 Claudia I. Rivas-Ortiz1
Claudia I. Rivas-Ortiz1 Yolanda Lopez-Vidal1
Yolanda Lopez-Vidal1 Luis Jose Rene Arredondo-Hernandez2
Luis Jose Rene Arredondo-Hernandez2 Gonzalo Castillo-Rojas1*
Gonzalo Castillo-Rojas1*
- 1Programa de Inmunología Molecular Microbiana, Departamento de Microbiología y Parasitología, Facultad de Medicina, Universidad Nacional Autónoma de México (UNAM), Mexico City, Mexico
- 2Programa de Microbioma, Facultad de Medicina, Universidad Nacional Autónoma de México (UNAM), Mexico City, Mexico
Gastric cancer is a world health problem and depicts the fourth leading mortality cause from malignancy in Mexico. Causation of gastric cancer is not only due to the combined effects of environmental factors and genetic variants. Recent molecular studies have transgressed a number of genes involved in gastric carcinogenesis. The aim of this review is to understand the recent basics of gene expression in the development of the process of gastric carcinogenesis. Genetic variants, polymorphisms, desoxyribonucleic acid methylation, and genes involved in mediating inflammation have been associated with the development of gastric carcinogenesis. Recently, these genes (interleukin 10, Il-17, mucin 1, β-catenin, CDX1, SMAD4, SERPINE1, hypoxia-inducible factor 1 subunit alpha, GSK3β, CDH17, matrix metalloproteinase 7, RUNX3, RASSF1A, TFF1, HAI-2, and COX-2) have been studied in association with oncogenic activation or inactivation of tumor suppressor genes. All these mechanisms have been investigated to elucidate the process of gastric carcinogenesis, as well as their potential use as biomarkers and/or molecular targets to treatment of disease.
Introduction
Gastric cancer is a health problem, because it is the sixth leading cause of death worldwide (1). In Mexico, gastric cancer is the fourth leading cause of death from malignancy in men and women (2). Among the risk factors associated with the development of gastric cancer are smoking, obesity, and consumption of foods high in nitrates (smoked foods and salt, for example). However, Helicobacter pylori (H. pylori) infection is the main risk factor in the development of gastric cancer, a Gram-negative bacillus that has a spiral morphology when it colonizes the gastric mucosa of human stomach. This bacillus is recognized their participation in the development of atrophic gastritis, peptic ulcer (gastric and duodenal ulcer), and intestinal type gastric cancer. World Health Organization through the International Agency for Research on Cancer recognized H. pylori as a carcinogen category I in humans, in 1994; later, in 2009, this was ratified by the collaborative group in cancer and Helicobacter (3).
Most gastric cancer patients are asymptomatic and may have advanced disease at diagnosis. These patients may receive potentially curative resection (total or subtotal gastrectomy and lymphadenectomy); therefore, the overall rate at 5 years of patient survival is ranges from 10 to 30% (4). Early gastric cancer is surgically curable, but only discovered during programs of screening for gastric cancer, that unfortunately not widely performed in the world, except for countries, such as Japan, China, Venezuela, and Chile that have a very high incidence of gastric cancer (5–7).
Molecular studies prove that gastric cancer emerges from the combination of environmental factors and a host genetic component. It is noteworthy that the accumulation of genetic and epigenetic alterations plays an important role in the process of carcinogenesis. This review focuses on recent aspects of gene expression in the development of gastric carcinogenesis.
Host Genetic Susceptibility
Molecular genetic studies have described some variants that are useful as biomarkers of genetic susceptibility involved in the development of gastric cancer. Single-nucleotide polymorphisms (SNPs) are a variation in desoxyribonucleic acid (DNA) sequence that affect a single base [adenine (A), thymine (T), cytosine (C), or guanine (G)] of a genome sequence. SNPs are considered a form of point mutation that has been evolutionarily successful enough to notice a significant part of the population of a species. Genetic variations may modulate the effects of exposure to environmental factors by regulating biological pathways during gastric carcinogenesis.
Genetic variants in genes from cytokines and their receptors associated with inflammation are considered to participate to tumor initiation and promotion. Considering genetic polymorphisms in gastric cancer, this approach has led to a growing interest in recent years. Recently, in the meta-analysis performed by Zhuang et al., found that a promoter polymorphism, in the interleukin 10 (-592C>), might be associated in Asians with gastric cancer development (8). Polymorphisms in the promoter of the gene encoding the tumor necrosis factor (TNFα-308) and interleukin 10 (IL-10) (IL-10-592 and -1082) also have an important role in the development of gastritis associated by H. pylori infection. They have been found to modify the risk for the development of gastric cancer in Caucasians (9–12). Wu et al. found an association between IL-17F A7488G and intestinal type gastric cancer (13). This is interesting because the polymorphism of IL-17F 7488 is associated with an increased inflammation by H. pylori infection (14). In a meta-analysis of a group of polymorphisms in inflammation-related genes, genes encoding proteins of interleukins [IL-1β, interleukin antagonist receptor, interleukin 8 (IL-8), and IL-10] and TNF-α were investigated. The results show that interleukin antagonist receptor 2 polymorphism represents an increased risk of intestinal cancer in non-Asian populations (15).
Among the genetic variants related to inflammation and its association with gastric cancer caused by H. pylori infection. It has been proposed that persistent chronic gastritis during H. pylori infection generates a microenvironment where inflammatory mediators activate signaling pathways that may lead to gastric tumorigenesis. The chronic infection of the stomach with H. pylori is associated with mutations in genes of epithelial cells, inhibition of apoptosis, stimulation of angiogenesis, and increased cell proliferation. Epithelial cells of the mucosa, including the gastric epithelium, express several cell surface receptors, which indicate the presence of mucosal pathogens and activate inflammatory pathways (16). Mucin (MUC), glycosylated proteins that play important roles protecting epithelial cells and pathogens, are involved in the process of epithelial renewal and differentiation. MUC6 and MUC1 are secreted in the stomach and can play an important role in the development of gastric cancer (17). Recently, studies of the genome-wide association in Japanese and Chinese populations identified chromosome 1q22 harboring the gene MUC1 as a locus that confers susceptibility to the development of gastric cancer. This gene encodes MUC bound to the cell membrane. MUC1 is known as an oncogene with anti-apoptotic function in cancer cells; however, in normal gastric mucosa, it is believed that this protein has a role in protecting gastric epithelial cells from attacks that cause inflammation and carcinogenesis (18).
In its role against cancer, MUC1 consists of two subunits: N- and C-terminal. MUC1-C has a transmembrane domain and a cytoplasmic tail that contains several phosphorylation sites and a binding site to β-catenin. Threonine phosphorylation is contained in the cytoplasmic tail interactions with MUC1 and promotes β-catenin, leading to gene regulation of p53 (19). It is proposed that the virulence factor cytotoxin-associated protein (CagA) destabilizes the β-catenin/E-cadherin complex located in the cytoplasm of epithelial cells and increases the accumulation of β-catenin in the nucleus. CagA activates β-catenin-dependent genes involved in intestinal differentiation, such as, caudal type homeobox 1 (CDX1) transcription factor expressed in the intestine, where it regulates of cell proliferation and differentiation. In areas of the stomach and esophagus with intestinal metaplasia aberrant expression of CDX1 has been found. Therefore, CDXI-induced CagA may play a role in the transdifferentiation of gastric mucosa to intestinal type. Indeed, induction of CDX1 by CagA–β-catenin complex is associated with ectopic expression of intestinal differentiation marker MUC2 in gastric epithelial cells. These genes CDX1 and MUC2 favor the development of intestinal metaplasia, which is a pre-neoplastic lesion (20). Also, the activation of the expression of IL-8, an inflammatory chemotactic cytokine, is induced by the nuclear accumulation of β-catenin. It has been shown that H. pylori regulate the increased expression of MUC1 in gastric cancer cells by CpG hypomethylation and signal transducer and activator of transcription 3. This cascade can exist in normal gastric epithelium as an anti-cancer mechanism against infection by H. pylori (21).
One of the most important signaling pathways of tumor suppressor genes is transforming growth factor beta (TGF-β), which plays a critical role in regulating cell growth and cell differentiation. Generally, signaling is started with the ligand-induced oligomerization of serine/threonine receptor kinases and the phosphorylation of cytoplasmic signaling molecule Smad. At present, the loss of the nuclear expression of SMAD4 has been described in the progression of gastric cancer (22, 23). Because of the function SMAD4 has in tumor suppression in gastric cancer, Wu et al. conducted a study of five SNPs, finding an association between the C allele (rs177663887) and G allele (rs12456284) with an increased expression of SMAD4 and low risk of gastric cancer (24).
The serpin peptidase inhibitor, clade E, member 1 (SERPINE1) plays a key role in tumorigenesis. It prevents the excessive proteolysis of the extracellular matrix, which is necessary to capillary morphogenesis, cell migration, and invasion. A study by Ju et al. found that a SERPINE1 polymorphism (c.1162+162C>T) in intron 7 is strongly associated with the susceptibility to diffuse gastric cancer (25). Many articles talk about the partnership that exists between genetic polymorphisms and the risk of developing gastric cancer; host genetic factors are very important to the development of cancer. The interaction of different polymorphisms with environmental factors is an effective formula that triggers cancer development and provides us with information to explain the various mechanisms of carcinogenesis.
Molecular Alterations in Gastric Cancer
Understanding which molecular mechanisms and alterations are carried out after the initiation and progression of gastric tumorigenesis is extremely important for the early detection of the disease and the identification of some therapeutic targets as well. We have identified certain genetic and epigenetic molecular abnormalities in gastric cancer that can help us to decipher the mechanisms of gastric carcinogenesis.
Genetic Abnormalities
The aberrant cellular metabolism is an important feature during tumorigenesis and cancer progression (26, 27). Programming the energy metabolism is an emerging hallmark of cancer (28); cancer cells reprogram their metabolism by increasing glycolysis instead of mitochondrial oxidative phosphorylation to generate cellular energy (29). Tissue hypoxia is a fundamental driving force that leads to the reprogramming of cell metabolism (30). In stomach cancer, a low percentage of cells are in the G0 phase of the cell cycle, producing accelerated cell proliferation. When the level of hypoxia increases, the activity level of oxidative phosphorylation as the main energy (ATP) producer decreases and glycolysis increases, allowing cancer cells to accelerate the uptake of glucose from the bloodstream to compensate for the loss of energy production, which is necessary for the cells to remain viable. This change in energy metabolisms accelerated cell proliferation and increased hypoxia, generating a vicious cycle of enhanced growth of cancer. This cycle is broken down when the angiogenesis occurs, and a high level of hypoxia triggers it. When this happens, there is a reduction in hypoxia levels and switches back to oxidative phosphorylation as the main energy producer. This process continues until the cells return to hypoxic and the levels of cellular hypoxia rise and fall periodically, which agrees with the increase in cancer mass and new angiogenesis, respectively. Under an atmosphere of hypoxia increased cell proliferation and cancer progression (31).
At a gene level, hypoxia-inducible factor 1 (HIF-1) is the primary transcriptional activator sensitive to oxygen that helps cells to adapt to low oxygen tension (hypoxia) (32). HIF-1 consists of a constitutively expressed β-subunit and a hypoxia-inducible α-subunit. The latter (HIF-1α) only stabilized in hypoxic conditions and regulated the transcriptional activity of HIF-1 (33). Overexpression of HIF-1α is involved in the activation of multiple target genes crucial in cancer biology, including erythropoiesis, angiogenesis, glucose metabolism, cell proliferation, survival, and apoptosis (34). It has been shown that the expression of over 20 genes is directly regulated by HIF-1α; among these genes is NFκB1, a regulatory molecule of inflammation and cancer (35).
Zheng et al. reported overexpression of the inactive form of glycogen synthase kinase 3 beta (GSK3β) and phospo-GSK3β (ser9) in gastric cancer compared to normal mucosa. This article addresses that overexpression of phospo-GSK3β (ser9) is positively correlated with prognosis of gastric cancer (36); phospo-GSK3β (ser9) is induced by gastrin and leads to GSK3β inhibition, increased Snail expression, nuclear translocation of β-catenin, and an increase in cell migration in gastric cancer (37).
Transmembrane proteins are proteins involved in signal communication between the extracellular and intracellular spaces. It has been described that the transmembrane protein in gastric cancer CD133 is overexpressed in 57% of gastric cancer and positively correlates with the expression of Ki-67 (38). A member of the cadherin superfamily, CDH17, was reported to be a marker for gastric cancer in early stage. CDH17 expression was positively associated with a good prognosis of gastric cancer (39). Another component of the extracellular matrix is hyaluronic acid (HA). The two known cellular receptors of HA are cluster of differentiation 168 (CD168) and cluster of differentiation 44 (CD44). The overexpression of CD168 was reported in a panel of gastric cancer cases, where CD168 expression was associated with the depth of invasion and cancer metastasis (40). In another study, Xie et al. described that tumor stem cell (SC) surface marker CD44 (CD44v6) is involved in gastric cancer metastasis. They observed the overexpression of CD44v6 in all cancer cell lines. The 5-year survival rate of patients with CD44v6 positive expression is significantly worse compared to the survival rate of patients with CD44v6 negative expression (41). Wang et al. performed a meta-analysis to quantitatively study the correlation of CD44 expression with the clinicopathological data of patients with gastric cancer. They found that CD44 expression was related to stage, tumor size, and lymph node metastasis of gastric cancer, whereas CD44v6 was related to lymph node metastasis, lymphatic invasion, and venous invasion (42).
Matrix metalloproteinases (MMPs) are enzymes that generate proteolysis, are zinc dependent, and are involved in diverse physiological and pathological processes, including degradation of extracellular matrix, tissue remodeling, inflammation, tumor invasion, and tumor cell metastasis. Due to the functioning of MMP their operation in cancer, Wang and colleagues performed a meta-analysis that showed the expression levels of MMP-7 were increased in gastric cancer. They associated it with lymph node metastasis, advanced tumor node metastasis (TNM) stage, and invasion of the tumor. Therefore, MMP-7 can be a useful biomarker for determining the progression of gastric cancer and prognosis (43). The increased expression of MMP-9, -12, and -21, is associated with decreased survival in gastric cancer (44).
Epithelial–mesenchymal transition (EMT) plays a significant role in tumor progression and invasion. Snail is a known regulator of EMT in various malignancies. After the analysis of gene expression and complementary DNA microarray, it was observed that the overexpression of Snail increases invasion, cell migration, and tumor progression in gastric cancer (45). This gene can also be used as a predictive biomarker for prognosis or aggressiveness in gastric cancer.
It is unclear whether H. pylori promote gastric cancer by direct invasion of the epithelium and interaction with oncoproteins. Although bacteria are not invasive and hide in the mucus layer to prevent stomach acid, we observed that about 10% of them adhere to the cells, and some intracellularly, but the cancer process is still unknown. There is evidence implicating the CagA as a promoting oncoprotein in human gastric cancer. CagA is a 120–145 kDa protein encoded by the cytotoxin-associated gene A (cagA) gene, located in the cag pathogenicity island (cag-PAI); it also encodes the type IV secretion system with which CagA is injected into the epithelial cells of the gastric guest (46). Infection with strains possessing this protein is associated with a greater degree of inflammation in gastric mucosa; in severe gastric atrophy, it is considered an important process in the development of gastric cancer (47–49).
CagA translocation to epithelial cells allows the interaction with host proteins in two different forms: phosphorylation-dependent and independent from each other. CagA is phosphorylated by the Src kinase (SFK) family and thus activates the tyrosine phosphatase (SHP-2) (50), altering cell adhesion and migration (51) and stimulating cell-signaling pathway Erk (52). CagA phosphorylated by SFK can be dependent and independent from Ras (53), whereby uncontrolled cell proliferation is promoted (49, 54). It also produces an aberrant epithelial cell adhesion and cytoskeletal rearrangement (55). Moreover, unphosphorylated CagA can deregulate different signaling cascades in epithelial cells, resulting in alterations that cause cell proliferation and the mobility of the cells with increased expression of genes involved in oncogenic transformation (56–58). CagA mediates the enhanced surface expression of epidermal growth factor receptor (EGFR) by inhibiting receptor endocytosis. The EGFR controls crucial cellular processes such as cell division or cell death as well as motility and matrix adhesion. Thus, a deregulation of the EGFR caused by increased receptor surface expression or mutations in the extracellular domain is linked to cancer development. In addition to its involvement in carcinogenesis, EGF signaling has been implicated in the inhibition of gastric acid secretion and liked to impaired wound healing of the gastric epithelial layer, two factors involved in the development of gastric ulcer (59).
Constitutive EGFR activity reduces acid secretion but also predisposes oncogenic transformation, supporting signaling deregulation as a direct cause of carcinogenesis. Furthermore, the association of CagA with protease active receptor (PAR1), a member of polarity complex, appears to disturb the epithelial cell polarity, promoting interaction between CagA and tyrosine phosphatase SHP-2, which activates extracellular regulated kinases by signals (ERK1/2) and promotes EMT (60). For all this, it seems that CagA houses adapter proteins and kidnaps signaling pathways to maintain a non-polarized phenotype with attenuated gastric acid secretion, which is favorable for bacterial survival (Table 1).
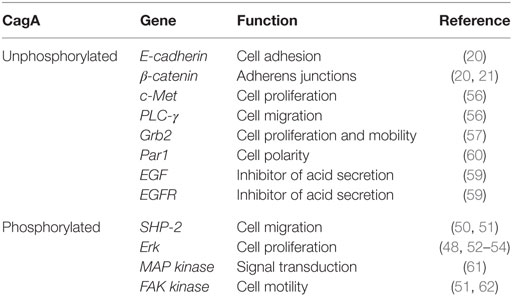
Table 1. Genes activated in the presence of CagA.
The Runt-related transcription factor 3 (RUNX3) gene is a tumor suppressor in many tissues and often inactive in gastric cancer. Tsang et al. showed that the virulence factor of H. pylori CagA is capable of binding RUNX3, inducing ubiquitination and degradation of RUNX3 by proteasome machinery (63). This tumor suppressor plays an important role in immune regulation, while its reduced expression is related to the development and progression of gastric carcinoma. The levels of expression of RUNX3, T-bet, and IFN-γ are lower in patients with gastric carcinoma than in control groups and may contribute to the development of cancer (64) (Figure 1).
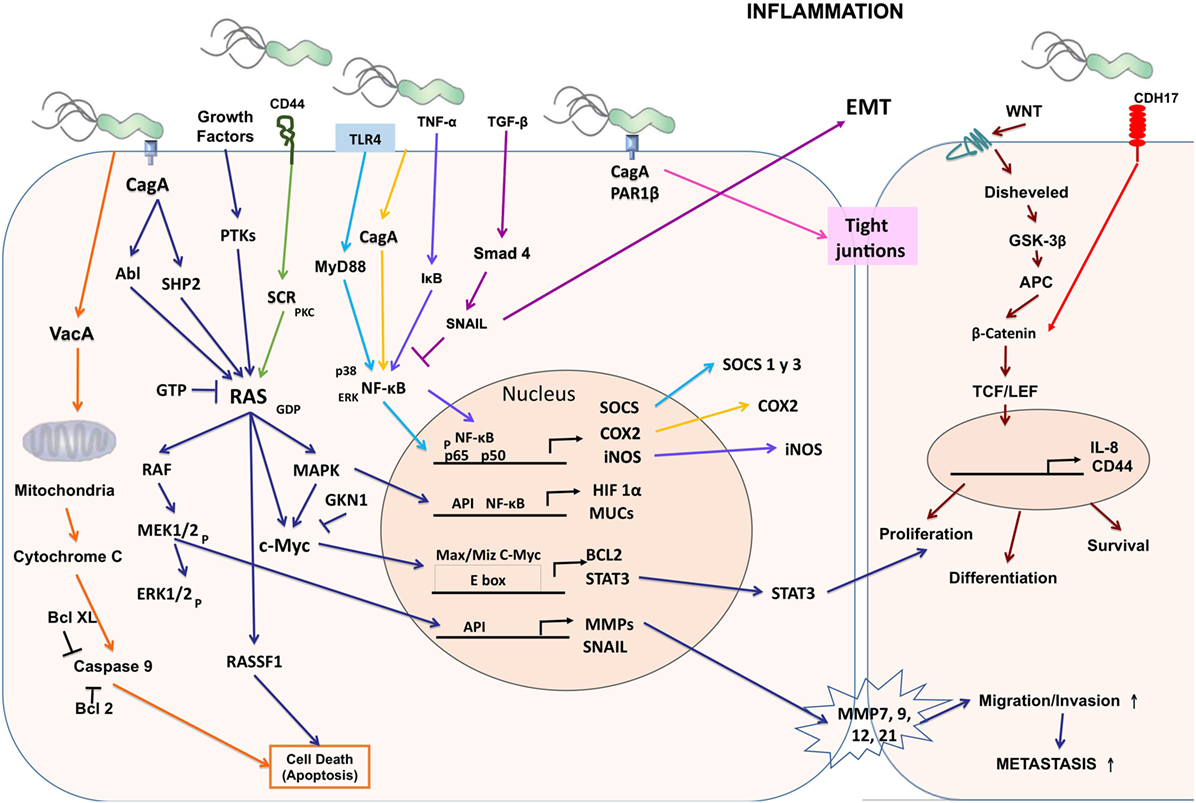
Figure 1. Cellular and molecular pathogenesis of Helicobacter pylori infection in gastric carcinogenesis. The phosphorylated CagA active in the SHP-2/MAPK pathway regulates MEK/extracellular signal-regulated kinase (ERK), RAS/cMyc, and NF-κB pathways as a result of the regulation of genes such as hypoxia-inducible factor 1 subunit alpha (HIF-1α), mucins (MUCs), suppressor of cytokine signaling (SOCS), COX-2, inducible nitric oxide synthase (iNOS), BCL2, signal transducer and activator of transcription 3 (STAT3), matrix metalloproteinases (MMPs), and SNAIL producing proliferation, differentiation, cell survival, and increased migration, invasion, and metastasis of cancer cells. CagA alters the tight junctions independently of phosphorylation. Vacuolating cytotoxin (VacA) alters the permeability of the mitochondrial membrane and favors apoptosis.
Epigenetic Changes Promote the Development of Gastric Cancer
Desoxyribonucleic acid methylation is an important epigenetic modification that is involved in the control of gene expression. It is a covalent chemical modification catalyzed by the DNA methyltransferase family of enzymes involved in the addition of a methyl group (CH3) on the fifth carbon of cytosine (65). DNA methylation is grouped in regions known as CpG islands, which are regions of DNA localized in up to 40% of promoters of genes. The methylation within a gene promoter is associated with the transcriptional inhibition of gene expression, particularly in tumor suppressor genes.
There is information regarding the roles of many tumor suppressor genes in cancer prevention; among those roles are DNA repair, cell adhesion, cell cycle control, and apoptosis. However, these genes are silenced by hypermethylation of the promoters during carcinogenesis. Numerous studies on DNA methylation report the existence of more than 100 tumor suppressor genes in gastric cancer, including E-cadherin, RASSF1A, p16, GSTP1, SOCS1, SFRP1, and PTEN (66).
E-cadherin is one of the most important suppressor genes in gastric cancer tumors. Inactivation of E-cadherin increases proliferation, invasion, and metastasis, which contribute to tumor progression. It is known that silencing of E-cadherin is due to CpG island methylation in undifferentiated-type gastric cancer (66). RASSF1A is part of the Ras association domain family, another group of tumor suppressor gene that plays a critical role in cell cycle regulation, apoptosis, and microtubule stability, by regulating signaling pathway Ras (67). Also, RASSF1A methylation is closely associated with advanced cancer, according to the TNM (68) and poor prognosis in patients with gastric cancer. Thus, it represents an objective diagnosis and a potential therapy for gastric cancer. H. pylori infection contributes to the loss of expression of the transcription factor related Runt-3 (RUNX3) in gastric cancer through its overmethylation (69).
Nakajima et al. demonstrated that methylation levels occurring in the gastric mucosa are increased in patients with H. pylori positive gastric cancer (70). The inflammation caused by infection with H. pylori is essential for DNA methylation of promoters of genes related to inflammation, as IL-1β, NOS2, and TNF (71, 72). Methylation induced by H. pylori infection in the promoter of E-cadherin, the component involved in maintaining stability of the epithelial extracellular matrix, is mediated by IL-1β (73). The eradication of H. pylori infection reduces the methylation of E-cadherin promoter, demonstrating that infection with H. pylori is strongly related to DNA methylation in gastric cancer (74). DNA methylation induced by H. pylori in precancerous lesions might be a new approach for the prevention of gastric cancer.
Zong et al. performed a genetic-level analysis of DNA methylation of gastric cancer cell lines, gastric cancer samples obtained from gastrectomies, normal gastric mucosa, and non-cancerous gastric mucosa where they confirmed that the OSR2, PPFIA3, and VAV3 genes are poorly methylated (5% or less) in non-cancerous and highly methylated (85% or more) cells in cancer cells. These three genes may be useful for estimating the fraction of cancer cells in gastric cancer (75).
Recent comprehensive analyses showed that many cancer-related pathways are further altered by aberrant DNA methylation than by mutations. It is known that in the non-cancerous gastric mucosa, DNA methylation occurs, which produces an epigenetic environment for the development of carcinogenesis. Also, the chronic inflammation induced by H. pylori infection induces aberrant DNA methylation. Expression of the IL-1β, NOS2, and TNF genes are highly associated with the induction of aberrant DNA methylation. The degree of aberrant DNA methylation is correlated with the risk of gastric cancer. It is known that the induction of DNA methylation is the main route by which H. pylori infection induces gastric cancer; this information may be useful to decrease gastric cancer (76).
Gene silencing in gastric cancer may occur mainly by point mutations, loss of heterozygosity, and promoter hypermethylation. A tumor suppressor gene whose expression is frequently deregulated in gastric cancer by hypermethylation of the Trefoil factor 1 gene (TFF1) whose function is to defend and repair of gastrointestinal mucosa. All normal gastric tissues express TFF1, but Im and co-workers have been shown to reduce the expression of this gene in gastric cancer (77). A recent study reported that polypeptide hormone gastrin is secreted by the glands of the pyloric antrum of the stomach, stimulating the secretion of hydrochloric acid and pepsinogen. It is worth noting that gastrin suppresses the TFF1 promoter (78).
The pancreatic duodenal homeobox transcription factor 1 (PDX1) is an important gene in cell differentiation and development of the pancreas, duodenum, and antrum. It is a tumor suppressor gene with an expression that is frequently deregulated in gastric cancer. PDX1 is often expressed in normal gastric glands but is absent in human gastric cancer cell lines and gastric cancer. PDX1 silencing is given by promoter hypermethylation and histone hypoacetylation (79, 80). In gastric cancer in several tumor suppressor, genes and tumor-related genes are silenced by promoter methylation of gene, generating loss of protein expression. Mikata et al. showed that BCL2L10, which belongs to the family of pro-apoptotic Bcl-2, is silenced by hypermethylation of the promoter. This study suggests that silencing BCL2L10 by methylation is a common feature in gastric cancer and may be involved in the early stages of gastric carcinogenesis (81).
In humans, X-ray repair cross-complementing protein 1 (XRCC1) is encoded by the XRCC1 gene involved in DNA repair due to promoter hypermethylation; the expression of this gene is regulated in gastric carcinogenesis (82). Serine protease inhibitor kunitz type 2 (SPINT2/HAI-2) has been implicated in tumor pathogenesis through its influence on cell cycle progression, tumor cell migration and invasion, angiogenesis, and protection from apoptotic stimuli. Promoter methylation and transcriptional silencing of HAI-2/SPINT2 is significantly linked to cell differentiation and metastasis in gastric cancer (83, 84).
Glutamate receptor ionotropic kainate 2 (GRIK2) is a protein that plays a tumor suppressor role in gastric cancer. GRIK2 is often highly methylated in cell lines and gastric cancer primary tumors, but not in normal adjacent tissues, leading to gene silencing (85). To date, many genes with different biological functions are found methylated in gastric cancer (Table 2).
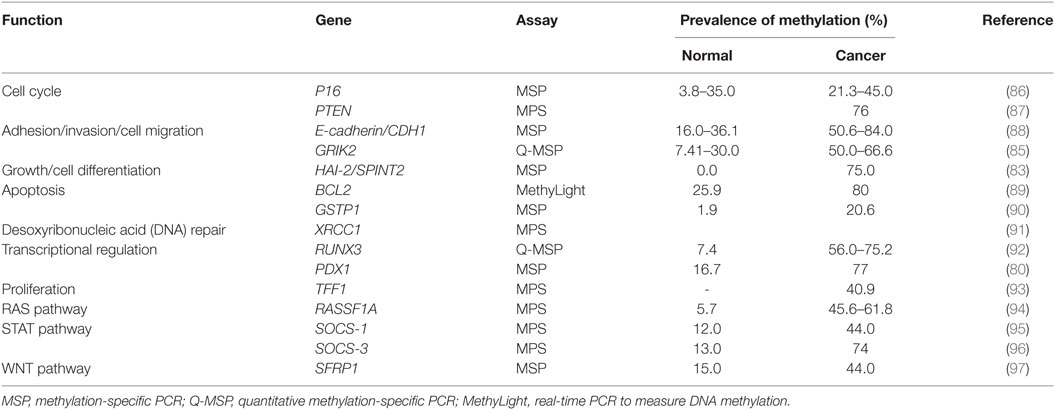
Table 2. Prevalence of gene methylation in gastric cancer.
Cancer Stem Cells (CSCs) and Gastric Carcinogenesis
In the gastrointestinal tract, there are normal SCs, which possess both self-renewal ability and asymmetric cleavage ability to generate progenitor cells that differentiate into epithelial cells. These SCs in the normal gastric mucosal are present in the proliferative region of the neck/isthmus region and experience a complex bipolar migration of the neck/isthmus region upward, thereby becoming differentiated normal epithelial cells (98).
Cancer stem cells are malignant cells with capacities or characteristics of SCs. There have been many studies of CSCs that demonstrate rapid growth and high metastatic potential. Recent evidence suggests the existence of cancerous SCs in solid tumors. This indicates that under conditions, the local microenvironment can promote the development of gastric cancer. H. pylori infection and accompanying chronic inflammatory processes provide the critical initiators that favor cell growth and tissue repair response, leading to gastric carcinogenesis (99).
The discovery of CSCs and their characteristics have favored the understanding of the molecular mechanism of tumorigenesis and tumor development, but above all generating new and effective strategies for the treatment of gastric cancer. Gastric CSCs are found in the base for the onset of gastric cancer. They can be derived from gastric SCs in gastric tissues or mesenchymal SCs from bone marrow. As with other SCs, gastric CSCs express drug resistance genes, which make them refractory to conventional therapies, explaining why cancer therapies are far from curative and why cancer relapses are frequent (100). Specific molecular markers have been studied as CD44 and CD133 for the identification and isolation of gastric CSCs. These CSC markers can help in early diagnosis in the classification of gastric cancer (101).
Maeda et al. reports that the aberrant methylation of the DNA that is induced in the SCs of a gastric gland is permanent because the methylation in the SCs would be conserved and reproduced, which would allow to determine the fraction of cells with methylation. Contrary to the methylation that is generated only in the differentiated cells will disappear when they are replaced with new cells without methylation that are derived from some SC without methylation, of important way this information indicates to us that the methylation induced in the differentiated cells will be a transient component (76).
Inflammation Induced by H. pylori is Important in Gastric Carcinogenesis
Several studies attempt to elucidate the mechanism by which H. pylori is able to recruit inflammatory cells. It has been shown that antigens extracted from H. pylori have in vitro chemotactic activity for monocytes and neutrophils (102). Infected gastric mucosa contains a significant population of macrophages that produce nitric oxide, interleukin 6 (IL-6), IL-1β, TNF-α, and interleukin 12 (IL-12), which help to establish a Th1 response responsible to produce IFN-γ and some interleukin 4 (IL-4) and interleukin 5. Smaller dendritic cells are also present and respond to H. pylori infection with the production of IL-6, IL-8, IL-10, and IL-12. They have a higher expression of cluster of differentiation 80, cluster of differentiation 83, cluster of differentiation 86, and human leukocyte antigens, because of stimulation with bacteria (103).
Urease antigen was detected in vivo in the lamina propria of gastric biopsies of patients infected with H. pylori. This led to examine immunomodulating substances secreted in response to H. pylori infection, as increased levels of IL-8 were present in H. pylori-associated gastritis (104). It is also known that components such as porin (105) and lipopolysaccharides (LPSs) can stimulate the expression of cytokine in polymorphonuclear cells. IL-8 is a chemokine secreted by several cell types, including monocytes, fibroblasts, endothelial cells, and epithelial cells. Its function is to serve as a potent inflammatory mediator for the attraction and activation of polymorphonuclear cells, particularly neutrophils. Since bacteria make contact only with the surface of the gastric epithelium, secretion of IL-8 is produced by the epithelium because of interaction and regulation of inflammatory and immune processes in response to the bacteria.
Helicobacter pylori also activate cyclooxygenase (COX), which is an enzyme that catalyzes proinflammatory key steps in the formation of inflammatory prostaglandins. COX-1 is constitutively expressed, whereas COX-2 is induced by cytokines such as TNF-α, interferon gamma (INF-γ), and IL-1 (106). The expression of COX-2 is increased in the gastric mucosa of individuals infected with H. pylori (107, 108). The expression of COX-2 is further increased in premalignant lesions (chronic atrophic gastritis and intestinal metaplasia) and malignant lesions induced by H. pylori (adenocarcinoma) (109). COX-2 is a key enzyme in the biosynthesis of prostanoids and is the primary target of non-steroidal anti-inflammatory drugs in neoplastic and inflammatory conditions. COX-2 is involved in the processes leading to tumor progression, including angiogenesis, survival, proliferation, invasion, and immunosuppression (91). For this reason, COX-2 is considered a potential therapeutic target in the prevention and treatment of gastric cancer.
The H. pylori-induced inflammatory response leads to the release of mutagenic substances, as metabolites of inducible nitric oxide synthase (iNOS), which promote oncogenesis (108, 110). Nitric oxide generated by iNOS may be converted into reactive nitrogen species that modify DNA and proteins. The superoxide anion radicals generated by neutrophils also induce DNA damage via formation of DNA adducts (111).
It has been shown that H. pylori can employ multiple mechanisms to degrade or destabilize host responses. Another mechanism used to regulate H. pylori decreased immune response is the vacuolating cytotoxin. This cytotoxin interferes with processing and presentation of antigens by antigen-presenting cells. It also inhibits the activation of T cells through interference associated with calcineurin signaling pathway of IL-2 (112).
Most persons infected with H. pylori develop specific antibodies found in serum, gastric aspirates, or extracts. High titers of IgG and IgA antibodies to membrane proteins, flagellin, urease, and the LPS of Helicobacter pylori adhesion A, were reported in patients infected with H. pylori (113). On the other hand, López-Vidal et al. found a progressive accumulation of a specific IgA response against H. pylori in patients with gastric cancer, suggesting that it participates in the damage and in the development of gastric cancer (114). Gastritis induced by H. pylori is produced by bacterial factors that stimulate epithelial cell, macrophage, and dendritic cell activation and induces the recruitment of T CD4+ and CD8+ cells in the gastric mucosa (115). Some studies have shown that the response of T helper cells to infection with H. pylori is polarized, as the T CD4+ cells in the gastric mucosa of infected individuals produce proinflammatory cytokines Th1 such as IL-12, and INF-γ, whereas Th2 regulatory cytokines such as IL-4 are absent in H. pylori infection (116, 117).
Amedei et al. suggested that neutrophil-activating protein of H. pylori (HP-NAP) contributes to Th1 response in the gastric mucosa of patients infected by H. pylori. Regulatory T cells are another important subset of cells producing IL-10 and TGF-β (118). There is an infiltration of CD4+ T cells in the gastric mucosa; therefore, several studies have tried to address the inefficiency of the host response in clearing the infection. They have shown that H. pylori infection may reduce T cell response and T cell anergy (115). CD45RO+ T cells of memory, and CD69+ and CD25+ activated cells are increased in the lamina propria of the antrum of infected subjects. This suggests that the presence of regulatory T cells, CD4+, and CD25+ in peripheral blood of individuals infected by H. pylori could inhibit the response of CD4+ T cells into the bacterium (115, 119). These findings help to explain the inability of the host response to clear the infection due to the activation of regulatory T cells. An increase of T CD4+, CD25high, and FOXP3+ cells was recently found in the gastric mucosa of infected individuals H. pylori. Such cells may reduce mucosal damage mediated by T cells as well as the response of specific T cells, possibly by reducing the activation of CD4+ T cells that produce INF-γ. These responses can be effective to protect against infection by this bacterium (115, 119). The role of CD8+ T cells in the gastric mucosa of H. pylori-infected individuals is less clear than that of CD4+ T cells. The favorable participation of CD8+ T cells in the immune response to H. pylori infection showed that H. pylori colonization is associated with a greater numbers of CD8+ lymphocytes in crypt of epithelium. Their role in the local responses is the production of IFN-γ, with increased expression of major histocompatibility complex class II molecules in adjacent cells (120). Despite these immune mechanisms, H. pylori infection is not eliminated because the bacteria have a number of mechanisms are evaded or regulate by host responses. Understanding these multiple mechanisms is a necessary step toward the development of immune strategies to protect from the initial infection and to remove the infections that are already established.
Inflammation is important in the development and progression of gastric cancer; suppressor of cytokine signaling (SOCS) 1 and 3 negatively regulate the signaling of proinflammatory cytokine. We analyzed the expression levels of SOCS-1 and SOCS-3 in gastric tumors and adjacent normal mucosa tissue; the results showed to be lower in the normal mucosa and in the tumor tissues. Also, the low expression of SOCS-1 and SOCS-3 is an indicator of a poor prognosis of gastric cancer (121), which might be of prognostic significance (Figure 2).
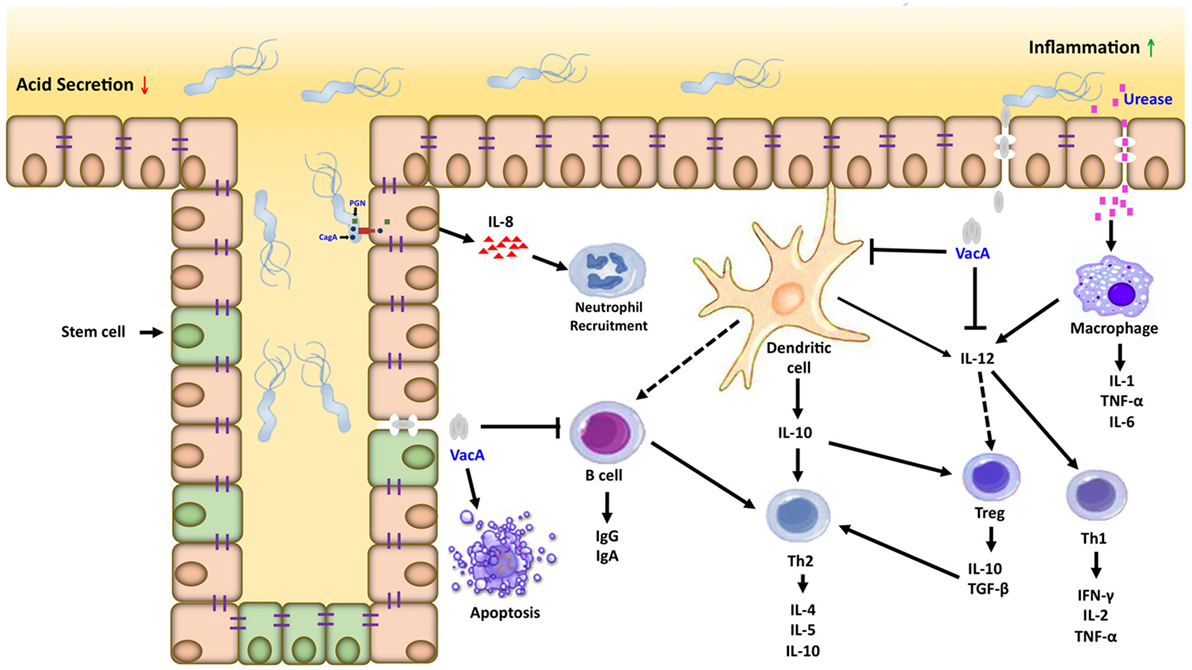
Figure 2. Inflammatory microenvironment induced by Helicobacter pylori (H. pylori) in gastric carcinogenesis. Dendritic cells and macrophages can serve as a bridge between the innate and adaptive immune response directed against H. pylori within the gastric mucosa. Interactions between H. pylori virulence factors and gastric epithelial cells can activate macrophages and recruit neutrophils, which amplifies the T cell response to this pathogen. Dendritic cells in contact with H. pylori antigens activate T cells in different ways, inducing the Th1, Th2/T regulatory response generating proinflammatory and anti-inflammatory cytokines, respectively.
Conclusion
Gastric cancer is a complex disease that emerges from the interaction of multiple processes and factors such as lifestyle, eating habits, and infections, along with genetic variants and molecular alterations acquired during the patient’s life. There are many research works that have been carried out to find molecular markers for gastric cancer; however, there are very few studies that include information about gene expression analysis of the stages of progression of gastric cancer. The real mechanisms are poorly known, and more work is needed to understand the causes of gastric cancer progression in order to obtain biomarkers for early diagnosis, prognosis, and effective treatment of the disease.
Author Contributions
CIRO, YLV, LJRAH and GCR have substantially contributed to the development of the manuscript outline and literature search strategy, have critically reviewed manuscript drafts for intellectual content, have approved the version to be published, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement
The authors have declared that no competing interests exist, and no relationships or commercial interests with Mexican or foreign companies are related to this review.
The reviewer, MS, and handling editor declared their shared affiliation, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
Funding
This review was supported by the grant CONACyT SALUD2009-C01-112588 and C0009-2014-02-239182. Rivas-Ortiz Claudia Ivette is a doctoral student from Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) and received fellowship, number 275851, from Consejo Nacional de Ciencia y Tecnología (CONACyT).
Abbreviations
DNA, desoxyribonucleic acid; H. pylori, Helicobacter pylori; IARC, International Agency for Research on Cancer; SNPs, single-nucleotide polymorphisms; TNF-α, tumor necrosis factor alpha; IL-1β, interleukin 1 beta; IL-1RN, interleukin antagonist receptor; IL-1RN2, interleukin antagonist receptor 2; IL-4, interleukin 4; IL-5, interleukin 5; IL-6, interleukin 6; IL-8, interleukin 8; IL-10, interleukin 10; IL-12, interleukin 12; IL-17F, interleukin 17F; Th, T helper cell; Th1, T helper cell type 1; Th2, T helper cell type 2; IgG, immunoglobulin G; IgA, immunoglobulin A; iNOS, nitric oxide synthases; MUC, mucin; MUC1, mucin 1; MUC1-C, mucin 1 subunit C-terminal; MUC2, mucin 2; MUC6, mucin 6; GWAS, genome-wide association study; β-catenin, beta-catenin; E-cadherin, cadherin epithelial tissue; cagA, cytotoxin-associated gene A; CagA, cytotoxin-associated protein; cag-PAI, cag pathogenicity island; T4SS, type IV secretion system; VacA, vacuolating cytotoxin; LPS, lipopolysaccharide; HpaA, Helicobacter pylori adhesion A; HP-NAP, Helicobacter pylori neutrophil-activating protein; CDX1, homeobox gene such as flow type 1; CpG, cytosine-phosphate-guanine; STAT3, signal transducer and activator of transcription 3; TGF-β, transforming growth factor beta; SMAD4 or DPC4, SMAD family member no 4 or mothers against decapentaplegic homolog 4, or deleted in pancreatic cancer-4; SERPINE1, serpin peptidase inhibitor clade E, member 1; HIF-1, hypoxia-inducible factor 1; HIF-1α, hypoxia-inducible factor 1 subunit alpha; NF-κB1, nuclear factor kappa-light-chain-enhancer of active B cells class I or p105 subunit; GSK3β, glycogen synthase kinase 3 beta; phospo-GSK3β (ser9), phospho-glycogen synthase kinase 3 beta-antibody (Ser9); Ki-67, marker of proliferation; CDH17, cadherin 17; CD4, cluster of differentiation 4; CD8, cluster of differentiation 8; CD25, cluster of differentiation 25; alpha chain of IL-2 receptor; CD44, cluster of differentiation 44, receptor for hyaluronic acid (HA); CD45RO, protein tyrosine phosphatase receptor type C isoform RO located on memory T cells; CD69, cluster of differentiation 69; CD80, cluster of differentiation 80; CD83, cluster of differentiation 83; CD86, cluster of differentiation 86; CD133, prominin-1; CD168, cluster of differentiation 168; hyaluronan-mediated motility receptor; FOXP3, forkhead box P3 or scurfin; MHC class II, major histocompatibility complex class II molecules; HLA-DR, human leukocyte antigens; GSH, glutathione; MMPs, matrix metalloproteinases; MMP-7, matrix metalloproteinase 7; MMP-9, matrix metalloproteinase 9; MMP-12, matrix metalloproteinase 12; MMP-21, matrix metalloproteinase 21; EMT, epithelial–mesenchymal transition; Snail, zinc finger protein; Src, proto-oncogene tyrosine-protein kinase; SFK, Src family kinase; SHP-2, tyrosine phosphatase 2; ERK, extracellular signal-regulated kinase; Ras, Ras superfamily; EGFR, epidermal growth factor receptor; PAR1, protease-activated receptors; RUNX3, runt-related transcription factor 3; T-bet, T-box transcription factor; IFN-γ, interferon gamma; CH3, methyl group; RASSF1A, Ras association domain-containing protein 1; p16, cyclin-dependent kinase inhibitor 2A, multiple tumor suppressor 1; GSTP1, glutathione S-transferase P; SOCS1, suppressor of cytokine signaling 1; SOCS3, suppressor of cytokine signaling 3; SFRP1, secreted frizzled-related protein 1; PTEN, phosphatase and tensin homolog; TNM, tumor node metastasis; NOS2, nitric oxide synthase 2 (inducible); TFF1, trefoil factor 1 gene; PDX1, pancreatic duodenal homeobox transcription factor 1; BCL2L10, Bcl-2-like protein 10; Bcl-2, B-cell lymphoma 2; XRCC1, X-ray repair cross-complementing protein 1; HAI-2 or SPINT2, serine peptidase inhibitor, kunitz type 2; GRIK2, glutamate receptor, ionotropic kainate 2; COX, cyclooxygenase; COX-1, cyclooxygenase-1 or prostaglandin-endoperoxide synthase 1; COX-2, prostaglandin-endoperoxide synthase 2.
References
1. Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. Lyon, France: International Agency for Research on Cancer (2013). Available from: http://globocan.iarc.fr
2. INEGI. Principales causas de mortalidad por residencia habitual, grupos de edad y sexo del fallecido: Consulta de resultados – Tabulados básicos. Mexico: Instituto Nacional de Estadística y Geografía (INEGI) (2016). Available from: http://www.inegi.org.mx/est/contenidos/proyectos/registros/vitales/mortalidad/tabulados/PC.asp?t=14&c=11817
3. IARC. Biological agents: a review of human carcinogens. IARC Monogr Eval Carcinog Risks Hum (2012) 100:1–441.
4. Dicken BJ, Bigam DL, Cass C, Mackey JR, Joy AA, Hamilton SM. Gastric adenocarcinoma: review and considerations for future directions. Ann Surg (2005) 241:27–39. doi: 10.1097/01.sla.0000149300.28588.23
5. Leung WK, Wu MS, Kakugawa Y, Kim JJ, Yeoh KG, Goh KL, et al. Screening for gastric cancer in Asia: current evidence and practice. Lancet Oncol (2008) 9:279–87. doi:10.1016/S1470-2045(08)70072-X
6. Llorens P. Gastric cancer mass survey in Chile. Semin Surg Oncol (1991) 7:339–43. doi:10.1002/ssu.2980070604
7. Pisani P, Oliver WE, Parkin DM, Alvarez N, Vivas J. Case-control study of gastric cancer screening in Venezuela. Br J Cancer (1994) 69:1102–5. doi:10.1038/bjc.1994.216
8. Zhuang W, Wu XT, Zhou Y, Liu L, Liu GJ, Wu TX, et al. Interleukin10 -592 promoter polymorphism associated with gastric cancer among Asians: a meta-analysis of epidemiologic studies. Dig Dis Sci (2010) 55:1525–32. doi:10.1007/s10620-009-0922-1
9. Lee SG, Kim B, Yook JH, Oh ST, Lee I, Song K. TNF/LTA polymorphisms and risk for gastric cancer/duodenal ulcer in the Korean population. Cytokine (2004) 28:75–82. doi:10.1016/j.cyto.2004.06.009
10. Kunstmann E, Epplen C, Elitok E, Harder M, Suerbaum S, Peitz U, et al. Helicobacter pylori infection and polymorphisms in the tumor necrosis factor region. Electrophoresis (1999) 20:1756–61. doi:10.1002/(SICI)1522-2683(19990101)20:8<1756::AID-ELPS1756>3.3.CO;2-2
11. Hurme M, Lahdenpohja N, Santtila S. Gene polymorphisms of interleukins 1 and 10 in infectious and autoimmune diseases. Ann Med (1998) 30:469–73. doi:10.3109/07853899809002488
12. Guo XF, Wang J, Yu SJ, Song J, Ji MY, Cao Z, et al. TNF-alpha-308 polymorphism and risk of digestive system cancers: a meta-analysis. World J Gastroenterol (2013) 19:9461–71. doi:10.3748/wjg.v19.i48.9461
13. Wu X, Zeng Z, Chen B, Yu J, Xue L, Hao Y, et al. Association between polymorphisms in interleukin-17A and interleukin-17F genes and risks of gastric cancer. Int J Cancer (2010) 127:86–92. doi:10.1002/ijc.25027
14. Arisawa T, Tahara T, Shibata T, Nagasaka M, Nakamura M, Kamiya Y, et al. Genetic polymorphisms of molecules associated with inflammation and immune response in Japanese subjects with functional dyspepsia. Int J Mol Med (2007) 20:717–23. doi:10.3164/jcbn.10-40
15. Persson C, Canedo P, Machado JC, El-Omar EM, Forman D. Polymorphisms in inflammatory response genes and their association with gastric cancer: a HuGE systematic review and meta-analyses. Am J Epidemiol (2011) 173:259–70. doi:10.1093/aje/kwq370
16. Guang W, Twaddell WS, Lillehoj EP. Molecular interactions between MUC1 epithelial mucin, beta-catenin, and CagA proteins. Front Immunol (2012) 3:105. doi:10.3389/fimmu.2012.00105
17. Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer (2009) 9:874–85. doi:10.1038/nrc2761
18. Saeki N, Sakamoto H, Yoshida T. Mucin 1 gene (MUC1) and gastric-cancer susceptibility. Int J Mol Sci (2014) 15:7958–73. doi:10.3390/ijms15057958
19. Behrens ME, Grandgenett PM, Bailey JM, Singh PK, Yi CH, Yu F, et al. The reactive tumor microenvironment: MUC1 signaling directly reprograms transcription of CTGF. Oncogene (2010) 29:5667–77. doi:10.1038/onc.2010.327
20. Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene (2007) 26:4617–26. doi:10.1038/sj.onc.1210251
21. Guang W, Czinn SJ, Blanchard TG, Kim KC, Lillehoj EP. Genetic regulation of MUC1 expression by Helicobacter pylori in gastric cancer cells. Biochem Biophys Res Commun (2014) 445:145–50. doi:10.1016/j.bbrc.2014.01.142
22. Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell (1998) 95:737–40. doi:10.1016/S0092-8674(00)81696-7
23. Wang LH, Kim SH, Lee JH, Choi YL, Kim YC, Park TS, et al. Inactivation of SMAD4 tumor suppressor gene during gastric carcinoma progression. Clin Cancer Res (2007) 13:102–10. doi:10.1158/1078-0432.CCR-06-1467
24. Wu DM, Zhu HX, Zhao QH, Zhang ZZ, Wang SZ, Wang ML, et al. Genetic variations in the SMAD4 gene and gastric cancer susceptibility. World J Gastroenterol (2010) 16:5635–41. doi:10.3748/wjg.v16.i44.5635
25. Ju H, Lim B, Kim M, Noh SM, Kim WH, Ihm C, et al. SERPINE1 intron polymorphisms affecting gene expression are associated with diffuse-type gastric cancer susceptibility. Cancer (2010) 116:4248–55. doi:10.1002/cncr.25213
26. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab (2008) 7:11–20. doi:10.1016/j.cmet.2007.10.002
27. Zhou W, Liotta LA, Petricoin EF. Cancer metabolism and mass spectrometry-based proteomics. Cancer Lett (2015) 356:176–83. doi:10.1016/j.canlet.2013.11.003
28. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
29. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi:10.1126/science.1160809
30. Liu X, Hajnoczky G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ (2011) 18:1561–72. doi:10.1038/cdd.2011.13
31. Cui J, Mao X, Olman V, Hastings PJ, Xu Y. Hypoxia and miscoupling between reduced energy efficiency and signaling to cell proliferation drive cancer to grow increasingly faster. J Mol Cell Biol (2012) 4:174–6. doi:10.1093/jmcb/mjs017
32. Solaini G, Baracca A, Lenaz G, Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys Acta (2010) 1797:1171–7. doi:10.1016/j.bbabio.2010.02.011
33. Bruick RK. Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia-inducible transcription factor. Genes Dev (2003) 17:2614–23. doi:10.1101/gad.1145503
34. Fan L, Li J, Yu Z, Dang X, Wang K. The hypoxia-inducible factor pathway, prolyl hydroxylase domain protein inhibitors, and their roles in bone repair and regeneration. Biomed Res Int (2014) 2014:239356. doi:10.1155/2014/239356
35. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature (2008) 453:807–11. doi:10.1038/nature06905
36. Zheng HC, Xu XY, Xia P, Yu M, Takahashi H, Takano Y. Involvement of inactive GSK3beta overexpression in tumorigenesis and progression of gastric carcinomas. Hum Pathol (2010) 41:1255–64. doi:10.1016/j.humpath.2010.02.003
37. Mishra P, Senthivinayagam S, Rana A, Rana B. Glycogen synthase kinase-3beta regulates snail and beta-catenin during gastrin-induced migration of gastric cancer cells. J Mol Signal (2010) 5:9. doi:10.1186/1750-2187-5-9
38. Zhao P, Li Y, Lu Y. Aberrant expression of CD133 protein correlates with Ki-67 expression and is a prognostic marker in gastric adenocarcinoma. BMC Cancer (2010) 10:218. doi:10.1186/1471-2407-10-218
39. Lee HJ, Nam KT, Park HS, Kim MA, Lafleur BJ, Aburatani H, et al. Gene expression profiling of metaplastic lineages identifies CDH17 as a prognostic marker in early stage gastric cancer. Gastroenterology (2010) 139:213–25 e3. doi:10.1053/j.gastro.2010.04.008
40. Ishigami S, Ueno S, Nishizono Y, Matsumoto M, Kurahara H, Arigami T, et al. Prognostic impact of CD168 expression in gastric cancer. BMC Cancer (2011) 11:106. doi:10.1186/1471-2407-11-106
41. Xie JW, Chen PC, Zheng CH, Li P, Wang JB, Lin JX, et al. Evaluation of the prognostic value and functional roles of CD44v6 in gastric cancer. J Cancer Res Clin Oncol (2015) 141:1809–17. doi:10.1007/s00432-015-1964-8
42. Wang W, Dong LP, Zhang N, Zhao CH. Role of cancer stem cell marker CD44 in gastric cancer: a meta-analysis. Int J Clin Exp Med (2014) 7:5059–66.
43. Long ZW, Wang JL, Wang YN. Matrix metalloproteinase-7 mRNA and protein expression in gastric carcinoma: a meta-analysis. Tumour Biol (2014) 35(11):11415–26. doi:10.1007/s13277-014-2441-8
44. Chang WJ, Du Y, Zhao X, Ma LY, Cao GW. Inflammation-related factors predicting prognosis of gastric cancer. World J Gastroenterol (2014) 20:4586–96. doi:10.3748/wjg.v20.i16.4586
45. Shin NR, Jeong EH, Choi CI, Moon HJ, Kwon CH, Chu IS, et al. Overexpression of snail is associated with lymph node metastasis and poor prognosis in patients with gastric cancer. BMC Cancer (2012) 12:521. doi:10.1186/1471-2407-12-521
46. Bagnoli F, Buti L, Tompkins L, Covacci A, Amieva MR. Helicobacter pylori CagA induces a transition from polarized to invasive phenotypes in MDCK cells. Proc Natl Acad Sci U S A (2005) 102:16339–44. doi:10.1073/pnas.0502598102
47. Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res (1995) 55:2111–5.
48. Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer (2004) 4:688–94. doi:10.1038/nrc1433
49. Hatakeyama M. Helicobacter pylori and gastric carcinogenesis. J Gastroenterol (2009) 44:239–48. doi:10.1007/s00535-009-0014-1
50. Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, et al. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science (2002) 295:683–6. doi:10.1126/science.1067147
51. Tsutsumi R, Takahashi A, Azuma T, Higashi H, Hatakeyama M. Focal adhesion kinase is a substrate and downstream effector of SHP-2 complexed with Helicobacter pylori CagA. Mol Cell Biol (2006) 26:261–76. doi:10.1128/MCB.26.1.261-276.2006
52. Maroun CR, Naujokas MA, Holgado-Madruga M, Wong AJ, Park M. The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol Cell Biol (2000) 20:8513–25. doi:10.1128/MCB.20.22.8513-8525.2000
53. Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, et al. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem (2004) 279:17205–16. doi:10.1074/jbc.M309964200
54. Suerbaum S, Josenhans C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat Rev Microbiol (2007) 5:441–52. doi:10.1038/nrmicro1658
55. Nguyen LT, Uchida T, Murakami K, Fujioka T, Moriyama M. Helicobacter pylori virulence and the diversity of gastric cancer in Asia. J Med Microbiol (2008) 57:1445–53. doi:10.1099/jmm.0.2008/003160-0
56. Churin Y, Al-Ghoul L, Kepp O, Meyer TF, Birchmeier W, Naumann M. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. J Cell Biol (2003) 161:249–55. doi:10.1083/jcb.200208039
57. Mimuro H, Suzuki T, Tanaka J, Asahi M, Haas R, Sasakawa C. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol Cell (2002) 10:745–55. doi:10.1016/S1097-2765(02)00681-0
58. Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol (2007) 19:150–8. doi:10.1016/j.ceb.2007.02.007
59. Bauer B, Bartfeld S, Meyer TF. H. pylori selectively blocks EGFR endocytosis via the non-receptor kinase c-Abl and CagA. Cell Microbiol (2009) 11:156–69. doi:10.1111/j.1462-5822.2008.01246.x
60. Bauer B, Meyer TF. The human gastric pathogen Helicobacter pylori and its association with gastric cancer and ulcer disease. Ulcers (2011) 340157:23. doi:10.1155/2011/340157
61. Watanabe T, Sato K, Kaibuchi K. Cadherin-mediated intercellular adhesion and signaling cascades involving small GTPases. Cold Spring Harb Perspect Biol (2009) 1:a003020. doi:10.1101/cshperspect.a003020
62. Schneider S, Weydig C, Wessler S. Targeting focal adhesions: Helicobacter pylori-host communication in cell migration. Cell Commun Signal (2008) 6:2. doi:10.1186/1478-811X-6-2
63. Tsang YH, Lamb A, Romero-Gallo J, Huang B, Ito K, Peek RM Jr, et al. Helicobacter pylori CagA targets gastric tumor suppressor RUNX3 for proteasome-mediated degradation. Oncogene (2010) 29:5643–50. doi:10.1038/onc.2010.304
64. Li Y, Ji X, Su Z, Tong J, Xia S, Chen X, et al. Downregulation of Runx3 is closely related to the decreased Th1-associated factors in patients with gastric carcinoma. Tumour Biol (2014) 35(12):12235–44. doi:10.1007/s13277-014-2532-6
65. Matsusaka K, Funata S, Fukayama M, Kaneda A. DNA methylation in gastric cancer, related to Helicobacter pylori and Epstein-Barr virus. World J Gastroenterol (2014) 20:3916–26. doi:10.3748/wjg.v20.i14.3916
66. Tamura G, Yin J, Wang S, Fleisher AS, Zou T, Abraham JM, et al. E-cadherin gene promoter hypermethylation in primary human gastric carcinomas. J Natl Cancer Inst (2000) 92:569–73. doi:10.1093/jnci/92.7.569
67. Byun DS, Lee MG, Chae KS, Ryu BG, Chi SG. Frequent epigenetic inactivation of RASSF1A by aberrant promoter hypermethylation in human gastric adenocarcinoma. Cancer Res (2001) 61:7034–8.
68. Yao D, Shi J, Shi B, Wang N, Liu W, Zhang G, et al. Quantitative assessment of gene methylation and their impact on clinical outcome in gastric cancer. Clin Chim Acta (2012) 413:787–94. doi:10.1016/j.cca.2012.01.013
69. Oh JH, Rhyu MG, Jung SH, Choi SW, Kim SI, Hong SJ. Slow overmethylation of housekeeping genes in the body mucosa is associated with the risk for gastric cancer. Cancer Prev Res (Phila) (2014) 7:585–95. doi:10.1158/1940-6207.CAPR-13-0320
70. Nakajima T, Maekita T, Oda I, Gotoda T, Yamamoto S, Umemura S, et al. Higher methylation levels in gastric mucosae significantly correlate with higher risk of gastric cancers. Cancer Epidemiol Biomarkers Prev (2006) 15:2317–21. doi:10.1158/1055-9965.EPI-06-0436
71. Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res (2010) 70:1430–40. doi:10.1158/0008-5472.CAN-09-2755
72. Hur K, Niwa T, Toyoda T, Tsukamoto T, Tatematsu M, Yang HK, et al. Insufficient role of cell proliferation in aberrant DNA methylation induction and involvement of specific types of inflammation. Carcinogenesis (2011) 32:35–41. doi:10.1093/carcin/bgq219
73. Qian X, Huang C, Cho CH, Hui WM, Rashid A, Chan AO. E-cadherin promoter hypermethylation induced by interleukin-1beta treatment or H. pylori infection in human gastric cancer cell lines. Cancer Lett (2008) 263:107–13. doi:10.1016/j.canlet.2007.12.023
74. Chan AO, Peng JZ, Lam SK, Lai KC, Yuen MF, Cheung HK, et al. Eradication of Helicobacter pylori infection reverses E-cadherin promoter hypermethylation. Gut (2006) 55:463–8. doi:10.1136/gut.2005.077776
75. Zong L, Hattori N, Yoda Y, Yamashita S, Takeshima H, Takahashi T, et al. Establishment of a DNA methylation marker to evaluate cancer cell fraction in gastric cancer. Gastric Cancer (2016) 19:361–9. doi:10.1007/s10120-015-0475-2
76. Maeda M, Moro H, Ushijima T. Mechanisms for the induction of gastric cancer by Helicobacter pylori infection: aberrant DNA methylation pathway. Gastric Cancer (2016) 20:8–15. doi:10.1007/s10120-016-0650-0
77. Im S, Yoo C, Jung JH, Choi HJ, Yoo J, Kang CS. Reduced expression of TFF1 and increased expression of TFF3 in gastric cancer: correlation with clinicopathological parameters and prognosis. Int J Med Sci (2013) 10:133–40. doi:10.7150/ijms.5500
78. Tomita H, Takaishi S, Menheniott TR, Yang X, Shibata W, Jin G, et al. Inhibition of gastric carcinogenesis by the hormone gastrin is mediated by suppression of TFF1 epigenetic silencing. Gastroenterology (2011) 140:879–91. doi:10.1053/j.gastro.2010.11.037
79. Ma J, Chen M, Wang J, Xia HH, Zhu S, Liang Y, et al. Pancreatic duodenal homeobox-1 (PDX1) functions as a tumor suppressor in gastric cancer. Carcinogenesis (2008) 29:1327–33. doi:10.1093/carcin/bgn112
80. Ma J, Wang JD, Zhang WJ, Zou B, Chen WJ, Lam CS, et al. Promoter hypermethylation and histone hypoacetylation contribute to pancreatic-duodenal homeobox 1 silencing in gastric cancer. Carcinogenesis (2010) 31:1552–60. doi:10.1093/carcin/bgq140
81. Mikata R, Fukai K, Imazeki F, Arai M, Fujiwara K, Yonemitsu Y, et al. BCL2L10 is frequently silenced by promoter hypermethylation in gastric cancer. Oncol Rep (2010) 23:1701–8. doi:10.3892/or_00000814
82. Wang P, Tang JT, Peng YS, Chen XY, Zhang YJ, Fang JY. XRCC1 downregulated through promoter hypermethylation is involved in human gastric carcinogenesis. J Dig Dis (2010) 11:343–51. doi:10.1111/j.1751-2980.2010.00459.x
83. Dong W, Chen X, Xie J, Sun P, Wu Y. Epigenetic inactivation and tumor suppressor activity of HAI-2/SPINT2 in gastric cancer. Int J Cancer (2010) 127:1526–34. doi:10.1002/ijc.25161
84. Kongkham PN, Northcott PA, Ra YS, Nakahara Y, Mainprize TG, Croul SE, et al. An epigenetic genome-wide screen identifies SPINT2 as a novel tumor suppressor gene in pediatric medulloblastoma. Cancer Res (2008) 68:9945–53. doi:10.1158/0008-5472.CAN-08-2169
85. Wu CS, Lu YJ, Li HP, Hsueh C, Lu CY, Leu YW, et al. Glutamate receptor, ionotropic, kainate 2 silencing by DNA hypermethylation possesses tumor suppressor function in gastric cancer. Int J Cancer (2010) 126:2542–52. doi:10.1002/ijc.24958
86. Sun Y, Deng D, You WC, Bai H, Zhang L, Zhou J, et al. Methylation of p16 CpG islands associated with malignant transformation of gastric dysplasia in a population-based study. Clin Cancer Res (2004) 10:5087–93. doi:10.1158/1078-0432.CCR-03-0622
87. Kang YH, Lee HS, Kim WH. Promoter methylation and silencing of PTEN in gastric carcinoma. Lab Invest (2002) 82:285–91. doi:10.1038/labinvest.3780422
88. Oue N, Mitani Y, Motoshita J, Matsumura S, Yoshida K, Kuniyasu H, et al. Accumulation of DNA methylation is associated with tumor stage in gastric cancer. Cancer (2006) 106:1250–9. doi:10.1002/cncr.21754
89. Kang GH, Lee S, Cho NY, Gandamihardja T, Long TI, Weisenberger DJ, et al. DNA methylation profiles of gastric carcinoma characterized by quantitative DNA methylation analysis. Lab Invest (2008) 88:161–70. doi:10.1038/labinvest.3700707
90. Ksiaa F, Ziadi S, Amara K, Korbi S, Trimeche M. Biological significance of promoter hypermethylation of tumor-related genes in patients with gastric carcinoma. Clin Chim Acta (2009) 404:128–33. doi:10.1016/j.cca.2009.03.044
92. Li WQ, Pan KF, Zhang Y, Dong CX, Zhang L, Ma JL, et al. RUNX3 methylation and expression associated with advanced precancerous gastric lesions in a Chinese population. Carcinogenesis (2011) 32:406–10. doi:10.1093/carcin/bgq259
93. Feng G, Zhang Y, Yuan H, Bai R, Zheng J, Zhang J, et al. DNA methylation of trefoil factor 1 (TFF1) is associated with the tumorigenesis of gastric carcinoma. Mol Med Rep (2014) 9:109–17. doi:10.3892/mmr.2013.1772
94. Agathanggelou A, Cooper WN, Latif F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res (2005) 65:3497–508. doi:10.1158/0008-5472.CAN-04-4088
95. Oshimo Y, Kuraoka K, Nakayama H, Kitadai Y, Yoshida K, Chayama K, et al. Epigenetic inactivation of SOCS-1 by CpG island hypermethylation in human gastric carcinoma. Int J Cancer (2004) 112:1003–9. doi:10.1002/ijc.20521
96. Tischoff I, Hengge UR, Vieth M, Ell C, Stolte M, Weber A, et al. Methylation of SOCS-3 and SOCS-1 in the carcinogenesis of Barrett’s adenocarcinoma. Gut (2007) 56:1047–53. doi:10.1136/gut.2006.111633
97. Zhao CH, Bu XM, Zhang N. Hypermethylation and aberrant expression of Wnt antagonist secreted frizzled-related protein 1 in gastric cancer. World J Gastroenterol (2007) 13:2214–7. doi:10.3748/wjg.v13.i15.2214
98. Zhao Y, Feng F, Zhou YN. Stem cells in gastric cancer. World J Gastroenterol (2015) 21:112–23. doi:10.3748/wjg.v21.i1.112
99. Saikawa Y, Fukuda K, Takahashi T, Nakamura R, Takeuchi H, Kitagawa Y. Gastric carcinogenesis and the cancer stem cell hypothesis. Gastric Cancer (2010) 13:11–24. doi:10.1007/s10120-009-0537-4
100. Rocco A, Compare D, Nardone G. Cancer stem cell hypothesis and gastric carcinogenesis: experimental evidence and unsolved questions. World J Gastrointest Oncol (2012) 4:54–9. doi:10.4251/wjgo.v4.i3.54
101. Li K, Dan Z, Nie YQ. Gastric cancer stem cells in gastric carcinogenesis, progression, prevention and treatment. World J Gastroenterol (2014) 20:5420–6. doi:10.3748/wjg.v20.i18.5420
102. Mai UE, Perez-Perez GI, Allen JB, Wahl SM, Blaser MJ, Smith PD. Surface proteins from Helicobacter pylori exhibit chemotactic activity for human leukocytes and are present in gastric mucosa. J Exp Med (1992) 175:517–25. doi:10.1084/jem.175.2.517
103. Kranzer K, Eckhardt A, Aigner M, Knoll G, Deml L, Speth C, et al. Induction of maturation and cytokine release of human dendritic cells by Helicobacter pylori. Infect Immun (2004) 72:4416–23. doi:10.1128/IAI.72.8.4416-4423.2004
104. Crabtree JE, Wyatt JI, Trejdosiewicz LK, Peichl P, Nichols PH, Ramsay N, et al. Interleukin-8 expression in Helicobacter pylori infected, normal, and neoplastic gastroduodenal mucosa. J Clin Pathol (1994) 47:61–6. doi:10.1136/jcp.47.1.61
105. Tufano MA, Rossano F, Catalanotti P, Liguori G, Capasso C, Ceccarelli MT, et al. Immunobiological activities of Helicobacter pylori porins. Infect Immun (1994) 62:1392–9.
106. Williams CS, Smalley W, DuBois RN. Aspirin use and potential mechanisms for colorectal cancer prevention. J Clin Invest (1997) 100:1325–9. doi:10.1172/JCI119651
107. Sawaoka H, Kawano S, Tsuji S, Tsuji M, Sun W, Gunawan ES, et al. Helicobacter pylori infection induces cyclooxygenase-2 expression in human gastric mucosa. Prostaglandins Leukot Essent Fatty Acids (1998) 59:313–6. doi:10.1016/S0952-3278(98)90079-5
108. Fu S, Ramanujam KS, Wong A, Fantry GT, Drachenberg CB, James SP, et al. Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology (1999) 116:1319–29. doi:10.1016/S0016-5085(99)70496-8
109. Sung JJ, Leung WK, Go MY, To KF, Cheng AS, Ng EK, et al. Cyclooxygenase-2 expression in Helicobacter pylori-associated premalignant and malignant gastric lesions. Am J Pathol (2000) 157:729–35. doi:10.1016/S0002-9440(10)64586-5
110. Mannick EE, Bravo LE, Zarama G, Realpe JL, Zhang XJ, Ruiz B, et al. Inducible nitric oxide synthase, nitrotyrosine, and apoptosis in Helicobacter pylori gastritis: effect of antibiotics and antioxidants. Cancer Res (1996) 56:3238–43.
111. Baik SC, Youn HS, Chung MH, Lee WK, Cho MJ, Ko GH, et al. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res (1996) 56:1279–82.
112. Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science (2003) 301:1099–102. doi:10.1126/science.1086871
113. Mattsson A, Quiding-Jarbrink M, Lonroth H, Hamlet A, Ahlstedt I, Svennerholm A. Antibody-secreting cells in the stomachs of symptomatic and asymptomatic Helicobacter pylori-infected subjects. Infect Immun (1998) 66:2705–12.
114. López-Vidal Y, Ponce-de-León S, Esquivel-Solís H, Amieva-Fernández R, Barreto-Zuñiga R, Torre-Delgadillo A, et al. Gastric cancer progression associated with local humoral immune responses. BMC Cancer (2015) 15:924. doi:10.1186/s12885-015-1858-9
115. Lundgren A, Suri-Payer E, Enarsson K, Svennerholm AM, Lundin BS. Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect Immun (2003) 71:1755–62. doi:10.1128/IAI.71.4.1755-1762.2003
116. Bamford KB, Fan X, Crowe SE, Leary JF, Gourley WK, Luthra GK, et al. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology (1998) 114:482–92. doi:10.1016/S0016-5085(98)70531-1
117. Haeberle HA, Kubin M, Bamford KB, Garofalo R, Graham DY, El-Zaatari F, et al. Differential stimulation of interleukin-12 (IL-12) and IL-10 by live and killed Helicobacter pylori in vitro and association of IL-12 production with gamma interferon-producing T cells in the human gastric mucosa. Infect Immun (1997) 65:4229–35.
118. Amedei A, Cappon A, Codolo G, Cabrelle A, Polenghi A, Benagiano M, et al. The neutrophil-activating protein of Helicobacter pylori promotes Th1 immune responses. J Clin Invest (2006) 116:1092–101. doi:10.1172/JCI27177
119. Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol (2001) 167:1245–53. doi:10.4049/jimmunol.167.3.1245
120. Azem J, Svennerholm AM, Lundin BS. B cells pulsed with Helicobacter pylori antigen efficiently activate memory CD8+ T cells from H. pylori-infected individuals. Clin Immunol (2006) 118:284–91. doi:10.1016/j.clim.2005.09.011
Keywords: gastric cancer, polymorphisms, desoxyribonucleic acid methylation, inflammation, deregulation and overexpression/gene inactivation
Citation: Rivas-Ortiz CI, Lopez-Vidal Y, Arredondo-Hernandez LJR and Castillo-Rojas G (2017) Genetic Alterations in Gastric Cancer Associated with Helicobacter pylori Infection. Front. Med. 4:47. doi: 10.3389/fmed.2017.00047
Received: 31 January 2017; Accepted: 07 April 2017;
Published: 02 May 2017
Edited by:
Akira Andoh, Shiga University of Medical Science, JapanReviewed by:
Mitsushige Sugimoto, Shiga University of Medical Science, JapanYuji Naito, Kyoto Prefectural University of Medicine, Japan
Copyright: © 2017 Rivas-Ortiz, Lopez-Vidal, Arredondo-Hernandez and Castillo-Rojas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gonzalo Castillo-Rojas, Z2Nhc3RpbGxvNTVAaG90bWFpbC5jb20=