 Pablo Granados-Durán1
Pablo Granados-Durán1 María D. López-Ávalos1
María D. López-Ávalos1 Jesús M. Grondona1
Jesús M. Grondona1 María del Carmen Gómez-Roldán1
María del Carmen Gómez-Roldán1 Manuel Cifuentes1,2
Manuel Cifuentes1,2 Margarita Pérez-Martín1
Margarita Pérez-Martín1 Martina Alvarez3
Martina Alvarez3 Fernando Rodríguez de Fonseca4
Fernando Rodríguez de Fonseca4 Pedro Fernández-Llebrez1*
Pedro Fernández-Llebrez1*- 1Departamento de Biología Celular, Genética y Fisiología, Instituto de Investigación Biomédica de Málaga (IBIMA), Facultad de Ciencias, Universidad de Málaga, Málaga, Spain
- 2Centro de Investigaciones Biomédicas en Red de Bioingeniería, Biomateriales y Nanomedicina, Facultad de Ciencias, Universidad de Málaga, Málaga, Spain
- 3Departamento de Anatomía Patológica, Facultad de Medicina, Universidad de Málaga, Málaga, Spain
- 4Unidad de Gestión Clínica de Salud Mental, Hospital Regional Universitario de Málaga, Instituto de Investigación Biomédica de Málaga (IBIMA), Málaga, Spain
In the present paper, we describe the facts that took place in the rat brain after a single injection of the enzyme neuraminidase from Clostridium perfringens into the right lateral ventricle. After injection, it diffused through the cerebrospinal fluid of the ipsilateral ventricle and the third ventricle, and about 400 μm into the periventricular brain parenchyma. The expression of ICAM1 in the endothelial cells of the periventricular vessels, IBA1 in microglia, and GFAP in astrocytes notably increased in the regions reached by the injected neuraminidase. The subependymal microglia and the ventricular macrophages begun to express IL1β and some appeared to cross the ependymal layer. After about 4 h of the injection, leukocytes migrated from large venules of the affected choroid plexus, the meninges and the local subependyma, and infiltrated the brain. The invading cells arrived orderly: first neutrophils, then macrophage-monocytes, and last CD8α-positive T-lymphocytes and B-lymphocytes. Leukocytes in the ventricles and the perivascular zones penetrated the brain parenchyma passing through the ependyma and the glia limitans. Thus, it is likely that a great part of the damage produced by microorganism invading the brain may be due to their neuraminidase content.
Introduction
In agreement to Graeber et al. (1) the term neuroinflammation implies not only microglial activation but also the invasion of the nervous tissue and/or the cerebrospinal fluid (CSF) by blood-circulating leukocytes. Inflammation of the brain can be triggered by the presence in the ventricle, the meninges, or the nervous parenchyma of biological agents such as bacteria or viruses, or by a plethora of chemical or mechanical stimuli (2–5). In any case, the clinical relevance of brain inflammation lies in part on the detrimental or even lethal effects of the invading defensive cells or proteins, which eventually can give rise to chronic neurodegenerative diseases (6, 7).
Among the many agents that trigger neuroinflammation some are located in the microbial envelope, such as lipopolysaccharides (8, 9) or the enzyme neuraminidase (NA) (10). Indeed, the abundance of NA in the outer surface of certain viruses and bacteria has been related to their pathogenicity (2, 11–14). So much so that a single mutation in the hemagglutinin-NA (HN) surface complex of the mump virus is enough as to greatly decrease its pathogenicity, an interesting feature in the design of vaccines (15). Moreover, inhibitors of the NA activity are among the most important antiviral compounds [i.e., oseltamivir; (16)]. Despite all evidences supporting a pre-eminent role of NA in neuroinflammation, little is known about its exact mechanism of action and the events that are triggered after its administration. Moreover, the molecular differences among the neuraminidases that can be found in the microbial covers, probably lead to differences in the pathogenesis that they provoke (17).
Clostridium perfringens is a ubiquitous bacterium that possesses NA at its surface (18). NA from C. perfringens is easily available, so we used it before to desialylate some molecules present in the ventricular CSF (19). We reported that a single intracerebroventricular (ICV) injection of a high dose of NA (about 10 μg) produced the massive loss of the multiciliated ependyma followed by acute inflammation of meninges and ventricles and the development of hydrocephalus (10). By decreasing the amount of injected NA, only a partial detachment of the ependymal layer occurred in the injected lateral ventricle, but not in the contralateral ventricle or the third ventricle. This model turned also useful to investigate the importance of the ependyma on postnatal neurogenesis (20). We investigated the time course of inflammatory events after ICV administration of low doses of NA from C. perfringens, what might prove to be a useful model to study neuroinflammation. The facts suggest the importance of NA in degenerative events known to occur after microbial invasion of the central nervous system.
Materials and Methods
Animals
Eighty-four adult male Wistar rats (body weight, bw, 250–300 g) were used in this study (Charles River Laboratories, Barcelona, Spain). All animals were housed under a 12 h light/dark cycle with food and water available ad libitum. Animal procedures were done according to the European Union (86/609/EEC) and Spanish (RD 1201/2005) legislations.
Intracerebroventricular Injection
Animals were anesthetized with 2,2,2-tribromoethanol (0.2 g/kg body weight, Fluka Chemika, St. Louis, MO, USA) and positioned in a stereotaxic frame. NA from C. perfringens (Roche Diagnostics, Basel, Switzerland, catalog No. 11 585 886 001) dissolved in 0.9% sterile saline (25 mU/μl) was administrated by stereotaxic surgery into the right lateral cerebral ventricle, in the following coordinates from Bregma: antero-posterior = −0.5 mm; medio-lateral = −1.4 mm; dorso-ventral = −3.5 mm (21). With the aid of a pump, a 500 mU (20 μl) of NA were injected at a rate of 2 μl/min during 10 min. Control rats were injected with saline. Some animals were sacrificed immediately after NA injection (time 0 h), and the rest were recovered from anesthesia and sacrificed at 2, 4, 12, 24 h, 2, 4, 7, 15, 30 days, 3, and 6 months after the administration of NA.
Histological Procedures
Animals were anesthetized with 2,2,2-tribromoethanol and transcardially perfused with saline followed by Bouin’s fixative. Brains were removed and immersed in the same fixative for 24 h at room temperature (RT), and later embedded in paraffin wax. Seven-micrometer sections were obtained from each brain, and series along the region of interest (from Bregma −0.3 to −1.2 mm approximately) were mounted in gelatine treated slides.
Lectin Binding
The Limax flavus agglutinin (LFA, Calbiochem, San Diego, CA, USA) was chosen for its exclusive affinity for sialic acid. Deparaffinized thin sections were incubated with 4 μg/ml LFA in lectin buffer (1 mM Cl2Ca, 1 mM Cl2Mg, 0.6% TRIS, pH 7.6) for 1 h at RT. The lectin bound to the tissue was later identified using a rabbit anti-LFA antibody [(22); see Immunohistochemistry]. The peanut (Arachis hypogea) agglutinin (PNA, Vector, Burlingame, CA, USA. l-1070) has affinity for terminal residues of galactose, which is the subterminal sugar residue in complex-type glycoproteins. After the removal of terminal sialic acid, galactose is exposed as terminal residue, and the PNA binding sites arise. To check for the removal of sialic acid by the action of NA, sections were incubated in 3 μg/ml peroxidase-labeled PNA in 0.1 M phosphate buffered saline (PBS) for 1 h at RT. The peroxidase activity was detected with 0.1 M PBS containing 0.05% diaminobenzidine (DAB; Sigma) and 0.03% hydrogen peroxide (Merck).
Immunohistochemistry
Immunohistochemistry was carried out on deparaffinized tissue sections using either immunoperoxidase or immunofluorescence techniques. The primary and secondary antibodies used are listed in Tables 1 and 2, respectively. All of them were diluted with PBT buffer (0.3% bovine serum albumin, 0.3% Triton X-100 in PBS pH 7.3). Primary antibodies were incubated overnight at RT. Negative controls for the immunostaining consisted in equivalent sections subjected to the same protocol but omitting the primary antibody.

Table 1. List of primary antibodies used for immunohistochemistry.
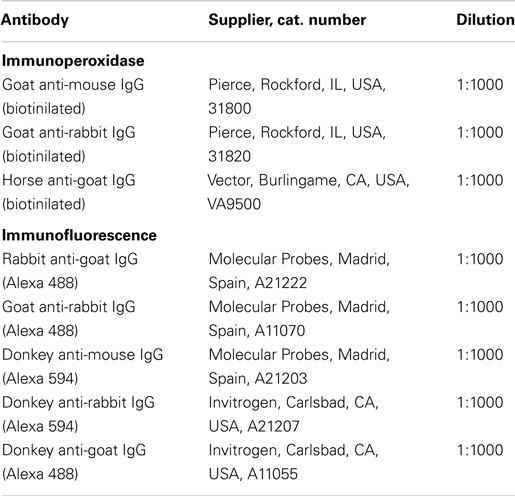
Table 2. List of secondary antibodies used for immunohistochemistry.
Sections for immunoperoxidase procedures were incubated in 3% hydrogen peroxide and 10% methanol in PB 0.1 M to quench endogenous peroxidase activity, prior to overnight incubation with the primary antibody. Biotinylated secondary antibodies were used (Table 2; 60 min incubation), along with the avidin-biotin-complex (ABC) amplification method. The ABC reagent was prepared according to manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA, USA) and incubated during 30 min. Peroxidase activity was revealed with 0.05% DAB and 0.03% hydrogen peroxide. All the incubations were performed in a moist chamber at RT.
To study the co-localization of particular markers, double immunofluorescence was performed using the following pairs of antibodies: ICAM1 and IBA1, IBA1, and IL1β, myeloperoxidase (MPO) and IL1β, and CD3ε and CD8α. The primary antibodies were mixed and incubated simultaneously (overnight at RT). Alexa fluor secondary antibodies (Table 2) were used. Coverslides were mounted with buffered glycerol containing 10% of the anti-fading agent Mowiol 4-88 (Calbiochem/EMD Chemicals, San Diego, CA, USA). The sections were then observed under a confocal microscope (Leica, SP5 II). Some antibodies required specific antigen retrieval pretreatment prior to immunostaining. Thus, the sections incubated with anti-CD3ε and anti-CD8α were pretreated with 10 mM TRIS-EDTA buffer pH 9.0, and the sections incubated with anti-PAX5 were pretreated with 10 mM citrate buffer pH 6.0. In both cases, pretreatment was done at 95°C in microwave for 8 months.
Quantitative Studies
Two different strategies were used depending on the staining properties of the cells or structures to be quantified. When the quantified resident cells displayed processes or irregular shapes (microglia, astrocytes, and endothelial cells) the immunostained area was measured. If the cells had well-defined profiles easily distinguishable from each other (infiltrating leukocytes), a direct count of the number of cells was preferred.
Quantification of ICAM1, IBA1, and GFAP immunostaining
To study the response of the resident cells to NA-injection, we quantified the area immunostained with ICAM1, IBA1, and GFAP at different times (0, 4, 24 h, 7, and 15 days) after the inoculation of NA. These times were determined after the qualitative observation of the differences between the injected and the contralateral sides over time.
A previous detailed study of immunostained serial sections allowed the selection of the levels to be studied. Thus, two sections from each animal (Bregma −0.3 and −0.92 mm approximately) were chosen for the quantification. In any case, the selected sections were close to the foramen of Monro but far enough from the site of injection as to avoid areas of surgically damaged tissue. In each section studied, micrographs were taken from the striatal ventricular wall, and from the septum-fimbria wall, both in the injected side and in the contralateral one (red squares in Figure 2A show the approximate location of the micrographs). Pictures were then processed using the image analysis software Visilog 6.3 (Noesis, France). To introduce a correction related to the basal level of expression of a particular marker in each animal, the immunostained area measured in the contralateral side was subtracted to the value obtained in the same location in the injected side. This correction was made to subtract the basal expression of a marker in each animal, which was found to vary from rat to rat. Afterwards, the mean value was obtained from the two locations and the two sections studied from each animal, which was considered the total stained area of that animal. A total of five animals were analyzed in each time post-injection studied.
Quantification of infiltrating leukocytes
Cell infiltration was evaluated by immunostaining with the following antibodies: anti-MPO for neutrophils, anti-IL1β for monocyte-macrophages, anti-CD3ε for T-lymphocytes, anti-CD8α for cytotoxic T-lymphocytes, and anti-PAX5 for B-lymphocytes. We realize that these substances could also be synthesized by other cell types. However, presently and in agreement to many researchers, these markers are used to identify these different kind of immunologic blood-borne cells with a high degree of confidence. However, a low possibility exists that some cell does not belong to the corresponding group.
Immunoreactive cells were counted in three different locations: (1) the choroid plexus within the injected ventricle; (2) the meninges close to the optic chiasm; and (3) the zone around the foramen of Monro containing large venules close to the origin of the choroid plexus and the subfornical organ. Serial sections from Bregma −0.85 to −1.13 mm approximately (the level of the foramen of Monro) were immunostained with each antibody. Four sections per animal were selected for counting of positive cells. Between three and five animals were studied in each time post NA-injection. The times post-injection studied were the same as those selected for ICAM1, IBA1, and GFAP quantification (0, 4, 24 h, 7, and 15 days). In the case of the choroid plexus, the area occupied by this structure in each section studied was measured with the Visilog 6.3 software; the number of positive cells counted in this location was expressed per millimeter square of choroid plexus. In the other two locations, the number of positive cells was expressed per section.
Statistical analysis
Differences between times post-injection of NA were evaluated by ANOVA (significance level of 0.05), followed by a Tukey post hoc test with a level of significance of 0.05. The values measured at time 0 were considered the baseline. The software SPSS® Statistics 20 (IBM®) was used for this analysis. In the histograms, the existence of significant differences between groups is indicated with different letters. Thus, two bars labeled with the same letter means that there is no significant difference between the two groups. Conversely, two bars labeled with different letters indicate that there is a significant difference between them.
Results
Animals were sacrificed immediately after the injection of NA (T: 0 h), LFA lectin exhibited a negative halo (~400 μm from the ventricle surface) around the injected lateral ventricle and the rostral third ventricle (Figures 1A,C). The ependymal surface of the injected lateral ventricle showed no staining at all (Figure 1G). Conversely, the contralateral ventricle showed no halo (Figures 1A,D) and a conspicuous staining of the subependyma and the surface of the ependymal cells (Figure 1H). On the contrary, when using PNA lectin (terminal galactose affinity) a positive label was found in the subependyma of the injected ventricle (Figures 1B,E) that was particularly intense in the surface of the ependymal cells (Figure 1I). The walls of the contralateral ventricle showed virtually no binding to PNA (Figures 1B,F), and the surface of the ependymal cells was only feebly positive (Figure 1J). After about a week, the ependymal cells that remained in the ventricular lining of the injected ventricle recovered the sialic acid and thus the LFA binding.
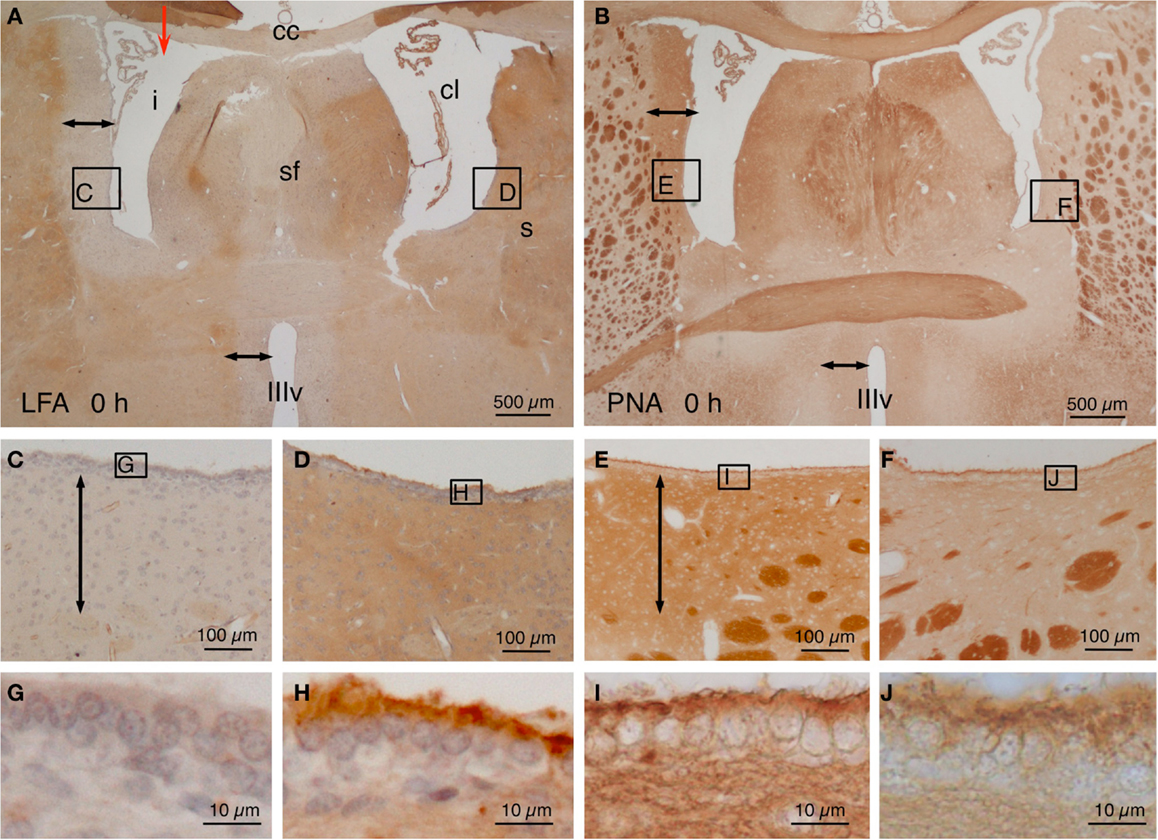
Figure 1. Lectin histochemistry in NA-injected rat brains immediately after the injection. (A,B) The site of injection is indicated with a red arrow in (A). Note a LFA-negative and PNA-positive halo around the injected lateral ventricle (i) and the third ventricle (IIIv) (double-headed arrow) but not the contralateral ventricle (cl). (C–F) Details of the zones squared in (A,B) showing the negative or positive halo (double-headed arrows). The squared areas are shown in detail in the following pictures. (G–J) The surface of the ependymal cells was LFA-negative (G) and PNA-positive (I) in the injected ventricle, whereas it was LFA-positive (H) and almost PNA-negative (J) in the contralateral ventricle. cc, corpus callosum; cl, contralateral ventricle; i, injected lateral ventricle; s, striatum; sf, septum-fimbria; IIIv, third ventricle. Sections in (A,C,D,G,H) were counterstained with hematoxilin–eosin.
Control rats, which were injected with the vehicle, did never present any sign of brain inflammation. Only the brain cortex at the level of the needle path showed a few signs of local inflammatory reaction that spontaneously healed as observed in brains examined at different times after the injection. No evidences of inflammation were observed in the brain ventricles, the meninges or the periventricular regions.
The elimination of sialic acid triggers a series of events resulting in inflammation. Some responses depend on changes in resident brain cells while others on infiltrating blood leukocytes. Therefore, both cell populations were quantified afterwards.
NA Induces Upregulation of IBA1, GFAP, and ICAM1 in Resident Cells
In NA-injected animals, the immunoreactivity to IBA1 antiserum increased notably in the affected periventricular zones (compare Figures 2E,F). We quantified the differences in two selected regions of the striatum and the fimbria-fornix at different post-injection times. IBA1 was upregulated from early times after NA injection, and the basal levels were recovered after about 2 weeks (Figure 2B). A notable increase in IBA1 immunoreactivity was also evident in the injected side in places located far from the site of injection, as is the optic tract (Figure 2K).
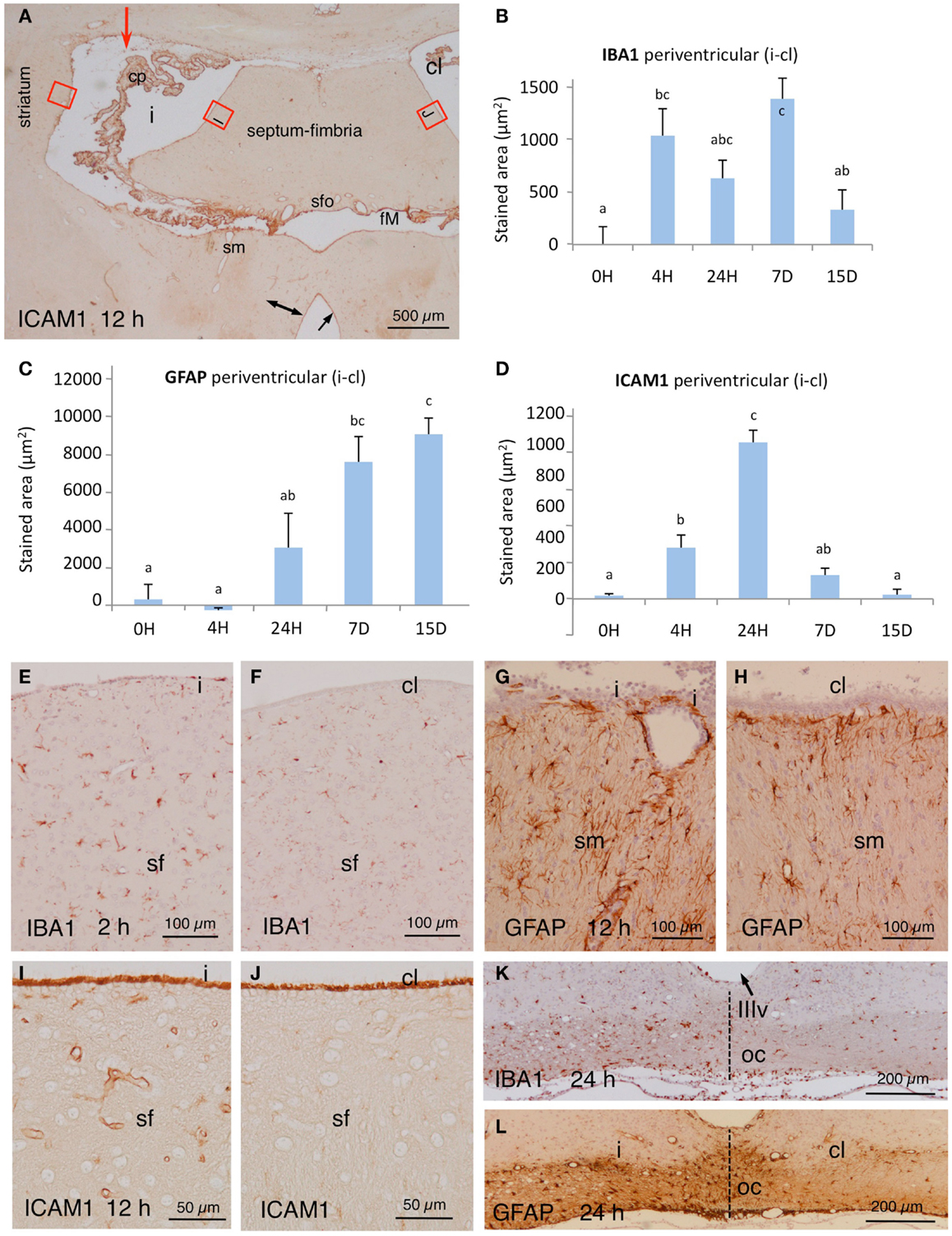
Figure 2. (A) Panoramic view at the level of the foramen of Monro (fM) of the brain of an animal sacrificed 12 h after NA injection (red arrow points injection site). The tissue was immunostained with ICAM1. Note an increase in the immunoreactivity in the vessels of the affected periventricular zones (double-headed arrow) and the strong reactivity of the choroid plexus (cp) and the surface of the ependymal cells (arrow). The red squares correspond roughly to the zones used for quantification; some are detailed in (I,J). (B–D) Quantification of the expression of IBA1, GFAP, and ICAM1 in the periventricular area at selected times post-injection. Red squares in (A) point approximately the areas where micrographs were taken in the striatum and the septum. The values represented are the difference of stained area between the injected lateral ventricle (i) minus de contralateral ventricle (cl). The bars are the mean ± SEM of five animals analyzed. Letters (a–c) on bars indicate the absence (same letter) or presence (different letter) of a significant difference between groups (α = 0.05). (E,F) Immunoreactivity to IBA1 in the periventricular region at the level of the septum-fimbria (sf) in the injected (i) and the contralateral (cl) ventricles, 2 h after NA injection. (G,H) Details of GFAP staining in the stria medullaris (sm) in the injected (i) and the contralateral (cl) sides 12 h after the administration of NA. (I,J) Details [red squares in (A)] of the immunoreactivity to ICAM1 in the septum-fimbria (sf) subependymal parenchyma of the injected (i) and the contralateral (cl) sides 12 h after the injection of NA. Note the strong immunoreactivity of the ependymal surface in both ventriculi. (K,L) Twenty-four hours after NA injection, the optic chiasm (oc) beneath the third ventricle (IIIv) showed a higher IBA1 and GFAP staining in the injected (i) than in the contralateral side (cl). cl, contralateral ventricle; cp, choroid plexus; fM, foramen of Monro; i, injected lateralventricle; sfo, subfornical organ; sm, stria medularis; sf, septum-fimbria; oc, optic chiasm; IIIv, third ventricle.
GFAP staining in astrocytes of control rats (non-injected or saline-injected) was as expected, with no staining detected in the ependyma or the choroid plexus. As for IBA1, quantification of the GFAP stained area was carried out in selected regions of the striatum and the fimbria-fornix area of the injected ventricle relative to the contralateral one. GFAP staining showed a higher expression 24 h after NA-injection, which further increased one week later. It remained overexpressed even 2 weeks post-injection (Figure 2C). The increase in immunoreactivity was clearly observed also in other region such as the stria medullaris (compare Figures 2G,H). Regions located farther from the site of injection also presented increased GFAP expression in the injected side with respect of the contralateral one, a fact particularly evident in the optic tract (Figure 2L).
In control rats, the wall of the large vessels of the meninges, the choroid plexus, the subependyma around the foramen of Monro, and the surface of the ependymal cells (especially of the choroid plexus) were immunoreactive to ICAM1. Parenchymal capillaries were virtually negative. Conversely, in the injected rats the wall of the parenchymal vessels located in the subependyma of the affected ventricles (up to ~400 μm from the ventricle surface) became ICAM1-immunoreactive (Figure 2A; compare Figures 2I,J). As for IBA1 and GFAP, the changes in immunoreactivity to ICAM1 were quantified in the same selected zones of the lateral ventricle wall. There was a progressive increase in immunoreactivity that was evident 4 h after the injection and peaked at 24 h (Figure 2D). ICAM1 expression decreased afterwards to reach control levels after about 2 weeks.
In control rats, IBA1 stained epiplexus cells in the choroid plexus and very few subependymal microglial cells (Figure 3A). In the experimental animals, a quite striking observation was that some IBA1-positive cells beneath the ependyma, seemed to penetrate between the ependymal cells even before the onset of blood cell infiltration (Figure 3B and inset). In similar locations, IL1β-positive cells could be found at early times after NA administration (Figures 3C,D). Moreover, double immunostaining demonstrated the existence of cells that were positive to both IBA1 and IL1β (Figures 3E,F).
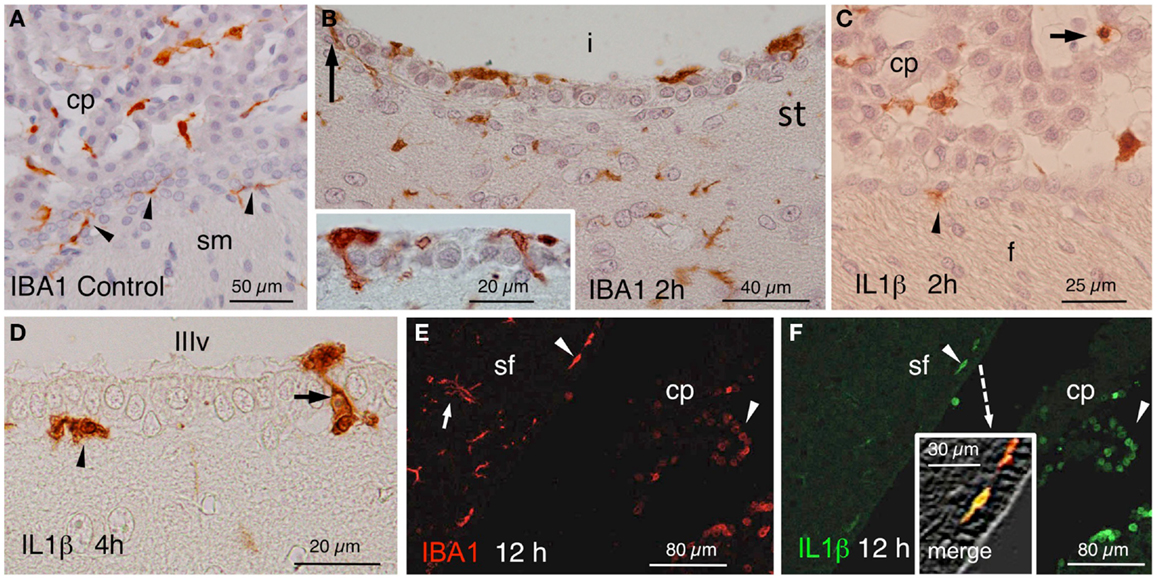
Figure 3. (A) IBA1-positive epiplexus and subependymal cells (arrowheads) in a vehicle-injected specimen sacrificed immediately after the injection. (B) Detail of the striatal (st) side of the injected lateral ventricle (i) near the inferior sulcus of the lateral ventricle 2 h after the injection. Note IBA1-positive cells over, beneath and among the ependymal cells (arrow and inset). (C) Two hours after the injection of NA, scattered IL1β-positive cells occurred inside (arrow) and outside the choroid plexus (cp) as well as in the subependyma (arrowhead) of the injected ventricle. (D) Detail of IL1β-stained cells in the subependyma (arrowhead) and crossing the ependymal wall (arrow) of the third ventricle (IIIv) 4 h after the injection of NA. (E,F) Double immunofluorescence with anti-IBA1 (red) and anti-IL1β (green). Double-stained cells appear in the ventricle and the subependyma (arrowheads, and merge detailed in the inset of (F) showing a double-stained subependymal cell). Many IBA1-positive cells in the brain parenchyma were not immunoreactive to IL1β [arrow in (E)]. cp, choroid plexus; sf, septum-fimbria; sm, stria medullaris; (i), injected lateral ventricle; st, striatal wall of the lateral ventricle; f, fornix; IIIv, third ventricle.
NA Induced Extravasation of Blood Leukocytes
The most noticeable observation after the ICV inoculation of NA was the extravasation of leukocytes in the injected, but not the contralateral side. From about 4 h after the injection, cellular infiltration took place in large blood vessels located in the meninges, choroid plexus, and subependymal parenchyma. Extravasation in smaller vessels was infrequent. The extravasated leukocytes penetrated the brain parenchyma directly from the vessels passing through the blood–brain barrier, the pial barrier, or the ependyma of the choroid plexus (Figure 4A) and of the ventricle walls (Figures 4B,C,E). To identify the nature of infiltrating cells, we used antibodies against MPO for neutrophils (Figures 4C,D), IL1β for monocyte-macrophages (Figures 3F and 4B,D), CD3ε and CD8α for T-lymphocytes (Figures 5A–G), and PAX5 for B-lymphocytes (Figures 5H–J). Some infiltrating cells, seemingly of the macrophagic-monocyte lineage, also stained for IBA1 (Figures 3E and 4A) and ICAM1 (Figures 4E–G). Double immunostaining with anti-MPO and anti-IL1β (Figure 4D) revealed no co-localization of these two markers, indicating that the antibodies discriminate quite well between neutrophils and macrophages. Moreover, no polymorphonucleated cell was stained using anti-IL1β, IBA1 or ICAM1 at any time post-injection (Figures 4A,B,F). It is likely that the monocyte-macrophage cells may be recognized by the IBA1 antiserum. Moreover, some of them (i.e., those that were activated) were also recognized by IL1β and ICAM1 antibodies (Figures 4B,D,E–G).
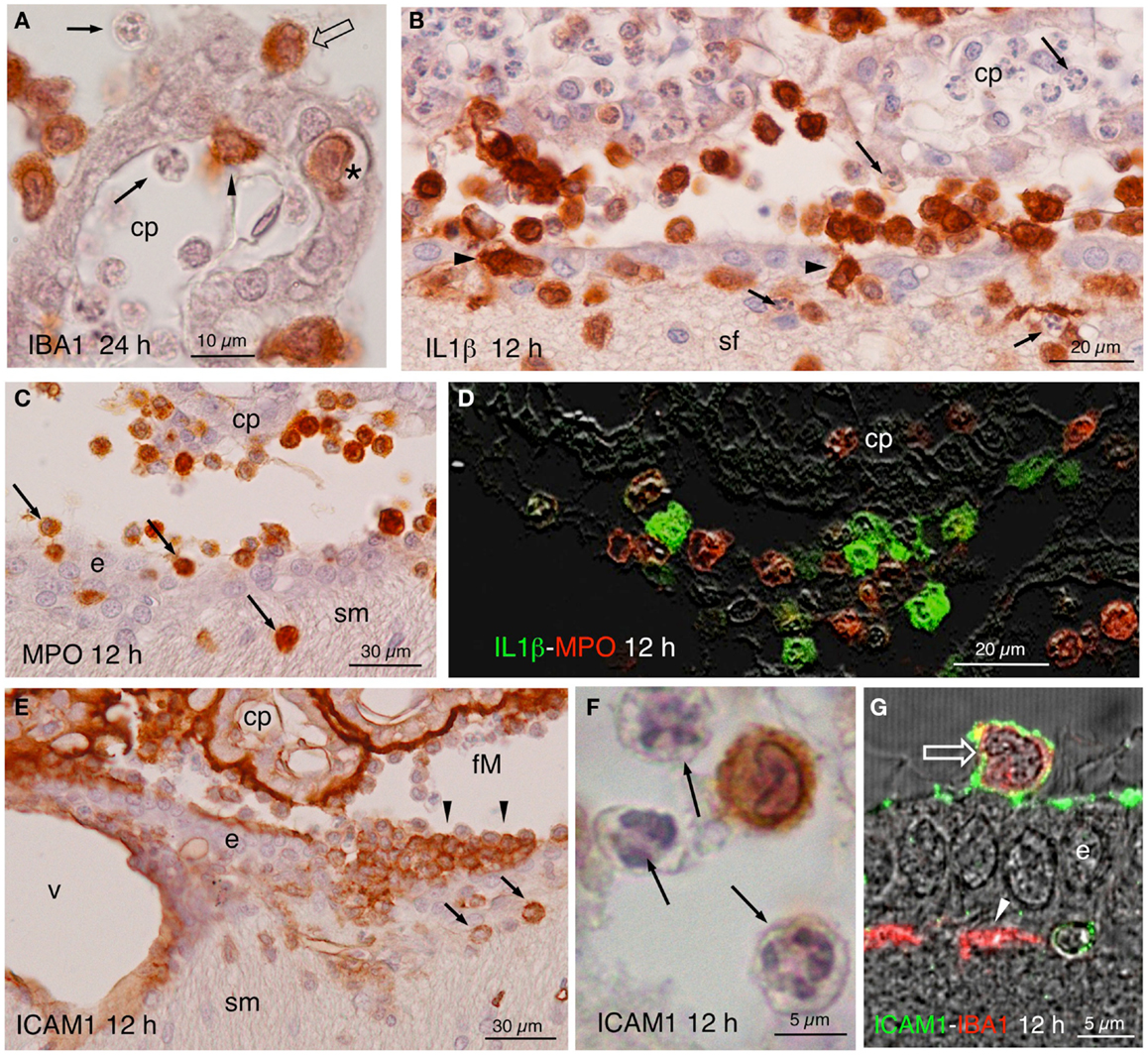
Figure 4. (A) Infiltrating IBA1-positive cells in the choroid plexus (cp) of the injected ventricle at 24 h post-injection. Polymorphonucleated cells inside and outside the choroid plexus were never stained (arrows); positive cells occurred inside (arrowhead) outside (open arrow), and in transit (asterisk). (B) Twelve hours after NA injection, abundant IL1β-positive cells were seen in the ventricle, penetrating the ependyma (arrowheads) and in the brain parenchyma, in the septum-fimbria (sf) area. Polymorphonuclear negative cells, present inside and outside choroid plexus (cp), the ventricle and the parenchyma (arrows), were also numerous. (C) MPO immunostaining in the region of the foramen of Monro, where positive cells were found near the choroid plexus (cp), the ependyma (e), and penetrating the brain parenchyma (arrows) in the region of the stria medullaris (sm). (D) IL1β (green) and MPO (red) double immunofluorescence in the region of the choroid plexus (cp) of the injected ventricle 12 h after NA administration. No double-stained cells were detected. (E) Detail of a zone of the foramen of Monro (fM) comprising a part of the stria medullaris (sm) 12 h after the injection of NA, immunostained with anti-NCAM1. The surface of the ependymal cells (e) and of the choroid plexus (cp) and the wall of the local vessels (v) was strongly stained. Some infiltrating cells in the ventricle (arrowheads) and in the brain parenchyma (arrows) were also positive. (F) Detail of an immunoreactive cell with lobed nucleus in the ventricle. The polymorphonucleated leukocytes (arrows) were never immunoreactive to ICAM1. (G) Some cells present in the ventricle were double-stained with ICAM1 and IBA1 (open arrow). The ICAM1-positive surface of the ependymal cells (e) can be observed, as well as some IBA1-positive subependymal cells (arrowhead). cp, choroid plexus; sf, septum-fimbria; e, ependyma; sm, stria medullaris; fM, foramen of Monro; v, vessels.
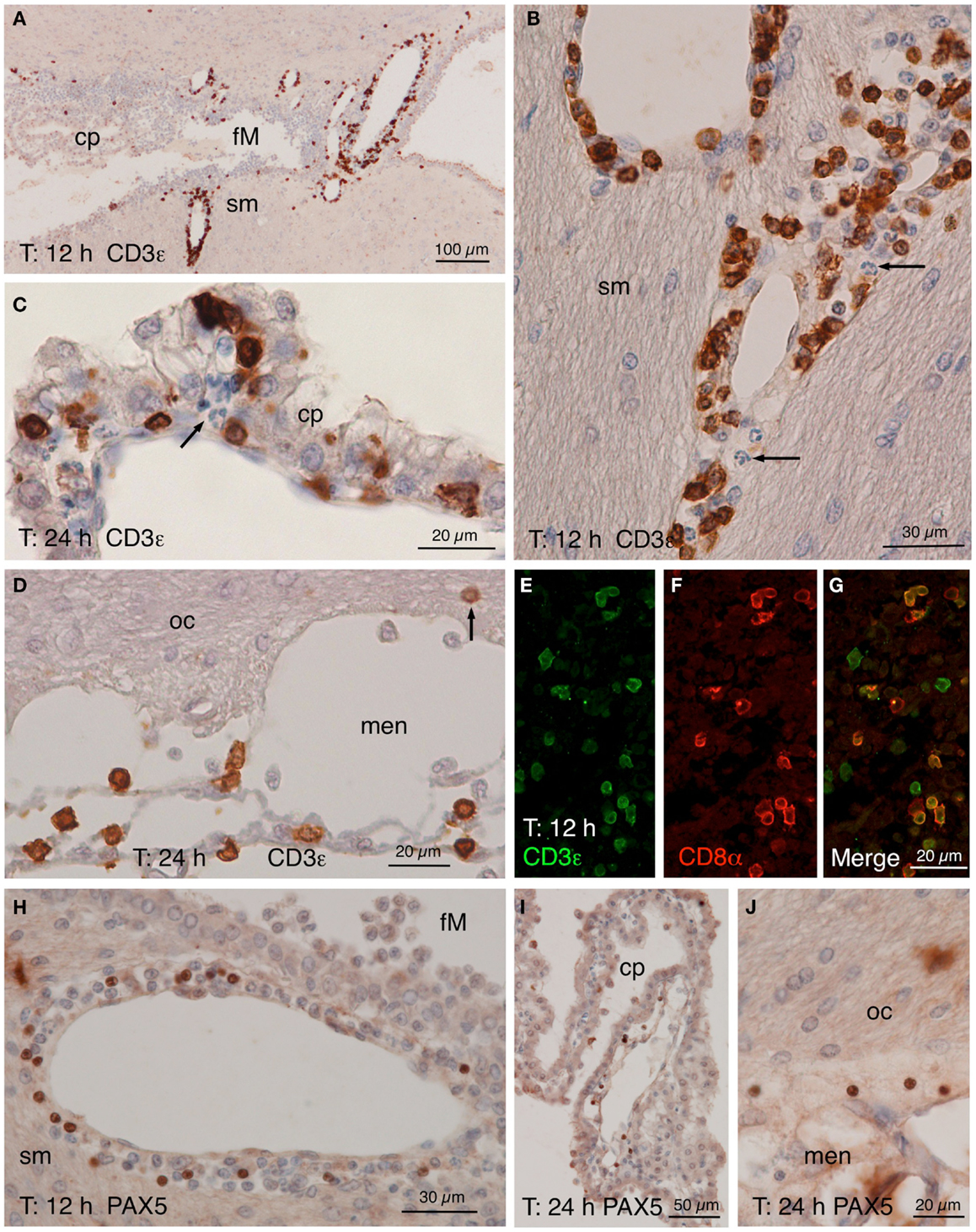
Figure 5. CD3ε, CD8α, and PAX5 immunostainings. (A–D) CD3ε staining in the zone of the foramen of Monro (fM) of the injected ventricle comprising a part of the stria medullaris [sm, (A)]. Many positive cells occurred in the perivascular spaces of the local large venules 12 h after NA injection. A detailed view (B) of the vessels in the stria medullaris (sm) shows that many positive cells have smooth nuclei while polymorphonucleated cells (arrow) are negative. (C) Twenty-four hours after the injection, some positive cells seemed to cross the ependyma of the choroid plexus (cp). (D) Positive cells were also present in the meninges (men) next to the optic chiasm (oc), and few appeared to penetrate the brain parenchyma (arrow). (E–G) Double immunofluorescence of lymphocytes in a 12 h post-injection brain section. Most CD3ε-positive cells [(E), green] found in the perivascular spaces of the large vessels were also positive for CD8α [(F), red]. (H–J) PAX5 immunostaining showing positive cells in the wall of the large vessels around the foramen of Monro [fM; (H)], although they were not as abundant as CD3ε-positive cells. Few PAX5-positive nuclei were also found in the choroid plexus (cp) of the injected lateral ventricle (I) as well as in the meninges (men) close to the optic chiasm [oc; (J)]. cp, choroid plexus; fM, foramen of Monro; men, meninges; oc, optic chiasm; sm, stria medullaris.
Lymphocytes infiltrated from the same vessels that neutrophils and macrophages (Figure 5), crossed the choroid plexus ependyma (Figures 5C,I), and invaded the ventricle (Figures 5A,I), the meningeal spaces (Figures 5D,J), and the brain parenchyma (Figures 5B,H). As revealed by double immunofluoresce using anti-CD3ε and anti-CD8α antibodies, most of them were cytotoxic T-lymphocytes (Figures 5E–G). B-lymphocytes (PAX5-positive) were also present, but they were much less abundant (about one-tenth) than T-lymphocytes (Figures 5H–J).
Infiltrating cells were counted at different post-injection times in three selected locations (Figure 6): (i) the choroid plexus of the injected ventricle (Choroid plexus in Figure 6); the contralateral plexus showed virtually no activation; (ii) the zone around the ipsilateral foramen of Monro, where large venules are found (Vessels in Figure 6); (iii) the region of the meninges close to the ipsilateral side of the optic chiasm (Optic chiasm in Figure 6).
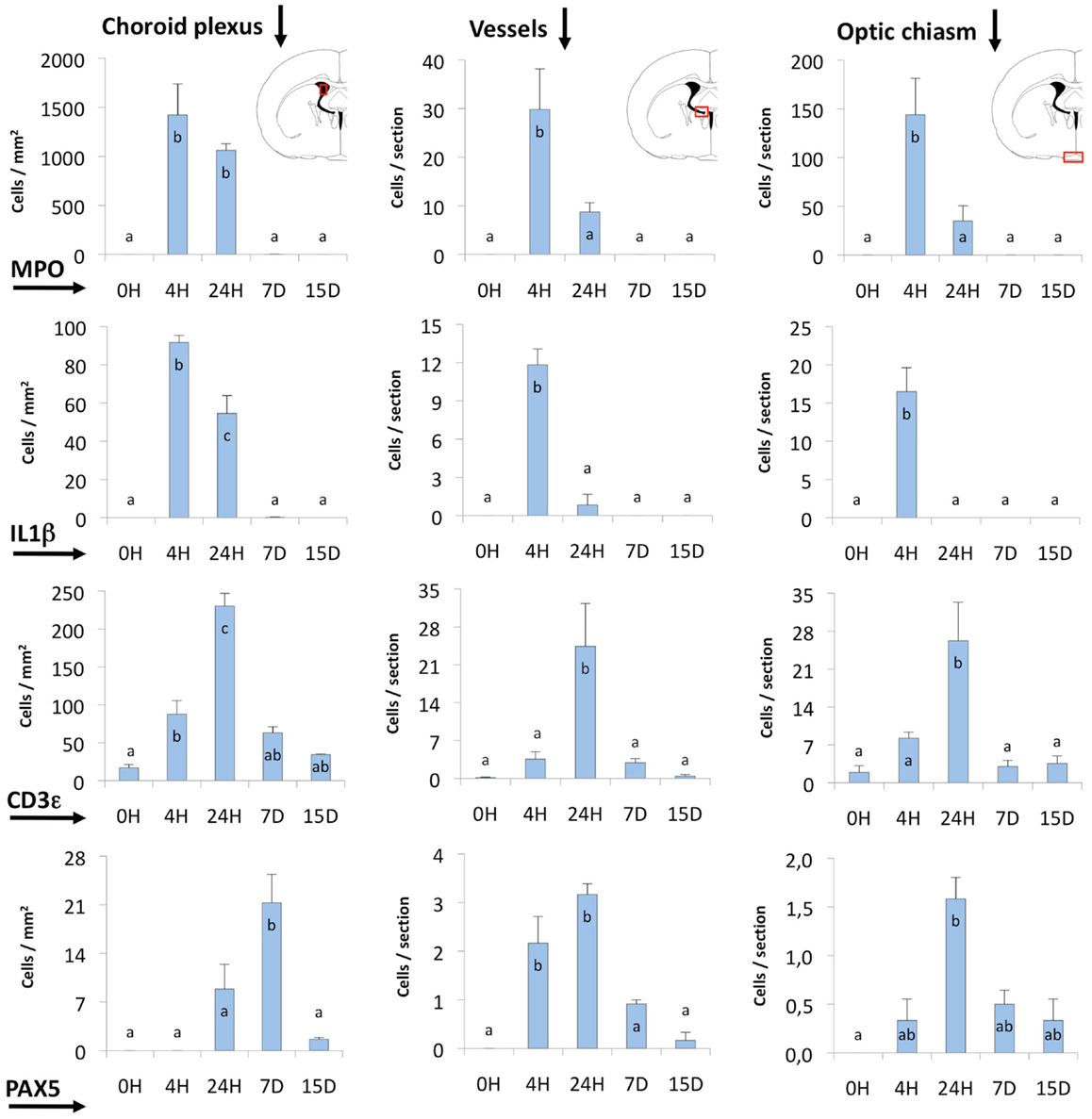
Figure 6. Quantification of MPO, IL1β, CD3ε, and PAX5 immunostained cells. The quantification was done in three different locations (indicated by red squares in the coronal section diagrams): (i) the choroid plexus of the injected lateral ventricle, (ii) the area comprising large vessels close to the foramen of Monro and near the subfornical organ (Vessels), and (iii) the zone of the meninges and the optic chiasm. Most of the cells stained and counted were infiltrating cells. MPO- and IL1β-positive cells (neutrophils and macrophagic cells, respectively) reached a maximum before 24 h. The peak of CD3ε and PAX5-positive cells (probably T- and B-lymphocytes) occurred later, and these cells remained longer (even up to 15 days) in some locations. The bars represent the mean ± SEM of five animals. Letters (a–c) on bars indicate the absence (same letter) or presence (different letter) of a significant difference between groups (α = 0.05). Time 0 post-injection of NA (0 h) is considered the baseline population of positive cells.
Neutrophils were by far the most numerous infiltrating cells, followed by T-lymphocytes and monocyte-macrophages, while B-lymphocytes were the least abundant. The choroid plexus was the richest source of blood cells, followed by the large vessels in the meninges and those around the foramen of Monro. The meningeal vessels, although situated far from the site of injection (i.e., the optic chiasm) were quite reactive.
Regarding the time course of infiltration, the earliest events were the invasion of neutrophils (MPO-positive; Figures 4C,D) and macrophages (IL1β-positive; Figures 4B,D), which reached a maximum only 4 h after the inoculation of NA; at 24 h these cells were already clearing (Figure 6). Lymphocytes (CD3ε-positive; Figures 5A–D) appeared later, and peaked at about 24 h (Figure 6). In the choroid plexus, these cells were more persistent and remained for up to 2 weeks.
Discussion
The present work describes a novel model of neuroinflammation based on the ICV administration of the enzyme NA, which specifically removes terminal sialic acid from carbohydrate chains. This model intends to demonstrate that the NA activity from brain-targeting pathogens might result in pleiotropic inflammatory changes. The events that follow NA administration closely resemble those of an inflammatory process, and could be divided into phases. The phase 1 comprises the first 4 h after injection; the events that take place during this period are (a) the cleavage of sialic acid from the ependymal surface and the periventricular parenchyma, (b) the detachment and death of a fraction of the ependymal cells; and (c) the response of resident cells such as ventricular macrophages, microglia, astrocytes and endothelial cells of the vessels. The phase 2 starts about 4 h after NA injection. Extravasation of leukocytes occurs from certain brain vessels, resulting in the invasion of the affected zones in an ordered sequence of neutrophils, macrophages, and lymphocytes. These cells invade mostly the ventricles, the meninges, and can even penetrate the brain parenchyma in the affected regions.
Toxicity of Neuraminidase
The enzyme NA from C. perfringens used in the present investigation contains <0.1% of unspecific contaminating protease activity. Thus, we think that the effects described may be mostly attributable to the specific cleavage of sialic acid. Moreover, we did also inject NA from other sources, named the bacteria Arthrobacter ureafaciens, the Newcastel virus, and the influenza virus and obtained similar results. The lectin study demonstrated that the NA from C. perfringens cleaves sialic acid and exposes galactose residues from the ependyma and the subependymal parenchyma to a depth of about 400 μm. The desialyzation affects the injected ventricle and (at least) the rostral part of the third ventricle but not the contralateral ventricle. In previous studies using a higher dose of NA (10), desialyzation extended up to the level of the foramina in the fourth ventricle. In this location, NA seems to escape from the ventricle toward the outer CSF compartments. By this route, the NA could exert its effects in the meninges and in places located farther from the injection site such as the optic tract.
Many microbes that provoke ependymal detachment, inflammation, and hydrocephalus posses NA in their envelopes (14, 23, 24). Kelly and Greiff (11, 12) demonstrated in mice the high toxic effects of bacterial and viral NA when injected directly into the brain. Also, in humans, NA activity seems to be associated with adverse prognosis in pneumococcal meningitis (13). So much so that mump viruses with low neurovirulence due to point mutations in hemagglutinin–neuraminidase capside proteins are being used to develop new harmless vaccines (15). Thus, it seems that the sole presence of NA in the CSF may trigger inflammation that gives rise to important brain damages.
Activation of Resident Cells
The activation of microglia, astrocytes, and endothelial vascular cells after the administration of NA was studied. IBA1 and GFAP, used here to identify microglia and astrocytes, respectively, are known to increase its expression after brain insults (25–27). The increase in IBA1 expression could be related to a higher mobility of microglia (28, 29). In this sense, we observed many IBA1-positive cells crossing the ependymal barrier after NA treatment and before the invasion of the brain by blood-borne cells. Interestingly, many of these cells also expressed IL1β, as observed by double immunostaining. Furthermore, we did not see any IL1β-positive cell in control specimens. This suggests that some IBA1-positive resident subependymal cells might have acquired IL1β positivity after the injection of NA, and that they move toward the lumen of the ventricle through the ependymal lining. However, since the ventricular macrophages (i.e., epiplexus cells in the choroid plexus) also expressed IBA1 and, after NA insults, begun to express IL1β, we do not have the definitive evidence to know whether the cells crossing the ependyma are indeed subependymal microglia or ventricular macrophages. IL1β is a known inflammatory mediator of the monocyte–macrophagic cell lineage (30). The capability of microglia to release IL1β was reported before in a variety of brain insults (31–33). It is likely that the increased expression of IBA1 and IL1β is related to the posterior pleocytosis, and reflects a tight relationship between the subependymal microglia and the resident ventricular macrophages. In this sense, both cell types share a common ontogenetic origin (34, 35) and may serve as sentinels of the ventricular system, probably acting as an important first-line defensive barrier to a variety of brain insults. It has been recently shown that may exists at least two subsets of microglial cells capable of increase the expression of IBA1 after experimental insults (36), one in the acute phase and the other in the chronic phase. We agree that different populations of microglial cells, as for macrophages, may play different roles in neuroinflammation after traumatic injuries. This subject deserves future investigation in our model too.
Regarding the increased expression of GFAP, the activation of astrocytes in those places more affected by the NA could be associated to the formation of a glial scar that takes place after ependymal denudation (20, 37). However, in structures located relatively far from the site of the injection, such as the stria medullaris or the optic chiasm, the activation of astrocytes and microglia is most probably related to the neuroinflammation process. It is quite evident that microglia and astrocytes play active roles in neuroinflammation but the exact mechanism of action is still controversial (38, 39).
ICAM1 is a key player in the recruitment of leukocytes during inflammation (40, 41). Its expression is increased in endothelial cells of vessels located in brain regions affected by an inflammatory insult (42–47). We also found a significant increase in the expression of ICAM1 in the cerebral blood vessels located within the regions affected by the injection of NA. In agreement to Alvarez and Teale (48), the vessels displaying a more prominent extravasation were the venules of the leptomeninges, the choroid plexus and the subependyma around the injected lateral ventricle and the foramen interventricularis. These same vessels showed the higher expression of ICAM1, which was detectable even in control non-injected specimens. Interestingly they also presented the highest expression of receptors for IL1β (49) and for other inflammatory mediators (44). This suggests that inflammatory mediators, such as IL1β, might preferentially elicited extravasation in these vessels. The functional relevance of the different permeability of parenchymal vessels versus large meningeal, choroidal, and subependymal vessels is still an open question (48, 50). On the other hand, ICAM1 was also expressed in the surface of the ependymal cells (especially in the choroid plexus) and of some ventricular cells. This could speak in favor of a role of ICAM1 in the penetration of some invading cells into the brain parenchyma (51–53).
Infiltration of Blood Leukocytes
In the model here described a profuse invasion of white blood cells takes place from about 4 h after the NA-injection. The time course of such neuroinflammation was similar to that of other models in rodents (23, 54–56). The first extravasated cells were neutrophils, followed by macrophages and then lymphocytes. In agreement to Ma et al. (57), most lymphocytes were T-lymphocytes (CD3ε-positive) and of these most were CD8α-positive, as occurred in other cases of brain inflammation (58). The leukocytes crossed both the blood–brain barrier and the blood–CSF barrier, and invaded the perivascular spaces and the ventricular or meningeal CSF (59). They also reached the brain parenchyma, either directly from vessels located in the parenchyma or from the ventricle after crossing through the ventricular ependyma. The production by the invading or the resident cells of metalloproteinases that degrade specific junctional proteins seems to be essential for the invasion process (60). The passage of cells (resident or infiltrative) through the ependymal lining is still an unresolved question (61, 62) and it should also require the production of metalloproteinases (63–65). The nature of the signals involved in this trafficking of cells, the fate of the amoeboid cells, and the consequences in the periventricular tissues are still not well understood and deserve future investigations.
In summary, we describe here a novel model, compatible with life, to study the time course of aseptic meningitis and ventriculitis in rodents. The pathology, which is induced by the ICV injection of the enzyme NA, is highly reversible, and rats cure almost completely after some weeks. Apparently, only minor long lasting consequences remain in certain zones, mostly nearby the site of injection. However, these minor consequences could make the NA-treated animals more prone to develop neurological disorders, a possibility that deserves further research.
Author Contributions
PD made the main laboratory works and hence contributed greatly to the acquisition of the information of the present paper. He drafted the work and approved the final form. They were in agreement with the contributions made by other authors. MR, JG, MM, MC, and MA supported technically all methods used in the present investigation; they also read critically the final form of the manuscript and approved it in agreement. JG also initiated the research. MA made a substantial contribution in the design and corrections of the final approved form of the manuscript, ensuring that any part of the work was appropriately investigated. Finally, FF and PL designed the research and conducted the group, making substantial contributions during all processes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was carried out with fundings from: Ministerio de Economía y Competitividad (SAF2010-19087) and Junta de Andalucía Consejería de Innovación Ciencia y Empleo (P-11-CVI-07637), Consejería de Salud SAS 08-0029. The Leica confocal microscope (SP5 II) was acquired with FEDER funds of the European Union. The authors are grateful to D. Navas Fernández for his help with confocal microscopy, the technical help of J. E. Casares-Mira and the skillfull immunohistochemistry support of E. Gil Lara from Centro Nacional de Investigaciones Oncológicas.
References
1. Graeber MB, Li W, Rodriguez ML. Role of microglia in CNS inflammation. FEBS Lett (2011) 585:3798–805. doi: 10.1016/j.febslet.2011.08.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Jang H, Boltz D, Sturm-Ramirez K, Shepherd KR, Jiao Y, Webster R, et al. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A (2009) 106:14063–8. doi:10.1073/pnas.0900096106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Gerber J, Nau R. Mechanisms of injury in bacterial meningitis. Curr Opin Neurol (2010) 23:312–8. doi:10.1097/WCO.0b013e32833950dd
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Majde JA. Neuroinflammation resulting from covert brain invasion by common viruses – a potential role in local and global neurodegeneration. Med Hypotheses (2010) 75:204–13. doi:10.1016/j.mehy.2010.02.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol (2010) 28:321–42. doi:10.1146/annurev-immunol-030409-101311
6. Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol (2006) 147:232–40. doi:10.1038/sj.bjp.0706400
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell (2010) 140:918–34. doi:10.1016/j.cell.2010.02.016
8. Ambrosini A, Louin G, Croci N, Plotkine M, Jafarian-Tehrani M. Characterization of a rat model to study acute neuroinflammation on histopathological, biochemical and functional outcomes. J Neurosci Methods (2005) 144:183–91. doi:10.1016/j.jneumeth.2004.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Dutta G, Zhang P, Liu B. The lipopolysaccharide Parkinson’s disease animal model: mechanistic studies and drug discovery. Fundam Clin Pharmacol (2008) 22:453–64. doi:10.1111/j.1472-8206.2008.00616.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Grondona JM, Pérez-Martín M, Cifuentes M, Pérez J, Jiménez-Lara AJ, Pérez-Fígares JM, et al. Ependymal denudation, aqueductal obliteration and hydrocephalus after a single injection of neuraminidase into the lateral ventricle of adult rats. J Neuropathol Exp Neurol (1996) 55:999–1008. doi:10.1097/00005072-199609000-00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Kelly RT, Greiff D. Neuraminidase and neuraminidase-labile substrates in experimental influenza virus encephalitis. Biochim Biophys Acta (1965) 110:548–53. doi:10.1016/S0926-6593(65)80068-6
13. O’Toole RD, Goode L, Howe C. Neuraminidase activity in bacterial meningitis. J Clin Invest (1971) 50:979–85. doi:10.1172/JCI106591
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Löve A, Rydbeck R, Kristensson K, Orvell C, Norrby E. Hemagglutinin-neuraminidase glycoprotein as a determinant of pathogenicity in mumps virus hamster encephalitis: analysis of mutants selected with monoclonal antibodies. J Virol (1985) 53:67–74.
15. Malik T, Wolbert C, Mauldin J, Sauder C, Carbone KM, Rubin SA. Functional consequences of attenuating mutations in the haemagglutinin neuraminidase, fusion and polymerase proteins of a wild-type mumps virus strain. J Gen Virol (2007) 88:2533–41. doi:10.1099/vir.0.82935-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Lew W, Chen X, Kim CU. Discovery and development of GS 4104 (oseltamivir): an orally active influenza neuraminidase inhibitor. Curr Med Chem (2000) 7:663–7. doi:10.2174/0929867003374886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Lewis AL, Lewis WG. Host sialoglycans and bacterial sialidases: a mucosal perspective. Cell Microbiol (2012) 14:1174–82. doi:10.1111/j.1462-5822.2012.01807
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Finsterer J, Hess B. Neuromuscular and central nervous system manifestations of Clostridium perfringens infections. Infection (2007) 35:396–405. doi:10.1007/s15010-007-6345-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Grondona JM, Pérez-Martín M, Cifuentes M, Pérez J, Estivill-Torrús G, Pérez-Fígares JM, et al. Neuraminidase injected into the cerebrospinal fluid impairs the assembly of the glycoproteins secreted by the subcommissural organ preventing the formation of Reissner’s fiber. Histochem Cell Biol (1998) 109:391–8. doi:10.1007/s004180050240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Del Carmen Gómez-Roldán M, Pérez-Martín M, Capilla-González V, Cifuentes M, Pérez J, García-Verdugo JM, et al. Neuroblast proliferation on the surface of the adult rat striatal wall after focal ependymal loss by intracerebroventricular injection of neuraminidase. J Comp Neurol (2008) 507:1571–87. doi:10.1002/cne.21618
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 2nd ed. Sydney, NSW: Academic Press (1986). 119 p.
22. Rodríguez EM, Garrido O, Oksche A. Lectin histochemistry of the human fetal subcommissural organ. Cell Tissue Res (1990) 262:105–13. doi:10.1007/BF00327751
23. Johnson RT, Johnson KP. Hydrocephalus as a sequela of experimental myxovirus infections. Exp Mol Pathol (1969) 10:68–80. doi:10.1016/0014-4800(69)90049-5
24. Margolis G, Kilham L. Hydrocephalus in hamsters, ferrets, rats, and mice following inoculations with reovirus type I. II. Pathologic studies. Lab Invest (1969) 21:189–98.
25. Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res (1998) 57:1–9. doi:10.1016/S0169-328X(98)00040-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Imai Y, Kohsaka S. Intracellular signaling in M-CSF-induced microglia activation: role of Iba1. Glia (2002) 40:164–74. doi:10.1002/glia.10149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Middeldorp J, Hol EM. GFAP in health and disease. Prog Neurobiol (2011) 93:421–43. doi:10.1016/j.pneurobio.2011.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Ohsawa K, Imai Y, Sasaki Y, Kohsaka S. Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J Neurochem (2004) 88:844–56. doi:10.1046/j.1471-4159.2003.02213.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Miller AM, Stella N. Microglial cell migration stimulated by ATP and C5a involve distinct molecular mechanisms: quantification of migration by a novel near-infrared method. Glia (2009) 57:875–83. doi:10.1002/glia.20813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Giulian D, Baker TJ, Shih LC, Lachman LB. Interleukin 1 of the central nervous system is produced by ameboid microglia. J Exp Med (1986) 164:594–604. doi:10.1084/jem.164.2.594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Hetier E, Ayala J, Denèfle P, Bousseau A, Rouget P, Mallat M, et al. Brain macrophages synthesize interleukin-1 and interleukin-1 mRNAs in vitro. J Neurosci Res (1988) 21:391–7. doi:10.1002/jnr.490210230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Bauer J, Berkenbosch F, Van Dam AM, Dijkstra CD. Demonstration of interleukin-1 beta in Lewis rat brain during experimental allergic encephalomyelitis by immunocytochemistry at the light and ultrastructural level. J Neuroimmunol (1993) 48:13–21. doi:10.1016/0165-5728(93)90053-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Sturrock RR. A developmental study of epiplexus cells and supraependymal cells and their possible relationship to microglia. Neuropathol Appl Neurobiol (1978) 4:307–22. doi:10.1111/j.1365-2990.1978.tb01345.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Jordan FL, Thomas WE. Brain macrophages: questions of origin and interrelationship. Brain Res (1988) 472:165–78. doi:10.1016/0165-0173(88)90019-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T. Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS One (2012) 7:e41892. doi:10.1371/journal.pone.0041892
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Roales-Buján R, Páez P, Guerra M, Rodríguez S, Vío K, Ho-Plagaro A, et al. Astrocytes acquire morphological and functional characteristics of ependymal cells following disruption of ependyma in hydrocephalus. Acta Neuropathol (2012) 124:531–46. doi:10.1007/s00401-012-0992-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Miljković D, Timotijević G, Mostarica Stojković M. Astrocytes in the tempest of multiple sclerosis. FEBS Lett (2011) 585:3781–8. doi:10.1016/j.febslet.2011.03.047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest (2012) 122:1164–71. doi:10.1172/JCI58644
40. Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol (1999) 163:5029–38.
41. Lyck R, Reiss Y, Gerwin N, Greenwood J, Adamson P, Engelhardt B. T-cell interaction with ICAM-1/ICAM-2 double-deficient brain endothelium in vitro: the cytoplasmic tail of endothelial ICAM-1 is necessary for transendothelial migration of T cells. Blood (2003) 102:3675–83. doi:10.1182/blood-2003-02-0358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Dustin ML, Rothlein R, Bhan AK, Dinarello CA, Springer TA. Induction by IL1 and interferon-gamma: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1). J Immunol (1986) 137:245–54.
43. Engelhardt B, Conley F, Butcher EC. Cell adhesion molecules on vessels during inflammation in the mouse central nervous system. J Neuroimmunol (1994) 51:199–208. doi:10.1016/0165-5728(94)90082-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Nadeau S, Rivest S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience (1999) 93:1449–64. doi:10.1016/S0306-4522(99)00225-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Dietrich JB. The adhesion molecule ICAM-1 and its regulation in relation with the blood-brain barrier. J Neuroimmunol (2002) 128:58–68. doi:10.1016/S0165-5728(02)00114-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Greenwood J, Etienne-Manneville S, Adamson P, Couraud PO. Lymphocyte migration into the central nervous system: implication of ICAM-1 signalling at the blood–brain barrier. Vascul Pharmacol (2002) 38:315–22. doi:10.1016/S1537-1891(02)00199-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Skeie JM, Fingert JH, Russell SR, Stone EM, Mullins RF. Complement component C5a activates ICAM-1 expression on human choroidal endothelial cells. Invest Ophthalmol Vis Sci (2010) 51:5336–42. doi:10.1167/iovs.10-5322
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Alvarez JI, Teale JM. Breakdown of the blood brain barrier and blood-cerebrospinal fluid barrier is associated with differential leukocyte migration in distinct compartments of the CNS during the course of murine NCC. J Neuroimmunol (2006) 173:45–55. doi:10.1016/j.jneuroim.2005.11.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Konsman JP, Vigues S, Mackerlova L, Bristow A, Blomqvist A. Rat brain vascular distribution of interleukin-1 type-1 receptor immunoreactivity: relationship to patterns of inducible cyclooxygenase expression by peripheral inflammatory stimuli. J Comp Neurol (2004) 472:113–29. doi:10.1002/cne.20052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol (2003) 3:569–81. doi:10.1038/nri1130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Steffen BJ, Breier G, Butcher EC, Schulz M, Engelhardt B. ICAM-1, VCAM-1, and MAdCAM-1 are expressed on choroid plexus epithelium but not endothelium and mediate binding of lymphocytes in vitro. Am J Pathol (1996) 148:1819–38.
52. López S, Prats N, Marco AJ. Expression of E-selectin, P-selectin, and intercellular adhesion molecule-1 during experimental murine listeriosis. Am J Pathol (1999) 155:1391–7. doi:10.1016/S0002-9440(10)65241-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Wolburg K, Gerhardt H, Schulz M, Wolburg H, Engelhardt B. Ultrastructural localization of adhesion molecules in the healthy and inflamed choroid plexus of the mouse. Cell Tissue Res (1999) 296:259–69. doi:10.1007/s004410051287
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Faustmann PM, Krause D, Dux R, Dermietzel R. Morphological study in the early stages of complement C5a fragment-induced experimental meningitis: activation of macrophages and astrocytes. Acta Neuropathol (1995) 89:239–47. doi:10.1007/BF00309339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Perry VH, Bell MD, Brown HC, Matyszak MK. Inflammation in the nervous system. Curr Opin Neurobiol (1995) 5:636–41. doi:10.1016/0959-4388(95)80069-7
56. Casarsa C, De Luigi A, Pausa M, De Simoni MG, Tedesco F. Intracerebroventricular injection of the terminal complement complex causes inflammatory reaction in the rat brain. Eur J Immunol (2003) 33:1260–70. doi:10.1002/eji.200323574
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Ma X, Foster J, Sakic B. Distribution and prevalence of leukocyte phenotypes in brains of lupus-prone mice. J Neuroimmunol (2006) 179:26–36. doi:10.1016/j.jneuroim.2006.06.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci (2002) 25:313–9. doi:10.1016/S0166-2236(02)02154-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Engelhardt B, Coisne C. Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS (2011) 8:4. doi:10.1186/2045-8118-8-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, et al. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med (2006) 203:1007–19. doi:10.1084/jem.20051342
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Amat-Peral G, Blazquez JL, Amat P, Pastor FE, Pelaez B, Sanchez A. Transependymal diapedesis: reality or speculation? J Submicrosc Cytol Pathol (1994) 26:449–51.
62. Man S, Ubogu EE, Ransohoff RM. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain Pathol (2007) 17:243–50. doi:10.1111/j.1750-3639.2007.00067.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Kuchler S, Graff MN, Gobaille S, Vincendon G, Roche AC, Delaunoy JP, et al. Mannose dependent tightening of the rat ependymal cell barrier. In vivo and in vitro study using neoglycoproteins. Neurochem Int (1994) 24:43–55. doi:10.1016/0197-0186(94)90128-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Alvarez JI, Teale JM. Differential changes in junctional complex proteins suggest the ependymal lining as the main source of leukocyte infiltration into ventricles in murine neurocysticercosis. J Neuroimmunol (2007) 187:102–13. doi:10.1016/j.jneuroim.2007.05.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Alvarez JI, Teale JM. Multiple expression of matrix metalloproteinases in murine neurocysticercosis: implications for leukocyte migration through multiple central nervous system barriers. Brain Res (2008) 1214:145–58. doi:10.1016/j.brainres.2008.03.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: inflammation, brain, ventricle, neuraminidase, ependyma
Citation: Granados-Durán P, López-Ávalos MD, Grondona JM, Gómez-Roldán MdC, Cifuentes M, Pérez-Martín M, Alvarez M, Rodríguez de Fonseca F and Fernández-Llebrez P (2015) Neuroinflammation induced by intracerebroventricular injection of microbial neuraminidase. Front. Med. 2:14. doi: 10.3389/fmed.2015.00014
Received: 28 July 2014; Accepted: 02 March 2015;
Published online: 17 March 2015.
Edited by:
Giulio Cesare Spagnoli, University of Basel, SwitzerlandReviewed by:
Pier Paolo Piccaluga, Bologna University, ItalyLiang-Yu Chen, Taipei Veterans General Hospital, Taiwan
Copyright: © 2015 Granados-Durán, López-Ávalos, Grondona, Gómez-Roldán, Cifuentes, Pérez-Martín, Alvarez, Rodríguez de Fonseca and Fernández-Llebrez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro Fernández-Llebrez, Departamento de Biología Celular Genética y Fisiología, Facultad de Ciencias, Universidad de Málaga, Málaga 29071, Spain e-mail:bGxlYnJlekB1bWEuZXM=