 Paul Zajac1
Paul Zajac1 Elke Schultz-Thater1
Elke Schultz-Thater1 Luigi Tornillo2Charlotte Sadowski1Emanuele Trella1
Luigi Tornillo2Charlotte Sadowski1Emanuele Trella1 Chantal Mengus1
Chantal Mengus1 Giandomenica Iezzi3
Giandomenica Iezzi3 Giulio C. Spagnoli1*
Giulio C. Spagnoli1*
- 1Oncology Surgery, Department of Biomedicine, University Hospital of Basel, Basel, Switzerland
- 2Department of Pathology, University Hospital of Basel, Basel, Switzerland
- 3Cancer Immunotherapy, Department of Biomedicine, University Hospital of Basel, Basel, Switzerland
MAGE-A antigens are expressed in a variety of cancers of diverse histological origin and germinal cells. Due to their relatively high tumor specificity, they represent attractive targets for active specific and adoptive cancer immunotherapies. Here, we (i) review past and ongoing clinical studies targeting these antigens, (ii) analyze advantages and disadvantages of different therapeutic approaches, and (iii) discuss possible improvements in MAGE-A-specific immunotherapies.
MAGE-A Tumor-Associated Antigens
MAGE-A were the first human tumor-associated antigens identified at the molecular level (1). They belong to the larger family of cancer/testis antigens (CTA), whose expression is consistently detected in cancers of different histological origin and germinal cells (2). The MAGE-A sub-family includes 12 highly homologous genes located on chromosome Xq28 (3, 4). Specific gene products have been identified by immunohistochemistry in cancers of different histological origin, including high percentages of non-small cell lung cancers (NSCLC), bladder cancers, esophageal and head and neck cancers, and sarcomas (5). These antigens are also frequently expressed in triple negative breast cancers (6), myeloma (7), and Reed–Sternberg cells (8) in Hodgkin’s disease, with the highest frequency being detected in synovial sarcoma (9). Among healthy tissues, the expression of specific members of the family has been observed in spermatogonia, placenta (10), and fetal ovary (11). However, recently, MAGE-A1 and -A12 genes have been shown to be expressed in CNS as well, as discussed below (12).
Functional Aspects of MAGE-A Antigens
Preferential intracellular location may be different for different antigens, e.g., mostly cytoplasmic for MAGE-A1, -A3, and -A4, but mostly nuclear for MAGE-A10 (13–16).
Functions are still unclear, although different studies have associated MAGE-A2, -A3/6, and -A9 expression with pro-tumorigenic activities such as p53 dysregulation (17–19), enhanced tumor cell proliferation potential, or maintenance of a cancer-stem cell-like functional profile (20).
In a variety of tumors of different histological origin, a clear correlation between expression of MAGE-A antigens and poor prognosis has been observed. In this context, data on bladder cancer (21, 22), NSCLC (23, 24), head and neck cancers (25–27), and ovarian cancer (28, 29) have consistently been reported. Indeed, MAGE-A antigen expression, at the gene and protein level, has repeatedly been shown to be associated with widespread DNA demethylation frequently observed in advanced cancers. On the same line, it has been shown to be inducible by demethylating agents, including chemotherapeutic compounds widely used in cancer treatment such as 5-aza-2′-deoxycytidine (30, 31), thus realistically envisaging the possibility of treatments combining chemotherapy and specific vaccination (32).
Immunogenicity of MAGE-A Antigens
Although peptides restricted by both HLA classes I and II have been identified (33), naturally occurring adaptive immune responses to MAGE-A antigens are usually characterized by a very low frequency of specific precursors (34) in both healthy donors and patients bearing cancers expressing them (35). However, responses to MAGE-A10 have been more frequently detected (36, 37). Responses in tumor-associated lymphocytes (TIL) have seldom been explored, but we have observed that MAGE-A10-specific CTL could be expanded from TIL infiltrating NSCLC displaying a high expression of the target antigen (38). On the other hand, CTL recognizing peptide motifs shared by multiple MAGE-A proteins may be generated from peripheral blood from patients and healthy donors (39). Most recently, tumor reactive CD8+ T cells, isolated based on their expression of activation marker (PD-1) from peripheral blood of melanoma patients, have been shown to relatively frequently target MAGE-A antigens (40).
Clinical Trials Targeting MAGE-A Antigens
In the past 10 years (2006–2016), a total of 44 clinical trials could be identified in “https://clinicaltrials.gov” database using “MAGE-A” as keyword: a total of 16 phase 0 or I, 13 phase I/II, 13 phase II, and 2 phase III studies. Regarding immunogen formulations, 16 studies utilized entire proteins in the presence or absence of adjuvants (41, 42), 11 used peptides (43–45), 6 used mRNA-transfected DC (46, 47), 1 was based on tumor cell lysate-pulsed DC, 2 took advantage of recombinant viral vectors (48, 49), and more recently, 6 and 2 trials, respectively, have focused on adoptive treatments by using specific T cell receptor (TCR)-transduced T cells (12) or expanded CTL (50).
Efficacy clinical data published so far, from patients immunized in the context of the 15 larger studies (phase II or III, Table S1 in Supplementary Material) mainly using MAGE-A protein (n = 11), do not appear to support significant clinical effectiveness (51).
Of interest, a chronological analysis of these 44 studies clearly underlines a strategy shift in the most recent years. Indeed, in the past 4 years, among the (only) 10 clinical studies initiated and including MAGE-A as antigens, there are no phase II or III studies. Moreover, the majority of the phase I or I/II studies are based on adoptive cell transfer. This “shift” in MAGE-A translational research strategy clearly results from the combined effect of “protein/peptide” efficacy failure and from the confidence generated by new approaches focusing on personalized effector T-cell treatment. In addition, one should also mention the shift in target paradigm from classical TAA to neo-antigens also contributing to the decreased use of MAGE-A antigens.
MAGE-A3 Protein as Immunogen
One of the most important clinical trials ever performed in MAGE-A cancer immunotherapy, involving thousands of patients with NSCLC, was focusing on the administration of recombinant MAGE-A3 protein together with adjuvants (52, 53). Despite promising initial data and the proven ability of the immunization protocol to induce detectable humoral responses in vaccinated patients (54), disease-free interval in patients with completely resected stage IB, II, and IIIA NSCLC did not appear to be significantly prolonged, as compared to patients of control group, in phase III studies in the context of an adjuvant therapy setting (41).
Why did these trials fail to reach efficacy? First, similar to MAGE-A antigens, a large majority of classical TAA-specific cancer vaccines clinically tested so far have been shown to induce heterogeneous immune responses rarely resulting in significant clinical effects.
However, specific issues should be considered for CTA-specific immunization. For instance, MAGE-A CTA expression, a pre-requisite for the eligibility of patients for treatment in these studies, has usually been assessed at the gene level by quantitative RT-PCR (RT-qPCR) (41, 54), which cannot provide insights into the actual numbers of CTA-positive tumor cells. Immunohistochemical studies using available MAGE-A-specific mAbs consistently underline that expression of these antigens might be highly heterogeneous in cancerous tissues with high expression often only detectable in relatively low percentages of tumor cells (10, 55). Remarkably, due to the high homology of sequences from different components of the MAGE-A family, a majority of currently available reagents do recognize multiple antigens. Our own experience based on the use of a MAGE-A10 highly specific mAb (Figure 1) suggests that expression of these antigens may be highly heterogeneous in a variety of tumors of different histological origin, with percentages of “positive” cells ranging between 5 and >60% (16). One could speculate that criteria based on the expression of target antigen(s), at the protein level, in high percentages of tumor cells and in multiple areas of primary and metastatic cancers could be applied for a more stringent selection of patients potentially eligible for MAGE-A-targeted antitumor immunization. Additionally, it might be of interest to verify the expression of the target MAGE-A antigen in recurrent tumors following specific immunization protocols, to verify possible selective immune editing (56). It is worth noting, however, that successful antigen-specific vaccination has also been shown to be able to promote responsiveness against unrelated antigens, the so-called “antigen spreading” phenomenon (57), thus potentially overcoming the requirement for a uniform expression of target antigens in tumors to be treated.
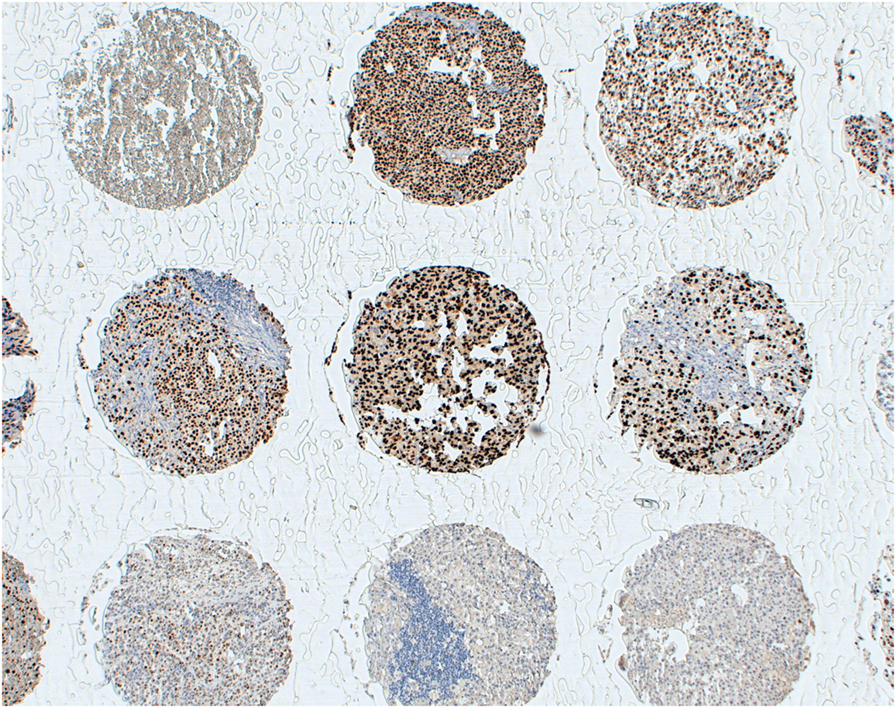
Figure 1. Heterogeneity of MAGE-A10 expression at the protein level. Melanoma tissues from a multi-tumor tissue microarray were stained with a MAGE-A10-specific reagent by standard techniques, as previously detailed (16). Antigen expression displays a high heterogeneity, regarding both percentages of antigen-positive tumor cells and staining intensity.
Importantly, the recombinant protein used in most efficacy studies was shown to induce humoral response and HLA class II-restricted lymphoproliferation, as expectable (41, 53, 54). However, the ability of these antigen formulations to promote class I-restricted responses appears to be more limited. One could speculate that libraries of overlapping “long” peptides (58), or highly immunogenic recombinant vectors (38, 59), could be more effective in this regard.
Heterogeneous Expression of MAGE-A Genes in Primary and Metastatic Cancers
Studies from our group clearly document the heterogeneity of MAGE-A antigens expression at the gene expression level as well. We tested by RT-qPCR the expression of Mage-A1, -A2, -A3, -A4, -A10, and -A12 genes in primary NSCLC from 33 patients (Table 1). In keeping with published data (23, 24), a total of 22 tumors (66%) showed evidence of expression of at least one of the antigens under investigation. Similar to recently published data in oral cancer (60), out of these patients with MAGE-A+ NSCLC, 10 (45%) had lymph nodes (LN) showing evidence of tumor metastasis, as compared with only 2 (18%) from the 11 MAGE-A(−) primary tumors. Interestingly, among the 10 metastatic LN from MAGE-A+ primary cancers, only half showed evidence of MAGE-A gene expression. Furthermore, in four LN, classified as non-metastatic, based on pathological evidence, expression of MAGE-A genes could be observed by RT-qPCR. Intriguingly, among LN associated with MAGE-A− primary cancers, 1/2 and 1/8 metastatic and non-metastatic samples, respectively, showed evidence of MAGE-A gene expression.
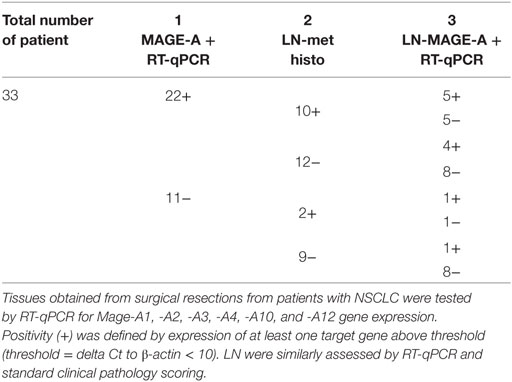
Table 1. MAGE-A gene expression, as detected by RT-qPCR in primary non-small cell lung cancers (NSCLC) and in corresponding lymph nodes (LN) showing evidence of metastatic outgrowth by standard clinical pathology techniques.
Taken together, these data suggest a higher sensitivity of RT-qPCR as compared to standard techniques for the detection of cancer cells within LN draining primary tumor tissues. Most importantly, however, they confirm the dynamic nature of MAGE-A antigens expression during cancer progression and may support the concept of combination therapies including treatments promoting MAGE-A antigen expression together with specific immunization procedures (61).
Adoptive Immunotherapies
In recent clinical studies, effector T cells, transduced with vectors encoding for specific TCRs recognizing peptides from MAGE-A3 or MAGE-4, have been adoptively transferred into patients bearing tumors expressing these antigens. Unfortunately, upon anti-MAGE-A3, HLA-A0201-restricted TCR gene therapy, despite measurable clinical responses in some patients, treatment-related severe adverse events and deaths were also reported. These events may possibly be due to the high affinity of these TCRs (see below) and to the recognition (“on-target/off-tumor”) of highly homologous peptide(s) from other MAGE-A proteins expressed in the CNS (12, 62). Similarly, myocardial toxicity, resulting in treatment related death, has also been observed following gene therapy with a MAGE-A3-specific HLA-A0101-restricted TCR (63, 64). In the latter case, the “off-target” effect was attributed to the high homology between the target peptide and a peptide from Titin muscle protein.
It is worth noting that the TCR transduced into T cells in the first study originally derived from “humanized” mice expressing HLA-A0201 and its affinity toward the target antigen was further improved by site-directed mutagenesis (65), thus increasing the chances of “on-target_off-tumor” adverse events affecting tissues characterized by low but detectable expression of defined MAGE-A antigens (12). The affinity of the TCR used in the second study, originally derived from a patient immunized with a recombinant viral vector (66), was also enhanced by site-directed mutagenesis.
By contrast, T cells expressing a MAGE-A4-specific TCR have been safely used in adoptive immunotherapy of patients with recurrent esophageal cancer (67).
Taken together, these data suggest that the clinical use of enhanced TCR effectors targeting MAGE-A antigens for cancer immunotherapy should be carefully evaluated in order to minimize potential “off-tumor” side effects.
However, natural MAGE-A-specific TCRs, from clones derived from tumor bearing patients or healthy donors, might also be of interest. Such CTLs would probably be characterized by a lower affinity for cognate HLA–class I peptide complex and possibly by a lower antitumor effector potential, but they would also likely have less toxic side effects. Considering the cumulative potency related to the high numbers of transduced cells usually infused into treated patients, and their ability to proliferate and generate “memory,” the effectiveness of this type of treatment should reasonably be further tested.
Conclusions
Taken together, published data may suggest that therapeutic strategies targeting MAGE-A antigens have so far failed to fulfill the promise of representing effective tools for cancer treatment. However, the understanding of mechanisms controlling immune response as a whole and cancer-specific immune responses in the tumor microenvironment in particular has made enormous progress in the past decade, generating an unprecedented “momentum” for cancer immunotherapy.
Successful utilization of therapeutic mAbs recognizing “immunological checkpoints” is currently generating enormous interest in clinical oncology. Their mechanisms of actions (MoA) are not fully clarified (68, 69). However, one of the main MoA is arguably represented by the “release of brakes” hampering T cell responses specific for tumor-specific or associated antigens. This hypothesis is supported, for instance, by the higher effectiveness of treatment with anti CTLA-4 therapeutic mAbs in cancers characterized by a high mutational load, likely to result in a higher expression of mutated proteins potentially recognized as “non-self” by the adaptive immune system (70). It is therefore reasonable to postulate that adequately timed combinations of vaccination procedures and administrations of therapeutic “checkpoint inhibitor” specific mAbs could be of high clinical relevance. Within this framework, a critical point might be represented by the choice of antigens of potential clinical use. Neo-antigens, e.g., tumor-specific mutated proteins have been successfully identified by whole exome sequencing (71–73), and the expression of defined antigenic “non-self” peptides associated with restricting HLA class I determinants may be detected by mass spectrometry techniques (74). Although highly appealing, the “personal” nature of neo-antigens might possibly also represent their Achilles’ heel, not only because of regulatory hurdles (75) but also because it would likely prevent the performance of conventional randomized trials, thereby complicating a reliable assessment of the effectiveness of innovative treatment procedures.
Based on these considerations, vaccination with tumor-associated or CTA could still realistically find an important place in cancer immunotherapy in the era of “immunological checkpoint” inhibitors (76). Considering that MAGE-A antigens are expressed in tumors with poor prognosis and a scarcity of therapeutic options, such as TNB, and lung and esophageal cancers, it is easy to predict that the interest of the scientific community in CTA might actually be revived in the light of the enormous advances in cancer immunotherapy of the last years.
Author Contributions
All authors participated in writing the manuscript and/or revising it critically for important intellectual content or providing the data mentioned in the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Part of this work was funded by the Swiss National Science Foundation; grant number: 310030_149745.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fmed.2017.00018/full#supplementary-material.
References
1. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science (1991) 254:1643–7. doi: 10.1126/science.1840703
2. Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer (2005) 5:615–25. doi:10.1038/nrc1669
3. Chomez P, De Backer O, Bertrand M, De Plaen E, Boon T, Lucas S. An overview of the MAGE gene family with the identification of all human members of the family. Cancer Res (2001) 61:5544–51.
4. De PE, Arden K, Traversari C, Gaforio JJ, Szikora JP, De SC, et al. Structure, chromosomal localization, and expression of 12 genes of the MAGE family. Immunogenetics (1994) 40:360–9. doi:10.1007/BF01246677
5. Juretic A, Spagnoli GC, Schultz-Thater E, Sarcevic B. Cancer/testis tumour-associated antigens: immunohistochemical detection with monoclonal antibodies. Lancet Oncol (2003) 4:104–9. doi:10.1016/S1470-2045(03)00982-3
6. Curigliano G, Viale G, Ghioni M, Jungbluth AA, Bagnardi V, Spagnoli GC, et al. Cancer-testis antigen expression in triple-negative breast cancer. Ann Oncol (2011) 22:98–103. doi:10.1093/annonc/mdq325
7. Van BN, Brasseur F, Godelaine D, Hames G, Ferrant A, Lehmann F, et al. Genes encoding tumor-specific antigens are expressed in human myeloma cells. Blood (1999) 94:1156–64.
8. Chambost H, Van Baren N, Brasseur F, Godelaine D, Xerri L, Landi SJ, et al. Expression of gene MAGE-A4 in Reed-Sternberg cells. Blood (2000) 95:3530–3.
9. Antonescu CR, Busam KJ, Iversen K, Kolb D, Coplan K, Spagnoli GC, et al. MAGE antigen expression in monophasic and biphasic synovial sarcoma. Hum Pathol (2002) 33:225–9. doi:10.1053/hupa.2002.31295
10. Jungbluth AA, Silva WA Jr, Iversen K, Frosina D, Zaidi B, Coplan K, et al. Expression of cancer-testis (CT) antigens in placenta. Cancer Immun (2007) 7:15.
11. Nelson PT, Zhang PJ, Spagnoli GC, Tomaszewski JE, Pasha TL, Frosina D, et al. Cancer/testis (CT) antigens are expressed in fetal ovary. Cancer Immun (2007) 7:1.
12. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36:133–51. doi:10.1097/CJI.0b013e3182829903
13. Kocher T, Schultz-Thater E, Gudat F, Schaefer C, Casorati G, Juretic A, et al. Identification and intracellular location of MAGE-3 gene product. Cancer Res (1995) 55:2236–9.
14. Landry C, Brasseur F, Spagnoli GC, Marbaix E, Boon T, Coulie P, et al. Monoclonal antibody 57B stains tumor tissues that express gene MAGE-A4. Int J Cancer (2000) 86:835–41. doi:10.1002/(SICI)1097-0215(20000615)86:6<835::AID-IJC12>3.0.CO;2-N
15. Schultz-Thater E, Juretic A, Dellabona P, Luscher U, Siegrist W, Harder F, et al. MAGE-1 gene product is a cytoplasmic protein. Int J Cancer (1994) 59:435–9. doi:10.1002/ijc.2910590324
16. Schultz-Thater E, Piscuoglio S, Iezzi G, Le MC, Zajac P, Carafa V, et al. MAGE-A10 is a nuclear protein frequently expressed in high percentages of tumor cells in lung, skin and urothelial malignancies. Int J Cancer (2011) 129:1137–48. doi:10.1002/ijc.25777
17. Marcar L, Maclaine NJ, Hupp TR, Meek DW. Mage-A cancer/testis antigens inhibit p53 function by blocking its interaction with chromatin. Cancer Res (2010) 70:10362–70. doi:10.1158/0008-5472.CAN-10-1341
18. Monte M, Simonatto M, Peche LY, Bublik DR, Gobessi S, Pierotti MA, et al. MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc Natl Acad Sci U S A (2006) 103:11160–5. doi:10.1073/pnas.0510834103
19. Yang B, O’Herrin SM, Wu J, Reagan-Shaw S, Ma Y, Bhat KM, et al. MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res (2007) 67:9954–62. doi:10.1158/0008-5472.CAN-07-1478
20. Costa FF, Le Blanc K, Brodin B. Concise review: cancer/testis antigens, stem cells, and cancer. Stem Cells (2007) 25:707–11. doi:10.1634/stemcells.2006-0469
21. Bergeron A, Picard V, LaRue H, Harel F, Hovington H, Lacombe L, et al. High frequency of MAGE-A4 and MAGE-A9 expression in high-risk bladder cancer. Int J Cancer (2009) 125:1365–71. doi:10.1002/ijc.24503
22. Kocher T, Zheng M, Bolli M, Simon R, Forster T, Schultz-Thater E, et al. Prognostic relevance of MAGE-A4 tumor antigen expression in transitional cell carcinoma of the urinary bladder: a tissue microarray study. Int J Cancer (2002) 100:702–5. doi:10.1002/ijc.10540
23. Bolli M, Kocher T, Adamina M, Guller U, Dalquen P, Haas P, et al. Tissue microarray evaluation of melanoma antigen E (MAGE) tumor-associated antigen expression: potential indications for specific immunotherapy and prognostic relevance in squamous cell lung carcinoma. Ann Surg (2002) 236:785–93. doi:10.1097/01.SLA.0000036266.09823.6C
24. Gure AO, Chua R, Williamson B, Gonen M, Ferrera CA, Gnjatic S, et al. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin Cancer Res (2005) 11:8055–62. doi:10.1158/1078-0432.CCR-05-1203
25. Cuffel C, Rivals JP, Zaugg Y, Salvi S, Seelentag W, Speiser DE, et al. Pattern and clinical significance of cancer-testis gene expression in head and neck squamous cell carcinoma. Int J Cancer (2011) 128:2625–34. doi:10.1002/ijc.25607
26. Figueiredo DL, Mamede RC, Spagnoli GC, Silva WA Jr, Zago M, Neder L, et al. High expression of cancer testis antigens MAGE-A, MAGE-C1/CT7, MAGE-C2/CT10, NY-ESO-1, and gage in advanced squamous cell carcinoma of the larynx. Head Neck (2011) 33:702–7. doi:10.1002/hed.21522
27. Laban S, Atanackovic D, Luetkens T, Knecht R, Busch CJ, Freytag M, et al. Simultaneous cytoplasmic and nuclear protein expression of melanoma antigen-A family and NY-ESO-1 cancer-testis antigens represents an independent marker for poor survival in head and neck cancer. Int J Cancer (2014) 135:1142–52. doi:10.1002/ijc.28752
28. Daudi S, Eng KH, Mhawech-Fauceglia P, Morrison C, Miliotto A, Beck A, et al. Expression and immune responses to MAGE antigens predict survival in epithelial ovarian cancer. PLoS One (2014) 9:e104099. doi:10.1371/journal.pone.0104099
29. Xu Y, Wang C, Zhang Y, Jia L, Huang J. Overexpression of MAGE-A9 is predictive of poor prognosis in epithelial ovarian cancer. Sci Rep (2015) 5:12104. doi:10.1038/srep12104
30. De SC, De BO, Faraoni I, Lurquin C, Brasseur F, Boon T. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc Natl Acad Sci U S A (1996) 93:7149–53. doi:10.1073/pnas.93.14.7149
31. Weber J, Salgaller M, Samid D, Johnson B, Herlyn M, Lassam N, et al. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-2’-deoxycytidine. Cancer Res (1994) 54:1766–71.
32. Karpf AR. A potential role for epigenetic modulatory drugs in the enhancement of cancer/germ-line antigen vaccine efficacy. Epigenetics (2006) 1:116–20. doi:10.4161/epi.1.3.2988
33. Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother (2005) 54:187–207. doi:10.1007/s00262-004-0560-6
34. Chaux P, Vantomme V, Coulie P, Boon T, van der Bruggen P. Estimation of the frequencies of anti-MAGE-3 cytolytic T-lymphocyte precursors in blood from individuals without cancer. Int J Cancer (1998) 77:538–42. doi:10.1002/(SICI)1097-0215(19980812)77:4<538::AID-IJC11>3.0.CO;2-2
35. Marchand M, Weynants P, Rankin E, Arienti F, Belli F, Parmiani G, et al. Tumor regression responses in melanoma patients treated with a peptide encoded by gene MAGE-3. Int J Cancer (1995) 63:883–5. doi:10.1002/ijc.2910630622
36. Bricard G, Bouzourene H, Martinet O, Rimoldi D, Halkic N, Gillet M, et al. Naturally acquired MAGE-A10- and SSX-2-specific CD8+ T cell responses in patients with hepatocellular carcinoma. J Immunol (2005) 174:1709–16. doi:10.4049/jimmunol.174.3.1709
37. Valmori D, Dutoit V, Rubio-Godoy V, Chambaz C, Lienard D, Guillaume P, et al. Frequent cytolytic T-cell responses to peptide MAGE-A10(254-262) in melanoma. Cancer Res (2001) 61:509–12.
38. Groeper C, Gambazzi F, Zajac P, Bubendorf L, Adamina M, Rosenthal R, et al. Cancer/testis antigen expression and specific cytotoxic T lymphocyte responses in non small cell lung cancer. Int J Cancer (2007) 120:337–43. doi:10.1002/ijc.22309
39. Graff-Dubois S, Faure O, Gross DA, Alves P, Scardino A, Chouaib S, et al. Generation of CTL recognizing an HLA-A*0201-restricted epitope shared by MAGE-A1, -A2, -A3, -A4, -A6, -A10, and -A12 tumor antigens: implication in a broad-spectrum tumor immunotherapy. J Immunol (2002) 169:575–80. doi:10.4049/jimmunol.169.1.575
40. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med (2016) 22:433–8. doi:10.1038/nm.4051
41. Vansteenkiste JF, Cho BC, Vanakesa T, De Pas TM, Zielinski M, Kim MS, et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol (2016) 17:822–35. doi:10.1016/S1470-2045(16)00099-1
42. Kruit WH, Suciu S, Dreno B, Mortier L, Robert C, Chiarion-Sileni V, et al. Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: results of a randomized phase II study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J Clin Oncol (2013) 31:2413–20. doi:10.1200/JCO.2012.43.7111
43. Germeau C, Ma W, Schiavetti F, Lurquin C, Henry E, Vigneron N, et al. High frequency of antitumor T cells in the blood of melanoma patients before and after vaccination with tumor antigens. J Exp Med (2005) 201:241–8. doi:10.1084/jem.20041379
44. Marchand M, Van BN, Weynants P, Brichard V, Dreno B, Tessier MH, et al. Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA-A1. Int J Cancer (1999) 80:219–30. doi:10.1002/(SICI)1097-0215(19990118)80:2<219::AID-IJC10>3.3.CO;2-J
45. Schuler-Thurner B, Dieckmann D, Keikavoussi P, Bender A, Maczek C, Jonuleit H, et al. Mage-3 and influenza-matrix peptide-specific cytotoxic T cells are inducible in terminal stage HLA-A2.1+ melanoma patients by mature monocyte-derived dendritic cells. J Immunol (2000) 165:3492–6. doi:10.4049/jimmunol.165.6.3492
46. Russo V, Pilla L, Lunghi F, Crocchiolo R, Greco R, Ciceri F, et al. Clinical and immunologic responses in melanoma patients vaccinated with MAGE-A3-genetically modified lymphocytes. Int J Cancer (2013) 132:2557–66. doi:10.1002/ijc.27939
47. Wilgenhof S, Corthals J, Heirman C, Van Baren N, Lucas S, Kvistborg P, et al. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. J Clin Oncol (2016) 34:1330–8. doi:10.1200/JCO.2015.63.4121
48. Batchu RB, Gruzdyn OV, Moreno-Bost AM, Szmania S, Jayandharan G, Srivastava A, et al. Efficient lysis of epithelial ovarian cancer cells by MAGE-A3-induced cytotoxic T lymphocytes using rAAV-6 capsid mutant vector. Vaccine (2014) 32:938–43. doi:10.1016/j.vaccine.2013.12.049
49. Lin L, Wei J, Chen Y, Huang A, Li KK, Zhang W. Induction of antigen-specific immune responses by dendritic cells transduced with a recombinant lentiviral vector encoding MAGE-A3 gene. J Cancer Res Clin Oncol (2014) 140:281–9. doi:10.1007/s00432-013-1552-8
50. Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Xu YY, Kalos M, et al. Combination immunotherapy after ASCT for multiple myeloma using MAGE-A3/Poly-ICLC immunizations followed by adoptive transfer of vaccine-primed and costimulated autologous T cells. Clin Cancer Res (2014) 20:1355–65. doi:10.1158/1078-0432.CCR-13-2817
51. Connerotte T, Van PA, Godelaine D, Tartour E, Schuler-Thurner B, Lucas S, et al. Functions of Anti-MAGE T-cells induced in melanoma patients under different vaccination modalities. Cancer Res (2008) 68:3931–40. doi:10.1158/0008-5472.CAN-07-5898
52. Brichard VG, Lejeune D. GSK’s antigen-specific cancer immunotherapy programme: pilot results leading to Phase III clinical development. Vaccine (2007) 25(Suppl 2):B61–71. doi:10.1016/j.vaccine.2007.06.038
53. Brichard VG, Godechal Q. MAGE-A3-specific anticancer immunotherapy in the clinical practice. Oncoimmunology (2013) 2:e25995. doi:10.4161/onci.25995
54. Vansteenkiste J, Zielinski M, Linder A, Dahabreh J, Gonzalez EE, Malinowski W, et al. Adjuvant MAGE-A3 immunotherapy in resected non-small-cell lung cancer: phase II randomized study results. J Clin Oncol (2013) 31:2396–403. doi:10.1200/JCO.2012.43.7103
55. Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev (2002) 188:22–32. doi:10.1034/j.1600-065X.2002.18803.x
56. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases – elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi:10.1016/j.coi.2014.01.004
57. Corbiere V, Chapiro J, Stroobant V, Ma W, Lurquin C, Lethe B, et al. Antigen spreading contributes to MAGE vaccination-induced regression of melanoma metastases. Cancer Res (2011) 71:1253–62. doi:10.1158/0008-5472.CAN-10-2693
58. Kenter GG, Welters MJ, Valentijn AR, Lowik MJ, Berends-van der Meer D, Vloon AP, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med (2009) 361:1838–47. doi:10.1056/NEJMoa0810097
59. Adamina M, Rosenthal R, Weber WP, Frey DM, Viehl CT, Bolli M, et al. Intranodal immunization with a vaccinia virus encoding multiple antigenic epitopes and costimulatory molecules in metastatic melanoma. Mol Ther (2010) 18:651–9. doi:10.1038/mt.2009.275
60. Brisam M, Rauthe S, Hartmann S, Linz C, Brands RC, Kubler AC, et al. Expression of MAGE-A1-A12 subgroups in the invasive tumor front and tumor center in oral squamous cell carcinoma. Oncol Rep (2016) 35:1979–86. doi:10.3892/or.2016.4600
61. Weiser TS, Ohnmacht GA, Guo ZS, Fischette MR, Chen GA, Hong JA, et al. Induction of MAGE-3 expression in lung and esophageal cancer cells. Ann Thorac Surg (2001) 71:295–301. doi:10.1016/S0003-4975(00)02421-8
62. Brichard VG, Louahed J, Clay TM. Cancer regression and neurological toxicity cases after anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36:79–81. doi:10.1097/CJI.0b013e3182829747
63. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med (2013) 5:197ra103. doi:10.1126/scitranslmed.3006034
64. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122:863–71. doi:10.1182/blood-2013-03-490565
65. Chinnasamy N, Wargo JA, Yu Z, Rao M, Frankel TL, Riley JP, et al. A TCR targeting the HLA-A*0201-restricted epitope of MAGE-A3 recognizes multiple epitopes of the MAGE-A antigen superfamily in several types of cancer. J Immunol (2011) 186:685–96. doi:10.4049/jimmunol.1001775
66. Karanikas V, Lurquin C, Colau D, VanBaren N, DeSmet C, Lethe B, et al. Monoclonal anti-MAGE-3 CTL responses in melanoma patients displaying tumor regression after vaccination with a recombinant canarypox virus. J Immunol (2003) 171:4898–904. doi:10.4049/jimmunol.171.9.4898
67. Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, et al. Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res (2015) 21:2268–77. doi:10.1158/1078-0432.CCR-14-1559
68. Furness AJ, Vargas FA, Peggs KS, Quezada SA. Impact of tumour microenvironment and Fc receptors on the activity of immunomodulatory antibodies. Trends Immunol (2014) 35:290–8. doi:10.1016/j.it.2014.05.002
69. Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol (2006) 90:297–339. doi:10.1016/S0065-2776(06)90008-X
70. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371:2189–99. doi:10.1056/NEJMoa1406498
71. Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature (2015) 520:692–6. doi:10.1038/nature14426
72. Mennonna D, Maccalli C, Romano MC, Garavaglia C, Capocefalo F, Bordoni R, et al. T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes. Gut (2017) 66(3):454–63. doi:10.1136/gutjnl-2015-309453
73. Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature (2016) 536:91–5. doi:10.1038/nature18945
74. Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med (2012) 18:1254–61. doi:10.1038/nm.2883
75. Britten CM, Singh-Jasuja H, Flamion B, Hoos A, Huber C, Kallen KJ, et al. The regulatory landscape for actively personalized cancer immunotherapies. Nat Biotechnol (2013) 31:880–2. doi:10.1038/nbt.2708
Keywords: MAGE-A, cancer–testis antigens, cancer immunotherapy, clinical trials, adoptive immunotherapy
Citation: Zajac P, Schultz-Thater E, Tornillo L, Sadowski C, Trella E, Mengus C, Iezzi G and Spagnoli GC (2017) MAGE-A Antigens and Cancer Immunotherapy. Front. Med. 4:18. doi: 10.3389/fmed.2017.00018
Received: 13 December 2016; Accepted: 09 February 2017;
Published: 08 March 2017
Edited by:
Diego Luigi Cortinovis, S Gerardo Hospital, ItalyReviewed by:
Pier Paolo Piccaluga, University of Bologna, ItalyFernando Schmitt, Porto University, Portugal
Copyright: © 2017 Zajac, Schultz-Thater, Tornillo, Sadowski, Trella, Mengus, Iezzi and Spagnoli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giulio C. Spagnoli, Z2l1bGlvLnNwYWdub2xpQHVzYi5jaA==