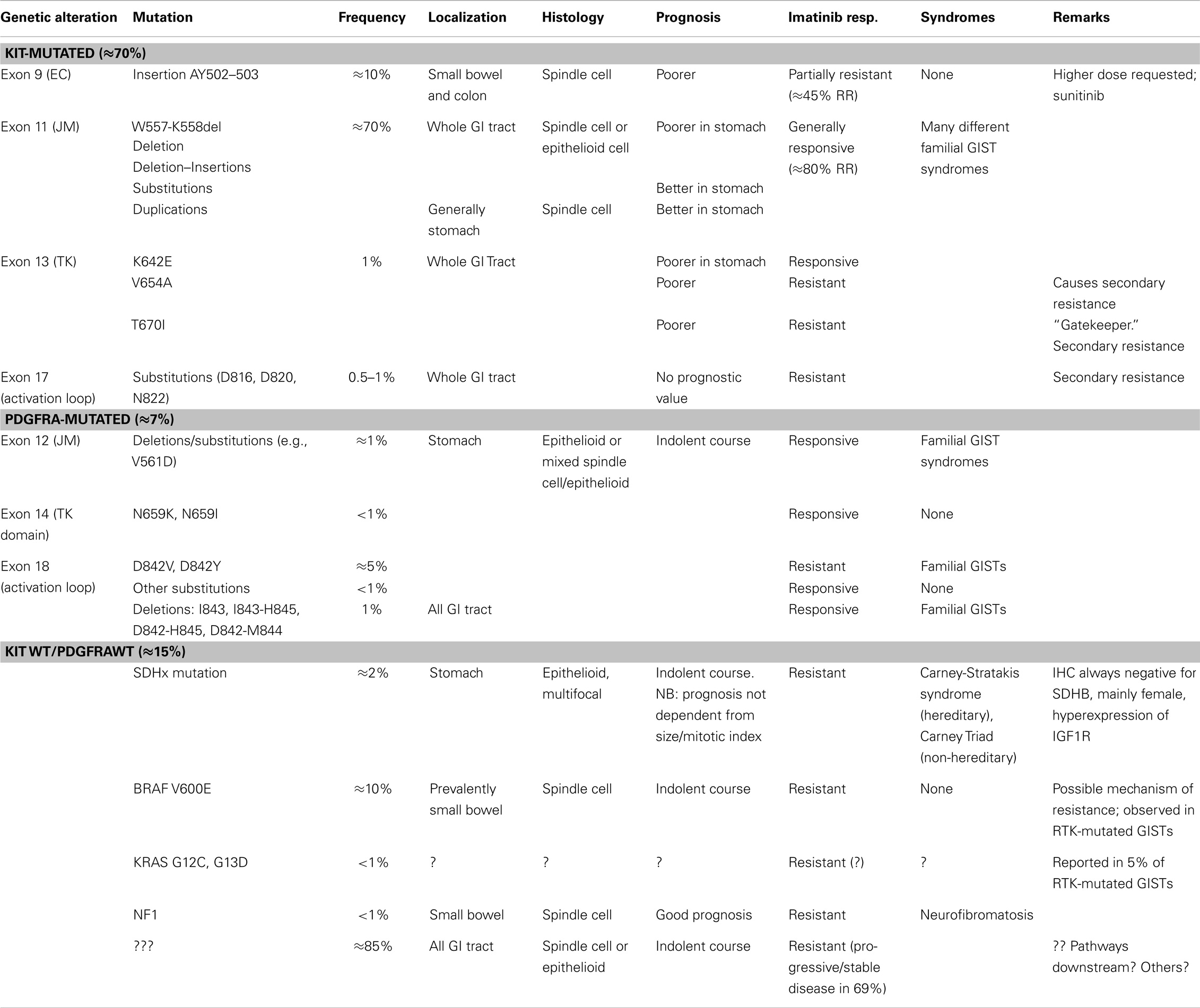
Table 1. Integration between clinicopathologic and molecular criteria in the classification of GISTs.
In the attempt to achieve a better response to therapy, many other different RTKI are in use or on trial. The best results have been achieved with sunitib and regorafenib that are at present used as second- and third-line treatments (138). A summary of the possible targets and targeted drugs is shown in Figure 3.
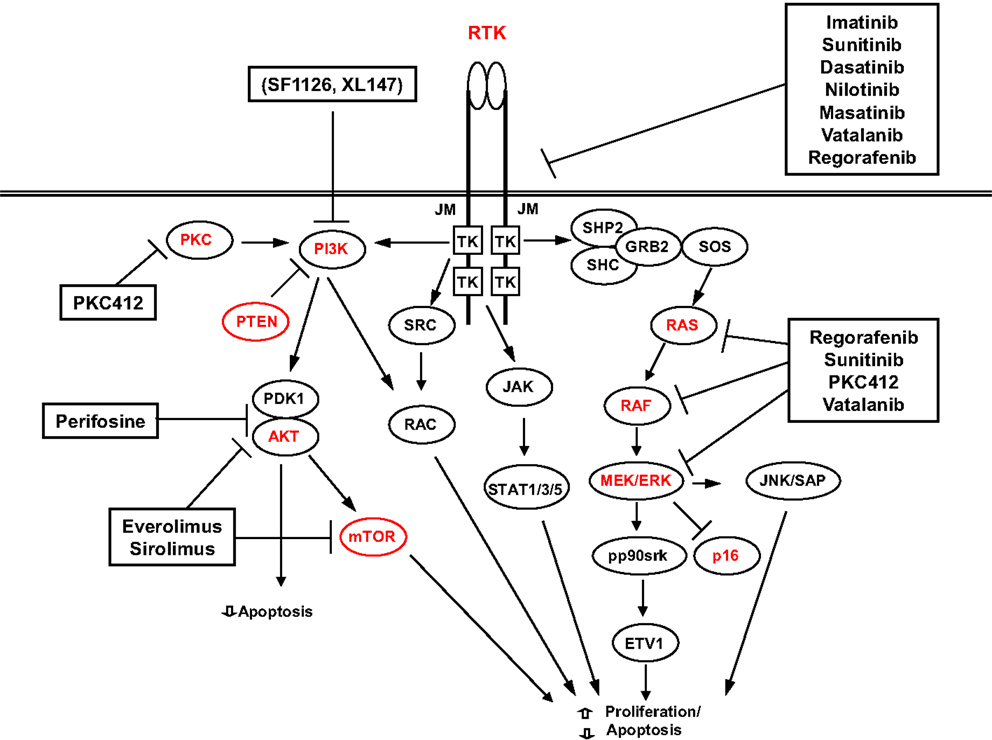
Figure 3. Possible therapeutic targets (red) and targeted drugs in GISTs.
Resistance to Therapy
In spite of the dramatic success of the targeted therapy with RTKI, there is evidence that also long-life imatinib treatment does not destroy completely GIST cells (139, 140) and that resistance to TKI therapy, both primary and secondary, arises in most of the cases (141).
Primary Resistance
Progression within the first 6 months of treatment with imatinib means primary resistance. The only predictor of primary resistance is the mutation in RTK (CKIT and PDGFRA) genes (29, 141). The probability of primary resistance is 5% for mutations in exon 11 of KIT, 16% for mutations in exon 9 of KIT, and 23% for wild-type GIST (59, 142). Tumors with mutation in exon 9 need a higher daily dose (800 mg instead of 400 mg) (29). Other CKIT mutations (EC domain, TK domain, or activation loop, exons 8, 13, and 17) of KIT are rare and mostly associated with resistance, although in some cases a response has been reported (e.g., K642E) (61, 143). PDGFRA D842V mutation in the exon 18 is strongly resistant to imatinib therapy, while other mutations in PDGFRA are usually sensitive (91, 141, 144). The mechanisms of primary resistance in “wild-type” KIT are probably multiple and not yet clarified. One possibility is represented by alterations in molecules “downstream” RTK, such as BRAF and KRAS, as hypothesized by Miranda et al. (26). In NF1-related GISTs, that are resistant to imatinib therapy, the activation of the MAPK-ERK axis due to silencing of the NF1 gene (see above) is probably the chief factor for imatinib resistance (105). For pediatric or SDH-mutated GISTs, that are almost invariably wild type for RTK, other possible targets may be KDR (VEGFR) or mTOR (41, 109).
Secondary Resistance
As recalled above, 50% of the patients treated with imatinib relapse after 2 years (23). It is interesting to note that progression is sometimes marked by an increase in density, with or without an increase in size. A sign of progression may also be the occurrence of an area of CT-hyperdensity within a responding (hypodense) lesion. This gives rise to the so-called “nodule within the nodule” pattern (141). This underlines the need of a revision of the classical oncologic criteria for progression (145). Secondary mutations in KIT or PDGFRA genes are the cause of most of the cases of secondary resistance (23, 142, 146–148). They generally occur in the same gene of the primary mutation. In KIT, they are localized in exon 13 or 17 of KIT, corresponding to the ATP-binding pocket and to the activation loop, respectively, the most frequent being T607I in the ATP-binding pocket (exon 13), followed by V654A (exon 13) and T823D (exon 17) (60, 137, 149). In most cases, the secondary mutation in PDGFRA is D842V. It is important to note that in case of relapse constituted by multiple nodes, these are often multiclonal, with different mutations in different nodes (23, 60, 142, 150). This molecular heterogeneity can also explain the relatively low efficacy of targeted therapy against relapsing GISTs.
Other Mechanisms of Resistance
Amplification of KIT or PDGFRA gene has been implicated in the development of resistance in RTK-wild-type GISTs (151). Activation of alternate oncogenic pathways is another possible resistance mechanism. This is the case of KRAS and BRAF (26) or PI3K/AKT pathway upstream of mTOR (152). IGF1R may represent another mechanism of resistance. It is expressed in subsets of RTK wild-type GISTs (58, 153). IGF1R-targeted therapy of wild-type GIST is being investigated in clinical trials (e.g., NCT01560260) (154).
These possible alternate mechanisms of resistance (primary or secondary) underline the necessity to develop schemes of therapy with a broad mechanism of action. Besides inhibitors combination and inhibitors with a broad spectrum (sorafenib, masitinib, vatalanib, nilotinib, or dasatinib that target also the VEGFR1 and VEGFR2), an important possibility is, therefore, aiming at other pathways. Promising targets are HSPs, histone deacetylases, signaling intermediates, or pathways, such as mTOR, PI3K, or MAPK [for review, see Ref. (138)].
Conclusion
The tremendous impact of molecular biology on modern medicine cannot be overestimated. In my opinion, the most important issue is the change of the paradigm of classification. GISTs represent a diagnostic category that changes its meaning and becomes more complex in parallel to the development of diagnostic tools and therapy (Figure 4). The non-committal term “GIST” probably, covers different “entities” (e.g., pediatric GISTs or SDH-deficient GISTs). A philosophical discussion on the actual meaning of the word “entity” in pathology and medicine goes beyond the purpose of this review. I think, however, that “entity” is an operative concept, whose content depends and is modified on the basis of the available diagnostic and therapeutic tools. This can be seen clearly in the case of GISTs. From the notion of a leiomyomatous tumor, based on HE staining, we are now dealing with a definition/classification that relies mainly on IHC stainings and molecular techniques, whereas the latter are essential for defining prognostic and therapeutic categories. The therapeutic strategy is the most important criterium for a medical classification, since the aim of medicine is the care of the patient. On the other hand, the behavior of GISTs can be defined only by integrating the molecular genetic findings with the “classical” clinicopathological parameter. A modern disease classification must, therefore, rely on the combination/integration of morphology and molecular biology (Table 1).
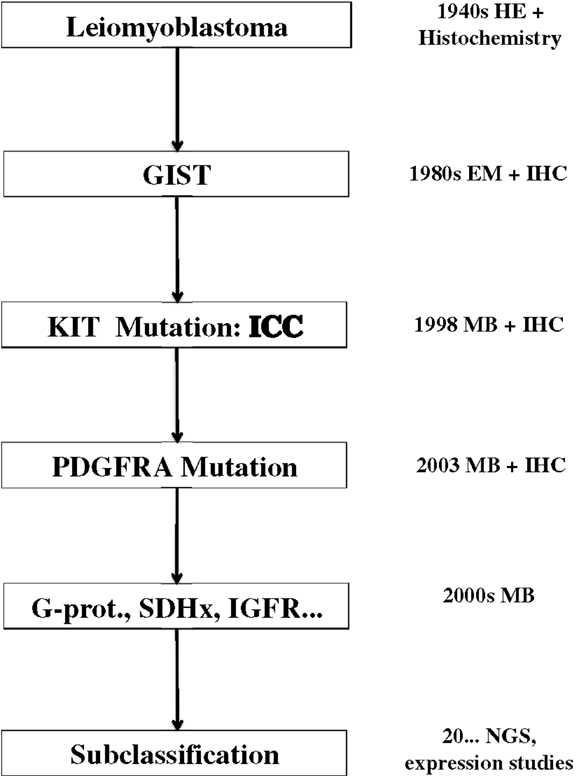
Figure 4. Evolution of the concept of GISTs since 70 years. EM, electron microscopy; IHC, immunohistochemistry; ICC, interstitial cells of Cajal; MB, molecular biology; SDH, succinil dehydrogenase; IGFR, insuline growth factor receptor; NGS, next generation sequencing.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Demetri GD, von Mehren M, Antonescu CR, DeMatteo RP, Ganjoo KN, Maki RG, et al. NCCN Task Force Report: update on the management of patients with gastrointestinal stromal tumors. J Natl Compr Canc Netw (2010) 8(Suppl 2):S1–41.
2. Nilsson B, Bumming P, Meis-Kindblom JM, Oden A, Dortok A, Gustavsson B, et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era – a population-based study in western Sweden. Cancer (2005) 103(4):821–9. doi: 10.1002/cncr.20862
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Agaimy A, Wunsch PH, Hofstaedter F, Blaszyk H, Rummele P, Gaumann A, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol (2007) 31(1):113–20. doi:10.1097/01.pas.0000213307.05811.f0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Corless CL, McGreevey L, Haley A, Town A, Heinrich MC. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol (2002) 160(5):1567–72. doi:10.1016/S0002-9440(10)61103-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Kawanowa K, Sakuma Y, Sakurai S, Hishima T, Iwasaki Y, Saito K, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol (2006) 37(12):1527–35. doi:10.1016/j.humpath.2006.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Muenst S, Thies S, Went P, Tornillo L, Bihl MP, Dirnhofer S. Frequency, phenotype, and genotype of minute gastrointestinal stromal tumors in the stomach: an autopsy study. Hum Pathol (2011) 42(12):1849–54. doi:10.1016/j.humpath.2011.01.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol (2006) 23(2):70–83. doi:10.1053/j.semdp.2006.09.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Stout AP. Bizarre smooth muscle tumors of the stomach. Cancer (1962) 15:400–9. doi:10.1002/1097-0142(196203/04)15:2<400::AID-CNCR2820150224>3.0.CO;2-P
10. Mazur MT, Clark HB. Gastric stromal tumors. Reappraisal of histogenesis. Am J Surg Pathol (1983) 7(6):507–19. doi:10.1097/00000478-198309000-00001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Miettinen M. Immunohistochemistry of soft tissue tumours – review with emphasis on 10 markers. Histopathology (2014) 64(1):101–18. doi:10.1111/his.12298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Robinson TL, Sircar K, Hewlett BR, Chorneyko K, Riddell RH, Huizinga JD. Gastrointestinal stromal tumors may originate from a subset of CD34-positive interstitial cells of Cajal. Am J Pathol (2000) 156(4):1157–63. doi:10.1016/S0002-9440(10)64984-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-KIT in human gastrointestinal stromal tumors. Science (1998) 279(5350):577–80. doi:10.1126/science.279.5350.577
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Nakahara M, Isozaki K, Hirota S, Miyagawa J, Hase-Sawada N, Taniguchi M, et al. A novel gain-of-function mutation of c-KIT gene in gastrointestinal stromal tumors. Gastroenterology (1998) 115(5):1090–5. doi:10.1016/S0016-5085(98)70079-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol (1998) 152(5):1259–69.
16. Sarlomo-Rikala M, Kovatich AJ, Barusevicius A, Miettinen M. CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol (1998) 11(8):728–34.
17. Huizinga JD, Thuneberg L, Kluppel M, Malysz J, Mikkelsen HB, Bernstein A. W/KIT gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature (1995) 373(6512):347–9. doi:10.1038/373347a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, Shinomura Y, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology (2003) 125(3):660–7. doi:10.1016/S0016-5085(03)01046-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology (2008) 53(3):245–66. doi:10.1111/j.1365-2559.2008.02977.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Agulnik M, Giel JL. Understanding rechallenge and resistance in the tyrosine kinase inhibitor era: imatinib in gastrointestinal stromal tumor. Am J Clin Oncol (2012) 37(4):417–22. doi:10.1097/COC.0b013e31824be3d6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Antonescu CR. The GIST paradigm: lessons for other kinase-driven cancers. J Pathol (2011) 223(2):251–61. doi:10.1002/path.2798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med (2001) 344(14):1052–6. doi:10.1056/NEJM200104053441404
23. Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res (2005) 11(11):4182–90. doi:10.1158/1078-0432.CCR-04-2245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Tornillo L. Biology of GISTs and mechanisms of imatinib resistance. Diagn Histopathol (2013) 19(6):203–10. doi:10.1016/j.mpdhp.2013.04.001
25. Agaimy A, Terracciano LM, Dirnhofer S, Tornillo L, Foerster A, Hartmann A, et al. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J Clin Pathol (2009) 62(7):613–6. doi:10.1136/jcp.2009.064550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Miranda C, Nucifora M, Molinari F, Conca E, Anania MC, Bordoni A, et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res (2012) 18(6):1769–76. doi:10.1158/1078-0432.CCR-11-2230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, Shamu T, et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature (2010) 467(7317):849–53. doi:10.1038/nature09409
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science (1988) 241(4861):42–52. doi:10.1126/science.3291115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer (2011) 11(12):865–78. doi:10.1038/nrc3143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene (2004) 23(22):3999–4006. doi:10.1038/sj.onc.1207525
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science (2003) 299(5607):708–10. doi:10.1126/science.1079666
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Kang HJ, Nam SW, Kim H, Rhee H, Kim NG, Kim H, et al. Correlation of KIT and platelet-derived growth factor receptor alpha mutations with gene activation and expression profiles in gastrointestinal stromal tumors. Oncogene (2005) 24(6):1066–74. doi:10.1038/sj.onc.1208358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Rossi F, Ehlers I, Agosti V, Socci ND, Viale A, Sommer G, et al. Oncogenic KIT signaling and therapeutic intervention in a mouse model of gastrointestinal stromal tumor. Proc Natl Acad Sci U S A (2006) 103(34):12843–8. doi:10.1073/pnas.0511076103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res (2001) 61(22):8118–21.
35. Nishikawa S, Kusakabe M, Yoshinaga K, Ogawa M, Hayashi S, Kunisada T, et al. In utero manipulation of coat color formation by a monoclonal anti-c-KIT antibody: two distinct waves of c-KIT-dependency during melanocyte development. EMBO J (1991) 10(8):2111–8.
36. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol (2006) 24(26):4340–6. doi:10.1200/JCO.2006.06.2984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Gari M, Goodeve A, Wilson G, Winship P, Langabeer S, Linch D, et al. c-KIT proto-oncogene exon 8 in-frame deletion plus insertion mutations in acute myeloid leukaemia. Br J Haematol (1999) 105(4):894–900. doi:10.1046/j.1365-2141.1999.01449.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Nagata H, Worobec AS, Oh CK, Chowdhury BA, Tannenbaum S, Suzuki Y, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-KIT in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci U S A (1995) 92(23):10560–4. doi:10.1073/pnas.92.23.10560
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Tian Q, Frierson HF Jr, Krystal GW, Moskaluk CA. Activating c-KIT gene mutations in human germ cell tumors. Am J Pathol (1999) 154(6):1643–7. doi:10.1016/S0002-9440(10)65419-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-KIT tyrosine kinase. J Biol Chem (2004) 279(30):31655–63. doi:10.1074/jbc.M403319200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Antonescu C. Gastrointestinal stromal tumors. Curr Top Microbiol Immunol (2012) 355:41–57. doi:10.1007/82_2011_161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Andersson J, Bumming P, Meis-Kindblom JM, Sihto H, Nupponen N, Joensuu H, et al. Gastrointestinal stromal tumors with KIT exon 11 deletions are associated with poor prognosis. Gastroenterology (2006) 130(6):1573–81. doi:10.1053/j.gastro.2006.01.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Martin J, Poveda A, Llombart-Bosch A, Ramos R, Lopez-Guerrero JA, Garcia del Muro J, et al. Deletions affecting codons 557-558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol (2005) 23(25):6190–8. doi:10.1200/JCO.2005.19.554
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Tzen CY, Mau BL. Analysis of CD117-negative gastrointestinal stromal tumors. World J Gastroenterol (2005) 11(7):1052–5.
45. Wardelmann E, Losen I, Hans V, Neidt I, Speidel N, Bierhoff E, et al. Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-KIT protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int J Cancer (2003) 106(6):887–95. doi:10.1002/ijc.11323
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Antonescu CR, Sommer G, Sarran L, Tschernyavsky SJ, Riedel E, Woodruff JM, et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res (2003) 9(9):3329–37.
47. Lasota J, Dansonka-Mieszkowska A, Stachura T, Schneider-Stock R, Kallajoki M, Steigen SE, et al. Gastrointestinal stromal tumors with internal tandem duplications in 3’ end of KIT juxtamembrane domain occur predominantly in stomach and generally seem to have a favorable course. Mod Pathol (2003) 16(12):1257–64. doi:10.1097/01.MP.0000097365.72526.3E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Tornillo L, Terracciano LM. An update on molecular genetics of gastrointestinal stromal tumours. J Clin Pathol (2006) 59(6):557–63. doi:10.1136/jcp.2005.031112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T, Demetri G, et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol (2000) 156(3):791–5. doi:10.1016/S0002-9440(10)64946-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Antonescu CR, Viale A, Sarran L, Tschernyavsky SJ, Gonen M, Segal NH, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res (2004) 10(10):3282–90. doi:10.1158/1078-0432.CCR-03-0715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Battochio A, Mohammed S, Winthrop D, Lefresne S, Mulder K, Chu Q, et al. Detection of c-KIT and PDGFRA gene mutations in gastrointestinal stromal tumors: comparison of DHPLC and DNA sequencing methods using a single population-based cohort. Am J Clin Pathol (2010) 133(1):149–55.
52. Lasota J, Corless CL, Heinrich MC, Debiec-Rychter M, Sciot R, Wardelmann E, et al. Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: a multicenter study on 54 cases. Mod Pathol (2008) 21(4):476–84. doi:10.1038/modpathol.2008.2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol (2006) 30(4):477–89. doi:10.1097/00000478-200604000-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Cho S, Kitadai Y, Yoshida S, Tanaka S, Yoshihara M, Yoshida K, et al. Deletion of the KIT gene is associated with liver metastasis and poor prognosis in patients with gastrointestinal stromal tumor in the stomach. Int J Oncol (2006) 28(6):1361–7. doi:10.3892/ijo.28.6.1361
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Kinoshita K, Isozaki K, Hirota S, Nishida T, Chen H, Nakahara M, et al. c-KIT gene mutation at exon 17 or 13 is very rare in sporadic gastrointestinal stromal tumors. J Gastroenterol Hepatol (2003) 18(2):147–51. doi:10.1046/j.1440-1746.2003.02911.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Steigen SE, Eide TJ, Wasag B, Lasota J, Miettinen M. Mutations in gastrointestinal stromal tumors – a population-based study from Northern Norway. APMIS (2007) 115(4):289–98. doi:10.1111/j.1600-0463.2007.apm_587.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Wardelmann E, Neidt I, Bierhoff E, Speidel N, Manegold C, Fischer HP, et al. c-KIT mutations in gastrointestinal stromal tumors occur preferentially in the spindle rather than in the epithelioid cell variant. Mod Pathol (2002) 15(2):125–36. doi:10.1038/modpathol.3880504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Agaram NP, Laquaglia MP, Ustun B, Guo T, Wong GC, Socci ND, et al. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res (2008) 14(10):3204–15. doi:10.1158/1078-0432.CCR-07-1984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer (2006) 42(8):1093–103. doi:10.1016/j.ejca.2006.01.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol (2008) 216(1):64–74. doi:10.1002/path.2382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Huss S, Kunstlinger H, Wardelmann E, Kleine MA, Binot E, Merkelbach-Bruse S, et al. A subset of gastrointestinal stromal tumors previously regarded as wild-type tumors carries somatic activating mutations in KIT exon 8 (p.D419del). Mod Pathol (2013) 26(7):1004–12. doi:10.1038/modpathol.2013.47
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Daniels M, Lurkin I, Pauli R, Erbstosser E, Hildebrandt U, Hellwig K, et al. Spectrum of KIT/PDGFRA/BRAF mutations and phosphatidylinositol-3-kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett (2011) 312(1):43–54. doi:10.1016/j.canlet.2011.07.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Duensing A, Joseph NE, Medeiros F, Smith F, Hornick JL, Heinrich MC, et al. Protein kinase C theta (PKCtheta) expression and constitutive activation in gastrointestinal stromal tumors (GISTs). Cancer Res (2004) 64(15):5127–31. doi:10.1158/0008-5472.CAN-04-0559
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Paner GP, Silberman S, Hartman G, Micetich KC, Aranha GV, Alkan S. Analysis of signal transducer and activator of transcription 3 (STAT3) in gastrointestinal stromal tumors. Anticancer Res (2003) 23(3B):2253–60.
65. Rubin BP, Antonescu CR, Scott-Browne JP, Comstock ML, Gu Y, Tanas MR, et al. A knock-in mouse model of gastrointestinal stromal tumor harboring KIT K641E. Cancer Res (2005) 65(15):6631–9. doi:10.1158/0008-5472.CAN-05-0891
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Zhu MJ, Ou WB, Fletcher CD, Cohen PS, Demetri GD, Fletcher JA. KIT oncoprotein interactions in gastrointestinal stromal tumors: therapeutic relevance. Oncogene (2007) 26(44):6386–95. doi:10.1038/sj.onc.1210464
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Kuzu G, Keskin O, Gursoy A, Nussinov R. Constructing structural networks of signaling pathways on the proteome scale. Curr Opin Struct Biol (2012) 22(3):367–77. doi:10.1016/j.sbi.2012.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets (2012) 16(Suppl 2):S17–27. doi:10.1517/14728222.2011.639361
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Johnston PA, Grandis JR. STAT3 signaling: anticancer strategies and challenges. Mol Interv (2011) 11(1):18–26. doi:10.1124/mi.11.1.4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Birner P, Beer A, Vinatzer U, Stary S, Hoftberger R, Nirtl N, et al. MAPKAP kinase 2 overexpression influences prognosis in gastrointestinal stromal tumors and associates with copy number variations on chromosome 1 and expression of p38 MAP kinase and ETV1. Clin Cancer Res (2012) 18(7):1879–87. doi:10.1158/1078-0432.CCR-11-2364
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Zhang Y, Gu ML, Zhou XX, Ma H, Yao HP, Ji F. Altered expression of ETV1 and its contribution to tumorigenic phenotypes in gastrointestinal stromal tumors. Oncol Rep (2014) 32(3):927–34. doi:10.3892/or.2014.3281
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res (2006) 66(18):9153–61. doi:10.1158/0008-5472.CAN-06-0165
73. Fumo G, Akin C, Metcalfe DD, Neckers L. 17-Allylamino-17-demethoxygeldanamycin (17-AAG) is effective in down-regulating mutated, constitutively activated KIT protein in human mast cells. Blood (2004) 103(3):1078–84. doi:10.1182/blood-2003-07-2477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Smyth T, Van Looy T, Curry JE, Rodriguez-Lopez AM, Wozniak A, Zhu M, et al. The HSP90 inhibitor, AT13387, is effective against imatinib-sensitive and -resistant gastrointestinal stromal tumor models. Mol Cancer Ther (2012) 11(8):1799–808. doi:10.1158/1535-7163.MCT-11-1046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Pauls K, Merkelbach-Bruse S, Thal D, Buttner R, Wardelmann E. PDGFRalpha- and c-KIT-mutated gastrointestinal stromal tumours (GISTs) are characterized by distinctive histological and immunohistochemical features. Histopathology (2005) 46(2):166–75. doi:10.1111/j.1365-2559.2005.02061.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Miettinen M, Furlong M, Sarlomo-Rikala M, Burke A, Sobin LH, Lasota J. Gastrointestinal stromal tumors, intramural leiomyomas, and leiomyosarcomas in the rectum and anus: a clinicopathologic, immunohistochemical, and molecular genetic study of 144 cases. Am J Surg Pathol (2001) 25(9):1121–33. doi:10.1097/00000478-200111000-00013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Miettinen M, Kopczynski J, Makhlouf HR, Sarlomo-Rikala M, Gyorffy H, Burke A, et al. Gastrointestinal stromal tumors, intramural leiomyomas, and leiomyosarcomas in the duodenum: a clinicopathologic, immunohistochemical, and molecular genetic study of 167 cases. Am J Surg Pathol (2003) 27(5):625–41. doi:10.1097/00000478-200305000-00006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Miettinen M, Sarlomo-Rikala M, Sobin LH, Lasota J. Gastrointestinal stromal tumors and leiomyosarcomas in the colon: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases. Am J Surg Pathol (2000) 24(10):1339–52. doi:10.1097/00000478-200010000-00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Penzel R, Aulmann S, Moock M, Schwarzbach M, Rieker RJ, Mechtersheimer G. The location of KIT and PDGFRA gene mutations in gastrointestinal stromal tumours is site and phenotype associated. J Clin Pathol (2005) 58(6):634–9. doi:10.1136/jcp.2004.021766
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Lasota J, Dansonka-Mieszkowska A, Sobin LH, Miettinen M. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest (2004) 84(7):874–83. doi:10.1038/labinvest.3700122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Lasota J, Stachura J, Miettinen M. GISTs with PDGFRA exon 14 mutations represent subset of clinically favorable gastric tumors with epithelioid morphology. Lab Invest (2006) 86(1):94–100. doi:10.1038/labinvest.3700360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Wardelmann E, Hrychyk A, Merkelbach-Bruse S, Pauls K, Goldstein J, Hohenberger P, et al. Association of platelet-derived growth factor receptor alpha mutations with gastric primary site and epithelioid or mixed cell morphology in gastrointestinal stromal tumors. J Mol Diagn (2004) 6(3):197–204. doi:10.1016/S1525-1578(10)60510-7
83. Subramanian S, West RB, Corless CL, Ou W, Rubin BP, Chu KM, et al. Gastrointestinal stromal tumors (GISTs) with KIT and PDGFRA mutations have distinct gene expression profiles. Oncogene (2004) 23(47):7780–90. doi:10.1038/sj.onc.1208056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Debiec-Rychter M, Wasag B, Stul M, De Wever I, Van Oosterom A, Hagemeijer A, et al. Gastrointestinal stromal tumours (GISTs) negative for KIT (CD117 antigen) immunoreactivity. J Pathol (2004) 202(4):430–8. doi:10.1002/path.1546
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Matei D, Satpathy M, Cao L, Lai YC, Nakshatri H, Donner DB. The platelet-derived growth factor receptor alpha is destabilized by geldanamycins in cancer cells. J Biol Chem (2007) 282(1):445–53. doi:10.1074/jbc.M607012200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Miettinen M, Wang ZF, Lasota J. DOG1 antibody in the differential diagnosis of gastrointestinal stromal tumors: a study of 1840 cases. Am J Surg Pathol (2009) 33(9):1401–8. doi:10.1097/PAS.0b013e3181a90e1a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Wozniak A, Sciot R, Guillou L, Pauwels P, Wasag B, Stul M, et al. Array CGH analysis in primary gastrointestinal stromal tumors: cytogenetic profile correlates with anatomic site and tumor aggressiveness, irrespective of mutational status. Genes Chromosomes Cancer (2007) 46(3):261–76. doi:10.1002/gcc.20408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol (2005) 23(23):5357–64. doi:10.1200/JCO.2005.14.068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Daum O, Grossmann P, Vanecek T, Sima R, Mukensnabl P, Michal M. Diagnostic morphological features of PDGFRA-mutated gastrointestinal stromal tumors: molecular genetic and histologic analysis of 60 cases of gastric gastrointestinal stromal tumors. Ann Diagn Pathol (2007) 11(1):27–33. doi:10.1016/j.anndiagpath.2006.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Haller F, Happel N, Schulten HJ, von Heydebreck A, Schwager S, Armbrust T, et al. Site-dependent differential KIT and PDGFRA expression in gastric and intestinal gastrointestinal stromal tumors. Mod Pathol (2007) 20(10):1103–11. doi:10.1038/modpathol.3800947
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol (2003) 21(23):4342–9. doi:10.1200/JCO.2003.04.190
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Holden JA, Willmore-Payne C, Coppola D, Garrett CR, Layfield LJ. High-resolution melting amplicon analysis as a method to detect c-KIT and platelet-derived growth factor receptor alpha activating mutations in gastrointestinal stromal tumors. Am J Clin Pathol (2007) 128(2):230–8. doi:10.1309/7TEH56K6WWXENNQY
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Medeiros F, Corless CL, Duensing A, Hornick JL, Oliveira AM, Heinrich MC, et al. KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am J Surg Pathol (2004) 28(7):889–94. doi:10.1097/00000478-200407000-00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Sakurai S, Hasegawa T, Sakuma Y, Takazawa Y, Motegi A, Nakajima T, et al. Myxoid epithelioid gastrointestinal stromal tumor (GIST) with mast cell infiltrations: a subtype of GIST with mutations of platelet-derived growth factor receptor alpha gene. Hum Pathol (2004) 35(10):1223–30. doi:10.1016/j.humpath.2004.07.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Wasag B, Debiec-Rychter M, Pauwels P, Stul M, Vranckx H, Oosterom AV, et al. Differential expression of KIT/PDGFRA mutant isoforms in epithelioid and mixed variants of gastrointestinal stromal tumors depends predominantly on the tumor site. Mod Pathol (2004) 17(8):889–94. doi:10.1038/modpathol.3800136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Hostein I, Faur N, Primois C, Boury F, Denard J, Emile JF, et al. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol (2010) 133(1):141–8. doi:10.1309/AJCPPCKGA2QGBJ1R
97. Lasota J, Xi L, Coates T, Dennis R, Evbuomwan MO, Wang ZF, et al. No KRAS mutations found in gastrointestinal stromal tumors (GISTs): molecular genetic study of 514 cases. Mod Pathol (2013) 26(11):1488–91. doi:10.1038/modpathol.2013.89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Ward BA, Gutmann DH. Neurofibromatosis 1: from lab bench to clinic. Pediatr Neurol (2005) 32(4):221–8. doi:10.1016/j.pediatrneurol.2004.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Nannini M, Biasco G, Astolfi A, Pantaleo MA. An overview on molecular biology of KIT/PDGFRA wild type (WT) gastrointestinal stromal tumours (GIST). J Med Genet (2013) 50(10):653–61. doi:10.1136/jmedgenet-2013-101695
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Kinoshita K, Hirota S, Isozaki K, Ohashi A, Nishida T, Kitamura Y, et al. Absence of c-KIT gene mutations in gastrointestinal stromal tumours from neurofibromatosis type 1 patients. J Pathol (2004) 202(1):80–5. doi:10.1002/path.1487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol (2006) 30(1):90–6. doi:10.1097/01.pas.0000176433.81079.bd
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Nemoto H, Tate G, Schirinzi A, Suzuki T, Sasaya S, Yoshizawa Y, et al. Novel NF1 gene mutation in a Japanese patient with neurofibromatosis type 1 and a gastrointestinal stromal tumor. J Gastroenterol (2006) 41(4):378–82. doi:10.1007/s00535-006-1772-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Takazawa Y, Sakurai S, Sakuma Y, Ikeda T, Yamaguchi J, Hashizume Y, et al. Gastrointestinal stromal tumors of neurofibromatosis type I (von Recklinghausen’s disease). Am J Surg Pathol (2005) 29(6):755–63. doi:10.1097/01.pas.0000163359.32734.f9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Yantiss RK, Rosenberg AE, Sarran L, Besmer P, Antonescu CR. Multiple gastrointestinal stromal tumors in type I neurofibromatosis: a pathologic and molecular study. Mod Pathol (2005) 18(4):475–84. doi:10.1038/modpathol.3800334
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Maertens O, Prenen H, Debiec-Rychter M, Wozniak A, Sciot R, Pauwels P, et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet (2006) 15(6):1015–23. doi:10.1093/hmg/ddl016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Rutter J, Winge DR, Schiffman JD. Succinate dehydrogenase – assembly, regulation and role in human disease. Mitochondrion (2010) 10(4):393–401. doi:10.1016/j.mito.2010.03.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Barletta JA, Hornick JL. Succinate dehydrogenase-deficient tumors: diagnostic advances and clinical implications. Adv Anat Pathol (2012) 19(4):193–203. doi:10.1097/PAP.0b013e31825c6bc6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Gill AJ. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology (2012) 44(4):285–92. doi:10.1097/PAT.0b013e3283539932
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A (2011) 108(1):314–8. doi:10.1073/pnas.1009199108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Miettinen M, Wang ZF, Sarlomo-Rikala M, Osuch C, Rutkowski P, Lasota J. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol (2011) 35(11):1712–21. doi:10.1097/PAS.0b013e3182260752
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Chou A, Chen J, Clarkson A, Samra JS, Clifton-Bligh RJ, Hugh TJ, et al. Succinate dehydrogenase-deficient GISTs are characterized by IGF1R overexpression. Mod Pathol (2012) 25(9):1307–13. doi:10.1038/modpathol.2012.77
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Doyle LA, Nelson D, Heinrich MC, Corless CL, Hornick JL. Loss of succinate dehydrogenase subunit B (SDHB) expression is limited to a distinctive subset of gastric wild-type gastrointestinal stromal tumours: a comprehensive genotype-phenotype correlation study. Histopathology (2012) 61(5):801–9. doi:10.1111/j.1365-2559.2012.04300.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Gaal J, Stratakis CA, Carney JA, Ball ER, Korpershoek E, Lodish MB, et al. SDHB immunohistochemistry: a useful tool in the diagnosis of Carney-Stratakis and Carney triad gastrointestinal stromal tumors. Mod Pathol (2011) 24(1):147–51. doi:10.1038/modpathol.2010.185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Gill AJ, Chou A, Vilain R, Clarkson A, Lui M, Jin R, et al. Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am J Surg Pathol (2010) 34(5):636–44. doi:10.1097/PAS.0b013e3181d6150d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Gill AJ, Chou A, Vilain RE, Clifton-Bligh RJ. “Pediatric-type” gastrointestinal stromal tumors are SDHB negative (“type 2”) GISTs. Am J Surg Pathol (2011) 35(8):1245–7; author reply 7–8. doi:10.1097/PAS.0b013e3182217b93
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Rege TA, Wagner AJ, Corless CL, Heinrich MC, Hornick JL. “Pediatric-type” gastrointestinal stromal tumors in adults: distinctive histology predicts genotype and clinical behavior. Am J Surg Pathol (2011) 35(4):495–504. doi:10.1097/PAS.0b013e31820e5f7d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet (2002) 108(2):132–9. doi:10.1002/ajmg.10235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Italiano A, Chen CL, Sung YS, Singer S, DeMatteo RP, LaQuaglia MP, et al. SDHA loss of function mutations in a subset of young adult wild-type gastrointestinal stromal tumors. BMC Cancer (2012) 12:408. doi:10.1186/1471-2407-12-408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Wardelmann E. Translational research and diagnosis in GIST. Pathologe (2012) 33(Suppl 2):273–7. doi:10.1007/s00292-012-1682-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Miettinen M, Lasota J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs) – A review. Int J Biochem Cell Biol (2014) 53:514–9. doi:10.1016/j.biocel.2014.05.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Briere JJ, Favier J, Benit P, El Ghouzzi V, Lorenzato A, Rabier D, et al. Mitochondrial succinate is instrumental for HIF1alpha nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Hum Mol Genet (2005) 14(21):3263–9. doi:10.1093/hmg/ddi359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell (2005) 7(1):77–85. doi:10.1016/j.ccr.2004.11.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Lasota J, Wang Z, Kim SY, Helman L, Miettinen M. Expression of the receptor for type I insulin-like growth factor (IGF1R) in gastrointestinal stromal tumors: an immunohistochemical study of 1078 cases with diagnostic and therapeutic implications. Am J Surg Pathol (2013) 37(1):114–9. doi:10.1097/PAS.0b013e3182613c86
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Beadling C, Patterson J, Justusson E, Nelson D, Pantaleo MA, Hornick JL, et al. Gene expression of the IGF pathway family distinguishes subsets of gastrointestinal stromal tumors wild type for KIT and PDGFRA. Cancer Med (2013) 2(1):21–31. doi:10.1002/cam4.57
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Rikhof B, van der Graaf WT, Suurmeijer AJ, van Doorn J, Meersma GJ, Groenen PJ, et al. “Big”-insulin-like growth factor-II signaling is an autocrine survival pathway in gastrointestinal stromal tumors. Am J Pathol (2012) 181(1):303–12. doi:10.1016/j.ajpath.2012.03.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Breiner JA, Meis-Kindblom J, Kindblom LG, McComb E, Liu J, Nelson M, et al. Loss of 14q and 22q in gastrointestinal stromal tumors (pacemaker cell tumors). Cancer Genet Cytogenet (2000) 120(2):111–6. doi:10.1016/S0165-4608(00)00212-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Derre J, Lagace R, Terrier P, Sastre X, Aurias A. Consistent DNA losses on the short arm of chromosome 1 in a series of malignant gastrointestinal stromal tumors. Cancer Genet Cytogenet (2001) 127(1):30–3. doi:10.1016/S0165-4608(00)00409-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. El-Rifai W, Sarlomo-Rikala M, Andersson LC, Knuutila S, Miettinen M. DNA sequence copy number changes in gastrointestinal stromal tumors: tumor progression and prognostic significance. Cancer Res (2000) 60(14):3899–903.
129. Gunawan B, von Heydebreck A, Sander B, Schulten HJ, Haller F, Langer C, et al. An oncogenetic tree model in gastrointestinal stromal tumours (GISTs) identifies different pathways of cytogenetic evolution with prognostic implications. J Pathol (2007) 211(4):463–70. doi:10.1002/path.2128
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Ylipaa A, Hunt KK, Yang J, Lazar AJ, Torres KE, Lev DC, et al. Integrative genomic characterization and a genomic staging system for gastrointestinal stromal tumors. Cancer (2011) 117(2):380–9. doi:10.1002/cncr.25594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Tornillo L, Duchini G, Carafa V, Lugli A, Dirnhofer S, Di Vizio D, et al. Patterns of gene amplification in gastrointestinal stromal tumors (GIST). Lab Invest (2005) 85(7):921–31. doi:10.1038/labinvest.3700284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Perrone F, Tamborini E, Dagrada GP, Colombo F, Bonadiman L, Albertini V, et al. 9p21 locus analysis in high-risk gastrointestinal stromal tumors characterized for c-KIT and platelet-derived growth factor receptor alpha gene alterations. Cancer (2005) 104(1):159–69. doi:10.1002/cncr.21113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Schneider-Stock R, Boltze C, Lasota J, Miettinen M, Peters B, Pross M, et al. High prognostic value of p16INK4 alterations in gastrointestinal stromal tumors. J Clin Oncol (2003) 21(9):1688–97. doi:10.1200/JCO.2003.08.101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Schneider-Stock R, Boltze C, Lasota J, Peters B, Corless CL, Ruemmele P, et al. Loss of p16 protein defines high-risk patients with gastrointestinal stromal tumors: a tissue microarray study. Clin Cancer Res (2005) 11(2 Pt 1):638–45.
135. Haller F, Lobke C, Ruschhaupt M, Cameron S, Schulten HJ, Schwager S, et al. Loss of 9p leads to p16INK4A down-regulation and enables RB/E2F1-dependent cell cycle promotion in gastrointestinal stromal tumours (GISTs). J Pathol (2008) 215(3):253–62. doi:10.1002/path.2352
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Dematteo RP, Heinrich MC, El-Rifai WM, Demetri G. Clinical management of gastrointestinal stromal tumors: before and after STI-571. Hum Pathol (2002) 33(5):466–77. doi:10.1053/hupa.2002.124122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol (2008) 26(33):5352–9. doi:10.1200/JCO.2007.15.7461
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Montemurro M, Bauer S. Treatment of gastrointestinal stromal tumor after imatinib and sunitinib. Curr Opin Oncol (2011) 23(4):367–72. doi:10.1097/CCO.0b013e3283477ac2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Gupta A, Roy S, Lazar AJ, Wang WL, McAuliffe JC, Reynoso D, et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc Natl Acad Sci U S A (2010) 107(32):14333–8. doi:10.1073/pnas.1000248107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Le Cesne A, Ray-Coquard I, Bui BN, Adenis A, Rios M, Bertucci F, et al. Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open-label multicentre randomised phase 3 trial. Lancet Oncol (2010) 11(10):942–9. doi:10.1016/S1470-2045(10)70222-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Casali PG. Successes and limitations of targeted cancer therapy in gastrointestinal stromal tumors. Prog Tumor Res (2014) 41:51–61. doi:10.1159/000355898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol (2006) 24(29):4764–74. doi:10.1200/JCO.2006.06.2265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Gounder MM, Maki RG. Molecular basis for primary and secondary tyrosine kinase inhibitor resistance in gastrointestinal stromal tumor. Cancer Chemother Pharmacol (2011) 67(Suppl 1):S25–43. doi:10.1007/s00280-010-1526-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Weisberg E, Wright RD, Jiang J, Ray A, Moreno D, Manley PW, et al. Effects of PKC412, nilotinib, and imatinib against GIST-associated PDGFRA mutants with differential imatinib sensitivity. Gastroenterology (2006) 131(6):1734–42. doi:10.1053/j.gastro.2006.09.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol (2007) 25(13):1753–9. doi:10.1200/JCO.2006.07.3049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Grimpen F, Yip D, McArthur G, Waring P, Goldstein D, Loughrey M, et al. Resistance to imatinib, low-grade FDG-avidity on PET, and acquired KIT exon 17 mutation in gastrointestinal stromal tumour. Lancet Oncol (2005) 6(9):724–7. doi:10.1016/S1470-2045(05)70321-1
147. Koyama T, Nimura H, Kobayashi K, Marushima H, Odaira H, Kashimura H, et al. Recurrent gastrointestinal stromal tumor (GIST) of the stomach associated with a novel c-KIT mutation after imatinib treatment. Gastric Cancer (2006) 9(3):235–9. doi:10.1007/s10120-006-0368-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Wakai T, Kanda T, Hirota S, Ohashi A, Shirai Y, Hatakeyama K. Late resistance to imatinib therapy in a metastatic gastrointestinal stromal tumour is associated with a second KIT mutation. Br J Cancer (2004) 90(11):2059–61. doi:10.1038/sj.bjc.6601819
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Roskoski R Jr. Structure and regulation of KIT protein-tyrosine kinase – the stem cell factor receptor. Biochem Biophys Res Commun (2005) 338(3):1307–15. doi:10.1016/j.bbrc.2005.09.150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Desai J, Shankar S, Heinrich MC, Fletcher JA, Fletcher CD, Manola J, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res (2007) 13(18 Pt 1):5398–405. doi:10.1158/1078-0432.CCR-06-0858
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Miselli FC, Casieri P, Negri T, Orsenigo M, Lagonigro MS, Gronchi A, et al. c-KIT/PDGFRA gene status alterations possibly related to primary imatinib resistance in gastrointestinal stromal tumors. Clin Cancer Res (2007) 13(8):2369–77. doi:10.1158/1078-0432.CCR-06-1745
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene (2007) 26(54):7560–8. doi:10.1038/sj.onc.1210558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Tarn C, Rink L, Merkel E, Flieder D, Pathak H, Koumbi D, et al. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc Natl Acad Sci U S A (2008) 105(24):8387–92. doi:10.1073/pnas.0803383105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Mahadevan D, Sutton GR, Arteta-Bulos R, Bowden CJ, Miller PJ, Swart RE, et al. Phase 1b study of safety, tolerability and efficacy of R1507, a monoclonal antibody to IGF-1R in combination with multiple standard oncology regimens in patients with advanced solid malignancies. Cancer Chemother Pharmacol (2014) 73(3):467–73. doi:10.1007/s00280-013-2372-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: gastrointestinal stromal tumors, gastrointestinal tract, CKIT, targeted therapy, receptor tyrosine kinase
Citation: Tornillo L (2014) Gastrointestinal stromal tumor – an evolving concept. Front. Med. 1:43. doi: 10.3389/fmed.2014.00043
Received: 19 August 2014; Accepted: 17 October 2014;
Published online: 11 November 2014.
Edited by:
Dolores Di Vizio, Harvard Medical School, USAReviewed by:
Rosa Marina Melillo, University of Naples Federico II, ItalyRenato Franco, Istituto Tumori Fondazione Giovanni Pascale, Italy
Copyright: © 2014 Tornillo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luigi Tornillo, Institute of Pathology, University of Basel, Schönbeinstrasse 40, CH-4003 Basel, Switzerland e-mail:dG9ybmlsbG9sQHVoYnMuY2g=