 Jan Willem van der Laan1,2*
Jan Willem van der Laan1,2* Wenny H. W. Buitenhuis1
Wenny H. W. Buitenhuis1 Laura Wagenaar3
Laura Wagenaar3 Ans E. M. F. Soffers4
Ans E. M. F. Soffers4 Eugene P. van Someren5
Eugene P. van Someren5 Cyrille A. M. Krul5
Cyrille A. M. Krul5 Ruud A. Woutersen4,5
Ruud A. Woutersen4,5
- 1Medicines Evaluation Board, Utrecht, Netherlands
- 2Division of Toxicology, Leiden Academic Center for Drug Research, Leiden, Netherlands
- 3Veterinary Faculty, IRAS, Utrecht, Netherlands
- 4Division of Toxicology, Wageningen University and Research Centre, Wageningen, Netherlands
- 5TNO Innovation for Life, Zeist, Netherlands
In an exercise designed to reduce animal use, we analyzed the results of rat subchronic toxicity studies from 289 pharmaceutical compounds with the aim to predict the tumor outcome of carcinogenicity studies in this species. The results were obtained from the assessment reports available at the Medicines Evaluation Board of the Netherlands for 289 pharmaceutical compounds that had been shown to be non-genotoxic. One hundred forty-three of the 239 compounds not inducing putative preneoplastic lesions in the subchronic study did not induce tumors in the carcinogenicity study [true negatives (TNs)], whereas 96 compounds were categorized as false negatives (FNs) because tumors were observed in the carcinogenicity study. Of the remaining 50 compounds, 31 showed preneoplastic lesions in the subchronic study and tumors in the carcinogenicity study [true positives (TPs)], and 19 only showed preneoplastic lesions in subchronic studies but no tumors in the carcinogenicity study [false positives (FPs)]. In addition, we then re-assessed the prediction of the tumor outcome by integrating the pharmacological properties of these compounds. These pharmacological properties were evaluated with respect to the presence or absence of a direct or indirect proliferative action. We found support for the absence of cellular proliferation for 204 compounds (TN). For 67 compounds, the presence of cellular hyperplasia as evidence for proliferative action could be found (TP). Therefore, this approach resulted in an ability to predict non-carcinogens at a success rate of 92% and the ability to detect carcinogens at 98%. The combined evaluation of pharmacological and histopathological endpoints eventually led to only 18 unknown outcomes (17 categorized as FN and 1 as FP), thereby enhancing both the negative and positive predictivity of an evaluation based upon histopathological evaluation only. The data show the added value of a consideration of the pharmacological properties of compounds in relation to potential class effects, both in the negative and positive direction. A high negative and a high positive predictivity will both result in waiving the need for conducting 2-year rat carcinogenicity studies, if this is accepted by Regulatory Authorities, which will save large numbers of animals and reduce drug development costs and time.
Introduction
Specific regulatory requirements for carcinogenicity assessment of new pharmaceuticals are described in International Conference (now Council) on Harmonisation (ICH) guidance documents, i.e., ICH Guidelines M3(R2) (1), S1A (2), S1B (3), S1C(R2) (4), S2 (5), and S6(R1) (6).
Carcinogenicity studies are generally required for new pharmaceuticals that would be administered for 6 months or longer, or in a frequent and intermittent manner. In general, a 2-year rat study, plus either an 18 (or 24)-month mouse study or an alternative 6- or 9-month study in transgenic mice, is requested for such compounds.
The current 2-year rodent carcinogenicity study design used since the mid-1960s has been the regulatory standard in the safety assessment of humans. These carcinogenicity studies are expensive and time-consuming animal tests required for the safety assessment of pharmaceutical compounds. However, there is considerable scientific doubt about the reliability of the rat bioassay. Too many compounds are positively enhancing a tumor response in these studies, which may simply be due to the long-term exposure of the animals to rather high doses of the test compound (7–9) rather than a true carcinogenic effect. Therefore, there is a continued and increasingly need to justify the 2-year rodent bioassay in an attempt to reduce animal numbers, time, and costs (10, 11). For this reason, pharmaceutical companies and regulatory bodies are aiming to find an alternative approach.
In light of concerns raised about the predictability of in vivo studies in general and the push for refinement, reduction, and replacement of animal studies, it is strongly recommended to re-evaluate the suitability of the 2-year rodent bioassay as the best approach to predict human disease (12–21).
We have undertaken a retrospective study of pharmaceuticals with available rodent subchronic (3- to 6-month studies) and carcinogenicity data to test the hypothesis that it is possible to replace the current 2-year bioassay with a weight-of-evidence approach, i.e., by using evidence from all the non-clinical data available at the stage of development of a compound, usually at the end of Clinical Phase II, when a decision for conducting a 2-year carcinogenicity study is usually taken. Data that contribute are the results of the subchronic toxicity studies, in combination with genotoxicity data and knowledge of pharmacodynamic properties relating to the mode of action.
Positive in vivo genotoxicity tests are generally considered as indicative for a carcinogenic potency of a compound. Under REACH (22), classification as a mutagen category 1A or 1B allows a waiving of the carcinogenicity study, since the default presumption is that a genotoxic mechanism for carcinogenicity is likely. The same is true for human pharmaceuticals, where the ICH S1A guideline indicates that in case of positive genotoxicity, no lifetime carcinogenicity studies are expected. Positivity in an assay for DNA reactivity will usually also preclude further development (23), unless the risk for genotoxicity is acceptable in view of the benefit of the compound.
Jacobs (24) examined the data from 13-week rat toxicity studies for the prediction of carcinogenicity outcome using 60 pharmaceutical compounds. The data were obtained from a USA Food and Drug Administration (FDA) database. She concluded that various short-term indicators of carcinogenicity, such as hyperplasia, hypertrophy, greater organ weight, tissue degeneration or atrophy, and mineralization in a tissue, did not always result in tumors in that tissue, although (some of) these indicators are considered signs of potential carcinogenicity. The tissues examined were limited to the liver, kidneys, mammary glands, adrenals, urinary bladder, and lung.
Reddy et al. (25) confirmed the conclusion of Jacobs (24) with a different dataset. They used a “whole animal response” instead of individual tissues, testing the hypothesis that evidence of absence of putative preneoplastic lesions in any tissue may accurately predict a compound’s lack of carcinogenic potential. In their view, the presence of treatment-related putative preneoplastic histopathological lesions is not a definitive indicator of a tumorigenic potential of a compound, but rather requires that a 24-month rat carcinogenicity study has to be run and all tissues and organs should be collected and examined.
Sistare et al. (16) further evaluated the predictivity of histopathological findings, considering risk factors for rat neoplasia (hypertrophy, hyperplasia, metaplasia, cell proliferation, foci of cellular alteration, and inflammation accompanied by recurrent cell necrosis and repair) that were observed microscopically in 6-month rat toxicity studies with the tumor outcomes in rat 2-year carcinogenicity studies for 182 pharmaceuticals derived from 13 pharmaceutical companies. They concluded that the absence, rather than the presence, of the aforementioned putative preneoplastic histopathological changes in rats was a reliable predictor of tumor outcome in the corresponding tissue. The authors proposed that compounds demonstrating no genotoxicity, no evidence for hormonal mechanisms, and no histopathological changes pointing to a risk factor for rat neoplasia in any tissue (called NEGCARC approach) are considered rat non-carcinogens and can be exempted from the requirement for testing in a 2-year carcinogenicity study.
Assessors from EU Regulatory Authorities on human pharmaceuticals started to evaluate this approach and this set of data by emphasizing the consideration of the pharmacological properties of each compound and relating these properties to the outcome of the rat carcinogenicity study. The dataset was extended with data from FDA and the Japanese Pharmaceutical Manufacturer’s Association, increasing the number of compounds to 255 (26). Pharmacological properties appeared to be well associated with the outcome of the rat carcinogenicity study, both in a positive and negative direction. Classes such as β2-agonists and dopamine D2 antagonists were rather strongly associated with induction of mesovarian leiomyoma and mammary gland tumors, respectively (26). In addition, compounds inducing liver-associated pathology appear to be important in predicting tumors in organs such as liver, thyroid, and testis. The enzymes responsible for metabolism are also important factors in this respect.
The present retrospective study is intended to gather information independently from the dataset of Sistare et al. (16) to further test the hypothesis that a weight-of-evidence approach is possible in predicting the carcinogenic potential of human pharmaceuticals based upon histopathological and pharmacological properties. We have used a more restrictive approach in the definition of putative preneoplastic changes as compared with Sistare et al. (16), which will be further discussed below.
Materials and Methods
Selection of Compounds
The rat subchronic toxicity and chronic carcinogenicity data used for this evaluation came from the assessment reports that are available at the Medicines Evaluation Board of the Netherlands. The compounds and studies that were included were those in the paper of Van Oosterhout et al. (27), which reviewed all of the carcinogenicity studies from 1979 when GLP was introduced, although it was not possible to retrieve all of the studies covered in that paper, those submitted for marketing authorization in the Netherlands between 1995 and 2004, and those authorized via the EU centralized procedures between 2004 and 2014. The criteria used to identify valid pairs of rat subchronic (3- and/or 6-month) studies and 2-year carcinogenicity studies were as described below.
ToxRefDB Database Structure
We have downloaded an empty version of ToxRefDB from the website of the US Environment Protection Agency (EPA), and we have used the structured organization to summarize all the data available for the carcinogenicity studies. We started our assessment based on the assessment report, but in cases where there was a lack of detail, we used the non-clinical expert report, non-clinical overview, or other parts from the dossier available on microfilm or electronically.
Comparison of Protocols of Subchronic Studies with Carcinogenicity Studies
Although similar criteria regarding overlap of dose range have been applied as previously reported by Reddy et al. (25) and Sistare et al. (16) for inclusion of the studies, we have included all compound datasets available, as the impact of the pharmacological properties is expected to be largely independent of the matching of doses.
The database contained the dose levels used in the subchronic and carcinogenicity studies and the effects of the pharmaceuticals on body weight, organ weight, histopathology of a large set of organs and tissues, and results of genotoxicity tests in vitro and in vivo.
Criteria for Positive Genotoxicity Test Result
Genotoxicity is usually tested with a battery of genotoxicity assays, as detailed in the ICH guidelines S2A and 2B revised in S2R2 (5). The outcome of these tests was taken from the assessment reports and was not re-assessed.
Compounds Classification and Description of Study Outcomes
The pharmaceuticals were classified for pharmacology, and subsequently, the histopathological outcome of the studies has been described. The results of genotoxicity studies were included to cover similar criteria as used by Sistare et al. (16).
Pharmacological Criteria
The pharmacotherapeutic areas were based on target organs, such as central nervous system (CNS), cardiovascular system (CVS), respiratory system (RS), metabolic system (MB), hormonal system (HM), gastrointestinal system (GIS), immunological system (IS), and antimicrobials (AM) (divided into antibacterials, antimalarials, antivirals, antifungals, and remaining compounds). Each pharmacotherapeutic category was subdivided into classes according to the primary drug target of the compounds in accordance with Stefansdottir et al. (28). Small molecules may exert additional pharmacological activity at a higher dose, which is termed secondary pharmacodynamics and could, as described by Keiser et al. (29), be responsible for the carcinogenic response.
Histopathological Criteria
Positive histopathology observations were scored if any of the selected histopathological changes were reported as being increased in the subchronic toxicity studies, i.e., cellular hypertrophy, cellular hyperplasia, presence of altered hyperplastic foci of cellular alteration (atypical) cell foci (basophilic; acidophilic foci), cellular proliferation, and dysplasia. Furthermore, any changes in body weights and organ weights changes were noted.
The compounds were scored as negative for histopathological evidence of potential preneoplasia when the aforementioned histopathological changes were absent or not considered as being increased by treatment. All incidences reported to be higher than the controls were considered positive; with no access to all of the original study reports, no attempts were made to re-evaluate them in terms of whether or not they were statistically significant. When the histopathological changes were incorporated in the database, they were considered to be related to treatment and significantly increased.
We have scored greater organ weights, hypertrophy, and hyperplastic findings separately in Table 1. Furthermore, we have listed the tumors observed by describing the organ system and the histopathological appearance.
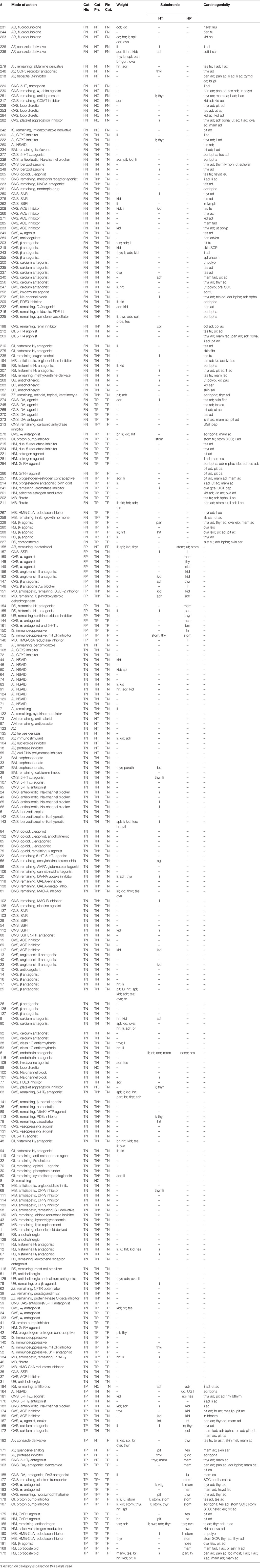
Table 1. Summary of the observations in the subchronic and carcinogenicity studies.
Step 1: Categorization Based on Histopathology
We have categorized all compounds on the basis of histopathology findings in chronic studies and their relation to tumor findings in the 2-year studies, similar, but not identical, to the criteria in the work of Sistare et al. (16). The following four categories were identified:
1. Compounds that were negative for histopathological findings considered to be putative preneoplastic in the subchronic study and in the carcinogenicity study were considered true negatives (TNs). Compounds inducing hypertrophy only in subchronic studies were scored as negative.
2. Compounds that were positive for histopathology findings considered to be putative preneoplastic in the subchronic study, but negative for carcinogenicity, were classified as false positives (FPs).
3. Compounds that were positive for both the presence of treatment-related putative preneoplastic histopathological lesions in the subchronic study and the presence of treatment-related benign and/or malignant tumors in the carcinogenicity study were considered true positives (TPs).
4. Compounds that were negative for histopathology findings in the subchronic study, but positive for carcinogenicity, were considered false negatives (FNs).
Step 2: Categorization Based on Pharmacology
For each pharmacological class, we have listed all compounds and the outcomes of the subchronic study and the carcinogenicity study. We have counted the number of compounds in a pharmacological class and the number of compounds inducing carcinogenicity.
• A class of compounds was called positive, when 75% of the compounds were associated with tumor induction. These classes are listed in Table 3.
• A class of compounds was called negative, when 75% of the compounds were not associated with tumor induction. These classes are listed in Table 4.
• A class of compounds was called with mixed outcome, when more than 25%, but less than 75%, of the compounds were associated with tumor induction. These classes are listed in Table 5.
After considering the mode of action (also based on publicly available literature) leading to induction of tumors, we made an evaluation of the probability that the pharmacology would be the main mode of action causing the carcinogenicity. We have discussed this and added to the tables as the proposed final categorization.
Results
A total of 366 pharmaceuticals have been evaluated in the present study, of which 289 met the criteria described in Section “Materials and Methods” for defining valid pairs of rat subchronic (3 and/or 6-month) and 2-year carcinogenicity studies.
Genotoxicity Evaluation
In our dataset, 21 compounds were assessed as positive or inconclusive with respect to genotoxicity. To give a detailed description is not relevant, since this assessment is conducted initially during the assessment for marketing authorization. After discussions with the sponsor, and before making a decision about the authorization, it was agreed that the genotoxicity findings do not influence the risk for patients. We have therefore decided not to give any weight to these data. None of those compounds is intended to be given to patients with a life-threatening disease, in which case a serious benefit would be judged to outweigh the genotoxicity risk as specified in ICH S1A (2).
Histopathological Classification
True Negative Compounds
One hundred forty-three (50% of the total) pharmaceuticals that did not induce putative preneoplastic lesions in the subchronic (3- and/or 6-month) study also did not cause treatment-related tumors in the carcinogenicity study (Table 1, third column). Ninety-nine of these 143 compounds (69%) did neither exhibit any effect on organ weight nor induce cellular hypertrophy in any organ (Table 1).
Ninety-one of the 143 TN compounds (31% of the total) demonstrated a greater weight of one or more organs, such as liver (n = 16); kidneys (n = 17); heart (n = 11); adrenals (n = 10); spleen, ovaries, and brain (n = 5); testes (n = 7); thyroid (n = 6); pituitary and lungs (n = 3), or incidentally (n = 1) other organs, either or not in combination with cellular hypertrophy (2 substances in liver and 1 substance in kidneys). Twenty-six pharmaceuticals showed cellular hypertrophy in the liver (n = 13), thyroid (n = 5), adrenals (n = 2), and incidentally in the kidneys, mammary glands, bone, salivary glands, or intestines.
Cellular hypertrophy was mainly observed in the liver (n = 17), in four cases accompanied by greater liver weight, but not accompanied by the development of benign or malignant hepatocellular tumors.
False Negative Compounds
Ninety-six (33% of the total) substances did not exhibit putative preneoplastic lesions in the subchronic study, whereas treatment-related benign and/or malignant tumors developed in the carcinogenicity study.
Fifty-two of these 96 compounds (54%) induced benign tumors in a single organ (39) or in multiple organs (13), mainly testes (Leydig cell adenomas), adrenals (pheochromocytomas), pancreas (acinar cell adenomas), thyroid (follicular cell adenomas), pituitary (pars distalis adenomas), and liver (hepatocellular adenomas). Incidental benign tumors were seen in mammary gland, skin, ovaries, urogenital tract, and spleen.
Thirty-five of the 96 compounds (36%) induced benign and malignant tumors in the same organ, mainly liver, thyroid, testes, mammary gland, and pancreas (9%), or in multiple organs (27%).
Nine of the 96 compounds (9%) induced malignant tumors only in a single or multiple organs. These malignant tumors comprised skin sarcomas; adenocarcinomas of the uterus, a localized lymphoma; adenocarcinomas in the kidneys; and leukemia.
Seventy-seven of the 96 compounds (80%) caused tumors that frequently occur spontaneously in rats of the age examined and most of them are not considered relevant for humans.
False Positive Compounds
Of the 50 compounds that induced putative preneoplastic (hyperplastic) histopathological lesions in the subchronic study, 19 of these (38%) failed to induce treatment-related tumors in the carcinogenicity study.
For these FP compounds, the site of histopathological evidence (cellular hyperplasia) of risk for rat neoplasia was the mammary glands, kidneys, and liver (three compounds each); thyroid and pancreas (two compounds each); and adrenals, stomach, uterus, lymph nodes, thymus, and bone marrow (one compound each).
True Positive Compounds
Thirty-one substances (11% of the total number) induced putative preneoplastic (hyperplastic) histopathological changes in the subchronic study and treatment-related neoplasms in the carcinogenicity study. In only 13 out of these 31 compounds (42%), the hyperplastic lesion and the tumor developed in the same organ, whereas for the other 18 compounds (58%), the hyperplastic lesions observed in the subchronic study did not develop in the same organ as the tumor in the carcinogenicity study.
Four compounds caused mammary gland hyperplasia in the subchronic study and mammary gland adenomas or carcinomas in the carcinogenicity study. One compound caused mammary gland hyperplasia without the development of mammary gland tumors. This compound induced follicular cell adenomas and carcinomas in the 24-month study but no follicular hyperplasia in the thyroid in the subchronic study. Six substances induced mammary gland (fibro)adenomas and/or adenocarcinomas in the mammary gland without mammary gland hyperplasia in the subchronic study.
Pharmacological Analysis of the Carcinogenic Response
The 289 human pharmaceuticals in the dataset were distributed over therapeutic areas as indicated above (Table 1, fourth column). Most of the therapeutic areas are divided over several pharmacological classes, and all compounds are distributed through these classes. However, the compounds are anonymized (see Table 1) due to intellectual-property reasons. A similar approach was followed as for the previous paper on the PhRMA–FDA–JPMA dataset (PFJ dataset) (26).
CNS Drugs
DA2 Agonists [1], Refers to the Class in Tables 3–5
All four (245, 265, 270, and 274) were found to induce tumors in the sexual organs. For three compounds (245, 270, and 274), Leydig cell adenomas were observed, while for the fourth compound (265), uterine carcinomas have been described. For one compound (274), skin fibromas were observed too, while for another (265), a decrease in spontaneous pituitary adenomas was seen. The dopaminergic DA2 agonists are associated with an increase of luteinizing hormone, and the fact that all four compounds induced either testis tumors or uterus tumors confirms the association. In the PFJ dataset (26), only two compounds were included, with one showing the same tumor profile. Based upon the literature (30), we could find support for a pharmacodynamic relationship and so we categorized this class as TP.
DA2 Antagonists [2]
Three out of four (59, 163, 188, and 273) showed mammary gland adenocarcinomas (two TP and one FN). For one compound, this was the only type of tumor observed (188). A second compound (273) induced in addition pancreatic acinar cell adenomas and pituitary adenomas. The third compound (163) induced adrenal pheochromocytomas, in addition to mammary gland carcinomas. In a 3-month study, mammary hyperplasia was observed for this compound making it a TP. The fourth compound (59) was a TN. It is well known that administration of dopaminergic DA2 antagonists is associated with an increase in prolactin, resulting in mammary adenocarcinoma in rodents. Based on this pharmacological effect, we categorized this class as TP.
5HT1b/d Agonists [43]
Four triptanes (4, 107, 181, and 277) showed a rather mixed response. Two compounds (4 and 107) did not induce either tumors or hyperplastic responses after 6 months (TN). Two other compounds induced a variety of tumors, one (277) with benign pheochromocytomas and Leydig cell adenomas (FN), while with the other (181), thyroid follicular cell adenomas and thymomas were observed. However, with the latter compound, Leydig cell hyperplasia was observed at 6 months (TP). Effects on thyroid and testis (Leydig cell hyperplasia and adenoma) are likely be related to liver enzyme induction. In both cases, these effects cannot be related to the direct pharmacodynamic action, which is in agreement with the absence of proliferative effects (31). A general category of TN is given to this class of compounds.
5HT3 Antagonists [44]
5HT3 antagonists (95 and 176) have an inhibitory effect on the growth of HT29 cells (32). Two 5HT3 antagonists showed different responses, one (95) was TN, while the other (176) induced liver adenocarcinomas. As this is unrelated to its pharmacology, the class was categorized as TN.
Serotonin and Noradrenaline Reuptake Inhibitors (SNRI) [47]
Four SNRIs (137, 203, 250, and 276) are included with two (137 and 103) as TN, while the other two compounds (250 and 276) as FNs showed thyroid adenomas or testicular adenomas, respectively. As these tumors were not associated with primary pharmacology, this class was categorized as TN.
μ-Opioid Antagonists [26]
Two are included (85 and 86), both TN, and we categorized the class in this way.
CNS, Na+ Channel Blockers [46]
From six antiepileptics sharing the property of being sodium channel blockers (24, 49, 65, 66, 183, and 239), four are TNs, one compound (183) showed liver hyperplasia at 6 months and hepatocellular adenocarcinomas after 2 years (TP). The sixth compound (239) showed only adrenal pheochromocytomas after 2 years (FN), which are not relevant for humans. The class is categorized as TN.
Selective Serotonin Reuptake Inhibitors [27]
Seven compounds (29, 35, 54, 112, 157, 262, and 88) are included. Five were TNs, with one of them (112) showing hepatic hypertrophy, but no hyperplasia, at 6 months. The sixth compound (262) induced lymphoreticulum cell tumors (FN), while the seventh (157) only showed liver hyperplasia (FP). There is no direct relation with pharmacology, and the relevance for humans is estimated to be negligible. Therefore, selective serotonin reuptake inhibitors (SSRIs) are categorized as TN.
μ-Opioid Agonists [42]
Of the three μ-opioid agonists, two (84 and 132) are TN, whereas one (205) compound showed testis tumors and leukemia (at a non-matching dose). As opiates are not associated with proliferative action, this group was categorized as TN.
Benzodiazepine(-Like) Compounds [45]
Three real benzodiazepines and two benzodiazepine-receptor agonists are in this class. Three (5, 142, and 143) are TN. One benzodiazepine (204) showed treatment-related thymus lymphomas, thyroid follicular cell adenomas, and uterus schwannomas, especially at high dosages, while another (248) showed only thyroid follicular cell adenomas. The latter compound also induced an increase in thyroid weight at 6 months. As these effects were associated with induction of liver metabolism rather than attributed to the primary pharmacology, this class was categorized as TN.
5HT2 Antagonists [16]
Two compounds (185 and 220) are included. Compound 185 induced mammary gland hyperplasia after 3–6 months, and mammary gland adenocarcinomas and thyroid follicular cell adenomas after 2 years (TP). Compound 220 induced only liver adenomas and no effects at 6 months. There are signals that 5HT2 antagonists induce an increase in prolactin, which might be responsible for the association with mammary gland tumors. This has also been discussed with the PFJ dataset (26). However, this relation between 5HT2-receptor blockade and prolactin is not without discussion, and in this case, compound 185 might have also anti-DA2 affinity. Because of this uncertainty with respect to the pharmacology, we did not apply a category based upon pharmacology for this class (NC).
Remaining 5HT Compounds
One compound (22), a 5HT1 agonist was TN. We maintained for the 5HT1 agonist the TN category.
Remaining Opioid Compounds
One κ-agonist (75) is a TN, which is what we categorized it too.
Remaining CNS Compounds
Seventeen compounds with a large variety of pharmacological targets remained. Nine TNs are a nicotine agonist (136), an acetylcholinesterase inhibitor (56), a GABA-enhancer (118), a GABA-metabolism inhibitor (138), a dopamine–noradrenaline reuptake inhibitor (20), a monoamine oxidase inhibitor A (MAO-A) (81), a MAO-B inhibitor (102), an AMPA glutamate antagonist (96), and a cannabinoid antagonist (106). Several FNs were also present. An α2δ agonist of the L-calcium channel (230) induced pancreatic acinar cell adenomas and acinar cell adenocarcinomas, and Leydig cell adenomas in males and uterine endometrial polyps in females (33). A direct pharmacological explanation could not be found in relation to this receptor (34). Therefore, we maintained a category FN. A melatonin receptor agonist (197) induced hepatocellular adenomas and carcinomas, and we gave a TN categorization because of the absence of an association with melatonin receptor stimulation. A COMT inhibitor (217) was associated with an increase in kidney tubular adenomas and carcinomas. We maintained category FN, as the kidney effect is likely to be an off-target effect (i.e., not related to pharmacology). An NMDA antagonist (223) induced Leydig cell tumors. However, NMDA are usually negative (26) and therefore were assigned a TN category. A nootropic drug (261) was associated with adrenal pheochromocytomas, but categorized as TN, as these tumors are not relevant to humans (see below). A tetracyclic antidepressant (251) induced liver adenomas and thyroid follicular cell adenomas based upon induction of metabolism, and also mammary gland tumors, and therefore, we categorized this compound as FN, as there was obvious pharmacological explanation for the latter tumor. A carbonic anhydrase inhibitor (213) induced urinary bladder papillomas. These compounds are known to be associated with crystallization in rat urinary bladder, and we applied a category TP for this compound. An electron transporter (177) is labeled as TP inducing forestomach hyperplasia in 6-month studies and forestomach squamous cell and basal cell carcinomas in the 2-year study.
Several CNS compounds were associated with adrenal pheochromocytoma, i.e., the DA2 antagonist 163, the NA-channel blocker 239, and the nootropic drug 261. Pheochromocytoma (a tumor developing from the chromaffin cells, which are the sites of synthesis and storage of catecholamines) is the most common neoplasia of the adrenal medulla in rodents. Pheochromocytomas are frequently found in a background of diffuse medullary hyperplasia. Compounds producing this feedback interference include lactose and sugar alcohols such as lactitol and Ca2+. High doses of low digestibility carbohydrates, such as mannitol, sorbitol, xylitol, and lactitol, have been reported to increase the absorption and urinary excretion of Ca2+ as well as the incidence of all types of proliferative lesions in the adrenal medulla. Hypercalcemia is known to increase catecholamine synthesis. Other compounds that might act via altered Ca2+ homeostasis and progressive nephrocalcinoses in aging rats include the retinoids. Vitamin D is the most potent in vivo stimulus, yet identified for chromaffin cell proliferation in the adrenal medulla. Vitamin D3 resulted in a fourfold to fivefold increase in bromodexoyuridine (BrdU) labeling (35) in the adrenal medulla (focal hyperplasia), leading to pheochromocytomas. In the PFJ dataset, we identified four vitamin D-analogs, all associated with adrenal pheochromocytoma (26).
Cardiovascular Drugs
ACE Inhibitors [33]
Eleven angiotensin-converting enzyme inhibitors showed a variety of classifications. Four (15, 37, 69, and 117) are TN. Two (174 and 186) are TP based on kidney hyperplasia at 6 months, and for one compound (174), pituitary gland adenocarcinomas and mesentery lipomas at 2 years, while for the other compound (186), benign hemangiomas were observed in the lymph node. Five compounds (208, 266, 271, 285, and 233) are FN with kidney adenomas in compound 271, mammary fibroadenomas in compound 285, Leydig cell tumors in compound 208, thyroid follicular cell adenomas with compound 266, and thyroid follicular cell adenomas and endometrial polyps with compound 233. Van der Laan et al. (26) mentioned seven negative compounds. The kidney as the target organ of compound 271 suggests a pharmacological effect that might be related to kidney hyperplasia (juxtaglomerular hyperplasia), which was seen with compounds 174 and 186, as well as with angiotensin II antagonists (26). Mammary gland fibroadenomas, as seen with compound 285, are the most common spontaneous tumors in female rats in almost all the routinely used rat strains with incidences of up to 70% in carcinogenicity studies. Fibroadenomas do not progress to malignancy and are not considered to be relevant for humans, whereas mammary gland adenocarcinomas in rats may be more relevant (35, 36). It is important to consider that the windows of mammary gland susceptibility or mammary gland sensitivity are missed when exposure starts in adult nulliparous rodents as is routinely the case in bioassays with pharmaceuticals. The tumors induced by 208 and 266 are likely to be associated with induction of liver metabolic enzymes. The variety of tumors seen with compound 174 is complex. We have categorized the ACE inhibitors as TN in accordance with Van der Laan et al. (26) overruling all other categories for individual compounds.
Ca Antagonists [14]
Twelve Ca antagonists are included in this dataset. Four are TN (9, 90, 92, and 93). Two (165 and 172) are TP, the first with lymph node hyperplasia at 6 months and thyroid follicular cell adenomas after 2 years, and the other showed colon hyperplasia of the muscularis mucosa at 6 months and after 2 years Leydig cell adenomas in the testis, mammary gland fibroadenomas, adrenal pheochromocytomas, and pituitary adenomas and carcinomas. Six others compounds (200, 235, 237, 240, 256, and 247) are FN. Two compounds (235 and 237) induced Leydig cell tumors, one induced thyroid follicular cell adenomas (256), and one compound (240) was associated with pituitary gland adenomas and mammary gland fibroadenomas. Compound 200 induced uterine polyps, while compound 247 induced uterine polyps and oral mucosa squamous cell carcinomas. Calcium antagonists, especially dihydropyridines, are not associated with tumor induction (37). Eight out of 12 compounds in our dataset are associated with the induction of tumors. Two compounds (165 and 256) induced thyroid tumors only, and two (235 and 237) only induced testis tumors. Two compounds (172 and 240) induced mammary fibroadenomas and pituitary tumors. Compound 247 induced uterine polyps (as did compound 200) and specifically gingival squamous cell carcinoma. The latter phenomenon is reported for mibefradil (38), as being due to the oral intake as diet mixture, and not directly related to the pharmacological effect. Van der Laan et al. (26) classified calcium antagonists as negative, in accordance with absence of induction of cancer in humans (39). We, therefore, categorized all Ca antagonists as TN, taking into account these considerations.
Angiotensin II Receptor Antagonists [21]
Five compounds (13, 40, 156, 23, and 162) were included. Three compounds (13, 23, and 40) are TN, and two (156 and 162) showed juxtoglomerular hyperplasia at 6 months (FP). The induction of juxtaglomerular hyperplasia does not predict further development to kidney tumors, which confirms the findings of Van der Laan et al. (26), with a slightly different sample set (only two compounds overlap). We categorized the class as TN.
Adrenergic α1 Antagonists [34]
Eight adrenergic α1 antagonists (19, 34, 133, 144, 192, 193, 278, and 161) showed different target organs, with three TN (19, 34, and 133). Three compounds showed mammary gland hyperplasia (144, 192, and 193) and two (193 and 278) mammary gland tumors. Two compounds (192 and 193) are TP with mammary gland hyperplasia at 6 months and mammary gland adenomas and mononuclear cell leukemia after 2 years. Two (144 and 161) are FP, with mammary gland acinar hyperplasia at 6 months for one (144) and bone marrow hyperplasia at 6 months for the other (161). One FN (278) showed after 2 years mammary gland adenocarcinomas and adrenal phaechromocytomas. A direct pharmacological explanation for this connection to the mammary system is unknown at this time. In addition, a connection between α1 antagonism and mammary tumor formation is also unknown. A choice for the final classification TP is based on the effects on the mammalian gland, both after 6 months and 2 years for several compounds, despite the fact that we have no clear molecular mechanism. Further research is, therefore, important to study this possible association of α1 antagonism and mammary tumor formation.
Adrenergic α1 Agonists [35]
Two compounds are included; one (249) is FN with only Leydig cell adenomas (related to enhanced liver metabolism) at 2 years, while the other (159) was FP with mammary gland acinar hyperplasia at 6 months but no tumors at 2 years. TN was chosen for the final categorization.
Adrenergic α2 Agonists [36]
Three compounds (145, 149, and 167) are included, of which one (145) is a TN. Compound 149 is FP with hyperplasia of the islets of Langerhans in the pancreas, while compound 167 is TP with small intestines hyperplasia at 6 months and pancreas acinar adenocarcinomas, thyroid follicular cell adenomas, and mammary gland adenomas after 2 years. Three adrenergic α2 agonists, therefore, showed variable effects. The islet cell hyperplasia seen with compound 149 might be related to the pancreatic adenocarcinoma seen with compound 167. A pharmacological target in the pancreas for this class is well known to be inhibitory, e.g., inhibiting insulin secretion. Other compounds in this class have been mentioned in the literature as negative (23, 26, 40), and therefore, this group is categorized as TN.
Adrenergic β-Antagonists [25]
Thirteen β-blockers (14, 16, 17, 25, 26, 126, 127, 147, 148, 203, 219, 243, and 255) are included, with seven categorized as TN. Two FP (147 and 148) showed thyroid follicular cell hyperplasia or hepatocellular hyperplasia but no tumors. Of the four FN, compound 201 showed hepatocellular hepatomas, compound 219 showed forestomach squamous cell papillomas, compound 243 showed pituitary gland tumors, and compound 255 showed spleen vascular neoplasia. The carcinogenicity potential of β-blockers has been a debate from their early existence, especially with respect to pronethalol (41).The tumors found were heterogeneous and therefore probably not related to pharmacology. Snyder and Green (23) also mentioned a low incidence of compounds associated with tumors for this class (2 out of 10). This class was therefore categorized as TN overruling the FN compounds.
Anticoagulants [37]
Two anticoagulants (10 and 289) showed slightly different outcomes. The tumor outcome of compound 289 was in fact not statistically relevant but was decided to be a safety signal. However, pancreatic acinar adenomas/carcinomas are usually not relevant for humans. Therefore, we applied a category TN.
Imidazoline Agonists [38]
Two compounds are included (105 and 252): one is TN, while the other is FN with adrenal phaechromocytomas and hind limb tumors after 2 years. The adrenal tumors seen with compound 252 might be reflected by a greater adrenal weight for compound 105, although this might be speculative. The human relevance is low anyway. Therefore, it was decided to categorize these as TN.
CVS, Na-Channel Blockers [39]
Of the three Na-channel blockers used in cardiac treatment (100, 101, and 272), two were TN, compound 227 showed thyroid and Leydig cell adenomas, as well as adrenal pheochromocytomas. As these tumors are more related to drug metabolism, and not pharmacology, we categorized this class as TN.
Loop Diuretics [16]
Three of the four (98, 229, 253, and 284) compounds are FN with compound 284 causing kidney carcinomas, compound 229 causing thyroid follicular cell carcinomas and pituitary gland adenomas, and compound 253 causing uterus adenocarcinomas and Leydig cell adenomas. Compound 284 is a FN, and its target organ suggests a pharmacological profile for the carcinogenesis. The target organs of the other loop diuretics are probably not associated with their pharmacology. As this positive relationship was found for only one compound, we decided to leave the categorization for this class as undecided (NC: non-categorizable based on pharmacological target), with no change of the histopathological categories.
Class 1C Channel Blockers [22]
Two compounds (38 and 53), are included, both are TNs, and we also applied this categorization to these based on their pharmacology.
Endothelin Antagonists [23]
Two compounds (6 and 115) are included, both are TNs. No proliferative effects are reported for these compounds, and we categorized them as TN based on pharmacology.
Vasopressin-2 Agonists [24]
Two compounds (110 and 131) are included and both were TN. No proliferative effects are reported for these compounds, and we categorized them as TN based on pharmacology.
Platelet Aggregation Inhibitors [40]
Two compounds (99 and 282) are included. One (99) was TN. The other (282) showed a variety of tumors, such as thyroid follicular cell adenomas, adrenal pheochromocytomas, hepatocellular adenomas, and endocrine-related tumors such as uterine adenocarcinomas, ovarian adenomas, and mammary gland adenomas. Two platelet aggregation inhibitors had a different tumor response; the first was TN, whereas the other compound was FN because of a dopaminergic action as a secondary pharmacological effect (42). These compounds were not categorized related to pharmacology (NC).
Phosphodiesterase 3 Inhibitors [41]
One compound (77) was TN, while with the other (209), adrenal pheochromocytomas were observed. Therefore, we decided to categorize the class as TN.
Remaining CVS Compounds
Eleven compounds with a variety on pharmacological targets remained to be categorized. A 5HT2 antagonist (63) is TN, as is a β1-partial agonist (141). Five other TNs are a nitrate agonist (89), a phosphodiesterase-5 inhibitor (113), a sodium-channel inhibitor, a hemostatic compound (36), and a vasodilator (nitric oxide agonist) (78). All compounds were maintained in the TN category. A hydrazine (169) is TP with thyroid follicular cell hyperplasia after 6 months and thyroid follicular cell adenomas and carcinomas after 2 years. Because of the thyroid hyperplasia (without any liver signal) after 6 months, we maintained the TP category in this case, although a relation with pharmacology needs to be substantiated. Four compounds are FN, i.e., a DA1/α1 agonist (232) that induced pancreatic acinar cell adenoma, an imidazole PDE inhibitor (216) that was associated with adrenal pheochromocytomas, a quinolone vasodilator (225), and a renin inhibitor (198) that was associated with colon adenoma and carcinoma. However, the colon is unlikely to be a pharmacological target, as Kochi et al. (43) described an antagonistic effect for this relation. Therefore, we decided to give a TN category to this class.
Gastrointestinal System
Proton Pump Inhibitors [12]
Four compounds (41, 178, 187, and 259) are included, with one TN. One TP (187) showed stomach hyperplasia after 3 months, and monocytic cell leukemia, adrenal pheochromocytomas, pituitary adenomas, Leydig cell adenomas, and squamous cell carcinomas in the forestomach. Another TP (178) showed stomach hyperplasia at 6 months and Leydig cell adenomas at 2 years. The third compound (259) was associated with hepatocellular adenomas and squamous cell carcinomas in the stomach. The fourth compound was TN. The stomach hyperplasia and squamous cell carcinoma are clearly related to the pharmacology of this class. Therefore, we decided that this class should be categorized as TP, overruling the TN case.
5HT4 Agonists [18]
From the three compounds (121, 269, and 212), one compound (121) is a TN, while the other two are FN with tumors in the 2-year studies. Compound 269 showed Leydig cell adenomas and pituitary adenomas, while compound 212 showed thyroid follicular cell adenomas, mammary gland fibroadenomas, pancreatic acinar cell adenomas, adrenal pheochromocytomas, hepatocellular adenomas, and pituitary adenomas. The GI 5HT4 agonists are likely to be associated with indirect metabolic effects on thyroid hormones (269) or testosterone (212). However, this can be debated for compound 269, but a direct pharmacological explanation is not known. We categorized all these compounds, therefore, as TN.
Histamine H2 Antagonists [49]
From the four compounds, two (48 and 94) are TN. The other two (210 and 275) are FN, with 275 associated with an increased number of skin fibromas, and the compound 210 with Leydig cell adenomas. Histamine H2 antagonists are used as gastric acid secretion inhibitors. Inhibition of gastric secretion might be associated with long-term induction of gastric carcinoids as associated with their pharmacological action. None of the compounds in this dataset showed this effect. Therefore, these were categorized as TNs, although in the previous paper, the H2 antagonists belonged to the positive class.
Gastrointestinal, Remaining Compounds
Five compounds remained in this class, and four were categorized as TN, i.e., a synthetic prostaglandin (80), a phosphate binder (30), an imaging/anti-osteoporose agent (119), and a Fe-chelator (32). A sugar alcohol (compound 238) is associated with Leydig cell adenomas (FN). As this is a rather unspecific effect, the compound was categorized as a TN.
Hormonal System
GnRH Agonist [3]
From the five compounds (21, 175, 180, 254, and 286), one is TN (21) while two are TP (175, 180), one with pituitary hyperplasia (180) after 3 months and pituitary adenomas after 2 years, and the other showed Leydig cell hyperplasia after 6 months and pituitary adenomas after 2 years. Two compounds (254 and 286) are FN, with no hyperplastic effects after 6 months, but both associated with pituitary adenomas and carcinomas after 2 years. One of these compounds (254) showed in addition Leydig cell adenomas and pancreatic islet cell adenomas, and adrenal benign and malignant pheochromocytomas. The relation between pharmacology of GnRH agonists and the pituitary tumors observed for four compounds is clear, as the target organ is the pituitary (44). Only one compound (21) is TN, but we categorize all as TP based upon the similar pharmacology as the other members of the class.
Estrogen Agonist [4]
Two compounds (221 and 281) are included; one compound (221) with pituitary gland adenomas, and the other (281) with mammary gland carcinomas and hepatocellular adenomas. For the estrogen agonists, the identity of the target organ supports a pharmacological relationship. Estrogen agonists are important as potential human-relevant carcinogens based upon IARC evaluations (45), and we categorized this class, therefore, as TP.
Selective Estrogen Receptor Modulators [5]
Two compounds (179 and 201) have a similar tumor profile, i.e., kidney adenocarcinomas and ovary adenomas. Only one (179) showed ovary hyperplasia after 6 months making it a TP, while the other (201) was an FN. We categorized this class as TP.
Dual 5-Reductase Inhibitors [6]
Two compounds (215 and 224) are included, and these are FN. One compound induced Leydig cell adenomas, while the other induced only thyroid follicular cell adenomas. Because of their hormonal action, we categorized these compounds as TP, although the possibility that the tumors might be the consequence of just the induction of liver metabolism cannot be discounted.
Progestogen–Estrogen Combinations [7]
From the two combinations (42 and 257), one (42) is a TN and the other is a FN (257), with mammary gland adenocarcinomas and pituitary adenomas after 2 years. A pharmacological effect on the basis of estrogenic activity is considered to be likely, and therefore, we applied a TP category.
Hormones, Remaining Compounds
An antiandrogen (166) is a TP, with Leydig cell hyperplasia and ovary hyperplasia observed at a 3- or 6-month study. Leydig cell adenomas and thyroid follicular cell adenomas and uterus adenocarcinomas were seen after 2 years. An aromatase inhibitor (241) is FN, with urinary bladder papillomas and ovary benign stromal cell tumors after 2 years.
For all pharmacological classes associated with sexual hormones, we conclude that such a relationship is likely to exist between pharmacological action and the induction of tumors, such as for the dual 5-reductase inhibitors (215 and 224), and the progestogen-containing combinations (42, 214, and 257). Therefore, we categorized all of these compounds as TP.
Immunological System
Immunosuppressives [29]
Six immunosuppressive compounds (120, 140, 150, 47, 152, and 52) are all negative. Four compounds are TN and two FP. One of the FPs, compound 150, showed lymph node hyperplasia at 6 months, and compound 152 showed stomach hyperplasia at 6 months. The fact that all compounds are negative is remarkable, as the immunosuppressive action is a well-known risk factor for the induction of cancer. Bugelski et al. (46) showed that around 50% of immunosuppressive compounds were associated with some type of cancer, probably based upon the spontaneous presence of oncogenic viruses. The absence of a carcinogenic effect for these immunosuppressive compounds could be explained by an absence of oncogenic viruses during these studies. It is known that immunosuppression is a real risk factor for human carcinogenicity. From that point of view, the ICH-S1 Expert Working Group indicated that compounds with an immunosuppressive risk could warrant a waiver in the future. Cyclosporin is also a class 1A (IARC) proven human carcinogen. Therefore, we decided to categorize the class of immunosuppressives as TP, despite the lack of tumors in these studies.
Immunomodulators [52]
Two compounds are included. Compound 8 is a TN, while compound 242 is a FN with pituitary adenomas noted at 2 years. Categorization of this class based on pharmacology remains uncertain. Therefore, we maintained the histopathological categorization for these compounds.
Metabolic System
Antidiabetics, α-Glycosidase Inhibitors [50]
Two compounds (76, 194) are included; one (76) is TN. The other has been studied in relation to glucose inclusion in the diet and as a pair-fed study. The study with glucose resulted in Leydig cell adenomas after 2 years, whereas without glucose, kidney tumors were observed. A final pair-fed study did not reveal any treatment-related increase in tumors. As the target of α-glycosidase inhibitors is rather the intestine than the kidney, we concluded that this should be considered an off-target effect. We categorized this class, therefore, as TN.
Dipeptidyl Peptidase Inhibitors (DPP4 Inhibitors) [28]
Four DPP4 inhibitors (68, 111, 114, and 139) were all TN. Apparently, the inhibition of the breakdown of GLP-1 is only low and does not lead to the induction of thyroid C-cell tumors, as is known from GLP-1 agonists such as liraglutide (47). We categorized this class as TN.
PPAR-α-Agonists (Fibrates) [8]
Three compounds are included (46, 202, and 211). One is TN (46). Two are FN, with both compounds inducing hepatocellular carcinomas, accompanied with pancreas acinar cell adenomas and forestomach squamous cell carcinomas for compound 211. Compounds 46 and 202 showed Leydig cell adenomas and adrenal pheochromocytomas. Fibrates are known to target the liver, but the pancreas can also be listed as a target organ based on their class properties related to peroxisome proliferation (48). Because of the pharmacological profile, the histopathological categorization TN and FN, respectively, was considered to be overruled by TP for this class.
HMG-CoA Reductase Inhibitors (Statins) [9]
Five compounds (27, 146, 190, 173, and 267) are included. One (27) is a TN and one (267) was a TN in Fisher rats but FN in Sprague-Dawley rats as hepatocellular adenocarcinomas and thyroid follicular cell adenomas were observed in this strain. Compound (146) was a FP with hepatocellular hyperplasia at 3 months, while compound (190) is a TP with hepatocellular hyperplasia at 3 months (although only hypertrophy was observed in a 6-month study) and uterus endometrial polyps after 2 years. Compound (173) induced forestomach hyperplasia after 6 months, and thyroid follicular cell adenomas and carcinomas and forestomach squamous cell papillomas after 2 years. Whether or not all these tumors are related to the pharmacological action of statins is not completely certain, as discussed in Ref. (26). In fact, the outcome is that statins can be expected to be carcinogenic anyway. Therefore, we categorized the complete class as TP.
Remaining Metabolic Compounds
Six remaining compounds are included. An antidiabetic sulfonylureum derivative (58), a lipid replacement (57), a nicotinic acid derivative (1), a triglyceride-lowering compound (43), a PPAR-gamma agonist (134), and an aldose reductase inhibitor (130), are all TN. We gave the category TN to all these compounds as no other evidence for a proliferative effect on the basis of receptor stimulation could be found. A 3-betahydroxy derivative (160) and an SGLT-2 inhibitor (151) are FP, with adrenal gland hyperplasia in the cortex or kidney hyperplasia after 6 months, respectively. Thus far, no SGLT-2 inhibitor was associated with renal effects in rats (49, 50). An inhibitor of growth hormone (258) is FN with skin sarcomas and uterus endometrium carcinomas after 2 years.
Respiratory System
Anticholinergics [30]
Two anticholinergic compound (61 and 128) intended to be administered via the inhalation route are TN. Therefore, we categorized these compounds as TN based upon their pharmacology.
Adrenergic β2-Agonists [10]
Five compounds are included (191, 199, 228, 280, and 288). The most common feature was ovarian leiomyomas as seen for four of the five compounds. For the RS, the association of mesovarian leiomyomas with β2-agonist is well described, although one compound (199) showed only thyroid adenoma. Even for recent long-acting β2-agonists such as indacaterol and vilanterol (51, 52), these tumors have been observed. A broader disturbance of the gender HM is clear from the mammary adenocarcinoma (228) and the effects on the pituitary (191 and 288). We categorized this class as TP based on pharmacology.
Corticosteroids [11]
Three compounds are included (168, 170, and 227). Two compounds (168 and 170) are TP. Compound 168 showed mammary gland acinar hyperplasia at 6 months, and at 2 years, mammary gland fibromas, brain astrocytomas, and hepatocellular adenomas were observed. Compound 170 showed lymph node hyperplasia and pancreas islet cell hyperplasia at 6 months, and hepatocellular adenomas, pancreas acinar cell adenomas, mammary gland adenomas and adenocarcinomas, and bone osteomas and osteosarcomas were observed in the 2-year study. Compound 227 is a FN and showed pheochromocytomas, pancreatic acinar cell tumors, and skin fibrosarcomas. The relationship between the pharmacology of corticosteroids and the target organs observed is not easy to understand, but the wide distribution of corticosteroid receptors is in accordance with the broad list of organs bearing tumors, suggesting a broad pharmacological perspective. We categorized the corticosteroids as TP.
Histamine H1 Antagonists [48]
Seven compounds (11, 12, 67, 154, 155, 195, and 207) are included, with three TN (11, 12, and 67). Two FPs (154 and 155) showed mammary gland acinar cell hyperplasia at 3 months, while compound 155 showed islet cell hyperplasia and hepatocellular hypertrophy at 6 months. Only two compounds (195 and 207) were associated with tumors (FN). Compound 195 showed pheochromocytomas, and compound 207 showed thyroid follicular cell adenomas, hepatocellular adenomas, and carcinomas. It is likely that these effects are associated with the induction of liver enzymes, based upon the target organs and the presence of pituitary adenomas and carcinomas. In general, the class is categorized as TN.
Remaining Respiratory Compounds
Compound 184, an antifibrotic agent, induced adrenal hyperplasia after 6 months, but liver adenoma and uterine adenocarcinoma after 2 years, making it histopathologically a TP. Pharmacologically, it could not be categorized, as no relationship is known to exist between the effects of the two endpoints. The effects are rather unspecific, and therefore, we finally decided to categorize it a TN. Compound 264 is a methylxanthine derivative is a FN with testis tumors and mammary fibroadenoma, rather than unspecific tumors, and therefore, we categorized this as TN. Compound 116 is a TN mast cell stabilizer.
Antimicrobial Agents
Antibiotics, Fluoroquinolones [13]
Three compounds (231, 244, and 263) are all FN. The first showed renal adenomas and carcinomas, the second showed leukemia, and the third was associated with pancreatic neoplasms. As the primary pharmacology is not directed to a mammalian target, we categorized these compounds as non-mammalian target (NT).
Remaining Antibiotics
One compound (158), a bactericidal agent, is FP with forestomach hyperplasia at 6 and 12 months.
Antifungal Agents, Conazole Derivatives [14]
Three compounds (182, 226, and 236) are included. The first (182) is a FN with skin, brain, testis, and mammary gland tumors. Compound 226 showed hepatocellular adenomas, and compound 236 showed a low incidence of soft tissue carcinomas. The classes of triazole antifungals do not have a mammalian target by definition, as discussed above. A specific effect on liver enzyme metabolism is described for related members of the class of antifungals (53). The first molecular event is not fully clear but might be the binding to a CYP450 subcategory. Conazoles induce hepatic cell proliferation in mice. We have categorized the antifungals as NT.
Remaining Antifungals
One compound (2) is a TN, while another (279) is a FN showing hepatocellular adenomas and carcinomas, as well as Leydig cell tumors. We categorized them all as NT.
Antimicrobials, Remaining Antimicrobials
Two AM are included, an antimalarial drug (73) and an antiparasital compound (97) (anti-lice). Both compounds are TN. As the primary pharmacology is not directed to a mammalian target, we categorized these compounds as NT.
Antivirals [32]
The antivirals (18, 55, 60, 104, 123, 135, 171, 189, 218, and 246) do not belong to the same class and have different types of therapeutic use and modes of actions. Six compounds, an immunostimulant (60), a nucleoside-analog (104), a viral DNA polymerase inhibitor (55), a protease inhibitor (18), a nucleoside combination (123), and an anti-herpes compound (135), are all TN. Four compounds (171, 189, 218, and 246) showed tumors. As antivirals do not have direct primary pharmacological targets in mammals, as discussed above, the variety of effects might be due to completely different mechanisms of action. For compound 246, liver induction might have led to increased T3 metabolism, eventually resulting in thyroid tumors. A similar explanation is not possible for compounds 171 and 218. In these individual cases, the potential human risk of the induction of tumors has been evaluated, and the relevance of these effects is negligible compared to the benefit of clinical treatment.
A CCR5 receptor antagonist (246) is FN with thyroid adenomas after 2 years. A guanosine analog (171) is FN with mammary gland adenocarcinomas after 2 years. A protease inhibitor (189) is TP and showed hepatocellular hyperplasia after 6 months and adrenal pheochromocytomas after 2 years. As none of the antiviral showed a clear connection to a mammalian pharmacological target, we just categorized them as NT.
Anti-inflammatory Compounds
Non-Steroidal Anti-inflammatory Drugs [19]
Twelve “classical” cyclooxygenase 1/2 inhibitors (44, 45, 50, 64, 71, 74, 83, 91, 124, 129, 164, and 260) are included with 10 being TN. One (164) compound is a TP with kidney hyperplasia at 6 months, and after 2 years, adrenal pheochromocytomas and hyperplasia in the urinary bladder were observed. The histopathological changes and tumors induced cannot be directly explained. The adrenal pheochromocytomas are assumed to be not relevant for humans. For another compound (260), it is assumed that induction of CYP450 would lead to an increased testosterone metabolism, which leads via a feedback mechanism to Leydig cell adenomas. We have categorized this class as TN.
Cyclooxygenase-2-Inhibitors [31]
Four compounds (108, 72, 206, and 222) are included. Two compounds (72 and 108) are TN. The two other compounds (206 and 222) are FN with hepatocellular adenomas and carcinomas. One compound (222) also showed thyroid follicular cell adenomas. The target organs suggest a relation with the liver-induced metabolism and not to a direct pharmacological response. This supports the negative properties of the NSAID (COX1/2-inhibitor) class, as discussed above. Based on this relationship, we categorized the class as TN.
Remaining Anti-inflammatory Compounds
Two compounds are included (7 and 122), which are both TN, and we categorized them the same.
Urinary Bladder
Anticholinergics [51]
Six compounds (31, 51, 125, 268, 283, and 287) are included, with three (31, 51, and 125) categorized as TN and three (268, 283, and 287) as FN. Compound 268 was associated with benign uterine polyps and kidney papillomas, compound 283 with kidney sarcomas, and compound 287 with skin sarcomas, all with high dosages or incidental findings. The class was categorized as TN.
Remaining Urinary Compounds
A xanthine oxidase inhibitor (153) is FP with thyroid hyperplasia at 6 months. A β3-agonist (79) is TN. We have placed both in the category TN.
Bone Metabolism
Bisphosphonates [20]
Three compounds (3, 33, and 87) are included and shown to be TN. Van der Laan et al. (26) described a pharmacological explanation for a positive finding, i.e., thyroid c-cell adenoma. This is not, however, confirmed in the present data, and we maintained the TN category.
Remaining Compounds Affecting Bone Metabolism
An isoflavone derivative (234) is a FN with pituitary gland adenomas and hepatocellular adenomas after 2 years. A calcium-mimetic (28) is TN. As the tumor targets for compound 234 are rather unspecific, we categorized both as TN.
Remaining Compounds
Four compounds (39, 62, 109, and 196) remained pharmacologically unclassified. Three were TNs, a prostaglandin E2-analog (39), a CFTR potentiator (62), and a protein kinase C-beta inhibitor (109). A retinoid for topical administration (196) is a FN, with adrenal phaechromocytomas and thyroid follicular cell adenomas after 2 years. As the tumor effects are rather unspecific, we assigned a TN category to this compound and maintained the TN for the others.
Discussion and Conclusion
The present retrospective study using data from rat subchronic (3- and/or 6-month) toxicity and 2-year carcinogenicity studies obtained from the non-clinical assessment reports, available at the Medicines Evaluation Board of the Netherlands, was performed to test independently a “weight-of-evidence approach,” expressed in the ICH Regulatory Notice Document (54) that was based on the “whole animal negative predictivity hypothesis” of Reddy et al. (25) and Sistare et al. (16). This might strengthen the assumption that the absence of any putative preneoplastic lesion in a subchronic study is associated with a negative outcome of a 2-year carcinogenicity study in rats, and therefore, no further long-term carcinogenicity study is needed. Furthermore, we have evaluated the role of the pharmacological properties in this respect.
In order to build evidence for the possibility of prediction, we used information that is commonly available at the end of Phase II in the development of human pharmaceuticals for designing a 2-year carcinogenicity study, including data on pharmacology and genotoxicity, and data from subchronic toxicity studies in rodents, as was undertaken earlier by Van der Laan et al. (26).
The dataset that we used has some overlap with the PFJ dataset as used by Van der Laan et al. (26), and we identified an overlap of 76 compounds. We decided to include these, as the application of more stringent criteria of putative neoplasm (see below) has led to differences in the categorization and to a higher number of FN.
Role of Liver Hypertrophy versus Hyperplasia
A large number of pharmaceutical compounds induced greater (relative) liver weight, and the question arises about the role of this property in the weight-of-evidence approach. A greater liver weight may result from a wide variety of causes such as hyperplasia (of any of the resident cell types), hypertrophy, inflammation, fibrosis, abnormal storage of metabolism or cleavage products, neoplasia, and congestion (55–57). Typically, these changes do not occur in isolation, so in the absence of overt adverse changes such as inflammation, necrosis, or degeneration, it is important to recognize that an increase in liver weight may be induced by hypertrophy, hyperplasia, or a combination of the two (58). A xenobiotic that induces an increase in liver weight of 150% in a subchronic study might be considered to induce adverse effects in the context of dose setting for longer term studies but would not be considered to be adverse in the context of safety evaluation (59, 60).
In a survey of 139 chemicals used in the agrochemical industry, Carmichael et al. (59) demonstrated that a relative greater liver weight of ≥150% of control values was correlated with the induction of liver tumors in mice. In a similar review of rat studies, a less statistically significant relationship between liver weight and hepatocarcinogenesis was noted, whereby greater liver weight alone correctly predicted 8 of 11 liver carcinogens (but falsely predicted 26 as positives) and failed to predict 3 TPs (59).
Our findings demonstrate that treatment-related changes in organ weights, observed in a subchronic study with rats, are most likely non-specific and therefore should not be considered as a risk factor for neoplasia.
The histopathological diagnosis of hypertrophy can have various connotations, including a greater weight of the organ and an increase in the average size of the cells and even enzyme induction (functional hypertrophy). Allen et al. (61) evaluated the results for 111 chemicals tested by the National Toxicology Program. If they applied hepatocellular necrosis, hepatocellular hypertrophy, hepatocellular cytomegaly, and greater liver weight as predictors for carcinogenicity, greater liver weight appeared to be the most sensitive parameter. However, chemicals that produced liver tumors frequently induced multiple morphological changes. They concluded that the best single predictor of liver cancer in mice was hepatocellular hypertrophy. They found no FNs, but numerous FPs, in their evaluation.
In the present study, 33 compounds with a TN label showed an increase in hepatocellular hypertrophy in the subchronic study that was not accompanied by development of hepatocellular adenomas and/or carcinomas in the carcinogenicity study, whereas in only 7 cases was liver hypertrophy accompanied by the development of hepatocellular adenomas (3) or hepatocellular adenomas and carcinomas (4).
These observations support our starting point to classify hyperthrophy as an adaptive, rather than a putative, preneoplastic lesion. If we had assessed hepatocellular hypertrophy as an indicator for the development of hepatocellular tumors, 33 compounds would have been overpredicted as potential liver carcinogens (FP substances). This confirms that liver hypertrophy observed in a subchronic study is an unreliable predictor of carcinogenicity. This is in agreement with the conclusion from the 3rd International ESTP Expert Workshop (62) that hepatomegaly as a consequence of hepatocellular hypertrophy without histological or clinical pathological alterations indicative of liver toxicity is an adaptive reaction.
Histopathological Evaluation
First, the predictivity was evaluated based upon the histopathological characterization.
The negative predictivity, the measure of the compounds evaluated that did not show any putative preneoplastic lesion in the subchronic studies and were negative in the carcinogenicity studies, was 60%, whereas the sensitivity, a measure of the subchronic study to predict positive carcinogenicity outcome, was only 24% (Table 2). In contrast, the specificity, the accuracy of the subchronic study to correctly identify non-carcinogens, was 88%. Based only on the absence of putative preneoplastic lesions in the subchronic study, 56% of the 2-year carcinogenicity studies could have been eliminated at the risk of 96 (33%) FN.
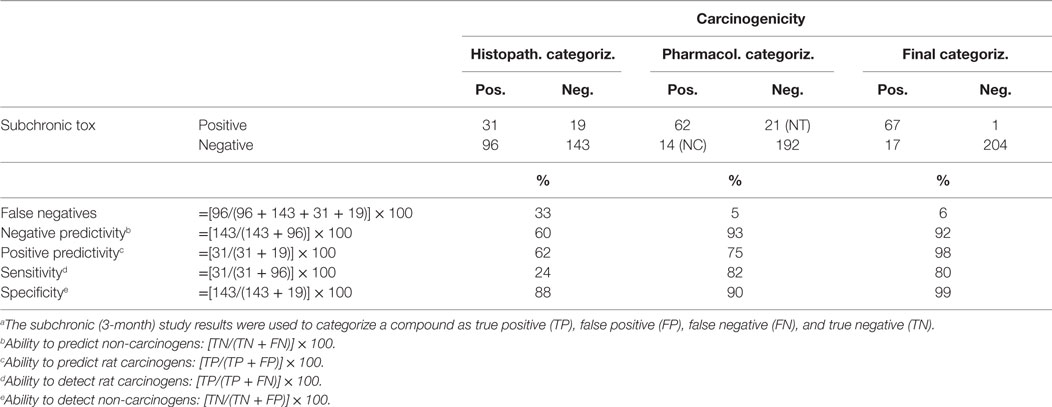
Table 2. Predictivity of the subchronic toxicity study for the carcinogenicity of non-genotoxic pharmaceuticals.a
The positive predictivity (62%) is the percentage of compounds that showed putative preneoplastic changes in the subchronic study and caused treatment-related tumors in the 24-month carcinogenicity study.
Thirty-one substances were classified as TP. However, for only 13 compounds, the putative preneoplastic lesions developed in the same organs as the tumors. Eighteen compounds induced hyperplastic lesions in another organ than the organ where the tumors occurred. This observation is in agreement with the conclusion of Reddy et al. (25) that the whole animal approach assumes that preneoplastic changes at any organ will be indicative for an increase in tumor incidence in that organ or in any organ at a distant site. A closer look at the organs (Table S1 in Supplementary Material: compounds 163–193) illustrates the importance of physiological relationships between organs. Several compounds induce their own metabolism in the liver and, as a consequence, enhance the enzymes responsible also for the metabolism of hormones, such as T3 and testosterone/estradiol, lowering their concentration. This decrease in hormone concentration leads to a feedback responses to the pituitary, resulting in enhanced secretion of thyroid-releasing hormone and/gonadotropin-releasing hormone, which in turn may lead to the development of tumors.
False Positive Compounds
Nineteen chemicals induced putative preneoplastic (hyperplastic) histopathological changes in the liver, thyroid, kidneys, mammary glands, pancreas, stomach, adrenals, and incidentally in other organs, whereas no tumors occurred in the carcinogenicity study, neither in the same organ nor in an organ at a distant site. This observation supports the conclusion of Jacobs (24), based on an evaluation of 60 pharmaceuticals, that various short-term indicators for carcinogenicity, such as hyperplasia, do not always result in tumors in that tissue, although such putative preneoplastic histopathological lesions are generally considered a sign of potential concern for carcinogenicity.
Woutersen et al. (63) performed a retrospective study and evaluated the subchronic (3 months) studies of 163 non-genotoxic chemicals with the aim to predict the tumor outcome of 24-month rat carcinogenicity studies. In this study, the negative predictivity, a measure to predict a negative carcinogenicity outcome, amounted to 97%, whereas the sensitivity, a measure to predict a positive carcinogenicity outcome, was only 5%. Overall, this study supports the concept that chemicals showing no histopathological risk factors for neoplasia in a subchronic study in rats may be considered non-carcinogenic and do not require further testing in a carcinogenicity study. The findings observed in the present paper with TP and FP compounds are in agreement with the conclusion of Reddy et al. (25) that more research is needed in order to achieve understanding of the biological links between putative preneoplastic lesions observed in a subchronic study and tumors developing at distant organ sites in the carcinogenicity study. The concept of adverse outcome pathways is helpful in this respect, defining as the first step the molecular event and then subsequent steps leading to the final outcome of tumors in different organs.
False Negative Compounds
Since it is generally accepted that the intention of screening assays should be conservative, it is most important that the number of FNs with respect to human carcinogens should be as low as possible. In the present study, 96 compounds were classified as FN because they did not show putative preneoplastic lesions in the 3- and/or 6-month study but caused treatment-related tumors in the carcinogenicity study. These compounds are of concern with regard to the acceptability of the negative predictivity of the whole animal approach stating that the absence of evidence of putative preneoplastic lesions in all tissues in the 3- and/or 6-month study may serve as a strong negative predictor of tumor outcome in the carcinogenicity study.
When we evaluate these FN substances histopathologically, 77 of the 96 FN substances appeared to induce benign tumors or benign and malignant tumors, which are considered not relevant for the human situation (36): acinar pancreatic tumors and islet cell neoplasia (36); pheochromocytomas of the adrenal medulla (64); forestomach tumors (65, 66); hepatocellular tumors induced by peroxisome proliferators (48, 67–69); fibroadenomas of the mammary gland (70); pituitary tumors (adenohypophysis tumors) (71); Leydig cell (interstitial cell) tumors of the testes (72, 73); thyroid follicular cell tumors (74–76); and urinary bladder tumors (75, 77–80) and uterus tumors (endometrial stromal polyps) (81). That means that 19 FN substances still remain using this approach.
Impact of Pharmacology
To evaluate further the remaining 19 FN substances, we compared this number with the outcome of an evaluation integrating the pharmacological properties of the compounds. We have tested the hypothesis that a pharmacological explanation, as known for several years (30), and integrated in the histopathological approach recently (26), will help to reduce the number of FNs.
To study the role of the pharmacology in relation to histopathology, we have evaluated whether the mode of action is related to a proliferative mechanism or only to non-proliferative mechanisms. We have selected those classes that are clearly related to induction of tumors as positive classes (Table 3) and classes not related to induction of tumors as negative classes (Table 4). A series of classes with mixed outcome have been evaluated in relation to literature, and for each pharmacological class, a clear outcome of “positive” or “negative” has been chosen (Table 5), meaning a high or low percentage of rat carcinogens, respectively. For the remaining individual compounds, we have taken into consideration the specificity of the tumors, i.e., those tumors likely to be associated with a change in metabolism of hormones (thyroid, or testosterone, or calciferol) are not related to a specific pharmacological mechanism. The compounds are, therefore, categorized as TN.
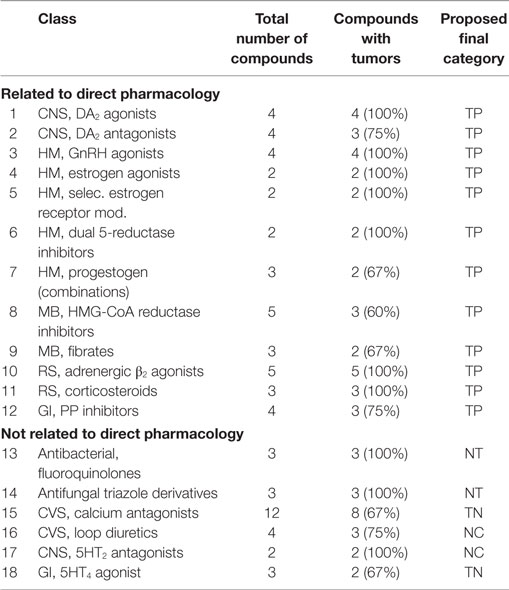
Table 3. Classes with high percentage of rat carcinogens (positive classes).
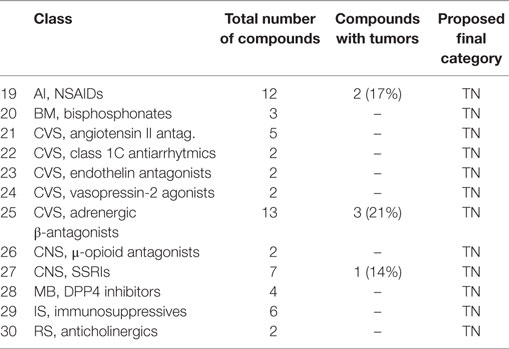
Table 4. Classes with low percentage of rat carcinogens (negative class).
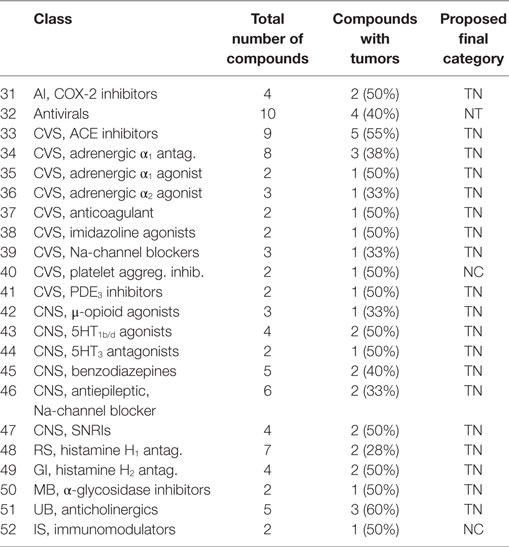
Table 5. Classes with medium percentage of rat carcinogens (mixed outcome class).
Based upon pharmacology, combined with a previous histopathological categorization, we have given a final category in a separate column. This final category is applicable only to those classes for which at least two compounds are present in the dataset.
We have separated the compounds without a direct pharmacological effect in mammalian tissue from the other compounds.
Table 3 contains a list of pharmacological classes with a high percentage of rat carcinogens (positive classes). In line with the earlier overview (26), we can differentiate between carcinogenicity directly related to pharmacology and carcinogenicity not related to pharmacology. Antibiotics, antifungals, and antivirals are developed to act against specific mechanisms in their target organism. Of course, it is possible that off-target effects exist in mammals. Apparently, this is true for the metabolic enzymes in liver. We have chosen, therefore, to give a separate classification to all compounds without a non-mammalian target (NT).
Based upon the identity of the organs bearing tumors (i.e., associated with liver, thyroid, adrenal, and testis), we have put the antibacterials, antifungals, and antivirals in the same category.
The calcium antagonists, the loop diuretics, and the GI 5HT4 agonists are in the list of “not related to pharmacology.”
Combined Evaluation of Histopathology and Pharmacology
In Table 1, we have incorporated the pharmacology-based categorization in the column labeled as Cat. Ph, by taking the pharmacology as an additional factor (Table 1, fifth column). In this way, we categorized most of the compounds as TP (if belonging to a positive class) or TN, as explained above.
A combination of the histopathological categorization of the compounds together with the pharmacological categorization of the pharmaceuticals gave the following overall results:
• Twenty-one pharmaceuticals that do not have a mammalian target (8FN, 1FP, 9TN, and 3TP) are included.
• Fifty-two of the 96 FN compounds were recategorized as TNs based on pharmacology; 27 were recategorized as TPs; in addition to the 8 without mammalian target, 9 were not recategorized due to an unknown relationship between pharmacology and carcinogenicity.
• Fourteen FP compounds were recategorized to TN based on pharmacology; five were recategorized as TP. One remained FP, as there is no pharmacological target.
• One hundred twenty-four TNs remained TN, while 14 were categorized as TP. Five were not categorized (NC).
• From the 41 TP compounds, 11 were recategorized as TN, while 17 remained TP on the basis of their pharmacology. Two could not be categorized, and three had no mammalian target.
• Finally, 14 out of 289 (5%) pharmaceuticals evaluated in the present retrospective study have not been recategorized. Nine are FN, and five are TN.
• After recategorization, based on both histopathology and pharmacology, the number of FN compounds was reduced to only 17 out of 289 (6%). The negative predictivity amounted to 92%; the positive predictivity to 98%, and the sensitivity was 80%, whereas the specificity amounted to 99% (TP: 67; FP: 1; FN: 17; and TN: 204) (see Table 6).
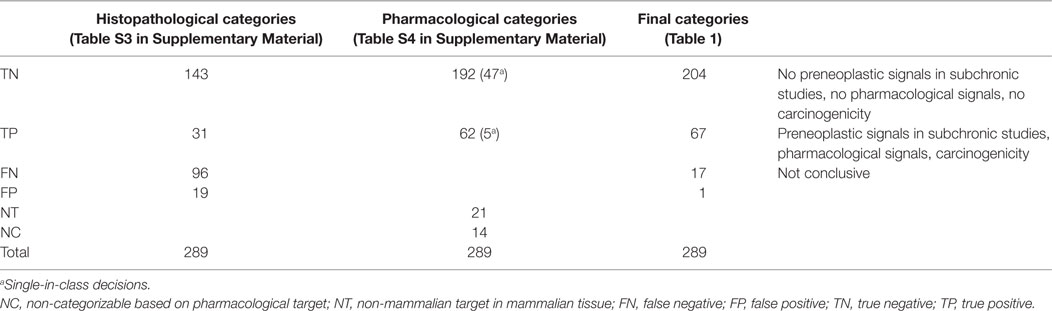
Table 6. Summarizing table based upon final categorization.
When the FN compounds that gave rise to tumors generally considered not relevant for human risk assessment, and those categorized as TN based on the pharmacological analyses, were moved from the FN category to the TN category, the negative predictivity of the 3- and/or 6-month study for the absence of carcinogenicity (the ability to predict non-carcinogens) amounts to 96% and the specificity (the ability to detect non-carcinogens) to 99% (Table 3).
The analysis of this paper clearly shows that adding pharmacological properties as an additional factor of potential carcinogenicity gives a good prediction, reducing the number of FN substantially, which has consequences for the risk assessment of these pharmaceuticals.
Conclusion
Our results demonstrated that subchronic (3- and/or 6-month) studies, in combination with knowledge of pharmacological properties, could appropriately categorize non-genotoxic pharmaceutical into two categories:
(i) unlikely to be carcinogenic in rats if (1) no histopathological risk factor for neoplastic lesions observed in the subchronic study in any tissue, and (2) general absence of systematic, specific carcinogenicity in the pharmacological class.
(ii) likely carcinogenic in rats if (1) putative preneoplastic lesions observed in the subchronic study may give rise to a type of tumor in the rat 24-month carcinogenicity study that is irrelevant for humans; therefore, a carcinogenicity study has no additional value; (2) this is confirmed by the results from the pharmacological class.
We should keep in mind that the real focus is on the prediction of carcinogenicity in humans, and we cannot quantify the full translational value of the rodent carcinogenicity study. However, overall, the results of this retrospective study support the whole animal approach as proposed by Reddy et al. (25) and Sistare et al. (16), especially with respect to the negative outcome of the subchronic studies as prediction for a negative outcome of a carcinogenicity study. Moreover, the results (predictivity) are consistent with the recent and similar investigation on chemicals (63). Furthermore, the data show the added value of the pharmacological evaluation of compounds in relation to potential class effects, both in the negative and positive direction. This evaluation strongly enhances the prediction of a possible impact for rodents and eventually for an extrapolation of the carcinogenic risk to humans. The outcome can be used to further prevent conducting unnecessary carcinogenicity studies. For most of the pharmacological mechanisms, it is well known that the non-genotoxic mode of action carries no risk of carcinogenicity for the human situation because of important species-related differences between rodents and humans.
In this way, the pharmacological analysis confirms the approach recently published by Van der Laan et al. (26) and reflected in the ICH Regulatory Notice Document to incorporate the pharmacological properties in predicting a positive and a negative outcome of a 2-year carcinogenicity study.
A high negative and a high positive predictivity of the carcinogenic potential of a pharmaceutical compound based on the findings in subchronic toxicity studies in rats, combined with knowledge of the pharmacological class, should result in waiving the need for conducting 2-year rat carcinogenicity studies, which will lead to a reduction in the numbers of animals used for scientific purposes and will save time and expense for drug development.
The dataset used for this analysis gives important opportunities for further research. As in addition to rat data, also mouse data are included, we can have a better understanding of the different outcomes between rats and mice, as discussed previously by Van Oosterhout et al. (27) and Friedrich and Olejniczak (15). Their reports point to the low regulatory relevance of lifetime mouse studies, and potential reduction of 2-year rat studies as proposed in the ICH RND (54), should at least be accompanied by measures with respect to 18- or 24-month mouse studies. Further discussion is recommended on the predictive value of the involvement of cytochrome p 450ies in the induction of cell proliferation in liver or other organs such as thyroid gland and testis. These and other questions require further analysis of the studies in this database.
Author Contributions
WB, LW, and AS conducted the data retrieval and filled the database. ES translated the database in an accessible spreadsheet. CK contributed to the design of the study and criticized the manuscript. RW and JL have equally contributed to the writing and the underlying concepts, with RW responsible for histopathological aspects and JL for the pharmacological and regulatory aspects.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The present study was supported initially by a grant from the Dutch Government, Department Economic Affairs, The Utrecht Province and the Utrecht City Administration (grant PID101063). Its completion was enabled by a grant received from the European Partnership for Alternative Approaches to Animal testing (EPAA) over 2013 and 2014. We thank David Jones and Tim Rowan for extensive linguistic correction of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fmed.2016.00045
Abbreviations
AB, antibiotics; ac, adenocarcinoma; ad, adenoma; AF, antifungal agents; adr, adrenals; astr, astrocytoma; AV, antivirals; bheam, benign hemangioma; bpha, benign pheochromocytoma; bm, bone marrow; bo, bone; br, brain; bthym, benign thymoma; ca, carcinoma; ce, cecum; col, colon; fad, fibroadenoma; FN, false negative; FP, false positive; gca, granulosa cell adenoma; hrt, heart; hsyst, hematopoietic system; islet, islet of Langerhans; kid, kidneys; leio, leiomyoma; leu, leukemia; li, liver; lip, lipoma; ln, lymph nodes; lu, lungs; lymph, lymphoma; mam, mammary glands; mel, melanoma; mes, mesentrium; most, malignant osteoma = osteosarcoma; mpha, malignant pheochromocytoma; NC, non-categorizable based on pharmacological target; NOS, not otherwise specified; NT, non-mammalian target in mammalian tissue; ova, ovaries; pan, pancreas; pap, papilloma; parathy, parathyroids; pit, pituitary; pros, prostate; sar, sarcoma; SCC, squamous cell carcinoma; schwan, schwannoma; SCP, squamous cell papilloma; soft t, soft tissue; spl, spleen; stom, stomach; tes, testes; thyr, thyroid; thym, thymus; TN, true negative; TP, true positive; tu, tumor; UGT, urogenital tract; ut, uterus; zymgl, Zymbal’s gland; ZZ, remaining compounds.
References
1. ICH. International Conference on Harmonization Guidelines M3: Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals. Geneva: ICH (2009).
2. ICH. International Conference on Harmonization Guidelines S1A; The Need for Carcinogenicity Studies of Pharmaceuticals. Geneva: ICH (1996).
3. ICH. International Conference on Harmonization Guidelines S1B: Carcinogenicity Testing for Carcinogenicity of Pharmaceuticals. Geneva: ICH (1998).
4. ICH. International Conference on Harmonization Guidelines S1C: Dose Selection for Carcinogenicity Studies. Geneva: ICH (2008).
5. ICH. International Conference on Harmonization Guidelines S2: Guidance for Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use. Geneva: ICH (2008).
6. ICH. International Conference on Harmonization Guidelines S6 (R1): Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals. (2009). Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf
7. Jacobson-Kram D, Sistare FD, Jacobs AC. Use of transgenic mice in carcinogenicity hazard assessment. Toxicol Pathol (2004) 32(Suppl 1):49–52. doi: 10.1080/01926230490424761
8. Anisimov VN, Ukraintseva SV, Yahin AI. Cancer in rodents: does it tell us about cancer in humans? Nat Rev Cancer (2005) 5:807–19.
9. Haseman JK, Hailey JR, Morris RW. Spontaneous neoplasm incidences in Fischer 344 rats and B6C3F1 mice in two-year carcinogenicity studies: a national toxicology programme update. Toxicol Pathol (1998) 26:428–41. doi:10.1177/019262339802600318
10. Annys E, Billington R, Clayton R, Bremm K-D, Graziano M, McKelvie J, et al. Advancing the 3Rs in regulatory toxicology – carcinogenicity testing: scope for harmonisation and advancing the 3Rs in regulated sectors of the European Union. Regul Toxicol Pharmacol (2014) 69:234–42. doi:10.1016/j.yrtph.2014.04.009
11. Billington R, Lewis RW, Mehta JM, Dewhurst I. The mouse carcinogenicity study is no longer a scientifically justifiable core data requirement for the safety assessment of pesticides. Crit Rev Toxicol (2010) 40:35–49. doi:10.3109/10408440903367741
12. Cohen SM. Human carcinogenic risk evaluation; an alternative approach to the two-year rodent bioassay. Toxicol Sci (2004) 80:225–9. doi:10.1093/toxsci/kfh159
13. Ward JM. The two-year rodent carcinogenesis bioassay – will it survive? J Toxicol Pathol (2007) 20:13–9. doi:10.1293/tox.20.13
14. Boobis AR, Cohen SM, Doerrer NG, Galloway SM, Haley PJ, Hard GC, et al. A data-based assessment of alternative strategies for identification of potential human cancer hazards. Toxicol Pathol (2009) 37:714–32. doi:10.1177/0192623309343779
15. Friedrich A, Olejniczak K. Evaluation of carcinogenicity studies of medical products for human use authorized via the European centralized procedure (1995-2009). Regul Toxicol Pharmacol (2011) 60:225–48. doi:10.1016/j.yrtph.2011.04.001
16. Sistare FD, Morton D, Alden C, Christensen J, Keller D, Jonghe SD, et al. An analysis of pharmaceutical experience with decades of rat carcinogenicity testing: support for proposal to modify current regulatory guidelines. Toxicol Pathol (2011) 39:716–44. doi:10.1177/0192623311406935
17. Benigni R. Alternatives to the carcinogenicity bioassay for toxicity prediction: are we there yet? Expert Opin Drug Metab Toxicol (2012) 8(4):407–17. doi:10.1517/17425255.2012.666238
18. Doktorova TY, Pauwels M, Vinken M, Vanhaseke T, Rogiers V. Opportunities for an alternative integrating testing strategy for carcinogen hazard assessment? Crit Rev Toxicol (2012) 42:91–106. doi:10.3109/10408444.2011.623151
19. Gori GB. Regulatory forum opinion piece: long-term animal bioassays: is the end near? Toxicol Pathol (2013) 41:805–7. doi:10.1177/0192623312467524
20. Osimitz TG, Droege W, Boobis AR, Lake BG. Evaluation of the utility of the lifetime mouse bioassay in the identification of cancer hazards for humans. Food Chem Toxicol (2013) 60:550–62. doi:10.1016/j.fct.2013.08.020
21. Marone PA, Hall WC, Hayes AW. Reassessing the two-year rodent carcinogenicity bioassay: a review of the applicability to human risk and current perspectives. Regul Toxicol Pharmacol (2014) 68:108–18. doi:10.1016/j.yrtph.2013.11.011
22. EC. Corrigendum to Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorization and Restriction of Chemicals (REACH), establishing a European Chemical Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC. Off J Eur Union (2007) L136:3–280.
23. Snyder RD, Green JW. A review of genotoxicity of marketed pharmaceuticals. Mutat Res (2001) 488:151–69. doi:10.1016/S1383-5742(01)00055-2
24. Jacobs A. Prediction of 2-year carcinogenicity study results for pharmaceutical products: how are we doing? Toxicol Sci (2005) 88:18–23. doi:10.1093/toxsci/kfi248
25. Reddy MV, Sistare FD, Christensen JS, DeLuca JG, Wollenberg GK, DeGeorge JJ. An evaluation of chronic six- and twelve-month rat toxicology studies as predictors of two-year tumor outcome. Vet Pathol (2010) 47:614–29. doi:10.1177/0300985810373242
26. van der Laan JW, Kasper P, Silva Lima B, Jones DR, Pasanen M. Critical analysis of carcinogenicity study outcomes. Relationship with pharmacological properties. Crit Rev Toxicol (2016) 46(7):587–614. doi:10.3109/10408444.2016.1163664
27. Van Oosterhout JPJ, Van der Laan JW, De Waal EJ, Olejniczak K, Hilgenfeld M, Schmidt V, et al. The utility of two rodent species in carcinogenic risk assessment of pharmaceuticals in Europe. Regul Toxicol Pharmacol (1997) 25:6–17. doi:10.1006/rtph.1996.1077
28. Stefansdottir G, Zoungas S, Chalmers J, Knol MJ, Leufkens HG, Woodward M, et al. The post hoc use of randomised controlled trials to explore drug associated cancer outcomes: methodological challenges. Curr Drug Saf (2013) 8(5):371–8. doi:10.2174/15748863113086660070
29. Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, et al. Predicting new molecular targets for known drugs. Nature (2009) 462:175–81. doi:10.1038/nature08506
30. Silva Lima BM, Van der Laan JW. Mechanisms of nongenotoxic carcinogenesis and assessment of the human hazard. Regul Toxicol Pharmacol (2000) 32:135–43. doi:10.1006/rtph.2000.1427
31. Rajamannan NM, Caplice N, Anthikad F, Sebo TJ, Orszulak TA, Edwards WD, et al. Cell proliferation in carcinoid valve disease: a mechanism for serotonin effects. J Heart Valve Dis (2001) 10(6):827–31.
32. Ataee R, Ajdary S, Rezayat M, Shokrgozar MA, Shahriari S, Zarrindast MR. Study of 5HT3 and HT4 receptor expression in HT29 cell line and human colon adenocarcinoma tissues. Arch Iran Med (2010) 13(2):120–5.
33. Sigler RE, Gough AW, de la Iglesia FA. Pancreatic acinar cell neoplasia in male Wistar rats following 2 years of gabapentin exposure. Toxicology (1995) 98:73–82. doi:10.1016/0300-483X(94)02966-X
34. Dethloff L, Barr B, Bestervelt L, Bulera S, Sigler R, LaGattuta M, et al. Gabapentin-induced mitogenic activity in rat pancreatic acinar cells. Toxicol Sci (2000) 55:52–9. doi:10.1093/toxsci/55.1.52
35. Haschek WM, Rousseaux CR, Wallig MA, Bolon B, Ochoa R, Mahler BW. Hascheck and Rousseaux’s Handbook of Toxicologic Pathology. 3rd ed. Amsterdam: Elsevier (2013). p. 2419–23. (phaeochromocytomas); 2672–2682 (mammary gland fibroadenomas).
36. Williams GM, Iatropoulos MJ, Enzmann HG, Deschl UF. Carcinogenicity of chemicals: assessment and human extrapolation. 6th ed. In: Hayes AW, Kruger CL, editors. Hayes’ Principles and Methods in Toxicology. Boca Raton: CRC Press (2014). p. 1251–304.
37. Ahr HJ, Bomhard E, Enzmann H, Karbe E, Mager H, Sander E, et al. Calcium channel blockers and the risk of cancer: a preclinical assessment. Cardiovasc Drugs Ther (1998) 12(2):157–69.
38. Lenz B, Crameri FM, Eichler DA, Schläppi B, Wiltshire HR, Wood J, et al. Modulation of oral squamous cell carcinoma incidence in rats via diet and a novel calcium channel antagonist. Toxicol Pathol (2005) 33:356–64. doi:10.1080/01926230590930119
39. Grimaldi-Bensouda L, Klungel O, Kurz X, de Groot MC, Maciel Afonso AS, de Bruin ML, et al. Calcium channel blockers and cancer: a risk analysis using the UK Clinical Practice Research Datalink (CPRD). BMJ Open (2016) 6(1):e009147. doi:10.1136/bmjopen-2015-009147
40. Tsai TH, Beitman RE, Gibson JP, Larson EJ, Friehe H, Fontaine R. Acute, subacute and chronic toxicity/Carcinogenicity of lofexidine. Arzneimittelforschung (1982) 32(8a):955–62.
41. Rauws AG, De Waal EJ, Van der Laan JW. Sense and non-sense in toxicity assessment of medicinal products. Adv Drug Res (1997) 30:15–72.
42. Brott DA, Andersson HAS, Stewart J, Ewart L, Christoph G, Harleman J, et al. A peripherally restricted P2Y12 receptor antagonist altered rat tumor incidences with no human relevance: mode of action consistent with dopamine agonism. Toxicol Rep (2014) 1:1202–12. doi:10.1016/j.toxrep.2014.11.010
43. Kochi T, Shimizu M, Ohno T, Baba A, Sumi T, Kubota M, et al. Preventive effects of the angiotensin-converting enzyme inhibitor, captopril, on the development of azoxymethane-induced colonic preneoplastic lesions in diabetic and hypertensive rats. Oncol Lett (2014) 8:223–9.
44. Berry PH. Effects of diet on reproductive status on the histology of spontaneous pituitary tumors in female Wistar rats. Vet Pathol (1986) 23:606–18.
45. Hernández LG, van Steeg H, Luijten M, van Benthem J. Mechanisms of non-genotoxic carcinogens and importance of a weight of evidence approach. Mutat Res (2009) 682(2–3):94–109. doi:10.1016/j.mrrev.2009.07.002
46. Bugelski PJ, Volk A, Walker MR, Krayer JH, Martin P, Descotes J. Critical review of preclinical approaches to evaluate the potential of immunosuppressive drugs to influence human neoplasia. Int J Toxicol (2010) 29(5):435–66. doi:10.1177/1091581810374654
47. Knudsen LB, Madsen LW, Andersen S, Almholt K, De Boer AS, Drucker DJ, et al. Glucagon-like peptide-1 receptor agonists activate rodent thyroid C-cells causing calcitonin release and C-cell proliferation. Endocrinology (2010) 151(4):1473–86. doi:10.1210/en.2009-1272
48. Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David R, et al. PPAR-alpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol (2003) 33:655–780. doi:10.1080/713608372
49. CHMP. European Public Assessment Report Forxiga. (2012). Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002322/WC500136026.pdf
50. CHMP. European Public Assessment Report Jardiance. (2014). Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002677/WC500168594.pdf
51. CHMP. European Public Assessment Report Onbrez. (2010). Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001114/WC500053735.pdf
52. CHMP. European Public Assessment Report Anoro. (2015). Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002751/WC500168425.pdf
53. Hester S, Moore T, Padgett WT, Murphy L, Wood CE, Nesnow S. The hepatocarcinogenic conazoles: cyproconazole, epoxiconazole, and propioconazole induce a common set of toxicological and transcriptional responses. Toxicol Sci (2012) 127:54–65. doi:10.1093/toxsci/kfs086
54. ICH. S1 Regulatory Notice Document. (2016). Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S1/S1_RND_Revisions_22Dec2015_Final.pdf
55. Carthew P, Nolan BM, Edwards RE, Smith LL. The role of cell death and cell proliferation in the promotion of rat liver tumours by tamoxifen. Cancer Lett (1996) 10:163–9. doi:10.1016/0304-3835(96)04310-8
56. Carthew P, Edwards RE, Nolan BM. The quantitative distinction of hyperplasia from hyperthrophy in hepatomegaly induced in the rat liver by phenobarbital. Toxicol Sci (1998) 44:46–51. doi:10.1093/toxsci/44.1.46
57. Greaves P. Histopathology of Preclinical Toxicity Studies. 4th ed. Amsterdam, The Netherlands: Elsevier (2012).
58. Maronpot RR, Yoshizawa K, Nyska A, Harada T, Flake G, Mueller G, et al. Hepatic enzyme induction. Histopathology. Toxicol Pathol (2010) 38:776–95. doi:10.1177/0192623310373778
59. Carmichael NG, Enzmann H, Pate I, Waechter F. The significance of mouse liver tumor formation for carcinogenic risk assessment. Results and conclusions from a survey of ten years of testing by the agrochemical industry. Environ Health Perspect (1997) 105:1196–203. doi:10.1289/ehp.971051196
60. Adams ET, Auerbach S, Blackshear PE, Bradley A, Gruebbel MM, Little PB, et al. Proceedings of the 2010 National Toxicology Program Satellite Symposium. Toxicol Pathol (2011) 39(1):240–66. doi:10.1177/0192623310391680
61. Allen DG, Pearse G, Haseman JK, Maronpot RR. Prediction of rodent carcinogenesis: an evaluation of prechronic liver lesions as forecasters of liver tumors in NTP carcinogenicity studies. Toxicol Pathol (2004) 32:393–401. doi:10.1080/01926230490440934
62. Hall AP, Elcombe CR, Foster JR, Harada T, Kaufmann W, Knippel A, et al. Liver hypertrophy: a review of adaptive (adverse and non-adverse) changes – conclusions from the 3rd International ESTP Expert Workshop. Toxicol Pathol (2012) 40:971–94. doi:10.1177/0192623312448935
63. Woutersen RA, Soffers AE, Kroese ED, Krul CAM, van der Laan JW, van Benthem J, et al. Prediction of carcinogenic potential of chemicals using repeated-dose (13-week) toxicity data. Regul Toxicol Pharmacol (2016) 81:242–9. doi:10.1016/j.yrtph.2016.09.003
64. Greim H, Hartwig A, Reuter U, Richter-Reichhelm HB, Thielmann HW. Chemically induced pheochromocytomas in rats: mechanisms and relevance for human risk assessment. Crit Rev Toxicol (2009) 39:695–718. doi:10.1080/10408440903190861
65. Kroes R, Wester PW. Forestomach carcinogens: possible mechanisms of action. Food Chem Toxicol (1986) 24:1083–9.
66. IARC. Forestomach tumors, pp 5-26, butylated hydroxyl anisole pp 13-40. IARC Monographs in the Predictive Value of Rodent Forestomach and Gastric Neuroendocrine Tumours in Evaluating Carcinogenic Risks to Humans. IARC Tech. Publ. No. 39. Lyon, France: International Agency for Research on Cancer (2003).
67. IARC. Peroxisome Proliferation and Its Role in Carcinogenesis: Views and Expert Opinion of an IARC Working Group, IARC Tech. Rep. No. 24. Lyon, France: International Agency for Research on Cancer (1995).
68. Williams GM, Perone C. Mechanism-based risk assessment of peroxisome proliferating rodent hepatocarcinogens. In: Reddy JK, Suga T, Mannaerts GP, Lazarow PB, Subramani S, editors. Peroxisomes: Biology and Role in Toxicology and Disease (Vol. 804), New York: The New York Academy of Sciences (1996). p. 554–72.
69. Cohen SM. Evaluation of possible carcinogenic risk to humans based on liver tumors in rodent assays; the two-year bioassay is no longer necessary. Toxicol Pathol (2010) 38:487–501. doi:10.1177/0192623310363813
70. Russo J, Russo IH, Rogers AE, van Zwieten MJ, Gusterson B. Tumours of the mammary gland. In: Turusov VS, Mohr U, editors. Pathology of Tumours in Laboratory Animals. Vol. I. Tumors of the Rat. Lyon, France: International Agency for Research on Cancer (1990). p. 47–78. IARC Publ. No. 99.
71. Gold LS, Manley NB, Slone TH, Ward JM. Compendium of chemical carcinogens by target organ: results of chronic bioassays in rats, mice, hamsters, dogs and monkeys. Toxicol Pathol (2001) 29:639–52. doi:10.1080/019262301753385979
72. Cook JC, Klinefelter GR, Hardisty JF, Sharpe RM, Foster PM. Rodent Leydig cell tumorigenesis: a review of the physiology, pathology, mechanisms and relevance to humans. Crit Rev Toxicol (1999) 29:169–261. doi:10.1080/10408449991349203
73. Prentice DE, Meikle AW. A review of drug-induced Leydig cell hyperplasia and neoplasia in the rat and comparisons with man. Hum Exp Toxicol (1995) 14:562–72. doi:10.1177/096032719501400703
74. Alison RH, Capen CC, Prentice DE. Neoplastic lesions of questionable significance to humans. Toxicol Pathol (1994) 22:179–86. doi:10.1177/019262339402200211
75. Rice JM, Baan RA, Bletter M, Genevois-Charneau C, Grosse Y, McGregor DB, et al. Rodent tumors of the urinary bladder, renal cortex, and thyroid gland in IARC monographs evaluation of carcinogenic risk to humans. Toxicol Sci (1999) 49:166–71. doi:10.1093/toxsci/49.2.166
76. Hill RN, Crisp TM, Hurley PM, Rosenthal SL, Singh DV. Risk assessment of thyroid follicular cell tumors. Environ Health Perspect (1998) 106:447–57. doi:10.1289/ehp.98106447
77. Cohen SM. Urinary bladder carcinogenesis. Toxicol Pathol (1998) 26:121–7. doi:10.1177/019262339802600114
78. Cohen SM, Ellwein LB. Genetic errors, cell proliferation and carcinogenesis. Cancer Res (1991) 51:6493–505.
79. Cohen SM, Klaunig J, Meek ME, Hill RN, Pastoor T, Lehman-McKeeman L, et al. Evaluating the human relevance of chemically induced animal tumors. Toxicol Sci (2004) 78:181–6. doi:10.1093/toxsci/kfh073
80. Ellwein LG, Cohen S. The health risk of saccharin revisited. CRC Crit Rev Toxicol (1990) 20:311–26. doi:10.3109/10408449009089867
Keywords: carcinogenicity, pharmacology, human pharmaceuticals, histopathology, predictivity
Citation: van der Laan JW, Buitenhuis WHW, Wagenaar L, Soffers AEMF, van Someren EP, Krul CAM and Woutersen RA (2016) Prediction of the Carcinogenic Potential of Human Pharmaceuticals Using Repeated Dose Toxicity Data and Their Pharmacological Properties. Front. Med. 3:45. doi: 10.3389/fmed.2016.00045
Received: 05 July 2016; Accepted: 20 September 2016;
Published: 14 October 2016
Edited by:
Per Spindler, University of Copenhagen, DenmarkCopyright: © 2016 van der Laan, Buitenhuis, Wagenaar, Soffers, van Someren, Krul and Woutersen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Willem van der Laan, ancudmQubGFhbkBjYmctbWViLm5s