 Célia von Siebenthal1
Célia von Siebenthal1 John-David Aubert2Periklis Mitsakis3Patrick Yerly4
John-David Aubert2Periklis Mitsakis3Patrick Yerly4 John O. Prior3
John O. Prior3 Laurent Pierre Nicod2*
Laurent Pierre Nicod2*
- 1University of Lausanne, Lausanne, Switzerland
- 2Pneumology, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland
- 3Nuclear Medicine and Molecular Imaging, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland
- 4Cardiology, Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland
Pulmonary hypertension (PH) is a rare disease, whose underlying mechanisms are not fully understood. It is characterized by pulmonary arterial vasoconstriction and vessels wall thickening, mainly intimal and medial layers. Several molecular pathways have been studied, but their respective roles remain unknown. Cardiac repercussions of PH are hypertrophy, dilation, and progressive right ventricular dysfunction. Multiple echocardiographic parameters are being used, in order to assess anatomy and cardiac function, but there are no guidelines edited about their usefulness. Thus, it is now recommended to associate the best-known parameters, such as atrial and ventricular diameters or tricuspid annular plane systolic excursion. Cardiac catheterization remains necessary to establish the diagnosis of PH and to assess pulmonary hemodynamic state. Concerning energetic metabolism, free fatty acids, normally used to provide energy for myocardial contraction, are replaced by glucose uptake. These abnormalities are illustrated by increased 18F-fluorodeoxyglucose (18F-FDG) uptake on positron emission tomography/computed tomography, which seems to be correlated with echocardiographic and hemodynamic parameters.
Introduction
Pulmonary hypertension (PH) is a serious disease, which evolves progressively to cardiac insufficiency and death (1, 2). It is characterized by increased pressures and resistances in pulmonary arteries. The diagnosis is made, by cardiac catheterization, by a mean pulmonary arterial pressure (PAPm) ≥25 mmHg. We talk about pulmonary arterial hypertension (PAH) when the pulmonary wedge pressure (PWP) is ≤15 mmHg and pulmonary vascular resistance (PVR) >3 WU (3, 4). We can classify the etiologies of PH in five different groups, according to DanaPoint classification (2008) and maintained with minor modifications after the fifth World symposium in Nice (2013), with the first group related to etiologies leading to PH of arterial origin and sharing similar histological pattern. All groups of PH are characterized by similar symptoms, especially dyspnea, cough, angina, fainting, or peripheral edemas (4, 5).
Without treatment, survival does not exceed 2–3 years with chronic PAH. Moreover, the diagnosis is usually made when cardiac repercussions and clinical symptoms are already severe [75% of patients in New York Heart Association (NYHA) class III or IV] (1, 5). Thus, it is necessary to develop new tools to make earlier diagnosis and to increase knowledge in medical caregivers about this poorly known disease.
The aim of this article is to review current knowledge about pathophysiology and cardiac repercussions of PAH, as well as the different clinical, biological, and imagery parameters used to assess PAH and right heart function.
Classification of Pulmonary Hypertension
Etiologies and forms of PH are classified in five groups, according Dana Point classification (2008). Different forms of PAH are joined in Group 1, to know idiopathic PAH, heritable PAH, PAH related to drugs and toxins, connective tissue disease, HIV, schistosomiasis, portal hypertension, and congenital heart disease. As previously mentioned, PWP is <15 mmHg, and PVR is >3 WU. Treatments for PAH are described below. Second group is PH related to left heart disease. Echocardiography and cardiac catheterization show left ventricular dysfunction. PWP is >15 mmHg, and PVR is >3 WU. These forms are mainly treated with usual treatments of left ventricular dysfunction or repair valvular diseases. If these are not sufficient, specific treatments for PH, such as vasodilators, can be added. Third group contains PH forms related to lung disease, such as chronic obstructive pulmonary disease, interstitial diseases, or sleep apneas. They are characterized by a PWP <15 mmHg and pathological pulmonary functional tests. First line treatment is an optimal therapy of the underlying disease and long-term oxygen therapy. Fourth group is chronic thromboembolic PH. As in PAH, PAPm is >25 mmHg, and PWP is <15 mmHg. Multiple thrombi can be found by pulmonary computed tomography or magnetic resonance angiography or by pulmonary ventilation/perfusion scan. Chronic thromboembolic PH can be reversed by endarterectomy, and long-term anticoagulation is recommended. In inoperable patients, or if PH persists after surgery, treatments of PAH can be useful. Last group contains PH forms from multifactorial and unclear mechanisms, such as hematologic diseases, sarcoidosis, or metabolic disease. PWP is a variable (3). After the fifth symposium in Nice (2013), minor modifications have been done. In Group 1, a few more mutations are described. Persistent PH of the newborn has been separated from other causes of PAH and some congenital cardiopathies have been added to Group 2 (4). The aim of this article is to review pathogenesis and evaluation of PAH forms.
Pathobiology and Therapeutic Treatment Options
Histology
Multiple abnormalities are found in histological analysis of lungs from PAH patients, mostly in small arteries. They all induce thickening of vessels wall. The first step in this process is thickening of intima, by increased proliferation and migration of local stem cells. Medial and adventitial layers are also altered, by medial smooth muscle cells hypertrophy, which invade the wall of small arteries, usually not muscularized, and by accumulation of fibroblasts from the adventitia. Extracellular matrix is produced and degraded in excess by metalloproteinases, which releases mitogenic factors for smooth muscle cells. Eventually, adventitial mesenchymatous cells called myofibroblasts migrate to the media and induce proliferation of smooth muscle cells. Later in the obstructive vascular process, hypertrophy and hyperplasia of endothelial cells, also known as neointima formation, continue and form plexiform lesions. Plexiform lesions are a monoclonal proliferation of endothelial cells forming multiple channels into one obliterated arteriole, surrounded by smooth muscle cells, extracellular matrix, and myofibroblastes (Figure 1). Beyond morphological abnormalities, they present many functional and metabolic alterations (6–8). As in many other proliferative diseases, several growth factors, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), or epidermal growth factor (EGF), are involved in vessels wall hyperplasia, through tyrosine kinase activation. Their overexpression in endothelial cells promotes inflammation, proliferation, migration, and survival of smooth muscle cells and fibroblasts. Greater serotonin sensitivity and increased transporter’s expression were found in smooth muscle cells, and a greater amount of serotonin, through its mitogenic effects, could take part to thickening of vascular wall (6, 7, 9, 10). The trigger for these changes in unknown, but some studies hypothesized that an endothelial injury could promote this proliferation. Indeed, Sakao et al. showed that inhibition of VEGF receptors and mechanical stress can provoke endothelial lesions by apoptosis. After a few days, resistance against apoptosis appears in surviving cells, probably induced by release of mediators from previously injured cells. Resistant cells express survivin, an apoptosis inhibitor frequently met in malignant tumors (11, 12). Parallel to hyperplasia of vessels wall, multiple microthromboses are frequently found in small arteries of PH lungs. Three components promote thromboses: activation of intravascular coagulation, platelets dysfunction, and release of vasoconstrictor and pro-coagulant mediators by endothelial cells. Platelets dysfunction and dysfunctional endothelium promote intravascular coagulation by releasing plasmatic pro-coagulants (von Willebrand factor, thromboxane, and serotonin) and anti-fibrinolytic molecules (plasminogen activator inhibitor). Platelets also store vasoconstrictor and mitotic substances. However, it is unclear if microthromboses are a consequence or a trigger for elevated PVRs and pressures (7–9).
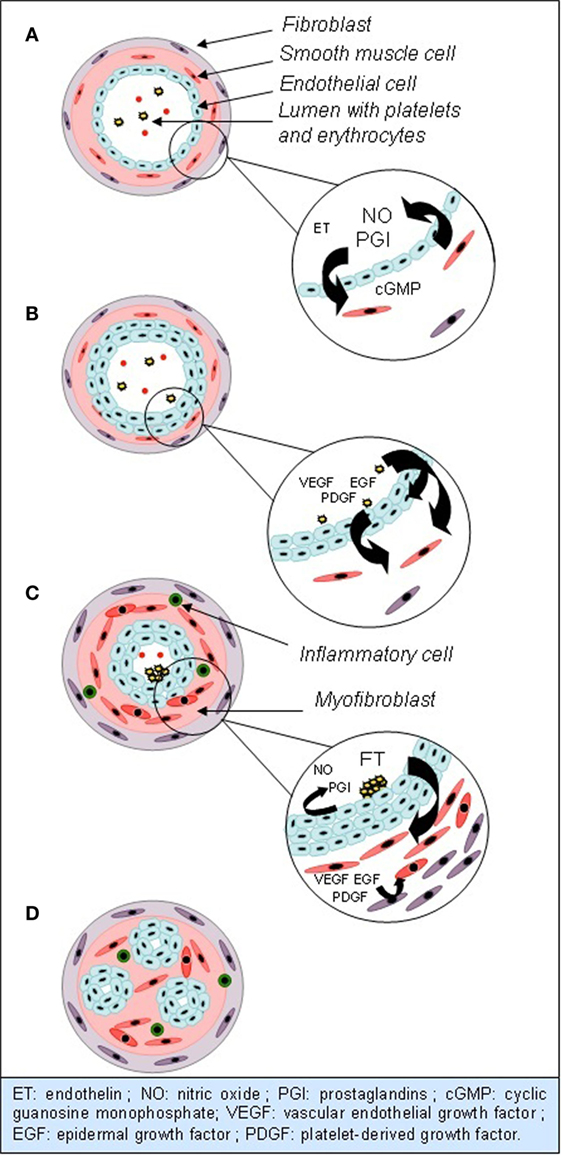
Figure 1. Evolution of anatomical and functional abnormalities in small arterioles in PAH. (A) Normal vessel: vasodilation is predominant, by NO and PGI. (B) Hypertrophy and hyperplasia of endothelial cells, mediated by growth factors. (C) Hypertrophy and hyperplasia of smooth muscle cells and fibroblasts. Extravasation of inflammatory cells. Endothelial dysfunction and clots formation. (D) Plexiform lesions.
Endothelial Dysfunction
Many studies identified the nitric oxide (NO) pathway to be linked to the emergence of PAH. NO is produced by NO synthase (eNOS) in endothelial cells. It is the predominant factor inducing vasodilation in normal conditions, via cyclic guanosine monophosphate (cGMP). It also inhibits proliferation of smooth muscle cells, platelets aggregation and activation, leukocytes migration, and endothelin (ET) secretion (9, 12). In mice, an inhibiting mutation of eNOS induces increased pulmonary arterial pressure (PAP) (13). In humans, it seems that a reduced amount of NO is also involved in pathogenesis of PAH, by increased degradation and reduced eNOS expression in pulmonary tissues (Figure 1). Giaid and Saleh showed a negative correlation between eNOS expression in immunohistochemical analysis, severity of vascular lesions and PVR (14).
Pathogenesis of PAH can be partly explained by increased ET secretion. ET is a neuropeptide released by vascular endothelium. It can bind, on vascular wall, to type A receptors, leading to vasoconstriction, or type B receptors, leading to vasodilation. In pathological conditions, vasoconstrictor effect is predominant and takes part to PAH development. ET seems to be acting partly by inhibition of Kv1.5 potassium channels synthesis. Increased potassium rate in cells induces depolarization and calcium influx through voltage-dependant calcium channels. Increased intracellular calcium and potassium rates promote vasoconstriction, proliferation, and apoptosis resistance. ET is thus responsible of both hyperplasia and endothelial dysfunction (Figure 1) (9, 10, 15).
Prostaglandins (PGI) pathway could also play a role in PAH development. PGI are derived from arachidonic acid and inhibit cellular proliferation, inflammation, coagulation, platelets aggregation, and vasoconstriction. A reduced rate is found in PAH patients (Figure 1) (7, 9, 12, 16).
This endothelial dysfunction is the target of current PAH treatments, such as ET receptors antagonists, PGI derivates, phosphodiesterases inhibitors, calcium-channel blockers, or cGMP kinase activators. However, it seems that remodeling and occlusion of small arteries contributes primarily to the increase of PAP and right atrial pressure (RAP). Thus, the therapeutic response is highly variable, frequently insufficient, and these treatments cannot reverse the underlying histological abnormalities (3, 8, 9, 15).
Inflammation and Immunity
Several studies identified an inflammatory component in pathogenesis of PAH. Indeed, different types of inflammatory cells are found on the surface and into the vessels wall. Furthermore, several cytokines and chemokines are excessively produced, by inflammatory and endothelial cells, and also take part to smooth muscle cells proliferation. Soon et al. demonstrated that blood levels of some cytokines, such as interleukin (IL)-2, IL-6, IL-8, or IL-10, could be correlated with survival, which support their implication in pathogenesis of PH. For many years, the presence of autoantibodies has been discovered in PH related to connective tissue disease and systemic sclerosis. Recent studies have also brought them out in idiopathic PAH cases. Indeed, immunoglobulin G (IgG) not only against endothelial cells components but also against fibroblasts and smooth muscle cells is found in about 60% of patients. Although little is known about the role of those antibodies, they could cause endothelial injuries and then trigger the excessive proliferation process (6, 10, 17–19).
BMPR2 and TGF-β
Mutations of bone morphogenetic protein receptor type II (BMPR2), a kinase receptor from transforming growth factor-beta (TGF-β) family, are not only found in 70% of patients with heritable PH but also in about 25% of patients with idiopathic PAH. BMPR2 is usually expressed in pulmonary endothelial cells and regulates cell growth, differentiation, and apoptosis, through a factor named Smad. Some studies showed that inactivation of BMPR2 promotes endothelial cell survival, proliferation, angiogenesis, and migration. A loss of BMPR2 could be one of the bases of PAH pathogenesis, leading to endothelial injuries, apoptosis resistance, and plexiform lesions. It could also be involved in remodeling and dysfunction of the right ventricle (RV). However, prevalence of PAH in patients with BMPR2 mutation is low, which suggests that those mutations remain predisposing factors and that other vascular injuries are necessary to develop the disease (6, 9, 20). Abnormalities of other members of TGF-β family, such as TGF-β1, which induce proliferation and remodeling of vascular wall, seem to be present. Thus, there could be an imbalance between BMPR2 and TGF-β1, in favor of an excessive proliferation (16, 20).
Survivin
Those different pathways have a common consequence: excessive proliferation and apoptosis resistance. As mentioned previously, survivin is involved in some proliferative diseases. McMurtry et al. showed that its expression is increased not only in pulmonary vascular wall, mainly in smooth muscle cells, but also in endothelium and fibroblasts. Survivin inhibits apoptosis and opening of Kv1.5 potassium channels. In rats, survivin inactivating treatments are useful to reduce wall remodeling, PAP, and RV hypertrophy. It seems that survivin expression can occur after endothelial injuries, which is consistent with results obtained about the signaling pathways presented above. This factor could be one of the links between those pathways and vascular wall remodeling (15).
micro-RNAs
Recent studies in rats showed that another link between these pathways could be related to micro-ribonucleid acids (miRNAs). miRNAs are a family of non-coding RNAs, which regulate expression of coding RNAs through binging and cleavage in the cytoplasm. Different mechanisms, such as hypoxia, hypoxia-inducible factor-1alpha (HIF-1α), inflammation, and growth factors, regulate miRNAs. It seems that the expression of miRNAs could be dysregulated in PAH, as some are increased and others decreased. We can mention the example of miR-204, which regulates cellular proliferation and apoptosis through B-cell lymphoma-2 (Bcl-2) and nuclear factor of activated T-cells (NFAT). This one seems to be decreased in smooth muscle cells. ET and PDGF could be involved in inhibition of miR-204 (21). Other miRNAs are probably involved in PH and right heart dysfunction (22, 23).
HIF-1α and Metabolism
Several studies demonstrated that endothelial cells and myocardial energetic metabolism is altered. Free fatty acids oxidation, used as energetic resource in normal conditions, is replaced by anaerobic glycolysis. Molecular analysis of endothelial and myocardial cells showed that this metabolic shift is based on normoxic activation of the transcription factor HIF-1α (24–26). This factor is normally activated in hypoxic conditions, in order to produce energy without oxygen, by anaerobic glycolysis. HIF-1α slows down Krebs cycle. Free fatty acids oxidation is thus replaced by aerobic or anaerobic glycolysis. The latter is predominant, explaining increased lactate production, which is observed in PAH. This is called Warburg effect and is found in several pathologies, especially in malignant tumors (24–28). This results in decreased energy production per glucose molecule and, thus increased glucidic needs. Adequate intake is provided by increased expression of glucose transporter 1 (GLUT-1) channels and hexokinase, which are found in greater amount, particularly in pulmonary vessels with plexiform lesions (24, 25). In rats and in humans, Bonnet et al. showed that all those elements reduce cellular metabolic dependence to oxygen and give an advantage in hypoxic conditions for cellular proliferation and survival. This process depends on the inhibition of Kv1.5 potassium channels by HIF-1α. The resulting cellular depolarization induces calcium inflow. Elevated intracellular potassium and calcium rates promote proliferation and apoptosis resistance (29). In pulmonary vessels, parallel to this metabolic shift, the reduced NO production observed in PAH leads to a decreased number of mitochondria per cell and inhibits mitochondrial genes expression. Mitochondrial function abnormalities are also found, specifically in PAH lungs. This results in a reduced production of reactive oxygen species and a decreased amount of superoxide dismutase (SOD). Hydrogen peroxide production (H2O2), normally responsible of HIF-1α inhibition, is inhibited. These signaling alterations by reactive oxygen species could be the basis of normoxic activation of HIF-1α (24, 26, 28, 29). In the myocardium, inhibition of aerobic metabolism probably takes part to development hypertrophy, in which a greater expression of GLUT-1 channels, decreased oxygen consumption and increased lactate production by anaerobic glycolysis are found. This could lead to reduced energetic reserves and further reduce right heart function (24, 26, 30). Experimental studies investigated the efficacy of treatments targeted on this metabolic shift, such as imatinib or dichloroactetate. They demonstrated beneficial effects on pulmonary remodeling, inflammation, and myocardial hypertrophy (24, 25, 27). Feasibility and usefulness of these treatments in humans remain to be determined.
Conclusion
In conclusion, several mechanisms have been proposed to explain the abnormal pulmonary vasoconstriction and hyperplasia, but links between these different pathways remain unclear. Current treatments are not always effective and cannot reverse the histological abnormalities. Experimental studies are looking for new therapeutic targets, in order to inhibit proliferation, reactivate apoptosis, and recover a normal organization in pulmonary vascular wall.
Right Ventricular Physiopathology
Hypertrophy and Dilation
In normal conditions, the RV is a complex structure with three different compartments: the filling chamber with the tricuspid valve, the apical chamber with the right ventricular free wall, and the outflow tract, near from the pulmonary valve. These three chambers play a different role in blood ejection toward the pulmonary artery (31). In PAH, the RV loses its contraction delay, which is necessary for blood diffusion through the ventricle toward the pulmonary artery. All the ventricular structure is altered, and the three-chambered structure evolves to a more cylindrical shape, which allows blood ejection despite simultaneous contraction of all compartments. Those contraction disturbances of ventricular wall can be explained by abnormalities of myocardial contractility, secondary to an alteration of muscular fibers orientation (31, 32).
In normal conditions, Kind et al. observed that the interventricular septum is made of oblique- and helical-shaped fibers, leading to a twisting contraction. In contrary, right ventricular free wall is mostly made of transverse fibers, leading to a circumferential contraction. In PAH, septal fibers orientation becomes transverse, which promotes a circumferential contraction, observed in the right ventricular remodeling (33).
When the afterload increases because of PAP elevation, RV hypertrophy maintains a normal RV ejection fraction (RVEF) and diminishes wall stress (34, 35). However, even if this mechanism first preserves cardiac function, it leads to diastolic dysfunction, ischemia, and dilation of the RV and is, furthermore, not sufficient to preserve cardiac function. Indeed, after pressure overload occurring in the beginning of PAH, a volume overload appears, probably secondary to tricuspid regurgitation, although all mechanisms are not well known yet. This dilation is mainly responsible of dysfunction of the RV (36–38). According to Archer et al., we can still differentiate two different phenotypes, depending on cardiac adaptation to pressure elevation: the adaptive phenotype, where the RV is less dilated and fibrotic, the RVEF is preserved and remodeling is concentric, and the maladaptive phenotype, where the RV is dilated, fibrotic, hypokinetic, and remodeling is eccentric (24). Little is known about the mechanisms leading to one or the other phenotype, but adaptive phenotype is rather observed in children with Eisenmenger syndrome, while maladaptive phenotype is seen in other types of PAH. There is also probably a continuum between these two patterns, as the elevated PAP becomes greater and perpetual (34). Indeed, studies showed a good correlation between cardiac remodeling and function, and pulmonary vascular abnormalities and hypertrophy. This has led to the conception of RV and pulmonary circulation as a unit, where one component cannot be considered alone, in order to get a better comprehension of their pathophysiology and relations (39–41).
Histology
It is well known that, following exposition to elevated PAP, the RV becomes hypertrophic. Histological examination of PAH hearts has demonstrated that the myocytes diameter is increased, in comparison with normal hearts. It seems that increased right ventricular mass is also secondary to cytoplasmic proteins and sarcomeres production (41, 42). Moreover, it has been demonstrated that the passive tension of sarcomeres is increased, by reduced phosphorylation of titin, which usually regulates tension of sarcomeres. This induces an impaired relaxation of myocytes. In parallel, deposition of collagen in the extracellular matrix induces fibrosis of the myocardium. Both impaired relaxation of myocytes and collagen deposition increase rigidity of the RV and induce a diastolic insufficiency, which could participate to the right ventricular dysfunction (37). Even if the myocardium has limited regenerative and proliferative properties, it seems that hyperplasia, by proliferation of progenitor cells, add to hypertrophy. After hypertrophy has occurred, these changes of structure and function of myocytes, followed by increased apoptosis, seem finally to lead to contractile dysfunction and dilation of the RV (38, 41, 42). Pathways determining those changes are not well known yet. However, it seems that they are probably triggered and maintained by inflammation, by neutrophils and ILs, oxidative stress, systemic and local neurohormonal factors, and ischemia (38, 42, 43). Despite many structural abnormalities happen, it seems that these modifications are, at least partially reversible, as it is well known that cardiac function improves after lungs transplantation or endarterectomy (42).
In the myocardium, alterations of energetic metabolism, similar to pulmonary vessels, participate to right ventricular hypertrophy and dysfunction. They lead to decreased oxygen consumption, increased lactate production, and reduced energetic reserves. Increased GLUT-1 channels and HIF-1α activation are found in PAH myocardium and seem to be associated with greater right ventricular dysfunction (24, 26, 30).
Septum and Left Ventricle
Interventricular septum acts like a passive membrane during diastole and systole, whose position depends on blood volume and pressure in the left ventricle (LV) and the RV. In PAH, when the right ventricular pressure becomes greater than the left ventricular pressure, septum is moved leftward, according to pressure gradient. We talk about abnormal eccentricity when the septum is moved leftward during systole (44, 45). The septum plays an important role in both ventricular contraction and even more in the RV in PAH. Indeed, according to López-Candales, it seems to be the “ventricular motor” in the right ventricular contraction against elevated afterload, which means that it provides most of the strength needed for blood ejection. Septal deviation is thus involved in progression of right ventricular dysfunction (45).
Right heart dysfunction, by several mechanisms, can also lead to a left cardiac dysfunction, related to interdependence of both ventricles. Septal abnormal eccentricity, as previously described, induces an abnormal left ventricular morphology. Furthermore, as the pericardium is not much extensible, the hypertrophied RV can compress the LV and diminish its volume. This results in left cardiac dysfunction, characterized by a reduced left ventricular filling, responsible for a diminished ejection volume (34, 45).
Finally, in PAH, we can find a dyssynchrony between both ventricles: right ventricular free wall is still contracting when the diastole has begun in the LV, which accentuates septal deviation and shortens diastole in the RV. Thus, filling is also reduced in the RV (32, 34, 46).
Blood Flow
Cardiac perfusion has been mostly studied in left cardiac hypertrophy. In the left coronary artery, in normal conditions, blood flow depends on arterial pressure, or aortic pressure, myocardial capillary pressure, determined by several neurohumoral and metabolic factors, and left ventricular pressure (47). In normal conditions, blood flow in the right coronary artery does not depend on ventricular contraction. It is a monophasic flow, which means that systolic flow is equal to diastolic flow. In contrary, in PAH, van Wolferen et al. demonstrated that the right coronary blood flow was diminished during systole, and increased during diastole. First, blood flow becomes biphasic, as seen in the left coronary artery in normal conditions, and thus depends on contraction and right ventricular systolic pressure (48). Second, the systolic coronary blood flow per mass unit decreases parallel to the progression of right ventricular hypertrophy, because of microvascular rarefaction compared to ventricular mass and by vessels compression during systole. This can explain the presence of sub-endocardial ischemia in severe right ventricular hypertrophy, as it is found in left ventricular hypertrophy (24, 38).
Clinical Parameters
Cardiopulmonary Exercise Test
Cardiopulmonary exercise test (CPET) has been widely used to assess exercise capacity in PAH patients. The patient undergoes ergometry with defined initial load, which is progressively increased until maximal symptom-limited intensity is reached. Several variables are measured: oxygen pulse which is oxygen consumption per heartbeat, maximal oxygen consumption (VO2), ventilatory equivalent for carbon dioxide (VE/VCO2), which reflects respiratory adaptation to exercise and carbon dioxide (CO2) production, heart rate, arterial oxygen saturation (SaO2), and systemic arterial tension. It has been showed that VO2 is decreased in PH and, as it reflects maximal cardiac output (CO), is a marker of impaired cardiac function. Indeed, several studies showed that decreased VO2 and increased VE/VCO2 are correlated with cardiac function. CPET seems to be a good test to evaluate functional capacity, as it is correlated with NYHA class, disease severity, clinical worsening, and survival in PAH. It is thus suitable for risk stratification. Studies also showed that it is a good test to evaluate treatment response. However, CPET is not suitable for most severe forms of PH and its realization needs technical expertise (49–52).
Six-Minute Walk Test
The six-minute walk test is a submaximal exercise test, frequently used in assessment of cardiovascular and pulmonary diseases, especially to measure the response to a medical intervention. The patient must walk during 6 min, choosing his walking velocity. He can use any walking help he usually needs and must not interrupt his medication. Dyspnea and tiredness are evaluated, before and after the test, with Borg scale. Even though the patient must provide a physical exertion, the test is well tolerated, even for severely disabled patients, because they can choose intensity and velocity of their walk and can stop at any time. Several contraindications must be taken in consideration, such as unstable angina or infarction in the past month. Incidence of complications, such as arrhythmias or cardiac arrest, is, for now, unknown, and it is thus necessary to perform this test in a place where emergency intervention is possible, where rapid administration of drugs and oxygen is feasible, and under surveillance of a physician certified in cardiopulmonary resuscitation (53).
Several studies demonstrated that it is a good, reproducible test to assess patients’ functional capacities. It represents the physical activity of daily living and thus estimates patients’ capacities in a more reliable way than with a self-reporting questionnaire. Although physical exertion is not maximal, it has been demonstrated that the results are correlated with maximal exertion tests (49). In PAH, results are strongly correlated with survival, quality of life, NYHA class, and hemodynamic parameters. For evaluation of treatment benefits, a modification of walk distance superior to 33 m is considered to be significant, even though the correlation between walk distance and survival remains controversial. However, walk distance reflects the global cardiovascular, pulmonary, and neuromuscular state and is thus not specific to the investigated disease. Despite a weaker sensitivity to assess treatment response in comparison with other exercise tests, it seems that, thanks to its good reproducibility and easy execution, it is most suitable to assess evolution of functional capacities and cardiovascular state after treatment (2, 5, 49–52, 54).
NYHA Class
New York Heart Association class is a self-assessment of dyspnea by the patient. This classification seems to be a good indicator of survival in PAH: survival is about 6 years in class I and II, 2.5 years in class III, and 6 month in class IV. Some studies showed a good correlation between NYHA class and hemodynamic parameters. Furthermore, treatment-induced changes can be easily measured and are also predictive of survival. Thus, NYHA class could be used to adapt treatment (5, 52, 55, 56). However, this classification has some limitations, such as important interobservers variability and the fact that it is a subjective and self-reported evaluation. Moreover, this classification lacks of sensitivity, as it is difficult to use it to detect precociously patients with PAH. Detection of improvement from a class II to a class I is difficult as well (52).
Equations
Eventually, in clinical practice, in order to improve patients’ classification according to their prognosis, equations combining different clinical, biological, and hemodynamic parameters have been developed. As their prognostic value relies on diagnostic and therapeutic advances, these equations must be regularly updated. The most recent equation results from the REVEAL study performed by Benza et al. It seems to allow a better risk stratification, in comparison with variables considered individually (1, 56).
Biological Parameters
N-Terminal of the Prohormone Brain Natriuretic Peptide
N-terminal of the prohormone brain natriuretic peptide (NT-proBNP) is a peptide released by myocardium when ventricular wall stress is increased by volume overload. It may be increased by any left or right cardiac pathology leading to reduced cardiac function, and its rate reflects changes of ventricular wall stress. In PAH, it has been demonstrated that a higher rate is associated with reduced survival (52, 54, 56–58). Indeed, Mauritz et al. showed that values above 1.256 pg/ml are associated with a worse prognosis. NT-proBNP rates are correlated with hemodynamic parameters, and negatively correlated with CO and RVEF (58–61). NT-proBNP was also an indicator of a long-term treatment response (61). Park et al. showed that decreased NT-proBNP rates after 3 months of treatment with epoprostenol were associated with a higher survival, particularly if they decreased more than 50% from baseline. Thus, it is a sensitive marker of right ventricular dysfunction’s severity and could allow an early assessment of treatment response (62). NT-proBNP is exclusively cleared by kidneys, and its rates can thus be overestimated in chronic renal impairment, frequently found in PAH. In this case, it seems that the correlation between NT-proBNP rates and hemodynamic parameters could be weaker (57, 58).
Diffusion Capacity for Carbon Monoxide
Diffusion capacity for carbon monoxide (DLCO) is used in a large number of pulmonary diseases, because it allows non-invasive detection of pathological processes inducing perturbations in gas exchanges in pulmonary vessels. It is frequently used in systemic sclerosis to reveal the presence of PH, as its diffusion is altered by arterial wall remodeling. In this pathology, DLCO is also strongly correlated with prognosis (63). In other types of PAH, although remodeling processes are similar, usefulness of DLCO as a prognostic factor remains controversial. Chandra et al. showed that a value inferior to 40% of predicted value is associated with a worse prognosis. In another study, a reduced DLCO was associated with increased mortality, while values above 80% of predicted were associated with increased survival. In contrary, Trip et al. could not demonstrate any association between DLCO and prognosis or hemodynamic parameters. Reduced DLCO in patients with PAH is correlated with smoking, which could mean that reduced values reflect rather an underlying pulmonary pathology than a severe vascular wall injury. Furthermore, no changes in DLCO were found after treatment (1, 56, 64, 65). In clinical practice, DLCO is a part of pulmonary function tests, which is mostly used in clinical practice in initial assessment of PH to detect connective tissue disease or interstitial lung diseases. However, the other variables of pulmonary function tests have not been included in studies and no correlation has been made with prognosis in PAH.
Markers of Cardiac Function
Cardiac Catheterization
Today, right heart catheterization remains the gold standard for the diagnosis of PH. We can measure different hemodynamic parameters, such as RAP, PVR, systolic pulmonary pressure (PAPs), PAPm, diastolic PAP (PAPd), PWP, cardiac index (CI), defined as the ratio between CO and body surface area, or SaO2, and venous oxygen saturation (SvO2). As previously mentioned, it is used in clinical practice to differentiate pre- and post-capillary PH: PWP < 15 mmHg and diastolic pressure gradient (PAPd-PWP) >7 mmHg suggest pre-capillary PH (3). A lot of studies showed a good correlation between prognosis, survival and RAP, PAPm, SvO2, CO, and CI. Correlation between CO and PVR is consistent with the concept of RV-pulmonary circulation unit (1, 2, 44, 52, 56, 66–70). PVR are also strongly correlated with survival, although association between their decrease after treatment and mortality is controversial. However, Sitbon et al. showed that a decrease of 30% or more in comparison with baseline values is associated with a better prognosis (55, 71). Evolution of CO after 16 weeks of treatment is also associated with prognosis, as an increase >0.22 l/min is predictive of a better survival (69). Nickel et al. demonstrated that CO values superior to 2.5 l/min/m2, and SvO2 superior to 65%, at the time of diagnosis, are associated with increased survival. Nevertheless, it is mainly their evolution that determines the patient’s prognosis: worsening despite satisfying baseline values is associated with a higher mortality rate. In contrary, improvement despite reduced baseline values predicts a lower mortality rate. These results evidence the importance of follow-up values (72). During stress, in normal conditions, CO and CI increase. Adaptation of CI during stress could allow a better stratification of patients, as an increase is predictive of a better survival. A positive correlation also exists with six-minute walk distance, and a negative correlation with NYHA class. This illustrates that, if the heart is capable to increase its output during stress, cardiac function is still preserved (73).
However, cardiac catheterization is not suitable for patients’ long-term follow-up, as it is quite invasive. Furthermore, values are most of the time, obtained at rest and cannot allow evaluation of cardiovascular adaptation to stress.
Echocardiography
Echocardiography is used as one of the first tool to estimate the presence of PH from any group, mainly by measuring PAPs. Tricuspid gradient is estimated from peak tricuspid regurgitation flow velocity (V). Pressure differential (ΔP) between the RV and the right atrium (RA) is obtained by Bernoulli equation: ΔP = 4V2. Without pulmonary stenosis, PAPs can then be estimated by adding RAP, obtained from the diameter of the vena cava and its respiratory variation: a diameter of vena cava >21 mm with a collapse <50% with deep inspiration suggests a pathological RAP value (>15 mmHg). According to the guidelines, this value cannot, itself, make the diagnosis of PAH. Indeed, in about 50% of patients, PAPs values estimated with ultrasound do not correspond with values obtained at cardiac catheterization. Furthermore, in certain situations, Bernoulli equation cannot be applied, because pressure gradient is underestimated (3, 74). Diagnosis of PAH should be considered, with a degree of probability, in the presence of several evocating signs at echocardiography. Even if PAP values measured by cardiac catheterization are correlated with survival, there is no evidence demonstrating such association with values obtained at ultrasound. Global evaluation of right and left cardiac chambers, as well as valvular state, helps to establish the probability of a pre- or post-capillary origin for PH.
The presence of right heart hypertrophy and dilation can be assessed with two-dimensional echocardiography (36). As previously mentioned, hypertrophy, even having deleterious effects, is an adaptive mechanism allowing the RV to function despite elevated PAP. It is mostly when right ventricular dilation occurs that right ventricular dysfunction appears. Indeed, it seems that dilation of RV, assessed by echocardiography, is better correlated with survival than hypertrophy. Ghio et al. demonstrated that increased ventricular diameter is associated with higher PVR, a higher RAP, a lower CI, and a worse survival (36).
RV ejection fraction, which reflects right ventricular function, can be measured by echocardiography but remains a visual estimation and is thus unreliable. It is better to use other indicators of ventricular function, which also allows an earlier recognition of right ventricular dysfunction. We can use the tricuspid annular plane systolic excursion (TAPSE). Several studies demonstrated that a reduced TAPSE is correlated with a greater right ventricular dysfunction and a worse survival (35, 70, 75, 76). Forfia et al. determined a cut-off of 1.8 cm below which is predicted a worse prognosis, and greater cardiac remodeling and dysfunction (76). TAPSE only illustrates longitudinal shortening of the RV. It seems that, during progression of right ventricular dysfunction, TAPSE reaches a limit and does not diminish further, even if right ventricular function and ejection fraction (EF) are still worsening. In contrary, transverse systolic shortening, measured by cardiac magnetic resonance imagery (cMRI) or echocardiography, illustrates right free wall and septal motion and is correlated with EF all along the progression of dysfunction. Even though free wall deviation also reaches a limit, septal displacement progresses with dysfunction. A reduced transverse shortening is thus mainly due septal deviation (33, 77). We can combine longitudinal and transverse shortening in one parameter: the right ventricular fractional area change (RVFAC), assessed by cMRI or two-dimensional echocardiography. We calculate RVFAC values through right ventricular end-diastolic and end-systolic areas like this: RVFAC = [(RV end-diastolic area − RV end-systolic area)/RV end-diastolic area]. RVFAC is correlated with RVEF and its evolution through the time and could thus be used as a marker of right ventricular function and its evolution. However, its assessment by two-dimensional echocardiography remains difficult and less sensitive than by cMRI (77, 78).
Septal position can also be assessed by the eccentricity index (EI), described first by Ryan et al., which seems to be a marker of both volume and pressure overload in the RV. It is calculated as the ratio between anteroposterior and septo-lateral diameters of the LV, on a short axis view in two-dimensional echocardiography. (Figure 2) (79). Several studies demonstrated that an increased EI, by septal displacement, is correlated with elevated RAP and PAP and is associated with a bad prognosis and a worse survival. The EI seems to be a very good indicator of right ventricular dysfunction and could thus be used as a follow-up tool (44, 45, 80). Moreover, Ghio et al. showed that association of TAPSE and EI allows detection of patients with global, diastolic and systolic dysfunction of the RV (80).
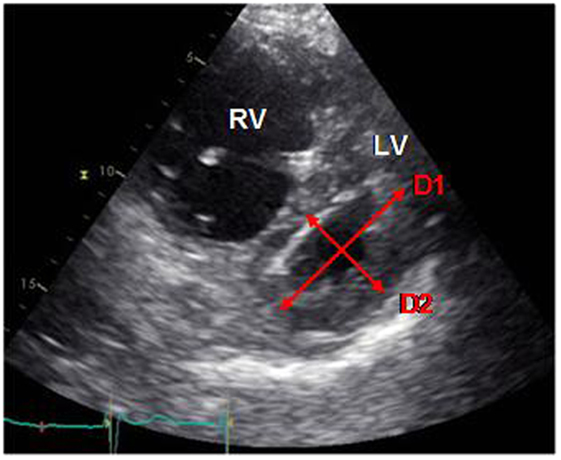
Figure 2. Measure of the EI (D1/D2) on a short axis view. D1, anteroposterior diameter; D2, septo-lateral diameter (own unpublished data).
Several studies agree about pericardial effusion to be an excellent predictive factor of mortality (35, 44, 66, 70). As it is due to lymphatic and venous drainage obstruction in the RA, secondary to elevated pressure in this one, it reflects diastolic dysfunction (35, 70).
Tei myocardial index is defined as the sum of isovolumetric contraction and relaxation, divided by ejection time. It is a good indicator of global right ventricular function and patient’s prognosis which is, furthermore, little influenced by ventricle geometry, tricuspid regurgitation, and right ventricular filling state. It is reproducible and easily calculated, by pulsed or tissular Doppler, from pulmonary and tricuspid valves. Increased Tei index is associated with a major right ventricular dysfunction (35, 70, 74).
Cardiac Magnetic Resonance Imaging
Cardiac magnetic resonance imaging is frequently used in PH to assess morphology and function of the RV. It is not the first tool used to diagnose PH, even if PH should be considered in presence of vascular late gadolinium enhancement and reduced pulmonary arterial distensibility (3). Quite the same morphological and functional parameters as echocardiography are studied with cMRI, i.e., right ventricular diameter and morphology, RVEF, RVFAC, and CO. cMRI can assess more precisely and more easily the complex three-dimensional shape of the RV as well as the RVEF. No surrogates to RVEF are needed, and thus it may be preferred to echocardiography. Moreover, it has been demonstrated that right ventricular diameter and RVEF are correlated with survival (30, 35, 39). It has also a better reproducibility than echocardiography and can be used to assess response to therapies (35, 39, 52). It is also possible to measure blood flow in coronary arteries to detect ischemic abnormalities, which participate to right ventricular dysfunction (52). Right ventricular mass can be easily measured and seems to be correlated with right ventricular function and survival (35, 39). Magnetic resonance angiography can be done simultaneously and can provide information about pulmonary vasculature. Indeed, vascular remodeling can be seen by loss of peripheral vascular enhancement and ramifications of vascular bed. Therefore, MRI allows evaluation of all RV-pulmonary circulation unit (39). However, despite major advantages, fewer studies have been done about usefulness of cMRI in PH evaluation in comparison with echocardiography, and some correlations with other clinical parameters are lacking. cMRI is also expensive and less available than echocardiography and can be limited by claustrophobia and ferromagnetic devices (35).
Conclusion
In conclusion, in PAH, a lot of markers of right ventricular function have been developed (Table 1). However, their predictive values remain, for most, to be determined. It is recommended to use the best studied markers, such as anatomical parameters, pericardial effusion, or TAPSE (70, 74).
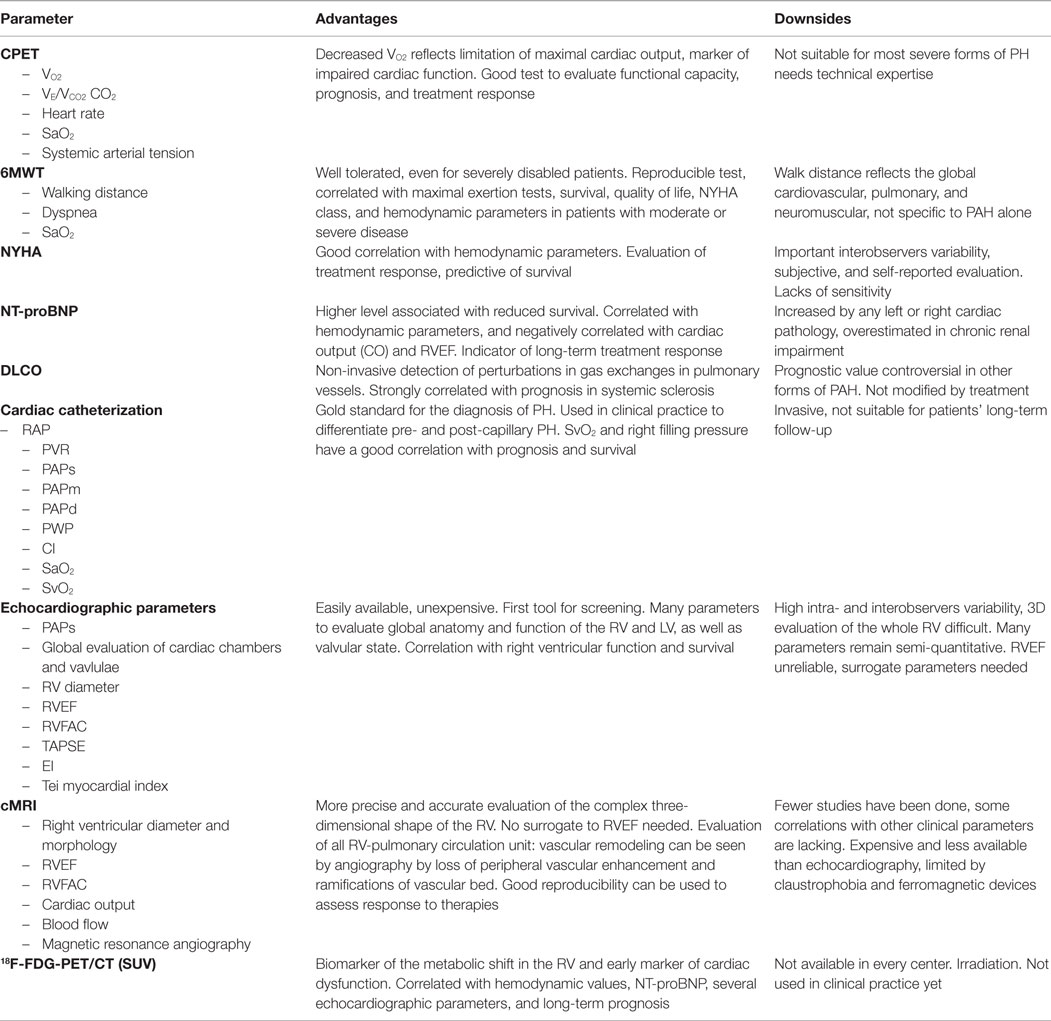
Table 1. Advantages and downsides of parameters for evaluation of PH.
Positron Emission Tomography/Computed Tomography
18F-FDG Uptake and Energetic Metabolism
It is possible to evidence these metabolic abnormalities, in rats and in humans, with 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography/computed tomography (PET/CT), both in myocardium and in pulmonary vessels (Figure 3) (25–27, 81). We usually describe 18F-FDG uptake by the standard uptake value (SUV), an index measured as: SUV = tracer concentration in a given place/(injected activity × body mass). In experimental studies, it has been shown that 18F-FDG uptake in PAH is secondary to increased GLUT-1 channels expression and anaerobic glycolysis activation (25, 81–83). In the myocardium, increased 18F-FDG uptake is also due to a greater cardiac work or myocardial stress, which is the strength that the heart must provide to eject a blood volume against elevated pulmonary pressure. It can also be due to ischemia present in hypertrophy, which can switch the metabolism toward anaerobic glycolysis (81, 84). In contrary, it has been demonstrated that increased uptake cannot be explained by hypertrophy itself or by increased right ventricular free wall thickness (81).
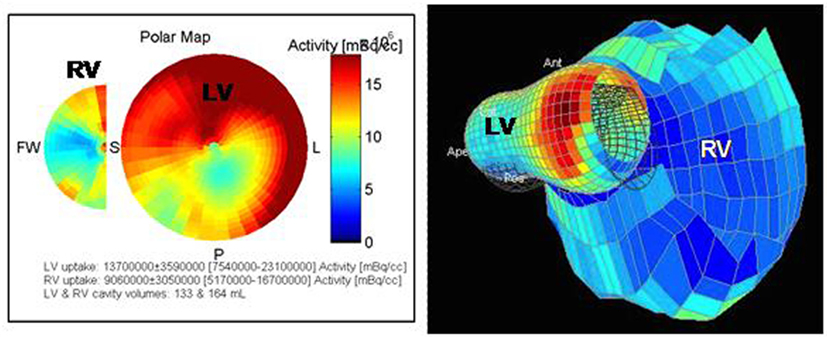
Figure 3. Pictures obtained from a 67-year-old man with PH of mixed origin (COPD, left heart dysfunction). 18F-FDG-PET/CT shows a dilation of the RV with an increased and heterogeneous uptake. A hypometabolism of the infero-lateral medial wall is found in the LV (own unpublished data).
Severity and Prognosis
A higher mortality rate has been observed when free fatty acids uptake was reduced in favor of glycolysis. Indeed, Tatebe et al. demonstrated that the SUV of the right ventricular free wall is correlated with long-term prognosis in patients with PAH, with a threshold of 8.3, below which survival is decreased. SUV could thus be useful as a biomarker of this metabolic shift and thus, as an early marker of cardiac dysfunction (25, 26, 30, 83). It could allow a better stratification of patients according to the severity of the disease and be used to define best their prognosis. Several studies evidenced that 18F-FDG uptake ratio between the RV and the LV is correlated with PAP (30). Kluge et al. demonstrated that RVEF and Tei index are correlated with uptake ratio, by reduced uptake in the LV. This could be explained by decreased energetic needs in the LV because of diminished parietal stress and pre-load (84). In contrary, other studies also showed the same correlation, but explained by increased uptake in the RV (82, 85). 18F-FDG uptake is thus correlated not only with hemodynamic values but also with NT-proBNP (85). Lundgrin et al. showed that the right myocardial SUV is correlated with several echocardiographic parameters, such as the right ventricular end-systolic diameter, the EI, and the RFVAC (26). A recent study performed by Li et al. confirmed the relationship between RV/LV 18F-FDG uptake ratio and cardiac dysfunction, as an increased uptake was an independent predictive factor of a long-term poor prognosis and mortality. Thus, PET/CT seems to have a prognostic value in assessment of right ventricular function (86).
Oikawa et al. showed that, in patients responding to treatment with epoprostenol, the decrease of right ventricular SUV was correlated with changes of PVR, PAPm, and parietal stress of the RV (81). 18F-FDG-PET/CT could also be a useful tool to assess treatment response.
82Rb-PET/CT
Studies about left ventricular hypertrophy showed that the PET/CT with rubidium-82 (82Rb), associated with a stress test and a cold pressor test, is a sensitive and non-invasive exam that can be used to evidence the presence of endothelial dysfunction and perfusion abnormalities in coronary arteries (87–90). However, it has never been used in the context of PH. Endothelial dysfunction has been demonstrated to be a part of the pathogenesis of PAH in pulmonary arteries, but only a few studies evidenced its presence in the systemic and coronary circulations (91–93). In the future, 82Rb-PET/CT could be used to study perfusion abnormalities of the RV and to better understand right ventricular dysfunction.
Conclusion
Despite major discoveries have been made these past years concerning the mechanisms underlying the development of PAH, mortality rates remain high and available treatments cannot reverse vessels wall injuries. Other treatments targeted on the pathogenesis are being studied. It will be necessary, in the coming years, to develop their clinical utilization in order to improve prognosis of patients with PAH.
Right ventricular dysfunction, mostly due to right ventricular dilation, is the main cause of death. More and more markers of right ventricular function have been studied, but clinical practices disagree concerning parameters that should be used. Currently, hemodynamic values, obtained by cardiac catheterization, remain decisive to establish, not only the diagnosis but also the prognosis, and to follow the evolution of the disease. It is recommended to associate biological, hemodynamic, and imagery parameters. However, it will be necessary to standardize patients’ care and to determine prognostic values of those parameters, in order to optimize follow-up and allow a better monitoring of right heart function.
Finally, nuclear cardiac imaging has been recently used to assess cardiac repercussions of PAH. Number of evidences demonstrated that metabolic abnormalities are present in the right myocardium and that they can be observed by 18F-FDG-PET/CT. Their participation in right heart dysfunction has also been highlighted. Even though these first results showed a good sensitivity to assess ventricular metabolism and function, further studies are necessary to confirm its usefulness and its adequacy for clinical patients’ follow-up.
Author Contributions
This article has been written mainly by CS, under supervision of Prof. LN, JP, and J-DA. Dr. PY and Dr. PM helped with information collection.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med (1991) 115(5):343–9. doi:10.7326/0003-4819-115-5-343
2. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation (2010) 122(2):156–63. doi:10.1161/CIRCULATIONAHA.109.911818
3. Galiè N, Humbert M, Vachiery J-L, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J (2016) 37(1):67–119. doi:10.1093/eurheartj/ehv317
4. Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol (2013) 62(25):D34–41. doi:10.1016/j.jacc.2013.10.029
5. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med (2006) 173(9):1023–30. doi:10.1164/rccm.200510-1668OC
6. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest (2012) 122(12):4306–13. doi:10.1172/JCI60658
7. Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol (2004) 43(12 Suppl S):13S–24S. doi:10.1016/j.jacc.2004.02.029
8. Archer S, Rich S. Primary pulmonary hypertension: a vascular biology and translational research “work in progress”. Circulation (2000) 102(22):2781–91. doi:10.1161/01.CIR.102.22.2781
9. Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol (2009) 54(1 Suppl):S20–31. doi:10.1016/j.jacc.2009.04.018
10. Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol (2009) 54(1 Suppl):S10–9. doi:10.1016/j.jacc.2009.04.006
11. Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J (2005) 19(9):1178–80.
12. Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation (2004) 109(2):159–65. doi:10.1161/01.CIR.0000102381.57477.50
13. Klinger JR, Abman SH, Gladwin MT. Nitric oxide deficiency and endothelial dysfunction in pulmonary arterial hypertension. Am J Respir Crit Care Med (2013) 188(6):639–46. doi:10.1164/rccm.201304-0686PP
14. Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med (1995) 333(4):214–21. doi:10.1056/NEJM199507273330403
15. McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, et al. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest (2005) 115(6):1479–91. doi:10.1172/JCI23203
16. Jasińska-Stroschein M, Orszulak-Michalak D. The current approach into signaling pathways in pulmonary arterial hypertension and their implication in novel therapeutic strategies. Pharmacol Rep (2014) 66(4):552–64. doi:10.1016/j.pharep.2014.04.001
17. Huertas A, Perros F, Tu L, Cohen-Kaminsky S, Montani D, Dorfmüller P, et al. Immune dysregulation and endothelial dysfunction in pulmonary arterial hypertension: a complex interplay. Circulation (2014) 129(12):1332–40. doi:10.1161/CIRCULATIONAHA.113.004555
18. Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation (2010) 122(9):920–7. doi:10.1161/CIRCULATIONAHA.109.933762
19. Arends SJ, Damoiseaux J, Duijvestijn A, Debrus-Palmans L, Boomars K, Broers B, et al. Prevalence of anti-endothelial cell antibodies in idiopathic pulmonary arterial hypertension. Eur Respir J (2010) 35(4):923–5. doi:10.1183/09031936.00164209
20. Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, et al. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation (2001) 104(7):790–5. doi:10.1161/hc3201.094152
21. Grant JS, White K, MacLean MR, Baker AH. microRNAs in pulmonary arterial remodeling. Cell Mol Life Sci (2013) 70(23):4479–94. doi:10.1007/s00018-013-1382-5
22. Potus F, Ruffenach G, Dahou A, Thebault C, Breuils-Bonnet S, Tremblay È, et al. Downregulation of microRNA-126 contributes to the failing right ventricle in pulmonary arterial hypertension. Circulation (2015) 132(10):932–43. doi:10.1161/CIRCULATIONAHA.115.016382
23. Drake JI, Bogaard HJ, Mizuno S, Clifton B, Xie B, Gao Y, et al. Molecular signature of a right heart failure program in chronic severe pulmonary hypertension. Am J Respir Cell Mol Biol (2011) 45(6):1239–47. doi:10.1165/rcmb.2010-0412OC
24. Archer SL, Fang Y-H, Ryan JJ, Piao L. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulm Circ (2013) 3(1):144–52. doi:10.4103/2045-8932.109960
25. Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, Morrow E, et al. Lung 18F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med (2012) 185(6):670–9. doi:10.1164/rccm.201108-1562OC
26. Lundgrin EL, Park MM, Sharp J, Tang WHW, Thomas JD, Asosingh K, et al. Fasting 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann Am Thorac Soc (2013) 10(1):1–9. doi:10.1513/AnnalsATS.201206-029OC
27. Zhao L, Ashek A, Wang L, Fang W, Dabral S, Dubois O, et al. Heterogeneity in lung (18)FDG uptake in pulmonary arterial hypertension: potential of dynamic (18)FDG positron emission tomography with kinetic analysis as a bridging biomarker for pulmonary vascular remodeling targeted treatments. Circulation (2013) 128(11):1214–24. doi:10.1161/CIRCULATIONAHA.113.004136
28. Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A (2007) 104(4):1342–7. doi:10.1073/pnas.0605080104
29. Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation (2006) 113(22):2630–41. doi:10.1161/CIRCULATIONAHA.105.609008
30. Bokhari S, Raina A, Rosenweig EB, Schulze PC, Bokhari J, Einstein AJ, et al. PET imaging may provide a novel biomarker and understanding of right ventricular dysfunction in patients with idiopathic pulmonary arterial hypertension. Circ Cardiovasc Imaging (2011) 4(6):641–7. doi:10.1161/CIRCIMAGING.110.963207
31. Calcutteea A, Chung R, Lindqvist P, Hodson M, Henein MY. Differential right ventricular regional function and the effect of pulmonary hypertension: three-dimensional echo study. Heart (2011) 97(12):1004–11. doi:10.1136/hrt.2010.208900
32. Calcutteea A, Lindqvist P, Soderberg S, Henein MY. Global and regional right ventricular dysfunction in pulmonary hypertension. Echocardiography (2014) 31(2):164–71. doi:10.1111/echo.12309
33. Kind T, Mauritz G-J, Marcus JT, van de Veerdonk M, Westerhof N, Vonk-Noordegraaf A. Right ventricular ejection fraction is better reflected by transverse rather than longitudinal wall motion in pulmonary hypertension. J Cardiovasc Magn Reson (2010) 12:35. doi:10.1186/1532-429X-12-35
34. Vonk-Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J, et al. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol (2013) 62(25 Suppl):D22–33. doi:10.1016/j.jacc.2013.10.027
35. Vonk Noordegraaf A, Galiè N. The role of the right ventricle in pulmonary arterial hypertension. Eur Respir Rev (2011) 20(122):243–53. doi:10.1183/09059180.00006511
36. Ghio S, Pazzano AS, Klersy C, Scelsi L, Raineri C, Camporotondo R, et al. Clinical and prognostic relevance of echocardiographic evaluation of right ventricular geometry in patients with idiopathic pulmonary arterial hypertension. Am J Cardiol (2011) 107(4):628–32. doi:10.1016/j.amjcard.2010.10.027
37. Rain S, Handoko ML, Trip P, Gan CT-J, Westerhof N, Stienen GJ, et al. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation (2013) 128(18):2016–25. doi:10.1161/CIRCULATIONAHA.113.001873
38. Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, et al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation (2009) 120(20):1951–60. doi:10.1161/CIRCULATIONAHA.109.883843
39. Champion HC, Michelakis ED, Hassoun PM. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit: state of the art and clinical and research implications. Circulation (2009) 120(11):992–1007. doi:10.1161/CIRCULATIONAHA.106.674028
40. Guihaire J, Haddad F, Boulate D, Decante B, Denault AY, Wu J, et al. Non-invasive indices of right ventricular function are markers of ventricular-arterial coupling rather than ventricular contractility: insights from a porcine model of chronic pressure overload. Eur Heart J Cardiovasc Imaging (2013) 14(12):1140–9. doi:10.1093/ehjci/jet092
41. Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, et al. Right ventricular function and failure: report of a national heart, lung, and blood institute working group on cellular and molecular mechanisms of right heart failure. Circulation (2006) 114(17):1883–91. doi:10.1161/CIRCULATIONAHA.106.632208
42. Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure. Chest (2009) 135(3):794–804. doi:10.1378/chest.08-0492
43. Guihaire J, Noly PE, Schrepfer S, Mercier O. Advancing knowledge of right ventricular pathophysiology in chronic pressure overload: insights from experimental studies. Arch Cardiovasc Dis (2015) 108(10):519–29. doi:10.1016/j.acvd.2015.05.008
44. Raymond RJ, Hinderliter AL, Willis PW, Ralph D, Caldwell EJ, Williams W, et al. Echocardiographic predictors of adverse outcomes in primary pulmonary hypertension. J Am Coll Cardiol (2002) 39(7):1214–9. doi:10.1016/S0735-1097(02)01744-8
45. López-Candales A. Determinants of an abnormal septal curvature in chronic pulmonary hypertension. Echocardiography (2015) 32(1):49–55.
46. López-Candales A, Dohi K, Rajagopalan N, Suffoletto M, Murali S, Gorcsan J, et al. Right ventricular dyssynchrony in patients with pulmonary hypertension is associated with disease severity and functional class. Cardiovasc Ultrasound (2005) 3:23. doi:10.1186/1476-7120-3-23
47. Knaapen P, Germans T, Camici PG, Rimoldi OE, ten Cate FJ, ten Berg JM, et al. Determinants of coronary microvascular dysfunction in symptomatic hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol (2008) 294(2):H986–93. doi:10.1152/ajpheart.00233.2007
48. van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KMJ, Bronzwaer JGF, et al. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J (2008) 29(1):120–7. doi:10.1093/eurheartj/ehm567
49. Miyamoto S, Nagaya N, Satoh T, Kyotani S, Sakamaki F, Fujita M, et al. Clinical correlates and prognostic significance of six-minute walk test in patients with primary pulmonary hypertension. Comparison with cardiopulmonary exercise testing. Am J Respir Crit Care Med (2000) 161(2 Pt 1):487–92. doi:10.1164/ajrccm.161.2.9906015
50. Deboeck G, Scoditti C, Huez S, Vachiéry J-L, Lamotte M, Sharples L, et al. Exercise testing to predict outcome in idiopathic versus associated pulmonary arterial hypertension. Eur Respir J (2012) 40(6):1410–9. doi:10.1183/09031936.00217911
51. Mainguy V, Malenfant S, Neyron A-S, Bonnet S, Maltais F, Saey D, et al. Repeatability and responsiveness of exercise tests in pulmonary arterial hypertension. Eur Respir J (2013) 42(2):425–34. doi:10.1183/09031936.00107012
52. Hassoun PM, Nikkho S, Rosenzweig EB, Moreschi G, Lawrence J, Teeter J, et al. Updating clinical endpoint definitions. Pulm Circ (2013) 3(1):206–16. doi:10.4103/2045-8932.109920
53. ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med (2002) 166(1):111–7. doi:10.1164/ajrccm.166.1.at1102
54. Fritz JS, Blair C, Oudiz RJ, Dufton C, Olschewski H, Despain D, et al. Baseline and follow-up 6-min walk distance and brain natriuretic peptide predict 2-year mortality in pulmonary arterial hypertension. Chest (2013) 143(2):315–23. doi:10.1378/chest.12-0270
55. Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Hervé P, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol (2002) 40(4):780–8. doi:10.1016/S0735-1097(02)02012-0
56. Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL). Circulation (2010) 122(2):164–72. doi:10.1161/CIRCULATIONAHA.109.898122
57. Sztrymf B, Souza R, Bertoletti L, Jaïs X, Sitbon O, Price LC, et al. Prognostic factors of acute heart failure in patients with pulmonary arterial hypertension. Eur Respir J (2010) 35(6):1286–93. doi:10.1183/09031936.00070209
58. Leuchte HH, El Nounou M, Tuerpe JC, Hartmann B, Baumgartner RA, Vogeser M, et al. N-terminal pro-brain natriuretic peptide and renal insufficiency as predictors of mortality in pulmonary hypertension. Chest (2007) 131(2):402–9. doi:10.1378/chest.06-1758
59. Mauritz G-J, Rizopoulos D, Groepenhoff H, Tiede H, Felix J, Eilers P, et al. Usefulness of serial N-terminal pro-B-type natriuretic peptide measurements for determining prognosis in patients with pulmonary arterial hypertension. Am J Cardiol (2011) 108(11):1645–50. doi:10.1016/j.amjcard.2011.07.025
60. Souza R, Bogossian HB, Humbert M, Jardim C, Rabelo R, Amato MBP, et al. N-terminal-pro-brain natriuretic peptide as a haemodynamic marker in idiopathic pulmonary arterial hypertension. Eur Respir J (2005) 25(3):509–13. doi:10.1183/09031936.05.00100504
61. Nagaya N, Nishikimi T, Okano Y, Uematsu M, Satoh T, Kyotani S, et al. Plasma brain natriuretic peptide levels increase in proportion to the extent of right ventricular dysfunction in pulmonary hypertension. J Am Coll Cardiol (1998) 31(1):202–8. doi:10.1016/S0735-1097(98)81321-1
62. Park MH, Scott RL, Uber PA, Ventura HO, Mehra MR. Usefulness of B-type natriuretic peptide as a predictor of treatment outcome in pulmonary arterial hypertension. Congest Heart Fail (2004) 10(5):221–5. doi:10.1111/j.1527-5299.2004.03881.x
63. Kawut SM, Taichman DB, Archer-Chicko CL, Palevsky HI, Kimmel SE. Hemodynamics and survival in patients with pulmonary arterial hypertension related to systemic sclerosis. Chest (2003) 123(2):344–50. doi:10.1378/chest.123.2.344
64. Chandra S, Shah SJ, Thenappan T, Archer SL, Rich S, Gomberg-Maitland M. Carbon monoxide diffusing capacity and mortality in pulmonary arterial hypertension. J Heart Lung Transplant (2010) 29(2):181–7. doi:10.1016/j.healun.2009.07.005
65. Trip P, Nossent EJ, de Man FS, van den Berk IAH, Boonstra A, Groepenhoff H, et al. Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: patient characteristics and treatment responses. Eur Respir J (2013) 42(6):1575–85. doi:10.1183/09031936.00184412
66. Eysmann SB, Palevsky HI, Reichek N, Hackney K, Douglas PS. Two-dimensional and Doppler-echocardiographic and cardiac catheterization correlates of survival in primary pulmonary hypertension. Circulation (1989) 80(2):353–60. doi:10.1161/01.CIR.80.2.353
67. Rich S, Levy PS. Characteristics of surviving and nonsurviving patients with primary pulmonary hypertension. Am J Med (1984) 76(4):573–8. doi:10.1016/0002-9343(84)90279-1
68. McLaughlin VV, Presberg KW, Doyle RL, Abman SH, McCrory DC, Fortin T, et al. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest (2004) 126(1 Suppl):78S–92S. doi:10.1378/chest.126.1_suppl.78S
69. Tiede H, Sommer N, Milger K, Voswinckel R, Bandorski D, Schermuly RT, et al. Short-term improvement in pulmonary hemodynamics is strongly predictive of long-term survival in patients with pulmonary arterial hypertension. Pulm Circ (2013) 3(3):523–32. doi:10.1086/674338
70. Howard LS. Prognostic factors in pulmonary arterial hypertension: assessing the course of the disease. Eur Respir Rev (2011) 20(122):236–42. doi:10.1183/09059180.00006711
71. Van de Veerdonk MC, Kind T, Marcus JT, Mauritz G-J, Heymans MW, Bogaard H-J, et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol (2011) 58(24):2511–9. doi:10.1016/j.jacc.2011.06.068
72. Nickel N, Golpon H, Greer M, Knudsen L, Olsson K, Westerkamp V, et al. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J (2012) 39(3):589–96. doi:10.1183/09031936.00092311
73. Chaouat A, Sitbon O, Mercy M, Ponçot-Mongars R, Provencher S, Guillaumot A, et al. Prognostic value of exercise pulmonary haemodynamics in pulmonary arterial hypertension. Eur Respir J (2014) 44(3):704–13. doi:10.1183/09031936.00153613
74. Howard LS, Grapsa J, Dawson D, Bellamy M, Chambers JB, Masani ND, et al. Echocardiographic assessment of pulmonary hypertension: standard operating procedure. Eur Respir Rev (2012) 21(125):239–48. doi:10.1183/09059180.00003912
75. López-Candales A, Dohi K, Rajagopalan N, Edelman K, Gulyasy B, Bazaz R. Defining normal variables of right ventricular size and function in pulmonary hypertension: an echocardiographic study. Postgrad Med J (2008) 84(987):40–5. doi:10.1136/pgmj.2007.059642
76. Forfia PR, Fisher MR, Mathai SC, Housten-Harris T, Hemnes AR, Borlaug BA, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med (2006) 174(9):1034–41. doi:10.1164/rccm.200604-547OC
77. Mauritz G-J, Kind T, Marcus JT, Bogaard H-J, van de Veerdonk M, Postmus PE, et al. Progressive changes in right ventricular geometric shortening and long-term survival in pulmonary arterial hypertension. Chest (2012) 141(4):935–43. doi:10.1378/chest.10-3277
78. Pica S, Ghio S, Tonti G, Camporotondo R, Turco A, Pazzano AS, et al. Analyses of longitudinal and of transverse right ventricular function provide different clinical information in patients with pulmonary hypertension. Ultrasound Med Biol (2014) 40(6):1096–103. doi:10.1016/j.ultrasmedbio.2013.12.005
79. Ryan T, Petrovic O, Dillon JC, Feigenbaum H, Conley MJ, Armstrong WF. An echocardiographic index for separation of right ventricular volume and pressure overload. J Am Coll Cardiol (1985) 5(4):918–27. doi:10.1016/S0735-1097(85)80433-2
80. Ghio S, Klersy C, Magrini G, D’Armini AM, Scelsi L, Raineri C, et al. Prognostic relevance of the echocardiographic assessment of right ventricular function in patients with idiopathic pulmonary arterial hypertension. Int J Cardiol (2010) 140(3):272–8. doi:10.1016/j.ijcard.2008.11.051
81. Oikawa M, Kagaya Y, Otani H, Sakuma M, Demachi J, Suzuki J, et al. Increased [18F]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol (2005) 45(11):1849–55. doi:10.1016/j.jacc.2005.02.065
82. Mielniczuk LM, Birnie D, Ziadi MC, deKemp RA, DaSilva JN, Burwash I, et al. Relation between right ventricular function and increased right ventricular [18F]fluorodeoxyglucose accumulation in patients with heart failure. Circ Cardiovasc Imaging (2011) 4(1):59–66. doi:10.1161/CIRCIMAGING.109.905984
83. Tatebe S, Fukumoto Y, Oikawa-Wakayama M, Sugimura K, Satoh K, Miura Y, et al. Enhanced [18F]fluorodeoxyglucose accumulation in the right ventricular free wall predicts long-term prognosis of patients with pulmonary hypertension: a preliminary observational study. Eur Heart J Cardiovasc Imaging (2014) 15(6):666–72. doi:10.1093/ehjci/jet276
84. Kluge R, Barthel H, Pankau H, Seese A, Schauer J, Wirtz H, et al. Different mechanisms for changes in glucose uptake of the right and left ventricular myocardium in pulmonary hypertension. J Nucl Med (2005) 46(1):25–31.
85. Hagan G, Southwood M, Treacy C, Ross RM, Soon E, Coulson J, et al. (18)FDG PET imaging can quantify increased cellular metabolism in pulmonary arterial hypertension: a proof-of-principle study. Pulm Circ (2011) 1(4):448–55. doi:10.4103/2045-8932.93543
86. Li W, Wang L, Xiong C-M, Yang T, Zhang Y, Gu Q, et al. The prognostic value of 18F-FDG uptake ratio between the right and left ventricles in idiopathic pulmonary arterial hypertension. Clin Nucl Med (2015) 40(11):859–63. doi:10.1097/RLU.0000000000000956
87. Lortie M, Beanlands RSB, Yoshinaga K, Klein R, Dasilva JN, DeKemp RA. Quantification of myocardial blood flow with 82Rb dynamic PET imaging. Eur J Nucl Med Mol Imaging (2007) 34(11):1765–74. doi:10.1007/s00259-007-0478-2
88. Yoshinaga K, Klein R, Tamaki N. Generator-produced rubidium-82 positron emission tomography myocardial perfusion imaging-from basic aspects to clinical applications. J Cardiol (2010) 55(2):163–73. doi:10.1016/j.jjcc.2010.01.001
89. Yoshinaga K, Manabe O, Tamaki N. Assessment of coronary endothelial function using PET. J Nucl Cardiol (2011) 18(3):486–500. doi:10.1007/s12350-011-9370-3
90. Schelbert HR. Quantification of myocardial blood flow: what is the clinical role? Cardiol Clin (2009) 27(2):277–89. doi:10.1016/j.ccl.2008.12.009
91. Gabrielli LA, Castro PF, Godoy I, Mellado R, Bourge RC, Alcaino H, et al. Systemic oxidative stress and endothelial dysfunction is associated with an attenuated acute vascular response to inhaled prostanoid in pulmonary artery hypertension patients. J Card Fail (2011) 17(12):1012–7. doi:10.1016/j.cardfail.2011.08.008
92. Peled N, Bendayan D, Shitrit D, Fox B, Yehoshua L, Kramer MR. Peripheral endothelial dysfunction in patients with pulmonary arterial hypertension. Respir Med (2008) 102(12):1791–6. doi:10.1016/j.rmed.2008.06.014
Keywords: pulmonary hypertension, right ventricle, PET/CT, echocardiography, cardiac catheterization
Citation: von Siebenthal C, Aubert J-D, Mitsakis P, Yerly P, Prior JO and Nicod LP (2016) Pulmonary Hypertension and Indicators of Right Ventricular Function. Front. Med. 3:23. doi: 10.3389/fmed.2016.00023
Received: 15 March 2016; Accepted: 10 May 2016;
Published: 03 June 2016
Edited by:
Anne Hilgendorff, Helmholtz Zentrum Muenchen, GermanyReviewed by:
Alexander Dietrich, Ludwig Maximilian University of Munich, GermanyDjuro Kosanovic, Justus-Liebig University, Germany
Copyright: © 2016 von Siebenthal, Aubert, Mitsakis, Yerly, Prior and Nicod. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurent Pierre Nicod, bGF1cmVudC5uaWNvZEBjaHV2LmNo