 Sandeep K. Mallipattu
Sandeep K. Mallipattu John Cijiang He
John Cijiang He- 1Division of Nephrology, Department of Medicine, Stony Brook University, New York, NY, USA
- 2Division of Nephrology, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 3Renal Section, James J. Peters VA Medical Center, New York, NY, USA
The primary etiology of CKD is a direct consequence of initial dysfunction and injury of the glomerulus, the main filtration system. Podocytes are terminally differentiated epithelial cells in the glomerulus, whose major function is the maintenance of this renal filtration barrier. Podocyte injury is implicated in many glomerular diseases including focal segmental glomerular sclerosis and HIV-associated nephropathy. In many of these diseased conditions, the podocyte can either undergo dedifferentiation and proliferation, apoptosis, or cell detachment. Regardless of the initial type of injury, the podocyte ultimately loses its functional capacity to maintain the glomerular filtration barrier. Significant injury resulting in a loss of the podocytes and failure to maintain the renal filtration barrier contributes to progressive kidney disease. Consequently, therapies that prevent podocyte injury and promote their regeneration will have a major clinical impact on glomerular disease. Retinoic acid (RA), which is a derivative of vitamin A, has many cellular functions including induction of cell differentiation, regulation of apoptosis, and inhibition of inflammation and proliferation. RA is required for kidney development and is essential for cellular differentiation in the setting of podocyte injury. The mechanism by which RA directs its beneficial effects is multifactorial, ranging from its anti-inflammatory and anti-fibrotic effects to a direct effect of upregulating podocyte differentiation markers in the podocyte. The focus of this review is to provide an overview of RA in kidney development and glomerular disease. We also highlight the key mechanism(s) by which RA restores podocyte differentiation markers and ameliorates glomerular disease.
Introduction
More than three decades have passed since the initial description of retinoids, synthetic derivatives of vitamin A (retinol), in human physiology (1). Vitamin A is a critical component of our diet since it cannot be synthesized by our body. It is essential for various cell processes, including kidney development, epithelial cell differentiation, and inhibiting inflammation and cell proliferation (1–5). The widespread use of vitamin A has been hindered by its toxicity, with therapeutic doses frequently resulting in complications of hypervitaminosis syndrome (1). Consequently, the synthesis of first generation derivatives of retinoids, all-trans-retinoic acid (atRA), proved to improve efficacy while minimizing toxicity (1). In the canonical retinoic acid (RA) synthesis pathway, retinol is taken up by retinol-binding protein (RBP) in circulation and transferred intracellularly, where it is initially metabolized to retinal by retinol dehydrogenases (RDHs) and transformed into retinaldehyde by retinaldehyde dehydrogenases (RALDHs), with eventual oxidation to RA (6). Subsequently, RA exerts its effect by binding to cytosolic retinoic acid receptors (RAR), which heterodimerizes with retinoid X receptors (RXR), leading to activation of RA response elements (RARE) on target genes (6). In the past two decades, the diverse and critical actions of RA have been clearly illustrated in several pathological processes, from cancer biology to skin treatment to kidney disease (3–5). Our objective in this mini-review is to demonstrate the essential role of retinoids in kidney disease and development as well as its therapeutic benefits in experimental models of glomerular disease. Furthermore, we hope to highlight the deep dichotomy between the numerous basic science studies and the relative dearth of clinical studies on the therapeutic benefits of retinoids in kidney disease.
Retinoic Acid in Kidney Development
During fetal life, the final nephron number is determined by the branching of the ureteric bud to develop the complete renal collecting system. Studies have clearly demonstrated that RA signaling is critical to this branching nephrogenesis and the final nephron number during kidney development (7–9). This arborization of the ureteric bud is specifically regulated by RA signaling from the stromal mesenchyme (9). Batourina et al. demonstrated that RA from the stromal mesenchyme regulates the expression of a tyrosine kinase receptor, Ret, in the ureteric bud during kidney development (9). Ret is a proto-oncogene that encodes for a tyrosine kinase receptor and is expressed from the initial stages of the Wolffian duct through the development of ureteric bud. Upon stimulation from RA, the epithelial cells with Ret expression initiate the outgrowth and subsequent branching of the ureteric bud (10). Impaired RA signaling directly limits the ureteric bud branching, thereby hindering the development of the collecting system (9). In addition, the authors observed that the deletion of Rarα and Rarβ2 (isoforms of receptors for RA) in the stromal mesenchyme downregulated the expression of Ret in the ureteric bud and impaired the ureteric bud outgrowth in mice (8, 9). Furthermore, others have confirmed that the expression of these receptors (Rarα and Rarβ2) is localized to the cells in the stromal mesenchyme, rather than in the cells of the developing nephron (8). Consequently, compound mutations in Rarα and Rarβ2 cause the stromal precursor cells to remain along the periphery, with few cells between the collecting ducts in the developing medullary zone (8). These mutations also contribute to the downregulation of Ret in the ureteric bud, thereby resulting in limited ureteric bud branching and small kidney size at birth (8). Thus, this paracrine-signaling pathway between RA and Ret expression is required for branching morphogenesis and the development of the renal collecting system.
Since RA is essential for branching morphogenesis and contributes to the final nephron number, the altered expression of genes involved in RA metabolism has been associated with impaired kidney development (11). For instance, mice lacking in Raldh2, enzyme required for the irreversible oxidation of retinaldehyde to RA, are embryonically lethal due to low RA levels and failed organogenesis (12, 13). Conversely, the authors observed that homozygosity for a common variant within the promoter region of the gene encoding for RALDH2, conferred an upregulation in RALDH2 with a subsequent increase in plasma RA levels and kidney volume in humans. The authors suggest that the presence of this variant may especially be protective in individuals that are nutritionally deficient for vitamin A during kidney development by maintaining RA signaling (11). Combined, these studies reveal the essential and protective role of RA signaling in kidney development.
Retinoic Acid in Glomerular Disease
Other than the critical role that RA plays in kidney development, RA has been demonstrated to restore differentiation markers in cellular injury as well as induce the differentiation of kidney progenitor cells. Furthermore, RA has also been shown to attenuate inflammation and apoptosis in models of podocyte injury. Finally, the progression in podocyte injury has been closely linked to RA metabolism.
Retinoic Acid Signaling in Podocyte Differentiation
Podocytes are terminally differentiated visceral epithelial cells in the glomerulus, whose function is critical to the maintenance of the glomerular filtration barrier. These highly specialized cells express distinct podocyte differentiation markers and lack the ability to proliferate. Previous studies have illustrated that in the setting of HIV-associated nephropathy (HIVAN) and collapsing focal segmental glomerular sclerosis (FSGS), the podocyte loses its terminal differentiation markers and reenters the cell cycle, leading to cell proliferation (14, 15) (Figure 1). RA has clearly been shown to exhibit anti-proliferative with pro-differentiation effects in multiple tissues, including the kidney and specifically in the podocyte (16–18).
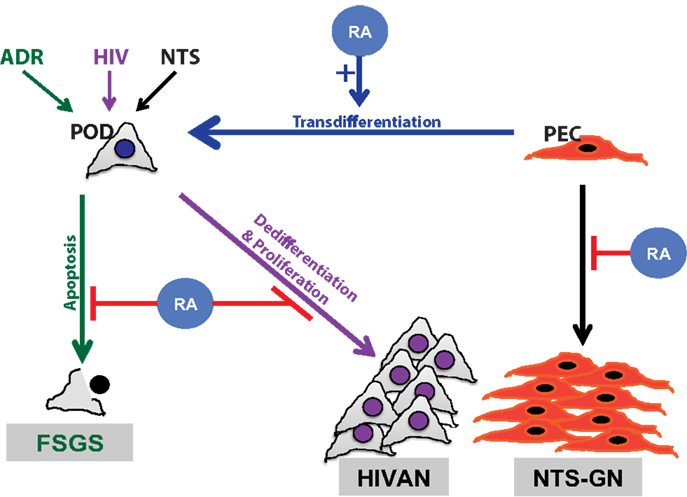
Figure 1. Schematic diagram of retinoids in podocyte injury. Retinoic acid (RA) has been demonstrated to play a critical role in attenuation of podocyte injury. The mechanism by which RA abrogates podocyte injury is dependent on the type of inciting injury. Podocyte apoptosis is improved with RA treatment in a murine model of adriamycin-induced nephropathy, focal segmental glomerulosclerosis (FSGS) model (green). Podocyte dedifferentiation is attenuated with RA treatment in models of HIV-associated nephropathy (HIVAN) (purple). In a murine model of crescentic glomerulonephritis, using nephrotoxic serum (NTS), RA treatment minimized parietal epithelial cell (PEC) proliferation (black) and restored podocytes by PEC trans differentiation (blue).
Earlier studies have demonstrated that treatment with RA reduced markers of proliferation and preserves podocyte-specific differentiation markers in in vitro and in vivo models of HIVAN (17). In models of HIVAN, the expression of HIV-1 transgene induces the podocyte to dedifferentiate and reenter the cell cycle (19). We have demonstrated that treatment with RA abrogates this process in a cyclic adenosine monophosphate (cAMP)-dependent manner (19). For instance, RARα antagonists impede the production of cAMP and the downstream anti-proliferative effects in the podocyte. Similarly, the use of cAMP inhibitors diminished RA-induced podocyte differentiation (19, 20). In contrast, the use of cAMP agonists restored the podocyte differentiation effects of RA. Furthermore, the use of phosphodiesterase-4 inhibitor, increased cAMP levels and enhanced the differentiation effects of RA in cultured human podocytes infected with HIV-1 (19). In addition, the loss of RARα in HIV-1 transgenic (Tg26) mice resulted in a significant increase in albuminuria and accelerated glomerulosclerosis (21). Conversely, RARα agonists, such as Am580, have shown to attenuate podocyte dedifferentiation in in vitro and in vivo models of podocyte injury (21). Interestingly, the concomitant use of phosphodiesterase inhibitor, Roflumilast, with Am580 resulted in a synergistic therapeutic effect on the podocyte (22). Combined, these findings suggest that treatment with RA inhibits podocyte dedifferentiation in a cAMP-dependent manner (18, 20).
Although RA-mediated podocyte differentiation is cAMP-dependent, many podocyte differentiation markers lack RARE or cAMP response element-binding protein (CREB) transcriptional binding sites. To address the mechanism by which RA-cAMP signaling mediates podocyte differentiation, recent microarray studies and comparative promoter analyses were performed (23, 24). These studies revealed that Krüppel-like factor 15 (KLF15) transcriptional binding sites exist on many podocyte-specific genes and may serve as a potential downstream target of CREB (23, 24). In general, KLFs are a subclass of zinc-finger family of DNA-binding transcriptional regulators that are involved in a broad range of cellular processes (i.e., cell differentiation, angiogenesis, erythropoiesis) (25). Specifically, KLF15 is a kidney-enriched zinc-finger binding transcription factor that has been shown to mediate differentiation in various cell types (26). Subsequently, it was demonstrated that KLF15 is downstream of RA and CREB and is a critical mediator of RA-induced podocyte differentiation (24). Furthermore, we observed that the glomerular expression of KLF15 is reduced in kidney biopsies from patients with HIVAN and FSGS as compared to healthy control subjects (24). Combined, these recent studies suggest that KLF15 is required for RA-cAMP-mediated podocyte differentiation.
Retinoic Acid Contributes to the Generation of Glomerular Transition Cells
In FSGS, podocyte injury is observed with proliferation of epithelial cells in Bowman’s space, a finding which has been defined as a pseudo-crescent (27). For years, it remained unclear if these were truly proliferating parietal epithelial cells or merely podocytes that have lost their differentiation markers and have reentered the cell cycle (28, 29). Regardless of the origin, RA has been demonstrated to be critical to this proliferative process (30). For instance, the mechanism by which RA may regulate the transition of parietal epithelial cells to podocytes has been closely examined in several models of podocyte injury (28–31). For instance, in a rat model of membranous nephropathy (where there is a loss of podocyte differentiation markers), RA was shown to restore podocyte number by increasing the number of epithelial cells expressing podocyte differentiation markers in the glomerulus (30). Specifically, the increase in these epithelial transition cells in the glomerulus colocalized to Paired box gene 2 (PAX2), parietal epithelial cell marker, and Wilms-Tumor 1 (WT1), podocyte-specific marker, thereby suggesting that RA treatment induced the transition of parietal epithelial cells to podocytes (30). Similarly, the authors confirmed this using an experimental model of FSGS to illustrate that the beneficial effect of RA in podocyte regeneration may go beyond a specific glomerular disease (30). Since podocytes are terminally differentiated cells with minimal capacity to self-replicate, restoration of these podocytes in the setting of cellular injury is critical to the recovery of glomerular function. This recent evidence demonstrates that RA may be essential to this regenerative process.
Impaired Retinoid Signaling Contributes to Podocyte Injury
In nephrotic range albuminuria, the sequestration of RA by albumin prevents podocyte regeneration in murine models of podocyte injury (31). For instance, it was shown that increasing albuminuria prevents the differentiation of progenitor cells toward a podocyte lineage (31). Specifically, Peired et al. demonstrated that sequestration of RA by increasing albuminuria prevented RARE-mediated regulation of podocyte differentiation markers, which was rescued with administration of RA (31) (Figure 1). In addition, in vitro studies revealed that albumin overload impairs podocyte differentiation markers without affecting podocyte survival. The authors also showed that RA-induced podocyte differentiation was attenuated in the setting of increasing albumin concentration. Their findings suggest that this process is mediated via the sequestration of RA by albumin, leading to downregulation of RARE and worsening podocyte injury (31).
Retinaldehyde dehydrogenase 2 is not only critical to the production of RA and kidney development, but RALDH2 levels are significantly upregulated as a consequence of injury to the podocyte (32). Using a rat model of podocytopathy (puromycin treatment), the authors demonstrated that Raldh2 levels increased with podocyte injury (32). Subsequent RA treatment restored podocyte differentiation markers in the setting of elevated Raldh2 levels. Conversely, vitamin A deficiency delayed podocyte recovery in this experimental model (32). This study revealed that both the substrates and the enzymes involved in RA signaling are required for podocyte repair after injury (32). Although RALDH2 is critical to RA production, the rate-limiting step of RA synthesis and its eventual downstream effects remains the conversion of retinol to retinal by retinol dehydrogenases (RDHs). Although many RDHs are critical for RA metabolism, RDH9 was recently demonstrated to rescue podocytes from injury using two murine models of FSGS, HIV-1 transgenic model and adriamycin-induced nephropathy model (33). We demonstrated that the overexpression of RDH9 in cultured podocytes induced the expression of podocyte-specific differentiation markers. In addition, podocyte-specific overexpression of RDH9, in both models of podocyte injury, attenuated glomerular injury and podocyte dedifferentiation in mice (33). Combined, these findings suggest that other than simply using RA for treatment, critical molecules in the RA synthesis pathway may serve as a potential therapeutic in the treatment of glomerular disease.
Anti-Inflammatory Effects of Retinoic Acid
The anti-inflammatory of RA has been demonstrated in multiple animal models of glomerular disease (19, 34). This is not surprising, considering the observed anti-inflammatory effects of RA are well-established in other tissues (1). For instance, in vitro and in vivo models of diabetic kidney disease revealed that treatment with RA ameliorates podocyte injury. Specifically, the treatment of cultured podocytes with RA reduced the activation of inflammatory pathways with inhibition of monocyte chemotactic protein-1 (MCP-1) synthesis, which is typically increased in diabetic conditions (34). In addition, the authors showed a reduction in proteinuria and inhibition of inflammation in RA-treated diabetic rats (34). Prior studies have also revealed that RA suppresses the transcripts of several pro-inflammatory cytokines and chemokines as well as the recruitment of macrophages (19). In addition, the anti-inflammatory role of RA was more evident with its inhibitory effect in an in vitro model of lipopolysaccharide (LPS)-induced macrophage production of IL-12 (35). Using this LPS model, the authors determined that the reduction in IL-12 production by RA was dependent on inhibition of NFκB activity, a potential mechanism by which RA reduced inflammation (35).
Retinoic acid has also been demonstrated to exhibit anti-inflammatory effects in the treatment of primary podocytopathies, such as MCD. Treatment with RA prevented foot process effacement and reversed albuminuria in puromycin aminoglycoside nephropathy (PAN) model, a rat model of minimal change disease (32, 36). In this study, the authors reveal that RA-treated PAN rats exhibited less interstitial mononuclear cell infiltration and reduced expression of fibronectin and MCP-1, which are critical factors for monocyte infiltration (36). RA has been demonstrated to abrogate kidney disease in these murine models of lupus nephritis. For instance, treatment with RA ameliorated kidney injury and prolonged survival in the NZB/W F1 mice, a model of lupus nephritis (37). Also, RA-treated mice exhibited a reduction in glomerular IgG deposits as well a reduction in cytokine expression in the lupus nephritis model (37). These similar effects were observed in another murine model of lupus nephritis, MRL/lpr mice. Specifically, RA treatment reduced albuminuria, improved glomerular lesions, and attenuated chemokine and cytokine expression in the kidney (38). In this model, the authors identified that the renal transforming-growth factor beta (TGFβ) was concurrently elevated with RA treatment, suggesting that TGFβ may potentially mediate the anti-inflammatory effects of RA (38). However, future studies are required to precisely define the mechanism by which TGFβ mediates RA-induced anti-inflammatory effects in the glomerulus. In addition, RA has therapeutic benefits in other models of glomerular disease independent of primary podocytopathies. For instance, treatment with RA reduced crescent formation and improved kidney function in animal models of anti-GBM disease, with a reduction in markers of inflammation and proliferation, such as PCNA, TNF-alpha, IL-1B, PDGF, and C/EBP sigma (16). Regardless of the specific experimental model of glomerular injury, these studies clearly demonstrate that the therapeutic benefits of RA in glomerular disease are at least, in part, mediated by its anti-inflammatory effects.
Other Potential Therapeutic Benefits of Retinoic Acid in Glomerular Disease
In addition to the anti-inflammatory effects of RA, treatment with RA has been demonstrated to reduce apoptosis in murine and cell culture models of podocyte injury (Figure 1). For instance, administration of RA to PAN-treated cultured podocytes resulted in inhibition of apoptosis (36). Furthermore, macrophage-specific loss of RXRα in mice demonstrated reduced clearance of apoptotic cells, resulting in glomerular injury resembling lupus nephritis (39).
Retinoic acid can also play a synergistic therapeutic role with other agents in the treatment of kidney disease. Nephrin, podocyte-specific differentiation marker, expression is induced by RA and activated vitamin D, 1,25-dihydroxyvitamin D3 (40). Interestingly, the synergistic increase in nephrin expression is mediated by selective cooperation between RAR and vitamin D receptor (VDR) in cell culture models (40). Therefore, future studies exploring the concomitant use of both these agents in treating glomerular disease may prove to be efficacious.
Other than the potential use of RA in the treatment of primary podocytopathies, RA may provide some therapeutic benefit in the treatment of other glomerular diseases. In a rat model of mesangial injury, anti-Thy1.1 nephritis, treatment with RA reduced albuminuria and improved glomerular lesions (19, 41). Specifically, administration of RA reduced endothelin-1 and endothelin Type A and B receptor expression levels, which are critical to the progression of renal injury (19). This suggests that a potential alternate mechanism by which RA treatment may abrogate glomerular injury.
Obstacles in the Clinical Use of Retinoic Acid for Treating Glomerular Disease
Retinoids have clearly been demonstrated to have a therapeutic benefit in experimental models of glomerular disease; however, its translation from the bench to the bedside has been hindered by its toxicity. For instance, the strong experimental evidence for the use of RA in kidney disease had prompted the initiation of NIH sponsored open-label randomized clinical trial with the use of Isotretinoin in the treatment of patients with FSGS and collapsing glomerulopathy (NCT00098020) in 2004. However, the recruitment of subjects has proved to be difficult due to the risk of toxicity associated with retinoids.
Toxicity of retinoids includes CNS abnormalities, craniofacial malformation, liver toxicity, hyperlipidemia, reduced spermiogenesis, myalgia, arthralgias, and mucocutaneous side effects (1). To minimize the toxicity and yet retain the therapeutic benefit of RA, we synthesized and demonstrated that a novel derivative of RARα agonist, boronic acid retinoid (BD4), attenuated kidney injury in the HIV-1 transgenic mice. BD4 retains the therapeutic benefit of RA, but with less toxicity due to metabolic oxidation (42). Specifically, we observed that BD4 binds to RARα and exerts its downstream effects with higher affinity and less toxicity as compared to atRA or Am580 (42). Future research is required to identify agents such as these with similar efficacy, but with lower toxicity than atRA. After establishing a safe therapeutic profile, we need to take the next step in implementing these agents in clinical trials.
Conclusion
In conclusion, we provide a brief overview on the essential role of retinoids in kidney disease. Studies extending for more than three decades have illustrated the therapeutic benefit of RA in models of kidney disease. Despite this large body of evidence, clinical studies demonstrating its therapeutic benefit are lacking. Consequently, there is a dire need for the synthesis of newer generation retinoids as well as novel agents involved in RA metabolism that maintain the therapeutic efficacy of retinoids while minimizing its toxicity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
SM is supported by NIH 1K08DK102519. JH is supported by NIH 1R01DK078897 and Chinese 973 fund 2012CB51760.
References
1. Orfanos CE, Ehlert R, Gollnick H. The retinoids. A review of their clinical pharmacology and therapeutic use. Drugs (1987) 34(4):459–503. doi: 10.2165/00003495-198734040-00003
2. Niederreither K, Dolle P. Retinoic acid in development: towards an integrated view. Nat Rev Genet (2008) 9(7):541–53. doi:10.1038/nrg2340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Mongan NP, Gudas LJ. Diverse actions of retinoid receptors in cancer prevention and treatment. Differentiation (2007) 75(9):853–70. doi:10.1111/j.1432-0436.2007.00206.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Xu Q, Lucio-Cazana J, Kitamura M, Ruan X, Fine LG, Norman JT. Retinoids in nephrology: promises and pitfalls. Kidney Int (2004) 66(6):2119–31. doi:10.1111/j.1523-1755.2004.66002.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Gronemeyer H, Miturski R. Molecular mechanisms of retinoid action. Cell Mol Biol Lett (2001) 6(1):3–52.
6. Evans TR, Kaye SB. Retinoids: present role and future potential. Br J Cancer (1999) 80(1–2):1–8. doi:10.1038/sj.bjc.6690312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Merlet-Benichou C, Vilar J, Lelievre-Pegorier M, Gilbert T. Role of retinoids in renal development: pathophysiological implication. Curr Opin Nephrol Hypertens (1999) 8(1):39–43. doi:10.1097/00041552-199901000-00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Mendelsohn C, Batourina E, Fung S, Gilbert T, Dodd J. Stromal cells mediate retinoid-dependent functions essential for renal development. Development (1999) 126(6):1139–48.
9. Batourina E, Gim S, Bello N, Shy M, Clagett-Dame M, Srinivas S, et al. Vitamin A controls epithelial/mesenchymal interactions through Ret expression. Nat Genet (2001) 27(1):74–8. doi:10.1038/83792
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Srinivas S, Wu Z, Chen CM, D’Agati V, Costantini F. Dominant effects of RET receptor misexpression and ligand-independent RET signaling on ureteric bud development. Development (1999) 126(7):1375–86.
11. El Kares R, Manolescu DC, Lakhal-Chaieb L, Montpetit A, Zhang Z, Bhat PV, et al. A human ALDH1A2 gene variant is associated with increased newborn kidney size and serum retinoic acid. Kidney Int (2010) 78(1):96–102. doi:10.1038/ki.2010.101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Niederreither K, Subbarayan V, Dolle P, Chambon P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet (1999) 21(4):444–8. doi:10.1038/7788
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Niederreither K, Vermot J, Schuhbaur B, Chambon P, Dolle P. Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development (2000) 127(1):75–85.
14. Barisoni L, Kriz W, Mundel P, D’Agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol (1999) 10(1):51–61.
15. Barisoni L, Bruggeman LA, Mundel P, D’Agati VD, Klotman PE. HIV-1 induces renal epithelial dedifferentiation in a transgenic model of HIV-associated nephropathy. Kidney Int (2000) 58(1):173–81. doi:10.1046/j.1523-1755.2000.00152.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Oseto S, Moriyama T, Kawada N, Nagatoya K, Takeji M, Ando A, et al. Therapeutic effect of all-trans retinoic acid on rats with anti-GBM antibody glomerulonephritis. Kidney Int (2003) 64(4):1241–52. doi:10.1046/j.1523-1755.2003.00219.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Vaughan MR, Pippin JW, Griffin SV, Krofft R, Fleet M, Haseley L, et al. ATRA induces podocyte differentiation and alters nephrin and podocin expression in vitro and in vivo. Kidney Int (2005) 68(1):133–44. doi:10.1111/j.1523-1755.2005.00387.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. He JC, Lu TC, Fleet M, Sunamoto M, Husain M, Fang W, et al. Retinoic acid inhibits HIV-1-induced podocyte proliferation through the cAMP pathway. J Am Soc Nephrol (2007) 18(1):93–102. doi:10.1681/ASN.2006070727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Lehrke I, Schaier M, Schade K, Morath C, Waldherr R, Ritz E, et al. Retinoid receptor-specific agonists alleviate experimental glomerulonephritis. Am J Physiol Renal Physiol (2002) 282(4):F741–51. doi:10.1152/ajprenal.00026.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Lu TC, Wang Z, Feng X, Chuang P, Fang W, Chen Y, et al. Retinoic acid utilizes CREB and USF1 in a transcriptional feed-forward loop in order to stimulate MKP1 expression in human immunodeficiency virus-infected podocytes. Mol Cell Biol (2008) 28(18):5785–94. doi:10.1128/MCB.00245-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Ratnam KK, Feng X, Chuang PY, Verma V, Lu TC, Wang J, et al. Role of the retinoic acid receptor-alpha in HIV-associated nephropathy. Kidney Int (2011) 79(6):624–34. doi:10.1038/ki.2010.470
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Zhong Y, Wu Y, Liu R, Deng Y, Mallipattu SK, Klotman PE, et al. Roflumilast enhances the renal protective effects of retinoids in an HIV-1 transgenic mouse model of rapidly progressive renal failure. Kidney Int (2012) 81(9):856–64. doi:10.1038/ki.2011.467
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Cohen CD, Klingenhoff A, Boucherot A, Nitsche A, Henger A, Brunner B, et al. Comparative promoter analysis allows de novo identification of specialized cell junction-associated proteins. Proc Natl Acad Sci U S A (2006) 103(15):5682–7. doi:10.1073/pnas.0511257103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Mallipattu SK, Liu R, Zheng F, Narla G, Ma’ayan A, Dikman S, et al. Kruppel-like factor 15 (KLF15) is a key regulator of podocyte differentiation. J Biol Chem (2012) 287(23):19122–35. doi:10.1074/jbc.M112.345983
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. McConnell BB, Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiol Rev (2010) 90(4):1337–81. doi:10.1152/physrev.00058.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Mori T, Sakaue H, Iguchi H, Gomi H, Okada Y, Takashima Y, et al. Role of Kruppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J Biol Chem (2005) 280(13):12867–75. doi:10.1074/jbc.M410515200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Albaqumi M, Barisoni L. Current views on collapsing glomerulopathy. J Am Soc Nephrol (2008) 19(7):1276–81. doi:10.1681/ASN.2007080926
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Dijkman HB, Weening JJ, Smeets B, Verrijp KC, van Kuppevelt TH, Assmann KK, et al. Proliferating cells in HIV and pamidronate-associated collapsing focal segmental glomerulosclerosis are parietal epithelial cells. Kidney Int (2006) 70(2):338–44. doi:10.1038/sj.ki.5001574
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Smeets B, Uhlig S, Fuss A, Mooren F, Wetzels JF, Floege J, et al. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol (2009) 20(12):2604–15. doi:10.1681/ASN.2009010122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Zhang J, Pippin JW, Vaughan MR, Krofft RD, Taniguchi Y, Romagnani P, et al. Retinoids augment the expression of podocyte proteins by glomerular parietal epithelial cells in experimental glomerular disease. Nephron Exp Nephrol (2012) 121(1–2):e23–37. doi:10.1159/000342808000342808
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Peired A, Angelotti ML, Ronconi E, la Marca G, Mazzinghi B, Sisti A, et al. Proteinuria impairs podocyte regeneration by sequestering retinoic acid. J Am Soc Nephrol (2013) 24(11):1756–68. doi:10.1681/ASN.2012090950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Suzuki A, Ito T, Imai E, Yamato M, Iwatani H, Kawachi H, et al. Retinoids regulate the repairing process of the podocytes in puromycin aminonucleoside-induced nephrotic rats. J Am Soc Nephrol (2003) 14(4):981–91. doi:10.1097/01.ASN.0000057857.66268.8F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Li X, Dai Y, Chuang PY, He JC. Induction of retinol dehydrogenase 9 expression in podocytes attenuates kidney injury. J Am Soc Nephrol (2014) 25(9):1933–41. doi:10.1681/ASN.2013111150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Han SY, So GA, Jee YH, Han KH, Kang YS, Kim HK, et al. Effect of retinoic acid in experimental diabetic nephropathy. Immunol Cell Biol (2004) 82(6):568–76. doi:10.1111/j.1440-1711.2004.01287.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Na SY, Kang BY, Chung SW, Han SJ, Ma X, Trinchieri G, et al. Retinoids inhibit interleukin-12 production in macrophages through physical associations of retinoid X receptor and NFkappaB. J Biol Chem (1999) 274(12):7674–80. doi:10.1074/jbc.274.12.7674
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Moreno-Manzano V, Mampaso F, Sepulveda-Munoz JC, Alique M, Chen S, Ziyadeh FN, et al. Retinoids as a potential treatment for experimental puromycin-induced nephrosis. Br J Pharmacol (2003) 139(4):823–31. doi:10.1038/sj.bjp.0705311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Kinoshita K, Yoo BS, Nozaki Y, Sugiyama M, Ikoma S, Ohno M, et al. Retinoic acid reduces autoimmune renal injury and increases survival in NZB/W F1 mice. J Immunol (2003) 170(11):5793–8. doi:10.4049/jimmunol.170.11.5793
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Perez de Lema G, Lucio-Cazana FJ, Molina A, Luckow B, Schmid H, de Wit C, et al. Retinoic acid treatment protects MRL/lpr lupus mice from the development of glomerular disease. Kidney Int (2004) 66(3):1018–28. doi:10.1111/j.1523-1755.2004.00850.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Roszer T, Menendez-Gutierrez MP, Lefterova MI, Alameda D, Nunez V, Lazar MA, et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol (2011) 186(1):621–31. doi:10.4049/jimmunol.1002230
40. Okamura M, Takano Y, Saito Y, Yao J, Kitamura M. Induction of nephrin gene expression by selective cooperation of the retinoic acid receptor and the vitamin D receptor. Nephrol Dial Transplant (2009) 24(10):3006–12. doi:10.1093/ndt/gfp243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Wagner J, Dechow C, Morath C, Lehrke I, Amann K, Waldherr R, et al. Retinoic acid reduces glomerular injury in a rat model of glomerular damage. J Am Soc Nephrol (2000) 11(8):1479–87.
42. Zhong Y, Wu Y, Liu R, Li Z, Chen Y, Evans T, et al. Novel retinoic acid receptor alpha agonists for treatment of kidney disease. PLoS One (2011) 6(11):e27945. doi:10.1371/journal.pone.0027945
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: retinoic acid, retinoic acid receptor alpha, podocytes, FSGS, HIVAN, retinol-binding proteins
Citation: Mallipattu SK and He JC (2015) The beneficial role of retinoids in glomerular disease. Front. Med. 2:16. doi: 10.3389/fmed.2015.00016
Received: 24 September 2014; Accepted: 08 March 2015;
Published online: 23 March 2015.
Edited by:
Raymond Clement Harris, Vanderbilt University, USAReviewed by:
Leslie Stuart Gewin, Vanderbilt University, USARichard Tyler Miller, University of Texas Southwestern Medical Center and Dallas VA Medical Center, USA
Copyright: © 2015 Mallipattu and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John Cijiang He, Department of Medicine/Nephrology, Mount Sinai School of Medicine, One Gustave L Levy Place, Box 1243, New York, NY 10029, USA e-mail:Y2lqaWFuZy5oZUBtc3NtLmVkdQ==