 Niels A. J. Cremers1,2
Niels A. J. Cremers1,2 Maarten Suttorp1†
Maarten Suttorp1† Marlous M. Gerritsen1†
Marlous M. Gerritsen1† Ronald J. Wong3Coby van Run-van Breda1Gooitzen M. van Dam4
Ronald J. Wong3Coby van Run-van Breda1Gooitzen M. van Dam4 Katrien M. Brouwer5,6
Katrien M. Brouwer5,6 Anne Marie Kuijpers-Jagtman7Carine E. L. Carels1Ditte M. S. Lundvig1
Anne Marie Kuijpers-Jagtman7Carine E. L. Carels1Ditte M. S. Lundvig1 Frank A. D. T. G. Wagener1*
Frank A. D. T. G. Wagener1*
- 1Department of Orthodontics and Craniofacial Biology, Radboud University Medical Center, Radboud Institute for Molecular Life Sciences, Nijmegen, Netherlands
- 2Experimental Rheumatology, Radboud University Medical Center, Radboud Institute for Molecular Life Sciences, Nijmegen, Netherlands
- 3Department of Pediatrics, Stanford University School of Medicine, Stanford, CA, USA
- 4Department of Surgery, University Medical Center Groningen, Groningen, Netherlands
- 5Department of Plastic, Reconstructive and Hand Surgery, VU University Medical Center, MOVE Research Institute Amsterdam, Amsterdam, Netherlands
- 6Association of Dutch Burn Centers, Beverwijk, Netherlands
- 7Department of Orthodontics and Craniofacial Biology, Cleft Palate Craniofacial Center, Radboud University Medical Center, Nijmegen, Netherlands
Mechanical stress following surgery or injury can promote pathological wound healing and fibrosis, and lead to functional loss and esthetic problems. Splinted excisional wounds can be used as a model for inducing mechanical stress. The cytoprotective enzyme heme oxygenase-1 (HO-1) is thought to orchestrate the defense against inflammatory and oxidative insults that drive fibrosis. Here, we investigated the activation of the HO-1 system in a splinted and non-splinted full-thickness excisional wound model using HO-1-luc transgenic mice. Effects of splinting on wound closure, HO-1 promoter activity, and markers of inflammation and fibrosis were assessed. After seven days, splinted wounds were more than three times larger than non-splinted wounds, demonstrating a delay in wound closure. HO-1 promoter activity rapidly decreased following removal of the (epi)dermis, but was induced in both splinted and non-splinted wounds during skin repair. Splinting induced more HO-1 gene expression in 7-day wounds; however, HO-1 protein expression remained lower in the epidermis, likely due to lower numbers of keratinocytes in the re-epithelialization tissue. Higher numbers of F4/80-positive macrophages, αSMA-positive myofibroblasts, and increased levels of the inflammatory genes IL-1β, TNF-α, and COX-2 were present in 7-day splinted wounds. Surprisingly, mRNA expression of newly formed collagen (type III) was lower in 7-day wounds after splinting, whereas, VEGF and MMP-9 were increased. In summary, these data demonstrate that splinting delays cutaneous wound closure and HO-1 protein induction. The pro-inflammatory environment following splinting may facilitate higher myofibroblast numbers and increase the risk of fibrosis and scar formation. Therefore, inducing HO-1 activity against mechanical stress-induced inflammation and fibrosis may be an interesting strategy to prevent negative effects of surgery on growth and function in patients with orofacial clefts or in patients with burns.
Introduction
Cleft lip with or without cleft palate (CL/P) is a developmental craniofacial disorder that is characterized by an opening in the upper lip and/or palate and alveolar bone (1). Patients with CL/P need multiple surgeries that inevitably result in scar formation (Figure 1A) (2, 3). In particular, scars on the palate may disrupt normal midfacial growth and impair dento-alveolar development (4, 5). Also, patients with severe burns can exhibit excessive scar formation (Figure 1B) (6). Scarring can be exaggerated by mechanical tension, such as during growth of the child and during wound repair (7). Overall, pathological wound healing following mechanical stress can result in hypertrophic scars, and subsequently lead to functional, psychosocial, and esthetical problems for patients (8, 9).
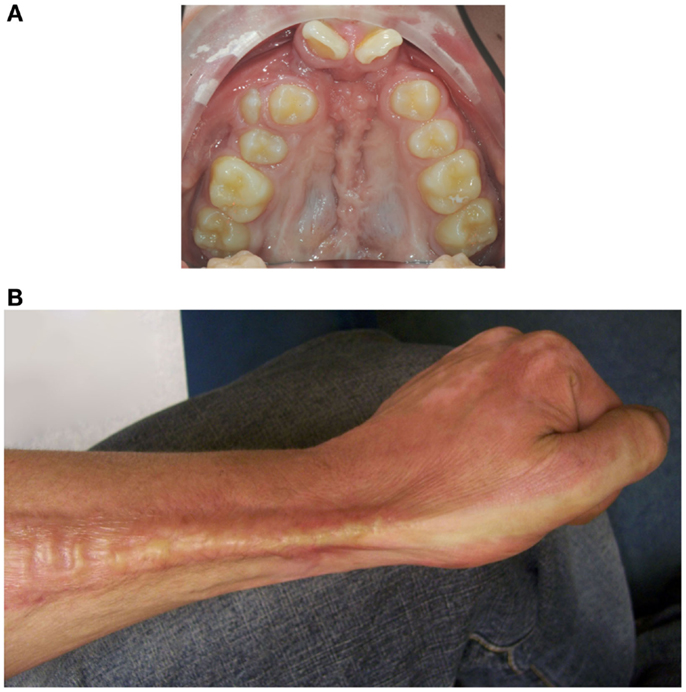
Figure 1. Mechanical stress may promote excessive scar formation following injury. (A) An intra-oral photo of the maxillary arch of a patient of our clinic with an operated complete bilateral cleft lip and palate is shown. Mechanical stress-induced scar formation impairs development of the upper jaw and the dentition. (B) Also, burns can result in excessive scar formation leading to cosmetic and functional problems as exemplified by scar formation near the wrist.
Mechanical load, together with cytokine expression and the composition of the extracellular matrix (ECM), promotes the differentiation of fibroblasts into myofibroblasts (10–12). During wound repair, myofibroblasts play a key role in the deposition of ECM and in wound contraction, thereby reducing wound size and preventing invasion by pathogens (10, 12). When a wound closes, myofibroblasts normally disappear by apoptosis. However, during pathologic wound healing, extended presence of myofibroblasts may result in excessive wound contracture and ECM deposition, leading to excessive scar formation and functional problems (5, 13). Mechanical stress during wound repair can trigger a continued expression of the myofibroblast marker alpha smooth muscle actin (α-SMA) and a prolonged survival of myofibroblasts (5, 14–17). Prolonged inflammatory and oxidative stress may also increase myofibroblast survival (18). A better understanding of the effects of mechanical stress during the wound healing process is warranted to develop novel adjuvant therapies.
Rodents, in contrast to humans, possess a subcutaneous muscle layer (m. panniculus carnosus), which can cause wound contraction (19). In these animals, the use of splinting can induce static mechanical stress to healing wounds (5, 14, 20), interfere with muscle contraction, and thus better simulate human wound healing that is mainly dependent on granulation and re-epithelialization of tissues (19). The effects of static mechanical stress on the different phases of wound healing, caused by splinting, remains to be unraveled. In addition, the involvement of cytoprotective mechanisms needs further exploration. Activation of the cytoprotective heme oxygenase (HO) system has shown protective effects in both inflammatory and fibrotic models (6, 18, 21).
Heme oxygenase is an enzyme that catabolizes heme, yielding the gasotransmitter carbon monoxide (CO), free iron, which is scavenged by co-induced ferritin, and biliverdin that is rapidly converted into the antioxidant bilirubin by biliverdin reductase (22). The HO system possesses antioxidative, anti-inflammatory, anti-fibrotic, and anti-apoptotic properties (18, 23), and can influence cell proliferation, differentiation, and migration. When these processes are disrupted, wound repair may be hampered (24). There are two isoforms of HO, the inducible HO-1 and the constitutively expressed HO-2. It has been shown that HO-1 is rapidly induced in wounded tissues (25, 26). Decreased HO-1 or HO-2 expression and enzyme activity in mice results in slower cutaneous wound closure; whereas, induction of HO-1 expression or administration of the HO effector molecule bilirubin attenuates the inflammatory response and accelerates wound healing in HO-1-deficient mice (27–29).
In this study, we investigated the expression of HO-1 during wound repair in both non-splinted and splinted wounds using transgenic HO-1-luc mice. Because mechanical stress has been shown to induce HO-1 expression in different experimental settings in a time- and force-dependent manner (30–32), we therefore postulated that mechanical stress by splinting would induce HO-1 expression during wound healing. In addition, we investigated the effects of mechanical stress on markers of inflammation, ECM remodeling, and fibrosis.
Materials and Methods
Animals
The Committee for Animal Experiments of Radboud University Nijmegen approved all procedures involving mice (RU-DEC 2010-248). Twelve mice (strain: HO-1-luc FVB/N-Tg background), 4–5 months of age and weighing 30 ± 5 g, were provided with food and water ad libitum. Mice were maintained on a 12-h light/dark cycle and specific pathogen-free housing conditions at the Central Animal Facility Nijmegen. More details on the housing conditions have been previously described (33). Mice were originally derived from Xenogen Corporation (Alameda, CA, USA) and generated as previously described (34).
The mice were euthanized with a standard CO2/O2 protocol 7 days after wounding, after which control skin, and wounded skin were isolated. Half of the tissue was fixed for 24h in 4% paraformaldehyde, and then embedded in paraffin following regular histosafe procedures and the other half was snap-frozen in liquid nitrogen and stored at −80°C until isolation of mRNA.
Excisional Non-Splinted and Splinted Wound Model
Splinted (n = 6) and non-splinted (n = 6) full-thickness excisional wounds 4 mm in diameter were created on the dorsum of the mice after shaving as previously described (35). In brief, excisional wounds were created using a sterile disposable 4-mm skin biopsy punch (Kai Medical, Seki City, Japan) on the dorsum to either side of the midline, and halfway between the shoulders and pelvis. Circular silicone splints of 6-mm inner- and 12-mm outer-diameter made from silicone sheets (3M, Saint Paul, MN, USA) were glued to the skin around the wound. Mice receiving splinted excisional wounds were wrapped with semi-permeable dressing (Petflex; Andover, Salisbury, MA, USA) around their torso, to cover the wound. One mouse in the splinted group died due to pulmonary failure as a result of respiratory obstruction by the bandage during the recovery from anesthesia.
Photographs of the wounds were taken immediately after wounding and then 1h and 1, 3, 5 and 7 days thereafter with a reference placed perpendicular next to the wounds for wound size normalization. The area of the wounds was blindly measured in triplicates using ImageJ (NIH) v1.44p software.
HO-1 Promoter Activity Measurements
Heme oxygenase-1 promoter activity was determined at baseline, immediately after wounding and 1h and 1, 3, and 7 days thereafter in the HO-1-luc-Tg mice by in vivo bioluminescence imaging using the IVIS Lumina system (Caliper Life Sciences, Hopkinton, MA, USA) as previously described (36). Images were quantified using Living Image 3.0 software (Caliper Life Sciences) by selecting regions of interest (ROI). The amount of emitted photons per second (total flux) per ROI was measured, and then calculated as fold change from baseline levels.
Immunohistochemical Staining
Immunohistochemical staining for HO-1, macrophages (F4/80), and myofibroblasts (α-SMA) were performed on paraffin sections of the wounds as previously described (35). In short, paraffin-embedded tissues were cut into 5-μm sections, which were then de-paraffinized, quenched for endogenous peroxidase activity with 3% H2O2 in methanol for 20 min, and rehydrated. Sections were post-fixed with 4% formalin, and washed with PBS containing 0.075 μg/mL glycine (PBSG). Antigens were retrieved with citrate buffer (0.01 M, pH 6.0) at 70°C for 10 min, followed by incubation in 0.075 g/mL trypsin in PBS at 37°C for 7 min. Next, the sections were pre-incubated with 10% normal donkey serum (NDS) in PBS-G. First antibodies (HO-1 from Stressgen #SPA-895 1:600 dilution, α-SMA from Sigma-Aldrich #A2547 1:600 dilution, and F4/80 from AbD Serotec #MCA497R 1:200 dilution) were diluted in 2% NDS in PBSG and incubated overnight at 4°C. After washing with PBSG, sections were incubated for 60 min with a biotin-labeled secondary antibody against host species (1:5000 dilution). Next, the sections were washed with PBSG and treated with avidin–biotin–peroxidase complex (ABC) for 45 min in the dark. After extensive washing with PBSG, diaminobenzidine–peroxidase (DAB) staining was performed for 10 min. After rinsing with water, staining was intensified with Cu2SO4 in 0.9% NaCl and rinsed with water again. Finally, the nuclei were stained with hematoxylin for 10s and sections were rinsed for 10 min in water, dehydrated and embedded in distyrene plasticizer xylene (DPX).
Immunoreactivity was evaluated by blindly scoring the wounds. For the HO-1 staining, we scored both the epidermal and dermal region of the wounds separately, since there were two different positively stained populations. A single section per wound was semi-quantitatively scored, by two assessors independently of each other, as previously described according to the following scale: 0 (minimal), 1 (mild), 2 (moderate), and 3 (marked) (35).
RNA Isolation and Quantitative-RT-PCR
Non-wounded control skin and wounds were pulverized in TRIzol (Invritrogen) using a micro-dismembrator (Sartorius BBI Systems GmbH, Melsungen, Germany) and RNA was further extracted as previously described (37). Quantitative-RealTime-PCR was performed. mRNA expression levels were calculated as minus delta delta Ct (−ΔΔCt) values, normalized to the reference gene GAPDH, and corrected for non-wounded control skin. Fold change were calculated by 2^(−ΔΔCt). The sequences of the mouse-specific primers are shown in Table 1.

Table 1. Mouse primers.
Statistics
Data were analyzed using GraphPad Prism 5.01 software (San Diego, CA, USA). No outliers were detected using the Grubbs’ test. Data was analyzed by a paired t-test for comparisons of two groups and a one- or two-way analysis of variance (ANOVA) for the comparison of multiple groups with a post hoc Bonferroni correction for multiple comparisons. The non-parametrical one-tailed Mann–Whitney U-test was used to compare the arbitrary scored immunohistological sections. Results were considered significant different when p < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001).
Results
Static Mechanical Stress Induced by Splinting Delays Excisional Wound Closure
Because mechanical stress can lead to excessive scar formation (Figures 1A,B), we used both splinted and non-splinted excisional wound healing to assess the effects of static mechanical stress on wound closure. Wound sizes of non-splinted and splinted wounds were monitored over time and representative photos are shown in Figure 2A. After quantification of the wound area the wound closure time in relation to t = 0 h was displayed (Figure 2B). All wounds closed gradually, but there were differences in wound closure between splinted and non-splinted wounds. After 3 days, significant differences between the groups were found. Non-splinted wounds were already closed by 45%; whereas, splinted wounds had only closed by 10%. After 7 days, closure of non-splinted wounds was 75% of the wound area, while that for splinted wounds was only 23%. This corresponds to a 3.3 times faster wound closure rate when no mechanical stress was applied. This demonstrates that splinting effectively delays wound closure.
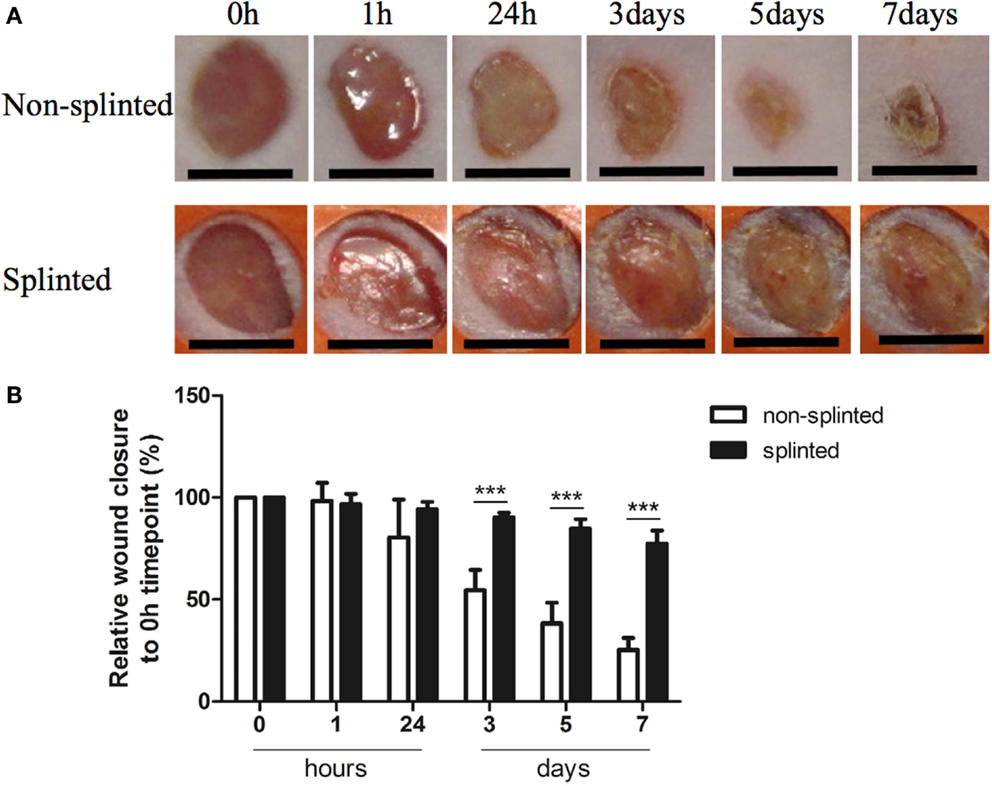
Figure 2. Splinted and non-splinted excisional wound closure over time. (A) Representative pictures of non-splinted and splinted wounds immediately following injury, and 1 h, and 1, 3, 5 and 7 days after wounding (black bars = 4 mm). (B) Quantification of closure of non-splinted (n = 6) and splinted (n = 5) wounds over time (0–7 days). Each mouse had two wounds. Time point 0 h was used as 100%. Data represent mean ± SD. *is significantly different between non-splinted and splinted wounds (***p < 0.001).
The HO System is Affected by Mechanical Stress During Excisional Wound Healing
Heme oxygenase-1 is thought to be a critical regulator and expressed in distinct cell types during the different phases of wound repair. In order to investigate the role of HO-1 following mechanical stress (splinting) during excisional wound healing, we used HO-1-luc mice to monitor HO-1 promoter activity (Figure 3A). HO-1 promoter activity was already present in the kidneys in non-injured HO-1-luc mice. In the wound area, HO-1 promoter activity was evident especially at days 3 and 7 after splinting.
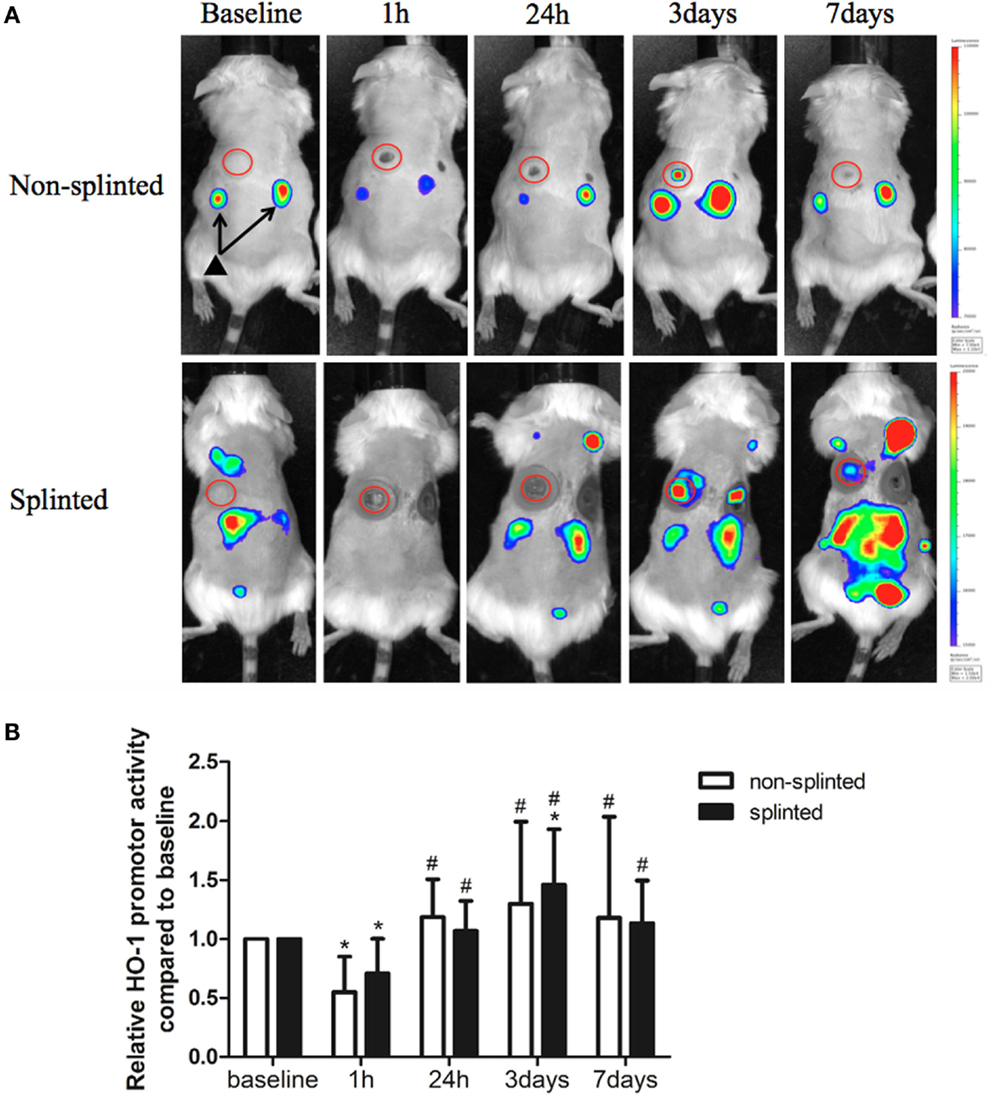
Figure 3. HO-1 promoter activity in splinted and non-splinted wounds. (A) Representative images of HO-1 promoter activity of non-splinted and splinted wounds over time using the IVIS system to measure in vivo bioluminescence in HO-1-luc mice. The rainbow-colored bar at the right side indicates the amount of luciferase signal represented in each group with a high signal in red and low signal in blue. The red circles surround the wounds in the mice and indicate the measured region of interest (ROI). Note: black triangle (▴) with black arrows shows that there is basal HO-1 promoter activity around the kidneys. (B) Quantification of HO-1 promoter activity during non-splinted (n = 6) and splinted (n = 5) wound healing. Each mouse had two wounds. Data represent mean ± SD. *is significantly different within the non-splinted or splinted group when compared to the baseline at the start of the experiment (*p < 0.05). #is significantly different within the non-splinted or splinted group when compared to 1 h after wounding (#p < 0.05).
Surprisingly, we found that following wounding, there was an initial significant decrease in HO-1 promoter activity after 1 h in both splinted as well as non-splinted wounds (Figure 3B). This decrease of HO-1 promoter activity returned to basal levels at day 1 and further increased at days 3–7, independent of splinting. Three days following wounding, the splinted wounds showed a significant increase of HO-1 promoter activity compared to basal levels.
To further elucidate the role of splinting on HO-1 expression during wound repair, we measured both HO-1 mRNA and protein expression in day-7 wounds. Using RT-PCR, HO-1 mRNA expression was assessed in the wounds and corrected for expression in non-wounded control skin (Figure 4A). Here, we found significantly higher (2.6 times) HO-1 mRNA expression in splinted wounds compared to non-splinted wounds. As expected, the constitutively expressed HO-2 levels were not significantly different after induction of mechanical stress.
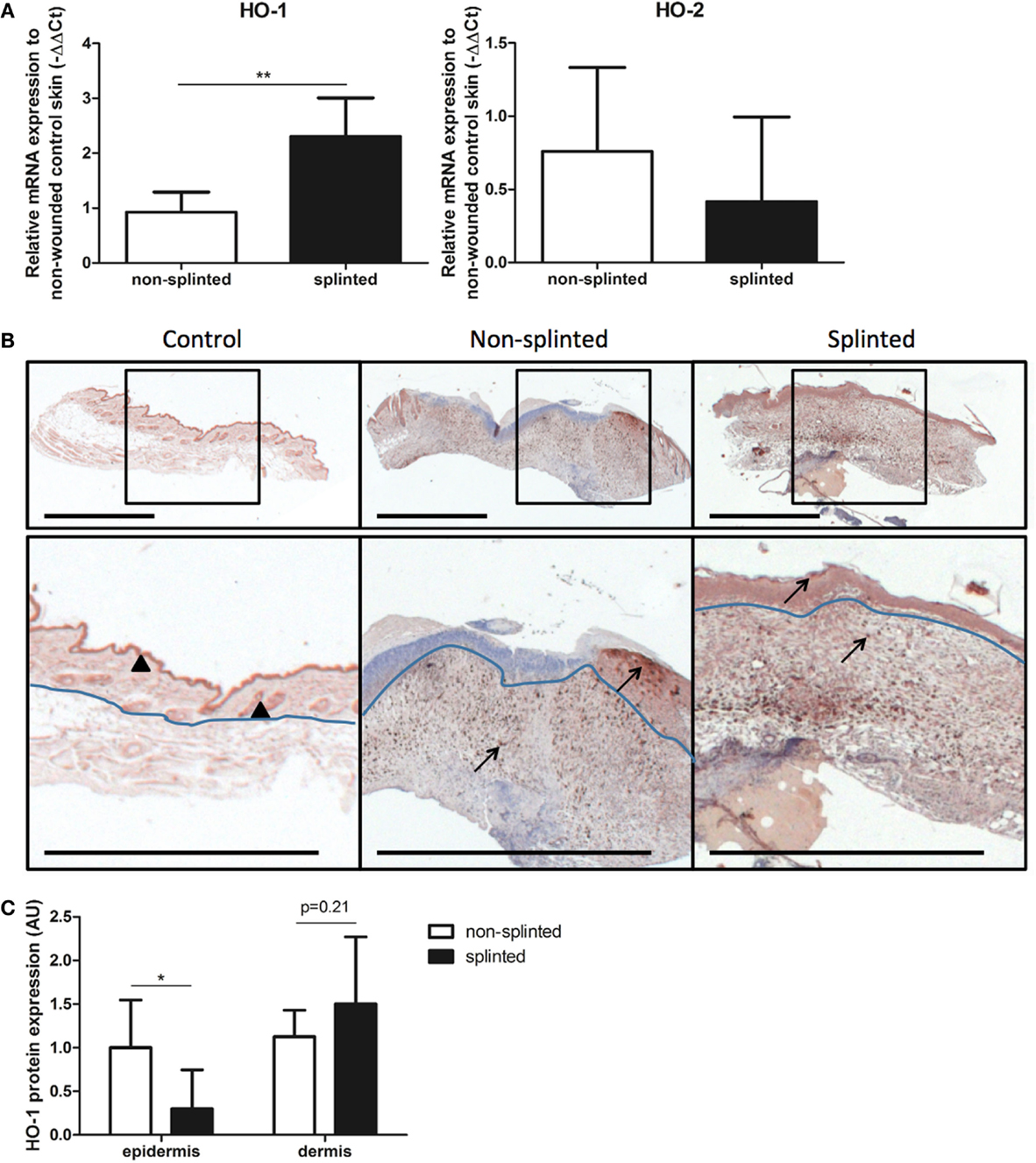
Figure 4. HO expression in wounds. (A) HO-1 and HO-2 mRNA expression levels in non-splinted and splinted wounds were determined in 7-day wounds and compared to control unwounded skin. Data represent mean ± SD. *is significantly different between non-splinted and splinted wounds (**p < 0.01). (B) HO-1 protein expression in control and non-splinted and splinted wounds after 7 days of healing. Region above the marked blue line is the epidermis and underneath the line is the dermal layer (bars = 1 mm). Note: black triangle (▴) in the non-wounded control skin shows HO-1-positive keratinocytes at the front of the epithelial layer, and HO-1 positive hair follicles. Black arrows (→) shows HO-1-positive cells in the epidermis and dermis of non-splinted and splinted wounds. (C) Quantification of scored HO-1 protein staining in epidermis and dermis of the wounds after 7 days in arbitrary units (AU). Data represent mean ± SD. *is significantly different between non-splinted and splinted wounds (*p < 0.05).
Immunohistochemical staining demonstrated that HO-1 levels were severely affected by wounding when compared to non-wounded skin. The number of HO-1-positive cells 7 days after wounding was increased in the dermal region of the wounds; also, more HO-1-positive cells were present in the epidermal region when compared to non-wounded skin.
Heme oxygenase-1 protein was particularly evident in the epithelial cells at the leading edge of the wound in the epidermis and in recruited inflammatory leukocytes in the dermis (Figure 4B). Since we observed two different populations of HO-1-positive cells depending on their region, the wounds were scored for the level of HO-1 protein expression in both the epidermal and dermal regions, which were compared between the different treatment groups (Figure 4C). Importantly, non-splinted wounds showed significantly higher HO-1 protein expression in the epidermal region compared to splinted wounds. In the dermal region, HO-1 was slightly higher after mechanical stress compared to the non-splinted wounds; however, this did not reach statistical significance (p = 0.21). Variation in HO-1 protein expression was found between animals, but was independent of the wound model.
Thus, wounding initially decreased HO-1 promoter activity in the wound, after which HO-1 levels were restored and further induced by recruitment of inflammatory cells and keratinocytes. Surprisingly, although splinted wounds delay HO-1 expression, as shown by an increased HO-1 promoter activity and HO-1 mRNA expression, HO-1 protein expression was, in contrast to in the dermis, lower in the epidermis when compared to non-splinted wounds.
Interplay Between HO-1 and Inflammation in Non-Splinted and Splinted Wound Healing
Since there was a clear delay in wound closure and HO-1 protein expression in splinted wounds, we investigated whether this delay correlated with altered levels of inflammatory gene expression. Because HO-1 has anti-inflammatory and antioxidative properties, we expected that the delayed HO-activity in the skin would result in increased levels of inflammation. Therefore, we quantitated the number of F4/80-positive macrophages using immunohistochemical staining in sections of non-splinted and splinted wounds (Figure 5A).
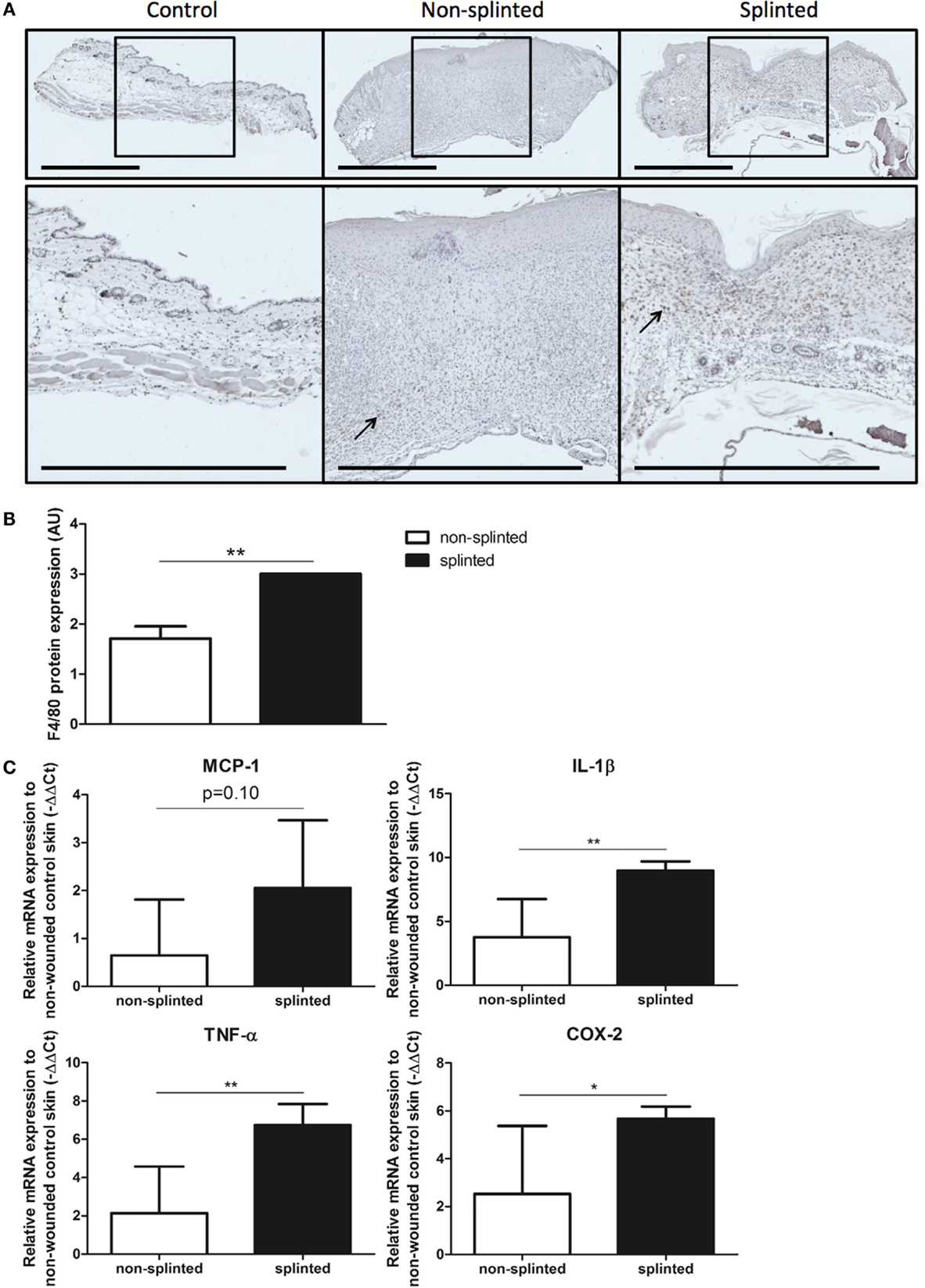
Figure 5. Effects of splinting on markers of inflammation. (A) Immunohistological staining of macrophages (F4/80) in control skin and non-splinted and splinted 7-day wounds (bars = 1 mm). Black arrow (→) shows F4/80-positive macrophages in non-splinted and splinted wounds. (B) Quantification of scored F4/80 protein staining of the wounds after 7 days in arbitrary units (AU). Data represent mean ± SD. *is significantly different between non-splinted and splinted wounds (**p < 0.01). (C) mRNA expression levels of inflammatory markers in 7-day non-splinted and splinted wounds normalized to levels in control unwounded skin (MCP-1, IL-1β, TNF-α, and COX-2). Data represent mean ± SD. *is significantly different between non-splinted and splinted wounds (*p < 0.05, **p < 0.01).
We found that there were indeed significantly more macrophages present in 7-day splinted wounds compared to non-splinted wounds (Figure 5B). Next, we investigated whether mRNA expression of monocyte chemotactic protein (MCP-1), the main chemokine to attract monocytes/macrophages, was altered by mechanical stress. We observed that MCP-1 was elevated 1.8-fold in splinted wounds compared to non-splinted wounds, however, this difference did not reach statistical significance (p = 0.10).
Finally, when we examined the gene expression of several other inflammatory markers, we found that mRNA expression levels of the pro-inflammatory cytokines IL-1β, TNF-α, and COX-2 in splinted wounds were significantly higher when compared to non-splinted wounds (36.9-, 24.4-, 8.8-fold, respectively) (Figure 5C).
In summary, we showed that delayed wound closure and reduced HO-1 protein expression following mechanical stress was clearly associated with elevated levels of macrophages and several other inflammatory genes.
Effects of Mechanical Stress on Remodeling and Fibrotic Genes
Because increased levels of inflammation by mechanical stress during wound repair may also affect markers of fibrosis, we investigated the effects of splinting on markers of ECM remodeling and fibrosis.
During wound healing, fibroblasts can transform into myofibroblasts. These myofibroblasts cause wound contraction and produce ECM. Normally, these myofibroblasts go into apoptosis after wound repair has finished. When these myofibroblasts fail to become apoptotic, ECM production continues, and pathologic wound healing with excessive scarring follows. The presence of myofibroblasts is dependent on the phase of wound healing. To investigate the role of splinting on myofibroblasts, we stained 7-day wound sections with the myofibroblast marker α-SMA. In non-wounded skin, α-SMA staining was already evident in the muscles of the vascular wall and the arrector pili muscles of the hair follicles (Figure 6A). Following injury, myofibroblasts were present both in non-splinted as well as splinted wounds. However, quantification of arbitrarily scored α-SMA-positive myofibroblasts showed significantly increased levels in splinted wounds when compared to non-splinted wounds (Figure 6B).
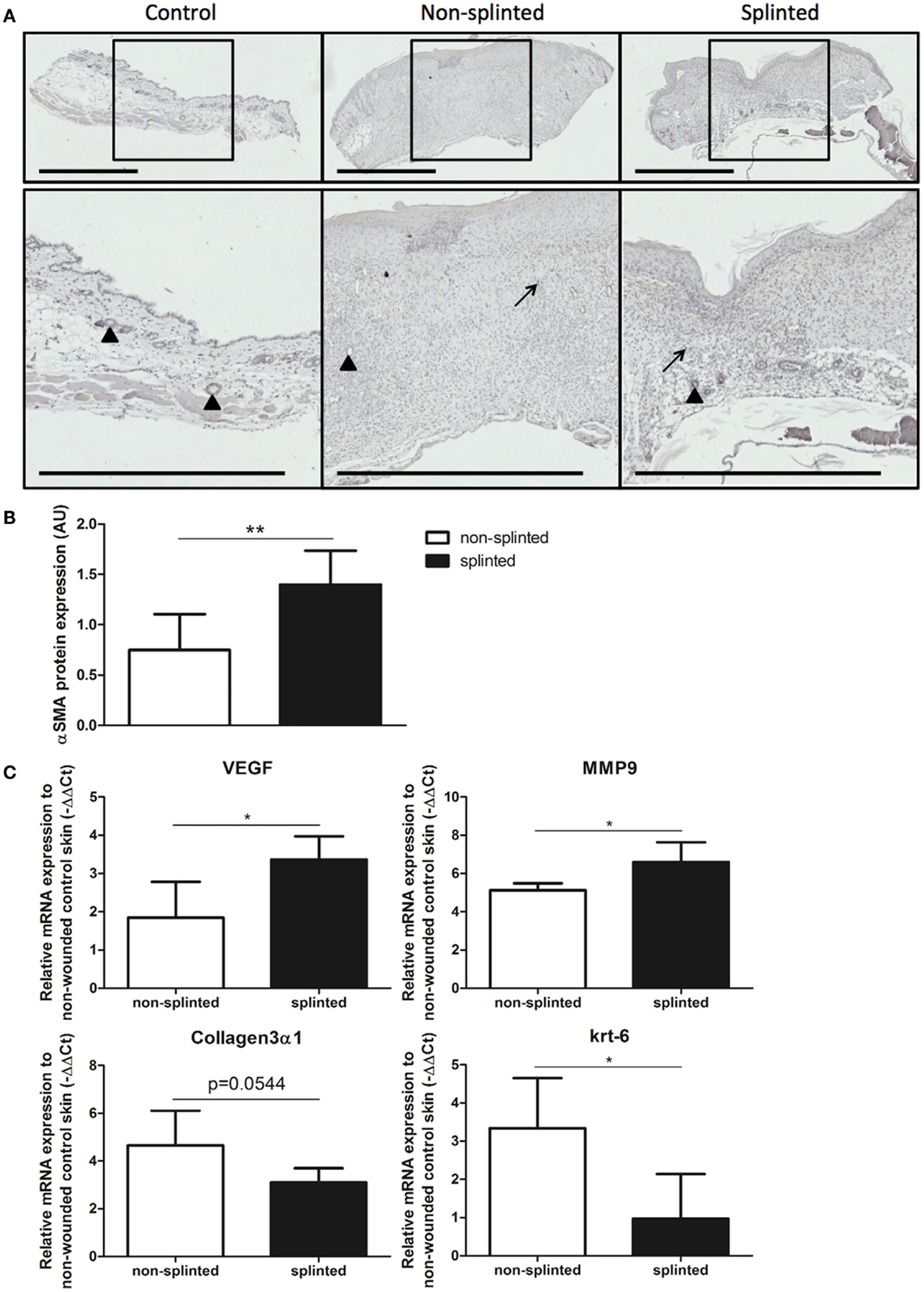
Figure 6. Effects of splinting on markers of fibrosis. (A) Immunohistological staining of α-SMA-positive myofibroblasts in control skin and non-splinted and splinted 7-day wounds (bars = 1 mm). Note: black triangle (▴) in the non-wounded control skin shows α-SMA-positive muscles in the vascular wall and the arrector pili muscles of the hair follicles. Black arrow (→) shows α-SMA-positive myofibroblasts. (B) Quantification of scored α-SMA protein staining of wounds after 7 days in arbitrary units (AU). Data represent mean ± SD. * is significantly different between non-splinted and splinted wounds (**p < 0.01). (C) mRNA expression levels of VEGF, α-SMA, MMP-9, collagen type 3, and krt-6 in 7-day non-splinted and splinted wounds normalized to levels in control unwounded skin. Data represent mean ± SD. *is significantly different between non-splinted and splinted wounds (*p < 0.05).
Next, we measured mRNA expression levels of genes involved in remodeling and fibrosis (Figure 6C). Vascular endothelial growth factor (VEGF) is an important regulator of angiogenesis, and was 2.9 times higher in splinted wounds compared to non-splinted wounds. Also matrix metalloproteinase (MMP)-9, an important enzyme that helps remodel the provisional ECM following wounding, was increased (four-fold).
Surprisingly, collagen type 3 gene expression, which is produced by (myo)fibroblasts, and forms the major collagen type expressed in granulation tissue, was almost three times lower in splinted wounds compared to non-splinted wounds (p = 0.0544).
Interestingly, we found significantly more (5.8-fold) gene expression of keratinocyte marker krt-6 in non-splinted wounds compared to splinted wounds.
Thus, there were more myofibroblasts and higher expression of VEGF and MMP-9 in day-7 wounds following mechanical stress.
Discussion
We found that splinting effectively delayed wound closure. Interestingly, HO-1 promoter activity initially decreased upon wounding, but was followed by induction of HO-1 mRNA and protein. However, HO-1 protein expression was delayed in the splinted wounds. Since HO activity attenuates inflammatory and oxidative stress and is thought to reduce fibrosis, we postulated that the splinting-mediated delay in HO-1 protein expression could result in a reduced defense against inflammatory and fibrotic processes. Indeed, more F4/80-positive macrophages, α-SMA-positive myofibroblasts, and pro-inflammatory cytokines were present in splinted wounds at day 7 when compared to non-splinted wounds. Normally, the wound healing cascade is characterized by three distinct phases: inflammation, proliferation, and remodeling (9, 38, 39). This suggests that mechanical stress causes delayed wound closure, a prolongation of the inflammatory phase, and an altered remodeling phase, and may promote fibrosis.
Mechanical Stress and HO-1
We found that mechanical stress by splinting delays wound closure 3.3 times in comparison to non-splinted wounds after 7 days. This was likely largely mediated by inhibiting the contraction of the subcutaneous muscle layer in mice; however, it may also be caused by interfering with other processes involved in wound healing, such as inflammation, proliferation or remodeling.
Previously, it was shown that in vitro administration of mechanical stress with a Flex-cell strain unit induces HO-1 mRNA and protein expression in periodontal ligament cells in a time- and force-dependent manner (30). HO-1 mRNA levels are also upregulated in vitro after mechanical stress in human aortic endothelial cells (40) and human dental pulp cells (41). Aortic smooth muscle cells also respond to mechanical stress, and induce HO-1 expression in a time-dependent response to laminar shear stress (32). In an in vivo model with inflammatory, oxidative, and mechanical stress, HO-1 mRNA was induced after 6 and 12 h as a result of unilateral urethral obstruction (31). We therefore postulated that splinting would induce HO-1 as a protective response.
We have shown that within the first hour of wounding, HO-1 promoter activity at the place of the wound decreases. This may be related to the removal of the HO-1-positive skin layer. This suggests that there is basal HO-1 promoter activity present in normal skin, which we previously validated in the epithelium of the skin (6). More precisely, keratinocytes in the hyperproliferative epithelium express high levels of HO-1 as normal physiology (25, 26). After the initial decrease in HO-1 promoter activity in both groups, HO-1 promoter activity significantly increased within one day, which may be due to the attraction of HO-1-positive macrophages during the inflammatory phase, and the influx of HO-1-positive epithelial cells during the re-epithelialization of the wounds (22, 26, 42). This rapid HO-1 induction is in line with a previous study in mouse excisional wounds, showing increased HO-1 mRNA and protein at day 1 after wounding (24). Additionally, the constitutively expressed HO-2 was also not affected by wounding in this previous study, which was similar to our current results. We found that effects on HO-1 promoter activity were independent of the wound model, since both splinted and non-splinted wounds showed similar activity. However, at day 3, there was only a significant increase in the splinted model compared to the baseline level. Increased HO-1 expression was also found for both models in 7-day wounds at the mRNA level when compared to non-wounded skin. This increase was significantly higher after mechanical stress.
On the protein level, we saw contradictory results with significantly more HO-1 protein staining in the epidermis of non-splinted wounds and a slight, non-statistically significant, increase of HO-1 protein in the dermis in splinted wounds after 7 days. Thus, although HO-1 induction was shown in diverse models of mechanical stress, we found only HO-1 promoter activation and HO-1 mRNA induction. HO-1 protein expression was, in fact, reduced by the static mechanical stress of the splint in the epidermal region.
When comparing protein levels in the epidermis, HO-1-positive cells were clustered in re-epithelialized tissue underneath the wound crust and were likely newly formed keratinocytes (25, 26). It is likely that the highly increased keratinocyte gene expression in non-splinted wounds account for the observed increase in HO-1 protein expression.
In the dermis, HO-1-positive cells in inflamed tissues were individually spread, and based on their location and morphology, appeared to be macrophages (25, 42). We and others demonstrated previously that HO-1-positive macrophages can be recruited during the wound repair process (6, 25, 26).
It is, however, striking that splinted wounds exhibited similar levels of HO-1-positive cells in the dermis when compared to non-splinted wounds, while there were higher levels of F4/80-positive macrophages in splinted wounds.
Mechanical Stress by Splinting Enhances Inflammation; The Role of HO-1
Macrophages promote strong inflammatory responses through secretion of cytokines, such as IL-1β and TNF-α (43). It is thought that during the wound repair process, first pro-inflammatory M1 macrophages enter the wound site to clear cellular debris and destroy invading pathogens. Besides pro-inflammatory M1 macrophages, there are other macrophage subsets, including the anti-inflammatory M2 macrophages and the Mhem, Mox, and M4 macrophages (44–46). To enter the proliferation phase, resolution of inflammation is necessary. Under the right conditions, M1 macrophages may therefore skew into other anti-inflammatory macrophage subsets (45, 46). The presence of large numbers of macrophages in the splinted wounds and the increased levels of pro-inflammatory mediators IL-1β, TNF-α, and COX-2 suggests that in the splinted wounds there were mainly M1 macrophages present. Prolonged inflammation and high levels of oxidative stress may result in excessive deposition of ECM, leading to fibrosis and excessive/hypertrophic scarring (18, 47). Cytokines that mediate inflammation, such as IL-1β and TNF-α, are well known to be involved in mechanical stretch-induced inflammation. For example, in vitro administration of mechanical stress induces both IL-1β and COX-2 in fibroblast-like synoviocyte cells (48), epithelial cells (49), chondrocytes (50), and cardiac muscle cells (51) in a time- and force-dependent manner. This increased expression of inflammatory markers corresponded to our findings. Moreover, we found a trend in MCP-1 mRNA induction (p = 0.10) after mechanical stress, matching the enhanced protein staining for macrophages after mechanical stress.
Heme oxygenase-1-positive macrophages are thought to protect the wound environment against oxidative stress (25, 52). Following injury, the HO substrate heme is abundantly released at the edges of the wound site and can stimulate recruitment of leukocytes (6, 25, 53). In contrast, HO activity inhibits leukocyte recruitment via down-regulation of vascular adhesion molecules (6). Moreover, HO-1 activity inhibits production of various pro-inflammatory cytokines, including IL-1β and TNF-α (54, 55). In 7-day wounds, the inflammation levels were high in both non-splinted and splinted wounds, but however were much higher after mechanical stress in the splinted wounds.
The expression of HO-1 may facilitate resolution of inflammatory and oxidative stress at the wound site. When mechanical stress delays HO-1 protein expression, this may also delay resolution of inflammation, and lead to fibrogenesis.
Mechanical Stress Results in More Fibrosis by a Prolonged Survival of Myofibroblasts; The Relation Between Inflammation, Remodeling, and HO-1
Mechanical stress, which is present in CL/P patients following palatal surgery and in patients with wounds in the vicinity of joints, increases the risk of excessive scar formation. In our splinting model, we demonstrated the increased presence of pro-inflammatory and pro-fibrotic cells and markers, suggesting that mechanical stress interferes with resolution of inflammation, which may ultimately lead to hampered wound repair and excessive scar formation. Application of mechanical loading to healing wounds in mice can cause hypertrophic scarring, through decreased apoptosis of myofibroblasts (56). It is tempting to speculate that the delayed HO-1 protein expression in the splinted model contributes to the delay in resolution of inflammation and allows the pro-inflammatory environment and the myofibroblast survival that we observed in the wound area at day 7.
When myofibroblasts fail to undergo apoptosis, this leads to continuation of contraction and production of ECM, and ultimately to fibrosis (5, 13, 17, 18). We found an increased presence of myofibroblasts after splinting, suggesting that either more myofibroblasts are formed upon mechanical stress, less myofibroblasts die, or splinting causes a delay in the myofibroblast formation when compared to non-splinted wounds. This corresponds to other reports, which showed that mechanical tension induces myofibroblast differentiation in wound granulation tissue in 6-day splinted wounds compared to non-splinted wounds (14, 20). These processes may be fine-tuned by HO-1 and its effector molecules as is shown in vitro by regulating the apoptosis of fibroblasts (18, 57).
Pathological cutaneous wound healing resulting in excessive scarring and fibrosis is the consequence of an imbalance between ECM synthesis by myofibroblasts and degradation and remodeling by MMPs (13). We found that remodeling marker MMP-9 was increased after mechanical stress, which was also observed by other groups (58–60). VEGF is an important promoter of angiogenesis and therefore microvessel density might be increased by mechanical tension. Chemokine and cytokine processing by MMPs affects the progression of inflammatory responses and leukocyte migration (61). For example, MMP-9 can inactivate/degrade CXCL1, CXCL4, CXCL9, and CXCL10, resulting in anti-inflammatory effects (62, 63). Increased MMP-9 may be indicative of inflammation and poor wound healing (64). The interplay between VEGF and HO-1 is complex and it was demonstrated that they can both inhibit or induce each other (65–68). Induction of HO-1 in a rat excisional wound model enhanced wound healing by increased cellular proliferation and collagen synthesis (28). HO-1 induction also reduced the inflammatory response by inhibition of pro-inflammatory molecules TNF-α, and ICAM-1 and an induction of the anti-inflammatory cytokine IL-10 (28).
In summary, mechanical stress leads to delayed wound closure, increased inflammation, and altered remodeling during the wound healing process. Since more myofibroblasts are present after splinting, mechanical stress may result in more scar formation. This was associated with a delayed anti-inflammatory and anti-fibrotic HO-1 protein expression in splinted wounds compared to non-splinted wounds. Since HO-1 attacks multiple targets that play an important role during fibrogenesis, pharmacologic induction of HO-1 may facilitate resolution of inflammation and attenuate fibrosis.
Conclusion
We demonstrated that splinting significantly delays wound closure and potentiates the influx of pro-inflammatory leukocytes and myofibroblasts in day-7 wounds, which may promote fibrosis. The cytoprotective HO-1 gene is increased upon wounding, but HO-1 protein was lower in the epidermis after mechanical stress, probably as a result of the increased number of HO-1-positive keratinocytes in the re-epithelialization tissue of non-splinted wounds. Therefore, targeted pharmacologic induction of cytoprotective mechanisms, including HO-1, as preventive therapy against mechanical stress-induced inflammation and fibrosis must be considered.
Author Contributions
NC, MS, MG, and CB performed the research. GD provided the original transgenic HO-1 luc mice for the study. NC, AK-J, CC, DL, and FW designed the study. NC, MS, MG, RW, KB, and FW analyzed the data. NC, RW, KB, AK-J, CC, DL, and FW wrote the manuscript. All authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Radboud University Medical Centre, European Commission (FP7-HEALTH-2011-1, Grant Agreement no. 279024), project EuroSkinGraft, and the Dutch Burns Foundation (# 09.110).
References
1. Bartzela TN, Carels CE, Bronkhorst EM, Ronning E, Rizell S, Kuijpers-Jagtman AM. Tooth agenesis patterns in bilateral cleft lip and palate. Eur J Oral Sci (2010) 118(1):47–52. doi:10.1111/j.1600-0722.2009.00698.x
2. Nollet PJ, Katsaros C, Van’t Hof MA, Kuijpers-Jagtman AM. Treatment outcome in unilateral cleft lip and palate evaluated with the GOSLON yardstick: a meta-analysis of 1236 patients. Plast Reconstr Surg (2005) 116(5):1255–62. doi:10.1097/01.prs.0000181652.84855.a3
3. Biggs LC, Goudy SL, Dunnwald M. Palatogenesis and cutaneous repair: a two-headed coin. Dev Dyn (2015) 244(3):289–310. doi:10.1002/dvdy.24224
4. Li J, Johnson CA, Smith AA, Salmon B, Shi B, Brunski J, et al. Disrupting the intrinsic growth potential of a suture contributes to midfacial hypoplasia. Bone (2014) 81:186–95. doi:10.1016/j.bone.2014.04.020
5. Brouwer KM, Lundvig DM, Middelkoop E, Wagener FA, Von den Hoff JW. Mechanical cues in orofacial tissue engineering and regenerative medicine. Wound Repair Regen (2015) 23(3):302–11. doi:10.1111/wrr.12283
6. Wagener FA, van Beurden HE, von den Hoff JW, Adema GJ, Figdor CG. The heme-heme oxygenase system: a molecular switch in wound healing. Blood (2003) 102(2):521–8. doi:10.1182/blood-2002-07-2248
7. van Beurden HE, Von den Hoff JW, Torensma R, Maltha JC, Kuijpers-Jagtman AM. Myofibroblasts in palatal wound healing: prospects for the reduction of wound contraction after cleft palate repair. J Dent Res (2005) 84(10):871–80. doi:10.1177/154405910508401002
8. Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med (2014) 6(265):265sr6. doi:10.1126/scitranslmed.3009337
9. Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature (2008) 453(7193):314–21. doi:10.1038/nature07039
10. Hinz B, Gabbiani G. Mechanisms of force generation and transmission by myofibroblasts. Curr Opin Biotechnol (2003) 14(5):538–46. doi:10.1016/j.copbio.2003.08.006
11. Eckes B, Zweers MC, Zhang ZG, Hallinger R, Mauch C, Aumailley M, et al. Mechanical tension and integrin alpha 2 beta 1 regulate fibroblast functions. J Investig Dermatol Symp Proc (2006) 11(1):66–72. doi:10.1038/sj.jidsymp.5650003
12. Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen (2005) 13(1):7–12. doi:10.1111/j.1067-1927.2005.130102.x
13. Micallef L, Vedrenne N, Billet F, Coulomb B, Darby IA, Desmouliere A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair (2012) 5(Suppl 1):S5. doi:10.1186/1755-1536-5-S1-S5
14. Hinz B, Mastrangelo D, Iselin CE, Chaponnier C, Gabbiani G. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am J Pathol (2001) 159(3):1009–20. doi:10.1016/S0002-9440(10)61776-2
15. van der Veer WM, Bloemen MC, Ulrich MM, Molema G, van Zuijlen PP, Middelkoop E, et al. Potential cellular and molecular causes of hypertrophic scar formation. Burns (2009) 35(1):15–29. doi:10.1016/j.burns.2008.06.020
16. Darby IA, Laverdet B, Bonte F, Desmouliere A. Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol (2014) 7:301–11. doi:10.2147/CCID.S50046
17. Van De Water L, Varney S, Tomasek JJ. Mechanoregulation of the myofibroblast in wound contraction, scarring, and fibrosis: opportunities for new therapeutic intervention. Adv Wound Care (New Rochelle) (2013) 2(4):122–41. doi:10.1089/wound.2012.0393
18. Lundvig DM, Immenschuh S, Wagener FA. Heme oxygenase, inflammation, and fibrosis: the good, the bad, and the ugly? Front Pharmacol (2012) 3:81. doi:10.3389/fphar.2012.00081
19. Galiano RD, Michaels JT, Dobryansky M, Levine JP, Gurtner GC. Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen (2004) 12(4):485–92. doi:10.1111/j.1067-1927.2004.12404.x
20. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol (2002) 3(5):349–63. doi:10.1038/nrm809
21. Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol (2010) 50:323–54. doi:10.1146/annurev.pharmtox.010909.105600
22. Wagener FA, Volk HD, Willis D, Abraham NG, Soares MP, Adema GJ, et al. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol Rev (2003) 55(3):551–71. doi:10.1124/pr.55.3.5
23. Wagener FA, Dankers AC, van Summeren F, Scharstuhl A, van den Heuvel JJ, Koenderink JB, et al. Heme oxygenase-1 and breast cancer resistance protein protect against heme-induced toxicity. Curr Pharm Des (2013) 19(15):2698–707. doi:10.2174/1381612811319150004
24. Schafer M, Werner S. Oxidative stress in normal and impaired wound repair. Pharmacol Res (2008) 58(2):165–71. doi:10.1016/j.phrs.2008.06.004
25. Hanselmann C, Mauch C, Werner S. Haem oxygenase-1: a novel player in cutaneous wound repair and psoriasis? Biochem J (2001) 353(Pt 3):459–66. doi:10.1042/bj3530459
26. Kampfer H, Kolb N, Manderscheid M, Wetzler C, Pfeilschifter J, Frank S. Macrophage-derived heme-oxygenase-1: expression, regulation, and possible functions in skin repair. Mol Med (2001) 7(7):488–98.doi:10.1.1.223.306
27. Grochot-Przeczek A, Lach R, Mis J, Skrzypek K, Gozdecka M, Sroczynska P, et al. Heme oxygenase-1 accelerates cutaneous wound healing in mice. PLoS One (2009) 4(6):e5803. doi:10.1371/journal.pone.0005803
28. Ahanger AA, Prawez S, Leo MD, Kathirvel K, Kumar D, Tandan SK, et al. Pro-healing potential of hemin: an inducer of heme oxygenase-1. Eur J Pharmacol (2010) 645(1–3):165–70. doi:10.1016/j.ejphar.2010.06.048
29. Wagener FA, Scharstuhl A, Tyrrell RM, Von den Hoff JW, Jozkowicz A, Dulak J, et al. The heme-heme oxygenase system in wound healing; implications for scar formation. Curr Drug Targets (2010) 11(12):1571–85. doi:10.2174/1389450111009011571
30. Cho JH, Lee SK, Lee JW, Kim EC. The role of heme oxygenase-1 in mechanical stress- and lipopolysaccharide-induced osteogenic differentiation in human periodontal ligament cells. Angle Orthod (2010) 80(4):552–9. doi:10.2319/091509-520.1
31. Carlsen I, Nilsson L, Frokiaer J, Norregaard R. Changes in phosphorylated heat-shock protein 27 in response to acute ureteral obstruction in rats. Acta Physiol (2013) 209(2):167–78. doi:10.1111/apha.12135
32. Wagner CT, Durante W, Christodoulides N, Hellums JD, Schafer AI. Hemodynamic forces induce the expression of heme oxygenase in cultured vascular smooth muscle cells. J Clin Invest (1997) 100(3):589–96.
33. Wever KE, Wagener FA, Frielink C, Boerman OC, Scheffer GJ, Allison A, et al. Diannexin protects against renal ischemia reperfusion injury and targets phosphatidylserines in ischemic tissue. PLoS One (2011) 6(8):e24276. doi:10.1371/journal.pone.0024276
34. Su H, van Dam GM, Buis CI, Visser DS, Hesselink JW, Schuurs TA, et al. Spatiotemporal expression of heme oxygenase-1 detected by in vivo bioluminescence after hepatic ischemia in HO-1/Luc mice. Liver Transpl (2006) 12(11):1634–9. doi:10.1002/lt.20852
35. Lundvig DM, Scharstuhl A, Cremers NA, Pennings SW, Te Paske J, van Rheden R, et al. Delayed cutaneous wound closure in HO-2 deficient mice despite normal HO-1 expression. J Cell Mol Med (2014) 18(12):2488–98. doi:10.1111/jcmm.12389
36. van den Brand BT, Vermeij EA, Waterborg CE, Arntz OJ, Kracht M, Bennink MB, et al. Intravenous delivery of HIV-based lentiviral vectors preferentially transduces F4/80+ and Ly-6C+ cells in spleen, important target cells in autoimmune arthritis. PLoS One (2013) 8(2):e55356. doi:10.1371/journal.pone.0055356
37. Cremers NA, Lundvig DM, van Dalen SC, Schelbergen RF, van Lent PL, Szarek WA, et al. Curcumin-induced heme oxygenase-1 expression prevents H2O2-induced cell death in wild type and heme oxygenase-2 knockout adipose-derived mesenchymal stem cells. Int J Mol Sci (2014) 15(10):17974–99. doi:10.3390/ijms151017974
38. Baum CL, Arpey CJ. Normal cutaneous wound healing: clinical correlation with cellular and molecular events. Dermatol Surg (2005) 31(6):674–86; discussion 686. doi:10.1111/j.1524-4725.2005.31612
40. Liu XM, Peyton KJ, Durante W. Physiological cyclic strain promotes endothelial cell survival via the induction of heme oxygenase-1. Am J Physiol Heart Circ Physiol (2013) 304(12):H1634–43. doi:10.1152/ajpheart.00872.2012
41. Lee SK, Lee CY, Kook YA, Lee SK, Kim EC. Mechanical stress promotes odontoblastic differentiation via the heme oxygenase-1 pathway in human dental pulp cell line. Life Sci (2010) 86(3–4):107–14. doi:10.1016/j.lfs.2009.11.013
42. Schurmann C, Seitz O, Klein C, Sader R, Pfeilschifter J, Muhl H, et al. Tight spatial and temporal control in dynamic basal to distal migration of epithelial inflammatory responses and infiltration of cytoprotective macrophages determine healing skin flap transplants in mice. Ann Surg (2009) 249(3):519–34. doi:10.1097/SLA.0b013e31819a8d6c
43. Rees P, Greaves N, Bayat A. Chemokines in wound healing and as potential therapeutic targets for reducing cutaneous scarring. Adv Wound Care (New Rochelle) (2015) 4(11):687–703. doi:10.1089/wound.2014.0568
44. Hull TD, Agarwal A, George JF. The mononuclear phagocyte system in homeostasis and disease: a role for heme oxygenase-1. Antioxid Redox Signal (2014) 20(11):1770–88. doi:10.1089/ars.2013.5673
45. Chistiakov DA, Bobryshev YV, Orekhov AN. Changes in transcriptome of macrophages in atherosclerosis. J Cell Mol Med (2015) 19(6):1163–73. doi:10.1111/jcmm.12591
46. Medbury HJ, Williams H, Fletcher JP. Clinical significance of macrophage phenotypes in cardiovascular disease. Clin Transl Med (2014) 3(1):63. doi:10.1186/s40169-014-0042-1
47. Sidgwick GP, Bayat A. Extracellular matrix molecules implicated in hypertrophic and keloid scarring. J Eur Acad Dermatol Venereol (2012) 26(2):141–52. doi:10.1111/j.1468-3083.2011.04200.x
48. Takao M, Okinaga T, Ariyoshi W, Iwanaga K, Nakamichi I, Yoshioka I, et al. Role of heme oxygenase-1 in inflammatory response induced by mechanical stretch in synovial cells. Inflamm Res (2011) 60(9):861–7. doi:10.1007/s00011-011-0346-1
49. Toshinaga A, Hosokawa R, Okinaga T, Masaki C. Inflammatory response in epithelial cells induced by mechanical stress is suppressed by hyaluronic acid. Jpn Soc Inflamm Regen (2010) 30(2):120–7. doi:10.2492/inflammregen.30.120
50. Huang J, Ballou LR, Hasty KA. Cyclic equibiaxial tensile strain induces both anabolic and catabolic responses in articular chondrocytes. Gene (2007) 404(1–2):101–9. doi:10.1016/j.gene.2007.09.007
51. Vandenburgh HH, Shansky J, Solerssi R, Chromiak J. Mechanical stimulation of skeletal muscle increases prostaglandin F2 alpha production, cyclooxygenase activity, and cell growth by a pertussis toxin sensitive mechanism. J Cell Physiol (1995) 163(2):285–94.
52. Ishii T, Itoh K, Sato H, Bannai S. Oxidative stress-inducible proteins in macrophages. Free Radic Res (1999) 31(4):351–5.
53. auf dem Keller U, Kumin A, Braun S, Werner S. Reactive oxygen species and their detoxification in healing skin wounds. J Investig Dermatol Symp Proc (2006) 11(1):106–11. doi:10.1038/sj.jidsymp.5650001
54. Morse D, Pischke SE, Zhou Z, Davis RJ, Flavell RA, Loop T, et al. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J Biol Chem (2003) 278(39):36993–8. doi:10.1074/jbc.M302942200
55. Lee TS, Tsai HL, Chau LY. Induction of heme oxygenase-1 expression in murine macrophages is essential for the anti-inflammatory effect of low dose 15-deoxy-Delta 12,14-prostaglandin J2. J Biol Chem (2003) 278(21):19325–30. doi:10.1074/jbc.M300498200
56. Aarabi S, Bhatt KA, Shi Y, Paterno J, Chang EI, Loh SA, et al. Mechanical load initiates hypertrophic scar formation through decreased cellular apoptosis. FASEB J (2007) 21(12):3250–61. doi:10.1096/fj.07-8218com
57. Scharstuhl A, Mutsaers HA, Pennings SW, Szarek WA, Russel FG, Wagener FA. Curcumin-induced fibroblast apoptosis and in vitro wound contraction are regulated by antioxidants and heme oxygenase: implications for scar formation. J Cell Mol Med (2009) 13(4):712–25. doi:10.1111/j.1582-4934.2008.00339.x
58. Beckmann R, Houben A, Tohidnezhad M, Kweider N, Fragoulis A, Wruck CJ, et al. Mechanical forces induce changes in VEGF and VEGFR-1/sFlt-1 expression in human chondrocytes. Int J Mol Sci (2014) 15(9):15456–74. doi:10.3390/ijms150915456
59. Kawata T, Kohno S, Kaku M, Fujita T, Ohtani J, Motokawa M, et al. Expression of vascular endothelial growth factor on neovascularization during experimental tooth movement by magnets. Biomed Res (2011) 22(2):249–54.
60. Huang D, Liu Y, Huang Y, Xie Y, Shen K, Zhang D, et al. Mechanical compression upregulates MMP9 through SMAD3 but not SMAD2 modulation in hypertrophic scar fibroblasts. Connect Tissue Res (2014) 55(5–6):391–6. doi:10.3109/03008207.2014.959118
61. Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol (2007) 82(6):1375–81. doi:10.1189/jlb.0607338
62. Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood (2000) 96(8):2673–81.
63. Van den Steen PE, Husson SJ, Proost P, Van Damme J, Opdenakker G. Carboxyterminal cleavage of the chemokines MIG and IP-10 by gelatinase B and neutrophil collagenase. Biochem Biophys Res Commun (2003) 310(3):889–96. doi:10.1016/j.bbrc.2003.09.098
64. Liu Y, Min D, Bolton T, Nube V, Twigg SM, Yue DK, et al. Increased matrix metalloproteinase-9 predicts poor wound healing in diabetic foot ulcers. Diabetes Care (2009) 32(1):117–9. doi:10.2337/dc08-0763
65. Bussolati B, Ahmed A, Pemberton H, Landis RC, Di Carlo F, Haskard DO, et al. Bifunctional role for VEGF-induced heme oxygenase-1 in vivo: induction of angiogenesis and inhibition of leukocytic infiltration. Blood (2004) 103(3):761–6. doi:10.1182/blood-2003-06-1974
66. Jozkowicz A, Huk I, Nigisch A, Weigel G, Weidinger F, Dulak J. Effect of prostaglandin-J(2) on VEGF synthesis depends on the induction of heme oxygenase-1. Antioxid Redox Signal (2002) 4(4):577–85. doi:10.1089/15230860260220076
67. Chao HM, Chuang MJ, Liu JH, Liu XQ, Ho LK, Pan WH, et al. Baicalein protects against retinal ischemia by antioxidation, antiapoptosis, downregulation of HIF-1alpha, VEGF, and MMP-9 and upregulation of HO-1. J Ocul Pharmacol Ther (2013) 29(6):539–49. doi:10.1089/jop.2012.0179
68. Jayasooriya RG, Park SR, Choi YH, Hyun JW, Chang WY, Kim GY. Camptothecin suppresses expression of matrix metalloproteinase-9 and vascular endothelial growth factor in DU145 cells through PI3K/Akt-mediated inhibition of NF-kappaB activity and Nrf2-dependent induction of HO-1 expression. Environ Toxicol Pharmacol (2015) 39(3):1189–98. doi:10.1016/j.etap.2015.04.011
Keywords: cleft palate, burns, mechanical stress, wound healing, heme oxygenase-1, inflammation, fibrosis
Citation: Cremers NAJ, Suttorp M, Gerritsen MM, Wong RJ, van Run-van Breda C, van Dam GM, Brouwer KM, Kuijpers-Jagtman AM, Carels CEL, Lundvig DMS and Wagener FADTG (2015) Mechanical Stress Changes the Complex Interplay Between HO-1, Inflammation and Fibrosis, During Excisional Wound Repair. Front. Med. 2:86. doi: 10.3389/fmed.2015.00086
Received: 11 September 2015; Accepted: 24 November 2015;
Published: 15 December 2015
Edited by:
Peter Olinga, University of Groningen, NetherlandsReviewed by:
Pier Paolo Piccaluga, University of Bologna, ItalyFrancesco Trapasso, University “Magna Græcia” of Catanzaro, Italy
Copyright: © 2015 Cremers, Suttorp, Gerritsen, Wong, van Run-van Breda, van Dam, Brouwer, Kuijpers-Jagtman, Carels, Lundvig and Wagener. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank A. D. T. G. Wagener, ZnJhbmsud2FnZW5lckByYWRib3VkdW1jLm5s
†Maarten Suttorp and Marlous M. Gerritsen have contributed equally to this work.