 Luca Ulianich
Luca Ulianich Luigi Insabato
Luigi Insabato- 1Istituto per l’Endocrinologia e l’Oncologia “Gaetano Salvatore”, Consiglio Nazionale delle Ricerche, Naples, Italy
- 2Section of Anatomical Pathology, Department of Advanced Biomedical Sciences, University Federico II of Naples, Naples, Italy
Endometrial cancer (EC) is a common gynecologic malignancy often diagnosed at early stage. In spite of a huge advance in our understanding of EC biology, therapeutic modalities do not have significantly changed over the past 40 years. A restricted number of genes have been reported to be mutated in EC, mediating cell proliferation and invasiveness. However, besides these alterations, few other groups and ourselves recently identified the activation of the unfolded protein response (UPR) and GRP78 increase following endoplasmic reticulum (ER) stress as mechanisms favoring growth and invasion of EC cells. Here, a concise update on currently available data in the field is presented, analyzing the crosstalk between the UPR and the main signaling pathways regulating EC cell proliferation and survival. It is evident that this is a rapidly expanding and promising issue. However, more data are very likely to yield a better understanding on the mechanisms through which EC cells can survive the low oxygen and glucose tumor microenvironment. In this perspective, the UPR and, particularly, GRP78 might constitute a novel target for the treatment of EC in combination with traditional adjuvant therapy.
Introduction
Endometrial cancer (EC) is the most frequent form of malignant tumor of the female reproductive tract, and overall the endometrium is the fourth most common cancer site, accounting for 6% of all women cancers (1, 2). EC is classified in type I and II (3). Type I tumors are low-grade estrogen-related endometrioid carcinomas (EEC) that usually develop in perimenopausal women. In contrast, type II tumors are aggressive non-endometrioid carcinomas (NEEC) (mainly serous and clear cell carcinomas) that develop in older women regardless to estrogen stimulation. The most frequent type of EC is endometrioid carcinoma, which accounts for more than 80% of all cases. It is often preceded by a precancerous condition, known as atypical endometrial hyperplasia (AEH) and it may progress, over time, to EC in 5–25% of patients (4). In addition, AEH is associated with a coexisting EC in approximately 20% of patients (5). Well recognized risk factors for endometrial carcinoma include polycystic ovarian syndrome, tamoxifen therapy, unopposed estrogen therapy, a history of nulliparity or infertility, irregular menstrual cycles, hypertension, obesity, and diabetes mellitus (6, 7). It has been shown that usually EEC (type I) carcinomas display microsatellite instability and alterations in the PTEN, K-RAS, PIK3CA, and CTNNB1 (beta-catenin) genes, whereas NEEC are characterized preferentially by mutations of p53, STK15, p16, E-cadherin, and c-erb-B2 genes (8–10). These molecular alterations promote cell proliferation and invasiveness. However, other emerging factors, such as the unfolded protein response (UPR) activation following endoplasmic reticulum (ER) stress and GRP78 (also known as BiP or HSPA5) overexpression and/or localization, have been recently described to affect not only EC cells growth and invasiveness but also their ability to survive both the hostile tumor microenvironment and the aggression of chemotherapeutic agents. Here, we report our data of UPR activation and GRP78 in EC, and will focus on the current literature about the role of ER stress in EEC.
ER Stress and UPR Activation
The ER is a complex and multifunctional organelle. It is the intracellular compartment of cargo protein folding. In order to accomplish its protein folding functions, the ER has high concentrations of chaperone proteins, which facilitate correct folding of nascent proteins. Many of these chaperones are Ca2+-dependent, and in fact the ER contains high concentrations of Ca2+ and plays an important role in intracellular Ca2+ homeostasis. The oxidizing environment that exists in the ER lumen is required for the formation of disulfide bonds during protein synthesis (11). Folded proteins will be then exported out of the ER along the secretory pathway, whereas misfolded proteins will eventually be disposed of by an endoplasmic reticulum-associated protein degradation pathway (ERAD). A wide variety of cellular stress stimuli can disrupt ER function. A number of these are tightly related to cancer and tumor development. They include changes to the redox environment, Ca2+ depletion, expression of mutant proteins, hypoxia, and/or glucose deprivation (12). As a consequence, the protein folding capacity is disrupted, leading to the accumulation of unfolded and misfolded proteins within the ER and, thus, to ER stress. When ER stress occurs, cells attempt to adjust the protein folding capacity to meet the new protein load or to counteract protein misfolding events through activation of signal transduction pathways collectively known as UPR (13). Three major classes of ER stress signal transducer are known, protein kinase RNA (PKR)-like endoplasmic reticulum kinase (PERK), inositol-requiring protein-1 (IRE1), and activating transcription factor-6 (ATF6). They are able, with their endoluminal domain, to sense the state of protein folding. Activation of these stress transducers results in an attempt to alleviate ER stress essentially by attenuating the general translation process, up-regulating the transcription of genes encoding for ER chaperones and folding enzymes, and enhancing the degradation of malfolded proteins by the ERAD machinery (14). Depending on the persistence and severity of ER stress, the UPR can ultimately result in cell death through the activation of apoptotic pathways specifically mediated by the ER, as well as coupling with the mitochondrial pathways (14, 15).
GRP78
Regulation of the UPR is mediated by members of the ER chaperone family. GRP78 is the more abundant ER chaperone and, besides its key role in the protein folding process, plays also a preeminent role in the regulation of the UPR (16). In absence of stress signals, indeed, PERK, IRE1, and ATF6 are maintained in an inactive state by physical interaction of their endoluminal domain with GRP78. When ER stress occurs, GRP78 dissociates from these sensor molecules, promoting their activation and subsequently coupling with accumulating misfolded proteins. As a result of UPR activation, GRP78 is upregulated in the attempt to increase the folding capacity of the ER (Figure 1). Thus, GRP78 represents both a regulator and a target of the UPR and is associated with pro-survival responses. GRP78 has been reported, indeed, to interact with components of ER related pro-apoptotic pathways. For example, GRP78 can bind and block caspase-12 and caspase-7 activation or pro-apoptotic proteins Bik and Bax preventing cytochrome c release from mitochondria and ultimately inhibiting apoptosis (17, 18). GRP78 expression has been reported to be increased in a number of human cancers, including breast (19, 20), lung (21, 22), prostate (23, 24), ovarian (25), gastric (26), hepatocellular (27, 28), esophageal (29), renal (30), endometrial (31, 32), melanoma (33), glioma (34), and fibrosarcoma (35) (Table 1). Furthermore, GRP78 overexpression in these cancers has been found to be strongly associated with increased malignancy, chemoresistance, and poor patient outcome. Furthermore, knockdown of GRP78 sensitizes tumor cells to drug treatment (18). Additionally, recent studies have identified GRP78 in compartments other than the ER, such as the cell surface, where it can interact with different molecular partners and mediate the transduction of additional cell growth and survival signals in different cancer cells (15, 36, 37) (Figure 1). It has been recently reported, indeed, that GRP78 present on prostate cancer cells can bind the prostate-specific antigen/activated a2-macroglobulin complex causing the activation of ERK1/2 (extracellular-signal-regulated kinase 1/2), p38 MAPK (mitogen-activated protein kinase), and PI3K (phosphoinositide 3-kinase) signaling pathways and promoting cell survival by the Akt and NF-κB (nuclear factor κB) signaling cascade (38). Furthermore, GRP78 has been reported to form a complex with Cripto, a multifunctional cell-surface protein that plays a key role in vertebrate embryogenesis and human tumor progression, enhancing tumor growth via inhibition of TGF-β (transforming growth factor-β) signaling (39). The localization of GRP78 mainly on the surface of tumor cells enables specific tumor targeting. The treatment of A375 melanoma cells and 1-LN and DU145 prostate cancer cells with polyclonal antibodies directed against the C-terminus of GRP78 has already been described to induce apoptosis via p53 upregulation, suppression of Ras/MAPK and PI3K/Akt signaling pathways, and inhibition of NF-κB1 and NF-κB2 activation (40).
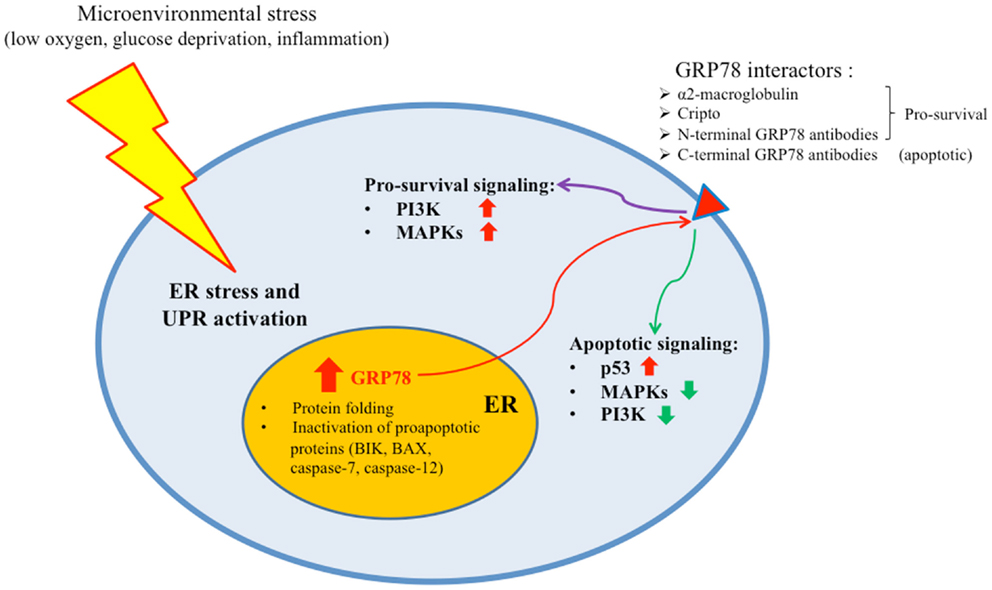
Figure 1. Microenvironmental stress causes ER stress and UPR activation in cancer cells. As a result, GRP78 is upregulated to enhance the folding capacity of the ER. A quote of the protein is transported to the cell membrane where it can bind different molecular partners and transduce either pro-survival or apoptotic signals.
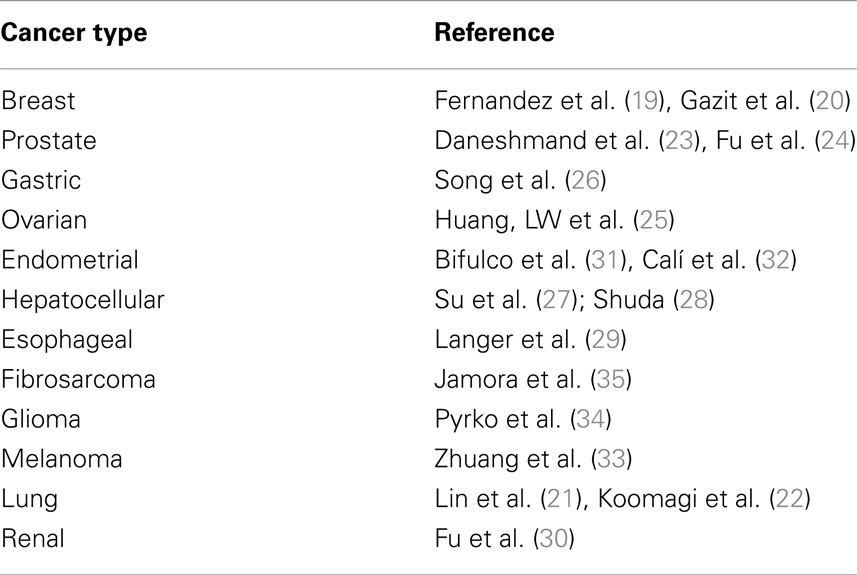
Table 1. Different cancer types where GRP78 has been reported to play a role in proliferation, invasiveness, and/or chemoresistance along with representative references are listed.
UPR Activation and GRP78 in Endometrial Cancer
We have recently reported for the first time that ER stress is activated in EC, as demonstrated by increased expression levels of ATF6, GRP78, and CHOP/GADD153 in EEC tissues (31). These data have been then confirmed by the studies of Luvsandagva (41) and Gray (42). To address the question whether GRP78 might play a role in EC cell proliferation, very recently we performed both salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and proliferation curve analysis in Ishikawa and AN3CA endometrial adenocarcinoma cell lines silenced for GRP78 expression. We demonstrated that, in silenced cells, the growth rate was significantly lower when compared to mock-transfected cells (31, 32). In addition, GRP78 attenuation resulted also in reduced invasion rate, suggesting that GRP78 also participates in the malignant phenotype of cell migration and invasiveness of EC cells (32). As reported in other cancer cells, such as melanoma and pancreatic cancer cells, besides the ER localization, we observed also a cell membrane localization of GRP78 in both EEC tissues and EC cell lines (32), suggesting that it might exert additional effects on cell growth and signaling. Interestingly, we also reported that, as already described in pancreatic cancer cells (40), the incubation of EC cells with a commercial polyclonal antibody directed against the C-terminus of GRP78 induces apoptosis only in cells that display GRP78 on their cell surface (32). Despite of the study by Misra in pancreatic cancer cells (40), apoptosis in EC cell was not accompanied by an increase of p53 protein, suggesting the involvement of other key mediators of the apoptotic pathway. However, we observed a significant decrease of the prosurvival PI3K/Akt signaling. Sustained AKT activity is a common feature of endometrioid ECs, due to PTEN loss and/or PI3K mutations, and initiates a cascade of downstream signals leading to proliferation, migration, survival, and angiogenesis (43). Whether the presence of GRP78 on the cell membrane of EC cells might further increase cell proliferation and/or invasiveness is still unclear and is under investigation in our laboratory. Another important question that should be addressed is whether the eventual advantage of presenting GRP78 on the cell membrane of EC cells could be related to the interaction with other key molecules mediating cell proliferation and/or invasiveness. It has been reported recently that GRP78 can interact with the epidermal growth factor receptor (EGFR) in human amnion FL cells following ER stress induction by the alkylating agent N-methyl-N-nitro-N-nitrosoguanidine (MNNG) (44). However, in this study emerged that the complex EGFR/GRP78 has rather an inhibitory activity on the EGFR-signaling pathway.
Role of GRP78 in Endometrial Cancer Chemoresistance and Therapeutic Implications
Endometrial cancer is often diagnosed at an early stage and, thus, has a favorable prognosis. However, 10–15% of these patients will experience a recurrence (45). Age, menopausal status, tumor stage and grade, and lymph-vascular space invasion, are known prognostic factors for the disease free survival of EC patients (46). Nevertheless, the number of recurrent EC patients tends to increase yearly and the prognosis for these patients is very poor (47). Thus, the identification of new biomarkers could help to better predict high-risk patients that need adjuvant therapy. Muinelo-Romay et al. have recently reported that TGF-β 1 plays as a key role in the initiation of tumor invasion in high-risk recurrence EC (47). Similarly, GRP78 has been described to play an important role in the progression of EC. GRP78 expression levels were, indeed, elevated in high-risk EC tissues compared with both the low-risk EC and normal endometrial tissues, suggesting that GRP78 may promote progression in EC and increase malignant potential (48). GRP78 might, thus, represent a potential biomarker to better predict high-risk EC and thereby guide clinical therapy. The combination of paclitaxel and cisplatin in chemotherapy is a standard regimen for adjuvant chemotherapy in advanced or recurrent EC (49, 50). However, very recently it has been reported that the treatment of EC cell lines with cisplatin is capable of inducing GRP78 expression (42). This is preceded by an increase of AKT phosphorylation but not by changes of MAPK activity (42). Use of the small molecule pan-AKT inhibitor MK2206, reducing AKT activity, blocked constitutive GRP78 expression, and cisplatin-mediated induction of GRP78 suggesting that GRP78’s antiapoptosis functions are part of the AKT survival pathway. On the other hand, GRP78 has been reported to affect optimal AKT activation since GRP78 knockdown by siRNA reduces AKT activity, suggesting that in EC cells, GRP78 may be both an upstream and downstream regulator of AKT (42). The attenuation of GRP78 expression significantly augments cisplatin-mediated cytotoxicity by enhancing the cleavage of apoptotic markers, Poly(ADP-ribose) polymerase (PARP) and caspase-3 (41, 42), highlighting the key role that GRP78 might play in the response of EC cells to chemotherapeutic treatments. A number of natural compounds have been found to block GRP78 transcription and/or inhibit its activity. One of these, epigallocatechin-3-gallate (EGCG), has been recently described to cause the arrest of Ishikawa EC cells in the G0/G1 phase of the cell cycle and to increase apoptosis by interfering with Akt activation and MAPK signals and modulating the expression of apoptosis related genes and proteins, such as caspases, Bcl-2, and Bax (51, 52). Several microbial metabolites, such as versipelostatin and verrucosidin, or plant product, such as arctigenin, or members of the biguanide class of compounds, such as metformin and phenformin, showed to inhibit GRP78 (53). However, many of these compounds exert additional biological functions besides inhibition of GRP78 and their action should be carefully evaluated on EC cells. Another promising opportunity of therapeutic intervention arises from the observation that GRP78 is often present on the membrane of different cancer cells, including EC cells (32, 40). As mentioned above, a commercial polyclonal antibody directed against the C-terminus of GRP78 induces apoptosis in AN3CA EC cells (32). Another opportunity of targeting GRP78 present on cell membrane of EC cells might be represented by synthetic chimeric ligand peptides containing programed cell death-inducing sequence, as they showed to suppress tumor growth in xenograft and isogenic mouse models of breast and prostate cancer (54).
Conclusion
The induction of the UPR pathways and GRP78 increase and/or localization following ER stress are increasingly recognized as important contributors to tumor survival and growth as well as to the development of resistance to chemotherapeutic agents in different types of cancer, including EC. While targeting ER stress and GRP78 as an anticancer strategy appears to be a very promising task also in EC, there are a number of limitations in our knowledge about the exact role of molecules involved in ER stress and how they might influence cell fate. Therefore, future studies in this area may further clarify whether GRP78 and/or other molecules involved in the UPR could really represent promising molecules to be investigated as target for EC therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Loos AH, Oostindier M, Weiderpass E. Geographic and temporal variations in cancer of the corpus uteri: incidence and mortality in pre-and postmenopausal women in Europe. Int J Cancer (2005) 117:123–31. doi: 10.1002/ijc.21099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Di Cristofano A, Hedrick Ellenson L. Endometrial carcinoma. Annu Rev Pathol (2007) 2:57–85. doi:10.1146/annurev.pathol.2.010506.091905
3. Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol (1983) 15:10–7. doi:10.1016/0090-8258(83)90111-7
4. D’Andrilli G, Bovicelli A, Paggi MG, Giordano A. New insights in endometrial carcinogenesis. J Cell Physiol (2012) 227:2842–6. doi:10.1002/jcp.24016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Masciullo V, Amadio G, Lo Russo D, Raimondo I, Giordano A, Scambia G. Controversies in the management of endometrial cancer. Obstet Gynecol Int (2010) 2010:638165. doi:10.1155/2010/638165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Nyholm HC, Nielsen AL, Norup P. Endometrial cancer in postmenopausal women with and without previous estrogen replacement treatment: comparison of clinical and histopathological characteristics. Gynecol Oncol (1993) 49:229–35. doi:10.1006/gyno.1993.1112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Kaaks R, Lukanova A, Kurzer MS. Obesity, endogenous hormones, and endometrial cancer risk: a synthetic review. Cancer Epidemiol Biomarkers Prev (2002) 11:1531–43.
8. Matias-Guiu X, Catasus L, Bussaglia E, Lagarda H, Garcia A, Pons C, et al. Molecular pathology of endometrial hyperplasia and carcinoma. Hum Pathol (2001) 32:569–77. doi:10.1053/hupa.2001.25929
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Prat J, Gallardo A, Cuatrecasas M, Catasús L. Endometrial carcinoma: pathology and genetics. Pathology (2007) 39:72–87. doi:10.1080/00313020601136153
10. Llobet D, Pallares J, Yeramian A, Santacana M, Eritja N, Velasco A, et al. Molecular pathology of endometrial carcinoma; practical aspects from the diagnostic and therapeutical view points. J Clin Pathol (2009) 62:777–85. doi:10.1136/jcp.2008.056101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Healy SJM, Gorman AM, Mousavi-Shafaei P, Gupta S, Samali A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur J Pharmacol (2009) 625:234–46. doi:10.1016/j.ejphar.2009.06.064
12. Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ (2006) 13:363–73. doi:10.1038/sj.cdd.4402296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Treglia AS, Turco S, Ulianich L, Ausiello P, Lofrumento DD, Nicolardi G, et al. Cell fate following ER stress: just a matter of “quo ante” recovery or death? Histol Histopathol (2012) 27(1):1–12.
14. Ma Y, Hendershot LM. The role of the unfolded protein response in tumor development: friend or foe? Nat Rev Cancer (2004) 4:966–77. doi:10.1038/nrc1505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med (2006) 6:45–54. doi:10.2174/156652406775574523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Pfaffenbach KT, Lee AS. The critical role of GRP78 in physiologic and pathologic stress. Curr Opin Cell Biol (2011) 23:150–6. doi:10.1016/j.ceb.2010.09.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res (2007) 67:3734–40. doi:10.1158/0008-5472.CAN-06-4594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem (2003) 278:20915–24. doi:10.1074/jbc.M212328200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Fernandez PM, Tabbara SO, Jacobs LK, Manning FC, Tsangaris TN, Schwartz AM, et al. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res Treat (2000) 59:15–26. doi:10.1023/A:1006332011207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Gazit G, Lu J, Lee AS. De-regulation of GRP stress protein expression in human breast cancer cell lines. Breast Cancer Res Treat (1999) 54:135–46. doi:10.1023/A:1006102411439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Lin Y, Wang Z, Liu L, Chen L. Akt is the downstream target of GRP78 in mediating cisplatin resistance in ER stress-tolerant human lung cancer cells. Lung Cancer (2011) 71:291–7. doi:10.1016/j.lungcan.2010.06.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Koomagi R, Mattern J, Volm M. Glucose-related protein (GRP78) and its relationship to the drug-resistance proteins P170, GST-pi, LRP56 and angiogenesis in non-small cell lung carcinomas. Anticancer Res (1999) 19:4333–6.
23. Daneshmand S, Quek ML, Lin E, Lee C, Cote RJ, Hawes D, et al. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum Pathol (2007) 38(10):1547–52. doi:10.1016/j.humpath.2007.03.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P, et al. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci USA (2008) 105:19444–9. doi:10.1073/pnas.0807691105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Huang LW, Lin CY, Lee CC, Liu TZ, Jeng CJ. Overexpression of GRP78 is associated with malignant transformation in epithelial ovarian tumors. Appl Immunohistochem Mol Morphol (2012) 20(4):381–5. doi:10.1097/PAI.0b013e3182434113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Song MS, Park YK, Lee JH, Park K. Induction of glucose-regulated protein 78 by chronic hypoxia in human gastric tumor cells through a protein kinase C-ε/ERK/AP-1 signaling cascade. Cancer Res (2001) 61:8322–30.
27. Su R, Li Z, Li H, Song H, Bao C, Wei J, et al. Grp78 promotes the invasion of hepatocellular carcinoma. BMC Cancer (2010) 10:20. doi:10.1186/1471-2407-10-20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Shuda M. Activation of the ATF6, XBP1 and GRP78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol (2003) 38:605–14. doi:10.1016/S0168-8278(03)00029-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Langer R, Feith M, Siewert JR, Wester HJ, Hoefler H. Expression and clinical significance of glucose regulated proteins GRP78 (BiP) and GRP94 (GP96) in human adenocarcinomas of the esophagus. BMC Cancer (2008) 8:70. doi:10.1186/1471-2407-8-70
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Fu W, Wu X, Li J, Mo Z, Yang Z, Huang W, et al. Upregulation of GRP78 in renal cell carcinoma and its significance. Urology (2010) 75:603–7. doi:10.1016/j.urology.2009.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Bifulco G, Miele C, Di Jeso B, Beguinot F, Nappi C, Di Carlo C, et al. Endoplasmic reticulum stress is activated in endometrial adenocarcinoma. Gynecol Oncol (2012) 125(1):220–5. doi:10.1016/j.ygyno.2011.11.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Calí G, Insabato L, Conza D, Bifulco G, Parrillo L, Mirra P, et al. GRP78 mediates cell growth and invasiveness in endometrial cancer. J Cell Physiol (2014) 229(10):1417–26. doi:10.1002/jcp.24578
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Zhuang L, Scolyer RA, Lee CS, McCarthy SW, Cooper WA, Zhang XD, et al. Expression of glucose-regulated stress protein GRP78 is related to progression of melanoma. Histopathology (2009) 54:462–70. doi:10.1111/j.1365-2559.2009.03242.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Pyrko P, Schönthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res (2007) 67:9809–16. doi:10.1158/0008-5472.CAN-07-0625
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Jamora C, Dennert G, Lee AS. Inhibition of tumor progression by suppression of stress protein GRP78/BiP induction in fibrosarcoma B/C10ME. Proc Natl Acad Sci USA (1996) 93:7690–4. doi:10.1073/pnas.93.15.7690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Kern J, Untergasser G, Zenzmaier C, Sarg B, Gastl G, Gunsilius E, et al. GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood (2009) 114:3960–7. doi:10.1182/blood-2009-03-209668
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signaling and therapeutic targeting. Biochem J (2011) 434:181–8. doi:10.1042/BJ20101569
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Misra UK, Deedwania R, Pizzo SV. Activation and cross-talk between Akt, NF-κB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J Biol Chem (2006) 281:13694–707. doi:10.1074/jbc.M511694200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Shani G, Fischer WH, Justice NJ, Kelber JA, Vale W, Gray PC. GRP78 and Cripto form a complex at the cell surface and collaborate to inhibit transforming growth factor beta signaling and enhance cell growth. Mol Cell Biol (2008) 28:666–77. doi:10.1128/MCB.01716-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Misra UK, Mowery Y, Kaczowka S, Pizzo SV. Ligation of cancer cell surface GRP78 with antibodies directed against its COOH-terminal domain up-regulates p53 activity and promotes apoptosis. Mol Cancer Ther (2009) 8:1350–62. doi:10.1158/1535-7163.MCT-08-0990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Luvsandagva B, Nakamura K, Kitahara Y, Aoki H, Murata T, Ikeda S, et al. GRP78 induced by estrogen plays a role in the chemosensitivity of endometrial cancer. Gynecol Oncol (2012) 126(1):132–9. doi:10.1016/j.ygyno.2012.04.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Gray MJ, Mhawech-Fauceglia P, Yoo E, Yang W, Wu E, Lee AS, et al. AKT inhibition mitigates GRP78 (glucose-regulated protein) expression and contribution to chemoresistance in endometrial cancers. Int J Cancer (2013) 133:21–30. doi:10.1002/ijc.27994
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Slomovitz BM, Coleman RL. The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clin Cancer Res (2012) 18:5856–64. doi:10.1158/1078-0432.CCR-12-0662
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Liu G, Shang Y, Yu Y. Induced endoplasmic reticulum (ER) stress and binding of over-expressed ER specific chaperone GRP78/BiP with dimerized epidermal growth factor receptor in mammalian cells exposed to low concentration of N-methyl-N-nitro-N-nitrosoguanidine. Mutat Res (2006) 596:12–21. doi:10.1016/j.mrfmmm.2005.10.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Creutzberg CL, van Putten WL, Koper PC, Lybeert ML, Jobsen JJ, Wárlám-Rodenhuis CC, et al. Surgery and postoperative radiotherapy versus surgery alone for patients with stage-1 endometrial carcinoma: multicentre randomized trial. PORTEC Study Group. Post operative radiation therapy in endometrial carcinoma. Lancet (2000) 355:1404–11. doi:10.1016/S0140-6736(00)02139-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Creasman WT, Odicino F, Maisonneuve P, Beller U, Benedet JL, Heintz AP, et al. Carcinoma of the corpus uteri. J Epidemiol Biostat (2001) 6(1):47–86.
47. Muinelo-Romay L, Colas E, Barbazan J, Alonso-Alconada L, Alonso-Nocelo M, Bouso M, et al. High-risk endometrial carcinoma profiling identifies TGF-b1 as a key factor in the initiation of tumor invasion. Mol Cancer Ther (2011) 10:1357–66. doi:10.1158/1535-7163.MCT-10-1019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Teng Y, Ai Z, Wang Y, Wang J, Luo L. Proteomic identification of PKM2 and HSPA5 as potential biomarkers for predicting high-risk endometrial carcinoma. J Obstet Gynaecol Res (2013) 39(1):317–25. doi:10.1111/j.1447-0756.2012.01970.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Akram T, Maseelall P, Fanning J. Carboplatin and paclitaxel for the treatment of advanced or recurrent endometrial cancer. Am J Obstet Gynecol (2005) 192:1365–7. doi:10.1016/j.ajog.2004.12.032
50. Michener CM, Peterson G, Kulp B, Webster KD, Markman M. Carboplatin plus paclitaxel in the treatment of advanced or recurrent endometrial carcinoma. J Cancer Res Clin Oncol (2005) 131:581–4. doi:10.1007/s00432-005-0676-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Manohar M, Fatima I, Saxena R, Chandra V, Sankhwar PL, Dwivedi A. (-)-Epigallocatechin-3-gallate induces apoptosis in human endometrial adenocarcinoma cells via ROS generation and p38 MAP kinase activation. J Nutr Biochem (2013) 24(6):940–7. doi:10.1016/j.jnutbio.2012.06.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Park SB, Bae JW, Kim JM, Lee SG, Han M. Antiproliferative and apoptotic effect of epigallocatechin-3-gallate on Ishikawa cells is accompanied by sex steroid receptor downregulation. Int J Mol Med (2012) 30(5):1211–8. doi:10.3892/ijmm.2012.1104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Schönthal AH. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem Pharmacol (2013) 85:653–66. doi:10.1016/j.bcp.2012.09.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W, et al. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell (2004) 6:275–84. doi:10.1016/j.ccr.2004.08.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: GRP78, endoplasmic reticulum, UPR, stress, endometrial cancer
Citation: Ulianich L and Insabato L (2014) Endoplasmic reticulum stress in endometrial cancer. Front. Med. 1:55. doi: 10.3389/fmed.2014.00055
Received: 25 September 2014; Accepted: 02 December 2014;
Published online: 22 December 2014.
Edited by:
Giulio Cesare Spagnoli, University of Basel, SwitzerlandReviewed by:
Renato Franco, Istituto Tumori Fondazione Giovanni Pascale, ItalySabrina Rossi, Treviso General Hospital, Italy
Copyright: © 2014 Ulianich and Insabato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luigi Insabato, Department of Advanced Biomedical Sciences, University Federico II of Naples, Via S. Pansini 5, Naples, 80131, Italy e-mail:Zy5pbnNhYmF0b0BnbWFpbC5jb20=