 Bram Verstockt
Bram Verstockt Isabelle Cleynen
Isabelle Cleynen- 1Department of Medicine and Cambridge Institute for Medical Research, University of Cambridge School of Clinical Medicine, Cambridge, UK
- 2Translational Research in Gastrointestinal Disorders (TARGID), Department of Clinical and Experimental Medicine, KU Leuven, Leuven, Belgium
- 3Laboratory of Complex Genetics, Department of Human Genetics, KU Leuven, Leuven, Belgium
Fibrostenotic strictures are an important complication in patients with Crohn’s disease (CD), very often necessitating surgery. This fibrotic process develops in a genetically susceptible individual and is influenced by an interplay with environmental, immunological, and disease-related factors. A deeper understanding of the genetic factors driving this fibrostenotic process might help to unravel the pathogenesis, and ultimately lead to development of new, anti-fibrotic therapy. Here, we review the genetic factors that have been associated with the development of fibrosis in patients with CD, as well as their potential pathophysiological mechanism(s). We also hypothesize on clinical implications, if any, and future research directions.
Introduction
Crohn’s disease (CD) is a chronic, inflammatory disease of the gastrointestinal tract, which predominantly affects the distal ileum and right colon. Although many patients express an inflammatory phenotype at diagnosis, the natural evolution of CD is commonly a progression toward a fibrostenotic or penetrating disease in 60% of patients (1, 2). The pathogenesis of CD is multifactorial, including a genetic background (3). Genome-wide association studies and meta-analysis have identified a total of 200 inflammatory bowel disease (IBD) risk loci thus far (4, 5).
Transmural inflammation as seen in CD can cause irreversible changes in the intestinal architecture, over time leading to fibrotic strictures necessitating surgery in 30–50% of patients (1, 2, 6). Recurrence of these fibrotic lesions occurs in 23–41% of CD patients, requiring additional resections (7), which in turn reinforce the recurrence rate (8). These fibrostenotic strictures are the result of a failure of physiological wound healing. The activation of mesenchymal cells by cytokines, growth factors, and other mediators released by immune cells, epithelial cells, and mesenchymal cells themselves are believed to play an important role (9–11). The development of fibrosis in CD is influenced by various genetic, environmental, immunological, and disease-related factors (12–15). So far, the relative contribution of each component in the pathogenesis is not clear. This review aims to clarify the genetic contribution in developing fibrosis in patients with CD.
Genetics and Fibrosis
There exists broad heterogeneity of published literature concerning the genetic background of fibrotic CD. Below, we give an overview of individual variants and genes that have been associated with fibrotic disease in CD (Table 1), and we hypothesize on the potential pathophysiological mechanisms.
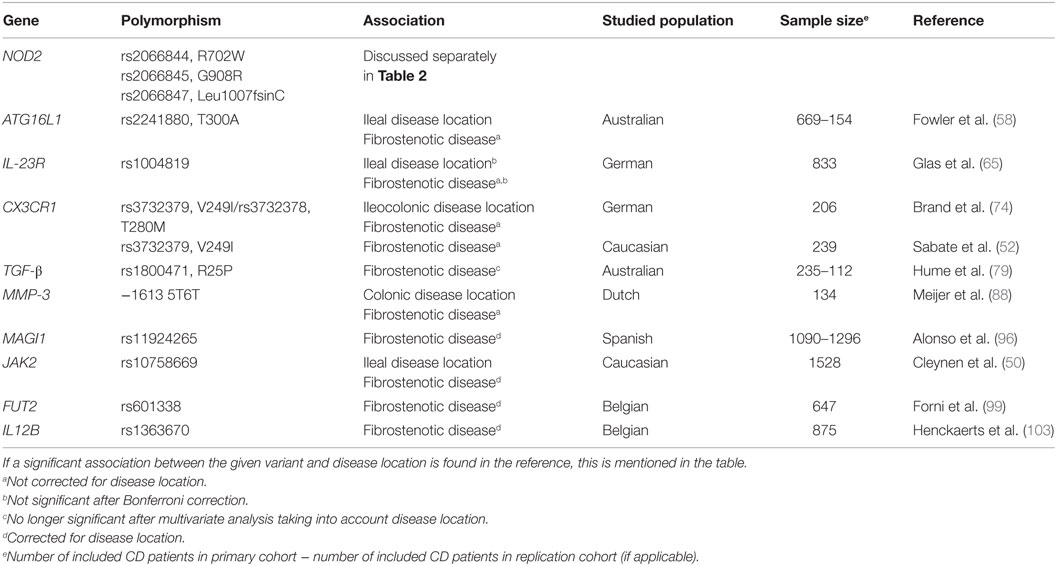
Table 1. Key gene polymorphisms and their significance in intestinal fibrosis in CD.
Nucleotide-Binding Oligomerization Domain-Containing Protein 2
The NOD2 gene is the most studied gene in relation to fibrostenotic disease in CD. Located in the IBD1 locus on chromosome 16q12, it encodes CARD15, a member of the Apaf-1/NOD1 family of CARD (caspase recruitment domain-containing protein) proteins (16, 17). NOD2/CARD15 is mainly expressed by monocytes and macrophages, where it acts as a cytosolic sensor for bacterial products, and is involved in apoptosis and activates NF-κB in response to lipopolysaccharide (LPS) binding at its leucine-rich repeating region (LRR) (18, 19). Moreover, the CARD-domain provides CARD15 the unique function to be able to induce interleukin 1-beta (IL-1β) processing and release (20). Importantly, NOD2 is also expressed in Paneth cells (21).
In the early 2000s, three NOD2 variants, including two amino acid substitutions (R702W in exon 4 and G908R in exon 8) and one frameshift mutation (Leu1007fsinC in exon 11), were identified as associated with CD (17, 22–25). Later on, several other NOD2 SNPs were found to be associated with CD, although the first three described still represent the strongest association signals. Ever since NOD2 was identified as a CD susceptibility gene, many genotype–phenotype studies were performed to find its role in defining CD disease location and behavior, but none of the three SNPs was uniformly found as an independent risk factor for developing fibrostenotic disease in CD (12, 15, 18, 19, 21, 24, 26–53). Some genotype–phenotype studies demonstrated strong associations between at least one of the three NOD2 variants and fibrostenotic disease (19, 32, 38, 52), often independent of an association with small bowel disease (18, 26, 31, 37, 50) (Table 2).
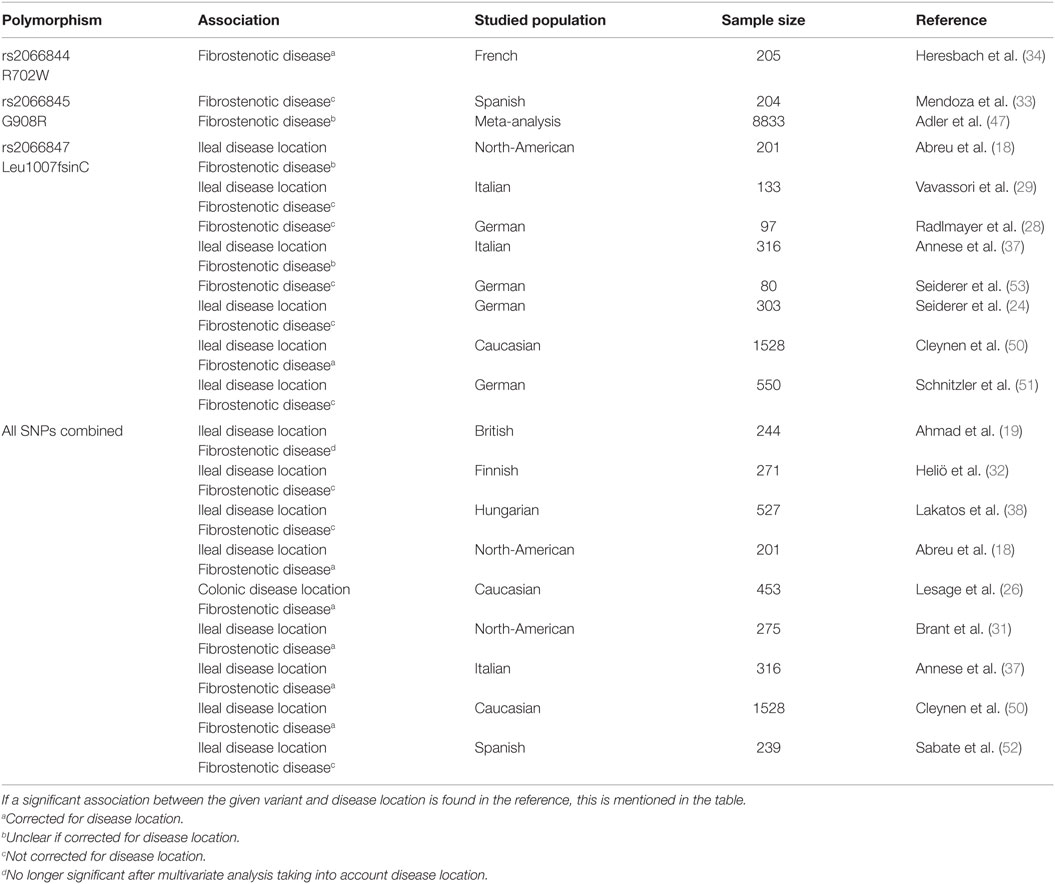
Table 2. Overview of original studies showing an association between NOD2 and fibrotic CD.
The lack of uniformity concerning this topic seems mainly based on the small sub-analyses done in the different studies (Table 2). In 2004, Heresbach et al. observed in a Northern-French population NOD2 R702W (rs2066844) as being a strong predictor of fibrostenotic disease, independently of ileal localization of the disease (34). No other group could confirm this association. An association of NOD2 G908R (rs2066845) and fibrostenotic disease was first reported in a Spanish CD cohort, although fibrostenotic disease was mainly dependent on location of disease in the terminal ileum (33). Later on, Adler et al. reported in their meta-analysis G908R as being associated with fibrostenotic disease [pooled relative risk (RR) = 1.90] (47). It is important to highlight that only 12 of all included studies in this meta-analysis had enough data to analyze individual NOD2 variants, and most included studies did not differentiate between G908R homo- and heterozygotes. Of the three NOD2 variants, the Leu1007fsinsC frameshift mutation (rs2066847) shows the strongest association with fibrostenotic disease (18, 37). The same finding was also published by Radlmayr et al., who moreover reported no association with ileal disease (28). Vavassori et al. also noticed an association between Leu1007fsinC and fibrostenotic disease, although no correction for ileal disease involvement was made (29). Seiderer et al. calculated a positive predictive value (PPV) of 80% and a negative predictive value (NPV) of 75% for the diagnosis of small bowel stenosis in clinically symptomatic patients with a Leu1007fsinC variant. Furthermore, they noticed 62% of their patients being Leu1007fsinC homo- or heterozygous needed surgery, whereas the need for surgical intervention in patients without this variant was remarkably low (53). A sub-analysis of another cohort with 19 patients, all Leu1007fsinC homozygous, identified a high-risk population, characterized by, for instance, long-segment stenosis, frequent need for surgery, and high risk for re-stenosis afterward (24). The same group confirmed these findings later on in a prospective study (53), whereafter the European IBD chip project reported the same in a retrospective study (n = 38) (50), as did Schnitzler et al. (51). Besides studying the association of individual NOD2 SNPs with a fibrostenotic CD phenotype, often the NOD2 SNPs are considered together. The pooled RR of stricturing disease with the presence of any NOD2 variant allele was 1.33 in a meta-analysis, including 35 studies by Adler et al. (47). Furthermore, Lesage et al. clearly described the “gene dosage effect” of NOD2 SNPs: patients carrying two SNPs have a higher incidence of stenosis compared to patients with one or two wild-type alleles (26), which was afterward confirmed by others (31, 40, 47). Although many groups, thus, reported an association between NOD2 variants and fibrostenotic disease, several studies could not find this association. Louis et al. found that only disease location and number of flares per year are significantly different between different CD phenotypes, and that ileal disease location was associated with a stricturing disease pattern (30). In addition, although NOD2 variants were associated with CD susceptibility in a Brazilian population, Baptista et al. could not find a genotype–phenotype correlation (42). The biggest study looking into genotype–phenotype associations in IBD to date, also did not find an association between NOD2 and fibrotic disease, when considering disease location. They conclude that while disease location is in part genetically determined, it is considered an intrinsic aspect of a patient’s clinical disease and the major driver to changes in disease behavior over time (15).
If there would be an independent association between NOD2 variants and fibrostenotic disease in CD, how could this be pathophysiologically explained? NOD2 variants might induce fibrostenotic disease by shifting T lymphocytes toward tissue growth factor beta (TGF-β) cytokine production and by increasing collagen deposition by smooth muscle cells and fibroblasts in the intestine (18). Of the three main variants, functional data are primarily available for Leu1007fsinC: Leu1007fsinC leads to a truncated CARD15 protein, resulting in an altered activation of NF-κB following bacterial triggers (23). Furthermore, it was previously thought that Leu1007fsinC was associated with an impaired IL-1β production and dendritic cell function, resulting in a dysregulation of the antibacterial host defense, increased intestinal permeability, and impaired regulation of innate and adaptive immunity in the intestinal tract (53). However, Maeda et al. later on reported Leu1007fsinC is associated with enhanced NF-κB activation and IL-1β secretion in mice (20). Additional mechanisms, such as diminished mucosal alpha-defensin expression, might also be involved (53). It is possible that the other two variants also alter the structure of LRR domain, resulting in abnormalities in bacterial recognition (35).
Autophagy-Related 16-Like 1
The ATG16L1 gene, member of a large family of genes involved in autophagocytosis, is located on chromosome 2q37. It encodes a protein in the autophagosome pathway that is essential in the targeting and destruction of pathogen-derived proteins in the innate immune response (54, 55). Furthermore, autophagy is important for degrading cytoplasmic components, sequestered within vesicles, by the lysosome (21).
After the ATG16L1 T300A variant (rs2241880) was identified as a susceptibility variant for CD (54, 56, 57), Prescott et al. were the first to associate this variant with ileal disease location, independent of NOD2 state or disease duration; they did not mention an association with stricturing disease (55). Later on, Fowler et al. reported a significant association between fibrostenotic disease, the GG risk genotype, and ileal disease, independent of NOD2 (although the number of NOD2 variants in their Australian CD population might be too small) (58). However, the European IBDchip Project could not confirm this association between ATG16L1 T300A and fibrostenotic disease (50).
The T300A amino acid substitution is a highly conserved residue that is located in the WD-repeat domain of autophagy-related 16-like 1 (ATG16L1) and which may therefore affect interactions of the protein with other components of the autophagosome (55). This variant plays an important role in pathogen clearance (59), resulting in imbalanced cytokine production (60). Moreover, presence of this ATG16L1 risk allele seems associated with a reduced ability to generate a specific type of macrophages (Mφind, phenotypically closely resembling the anti-inflammatory CD206+ M2-macrophages), also implying an impaired anti-inflammatory functioning (61). The resulting inflammatory signals could eventually stimulate mesenchymal cells to make enormous amounts of collagen and other fibrogenic molecules (62). Importantly, a link between NOD2 and ATG16L1 in the activation of autophagy could also be relevant for intestinal fibrogenesis: it is possible that NOD2 and/or ATG16L1 variants jointly can alter the responsiveness of immune cells to bacterial components, thereby amplifying inflammatory signals leading to fibrosis (62). Moreover, the ATG16L1 T300A variant enhances NOD2-driven cytokine production in an autophagy-independent manner (60, 63).
Interleukin-23 Receptor
IL-23R is found on chromosome 1p31 and encodes a subunit of the receptor for the proinflammatory cytokine interleukin-23 (64). It is highly expressed on the cell membrane of memory T cells and other immune cells, such as natural killer cells, monocytes, and dendritic cells, which identify foreign substances to defend the body against infection. Interleukin-23 receptor (IL-23R) is involved in the mediation of proinflammatory activities by the production of interleukin 17 via the activation of Th17 lymphocytes (21).
After Duerr et al. described IL-23R as a susceptibility gene to CD (64), Glas et al. published a genotype–phenotype correlation for the rs1004819 SNP within IL-23R. This group noticed an increased incidence of ileal involvement and fibrostenotic disease in TT homozygous carriers compared to CC wild-type carriers, which, both however, lost significance after Bonferroni correction (65). We did not find evidence of an association of the main CD-associated SNP in IL-23R, rs11202926 (64), with intestinal fibrosis in existing literature.
Major Histocompatibility Complex
The major histocompatibility complex (MHC) region encodes a large number of immunological proteins, including the antigen-presenting classical human leukocyte antigen (HLA) molecules. Genome-wide association studies of IBD have shown strong evidence of association with the MHC complex (66). Because of the complexity of the region, many researchers avoid including this region into their analysis. One study by Ahmad et al. studied 340 SNPs in 24 genes from the HLA region in relation with fibrotic CD, but did not find an association (19). The IIBDGC genotype–phenotype study found a genome-wide significant association with rs77005575 located in the MHC region and disease behavior, independent of disease location (15). None of the included classical HLA alleles were independently associated with disease behavior in this study.
Toll-Like Receptors
Toll-like receptors (TLRs) are transmembrane domain protein with a tripartite structure: they contain an extracellular domain (including LRRs) responsible for ligand recognition, a single transmembrane-spanning region, and a globular cytoplasmic Toll/IL-1 receptor (TIR)-signaling domain. Currently, 10 TLRs are described in humans (67). They are expressed in myeloid cells and play a major role both in detecting microbes and in initiating innate immune responses. TLR4, expressed in the Golgi apparatus of intestinal epithelial cells (IECs), interacts with LPS, contributing to the perpetuation of inflammatory epithelial cell injury via tumor necrosis factor alpha (TNF-α)-induced alterations of enterocyte turnover in an (auto)paracrine matter (38).
In 2004, Franchimont et al. identified rs4986790 (Asp299Gly), within TLR4 as a susceptibility variant to CD, regardless of association with phenotype (68). This variant is associated with decreased responsiveness to endotoxins in humans (69, 70). Lakatos et al. could not find this association in a Hungarian cohort (possibly because the variant allele is more present in their control population compared to Franchimont et al.), and they also did not find a genotype–phenotype correlation for this SNP (38). Although there is no strong evidence for a role for TLR4 in the pathogenesis of fibrostenotic disease in CD, Rieder et al. suggested the first direct link between innate immunity to bacteria (via TLRs) and fibrosis in humans (71). Furthermore, in other diseases, such as systemic sclerosis and liver fibrosis, TLR4 might have a pathophysiological contribution (72, 73).
Fractalkine Receptor 1, CX3CR1
CX3CR1 (previously termed V28) is a leukocyte chemotactic and adhesion receptor that binds fractalkine (CX3CL1 or neurotactin, expressed in epithelial and endothelial cells), a CX3C chemokine that exhibits properties of both traditional chemokines and adhesion molecules (74). CX3CR1 is expressed on natural killer cells, monocytes, CD8+, and some CD4+ T cells. By binding fractalkine, it regulates the migration of a subpopulation of CD8+ intraepithelial lymphocytes into the intestinal lamina propria and their interaction with IECs (74). After stimulation by bacteria (or bacterial degradation products), CX3CR1-expressing cells rapidly adhere to the inflamed vascular endothelium and may play a role as a vascular gateway for cytotoxic effector cells (52).
After two strongly correlated (D′ = 0.99) CX3CR1 polymorphisms (V249I, rs3732379; T280M, rs3732378) were identified in HIV-positive patients (75), Brand et al. investigated these SNPs in the context of CD. They observed an association between both SNPs and fibrostenotic disease (without Bonferroni correction), but this was not independent of ileocolonic disease location (74). Later, Sabate et al. again noticed a trend toward fibrostenotic behavior in V249I carriers (not statistically significant after Bonferroni correction), especially in smokers, independent of NOD2 Leu1007fsinC carriage and ileal involvement (52). Although the two SNPs are strongly correlated (75), Sabate et al. did not see a similar trend for T280M (52).
Several findings point toward CX3CR1 as a critical component in maintaining homeostasis of lamina propria macrophages, and master regulators of inflammation and fibrosis (76). Importantly, specifically for the described variants, it was shown in vitro that peripheral blood mononuclear cells (PBMCs) from individuals with wild-type CX3CR1 genotype adhere more potently to membrane-bound fractalkine than do PBMCs from homozygous V249I–T280M donors (74, 77). Despite the limited data about an association between CX3CR1 and fibrostenotic disease, these functional data could point toward a true role for the CX3CR1/fractalkine axis in fibrosis in CD.
Tissue Growth Factor Beta
Tissue growth factor beta is encoded by a gene on chromosome 19q13. It is a regulatory protein that plays a key role in inflammatory, fibrotic, and immunological events in the intestinal mucosa (78, 79). Enhanced expression of TGF-β and its receptors seems to be involved in the pathogenesis of CD, and might contribute to fibrosis (80, 81). After some SNPs (including C509T) in the TGF-β1-gene were described to lead to variations in the production of TGF-β serum levels in women (82, 83), some groups looked in vain for an association with susceptibility to CD (79, 81, 84). However, Hume et al. observed a significant association between the AA genotype of a SNP in codon 25 in the TGF-β1 gene and a fibrostenotic phenotype. CD patients homozygous for the profibrotic A allele also tended to have a shorter time to intestinal resection (79).
Angiotensinogen
Angiotensinogen, mapped to chromosome 1q42, is meant to function locally as a cytokine in several organ systems, participating in the regulation of inflammation and fibrosis. After being cleaved by renin into angiotensin I and processed to angiotensin II, it may increase the production of TGF-β1 (79).
After a gain of function SNP 6 bp from the transcription site was described in the angiotensinogen gene (85), Hume et al. studied its association with CD and CD phenotype. They reported a positive association for the A allele and CD, although without any genotype–phenotype association at the univariate or multivariate level (79).
Matrix Metalloproteinases and Tissue Inhibitors of MMPs
Matrix metalloproteinases (MMPs), all Zn-activated endoproteinases, are subdivided into four groups, depending on their structure and substrate specificity: collagenases, gelatinases, stromelysins, and membrane-type MMPs (86–88). They mediate degradation of essentially all components of the extracellular matrix. The enzymatic activity of these potentially harmful proteinases is tightly controlled and counterbalanced by endogenous inhibitors, such as alpha 2 macroglobulin, and specific tissue inhibitors of MMPs, the so-called TIMPs. TIMPs are produced by the same cell types that produce MMPs, primarily in cells resembling macrophages and fibroblasts (87, 89).
In the last decade, many different SNPs in these genes were described, related to processes such as fetal development (90), primary sclerosing cholangitis (91), and coronary atherosclerosis (92). Meijer et al. also studied their role in relation to CD susceptibility and CD phenotype. They found that the 5T5T genotype at the MMP-3 locus (an additional thymidine insertion at −1613 of the MMP-3 promoter) was associated with fibrostenotic CD (88). Furthermore, expression data of Warnaar et al. demonstrated increased levels of MMP-3 in stenotic and prestenotic resected CD ileum, pointing to an MMP-3 (stromelysin-1)-mediated altered clinical course of CD patients. These findings might explain the high recurrence rate of intestinal strictures, as in non-resected, prestenotic CD ileum in which the anastomosis is made, tissue turnover is present (89). Conflicting evidence exists regarding the consequences of the 5T5T genotype: some groups reported upregulation of MMP-3 expression (93, 94), whereas others reported a downregulation (95). In the study by Meijer et al., patients stratified according to MMP-3 genotype had similar MMP-3 total activity (88).
Membrane-Associated Guanylate Kinase, WW and PDZ Domain-Containing 1
MAGI1 is located on chromosome 3p14 and encodes the membrane-associated guanylate kinase WW and PDZ domain-containing protein 1 (96). This protein plays an important role in the tight junction of IECs through interaction with JAM4, a junctional adhesion transmembrane molecule. Disruption of this epithelial barrier can have dramatic effect on the mucosal integrity, which has been shown to contribute to the development of CD (96).
Alonso et al. recently published an interesting association between fibrostenotic CD and rs11924265, located in a 46.5-kb haplotype block inside a MAGI1 intron. They validated this association in an independent replication cohort (96). Previously, other groups have shown a significant increase in intestinal permeability in patients with stricturing disease (97). Rs11924265 might induce an alteration in the membrane-associated guanylate kinase, WW and PDZ domain-containing 1 (MAGI1) protein function, contributing to an exaggerated immune response and to the subsequent transmural inflammation of the gastrointestinal tract (96).
Janus Kinase 2
JAK2, located on chromosome 9, encodes for an intracellular tyrosine kinase that transduces cytokine-mediated signals via the JAK-STAT pathway (50, 70). The large, retrospective, and multicentre IBDchip study found that rs10758669 (C allele), within the JAK2 gene, is associated with an increased risk for ileal involvement and stenosing disease behavior. One mechanism by which Janus kinase 2 (JAK2) contributes to this fibrostenotic disease could be by altering intestinal permeability (50). Indeed, Prager et al. previously demonstrated that patients carrying the rs10758669 C risk allele significantly more often had an increased permeability compared with patients without the C allele (98).
Fucosyltransferase 2
FUT2, located on chromosome 19 (70), encodes the secretor enzyme alpha(1,2)-fucosyltransferase (Lewis blood group system), which allows expression of ABO antigens on the gastrointestinal mucosa and in bodily secretions (secretor phenotype) (99). After a nonsense allele in FUT2, rs601338 (W143X), was identified as a susceptibility variant for CD (100, 101), Forni et al. found non-secretors to be at slightly higher risk of a stricturing/penetrating behavior (OR 1.51, p = 0.046). Additionally, their analysis revealed patients with blood group O are less likely to develop a stricturing disease (OR 0.70, p = 0.038) (99). Although it is known that fucosyltransferase 2 (FUT2) expression affects the composition of the gut microbiota (102), the pathophysiological link between this specific SNP and fibrostenotic disease has not been unraveled yet. Theoretically, an altered microbial environment might induce more severe inflammation, leading to a more aggressive phenotype.
Other Genes
In 2009, Henckaerts et al. examined the influence of some CD-associated susceptibility loci on changes in disease behavior. They found that homozygosity for the rs1363670 G-allele in a gene encoding a hypothetical protein near the IL12B gene, located on chromosome 5, was independently associated with stricturing disease behavior, especially in patients with ileal involvement (70, 103). So far, the pathophysiological consequences of this SNP, leading to a non-coding transcript variant, are not fully understood (70).
Because inherited risk factors [factor V Leiden, methylenetetrahydrofolate reductase (MTHFR) C677T] have been reported to be associated with fibrosis in other chronic inflammatory diseases, Novacek et al. performed a retrospective study in CD patients aiming to identify these risk factors in fibrostenotic CD. They concluded that the MTHFR 6777TT variant, factor V Leiden, and the prothrombin G20210A variant are not associated with fibrostenosis in CD (104).
As TNFα plays a pivotal role in the pathophysiology of IBD, confirmed by the efficacy of anti-TNF drugs, such as infliximab and adalimumab (105), Meijer et al. investigated the association between a SNP (G308A) in TNFα and fibrostenotic disease (88). In line with other reports (106, 107), they could not find an association between this SNP and fibrostenotic CD (88).
The Combined Action of the Known Susceptibility Variants
Crohn’s disease is a complex multigenic disease, where several small-effect risk variants combined influence disease onset. It is more and more suggested that combining the many individually weak signals into a genetic risk score might be a more powerful approach to study the genetic association with subphenotypes or to improve predictive ability of disease (15, 108). Such a genetic risk score was calculated in the IIBDGC genotype–phenotype study and tested for association with several disease subphenotypes. A strong association with disease behavior was found (p = 9.23 × 10−18), indicating that the known susceptibility loci combined can be a useful measurement of CD subtypes, but still do not have enough predictive ability to distinguish between the different subtypes (15).
Genetics and Fibrosis in Pediatric CD
Currently, not much is known about the genotype–phenotype association in pediatric CD. Russell et al. studied NOD2 variants in the Scottish early onset CD population (aged <16 years) and noticed a relatively small contribution to CD susceptibility, but a major impact on phenotype. Presence of stricturing disease behavior at diagnosis showed a trend toward an increase in carriers of NOD2 variant alleles, which became significant by 2 years of follow-up (39). The association of NOD2 variants and fibrostenotic pediatric CD was previously already reported by two other groups (109, 110).
In contrast with a study in adult CD (79), Liberek et al. could not find any significant correlation between the four common SNPs in TGF-β and any specific clinical parameter (111).
In 2014, Strisciuglio et al. performed a genotype–phenotype correlation study, focusing on autophagy gene variants. They observed a trend toward switching to a fibrostenotic disease in children homozygous for the ATG16L1 T300A risk allele. They did not find an association between NOD2 variants and stricturing CD (112).
Genetics and Fibrosis Around the World
Although the incidence of IBD is rising in developing countries (113, 114), epidemiological data on the clinical phenotype of disease, and genotype–phenotype association studies, in non-European populations are limited. Similar as for Caucasian populations though, several smaller genotype–phenotype studies have been performed in non-Caucasian populations (115–120). These usually study the same variants as those considered in Caucasian populations (NOD2, IL-23R, etc.), but only one Korean study found the IL23R variant rs1004819 associated with stricturing and penetrating disease (119). It is possibly not surprising that NOD2 variants are not found to be associated with disease (subtypes) in different populations, as NOD2 variants have been seen with different frequencies in geographically diverse populations. Whereas the prevalence of CD patients who carry at least one NOD2 susceptibility variant varies from 27 to 50% in most Caucasian European populations, observed frequencies are much lower (15–21%) in Scandinavian countries (121, 122), which are generally characterized by more homogenous study populations. Caucasian populations, relatively far from Europe, but with European ancestry with hardly no racial mixing, such as the United States, Canada, and Australia, have NOD2 variant frequencies comparable with those found across the rest of Europe (122). In Asians (Japanese, Chinese, and Korean), Arabs, Africans, and African-Americans, the NOD2 variants are rare or even absent (5, 21, 116).
Recently, the first trans-ancestry association study of IBD was published by the IIBDGC (5). They collected subphenotype data on 1991 patients with CD from East Asia, India, and Iran, and compared these data with available clinical phenotypes for 19,290 Europeans (15). They showed some demographic differences, with, for example, more stricturing behavior and perianal and less inflammatory CD in the non-European population compared to the European population, in line with the previously reported prospectively collected clinical findings in incident cases of IBD in non-Europeans (114). It will be interesting to see if these differences are explained by genetic factors that differ between populations, or rather by environmental factors (including different health-care systems), ascertainment bias, or a combination of these. The trans-ancestry association study showed that although for the majority of the IBD risk loci, the direction and magnitude of effect are consistent in European and non-European cohorts, genetic heterogeneity was seen between divergent populations at several established risk loci, driven by differences in allele frequency (NOD2), effect size (TNFSF15 and ATG16L1), or a combination of both (IL23R and IRGM). A large trans-ancestry genotype–phenotype study is under way, undoubtedly shedding light on possible genetic heterogeneity of disease subphenotypes in different populations.
The Promise of Epigenetics
So far, the epigenetic control of inflammation and fibrosis in IBD is not fully understood (12). But, as only a modest fraction (ca. 15%) of the 200 association signals is driven by missense mutations, the large majority of causal alleles is likely to be related to regulatory functions such as modulation of gene expression (123). Intestinal disease-associated DNA methylations in IBD do occur, leading to changes in gene expression (124), such as in several loci within the interleukin 12/interleukin 23 pathway (125). As epigenetic changes are dynamically responsive to the environment, they are likely to play a key role in the pathogenesis of fibrosis and offer a molecular explanation for how the intestine becomes profibrotic (126).
Sadler et al. recently published their genome-wide analysis of DNA methylation and gene expression in the context of fibrotic CD (126). They found three functional candidate genes to be differentially methylated and expressed in fibrotic CD: wingless-type mouse mammary tumor virus integration site family member 2B (WNT2B), prostacyclin synthase (PTGIS), and prostaglandin D2 synthase (PTGDS) (126). The decreased expression of hypermethylated WNT2B and PTGIS are novel findings both in the context of fibrosis and CD, though hypermethylation of the PTGIS promoter has been described as a feature of colorectal cancer (127). Interestingly, increased expression of WNT2B has been detected in the intestinal mucosa of UC patients (128), suggesting that WNT2B may perform distinct functions in CD and UC (126). Hypomethylation of the PTGDS gene, together with increased gene expression levels, was previously noticed in a murine UC model (129). In future, both epigenetic and transcriptomic analyses will undoubtedly reveal novel insights in the pathogenesis of stricturing CD, potentially leading to new targeted therapies.
Clinical Implications
Based on current evidence, it is too early to adjust treatment in CD patients according to genetic profiles, in order to personalize treatment in CD (15). NOD2 is by far the most studied genetic predictor for fibrostenotic disease in CD, and many studies suggested an important role for NOD2 variants in developing fibrostenotic CD. Still, the low sensitivity of a single NOD2 variant for predicting fibrostenotic disease does not justify NOD2 genotyping in all patients (48), and there is no adequate scientific evidence for a top-down medical therapy based solely on NOD2 variants. It has been suggested that targeted early-intensive therapy for high-risk patients with two NOD2 mutations might be beneficial, if proven by prospective trials (47), but so far this evidence does not exist. Importantly, based on the biggest genotype–phenotype study ever done including over 19,000 CD patients, it was found that no NOD2 variants are associated with fibrostenotic disease after conditioning for disease location. Disease location thus seems to be the major driver to changes in disease behavior over time (15). Preferential involvement of the terminal ileum could be explained by NOD2 variants abrogating normal Paneth cell behavior, as Paneth cells express NOD2/CARD15 throughout the small intestine, with maximal expression in the terminal ileum (35, 130).
Conclusion and Future Directions
As outlined above, several genotype–phenotype studies have been performed to find which genetic variants play a role in defining CD disease location and behavior, but hardly any variants were uniformly found as independent risk factors for developing fibrostenotic disease in CD. Different reasons can be put forward. The first one is related to power of the individual studies. Many studies indeed included relatively small patient numbers (Table 1), and looking into subgroups of patients makes the sample sizes even smaller. It should also be noted that various studies might include patient groups from either population-based registries and/or from secondary or tertiary referral centers. This has a direct influence on the proportion of patients with more severe disease as opposed to inflammatory disease, which in turn could lead to over or under presentation of certain genetic associations. An example is the Scandinavian registries that are population-based and where indeed a lower proportion of stenosing and penetrating CD is seen (15). NOD2 frequencies in these populations are also lower (see above) (121), but this could be linked to the population-based character of the study population. Third, most susceptibility variants are not the pathophysiological causal ones, but are in LD with the true causal variant(s) at that locus, which might have more qualitative or quantitative effects and explain the association with a certain clinical feature. Fourth, many studies apply different definitions for stenosing disease or use a limited number of variables given in the Vienna Classification (41). Another reason could be the dramatic change in disease behavior over the course of the disease, implying disease behavior of CD cannot be analyzed without taking into account the duration of disease (2, 30). Also, because of the importance of disease location in driving changes of disease behavior over time (15), disease location should always be considered when analyzing risk factors for stenosing disease. In the case of, for example, NOD2, there is a strong correlation of NOD2 and ileal disease location (131), which might induce a false, confounded association between NOD2 variants and fibrostenotic disease in those cases where disease location is not considered in the genotype–phenotype analysis. Finally, disease behavior is influenced by environmental factors (132), which can be dramatically different in the different studies. Examples not only include smoking and NSAIDS use but also specific treatments may hide patients at risk to develop certain subtypes of disease. Any disease behavior and severity analysis should be interpreted with caution, when there is no access to medication use and response to medications, especially for patients in the biologics era.
Among the 163 genome-wide significant IBD susceptibility loci as identified in the study by Jostins et al. (4), genetic variants in immune system components (NOD2, IL-23R, IL-12B, JAK2, and FUT2) and autophagy [ATG16L1, leucine-rich repeat kinase 2 (LRRK2)] could (jointly) contribute to the activation of mesenchymal cells and pathogenesis of fibrosis (4, 9, 133). Although these susceptibility genes might pathophysiologically contribute to fibrostenotic processes, not all have been found to be associated with stricturing CD. For example, the LRRK2 CD-associated M2397 allele inhibits nuclear factor of activated T cells (NFAT) (134), which is known to control fibroblast plasticity in the heart (135). LRRK2 might thus also be involved in fibrosis in the gut, although so far this has not been reported. We did not find any studies where a genetic association was shown between LRRK2 and fibrostenotic CD. The IIBDGC study by Cleynen et al. in addition found that while many of the known 163 risk loci showed nominal evidence of association with CD behavior, a combined genetic risk score of these individual weak signals was strongly associated with CD disease behavior (15). Still, the development of fibrosis is preceded by a period of initial inflammation, and not all patients with CD express a fibrostenotic phenotype (1, 2, 6). This highlights the possible difference between loci predisposing to overall disease (CD or UC) and loci predisposing to clinical phenotypes (4, 9, 136). It is thus important to consider the idea of different genes driving susceptibility on the one hand and disease behavior on the other. The IIBDGC study for the first time does this on a large scale, but hardly finds any genome-wide significant loci for disease behavior independent from disease location, except from rs77005575 (MHC) (15).
Figure 1 gives an overview of different interacting aspects we believe are important in fibrotic CD pathogenesis and summarizes the genetic factors identified thus far. In future studies aimed at finding the genetic contributions to fibrotic CD, it will be important to consider as much as possible environmental risk factors (smoking, diet, microbiota, medication use, etc.), as well as non-Caucasian populations. To gain additional insights into the pathophysiology of fibrotic CD, and find predictive markers, an integrated analysis, including genetics, epigenetics, transcriptomics, microbiomics, etc., and prospective study setups, will be the next steps.
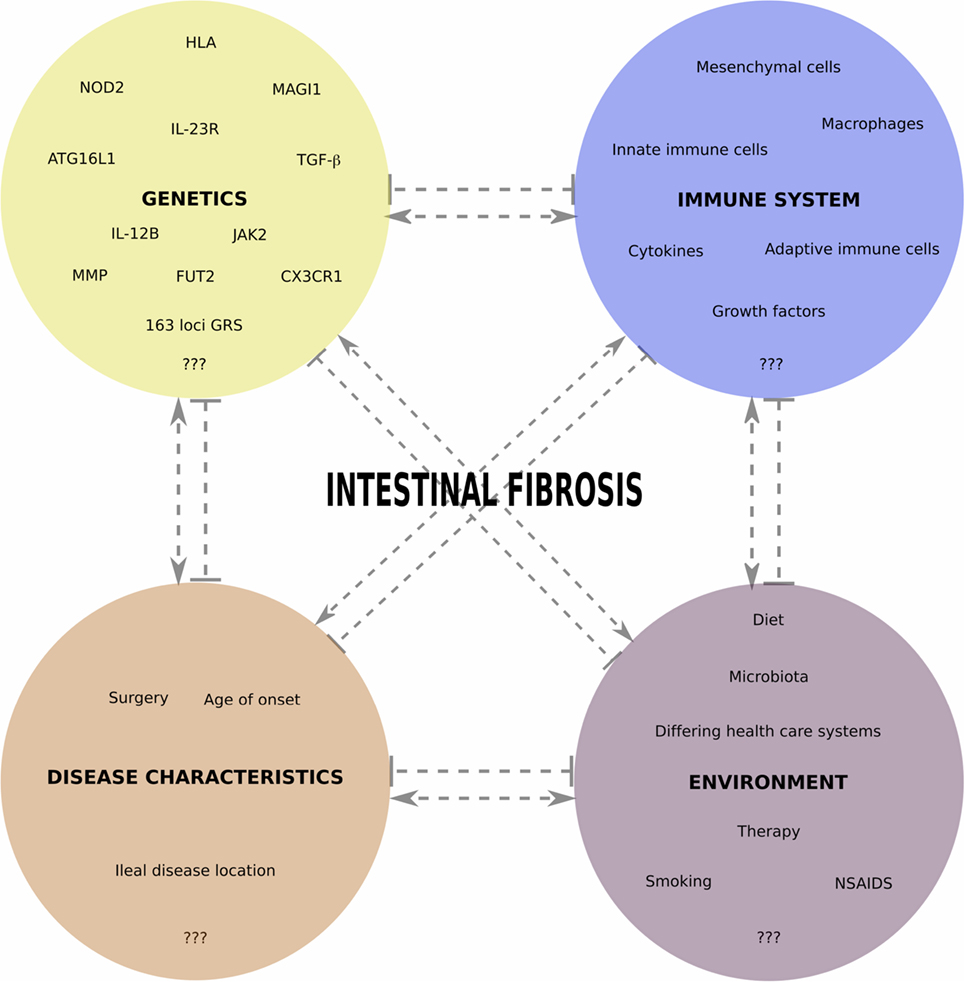
Figure 1. Overview of different interacting aspects important in fibrotic CD pathogenesis. In each of the four main categories, we indicate some factors for which a role in fibrotic CD has been shown, with an emphasis on the genetic factors reviewed in this paper. Pointed and barred arrows denote a positive and negative interaction, respectively.
Author Contributions
BV: literature search and drafting of the manuscript. IC: drafting of the manuscript and critical revision of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
BV is a doctoral fellow funded by the Research Foundation Flanders, Belgium.
References
1. Cosnes J, Cattan S, Blain A, Beaugerie L, Carbonnel F, Parc R, et al. Long-term evolution of disease behavior of Crohn’s disease. Inflamm Bowel Dis (2002) 8(4):244–50. doi:10.1097/00054725-200207000-00002
2. Louis E, Collard A, Oger AF, Degroote E, Aboul Nasr El Yafi FA, Belaiche J. Behaviour of Crohn’s disease according to the Vienna classification: changing pattern over the course of the disease. Gut (2001) 49(6):777–82. doi:10.1136/gut.49.6.777
3. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature (2007) 448(7152):427–34. doi:10.1038/nature06005
4. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491(7422):119–24. doi:10.1038/nature11582
5. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet (2015) 47:979–86. doi:10.1038/ng.3359
6. Van Assche G, Geboes K, Rutgeerts P. Medical therapy for Crohn’s disease strictures. Inflamm Bowel Dis (2004) 10(1):55–60. doi:10.1097/00054725-200401000-00009
7. Rieder F, Zimmermann EM, Remzi FH, Sandborn WJ. Crohn’s disease complicated by strictures: a systematic review. Gut (2013) 62(7):1072–84. doi:10.1136/gutjnl-2012-304353
8. Greenstein AJ, Zhang LP, Miller AT, Yung E, Branco BC, Sachar DB, et al. Relationship of the number of Crohn’s strictures and strictureplasties to postoperative recurrence. J Am Coll Surg (2009) 208(6):1065–70. doi:10.1016/j.jamcollsurg.2009.01.039
9. Li C, Kuemmerle JF. Mechanisms that mediate the development of fibrosis in patients with Crohn’s disease. Inflamm Bowel Dis (2014) 20(7):1250–8. doi:10.1097/MIB.0000000000000043
10. Speca S, Giusti I, Rieder F, Latella G. Cellular and molecular mechanisms of intestinal fibrosis. World J Gastroenterol (2012) 18(28):3635–61. doi:10.3748/wjg.v18.i28.3635
11. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis (2010) 30(3):245–57. doi:10.1055/s-0030-1255354
12. Latella G, Rogler G, Bamias G, Breynaert C, Florholmen J, Pellino G, et al. Results of the 4th scientific workshop of the ECCO (I): pathophysiology of intestinal fibrosis in IBD. J Crohns Colitis (2014) 8(10):1147–65. doi:10.1016/j.crohns.2014.03.008
13. Latella G, Di Gregorio J, Flati V, Rieder F, Lawrance IC. Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand J Gastroenterol (2015) 50(1):53–65. doi:10.3109/00365521.2014.968863
14. Chang CW, Wong JM, Tung CC, Shih IL, Wang HY, Wei SC. Intestinal stricture in Crohn’s disease. Intestinal Res (2015) 13(1):19–26. doi:10.5217/ir.2015.13.1.19
15. Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet (2016) 387(10014):156–67. doi:10.1016/S0140-6736(15)00465-1
16. Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem (2001) 276(7):4812–8. doi:10.1074/jbc.M008072200
17. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411(6837):599–603. doi:10.1038/35079107
18. Abreu MT, Taylor KD, Lin YC, Hang T, Gaiennie J, Landers CJ, et al. Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn’s disease. Gastroenterology (2002) 123(3):679–88. doi:10.1053/gast.2002.35393
19. Ahmad T, Armuzzi A, Bunce M, Mulcahy-Hawes K, Marshall SE, Orchard TR, et al. The molecular classification of the clinical manifestations of Crohn’s disease. Gastroenterology (2002) 122(4):854–66. doi:10.1053/gast.2002.32413
20. Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, et al. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science (2005) 307(5710):734–8. doi:10.1126/science.1103685
21. Naser SA, Arce M, Khaja A, Fernandez M, Naser N, Elwasila S, et al. Role of ATG16L, NOD2 and IL23R in Crohn’s disease pathogenesis. World J Gastroenterol (2012) 18(5):412–24. doi:10.3748/wjg.v18.i5.412
22. Hampe J, Cuthbert A, Croucher PJ, Mirza MM, Mascheretti S, Fisher S, et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet (2001) 357(9272):1925–8. doi:10.1016/S0140-6736(00)05063-7
23. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature (2001) 411(6837):603–6. doi:10.1038/35079114
24. Seiderer J, Schnitzler F, Brand S, Staudinger T, Pfennig S, Herrmann K, et al. Homozygosity for the CARD15 frameshift mutation 1007fs is predictive of early onset of Crohn’s disease with ileal stenosis, entero-enteral fistulas, and frequent need for surgical intervention with high risk of re-stenosis. Scand J Gastroenterol (2006) 41(12):1421–32. doi:10.1080/00365520600703900
25. Cuthbert AP, Fisher SA, Mirza MM, King K, Hampe J, Croucher PJ, et al. The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology (2002) 122(4):867–74. doi:10.1053/gast.2002.32415
26. Lesage S, Zouali H, Cézard JP, Colombel JF, Belaiche J, Almer S, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet (2002) 70(4):845–57. doi:10.1086/339432
27. Hampe J, Grebe J, Nikolaus S, Solberg C, Croucher PJ, Mascheretti S, et al. Association of NOD2 (CARD 15) genotype with clinical course of Crohn’s disease: a cohort study. Lancet (2002) 359(9318):1661–5. doi:10.1016/S0140-6736(02)08590-2
28. Radlmayr M, Török HP, Martin K, Folwaczny C. The c-insertion mutation of the NOD2 gene is associated with fistulizing and fibrostenotic phenotypes in Crohn’s disease. Gastroenterology (2002) 122(7):2091–2. doi:10.1053/gast.2002.34020
29. Vavassori P, Borgiani P, D’Apice MR, De Negris F, Del Vecchio Blanco G, Monteleone I, et al. 3020insC mutation within the NOD2 gene in Crohn’s disease: frequency and association with clinical pattern in an Italian population. Dig Liver Dis (2002) 34(2):153. doi:10.1016/S1590-8658(02)80249-3
30. Louis E, Michel V, Hugot JP, Reenaers C, Fontaine F, Delforge M, et al. Early development of stricturing or penetrating pattern in Crohn’s disease is influenced by disease location, number of flares, and smoking but not by NOD2/CARD15 genotype. Gut (2003) 52(4):552–7. doi:10.1136/gut.52.4.552
31. Brant SR, Picco MF, Achkar JP, Bayless TM, Kane SV, Brzezinski A, et al. Defining complex contributions of NOD2/CARD15 gene mutations, age at onset, and tobacco use on Crohn’s disease phenotypes. Inflamm Bowel Dis (2003) 9(5):281–9. doi:10.1097/00054725-200309000-00001
32. Heliö T, Halme L, Lappalainen M, Fodstad H, Paavola-Sakki P, Turunen U, et al. CARD15/NOD2 gene variants are associated with familially occurring and complicated forms of Crohn’s disease. Gut (2003) 52(4):558–62. doi:10.1136/gut.52.4.558
33. Mendoza JL, Murillo LS, Fernández L, Peña AS, Lana R, Urcelay E, et al. Prevalence of mutations of the NOD2/CARD15 gene and relation to phenotype in Spanish patients with Crohn disease. Scand J Gastroenterol (2003) 38(12):1235–40. doi:10.1080/00365520310006612
34. Heresbach D, Gicquel-Douabin V, Birebent B, D’halluin PN, Heresbach-Le Berre N, Dreano S, et al. NOD2/CARD15 gene polymorphisms in Crohn’s disease: a genotype-phenotype analysis. Eur J Gastroenterol Hepatol (2004) 16(1):55–62. doi:10.1097/00042737-200401000-00009
35. Economou M, Trikalinos TA, Loizou KT, Tsianos EV, Ioannidis JP. Differential effects of NOD2 variants on Crohn’s disease risk and phenotype in diverse populations: a metaanalysis. Am J Gastroenterol (2004) 99(12):2393–404. doi:10.1111/j.1572-0241.2004.40304.x
36. Alvarez-Lobos M, Arostegui JI, Sans M, Tassies D, Plaza S, Delgado S, et al. Crohn’s disease patients carrying Nod2/CARD15 gene variants have an increased and early need for first surgery due to stricturing disease and higher rate of surgical recurrence. Ann Surg (2005) 242(5):693–700. doi:10.1097/01.sla.0000186173.14696.ea
37. Annese V, Lombardi G, Perri F, D’Incà R, Ardizzone S, Riegler G, et al. Variants of CARD15 are associated with an aggressive clinical course of Crohn’s disease – an IG-IBD study. Am J Gastroenterol (2005) 100(1):84–92. doi:10.1111/j.1572-0241.2005.40705.x
38. Lakatos PL, Lakatos L, Szalay F, Willheim-Polli C, Osterreicher C, Tulassay Z, et al. Toll-like receptor 4 and NOD2/CARD15 mutations in Hungarian patients with Crohn’s disease: phenotype-genotype correlations. World J Gastroenterol (2005) 11(10):1489–95. doi:10.3748/wjg.v11.i10.1489
39. Russell RK, Drummond HE, Nimmo EE, Anderson N, Smith L, Wilson DC, et al. Genotype-phenotype analysis in childhood-onset Crohn’s disease: NOD2/CARD15 variants consistently predict phenotypic characteristics of severe disease. Inflamm Bowel Dis (2005) 11(11):955–64. doi:10.1097/01.MIB.0000183423.38037.f3
40. Schnitzler F, Brand S, Staudinger T, Pfennig S, Hofbauer K, Seiderer J, et al. Eight novel CARD15 variants detected by DNA sequence analysis of the CARD15 gene in 111 patients with inflammatory bowel disease. Immunogenetics (2006) 58(2–3):99–106. doi:10.1007/s00251-005-0073-2
41. Oostenbrug LE, Nolte IM, Oosterom E, van der Steege G, te Meerman GJ, van Dullemen HM, et al. CARD15 in inflammatory bowel disease and Crohn’s disease phenotypes: an association study and pooled analysis. Dig Liver Dis (2006) 38(11):834–45. doi:10.1016/j.dld.2006.06.042
42. Baptista ML, Amarante H, Picheth G, Sdepanian VL, Peterson N, Babasukumar U, et al. CARD15 and IL23R influences Crohn’s disease susceptibility but not disease phenotype in a Brazilian population. Inflamm Bowel Dis (2008) 14(5):674–9. doi:10.1002/ibd.20372
43. Glas J, Seiderer J, Tillack C, Pfennig S, Beigel F, Jürgens M, et al. The NOD2 single nucleotide polymorphisms rs2066843 and rs2076756 are novel and common Crohn’s disease susceptibility gene variants. PLoS One (2010) 5(12):e14466. doi:10.1371/journal.pone.0014466
44. Jürgens M, Brand S, Laubender RP, Seiderer J, Glas J, Wetzke M, et al. The presence of fistulas and NOD2 homozygosity strongly predict intestinal stenosis in Crohn’s disease independent of the IL23R genotype. J Gastroenterol (2010) 45(7):721–31. doi:10.1007/s00535-010-0231-7
45. Ippoliti A, Devlin S, Mei L, Yang H, Papadakis KA, Vasiliauskas EA, et al. Combination of innate and adaptive immune alterations increased the likelihood of fibrostenosis in Crohn’s disease. Inflamm Bowel Dis (2010) 16(8):1279–85. doi:10.1002/ibd.21196
46. Rieder F, Lawrance IC, Leite A, Sans M. Predictors of fibrostenotic Crohn’s disease. Inflamm Bowel Dis (2011) 17(9):2000–7. doi:10.1002/ibd.21627
47. Adler J, Rangwalla SC, Dwamena BA, Higgins PD. The prognostic power of the NOD2 genotype for complicated Crohn’s disease: a meta-analysis. Am J Gastroenterol (2011) 106(4):699–712. doi:10.1038/ajg.2011.19
48. Brand S. Homozygosity for the NOD2 p.Leu1007fsX1008 variant is the main genetic predictor for fibrostenotic Crohn’s disease. Inflamm Bowel Dis (2012) 18(2):393–4. doi:10.1002/ibd.21914
49. Brand S. Moving the genetics of inflammatory bowel diseases from bench to bedside: first steps towards personalised medicine. Gut (2013) 62(11):1531–3. doi:10.1136/gutjnl-2012-304151
50. Cleynen I, González JR, Figueroa C, Franke A, McGovern D, Bortlík M, et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: results from the IBDchip European Project. Gut (2013) 62(11):1556–65. doi:10.1136/gutjnl-2011-300777
51. Schnitzler F, Friedrich M, Wolf C, Angelberger M, Diegelmann J, Olszak T, et al. The NOD2 p.Leu1007fsX1008 mutation (rs2066847) is a stronger predictor of the clinical course of Crohn’s disease than the FOXO3A intron variant rs12212067. PLoS One (2014) 9(11):e108503. doi:10.1371/journal.pone.0108503
52. Sabate JM, Ameziane N, Lamoril J, Jouet P, Farmachidi JP, Soulé JC, et al. The V249I polymorphism of the CX3CR1 gene is associated with fibrostenotic disease behavior in patients with Crohn’s disease. Eur J Gastroenterol Hepatol (2008) 20(8):748–55. doi:10.1097/MEG.0b013e3282f824c9
53. Seiderer J, Brand S, Herrmann KA, Schnitzler F, Hatz R, Crispin A, et al. Predictive value of the CARD15 variant 1007fs for the diagnosis of intestinal stenoses and the need for surgery in Crohn’s disease in clinical practice: results of a prospective study. Inflamm Bowel Dis (2006) 12(12):1114–21. doi:10.1097/01.mib.0000235836.32176.5e
54. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet (2007) 39(2):207–11. doi:10.1038/ng1954
55. Prescott NJ, Fisher SA, Franke A, Hampe J, Onnie CM, Soars D, et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn’s disease and is independent of CARD15 and IBD5. Gastroenterology (2007) 132(5):1665–71. doi:10.1053/j.gastro.2007.03.034
56. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet (2007) 39(5):596–604. doi:10.1038/ng2032
57. Cummings JR, Cooney R, Pathan S, Anderson CA, Barrett JC, Beckly J, et al. Confirmation of the role of ATG16L1 as a Crohn’s disease susceptibility gene. Inflamm Bowel Dis (2007) 13(8):941–6. doi:10.1002/ibd.20162
58. Fowler EV, Doecke J, Simms LA, Zhao ZZ, Webb PM, Hayward NK, et al. ATG16L1 T300A shows strong associations with disease subgroups in a large Australian IBD population: further support for significant disease heterogeneity. Am J Gastroenterol (2008) 103(10):2519–26. doi:10.1111/j.1572-0241.2008.02023.x
59. Begun J, Lassen KG, Jijon HB, Baxt LA, Goel G, Heath RJ, et al. Integrated genomics of Crohn’s disease risk variant identifies a role for CLEC12A in antibacterial autophagy. Cell Rep (2015) 11(12):1905–18. doi:10.1016/j.celrep.2015.05.045
60. Salem M, Ammitzboell M, Nys K, Seidelin JB, Nielsen OH. ATG16L1: a multifunctional susceptibility factor in Crohn disease. Autophagy (2015) 11(4):585–94. doi:10.1080/15548627.2015.1017187
61. Levin AD, Koelink PJ, Bloemendaal FM, Vos AC, D’Haens GR, van den Brink GR, et al. Autophagy contributes to the induction of anti-TNF induced macrophages. J Crohns Colitis (2016) 10(3):323–9. doi:10.1093/ecco-jcc/jjv174
62. Zorzi F, Calabrese E, Monteleone G. Pathogenic aspects and therapeutic avenues of intestinal fibrosis in Crohn’s disease. Clin Sci (Lond) (2015) 129(12):1107–13. doi:10.1042/CS20150472
63. Sorbara MT, Ellison LK, Ramjeet M, Travassos LH, Jones NL, Girardin SE, et al. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity (2013) 39(5):858–73. doi:10.1016/j.immuni.2013.10.013
64. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science (2006) 314(5804):1461–3. doi:10.1126/science.1135245
65. Glas J, Seiderer J, Wetzke M, Konrad A, Török HP, Schmechel S, et al. rs1004819 is the main disease-associated IL23R variant in German Crohn’s disease patients: combined analysis of IL23R, CARD15, and OCTN1/2 variants. PLoS One (2007) 2(9):e819. doi:10.1371/journal.pone.0000819
66. Goyette P, Boucher G, Mallon D, Ellinghaus E, Jostins L, Huang H, et al. High-density mapping of the MHC identifies a shared role for HLA-DRB1*01:03 in inflammatory bowel diseases and heterozygous advantage in ulcerative colitis. Nat Genet (2015) 47:172–9. doi:10.1038/ng.3176
67. De Nardo D. Toll-like receptors: activation, signalling and transcriptional modulation. Cytokine (2015) 74(2):181–9. doi:10.1016/j.cyto.2015.02.025
68. Franchimont D, Vermeire S, El Housni H, Pierik M, Van Steen K, Gustot T, et al. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis. Gut (2004) 53(7):987–92. doi:10.1136/gut.2003.030205
69. Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet (2000) 25(2):187–91. doi:10.1038/76048
70. Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, et al. Ensembl 2016. Nucleic Acids Res (2016) 44(D1):D710–6. doi:10.1093/nar/gkv1157
71. Rieder F, Schirbel A, Ouyang Z, West G, Rho H, de la Motte C, et al. Pro-fibrogenic activity of Toll-like receptor (TLR) and NOD-like receptor (NLR) ligands on human intestinal myofibroblasts (HIF) – linking bacterial innate immunity to intestinal fibrosis. Gastroenterology (2010) 38(5):S35. doi:10.1016/S0016-5085(10)60163-1
72. Bhattacharyya S, Varga J. Emerging roles of innate immune signaling and toll-like receptors in fibrosis and systemic sclerosis. Curr Rheumatol Rep (2015) 17(1):474. doi:10.1007/s11926-014-0474-z
73. Petrasek J, Csak T, Szabo G. Toll-like receptors in liver disease. Adv Clin Chem (2013) 59:155–201. doi:10.1016/B978-0-12-405211-6.00006-1
74. Brand S, Hofbauer K, Dambacher J, Schnitzler F, Staudinger T, Pfennig S, et al. Increased expression of the chemokine fractalkine in Crohn’s disease and association of the fractalkine receptor T280M polymorphism with a fibrostenosing disease Phenotype. Am J Gastroenterol (2006) 101(1):99–106. doi:10.1111/j.1572-0241.2005.00361.x
75. Faure S, Meyer L, Costagliola D, Vaneensberghe C, Genin E, Autran B, et al. Rapid progression to AIDS in HIV+ individuals with a structural variant of the chemokine receptor CX3CR1. Science (2000) 287(5461):2274–7. doi:10.1126/science.287.5461.2274
76. Medina-Contreras O, Geem D, Laur O, Williams IR, Lira SA, Nusrat A, et al. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J Clin Invest (2011) 121(12):4787–95. doi:10.1172/JCI59150
77. Daoudi M, Lavergne E, Garin A, Tarantino N, Debré P, Pincet F, et al. Enhanced adhesive capacities of the naturally occurring Ile249-Met280 variant of the chemokine receptor CX3CR1. J Biol Chem (2004) 279(19):19649–57. doi:10.1074/jbc.M313457200
78. Schulte CM, Dignass AU, Goebell H, Röher HD, Schulte KM. Genetic factors determine extent of bone loss in inflammatory bowel disease. Gastroenterology (2000) 119(4):909–20. doi:10.1053/gast.2000.18158
79. Hume GE, Fowler EV, Lincoln D, Eri R, Templeton D, Florin TH, et al. Angiotensinogen and transforming growth factor beta1: novel genes in the pathogenesis of Crohn’s disease. J Med Genet (2006) 43(10):e51. doi:10.1136/jmg.2005.040477
80. di Mola FF, Friess H, Scheuren A, Di Sebastiano P, Graber H, Egger B, et al. Transforming growth factor-betas and their signaling receptors are coexpressed in Crohn’s disease. Ann Surg (1999) 229(1):67–75. doi:10.1097/00000658-199901000-00009
81. Schulte CM, Goebell H, Röher HD, Schulte KM. C-509T polymorphism in the TGFB1 gene promoter: impact on Crohn’s disease susceptibility and clinical course? Immunogenetics (2001) 53(2):178–82. doi:10.1007/s002510100309
82. Yamada Y, Miyauchi A, Goto J, Takagi Y, Okuizumi H, Kanematsu M, et al. Association of a polymorphism of the transforming growth factor-beta1 gene with genetic susceptibility to osteoporosis in postmenopausal Japanese women. J Bone Miner Res (1998) 13(10):1569–76. doi:10.1359/jbmr.1998.13.10.1569
83. Grainger DJ, Heathcote K, Chiano M, Snieder H, Kemp PR, Metcalfe JC, et al. Genetic control of the circulating concentration of transforming growth factor type beta1. Hum Mol Genet (1999) 8(1):93–7. doi:10.1093/hmg/8.1.93
84. García-González MA, Crusius JB, Strunk MH, Bouma G, Pérez-Centeno CM, Pals G, et al. TGFB1 gene polymorphisms and inflammatory bowel disease. Immunogenetics (2000) 51(10):869–72. doi:10.1007/s002510000211
85. Inoue I, Nakajima T, Williams CS, Quackenbush J, Puryear R, Powers M, et al. A nucleotide substitution in the promoter of human angiotensinogen is associated with essential hypertension and affects basal transcription in vitro. J Clin Invest (1997) 99(7):1786–97. doi:10.1172/JCI119343
86. Huppertz B, Kertschanska S, Demir AY, Frank HG, Kaufmann P. Immunohistochemistry of matrix metalloproteinases (MMP), their substrates, and their inhibitors (TIMP) during trophoblast invasion in the human placenta. Cell Tissue Res (1998) 291(1):133–48. doi:10.1007/s004410050987
87. Matrisian LM. Metalloproteinases and their inhibitors in matrix remodeling. Trends Genet (1990) 6(4):121–5. doi:10.1016/0168-9525(90)90126-Q
88. Meijer MJ, Mieremet-Ooms MA, van Hogezand RA, Lamers CB, Hommes DW, Verspaget HW. Role of matrix metalloproteinase, tissue inhibitor of metalloproteinase and tumor necrosis factor-alpha single nucleotide gene polymorphisms in inflammatory bowel disease. World J Gastroenterol (2007) 13(21):2960–6. doi:10.3748/wjg.v13.i21.2960
89. Warnaar N, Hofker HS, Maathuis MH, Niesing J, Bruggink AH, Dijkstra G, et al. Matrix metalloproteinases as profibrotic factors in terminal ileum in Crohn’s disease. Inflamm Bowel Dis (2006) 12(9):863–9. doi:10.1097/01.mib.0000231568.43065.ed
90. Fujimoto T, Parry S, Urbanek M, Sammel M, Macones G, Kuivaniemi H, et al. A single nucleotide polymorphism in the matrix metalloproteinase-1 (MMP-1) promoter influences amnion cell MMP-1 expression and risk for preterm premature rupture of the fetal membranes. J Biol Chem (2002) 277(8):6296–302. doi:10.1074/jbc.M107865200
91. Satsangi J, Chapman RW, Haldar N, Donaldson P, Mitchell S, Simmons J, et al. A functional polymorphism of the stromelysin gene (MMP-3) influences susceptibility to primary sclerosing cholangitis. Gastroenterology (2001) 121(1):124–30. doi:10.1053/gast.2001.25527
92. Zhi H, Wang H, Ren L, Shi Z, Peng H, Cui L, et al. Functional polymorphisms of matrix metallopeptidase-9 and risk of coronary artery disease in a Chinese population. Mol Biol Rep (2010) 37(1):13–20. doi:10.1007/s11033-009-9482-x
93. Borghaei RC, Rawlings PL, Javadi M, Woloshin J. NF-kappaB binds to a polymorphic repressor element in the MMP-3 promoter. Biochem Biophys Res Commun (2004) 316(1):182–8. doi:10.1016/j.bbrc.2004.02.030
94. Medley TL, Kingwell BA, Gatzka CD, Pillay P, Cole TJ. Matrix metalloproteinase-3 genotype contributes to age-related aortic stiffening through modulation of gene and protein expression. Circ Res (2003) 92(11):1254–61. doi:10.1161/01.RES.0000076891.24317.CA
95. Samnegård A, Silveira A, Lundman P, Boquist S, Odeberg J, Hulthe J, et al. Serum matrix metalloproteinase-3 concentration is influenced by MMP-3 -1612 5A/6A promoter genotype and associated with myocardial infarction. J Intern Med (2005) 258(5):411–9. doi:10.1111/j.1365-2796.2005.01561.x
96. Alonso A, Domènech E, Julià A, Panés J, García-Sánchez V, Mateu PN, et al. Identification of risk loci for Crohn’s disease phenotypes using a genome-wide association study. Gastroenterology (2015) 148(4):794–805. doi:10.1053/j.gastro.2014.12.030
97. Benjamin J, Makharia GK, Ahuja V, Kalaivani M, Joshi YK. Intestinal permeability and its association with the patient and disease characteristics in Crohn’s disease. World J Gastroenterol (2008) 14(9):1399–405. doi:10.3748/wjg.14.1399
98. Prager M, Büttner J, Haas V, Baumgart DC, Sturm A, Zeitz M, et al. The JAK2 variant rs10758669 in Crohn’s disease: altering the intestinal barrier as one mechanism of action. Int J Colorectal Dis (2012) 27(5):565–73. doi:10.1007/s00384-011-1345-y
99. Forni D, Cleynen I, Ferrante M, Cassinotti A, Cagliani R, Ardizzone S, et al. ABO histo-blood group might modulate predisposition to Crohn’s disease and affect disease behavior. J Crohns Colitis (2014) 8(6):489–94. doi:10.1016/j.crohns.2013.10.014
100. McGovern DP, Jones MR, Taylor KD, Marciante K, Yan X, Dubinsky M, et al. Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn’s disease. Hum Mol Genet (2010) 19(17):3468–76. doi:10.1093/hmg/ddq248
101. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet (2010) 42(12):1118–25. doi:10.1038/ng.717
102. Mäkivuokko H, Lahtinen SJ, Wacklin P, Tuovinen E, Tenkanen H, Nikkilä J, et al. Association between the ABO blood group and the human intestinal microbiota composition. BMC Microbiol (2012) 12:94. doi:10.1186/1471-2180-12-94
103. Henckaerts L, Van Steen K, Verstreken I, Cleynen I, Franke A, Schreiber S, et al. Genetic risk profiling and prediction of disease course in Crohn’s disease patients. Clin Gastroenterol Hepatol (2009) 7(9):972–980.e2. doi:10.1016/j.cgh.2009.05.001
104. Novacek G, Papay P, Miehsler W, Reinisch W, Lichtenberger C, Sunder-Plassmann R, et al. Are inherited thrombotic risk factors associated with fibrostenosis in Crohn’s disease? Inflamm Bowel Dis (2011) 17(12):2505–11. doi:10.1002/ibd.21648
105. Van Deventer SJ. Tumour necrosis factor and Crohn’s disease. Gut (1997) 40(4):443–8. doi:10.1136/gut.40.4.443
106. Cantor MJ, Nickerson P, Bernstein CN. The role of cytokine gene polymorphisms in determining disease susceptibility and phenotype in inflammatory bowel disease. Am J Gastroenterol (2005) 100(5):1134–42. doi:10.1111/j.1572-0241.2005.40979.x
107. Zipperlen K, Peddle L, Melay B, Hefferton D, Rahman P. Association of TNF-alpha polymorphisms in Crohn disease. Hum Immunol (2005) 66(1):56–9. doi:10.1016/j.humimm.2004.10.004
108. Sleegers K, Bettens K, De Roeck A, Van Cauwenberghe C, Cuyvers E, Verheijen J, et al. A 22-single nucleotide polymorphism Alzheimer’s disease risk score correlates with family history, onset age, and cerebrospinal fluid Abeta42. Alzheimers Dement (2015) 11(12):1452–60. doi:10.1016/j.jalz.2015.02.013
109. Kugathasan S, Collins N, Maresso K, Hoffmann RG, Stephens M, Werlin SL, et al. CARD15 gene mutations and risk for early surgery in pediatric-onset Crohn’s disease. Clin Gastroenterol Hepatol (2004) 2(11):1003–9. doi:10.1016/S1542-3565(04)00452-5
110. Sun L, Roesler J, Rösen-Wolff A, Winkler U, Koch R, Thürigen A, et al. CARD15 genotype and phenotype analysis in 55 pediatric patients with Crohn disease from Saxony, Germany. J Pediatr Gastroenterol Nutr (2003) 37(4):492–7. doi:10.1097/00005176-200310000-00017
111. Liberek A, Jakóbkiewicz-Banecka J, Kloska A, Świderska J, Kmieć Z, Łuczak G, et al. Clinical parameters of inflammatory bowel disease in children do not correlate with four common polymorphisms of the transforming growth factor β1 gene. Acta Biochim Pol (2011) 58(4):641–4.
112. Strisciuglio C, Auricchio R, Martinelli M, Staiano A, Giugliano FP, Andreozzi M, et al. Autophagy genes variants and paediatric Crohn’s disease phenotype: a single-centre experience. Dig Liver Dis (2014) 46(6):512–7. doi:10.1016/j.dld.2014.02.016
113. Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology (2012) 142(1):46–54.e42. doi:10.1053/j.gastro.2011.10.001 quiz e30.
114. Ng SC, Tang W, Ching JY, Wong M, Chow CM, Hui AJ, et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-pacific Crohn’s and colitis epidemiology study. Gastroenterology (2013) 145(1):158–165.e2. doi:10.1053/j.gastro.2013.04.007
115. Mahurkar S, Banerjee R, Rani VS, Thakur N, Rao GV, Reddy DN, et al. Common variants in NOD2 and IL23R are not associated with inflammatory bowel disease in Indians. J Gastroenterol Hepatol (2011) 26(4):694–9. doi:10.1111/j.1440-1746.2010.06533.x
116. Adeyanju O, Okou DT, Huang C, Kumar A, Sauer C, Galloway C, et al. Common NOD2 risk variants in African Americans with Crohn’s disease are due exclusively to recent Caucasian admixture. Inflamm Bowel Dis (2012) 18(12):2357–9. doi:10.1002/ibd.22944
117. Zouiten-Mekki L, Kharrat M, Karoui S, Serghimi M, Fekih M, Matri S, et al. Tolllike receptor 4 (TLR4) polymorphisms in Tunisian patients with Crohn’s disease: genotype-phenotype correlation. BMC Gastroenterol (2009) 9:62. doi:10.1186/1471-230X-9-62
118. Meddour Y, Chaib S, Bousseloub A, Kaddache N, Kecili L, Gamar L, et al. NOD2/CARD15 and IL23R genetic variability in 204 Algerian Crohn’s disease. Clin Res Hepatol Gastroenterol (2014) 38(4):499–504. doi:10.1016/j.clinre.2014.02.003
119. Yang SK, Park M, Lim J, Park SH, Ye BD, Lee I, et al. Contribution of IL23R but not ATG16L1 to Crohn’s disease susceptibility in Koreans. Inflamm Bowel Dis (2009) 15(9):1385–90. doi:10.1002/ibd.20921
120. Yamazaki K, Takahashi A, Takazoe M, Kubo M, Onouchi Y, Fujino A, et al. Positive association of genetic variants in the upstream region of NKX2-3 with Crohn’s disease in Japanese patients. Gut (2009) 58(2):228–32. doi:10.1136/gut.2007.140764
121. Ernst A, Jacobsen B, Østergaard M, Okkels H, Andersen V, Dagiliene E, et al. Mutations in CARD15 and smoking confer susceptibility to Crohn’s disease in the Danish population. Scand J Gastroenterol (2007) 42(12):1445–51. doi:10.1080/00365520701427102
122. Barreiro-de-Acosta M, Mendoza JL, Lana R, Domínguez-Muñoz JE, Díaz-Rubio M. NOD2/CARD15: geographic differences in the Spanish population and clinical applications in Crohn’s disease. Rev Esp Enferm Dig (2010) 102(5):321–6. doi:10.4321/S1130-01082010000500006
123. Cho JH. The promise of epigenetics. Has it delivered new insights? Digest Dis (2016) 34(1–2):12–9. doi:10.1159/000442919
124. Lin Z, Hegarty JP, Cappel JA, Yu W, Chen X, Faber P, et al. Identification of disease-associated DNA methylation in intestinal tissues from patients with inflammatory bowel disease. Clin Genet (2011) 80(1):59–67. doi:10.1111/j.1399-0004.2010.01546.x
125. Lin Z, Hegarty JP, Yu W, Cappel JA, Chen X, Faber PW, et al. Identification of disease-associated DNA methylation in B cells from Crohn’s disease and ulcerative colitis patients. Dig Dis Sci (2012) 57(12):3145–53. doi:10.1007/s10620-012-2288-z
126. Sadler T, Bhasin JM, Xu Y, Barnholz-Sloan J, Chen Y, Ting AH, et al. Genome-wide analysis of DNA methylation and gene expression defines molecular characteristics of Crohn’s disease-associated fibrosis. Clin Epigenet (2016) 8:30. doi:10.1186/s13148-016-0193-6
127. Frigola J, Muñoz M, Clark SJ, Moreno V, Capellà G, Peinado MA. Hypermethylation of the prostacyclin synthase (PTGIS) promoter is a frequent event in colorectal cancer and associated with aneuploidy. Oncogene (2005) 24(49):7320–6. doi:10.1038/sj.onc.1208883
128. You J, Nguyen AV, Albers CG, Lin F, Holcombe RF. Wnt pathway-related gene expression in inflammatory bowel disease. Dig Dis Sci (2008) 53(4):1013–9. doi:10.1007/s10620-007-9973-3
129. Hokari R, Kurihara C, Nagata N, Aritake K, Okada Y, Watanabe C, et al. Increased expression of lipocalin-type-prostaglandin D synthase in ulcerative colitis and exacerbating role in murine colitis. Am J Physiol Gastrointest Liver Physiol (2011) 300(3):G401–8. doi:10.1152/ajpgi.00351.2010
130. Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, et al. Crohn’s disease and the NOD2 gene: a role for Paneth cells. Gastroenterology (2003) 125(1):47–57. doi:10.1016/S0016-5085(03)00661-9
131. Vermeire S, Wild G, Kocher K, Cousineau J, Dufresne L, Bitton A, et al. CARD15 genetic variation in a Quebec population: prevalence, genotype-phenotype relationship, and haplotype structure. Am J Hum Genet (2002) 71(1):74–83. doi:10.1086/341124
132. Dotan I. Disease behavior in adult patients: are there predictors for stricture or fistula formation? Dig Dis (2009) 27(3):206–11. doi:10.1159/000228551
133. Cleynen I, Vermeire S. The genetic architecture of inflammatory bowel disease: past, present and future. Curr Opin Gastroenterol (2015) 31(6):456–63. doi:10.1097/MOG.0000000000000215
134. Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol (2011) 12(11):1063–70. doi:10.1038/ni.2113
135. Lighthouse JK, Small EM. Transcriptional control of cardiac fibroblast plasticity. J Mol Cell Cardiol (2016) 91:52–60. doi:10.1016/j.yjmcc.2015.12.016
Keywords: stricturing disease, fibrosis, Crohn’s disease, genetics, NOD2
Citation: Verstockt B and Cleynen I (2016) Genetic Influences on the Development of Fibrosis in Crohn’s Disease. Front. Med. 3:24. doi: 10.3389/fmed.2016.00024
Received: 07 April 2016; Accepted: 13 May 2016;
Published: 30 May 2016
Edited by:
Jennifer Bailey, University of Bristol, UKReviewed by:
Yuji Naito, Kyoto Prefectural University of Medicine, JapanTaro Takami, Yamaguchi University, Japan
Copyright: © 2016 Verstockt and Cleynen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabelle Cleynen, aXNhYmVsbGUuY2xleW5lbkBrdWxldXZlbi5iZQ==