 Rita Cascão
Rita Cascão João E. Fonseca
João E. Fonseca Luis F. Moita
Luis F. Moita- 1Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Lisbon, Portugal
- 2Rheumatology Department, Centro Hospitalar de Lisboa Norte, EPE, Hospital de Santa Maria, Lisbon Academic Medical Centre, Lisbon, Portugal
- 3Instituto Gulbenkian de Ciência, Oeiras, Portugal
The identification of new bioactive compounds derived from medicinal plants with significant therapeutic properties has attracted considerable interest in recent years. Such is the case of the Tripterygium wilfordii (TW), an herb used in Chinese medicine. Clinical trials performed so far using its root extracts have shown impressive therapeutic properties but also revealed substantial gastrointestinal side effects. The most promising bioactive compound obtained from TW is celastrol. During the last decade, an increasing number of studies were published highlighting the medicinal usefulness of celastrol in diverse clinical areas. Here we systematically review the mechanism of action and the therapeutic properties of celastrol in inflammatory diseases, namely, rheumatoid arthritis, systemic lupus erythematosus, inflammatory bowel diseases, osteoarthritis and allergy, as well as in cancer, neurodegenerative disorders and other diseases, such as diabetes, obesity, atherosclerosis, and hearing loss. We will also focus in the toxicological profile and limitations of celastrol formulation, namely, solubility, bioavailability, and dosage issues that still limit its further clinical application and usefulness.
Introduction
The identification of bioactive compounds derived from medicinal plants has attracted considerable interest in recent years for their strong and in some cases unique, anti-inflammatory, anticancer, and neuroprotective properties. One representative example is the Tripterygium wilfordii (TW) plant, used in Chinese medicine to treat an array of immunological disorders, including rheumatoid arthritis (RA), with promising results in a series of clinical trials. TW is a perennial vine of the Celastraceae family, also called Thunder God Vine or “lei gong teng” (Chinese name). This plant is poisonous but its root pulp contains several therapeutic compounds, including terpenoids, alkaloids, and steroids. The chemical structure of its purified compounds has been determined by nuclear magnetic resonance and mass spectroscopy. More than 46 diterpenoids (such as triptolide), 20 triterpenoids (e.g., celastrol), 21 alkaloids (like euonine), and other small molecules have been identified from TW. The most abundant and promising bioactive compound derived from the root of this plant is celastrol, also called tripterine, which possess a broad range of biological activities. Celastrol (3-hydroxy-9β,13α-dimethyl-2-oxo-24,25,26-trinoroleana-1(10),3,5,7-tetraen-29-oic acid) is a pentacyclic triterpenoid (Figure 1) that belongs to a small category of natural products of triterpene quinine methides.
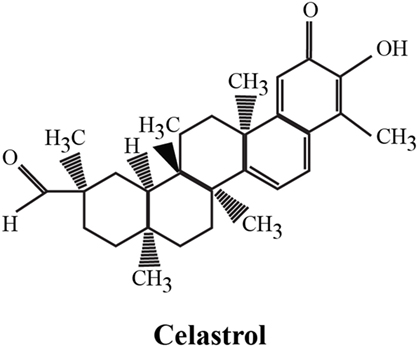
Figure 1. Chemical structure of celastrol. The chemical formula of celastrol is C29H38O4 [adapted from Ref. (1)].
Here, we systematically review the therapeutic properties of celastrol in chronic diseases and its toxicological profile, illustrating its potential clinical application.
Clinical Experience with the Use of TW
Clinical trials using TW plant extracts have already been conducted in inflammatory diseases.
TW is generally used in the treatment of Crohn’s disease (CD) in China. Its therapeutic benefits have been explored in an open-label clinical trial conducted in 20 CD patients treated with TW tablets (120 mg daily) for a period of 12 weeks. CD Activity Index diminished during the first 8 weeks, and the endoscopic improvement was observed after 12 weeks. Inflammatory parameters, including c-reactive protein (CRP), also decreased (2). In addition, two placebo-controlled trials and one prospective single-blind clinical trial have studied the therapeutic potential of polyglycoside TW (1 mg/kg daily) in the prevention of postsurgical relapses in patients with CD. Results from these studies suggest that this is an effective and well-tolerated drug (3–5). Recently, a randomized clinical trial has shown that TW (1.5 mg/kg/day) was comparable to azathioprine to prevent postoperative clinical recurrence of CD, although less efficient in preserving endoscopic remission at week 52 (6). An additional clinical trial enrolled 198 patients with CD, which were randomized to receive mesalazin (3 g/day), low-dose TW (1.5 mg/kg/day) or high-dose TW (2.0 mg/kg/day) over a 52-week period (7). Importantly, data have shown that less patients in the high-dose group (7/71) had clinical recurrence in comparison with patients in the low-dose (15/68, p = 0.047) or patients treated with mesalazin (17/59, p = 0.006). However, patients under mesalazin treatment had less adverse effects than those treated with high-dose (p = 0.029) and low-dose of TW (p = 0.048) (7).
The benefits of TW plant extracts have also been tested in psoriatic patients. A randomized clinical trial has shown equal efficacy of TW (20 mg, three times a day) and acitretin for the treatment of psoriasis vulgaris for 8 weeks in a total of 115 patients (8).
Additionally, several clinical trials with TW have already been conducted in RA. Uncontrolled trials from 1980s enrolling more than 100 RA patients have shown 87% response rates. To evaluate these claims, in 2002, a study recruited 35 patients and randomized them to placebo, low (180 mg/day) or high (360 mg/day) dose of an ethanol/ethyl acetate extract of TW (9). After 5 months, 80% of the high-dose and 40% of the low-dose groups had achieved an American College of Rheumatology 20% (ACR20) improvement response criteria, compared with none of the patients taking placebo. Both physical function and inflammation were improved. Diarrhea was the most common adverse event followed by nausea. A systematic review of a total of seven randomized controlled trials was done in 2009 by Jiang and colleagues to evaluate the efficacy and safety of TW in the treatment of RA (10). An interesting conclusion was that although TW was clinically as effective as disease-modifying anti-rheumatic drugs (DMARDs), no effect was observed in delaying bone erosions. However, these are small sample size trials and in six of them there were some methodological limitations that can have produced relevant biases. Subsequently, Goldbach-Mansky’s group enrolled 121 RA patients and randomly assigned them to receive TW extract (60 mg, three times daily) or sulfasalazine (11). After 6 months, 68% of those treated with TW and only 36% of those under sulfasalazine reached an ACR20 response. More patients in the sulfasalazine group experienced moderate or severe adverse effects. The most frequent side effects in patients receiving TW were diarrhea, nausea, dyspepsia, abdominal pain, and upper respiratory tract infection (11). In 2012, an interesting concept was tested in a randomized controlled, single-blind clinical trial that used external application of TW extracts in 67 active RA patients with positive results and only referring two cases of mild skin allergy (12). This result has been also reported by Cibere et al. (13). In 2013, due to inconsistency in some clinical trial data, Liu et al. performed a meta-analysis of randomized controlled trials. In this review, authors have concluded that TW extracts are a sort of "herbal DMARD" with a similar efficacy to synthetic DMARDs in RA treatment and that well-designed confirmatory randomized controlled trials should be done (14). Recently, Zhang and co-workers have shown in a multicenter, open-label, randomized controlled trial that combined therapy of methotrexate (MTX) and TW was more effective than MTX monotherapy in the treatment of active RA patients (15, 16). In this study, about 52.7% of the 207 patients experienced adverse events, which were seen in 46.4, 62.3, and 49.3% of patients receiving TW (20 mg, three times per day), MTX (12.5 mg/week), and TW + MTX, respectively (p = 0.136). The most common adverse event was mild gastrointestinal side effect, reported by 43.5% of patients on MTX and by 34.8% of those receiving the combination (16). According with this reported experience, combining both medications might be a good strategy to decrease the amount of MTX needed, which may reduce the toxic effects that can limit MTX long-term treatment (17). This report is in accordance with an observation from 2001, where 70 RA patients receiving MTX combined with small doses of TW polyglycoside (10 mg, three times a day) had a better effect and less adverse reactions than monotherapy with MTX (18). Finally, an interesting clinical trial has been conducted in order to evaluate the efficacy and safety of etanercept plus TW (10 mg, three times per day) in elderly patients with active RA. Etanercept plus TW had an equivalent therapeutic effect to that of Etanercept plus MTX and were both well tolerated (19). Altogether, these clinical trials show relevant data regarding the use of TW in RA treatment; however, there are limitations in their design, including the open-label design, short duration (6 months), and lack of radiological assessment. Although TW extracts showed efficacy in the symptomatic treatment of RA patients, it is still unclear if it provides structural damage control.
Regarding the clinical experience with TW in cancer, two water-soluble derivatives of triptolide (TW diterpenoid, a different class from celastrol) have been synthesized (PG490-88 and F60008) and approved for entry into a Phase I clinical trial for the treatment of solid tumors. PG490-88 will be tested in a Phase I clinical trial for prostate cancer in USA (20). Also, a phase I trial was performed with F60008 given intravenously in 20 advanced solid tumors patients in a total of 35 cycles. The most frequent side effects were mild anemia, fatigue, nausea, vomiting, diarrhea, and constipation. Two lethal events were observed and the high inter-individual variability rendered this derivative far from optimal (21).
In the case of neurodegenerative disorders, no clinical data are available about the use of TW. A study regarding the use of celastrol in amyotrophic lateral sclerosis concludes that further preclinical data, human toxicity, and pharmacokinetic results are required to proceed with trials (22).
The therapeutic effect of TW extracts has also been evaluated in diabetic patients. A prospective clinical trial enrolled 45 patients with type 2 diabetic kidney disease, randomly divided into three groups: TW (1–2 mg/kg/day), irbesartan (150–300 mg/day), and TW combined with irbesartan. Data have shown that treatment with TW for 12 weeks may be effective in preventing podocyte injury with a synergistic protective effect with irbesartan (23). Another clinical trial has been performed to evaluate the efficacy of TW in the treatment of type 2 diabetes mellitus (DM)-induced nephropathy. A total of 65 patients were enrolled in this 6-month, prospective, controlled study, and randomized into treatment groups: 120 mg/day of TW extract for 3 months, followed by 60 mg/day for 3 more months, or 160 mg/day of valsartan for 6 months. It was found that TW can significantly reduce the urine protein levels (24). Similar nephroprotective effects for TW preparations have been described in a meta-analysis of randomized controlled trials of chronic kidney disease patients (25).
A clinical trial, in China, also tested the use of TW in human kidney transplantation (26). Rejection occurred in 4.1% of patients treated with TW versus 24.5% of control patients, showing efficacy in the prevention of renal allograph rejection. All patients tolerated well TW administration during the 5 years of the study (26). This interesting potential of TW, which was already demonstrated in different in vitro and in vivo experimental set-ups, may be of interest in several medical areas.
In summary, clinical trials have only tested TW plant extracts. Despite its potential clinical usefulness, the sale of TW has been prohibited in many countries because the misuse of the herb can cause severe consequences, including diarrhea, nausea, and infertility. Even an apparent non-toxic dose may cause antifertility effects in men, male rats, and guinea pigs due to oral administration of some toxic components of TW extracts. Unfortunately, in men, this dose is only one-third of the recommended dose for the treatment of RA or skin diseases (27–29). As such, treating patients with TW bioactive compounds with known pharmacological properties may circumvent toxicological limitations. As stated before, celastrol is the most abundant bioactive compound existing in TW and these impressive therapeutic properties support the growing interest on this compound.
Anti-Inflammatory Properties of Celastrol
The therapeutic usefulness and anti-inflammatory properties of celastrol have been studied in several inflammatory diseases, including RA, ankylosing spondylitis, systemic lupus erythematosus (SLE), inflammatory bowel disease, osteoarthritis (OA), allergy, and skin inflammation.
Rheumatoid Arthritis
RA is a chronic inflammatory immune-mediated disease characterized by polyarthritis and joint damage. Both immune cells and cytokines play crucial roles in the pathogenesis of this disease. The anti-inflammatory properties of celastrol in this condition have so far been attributed to the: (i) regulation of cytokine and chemokine production, (ii) regulation of inflammatory mediators expression; (iii) modulation of inflammatory cell functions, and (iv) osteoclast modulation and bone damage control (Figure 2).
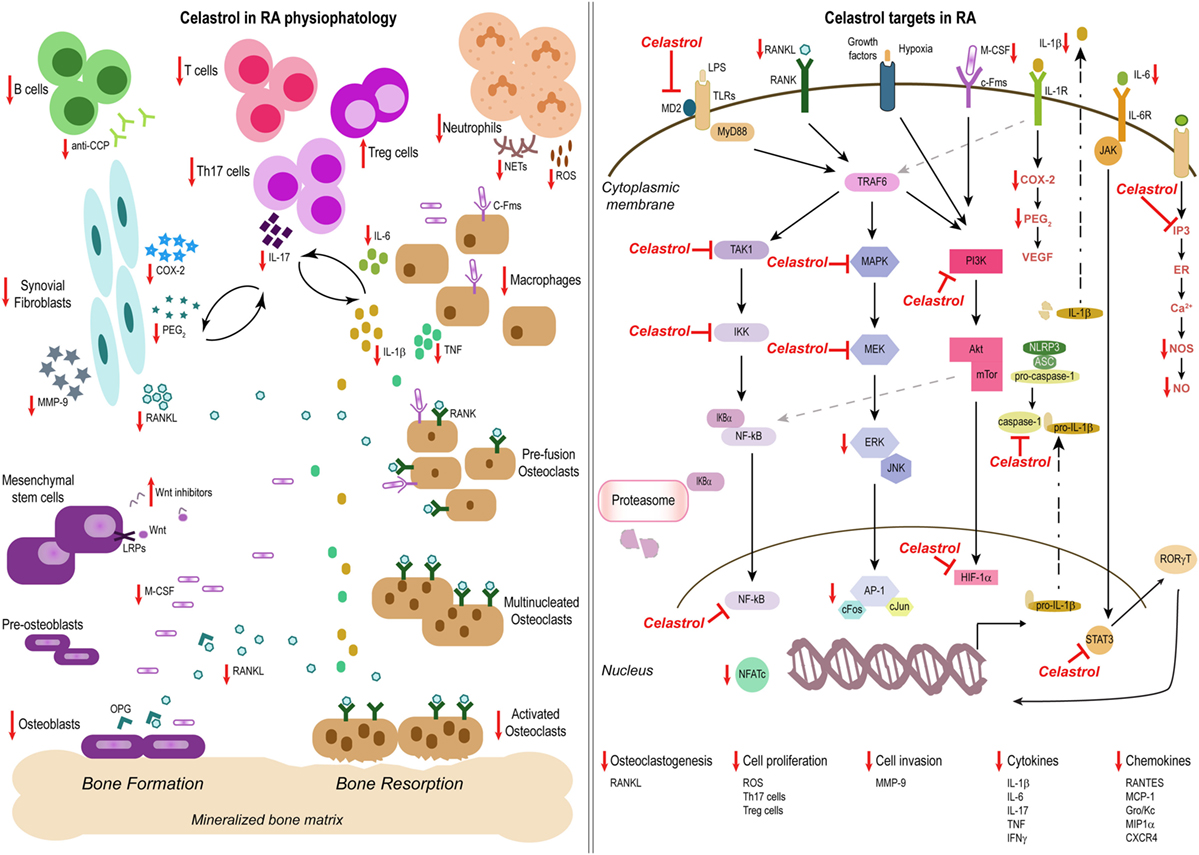
Figure 2. Celastrol in the treatment of rheumatoid arthritis (RA). Schemes illustrate the main anti-inflammatory properties and molecular targets of celastrol in the physiopathology of RA, as a prototype of an inflammatory disease. Celastrol has several cellular targets, interfering with the production of cytokines, chemokines, and inflammatory mediators; inhibiting cell invasion and proliferation; and suppressing bone resorption and thus constitutes a potential candidate for the treatment of inflammatory diseases.
Regulation of Cytokine, Chemokine, and Inflammatory Mediators’ Production
In 1991, Xu et al. (30) have shown in vitro that tripterine (celastrol) inhibits lipopolysaccharide (LPS)-induced interleukin (IL)-1β production from murine peritoneal macrophages and human monocytes, concanavaline A-activated IL-2 production by murine splenocytes, and prostaglandin E2 (PEG2) release from synovial cells. Moreover, in vivo it possess disease-modifying activities in streptococcus cell wall-induced (SCW) and collagen-induced (CIA) arthritis mouse models (31). Accordingly, Li et al. (32) have reported that tripterine inhibits paw swelling and bone destruction in adjuvant-induced arthritis (AIA) rats associated with a decrease in IL-1β mRNA in the synovial membrane and tumor necrosis factor (TNF) mRNA in hind paw tissue. Later, in vitro data have revealed that low concentrations (0.05–1 μM) of celastrol inhibit the production of nitric oxide (NO) and PEG2, accompanied by the decrease in iNOS and cyclooxygenase (COX)-2 protein, inhibit TNF and IL-6 release, and suppress the activity of nuclear factor kappa B (NF-kB) and activator protein (AP)-1 in LPS-stimulated macrophages (33, 34). In vivo, Venkatesha et al. (35) have shown that celastrol possess antiarthritic activities in AIA rats. It inhibits IL-6, IL-17, and interferon (IFN)-γ mRNA in lymph node (LN) and spleen adherent (SAC) cells isolated from arthritic rats. In LN cells celastrol induces a reduction in pSTAT3 but not in RORγt, suggesting that it may interfere with IL-17 production by T helper (Th)17 cells rather than with Th17 cells differentiation. In addition, celastrol reduces IL-6 production, metalloproteinase (MMP)-9 and pERK in AIA-derived fibroblasts, with no effect on VEGF and MMP-2. Interestingly, it also reduces anti-CCP serum levels in AIA rats (35). The same group has reported that celastrol inhibits the production of proinflammatory cytokines (TNF and IL-1β) and chemokines (RANTES, MCP-1, and GRO/KC) by SAC cells isolated from AIA rats after treatment. It also inhibits the migration capability of these cells (36). Except for MCP-1, serum levels of these cytokines and chemokines were also decreased in celastrol-treated AIA rats. Additionally, the same results, together with a reduction in MIP-1α, were observed in synovial fibroblasts isolated from arthritic rats and cultured with celastrol. Regarding chemokine receptors, celastrol does not seem to have much effect on their cellular expression, besides CCR1 (36). Of note, CCR1 correlates with the level of inflammation in experimental arthritis (37, 38).
Accordingly, in 2012 our group showed that celastrol decreases the secretion of both IL-1β and TNF in the THP-1 macrophage-like cell line, associated not only with NF-kB inhibition but also with caspase-1 inactivation (39). In vivo, we observed that celastrol has anti-inflammatory effects both in an early and established phase of arthritis development, suppressing ankle swelling, joint inflammatory cell infiltration and proliferation, and serum IL-6 levels in AIA rats (39, 40). This inhibitory effect in cellular infiltration and proliferation may prevent and treat synovial pannus tissue development characteristic of RA patients and responsible for bone damage.
Recently, it was found that celastrol inhibits LPS-induced cytokine production in macrophages by blocking LPS binding to MD2 from the MD2/TLR4 complex, inhibiting TLR4 signaling activation and thus the initiation of inflammatory responses (41).
This ability of celastrol to suppress the production of cytokines and chemokines might lead to the inhibition of leukocyte migration into the inflamed joints, contributing to its anti-inflammatory activity.
Modulation of Inflammatory Cell Functions
In human fibroblasts isolated from RA patients (RA-FLS), it has been shown that celastrol inhibits LPS-induced migration and invasion by inhibiting MMP-9 expression and activity (42). It inhibits MMP-9 transcription through the suppression of the binding activity of NF-kB in the promoter and the TLR4/MyD88/NF-kB pathway signaling (42). Previously, these authors have also reported that celastrol inhibits IL-17-induced migration and invasion of human RA-FLS by suppression of NF-kB-mediated MMP-9 expression and activity (43). In addition, under hypoxic conditions mimicking the synovial hypoxia of arthritic joints, it has been found that celastrol decreases hypoxia-induced FLS invasion by inhibiting HIF-1α-mediated CXCR4 transcription (44).
In 2015, our group has reported that celastrol restores synovial homeostasis in AIA rats, with a reduction in joint synovial CD3+ and CD19+ lymphocytes, associated with a suppression of bone erosions and a remarkable reduction in CD68+ macrophages, a marker of therapeutic efficacy in human RA and experimental arthritis. Importantly, neither blood biochemical parameters nor tissue histological structures revealed drug-induced toxicity associated with the intraperitoneally administration of 1 mg/kg/day of celastrol for 18 days (40).
Interestingly, Yu et al. have shown in vitro that non-cytotoxic concentrations (≤10μM) of celastrol inhibit neutrophil oxidative burst and extracellular trap (NET) formation through the inhibition of SYK/MEK/ERK and IkBα signaling cascade (45). Moreover, authors have shown that it decreases the levels of citrullinated histones, which are autoantigens in RA patients (45). Recently, Astry and colleagues have described, in synovium-infiltrating cells (SIC) from AIA rats, that celastrol reduces Th17 and increases T regulatory (Treg) cell frequencies, possibly favoring an anti-inflammatory/immunomodulatory local environment in the inflamed joints (46). In vitro, it inhibits Th17 and promotes Treg differentiation through the decrease of pSTAT3 as well as of IL-1β and IL-6 production (46). These data, together with our results showing that celastrol is more effective than Digoxin (specific RORγt inhibitor and hence suppressor of Th17 cells differentiation) in suppressing arthritis in AIA model (39), suggest that the combined effect of celastrol in both Th17 and Treg cells is an advantage in the treatment of arthritis.
Osteoclast Modulation and Bone Damage Control
In 2010, Idris et al. have shown in osteoblast/bone marrow co-cultures that celastrol inhibits osteoclast formation and bone resorption, and suppresses IL-1β-induced receptor activator of nuclear factor kappa-B ligand (RANKL) expression in osteoblasts by NF-kB signaling inhibition (47). In bone marrow cultures, it suppresses M-CSF and RANKL-induced osteoclast formation and induces apoptosis, via inhibition of NF-kB, ERK, and c-Fos activation. In osteoblasts, it prevents TAK1 activation upstream of NF-kB and reduces cell viability and activity (47). These results suggest that, at least in vitro, celastrol suppresses both osteoclastic and osteoblastic activities, affecting not only bone resorption but also bone formation. Nanjundaiah and colleagues have found that celastrol suppresses inflammation and bone erosions in rats with AIA, with an increase of bone volume coupled with a decrease of osteoclast numbers (48). Authors have shown that it decreases the production of IL-1β, IL-6, IL-17, IL-18, and TNF by SIC cells harvested from arthritic rats, inhibiting RANKL production and decreasing RANKL/OPG ratio, consequently inhibiting osteoclastogenesis. Also, celastrol reduces MMP-9 production, which limits bone damage (48). More recently, Gan et al. have described that celastrol not only directly suppresses osteoclast formation and function, but also reduces the RANKL-induced expression of osteoclastic genes and transcriptional factors (49). Authors have demonstrated that it inhibits osteoclast differentiation and function in RAW264.7 cells, with a reduction in osteoclastic genes (MMP-9, Trap, and Ctsk) and in transcriptional factors (c-Fos, c-Jun, and NFATc1), possibly due to NF-kB and MAPK inhibition (49). These results suggest that celastrol acts both downstream of RANKL at the levels of NF-kB activation and upstream targeting proinflammatory cytokine signaling. In vivo, these authors have also observed that it suppresses arthritis and ankle joint destruction in CIA mice, with a decrease in osteoclast numbers, as well as in osteoclastic genes and transcriptional factors (49).
Furthermore, data from our lab have revealed that celastrol is able to control inflammation-induced bone damage, with a reduction in articular cartilage degradation and bone erosions (40). In accordance, Liu and colleagues have observed in a mouse model of dexamethasone-induced secondary osteoporosis that celastrol not only improves lipid metabolism and reduces hypercalciuria, but also mitigates articular cartilage lesions, decreases NF-kB, MMP-1, and MMP-9 expression, and reduces serum PTH, tartrate-resistant acid phosphatase (TRACP)5b, CTX-I, as well as deoxypyridinoline (DPD), suggesting that it ameliorates abnormal bone metabolism (50).
In contrast to RA, ankylosing spondylitis (AS) is an inflammatory and autoimmune disease mainly characterized by new bone formation in axial joints (51, 52). PGE2 modulates the anabolic/catabolic process of bone, promoting bone remodeling through osteoblastic cell differentiation (53–55). It has been already reported that celastrol suppresses LPS-induced expression of PEG2 via the downregulation of COX-1 and COX-2 activation (33, 56). Recently, Zou et al. (57) have pointed out in vitro that celastrol inhibits the proliferation of PEG2-induced AS fibroblasts and their differentiation into an osteogenic phenotype, associated with a decrease in PI3K/Akt pathway and increase in Wnt inhibitors.
Therefore, data show that celastrol is effective in treating persistent synovitis and preventing cartilage and bone damage, which are the hallmarks of RA physiopathology. Remarkably, this bone-protective property of celastrol in arthritic models is further supported by studies performed in cancer models, as described next. Although the three main pharmacological mechanisms of celastrol that contribute to its efficacy are already extensively studied in arthritis, most of the data results from in vitro experiments, which may not completely reflect the in vivo complexity of the disease. Specifically, the protective property of celastrol against bone damage needs additional research in in vivo models with the inclusion of morphologic and mechanical testing.
Finally, the major complaint of patients with RA, or other forms of arthritis, is joint pain derived from joint inflammation and damage. Interestingly, Yang and co-workers have demonstrated in animal models of inflammatory pain that celastrol not only reduces the mRNA expression of IL-1β, IL-6, and TNF in mice paws but also has antihyperalgesic effects, possible mediated by the activation of cannabinoid receptor-2 (CB2) signaling (58). CB2 inhibits proinflammatory factors release from inflammatory cells near nociceptive neuron terminals, reducing pain perception without centrally mediated side effects (59). This new interesting capability of celastrol should be further investigated in animal models of other diseases because it could be useful as an adjuvant therapy in different medical areas.
Systemic Lupus Erythematosus
SLE is a chronic autoimmune inflammatory disease that affects multiple organ systems, prototypically characterized by high levels of circulating autoantibodies and glomerulonephritis. In 2003, a study using the spontaneous (NZBxW)F1 mice model of experimental SLE has shown that treatment with 3 and 6 mg/kg/day of celastrol reduces urinary protein excretion and serum anti-ds DNA autoantibodies, ameliorating clinical symptoms and survival rate (60). In agreement, Li et al. have also found in an experimental SLE mice model induced by active chromatin that administration of 12 mg/kg/day of celastrol decreases circulating anti-ss DNA, anti-ds DNA, and IgG antibodies, reduces serum NO and IL-10 production, and improves splenocyte proliferation, associated with amelioration of proteinuria and renal histological changes (61). Of note, these effects were comparable to 5 mg/kg prednisone in the treatment of experimental SLE (61).
Inflammatory Bowel Disease
CD and ulcerative colitis (UC) are multifactorial chronic relapsing inflammatory bowel diseases (62), both characterized by an imbalance between pro- and anti-inflammatory cytokines (63). In 2004, a work from Pinna and colleagues has evaluated the effect of celastrol in CD patient’s biopsies of inflamed intestinal mucosa and peripheral blood mononuclear cells (PBMCs). Authors have described that celastrol inhibits proinflammatory cytokine production (IL-1β, IL-6, IL-8, and TNF) in LPS-activated PBMCs and biopsies from CD patients, possibly via NF-kB and p38 MAPK inhibition (64). In mice models of DSS-induced colitis, it has been found that celastrol ameliorates acute intestinal injury and prevents the loss of intestinal epithelial homeostasis through the reduction of colonic oxidative stress, inhibition of NLRP3-inflammasome and IL-23/IL-17 pathway, reduction of inflammatory cytokines and increase in IL-10 and TNF levels, attenuation of neutrophil infiltration and upregulation of E-cadherin expression (65, 66). It also suppresses necroptosis death of colonic epithelial cells by upregulating caspase-8 and thus inhibiting RIP3/MLKL axis, avoiding the breakdown of the intestinal barrier and the chronic inflammatory process (66). In this study authors have suggested that the unexpected increase in TNF levels may be induced by the LPS-induced-TNF-α-factor (LITAF), independently of NF-kB (66, 67). More recently, it has been shown that celastrol ameliorates experimental colitis in IL-10 deficient mice via the upregulation of autophagy of the colon tissue cells by PI3K/Akt/mTOR signaling downregulation (68).
UC is one of the three highest risk factors for developing colorectal cancer. Importantly, a study from Lin et al. have demonstrated in a mice model of UC-related colorectal cancer (AOM/DSS mice model) that celastrol increases survival rate associated with a reduction in colonic neoplasms, prevents the upregulation of oncogenic markers namely, β-catenin, proliferating cell nuclear antigen (PCNA), and dysfunctional p53, inhibits proinflammatory mediators and NF-kB activation and suppresses epithelial mesenchymal transition (EMT), through E-cadherin upregulation and N-cadherin, vimentin, and Snail downregulation (69). The same group has also found that celastrol inhibits cell proliferation in colorectal cancer cell lines and inhibits tumor growth by reversing EMT in colonic xenografts (69). Finally, an interesting lipidomics report unrevealed that celastrol recovers lysophosphatidylcholine and sphingomyelin metabolism of DSS-induced colitis mice, partially by upregulating stearoyl-CoA desaturase-1 (SCD1) (an enzyme responsible for fatty acids desaturation) expression and restoring the altered balance between steric acid- and oleic acid-derived lipid species against proinflammatory signaling (70).
Osteoarthritis
OA is a multifactorial joint disease characterized by joint cartilage degradation. This pathology results from an imbalance between anabolic and catabolic activities of chondrocytes, with matrix MMPs, COX-2, and iNOS playing a central role in cartilage degradation. Inflammation of the synovium also occurs, though often mild compared to RA, which is primarily an inflammatory condition. This can happen as breakdown products from the cartilage are released into the synovial space and as cells lining the joint attempt to remove them. Interestingly, it has been found in human osteoarthritic chondrocytes that celastrol suppresses the expression of the catabolic mediators, MMP-1, MMP-3, MMP-13, COX-2, iNOS, and HSP90β, possibly through the inhibition of NF-kB activation (71).
Allergy
Allergic asthma is a common chronic inflammatory disease of the airways of the lung which is characterized by inflammatory cells infiltration in lung tissues, hypersecretion of mucus by goblet cells, obstruction of the lung airway, and higher expression of Th2 cytokines (72). MMPs mediate airway tissue remodeling through regulation of basal lamina integrity and of the infiltration by inflammatory cells (73, 74). In 2004, Liu et al. have shown that celastrol suppresses airway inflammation in an allergic asthma mice model through the inhibition of histamine and eotaxin production in mast cells (75). Later, in the same animal model, it has been found that celastrol suppresses the ovalbumin-induced airway inflammation through the decrease in airway hyper responsiveness, inflammatory cell infiltration and tissue remodeling. In addition, celastrol also regulates the imbalance of MMP-2, MMP-9, and tissue inhibitor of metalloproteinase 1 and 2 (TIMP-1 and TIMP-2), which is caused by inflammatory cytokines produced via MAPK/NF-kB pathway in inflammatory cells (76). Furthermore, in vitro data have revealed that celastrol regulates the expression of EMT-related proteins, by the inhibition of immunoglobulin Fc epsilon receptor I (FcεRI) signaling, involving protein kinase C (PKC), Rac1, ERK, and HSP90 (77). Also, it reduces skin inflammation in a Nc/Nga mice model of allergic atopic dermatitis and in a Balb/c mice model of skin inflammation, exerting antiallergic effects (77).
Altogether, data have highlighted that celastrol has great potential as an anti-inflammatory compound mainly due to its ability to inhibit the NF-kB pathway, interfering with the production of proinflammatory cytokines and chemokines, and also with inflammatory cells migration, proliferation, and activation.
Anticancer Properties of Celastrol
Since 2003 several publications have demonstrated the capacity of celastrol to contribute to the treatment of many types of cancer. Data from different tumor cell lines and animal cancer models have suggested that the anticancer properties of celastrol can be attributed to: (i) cell death activation, (ii) angiogenesis inhibition, (iii) treatment and radiotherapy sensitizing action, and (iv) anti-invasive effect.
Cell Death Activation
Celastrol inhibits cancer cell progression and induces cell death in a broad range of cancer cell lines such as lung, breast, esopharyngeal, glioblastoma, prostate, hepatoma, myeloma, pancreas, colon, liver, melanoma, leukemia, osteosarcoma, and gastric cancer. Namely, it has been found that celastrol induces cell cycle arrest, apoptosis, and autophagy by the activation of reactive oxygen species (ROS)/c-Jun N-terminal kinases (JNK) signaling pathway (78) in osteosarcoma and by downregulation of miR-21 expression (79–81) in gastric cancer cell lines. This negative regulatory effect of celastrol on microRNAs to induce autophagy was described as well in androgen receptor (AR)-positive prostate cancer cells. In these cells it induces autophagy by inhibiting the AR/miR-101 axis (82). Celastrol also suppresses AR signaling due to receptor degradation. This target to AR signaling occurs via HSP90 inhibition or calpain activation (83, 84). Interestingly, in AR-negative prostate cancer cells, celastrol is still able to induce autophagy through HIF/BNIP3 activation (85). The inhibition of HSP90 by celastrol occurs due to its capacity to suppress the interaction of HSP90 with its co-chaperone Cdc37 in pancreatic cancer cells (86). It dissociates the HSP90-Cdc37 complex by interacting with Cdc37 (87), potentially inhibiting some oncogenic proteins. Also, Chadli et al. have shown that celastrol inhibits p23, a HSP90 co-chaperon specific for steroid receptors, which is relevant for steroid-mediated cancers (88). Contradicting data propose that in fact it binds directly to the C-terminal region of HSP90α, inducing oligomerization and affecting some of its functions (89).
In addition to autophagy, celastrol is able to eliminate cancer cells by different apoptotic pathways, including (1) upregulation of death receptors in breast and colon cancer, enhancing TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis (90, 91); (2) activation of Fas/Fas ligand pathway in non-small-cell lung cancer (92); (3) inhibition of mitochondrial respiratory chain (MRC) complex I, and consequently ROS accumulation inside cancer cells, in non-small-cell lung carcinoma, liver cancer (93), osteosarcoma (94) and hepatocellular carcinoma (95) cell lines; (4) mitochondrial dysfunction and PI3K/Akt/mTOR pathway inhibition in triple negative breast cancer (96), melanoma cells (97) and several other types of cancer (98); (5) reduction in phosphorylated Akt, mTOR, and S6K and increase in AMP-activated protein kinase (AMPK) phosphorylation in gastric cancer cell lines and xenografts (79); (6) AMPK-induced PLK-2 pathway in breast cancer cell line (99); (7) destabilization of the ErbB2 and estrogen receptors in breast cancer cells (100, 101); (8) activation of caspase-dependent and independent pathways in breast cancer cells (102); (9) inhibition of topoisomerase II in HL-60 leukemia cells (103); (10) mitochondrial instability, activation of caspases and downregulation AML1-ETO/C-KIT oncoprotein, thus inhibiting the Akt, STAT3 and Erk1/2 downstream pathways in acute myeloid leukemia (AML) t(8:21) translocation cell line (104); (11) inhibition of STAT3/Janus kinase 2 (JAK2) in hepatocellular carcinoma (105); (12) inhibition of Myb in AML cells, while not affecting normal hematopoietic progenitor cells (106); (13) induction of the unfolded protein response-dependent cell death, endoplasmic reticulum (ER) stress, and PERK-eukaryotic initiation factor 2 (eIF2)–activating transcription factor (ATF4)-C/EBP homology protein (CHOP) signaling in oral squamous cell carcinoma cell lines (107); (14) reduction of GSK3β levels in HeLa cells (108); (15) target proteostasis in human glioblastoma cells, potentiating the proteotoxic stress response of HSP inhibitors (109); (16) targeting AR, ERG, and NF-kB signaling pathways in prostate cancer (110, 111); (17) upregulation of miR-146a expression, suppressing the NF-kB activity in gastric cancer (112); (18) inhibition of NF-kB in multiple myeloma (113–115), prostate cancer (116, 117) and leukemia cells (118); and (19) downregulation of IL-6 gene expression via NF-kB inhibition in prostate carcinoma cells (119).
As in inflammatory diseases, one key target of celastrol in cancer physiopathology is the NF-kB pathway. It has been demonstrated in prostate cancer, in vitro and in vivo, that celastrol can induce proteasomal inhibition. This inhibition leads to the accumulation of ubiquitinated Ikβ-α proteins that sequester the cytoplasmic components of the NF-kB complex, consequently causing apoptotic cell death (116, 120). This ability of celastrol to inhibit NF-kB was also observed in breast cancer xenografts, with a tumor growth decrease of approximately 60% (34). It could also augment the apoptotic effects driven by TNF and chemotherapeutic agents, through the suppression of both inducible and constitutive NF-kB activation (34).
In addition to autophagy and apoptosis, celastrol may also induce paraptosis, another form of programmed cell death. The inositol trisphosphate receptor (IP3R)-mediated release of Ca2+ from the ER induced by celastrol and its subsequent mitochondrial Ca2+ uniporter-mediated influx might lead to a major expansion of mitochondria and ER, and thus to paraptotic cell death. This mechanism has been already descried in breast and colon cancer cell lines (121).
Angiogenesis Inhibition
Besides cell death, the anticancer properties of celastrol can be also attributed to tumor angiogenesis inhibition. The inhibitory effect of celastrol on angiogenesis is mediated by the suppression of HIF-1α, through HSP90 (122) and mTOR/p70S6K/eIF4E pathway inhibition and ERK1/2 phosphorylation (123, 124). This inhibition of HIF-1α leads to the decrease of its target genes, such as the VEGF, that are crucial in the angiogenic process. Nonetheless, it has been shown in human glioma in vitro and in xenografts that celastrol does not seem to have an effect on VEGF, but rather in the expression of VEGF receptors (125, 126). On the contrary, a recent study has demonstrated that short-time exposure to celastrol did not alter HIF-1α mRNA levels. Alternatively, it induces HIF-1α protein accumulation in different cancer cell lines in an oxygen-independent manner via ROS and Akt/p70S6K signaling activation, promoting the transcription of VEGF and Glut-1 genes (85). These results are contradictory and raise concerns of radiotherapy resistance triggered by low-dose radiation-induced HIF-1α (127, 128). As described above, data also seems to support the inhibitory effect of celastrol upon HIF-1α in the context of arthritis.
Treatment and Radiotherapy Sensitizing Action
Another interesting anticancer mechanism of celastrol is its radiosensitizing effect. Specifically, celastrol can overcome tumor resistance to radiotherapy in prostate (129) and lung cancer cells (130, 131). This radiosensitizing effect was correlated with significant decreases in HSP90 clients, including epidermal growth factor receptor (EGFR), ErbB2, and survivin, and with an increase in p53 (130). The enhanced cytotoxic effects of ionizing radiation induced by celastrol on human lung tumor cells were also mediated by ROS production (131). Moreover, Wang and colleagues have found that incubation with celastrol after chemo-drug exposure causes persistent DNA damage and apoptosis of lung cancer cells (132). This data was confirmed in vitro and in vivo using the PC-3 human prostate cancer model (129), in which celastrol induced a reduction of DNA repair capacity, thus enhancing therapeutic efficacy. Recently, results from an in vitro study in non-small-cell lung cancer indicate a potential increase in treatment sensitivity due to celastrol-mediated ATF2/cJUN inhibition (133). In leukemia cells, a study has highlighted the ability of celastrol to induce chemotherapy sensitization, associated with caspase-3 activation, PARP cleavage, and decrease in oncoprotein Bcr-Abl (134). Likewise, it depletes Bcr-Abl protein and induces mitochondrial-dependent apoptosis in imatinib-resistant chronic myelogenous leukemia cells (135). Also, in temozolomide-resistant melanoma cells, combined therapy with celastrol has increased treatment sensitization, increasing cell death possibly via NF-kB and MAPK pathways (136). Additionally, in some types of cancer, including lung, hepatocellular, and breast cancer, treatment resistance may be overcome by celastrol mainly due to its proapoptotic properties (100, 137–139).
Invasion Inhibition
Finally, studies have also suggested that celastrol inhibits tumor invasion. In lung adenocarcinoma cells, it has been shown that this compound inhibits TNF-induced invasive activity, which was correlated with the downregulation of NF-kB-mediated gene products and inhibition of MMP-9 (140). Similar results were also described in breast cancer cells (141). This enzymatic breakdown of the extracellular matrix constituents by MMPs is one critical early step for the metastatic process. In accordance, a study published by Mi et al. have demonstrated that celastrol induces breast tumor cells apoptosis and inhibits their invasion via downregulating TNF-induced MMP-9 expression, with no effect on MMP-1 and MMP-2 (142). Additionally, it has been suggested that, apart from NF-kB inhibition, celastrol also inhibits invasion of hepatocellular carcinoma cells through the reduction of miR-224 expression, decreasing MMP-2 and MMP-9 protein levels (143). More recently, data have proposed that the inhibition of metastasis in lung cancer cells occurs through the suppression of Akt signaling pathway and integrin expression (144). Also, in esophageal cancer cells, the antimetastatic effect of celastrol was attributed to the inhibition of Wnt signaling pathway and integrin expression (145). The role of integrin inhibition in this process was further pointed out in melanoma cancer cells. In these cells the inhibition of migration and invasion by celastrol was attributed to the regulation of integrin function and cell adhesion, partly via p38 MAPK activation (146). Likewise, in colon and pancreatic cancer cells, celastrol reduces tumor invasiveness by downregulation of the chemokine receptor CXCR4 expression (147). Importantly, Idris et al. have proven that celastrol can prevent osteolytic bone metastasis, inhibiting osteoclastic formation and survival mainly due to the suppression of TAK1 and IkappaB kinase (IKK) complex activation (148).
Collectively these reports demonstrate that celastrol has potential to be used in the treatment of different types of cancer, mainly due to its capacity to inhibit transcription factors, such as NF-kB and HIF-1α, and mediators of protein homeostasis, like HSP90 chaperon. Moreover, the ability of celastrol to pass the brain blood barrier makes this compound an attractive therapeutic option in brain tumors and brain metastasis, which are associated with poor prognosis due to ineffective treatments and long-term toxicity.
Neuroprotective Properties of Celastrol
In the last decade, some studies have reported that celastrol is a promising neuroprotective agent in animal models of neurodegenerative diseases, such as Parkinson disease (149), Huntington disease (149–151), Alzheimer disease (152), and amyotrophic lateral sclerosis (153–155). Neurodegenerative diseases have been termed “protein misfolding disorders” and are characterized by the neuronal accumulation of protein aggregates (156–158). HSPs are repair agents which provide a line of defense against these misfolded, aggregation-prone proteins (158). Importantly, celastrol leads to the induction of HSPs (159). It is capable of inducing a set of HSPs in differentiated neurons in humans and in rodent cell lines (160). Interestingly, celastrol induces a wider set of potentially neuroprotective HSPs, including HSP70B’, in differentiated human neurons compared to differentiated rodent neurons (160), which may suggest that it confers greater protective effects against neurodegenerative diseases in the human brain (149, 150, 153). The induction of HSP70 confers several important therapeutic benefits: (i) maintain cellular protein quality status; (ii) inhibit inflammatory responses by binding to the regulatory NF-kappa-B essential modulator (NEMO) unit in the IKK complex, reducing its activation (161, 162); and (iii) bind TNF receptor associated factor 6 (TRAF6) and suppress several immune responses (163). Therefore, this induction of HSP70 by celastrol explains its beneficial effects not only in neurodegenerative disorders but also in inflammatory diseases.
In a Parkinson’s mouse model (MPTP neurotoxin induced), it has been shown that celastrol attenuates dopaminergic neurons loss and dopamine concentration deficiency, accompanied by an increase in HSP70 expression and a reduction in inflammation with a decline in TNF and NF-kB production (149). Similarly, in a Huntington rat model (3-NP neurotoxin-induced), studies have demonstrated that celastrol protects from striatal damage and astrogliosis through the induction of HSP70 in dopaminergic neurons via heat shock transcription factor (HSF)-1 activation (149, 151). Also, using an experimental model of Parkinson’s disease in vitro, Choi et al. have revealed that celastrol protects human dopaminergic cells from injury and apoptosis and prevents ROS generation and mitochondrial membrane potential loss (164). It inhibits cytochrome c release, Bax/Bcl-2 alterations, caspase-9/3 activation, and p38 MAPK activation (164). These data propose that celastrol protects dopaminergic cells through the inhibition of mitochondrial-dependent apoptotic pathway and preservation of mitochondria functions, as well as by the inhibition of p38 MAPK activation. Furthermore, using an in vitro cadmium-induced model of neurodegenerative diseases, it has been shown that celastrol prevents apoptosis in neuronal cells by inhibition of JNK and Akt/mTOR signaling pathways partly through the increase in their negative regulator PTEN (165). Surprisingly, Konieczny and colleagues have shown in in vitro and in vivo models of Parkinson disease that celastrol has no neuroprotective effects (166). Authors discuss that it seems to have a narrow therapeutic window, and suggest that it may have a biphasic effect with protective properties at low concentrations and toxic effects at higher concentrations. In accordance, in proteasome inhibitor-lactacystin and mitochondrial toxin-rotenone in vitro models of Parkinson’s disease, low concentrations of celastrol are able to partially attenuate cell damage (167). These discrepancies maybe a result of methodological differences among publications, namely cell culture conditions and animal models specificities.
Similar to what is observed in a transgenic mouse model of Huntington disease (151), it has been demonstrated that celastrol reduces the β-amyloid amount in an Alzheimer’s disease model (152). In vivo administration of celastrol in a transgenic model of Alzheimer’s disease reduces β-amyloid by inhibiting β-site amyloid precursor protein cleaving enzyme 1 (BACE-1) via NF-kB (152). Recently, Chow and co-workers have demonstrated that celastrol induces a set of HSPs (HSP27, 32, and 70) in rat cerebral cortical cultures, which are selectively impacted during the progression of this disease (168). It induces HSP70 in neurons, and HSP27 and HSP32 in glial cells at dosages that do not affect cell viability (169). Additionally, it has been proven in vitro that celastrol reduces both LPS-induced cell death and β-amyloid production mainly through the increase in HSP70 but also by increasing Bcl-2 expression and reducing NF-kB, COX-2, and GSK-3β expression and oxidative stress (170). Furthermore, the activated microglia-derived proinflammatory factors, ROS and NO, have long been believed to be involved with neuroinflammation in neurodegenerative diseases, including Parkinson and Alzheimer’s disease (171). Thus, intervention of microglial activation has become an interesting therapeutic target for the treatment of these conditions (172). Of interest, celastrol exhibits anti-inflammatory activity in LPS-activated BV-2 microgial cells through the downregulation of ERK/MAPK phosphorylation and NF-kB activation, which results in the inhibition of inflammatory mediators, such as IL-1β, TNF, and NO (173).
The therapeutic benefits of celastrol have also been elucidated in an amyotrophic lateral sclerosis animal model (G93A SOD1 transgenic mice). It increases mice survival with an augment in neuronal numbers, a reduction in TNF and iNOS levels, and a rise of HSP70 levels in lumbar spinal cord (153). In addition, it has been demonstrated in the experimental autoimmune encephalomyelitis (EAE) mice model that celastrol inhibits pathogenic Th17 cell responses in peripheral LNs (154). This ability of celastrol to interfere with Th17 cells in favor of an anti-inflammatory response was also found in the context of arthritis, as described above. In the EAE mice model, Abdin et al. (155) have further proven that celastrol ameliorates disease signs and relapse and causes a shift in the cytokine profile from Th1 towards Th2 cell pattern. Authors have also shown that it reduces NF-kB expression, nitrites levels, TLR2 expression, and CD3+ T cell count (155).
Celastrol is thus a promising therapeutic candidate for the treatment of neurodegenerative diseases mainly via NF-kB and HSPs inhibition. These new highlights in the potential therapeutic applications of celastrol still need intensive investigation due to dosage and toxicological limitations, especially concerning brain-related diseases.
Therapeutic Properties of Celastrol in Other Diseases
The most recent data regarding the therapeutic applications of celastrol has emerged in diabetes, obesity, atherosclerosis, and hearing loss.
Diabetes
The exact mechanism underlying type 2 diabetes is still unclear despite the implication of processes such as mitochondrial dysfunction and inflammation (174). In an in vitro model of insulin resistance on 3T3-L1 adipocytes with mitochondrial dysfunction, it has been revealed that celastrol markedly improves metabolic functions with a reduction in ROS production and an increase in mitochondrial membrane potential via NF-kB pathway inhibition (1). Additionally, in an in vitro model of mitochondrial dysfunction and insulin resistance using human skeletal muscle cells, it has been shown that celastrol treatment improves insulin-stimulated glucose uptake activity, apparently via PI3K/Akt pathway, with significant enhancement of mitochondrial activities (175). It has also been demonstrated in these cell cultures that it amplifies the expression of AMPK protein and attenuates oxidative damage, PKC θ and NF-kB activation, leading to the reduction of IL-1β, IL-6, and TNF levels (175).
Type 2 diabetes is the leading cause of end-stage renal disease. In this context, it has been recently shown in vivo that celastrol treatment not only improved insulin sensitivity and glycemic control but also improved kidney structure and function, through both metabolic and anti-inflammatory effects, possibly via NF-kB inhibition (176). Additionally, diabetes often coexists with different metabolic-related syndromes, such as dyslipidemia, hypertension, and liver damage. In a rat model of type 2 diabetes, it has been demonstrated that celastrol reduces macrophage infiltration and downregulates the expression of TLR4, MyD88, and NF-kB, thus decreasing IL-1β and TNF in the hepatic tissue, which can delay the progression of diabetic liver inflammation and injury (177). Another chronic complication of diabetes is the reduction in muscle mass, strength, and physical capacity. Importantly, Guan et al. (178) have shown that celastrol exerts antioxidant effects on skeletal muscle, partly by activating the AMPK/PGC1α/Sirt3 signaling pathway, attenuating diabetic myopathy.
In type 1 diabetes, a study has suggested that celastrol is not effective in reducing disease incidence in NOD mice (179). Using a 25 mg/kg twice a week regimen, celastrol was found to slightly reduce blood glucose levels on the day after dosing, but not at 2 days post administration (179), suggesting that it lowers blood glucose levels acutely. This might also indicate that this is not an effective dose or regimen. In fact, a study of celastrol pharmacokinetics has shown that its half-life is about 10 h in healthy rats (180). Contrarily, an in vitro study using RINm5F rat pancreatic β-cell line showed that celastrol regulates cytokine-induced cell death and proinflammatory responses by downregulating iNOS, COX-2, and chemokine (C-C motif) ligand 2 (CCL2) chemokine through NF-kB inhibition, exerting cytoprotective effects (181), which suggests that indeed it may be a therapeutic agent against type 1 diabetes.
Obesity
In obese condition, hyperleptinemia coexists with the loss of response to leptin, an inhibitor of food intake and inducer of energy expenditure. This phenomenon has been defined as leptin resistance and the restoration of its sensitivity is a useful strategy to treat obesity. Recently, in a hyperleptinemic diet-induced obese mice, celastrol has shown the ability to increase leptin sensitivity (182). It can restore the leptin signaling in neurons by overexpressing anorexigenic peptides pro-opiomelanocortin (POMC) and/or repressing orexigenic peptides [neuropeptide Y (NPY)/AgRP] (182, 183). Also, celastrol ability to increase mitochondrial function and HSF-1, a regulator of energy expenditure, through the activation of a PGC1α-dependent metabolic program in adipose tissues and muscles leads to an augment in energy expenditure and represents a possible therapeutic strategy to treat obesity and its metabolic consequences (184).
Atherosclerosis
Atherosclerosis is a multifocal, smoldering, chronic immunoinflammatory disease, which pathogenesis involves imbalanced lipid metabolism and a maladaptive immune response that lead to a chronic inflammation of the arterial wall. Importantly, in a rabbit experimental carotid atherosclerosis model, it has been recently shown that celastrol can effectively reduce the plaque ratio, the serum levels of low-density lipoprotein (LDL), and the expression of VEGF, suggesting an antiatherosclerotic effect (185). Another study has also found in a apoE(−/−) mouse fed with a high-fat/high-cholesterol diet that celastrol inhibits lectin-like oxidized LDL receptor-1 (LOX-1) and ROS, preventing atherosclerosis (186). In addition, it has been highlighted that the inhibition of NF-kB pathway was also at least partially involved in this protective effect of celastrol (186). Atherosclerosis is also associated with a dysregulation of endothelial progenitor cells (EPCs). Importantly, in vitro and in vivo data recently showed that celastrol improves the functional integrity of EPCs, which allows an effective EPC transplantation, used in the treatment of cardiovascular and ischemic diseases (187, 188).
Hearing Loss
Hearing impairment can be temporary or permanent. It is commonly caused by mechanosensory hair cell death in the inner ear due to aging, noise trauma, chemicals, and therapeutics. Aminoglycoside antibiotics, one of the most used antibiotics, may cause ototoxicity (189). Considering that aminoglycoside-induced hearing loss is irreparable and that its incidence is up to 33% (189, 190), finding a therapeutic strategy has great interest. In this context, it has been recently found that celastrol provides protection against aminoglycoside-induced ototoxicity in vitro in utricles and in vivo in mice receiving systemic kanamycin, via HSP32/HO-1 induction (191). Similar results were also observed in cisplatin-induced ototoxicity (192).
Altogether, the available data suggest that celastrol is a potential therapeutic molecule for the treatment of several chronic conditions but further studies are necessary to substantiate and deepen these conclusions.
Toxicity and Limitations of Celastrol Formulation
Despite the therapeutic potential of celastrol, further clinical application is still limited by low water solubility, reduced oral bioavailability, narrow window of dosage, and side effects.
Celastrol has poor water solubility (13.25 ± 0.83 mg/ml at 37°C). Its solubility was studied by Qi et al. (193) in various vehicles, and authors have shown that ethyl oleate, olive oil, the surfactants Labrasol and OP-10, and the cosurfactants PEG200, ethanol, butanol, and specially Transcutol P, were adequate solvents for celastrol with a solubility >20 mg/ml.
Due to this poor water solubility, celastrol has low bioavailability. A study from Zhang et al. (180) have demonstrated that oral administration of celastrol in rats results in ineffective absorption into the systemic circulation, with an absolute bioavailability of 17.06%. Li et al. (194). suggest that besides low aqueous solubility in vivo metabolism and/or tissue distribution might also cause this poor bioavailability.
One great concern regarding the clinical use of celastrol is its narrow therapeutic window of dose together with the occurrence of adverse effects. Our own data showed in vivo that the doses of 2.5 and 5 μg/g/day are effective and non-toxic in the treatment of arthritis in rats; however, lower concentrations immediately lose efficacy and higher concentrations show signs of toxicity (195). The same result has been recently described in Parkinson’s disease models (166). Similarly, in an osteosarcoma xenograft mouse model, it was described that treatment with celastrol at 1 and 2 mg/kg reduced tumor growth (42.9–50.2%), but it caused 5.7–9% weight loss in animals (78). Discrepancies exist in celastrol dosing and toxicity, with data in rodents showing that at 3 mg/kg there are adverse events and 27% mortality but other studies showing no toxic effects at this dose. In addition, there are reports showing an LD50 dose of 20.5 mg/kg and others suggesting a 40% mortality at 4 mg/kg (42, 100, 116, 149, 166). One major side effect of celastrol administration might be infertility (29, 196). In fact, a study using mice spermatogenic cells suggests that Ca2+ currents inhibition by celastrol may cause antifertility effects (196). In addition, celastrol blocks ion conduction of cardiac Kir2.1 and hERG potassium channels and reduces channel density on cell surface upon chronic treatment (197), which may predict cardiotoxicity. Therefore, new extensive studies on celastrol in vivo regimen and toxicity are still needed.
Celastrol may have a dual effect, suppressing oxidative stress at nanomolar concentrations and inducing detectable ROS above 1 μM. Specifically, in contrast to the ROS generation effect of celastrol in cancer cells, some studies have reported that celastrol has antioxidant properties on microglia and endothelial cells and attenuates hypertension-induced oxidative stress in vascular smooth muscle cells (198, 199). This discrepancy may be attributed not only to the difference between non-cancerous cells and cancer cells but also to dosage.
New Strategies for the Use of Celastrol
To surpass the physicochemical and pharmacokinetic limitations of celastrol and to diminish the effective dose, several methodologies have been tested that can represent useful strategies, such as exosomes (200), lipid nanospheres (201), nanoencapsulation (202), polyamidoamine dendrimer nanocarriers (203), liposomes (204–206), polymeric micelles (207, 208), cell-penetrating peptides-coated nanostructured lipid carriers (194, 209–211), sugar-silica nanoparticles (212), and self-microemulsifying drug delivery system (193). For instance, recent data have shown that celastrol-loaded exosomes enhance free celastrol efficacy and reduce dose-related toxicity in lung cancer (200). Also, celastrol-loaded sugar-decorated mesoporous silica nanoparticles have shown an increased specific anticancer activity with no induced toxicity in HeLa and A549 cells (212). In vivo, it has been shown that celatrol-loaded lipid nanospheres, liposomal celastrol, and solid self-microemulsifying dispersible tablets of celastrol significantly increase its oral bioavailability, increase efficacy, and diminishes the occurrence of side effects (193, 201, 205). Although these strategies have demonstrated encouraging results both in vitro and in vivo, no tripterine preparation have completed the research phase and reached the market.
Depending on the desired therapeutic effect, the concentration range of celastrol is also highly variable. For this reason, it would be interesting to modify its chemical structure producing an effective and less toxic derivative or analog. Some studies have found correlations between celastrol domains and its properties. Structurally, atomic orbital energy of celastrol′s carbons C2 on A-ring and C6 on B-ring possess a great susceptibility towards a nucleophilic attack that gives celastrol antitumor activity against a vast spectrum of tumors (213, 214). Also, it was observed that the celastrol’s acidic carboxylate group was not required for its cytotoxic effect in several cancer cell lines, but instead its quinone methide moiety (215). Different analogs of celastrol were already synthesized, and their neuroprotective properties against t-BHP-induced cytotoxicity were studied in neuronal PC12 cells by Sun and colleagues. Authors have found that the compound CL12 (celastrol coupled with tetramethylpyrazine) is more effective than the parent celastrol (216). In addition, celastrol derivatives with aromatic phenyl substituents appear to inhibit the growth of hepatocellular carcinoma patient-derived xenografts with reduced toxicity (217). Some structure modifications of celastrol were also carried out focusing on either the esterification or amidification of 20-carboxylic acid or the reduction products of A/B rings (215, 216, 218, 219). Structural modifications at the C-2, 3 positions were described as inducers of heat shock response, while those at C-6 position lead to anticancer properties. However, previous studies also suggested that the intact quinone methide moiety was essential for its cytotoxic activity in cancer cells and neuroprotective effect (215, 216, 220).
Conclusion
Accumulating evidence indicates that the anti-inflammatory, anticancer, and neuroprotective properties of celastrol can be mainly attributed to its ability to inhibit NF-kB, a central player in inflammation, cancer and neurodegenerative diseases. However, it also targets several other molecules, which allows celastrol to have a broad array of pharmacological mechanisms and potential therapeutic applications (Table 1). Celastrol is one of the main components of TW plant that has shown in clinical trials to be efficient in the treatment of different diseases, being generally well-tolerated, but with mild gastrointestinal side effects and possibly fertility issues. Despite the great therapeutic potential of celastrol, there are still some drawbacks in the process of developing it as a new drug. Hopefully, future preclinical studies will provide crucial information regarding celastrol formulation, pharmacokinetics, dosage, and toxicity for further optimization of this compound. The development of new celastrol derivatives and analogs, with higher pharmacological activities and lower toxicological issues, seems to be the next logic step in the development of this therapeutic concept.
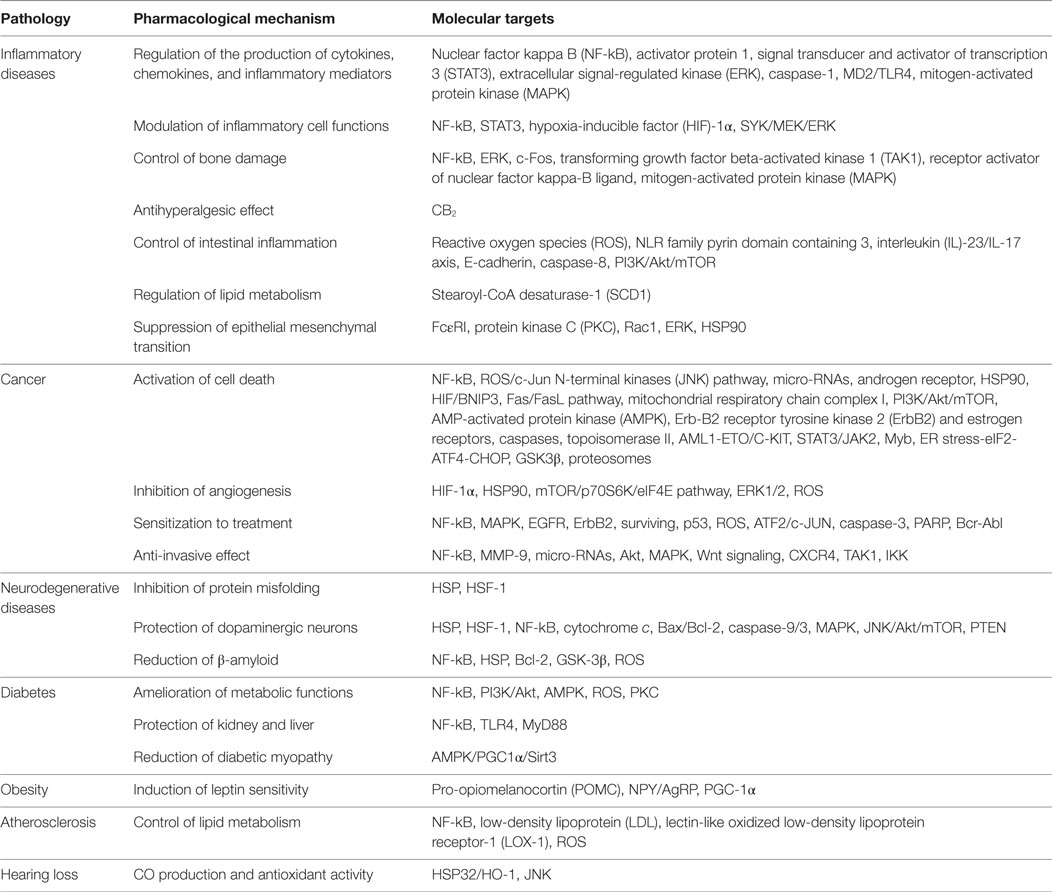
Table 1. Overview of the mechanisms of action of celastrol in chronic diseases.
Author Contributions
Drafting manuscript: RC. Revising manuscript content: LFM and JEF. Approving final version of manuscript: RC, LFM and JEF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
RC was supported by a fellowship from Fundação para a Ciência e a Tecnologia (FCT, SFRH/BPD/92860/2013). LFM is an FCT Investigator and receives financial support from the European Research Council (ERC-2014-CoG 647888-iPROTECTION) and FCT (PTDC/BIM-MEC/4665/2014). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
TW, Tripterygium wilfordii; RA, rheumatoid arthritis; CD, Crohn’s disease; CRP, C-reactive protein; ACR, American College of Rheumatology; DMARD, disease-modifying antirheumatic drug; MTX, methotrexate; DM, diabetes mellitus; LPS, lipopolysaccharide; IL, interleukin; PEG2, prostaglandin E2; SCW, Streptococcus cell wall-induced arthritis; CIA, collagen-induced arthritis; AIA, adjuvant-induced arthritis; TNF, tumor necrosis factor; NO, nitric oxide; iNOS, inducible nitric oxide synthase; COX, cyclooxygenase; NF-kB, nuclear factor kappa B; AP-1, activator protein 1; IFN-γ, interferon gamma; LN, lymph node; SA, spleen adherent; STAT3, signal transducer and activator of transcription 3; RORγ, RAR-related orphan receptor gamma; Th, T helper; MMP, metalloproteinase; ERK, extracellular signal-regulated kinase; VEGF, vascular endothelial growth factor; Anti-CCP, anti-cyclic citrullinated peptide; RANTES, regulated on activation, normal T cell expressed and secreted; MCP-1, monocyte chemoattractant protein-1; GRO/KC, chemokine ligand 1; MIP-1α, macrophage inflammatory protein 1-alpha; CCR1, C-C chemokine receptor type 1; MD2, lymphocyte antigen 96; TLR, Toll-like receptor; FLS, fibroblasts; MyD88, myeloid differentiation primary response gene 88; HIF, hypoxia-inducible factor; CXCR, C-X-C chemokine receptor; NET, extracellular trap; SYK, spleen tyrosine kinase; MEK, serine/tyrosine/threonine kinase; IkBα, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha; SIC, synovium-infiltrating cells; Treg, T regulatory; RANKL, receptor activator of nuclear factor kappa-B ligand; M-CSF, Macrophage colony-stimulating factor; TAK1, transforming growth factor beta-activated kinase 1; OPG, osteoprotegerin; Trap, tartrate-resistant acid phosphatase; Ctsk, Cathepsin k; NFATc1, nuclear factor of activated T-cells 1; MAPK, mitogen-activated protein kinase; PTH, parathyroid hormone; TRACP5b, tartrate-resistant acid phosphatase 5b; CTX-I, cross-linked C-telopeptide of type I collagen; DPD, deoxypyridinoline; AS, ankylosing spondylitis; PI3K, phosphoinositide 3-kinase; Akt, protein kinase B; CB2, cannabinoid receptor-2; SLE, systemic lupus erythematosus; UC, ulcerative colitis; PBMCs, peripheral blood mononuclear cells; NLRP3, NLR family pyrin domain containing 3; RIP3, receptor-interacting serine-threonine kinase 3; MLKL, mixed lineage kinase domain-like pseudokinase; LITAF, LPS-induced-TNF-α-factor; mTOR, mechanistic target of rapamycin; PCNA, proliferating cell nuclear antigen; EMT, mesenchymal transition; SCD1, stearoyl-CoA desaturase-1; OA, osteoarthritis; HSP, heat shock protein; TIMP, inhibitor of metalloproteinase; FcεRI, Fc epsilon receptor I; PKC, protein kinase C; ROS, reactive oxygen species; JNK, c-Jun N-terminal kinases; AR, androgen receptor; TRAIL, TNF-related apoptosis-inducing ligand; MRC, mitochondrial respiratory chain; AMPK, AMP-activated protein kinase; ErbB2, Erb-B2 receptor tyrosine kinase 2; AML, acute myelogenous leukemia; JAK, Janus kinase; ER, endoplasmic reticulum; PERK, ER stress; eIF2, eukaryotic initiation factor 2; ATF, activating transcription factor; CHOP, C/EBP homology protein; GSK3β, glycogen synthase kinase 3 beta; IP3R, inositol trisphosphate receptor; EGFR, epidermal growth factor receptor; PARP, poly (ADP-ribose) polymerase; IKK, IkappaB kinase; HSF, heat shock transcription factor; NEMO, NF-kappa-B essential modulator; TRAF, TNF receptor associated factor; BACE-1, β-site amyloid precursor protein cleaving enzyme 1; EAE, experimental autoimmune encephalomyelitis; PGC1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; Sirt3, NAD-dependent deacetylase sirtuin-3; CCL2, chemokine (C-C motif) ligand 2; POMC, pro-opiomelanocortin; AgRP, Agouti-related protein; NPY, Neuropeptide Y; LDL, low-density lipoprotein; apoE, apolipoprotein E; LOX-1, lectin-like oxidized low-density lipoprotein receptor-1; EPC, endothelial progenitor cell; LD, lethal dose.
References
1. Bakar MH, Sarmidi MR, Kai CK, Huri HZ, Yaakob H. Amelioration of mitochondrial dysfunction-induced insulin resistance in differentiated 3T3-L1 adipocytes via inhibition of NF-kappaB pathways. Int J Mol Sci (2014) 15(12):22227–57. doi: 10.3390/ijms151222227
2. Ren J, Tao Q, Wang X, Wang Z, Li J. Efficacy of T2 in active Crohn’s disease: a prospective study report. Dig Dis Sci (2007) 52(8):1790–7. doi:10.1007/s10620-007-9747-y
3. Tao QS, Ren JA, Ji ZL, Li JS, Wang XB, Jiang XH. [Maintenance effect of polyglycosides of Tripterygium wilfordii on remission in postoperative Crohn disease]. Zhonghua Wei Chang Wai Ke Za Zhi (2009) 12(5):491–3.
4. Liao NS, Ren JA, Fan CG, Wang GF, Zhao YZ, Li JS. [Efficacy of polyglycosides of Tripterygium wilfordii in preventing postoperative recurrence of Crohn disease]. Zhonghua Wei Chang Wai Ke Za Zhi (2009) 12(2):167–9.
5. Ren J, Wu X, Liao N, Wang G, Fan C, Liu S, et al. Prevention of postoperative recurrence of Crohn’s disease: Tripterygium wilfordii polyglycoside versus mesalazine. J Int Med Res (2013) 41(1):176–87. doi:10.1177/0300060512474744
6. Zhu W, Li Y, Gong J, Zuo L, Zhang W, Cao L, et al. Tripterygium wilfordii Hook. F. versus azathioprine for prevention of postoperative recurrence in patients with Crohn’s disease: a randomized clinical trial. Dig Liver Dis (2015) 47(1):14–9. doi:10.1016/j.dld.2014.09.008
7. Sun J, Shen X, Dong J, Wang H, Zuo L, Zhao J, et al. Tripterygium wilfordii Hook F as maintenance treatment for Crohn’s disease. Am J Med Sci (2015) 350(5):345–51. doi:10.1097/MAJ.0000000000000591
8. Wu C, Jin HZ, Shu D, Li F, He CX, Qiao J, et al. Efficacy and safety of Tripterygium wilfordii Hook F versus acitretin in moderate to severe psoriasis vulgaris: a randomized clinical trial. Chin Med J (Engl) (2015) 128(4):443–9. doi:10.4103/0366-6999.151069
9. Tao X, Younger J, Fan FZ, Wang B, Lipsky PE. Benefit of an extract of Tripterygium wilfordii Hook F in patients with rheumatoid arthritis: a double-blind, placebo-controlled study. Arthritis Rheum (2002) 46(7):1735–43. doi:10.1002/art.10411
10. Jiang Q, Cao W, Tang X, Jiao J. [Tripterygium wilfordii extract for treating rheumatoid arthritis: systematic review]. Zhongguo Zhong Yao Za Zhi (2009) 34(20):2637–43.
11. Goldbach-Mansky R, Wilson M, Fleischmann R, Olsen N, Silverfield J, Kempf P, et al. Comparison of Tripterygium wilfordii Hook F versus sulfasalazine in the treatment of rheumatoid arthritis: a randomized trial. Ann Intern Med (2009) 151(4):229–40. doi:10.7326/0003-4819-151-4-200908180-00005W49-51
12. Jiao J, Jiang Q. [External application of compound Tripterygium wilfordii decreased the activity of rheumatoid arthritis]. Zhongguo Zhong Xi Yi Jie He Za Zhi (2012) 32(11):1470–2. 6
13. Cibere J, Deng Z, Lin Y, Ou R, He Y, Wang Z, et al. A randomized double blind, placebo controlled trial of topical Tripterygium wilfordii in rheumatoid arthritis: reanalysis using logistic regression analysis. J Rheumatol (2003) 30(3):465–7.
14. Liu Y, Tu S, Gao W, Wang Y, Liu P, Hu Y, et al. Extracts of Tripterygium wilfordii Hook F in the treatment of rheumatoid arthritis: a systemic review and meta-analysis of randomised controlled trials. Evid Based Complement Alternat Med (2013) 2013:410793. doi:10.1155/2013/410793
15. Zhang C, Jiang M, He XJ, Lu AP. Clinical trials of integrative medicine for rheumatoid arthritis: issues and recommendations. Chin J Integr Med (2015) 21(6):403–7. doi:10.1007/s11655-015-2041-5
16. Lv QW, Zhang W, Shi Q, Zheng WJ, Li X, Chen H, et al. Comparison of Tripterygium wilfordii Hook F with methotrexate in the treatment of active rheumatoid arthritis (TRIFRA): a randomised, controlled clinical trial. Ann Rheum Dis (2015) 74(6):1078–86. doi:10.1136/annrheumdis-2013-204807
17. Alarcon GS, Tracy IC, Blackburn WD Jr. Methotrexate in rheumatoid arthritis. Toxic effects as the major factor in limiting long-term treatment. Arthritis Rheum (1989) 32(6):671–6. doi:10.1002/anr.1780320603
18. Wu YJ, Lao ZY, Zhang ZL. [Clinical observation on small doses Tripterygium wilfordii polyglycoside combined with methotrexate in treating rheumatoid arthritis]. Zhongguo Zhong Xi Yi Jie He Za Zhi (2001) 21(12):895–6.
19. He WZ, Yin ZH, Gao JH, Ye ZZ, Xie Y, Kong WH, et al. [Etanercept combined with Tripterygium wilfordii polyglycoside for treatment of rheumatoid arthritis in the elderly: a clinical study]. Zhongguo Zhong Xi Yi Jie He Za Zhi (2014) 34(3):267–71.
20. Kiviharju TM, Lecane PS, Sellers RG, Peehl DM. Antiproliferative and proapoptotic activities of triptolide (PG490), a natural product entering clinical trials, on primary cultures of human prostatic epithelial cells. Clin Cancer Res (2002) 8(8):2666–74.
21. Kitzen JJ, de Jonge MJ, Lamers CH, Eskens FA, van der Biessen D, van Doorn L, et al. Phase I dose-escalation study of F60008, a novel apoptosis inducer, in patients with advanced solid tumours. Eur J Cancer (2009) 45(10):1764–72. doi:10.1016/j.ejca.2009.01.026
22. Traynor BJ, Bruijn L, Conwit R, Beal F, O’Neill G, Fagan SC, et al. Neuroprotective agents for clinical trials in ALS: a systematic assessment. Neurology (2006) 67(1):20–7. doi:10.1212/01.wnl.0000223353.34006.54
23. Ma RX, Zhao N, Zhang W. [The effects and mechanism of Tripterygium wilfordii Hook F combination with irbesartan on urinary podocyte excretion in diabetic nephropathy patients]. Zhonghua Nei Ke Za Zhi (2013) 52(6):469–73.
24. Ge Y, Xie H, Li S, Jin B, Hou J, Zhang H, et al. Treatment of diabetic nephropathy with Tripterygium wilfordii Hook F extract: a prospective, randomized, controlled clinical trial. J Transl Med (2013) 11:134. doi:10.1186/1479-5876-11-134
25. Zhu B, Wang Y, Jardine M, Jun M, Lv JC, Cass A, et al. Tripterygium preparations for the treatment of CKD: a systematic review and meta-analysis. Am J Kidney Dis (2013) 62(3):515–30. doi:10.1053/j.ajkd.2013.02.374
26. Ji SM, Wang QW, Chen JS, Sha GZ, Liu ZH, Li LS. Clinical trial of Tripterygium Wilfordii Hook F. in human kidney transplantation in China. Transplant Proc (2006) 38(5):1274–9. doi:10.1016/j.transproceed.2006.03.017
27. Qian SZ. Tripterygium wilfordii, a Chinese herb effective in male fertility regulation. Contraception (1987) 36(3):335–45. doi:10.1016/0010-7824(87)90104-1
28. Matlin SA, Belenguer A, Stacey VE, Qian SZ, Xu Y, Zhang JW, et al. Male antifertility compounds from Tripterygium wilfordii Hook F. Contraception (1993) 47(4):387–400. doi:10.1016/0010-7824(93)90036-7
29. Yuan YY, Gu ZP, Shi QX, Qin GW, Xu RS, Cao L. [In vitro inhibition of celastrol on spermatozoa fertilization ability of guinea pig]. Yao Xue Xue Bao (1995) 30(5):331–5.
30. Xu WM, Zhang LX, Cheng ZH, Cai WZ, Miao HH, Pan DJ. [Inhibitory effect of tripterine on activities of IL-1, IL-2 and release of PGE2]. Yao Xue Xue Bao (1991) 26(9):641–5.
31. Li H, Jia YF, Pan Y, Pan DJ, Li D, Zhang LX. Effect of tripterine on collagen-induced arthritis in rats. Zhongguo Yao Li Xue Bao (1997) 18(3):270–3.
32. Li H, Zhang YY, Tan HW, Jia YF, Li D. Therapeutic effect of tripterine on adjuvant arthritis in rats. J Ethnopharmacol (2008) 118(3):479–84. doi:10.1016/j.jep.2008.05.028
33. Kim DH, Shin EK, Kim YH, Lee BW, Jun JG, Park JH, et al. Suppression of inflammatory responses by celastrol, a quinone methide triterpenoid isolated from Celastrus regelii. Eur J Clin Invest (2009) 39(9):819–27. doi:10.1111/j.1365-2362.2009.02186.x
34. Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K, et al. Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol (2006) 72(10):1311–21. doi:10.1016/j.bcp.2006.08.014
35. Venkatesha SH, Yu H, Rajaiah R, Tong L, Moudgil KD. Celastrus-derived celastrol suppresses autoimmune arthritis by modulating antigen-induced cellular and humoral effector responses. J Biol Chem (2011) 286(17):15138–46. doi:10.1074/jbc.M111.226365
36. Venkatesha SH, Astry B, Nanjundaiah SM, Yu H, Moudgil KD. Suppression of autoimmune arthritis by Celastrus-derived celastrol through modulation of pro-inflammatory chemokines. Bioorg Med Chem (2012) 20(17):5229–34. doi:10.1016/j.bmc.2012.06.050
37. Haas CS, Martinez RJ, Attia N, Haines GK III, Campbell PL, Koch AE. Chemokine receptor expression in rat adjuvant-induced arthritis. Arthritis Rheum (2005) 52(12):3718–30. doi:10.1002/art.21476
38. Shahrara S, Amin MA, Woods JM, Haines GK, Koch AE. Chemokine receptor expression and in vivo signaling pathways in the joints of rats with adjuvant-induced arthritis. Arthritis Rheum (2003) 48(12):3568–83. doi:10.1002/art.11344
39. Cascao R, Vidal B, Raquel H, Neves-Costa A, Figueiredo N, Gupta V, et al. Effective treatment of rat adjuvant-induced arthritis by celastrol. Autoimmun Rev (2012) 11(12):856–62. doi:10.1016/j.autrev.2012.02.022
40. Cascao R, Vidal B, Lopes IP, Paisana E, Rino J, Moita LF, et al. Decrease of CD68 synovial macrophages in celastrol treated arthritic rats. PLoS One (2015) 10(12):e0142448. doi:10.1371/journal.pone.0142448
41. Lee JY, Lee BH, Kim ND, Lee JY. Celastrol blocks binding of lipopolysaccharides to a toll-like receptor4/myeloid differentiation factor2 complex in a thiol-dependent manner. J Ethnopharmacol (2015) 172:254–60. doi:10.1016/j.jep.2015.06.028
42. Li G, Liu D, Zhang Y, Qian Y, Zhang H, Guo S, et al. Celastrol inhibits lipopolysaccharide-stimulated rheumatoid fibroblast-like synoviocyte invasion through suppression of TLR4/NF-kappaB-mediated matrix metalloproteinase-9 expression. PLoS One (2013) 8(7):e68905. doi:10.1371/journal.pone.0068905
43. Li GQ, Zhang Y, Liu D, Qian YY, Zhang H, Guo SY, et al. Celastrol inhibits interleukin-17A-stimulated rheumatoid fibroblast-like synoviocyte migration and invasion through suppression of NF-kappaB-mediated matrix metalloproteinase-9 expression. Int Immunopharmacol (2012) 14(4):422–31. doi:10.1016/j.intimp.2012.08.016
44. Li GQ, Liu D, Zhang Y, Qian YY, Zhu YD, Guo SY, et al. Anti-invasive effects of celastrol in hypoxia-induced fibroblast-like synoviocyte through suppressing of HIF-1alpha/CXCR4 signaling pathway. Int Immunopharmacol (2013) 17(4):1028–36. doi:10.1016/j.intimp.2013.10.006
45. Yu Y, Koehn CD, Yue Y, Li S, Thiele GM, Hearth-Holmes MP, et al. Celastrol inhibits inflammatory stimuli-induced neutrophil extracellular trap formation. Curr Mol Med (2015) 15(4):401–10. doi:10.2174/1566524015666150505160743
46. Astry B, Venkatesha SH, Laurence A, Christensen-Quick A, Garzino-Demo A, Frieman MB, et al. Celastrol, a Chinese herbal compound, controls autoimmune inflammation by altering the balance of pathogenic and regulatory T cells in the target organ. Clin Immunol (2015) 157(2):228–38. doi:10.1016/j.clim.2015.01.011
47. Idris AI, Krishnan M, Simic P, Landao-Bassonga E, Mollat P, Vukicevic S, et al. Small molecule inhibitors of IkappaB kinase signaling inhibit osteoclast formation in vitro and prevent ovariectomy-induced bone loss in vivo. FASEB J (2010) 24(11):4545–55. doi:10.1096/fj.10-164095
48. Nanjundaiah SM, Venkatesha SH, Yu H, Tong L, Stains JP, Moudgil KD. Celastrus and its bioactive celastrol protect against bone damage in autoimmune arthritis by modulating osteoimmune cross-talk. J Biol Chem (2012) 287(26):22216–26. doi:10.1074/jbc.M112.356816
49. Gan K, Xu L, Feng X, Zhang Q, Wang F, Zhang M, et al. Celastrol attenuates bone erosion in collagen-Induced arthritis mice and inhibits osteoclast differentiation and function in RANKL-induced RAW264.7. Int Immunopharmacol (2015) 24(2):239–46. doi:10.1016/j.intimp.2014.12.012
50. Liu X, Cai F, Zhang Y, Yang A, Liu L. Celastrol, an NF-kappaB inhibitor, ameliorates hypercalciuria and articular cartilage lesions in a mouse model of secondary osteoporosis. J Pharmacol Sci (2016) 130(4):204–11. doi:10.1016/j.jphs.2016.02.001
51. Braun J, Sieper J. Ankylosing spondylitis. Lancet (2007) 369(9570):1379–90. doi:10.1016/S0140-6736(07)60635-7
52. Sieper J, Appel H, Braun J, Rudwaleit M. Critical appraisal of assessment of structural damage in ankylosing spondylitis: implications for treatment outcomes. Arthritis Rheum (2008) 58(3):649–56. doi:10.1002/art.23260
53. Kawaguchi H, Pilbeam CC, Harrison JR, Raisz LG. The role of prostaglandins in the regulation of bone metabolism. Clin Orthop Relat Res (1995) 313:36–46.
54. Ramirez-Yanez GO, Seymour GJ, Walsh LJ, Forwood MR, Symons AL. Prostaglandin E2 enhances alveolar bone formation in the rat mandible. Bone (2004) 35(6):1361–8. doi:10.1016/j.bone.2004.08.006
55. Flanagan AM, Chambers TJ. Stimulation of bone nodule formation in vitro by prostaglandins E1 and E2. Endocrinology (1992) 130(1):443–8. doi:10.1210/endo.130.1.1309342
56. Tao X, Schulze-Koops H, Ma L, Cai J, Mao Y, Lipsky PE. Effects of Tripterygium wilfordii Hook F extracts on induction of cyclooxygenase 2 activity and prostaglandin E2 production. Arthritis Rheum (1998) 41(1):130–8. doi:10.1002/1529-0131(199801)41:1<130::AID-ART16>3.3.CO;2-W
57. Zou YC, Yang XW, Yuan SG, Zhang P, Li YK. Celastrol inhibits prostaglandin E2-induced proliferation and osteogenic differentiation of fibroblasts isolated from ankylosing spondylitis hip tissues in vitro. Drug Des Devel Ther (2016) 10:933–48. doi:10.2147/DDDT.S97463
58. Yang L, Li Y, Ren J, Zhu C, Fu J, Lin D, et al. Celastrol attenuates inflammatory and neuropathic pain mediated by cannabinoid receptor type 2. Int J Mol Sci (2014) 15(8):13637–48. doi:10.3390/ijms150813637
59. Rice AS, Farquhar-Smith WP, Nagy I. Endocannabinoids and pain: spinal and peripheral analgesia in inflammation and neuropathy. Prostaglandins Leukot Essent Fatty Acids (2002) 66(2–3):243–56. doi:10.1054/plef.2001.0362
60. Xu X, Wu Z, Xu C, Ren Y, Ge Y. Observation on serum anti-double stranded DNA antibodies of tripterine in systemic lupus erythematosus of (NZBxW)F1 mice. Ann Rheum Dis (2003) 62(4):377–8. doi:10.1136/ard.62.4.377
61. Li H, Zhang YY, Huang XY, Sun YN, Jia YF, Li D. Beneficial effect of tripterine on systemic lupus erythematosus induced by active chromatin in BALB/c mice. Eur J Pharmacol (2005) 512(2–3):231–7. doi:10.1016/j.ejphar.2005.02.030
62. Elson CO. Genes, microbes, and T cells – new therapeutic targets in Crohn’s disease. N Engl J Med (2002) 346(8):614–6. doi:10.1056/NEJM200202213460812
63. Neurath MF, Finotto S, Glimcher LH. The role of Th1/Th2 polarization in mucosal immunity. Nat Med (2002) 8(6):567–73. doi:10.1038/nm0602-567
64. Pinna GF, Fiorucci M, Reimund JM, Taquet N, Arondel Y, Muller CD. Celastrol inhibits pro-inflammatory cytokine secretion in Crohn’s disease biopsies. Biochem Biophys Res Commun (2004) 322(3):778–86. doi:10.1016/j.bbrc.2004.07.186
65. Shaker ME, Ashamallah SA, Houssen ME. Celastrol ameliorates murine colitis via modulating oxidative stress, inflammatory cytokines and intestinal homeostasis. Chem Biol Interact (2014) 210:26–33. doi:10.1016/j.cbi.2013.12.007
66. Jia Z, Xu C, Shen J, Xia T, Yang J, He Y. The natural compound celastrol inhibits necroptosis and alleviates ulcerative colitis in mice. Int Immunopharmacol (2015) 29(2):552–9. doi:10.1016/j.intimp.2015.09.029
67. Bushell KN, Leeman SE, Gillespie E, Gower AC, Reed KL, Stucchi AF, et al. LITAF mediation of increased TNF-alpha secretion from inflamed colonic lamina propria macrophages. PLoS One (2011) 6(9):e25849. doi:10.1371/journal.pone.0025849
68. Zhao J, Sun Y, Shi P, Dong JN, Zuo LG, Wang HG, et al. Celastrol ameliorates experimental colitis in IL-10 deficient mice via the up-regulation of autophagy. Int Immunopharmacol (2015) 26(1):221–8. doi:10.1016/j.intimp.2015.03.033
69. Lin L, Sun Y, Wang D, Zheng S, Zhang J, Zheng C. Celastrol ameliorates ulcerative colitis-related colorectal cancer in mice via suppressing inflammatory responses and epithelial-mesenchymal transition. Front Pharmacol (2015) 6:320. doi:10.3389/fphar.2015.00320
70. Wang R, Gu X, Dai W, Ye J, Lu F, Chai Y, et al. A lipidomics investigation into the intervention of celastrol in experimental colitis. Mol Biosyst (2016) 12(5):1436–44. doi:10.1039/c5mb00864f
71. Ding QH, Cheng Y, Chen WP, Zhong HM, Wang XH. Celastrol, an inhibitor of heat shock protein 90beta potently suppresses the expression of matrix metalloproteinases, inducible nitric oxide synthase and cyclooxygenase-2 in primary human osteoarthritic chondrocytes. Eur J Pharmacol (2013) 708(1–3):1–7. doi:10.1016/j.ejphar.2013.01.057
72. Zimmermann N, Hershey GK, Foster PS, Rothenberg ME. Chemokines in asthma: cooperative interaction between chemokines and IL-13. J Allergy Clin Immunol (2003) 111(2):227–42. doi:10.1067/mai.2003.139 quiz 43,
73. Kumagai K, Ohno I, Okada S, Ohkawara Y, Suzuki K, Shinya T, et al. Inhibition of matrix metalloproteinases prevents allergen-induced airway inflammation in a murine model of asthma. J Immunol (1999) 162(7):4212–9.
74. Lanone S, Zheng T, Zhu Z, Liu W, Lee CG, Ma B, et al. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and -12 in IL-13-induced inflammation and remodeling. J Clin Invest (2002) 110(4):463–74. doi:10.1172/JCI0214136
75. Liu RL, Liu ZL, Li Q, Qiu ZM, Lu HJ, Yang ZM, et al. [The experimental study on the inhibitory effect of tripterine on airway inflammation in asthmatic mice]. Zhonghua Jie He He Hu Xi Za Zhi (2004) 27(3):165–8.
76. Kim DY, Park JW, Jeoung D, Ro JY. Celastrol suppresses allergen-induced airway inflammation in a mouse allergic asthma model. Eur J Pharmacol (2009) 612(1–3):98–105. doi:10.1016/j.ejphar.2009.03.078
77. Kim Y, Kim K, Lee H, Han S, Lee YS, Choe J, et al. Celastrol binds to ERK and inhibits FcepsilonRI signaling to exert an anti-allergic effect. Eur J Pharmacol (2009) 612(1–3):131–42. doi:10.1016/j.ejphar.2009.03.071
78. Li HY, Zhang J, Sun LL, Li BH, Gao HL, Xie T, et al. Celastrol induces apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells: an in vitro and in vivo study. Cell Death Dis (2015) 6:e1604. doi:10.1038/cddis.2014.543
79. Lee HW, Jang KS, Choi HJ, Jo A, Cheong JH, Chun KH. Celastrol inhibits gastric cancer growth by induction of apoptosis and autophagy. BMB Rep (2014) 47(12):697–702. doi:10.5483/BMBRep.2014.47.12.069
80. Sha M, Ye J, Zhang LX, Luan ZY, Chen YB, Huang JX. Celastrol induces apoptosis of gastric cancer cells by miR-21 inhibiting PI3K/Akt-NF-kappaB signaling pathway. Pharmacology (2014) 93(1–2):39–46. doi:10.1159/000357683
81. Sha M, Ye J, Luan ZY, Guo T, Wang B, Huang JX. Celastrol induces cell cycle arrest by MicroRNA-21-mTOR-mediated inhibition p27 protein degradation in gastric cancer. Cancer Cell Int (2015) 15:101. doi:10.1186/s12935-015-0256-3
82. Guo J, Huang X, Wang H, Yang H. Celastrol induces autophagy by targeting AR/miR-101 in prostate cancer cells. PLoS One (2015) 10(10):e0140745. doi:10.1371/journal.pone.0140745
83. Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell (2006) 10(4):321–30. doi:10.1016/j.ccr.2006.09.005
84. Yang H, Murthy S, Sarkar FH, Sheng S, Reddy GP, Dou QP. Calpain-mediated androgen receptor breakdown in apoptotic prostate cancer cells. J Cell Physiol (2008) 217(3):569–76. doi:10.1002/jcp.21565
85. Han X, Sun S, Zhao M, Cheng X, Chen G, Lin S, et al. Celastrol stimulates hypoxia-inducible factor-1 activity in tumor cells by initiating the ROS/Akt/p70S6K signaling pathway and enhancing hypoxia-inducible factor-1alpha protein synthesis. PLoS One (2014) 9(11):e112470. doi:10.1371/journal.pone.0112470
86. Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG, et al. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther (2008) 7(1):162–70. doi:10.1158/1535-7163.MCT-07-0484
87. Sreeramulu S, Gande SL, Gobel M, Schwalbe H. Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew Chem Int Ed Engl (2009) 48(32):5853–5. doi:10.1002/anie.200900929
88. Chadli A, Felts SJ, Wang Q, Sullivan WP, Botuyan MV, Fauq A, et al. Celastrol inhibits Hsp90 chaperoning of steroid receptors by inducing fibrillization of the co-chaperone p23. J Biol Chem (2010) 285(6):4224–31. doi:10.1074/jbc.M109.081018
89. Zanphorlin LM, Alves FR, Ramos CH. The effect of celastrol, a triterpene with antitumorigenic activity, on conformational and functional aspects of the human 90kDa heat shock protein Hsp90alpha, a chaperone implicated in the stabilization of the tumor phenotype. Biochim Biophys Acta (2014) 1840(10):3145–52. doi:10.1016/j.bbagen.2014.06.008
90. Sung B, Park B, Yadav VR, Aggarwal BB. Celastrol, a triterpene, enhances TRAIL-induced apoptosis through the down-regulation of cell survival proteins and up-regulation of death receptors. J Biol Chem (2010) 285(15):11498–507. doi:10.1074/jbc.M109.090209
91. Zhu H, Liu XW, Ding WJ, Xu DQ, Zhao YC, Lu W, et al. Up-regulation of death receptor 4 and 5 by celastrol enhances the anti-cancer activity of TRAIL/Apo-2L. Cancer Lett (2010) 297(2):155–64. doi:10.1016/j.canlet.2010.04.030
92. Mou H, Zheng Y, Zhao P, Bao H, Fang W, Xu N. Celastrol induces apoptosis in non-small-cell lung cancer A549 cells through activation of mitochondria- and Fas/FasL-mediated pathways. Toxicol In Vitro (2011) 25(5):1027–32. doi:10.1016/j.tiv.2011.03.023
93. Chen G, Zhang X, Zhao M, Wang Y, Cheng X, Wang D, et al. Celastrol targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent cytotoxicity in tumor cells. BMC Cancer (2011) 11:170. doi:10.1186/1471-2407-11-170
94. Yu X, Zhou X, Fu C, Wang Q, Nie T, Zou F, et al. Celastrol induces apoptosis of human osteosarcoma cells via the mitochondrial apoptotic pathway. Oncol Rep (2015) 34(3):1129–36. doi:10.3892/or.2015.4124
95. Li PP, He W, Yuan PF, Song SS, Lu JT, Wei W. Celastrol induces mitochondria-mediated apoptosis in hepatocellular carcinoma Bel-7402 cells. Am J Chin Med (2015) 43(1):137–48. doi:10.1142/S0192415X15500093
96. Shrivastava S, Jeengar MK, Reddy VS, Reddy GB, Naidu VG. Anticancer effect of celastrol on human triple negative breast cancer: possible involvement of oxidative stress, mitochondrial dysfunction, apoptosis and PI3K/Akt pathways. Exp Mol Pathol (2015) 98(3):313–27. doi:10.1016/j.yexmp.2015.03.031
97. Lee JH, Won YS, Park KH, Lee MK, Tachibana H, Yamada K, et al. Celastrol inhibits growth and induces apoptotic cell death in melanoma cells via the activation ROS-dependent mitochondrial pathway and the suppression of PI3K/AKT signaling. Apoptosis (2012) 17(12):1275–86. doi:10.1007/s10495-012-0767-5
98. Kannaiyan R, Manu KA, Chen L, Li F, Rajendran P, Subramaniam A, et al. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis (2011) 16(10):1028–41. doi:10.1007/s10495-011-0629-6
99. Kim JH, Lee JO, Lee SK, Kim N, You GY, Moon JW, et al. Celastrol suppresses breast cancer MCF-7 cell viability via the AMP-activated protein kinase (AMPK)-induced p53-polo like kinase 2 (PLK-2) pathway. Cell Signal (2013) 25(4):805–13. doi:10.1016/j.cellsig.2012.12.005
100. Raja SM, Clubb RJ, Ortega-Cava C, Williams SH, Bailey TA, Duan L, et al. Anticancer activity of celastrol in combination with ErbB2-targeted therapeutics for treatment of ErbB2-overexpressing breast cancers. Cancer Biol Ther (2011) 11(2):263–76. doi:10.4161/cbt.11.2.13959
101. Jang SY, Jang SW, Ko J. Celastrol inhibits the growth of estrogen positive human breast cancer cells through modulation of estrogen receptor alpha. Cancer Lett (2011) 300(1):57–65. doi:10.1016/j.canlet.2010.09.006
102. Yang HS, Kim JY, Lee JH, Lee BW, Park KH, Shim KH, et al. Celastrol isolated from Tripterygium regelii induces apoptosis through both caspase-dependent and -independent pathways in human breast cancer cells. Food Chem Toxicol (2011) 49(2):527–32. doi:10.1016/j.fct.2010.11.044
103. Nagase M, Oto J, Sugiyama S, Yube K, Takaishi Y, Sakato N. Apoptosis induction in HL-60 cells and inhibition of topoisomerase II by triterpene celastrol. Biosci Biotechnol Biochem (2003) 67(9):1883–7. doi:10.1271/bbb.67.1883
104. Yu X, Ruan X, Zhang J, Zhao Q. Celastrol induces cell apoptosis and inhibits the expression of the AML1-ETO/C-KIT oncoprotein in t(8;21) leukemia. Molecules (2016) 21:5. doi:10.3390/molecules21050574
105. Rajendran P, Li F, Shanmugam MK, Kannaiyan R, Goh JN, Wong KF, et al. Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of STAT3/JAK2 signaling cascade in vitro and in vivo. Cancer Prev Res (Phila) (2012) 5(4):631–43. doi:10.1158/1940-6207.CAPR-11-0420
106. Uttarkar S, Dasse E, Coulibaly A, Steinmann S, Jakobs A, Schomburg C, et al. Targeting acute myeloid leukemia with a small molecule inhibitor of the Myb/p300 interaction. Blood (2016) 127(9):1173–82. doi:10.1182/blood-2015-09-668632
107. Fribley AM, Miller JR, Brownell AL, Garshott DM, Zeng Q, Reist TE, et al. Celastrol induces unfolded protein response-dependent cell death in head and neck cancer. Exp Cell Res (2015) 330(2):412–22. doi:10.1016/j.yexcr.2014.08.014
108. Feng L, Zhang D, Fan C, Ma C, Yang W, Meng Y, et al. ER stress-mediated apoptosis induced by celastrol in cancer cells and important role of glycogen synthase kinase-3beta in the signal network. Cell Death Dis (2013) 4:e715. doi:10.1038/cddis.2013.222
109. Boridy S, Le PU, Petrecca K, Maysinger D. Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells. Cell Death Dis (2014) 5:e1216. doi:10.1038/cddis.2014.182
110. Ji N, Li J, Wei Z, Kong F, Jin H, Chen X, et al. Effect of celastrol on growth inhibition of prostate cancer cells through the regulation of hERG channel in vitro. Biomed Res Int (2015) 2015:308475. doi:10.1155/2015/308475
111. Shao L, Zhou Z, Cai Y, Castro P, Dakhov O, Shi P, et al. Celastrol suppresses tumor cell growth through targeting an AR-ERG-NF-kappaB pathway in TMPRSS2/ERG fusion gene expressing prostate cancer. PLoS One (2013) 8(3):e58391. doi:10.1371/journal.pone.0058391
112. Sha M, Ye J, Zhang LX, Luan ZY, Chen YB. Celastrol induces apoptosis of gastric cancer cells by miR-146a inhibition of NF-kappaB activity. Cancer Cell Int (2013) 13(1):50. doi:10.1186/1475-2867-13-50
113. Kannaiyan R, Hay HS, Rajendran P, Li F, Shanmugam MK, Vali S, et al. Celastrol inhibits proliferation and induces chemosensitization through down-regulation of NF-kappaB and STAT3 regulated gene products in multiple myeloma cells. Br J Pharmacol (2011) 164(5):1506–21. doi:10.1111/j.1476-5381.2011.01449.x
114. Ni H, Zhao W, Kong X, Li H, Ouyang J. NF-kappa B modulation is involved in celastrol induced human multiple myeloma cell apoptosis. PLoS One (2014) 9(4):e95846. doi:10.1371/journal.pone.0095846
115. Tozawa K, Sagawa M, Kizaki M. Quinone methide tripterine, celastrol, induces apoptosis in human myeloma cells via NF-kappaB pathway. Int J Oncol (2011) 39(5):1117–22. doi:10.3892/ijo.2011.1161
116. Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res (2006) 66(9):4758–65. doi:10.1158/0008-5472.CAN-05-4529
117. Dai Y, Desano J, Tang W, Meng X, Meng Y, Burstein E, et al. Natural proteasome inhibitor celastrol suppresses androgen-independent prostate cancer progression by modulating apoptotic proteins and NF-kappaB. PLoS One (2010) 5(12):e14153. doi:10.1371/journal.pone.0014153
118. Wang XN, Wu Q, Yang X, Zhang LS, Wu YP, Lu C. Effects of Celastrol on growth inhibition of U937 leukemia cells through the regulation of the Notch1/NF-kappaB signaling pathway in vitro. Chin J Cancer (2010) 29(4):385–90. doi:10.5732/cjc.009.10526
119. Chiang KC, Tsui KH, Chung LC, Yeh CN, Chen WT, Chang PL, et al. Celastrol blocks interleukin-6 gene expression via downregulation of NF-kappaB in prostate carcinoma cells. PLoS One (2014) 9(3):e93151. doi:10.1371/journal.pone.0093151
120. Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol (2009) 27:693–733. doi:10.1146/annurev.immunol.021908.132641
121. Yoon MJ, Lee AR, Jeong SA, Kim YS, Kim JY, Kwon YJ, et al. Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget (2014) 5(16):6816–31. doi:10.18632/oncotarget.2256
122. Huang L, Zhang Z, Zhang S, Ren J, Zhang R, Zeng H, et al. Inhibitory action of Celastrol on hypoxia-mediated angiogenesis and metastasis via the HIF-1alpha pathway. Int J Mol Med (2011) 27(3):407–15. doi:10.3892/ijmm.2011.600
123. Ma J, Han LZ, Liang H, Mi C, Shi H, Lee JJ, et al. Celastrol inhibits the HIF-1alpha pathway by inhibition of mTOR/p70S6K/eIF4E and ERK1/2 phosphorylation in human hepatoma cells. Oncol Rep (2014) 32(1):235–42. doi:10.3892/or.2014.3211
124. Pang X, Yi Z, Zhang J, Lu B, Sung B, Qu W, et al. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res (2010) 70(5):1951–9. doi:10.1158/0008-5472.CAN-09-3201
125. Huang Y, Zhou Y, Fan Y, Zhou D. Celastrol inhibits the growth of human glioma xenografts in nude mice through suppressing VEGFR expression. Cancer Lett (2008) 264(1):101–6. doi:10.1016/j.canlet.2008.01.043
126. Zhou YX, Huang YL. Antiangiogenic effect of celastrol on the growth of human glioma: an in vitro and in vivo study. Chin Med J (Engl) (2009) 122(14):1666–73.
127. Lerman OZ, Greives MR, Singh SP, Thanik VD, Chang CC, Seiser N, et al. Low-dose radiation augments vasculogenesis signaling through HIF-1-dependent and -independent SDF-1 induction. Blood (2010) 116(18):3669–76. doi:10.1182/blood-2009-03-213629
128. Harada H, Kizaka-Kondoh S, Li G, Itasaka S, Shibuya K, Inoue M, et al. Significance of HIF-1-active cells in angiogenesis and radioresistance. Oncogene (2007) 26(54):7508–16. doi:10.1038/sj.onc.1210556
129. Dai Y, DeSano JT, Meng Y, Ji Q, Ljungman M, Lawrence TS, et al. Celastrol potentiates radiotherapy by impairment of DNA damage processing in human prostate cancer. Int J Radiat Oncol Biol Phys (2009) 74(4):1217–25. doi:10.1016/j.ijrobp.2009.03.057
130. Lee JH, Choi KJ, Seo WD, Jang SY, Kim M, Lee BW, et al. Enhancement of radiation sensitivity in lung cancer cells by celastrol is mediated by inhibition of Hsp90. Int J Mol Med (2011) 27(3):441–6. doi:10.3892/ijmm.2011.601
131. Seo HR, Seo WD, Pyun BJ, Lee BW, Jin YB, Park KH, et al. Radiosensitization by celastrol is mediated by modification of antioxidant thiol molecules. Chem Biol Interact (2011) 193(1):34–42. doi:10.1016/j.cbi.2011.04.009
132. Wang GZ, Liu YQ, Cheng X, Zhou GB. Celastrol induces proteasomal degradation of FANCD2 to sensitize lung cancer cells to DNA crosslinking agents. Cancer Sci (2015) 106(7):902–8. doi:10.1111/cas.12679
133. Lo Iacono M, Monica V, Vavala T, Gisabella M, Saviozzi S, Bracco E, et al. ATF2 contributes to cisplatin resistance in non-small cell lung cancer and celastrol induces cisplatin resensitization through inhibition of JNK/ATF2 pathway. Int J Cancer (2015) 136(11):2598–609. doi:10.1002/ijc.29302
134. Davenport A, Frezza M, Shen M, Ge Y, Huo C, Chan TH, et al. Celastrol and an EGCG pro-drug exhibit potent chemosensitizing activity in human leukemia cells. Int J Mol Med (2010) 25(3):465–70.
135. Lu Z, Jin Y, Qiu L, Lai Y, Pan J. Celastrol, a novel HSP90 inhibitor, depletes Bcr-Abl and induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation. Cancer Lett (2010) 290(2):182–91. doi:10.1016/j.canlet.2009.09.006
136. Chen M, Rose AE, Doudican N, Osman I, Orlow SJ. Celastrol synergistically enhances temozolomide cytotoxicity in melanoma cells. Mol Cancer Res (2009) 7(12):1946–53. doi:10.1158/1541-7786.MCR-09-0243
137. Fan XX, Li N, Wu JL, Zhou YL, He JX, Liu L, et al. Celastrol induces apoptosis in gefitinib-resistant non-small cell lung cancer cells via caspases-dependent pathways and Hsp90 client protein degradation. Molecules (2014) 19(3):3508–22. doi:10.3390/molecules19033508
138. Jiang HL, Jin JZ, Wu D, Xu D, Lin GF, Yu H, et al. Celastrol exerts synergistic effects with PHA-665752 and inhibits tumor growth of c-Met-deficient hepatocellular carcinoma in vivo. Mol Biol Rep (2013) 40(7):4203–9. doi:10.1007/s11033-013-2501-y
139. Zhu H, Yang W, He LJ, Ding WJ, Zheng L, Liao SD, et al. Upregulating Noxa by ER stress, celastrol exerts synergistic anti-cancer activity in combination with ABT-737 in human hepatocellular carcinoma cells. PLoS One (2012) 7(12):e52333. doi:10.1371/journal.pone.0052333
140. Sethi G, Ahn KS, Pandey MK, Aggarwal BB. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB activation. Blood (2007) 109(7):2727–35. doi:10.1182/blood-2006-10-050807
141. Kim Y, Kang H, Jang SW, Ko J. Celastrol inhibits breast cancer cell invasion via suppression of NF-kB-mediated matrix metalloproteinase-9 expression. Cell Physiol Biochem (2011) 28(2):175–84. doi:10.1159/000331729
142. Mi C, Shi H, Ma J, Han LZ, Lee JJ, Jin X. Celastrol induces the apoptosis of breast cancer cells and inhibits their invasion via downregulation of MMP-9. Oncol Rep (2014) 32(6):2527–32. doi:10.3892/or.2014.3535
143. Li H, Li Y, Liu D, Sun H, Liu J. miR-224 is critical for celastrol-induced inhibition of migration and invasion of hepatocellular carcinoma cells. Cell Physiol Biochem (2013) 32(2):448–58. doi:10.1159/000354450
144. Xu J, Wu CL, Huang J. [Effect of celastrol in inhibiting metastasis of lung cancer cells by influencing Akt signaling pathway and expressing integrins]. Zhongguo Zhong Yao Za Zhi (2015) 40(6):1129–33.
145. Xu J, Wu CL. [Anti-metastasis of celastrol on esophageal cancer cells and its mechanism]. Sheng Li Xue Bao (2015) 67(3):341–7.
146. Zhu H, Liu XW, Cai TY, Cao J, Tu CX, Lu W, et al. Celastrol acts as a potent antimetastatic agent targeting beta1 integrin and inhibiting cell-extracellular matrix adhesion, in part via the p38 mitogen-activated protein kinase pathway. J Pharmacol Exp Ther (2010) 334(2):489–99. doi:10.1124/jpet.110.165654
147. Yadav VR, Sung B, Prasad S, Kannappan R, Cho SG, Liu M, et al. Celastrol suppresses invasion of colon and pancreatic cancer cells through the downregulation of expression of CXCR4 chemokine receptor. J Mol Med (Berl) (2010) 88(12):1243–53. doi:10.1007/s00109-010-0669-3
148. Idris AI, Libouban H, Nyangoga H, Landao-Bassonga E, Chappard D, Ralston SH. Pharmacologic inhibitors of IkappaB kinase suppress growth and migration of mammary carcinosarcoma cells in vitro and prevent osteolytic bone metastasis in vivo. Mol Cancer Ther (2009) 8(8):2339–47. doi:10.1158/1535-7163.MCT-09-0133
149. Cleren C, Calingasan NY, Chen J, Beal MF. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J Neurochem (2005) 94(4):995–1004. doi:10.1111/j.1471-4159.2005.03253.x
150. Wang J, Gines S, MacDonald ME, Gusella JF. Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci (2005) 6:1. doi:10.1186/1471-2202-6-1
151. Zhang YQ, Sarge KD. Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J Mol Med (Berl) (2007) 85(12):1421–8. doi:10.1007/s00109-007-0251-9
152. Paris D, Ganey NJ, Laporte V, Patel NS, Beaulieu-Abdelahad D, Bachmeier C, et al. Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation (2010) 7:17. doi:10.1186/1742-2094-7-17
153. Kiaei M, Kipiani K, Petri S, Chen J, Calingasan NY, Beal MF. Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neurodegener Dis (2005) 2(5):246–54. doi:10.1159/000090364
154. Wang Y, Cao L, Xu LM, Cao FF, Peng B, Zhang X, et al. Celastrol ameliorates EAE induction by suppressing pathogenic T cell responses in the peripheral and central nervous systems. J Neuroimmune Pharmacol (2015) 10(3):506–16. doi:10.1007/s11481-015-9598-9
155. Abdin AA, Hasby EA. Modulatory effect of celastrol on Th1/Th2 cytokines profile, TLR2 and CD3+ T-lymphocyte expression in a relapsing-remitting model of multiple sclerosis in rats. Eur J Pharmacol (2014) 742:102–12. doi:10.1016/j.ejphar.2014.09.001
156. Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer’s and Parkinson’s diseases. Nat Cell Biol (2004) 6(11):1054–61. doi:10.1038/ncb1104-1054
157. Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med (2004) 10(10):1055–63. doi:10.1038/nm1113
158. Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci (2005) 6(1):11–22. doi:10.1038/nrn1587
159. Salminen A, Lehtonen M, Paimela T, Kaarniranta K. Celastrol: molecular targets of thunder god vine. Biochem Biophys Res Commun (2010) 394(3):439–42. doi:10.1016/j.bbrc.2010.03.050
160. Chow AM, Brown IR. Induction of heat shock proteins in differentiated human and rodent neurons by celastrol. Cell Stress Chaperones (2007) 12(3):237–44. doi:10.1379/CSC-269.1
161. Ran R, Lu A, Zhang L, Tang Y, Zhu H, Xu H, et al. Hsp70 promotes TNF-mediated apoptosis by binding IKK gamma and impairing NF-kappa B survival signaling. Genes Dev (2004) 18(12):1466–81. doi:10.1101/gad.1188204
162. Weiss YG, Bromberg Z, Raj N, Raphael J, Goloubinoff P, Ben-Neriah Y, et al. Enhanced heat shock protein 70 expression alters proteasomal degradation of IkappaB kinase in experimental acute respiratory distress syndrome. Crit Care Med (2007) 35(9):2128–38. doi:10.1097/01.CCM.0000278915.78030.74
163. Chen H, Wu Y, Zhang Y, Jin L, Luo L, Xue B, et al. Hsp70 inhibits lipopolysaccharide-induced NF-kappaB activation by interacting with TRAF6 and inhibiting its ubiquitination. FEBS Lett (2006) 580(13):3145–52. doi:10.1016/j.febslet.2006.04.066
164. Choi BS, Kim H, Lee HJ, Sapkota K, Park SE, Kim S, et al. Celastrol from ‘Thunder God Vine’ protects SH-SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson’s disease. Neurochem Res (2014) 39(1):84–96. doi:10.1007/s11064-013-1193-y
165. Chen S, Gu C, Xu C, Zhang J, Xu Y, Ren Q, et al. Celastrol prevents cadmium-induced neuronal cell death via targeting JNK and PTEN-Akt/mTOR network. J Neurochem (2014) 128(2):256–66. doi:10.1111/jnc.12474
166. Konieczny J, Jantas D, Lenda T, Domin H, Czarnecka A, Kuter K, et al. Lack of neuroprotective effect of celastrol under conditions of proteasome inhibition by lactacystin in in vitro and in vivo studies: implications for Parkinson’s disease. Neurotox Res (2014) 26(3):255–73. doi:10.1007/s12640-014-9477-9
167. Jantas D, Roman A, Kusmierczyk J, Lorenc-Koci E, Konieczny J, Lenda T, et al. The extent of neurodegeneration and neuroprotection in two chemical in vitro models related to Parkinson’s disease is critically dependent on cell culture conditions. Neurotox Res (2013) 24(1):41–54. doi:10.1007/s12640-012-9374-z
168. Chow AM, Tang DW, Hanif A, Brown IR. Induction of heat shock proteins in cerebral cortical cultures by celastrol. Cell Stress Chaperones (2013) 18(2):155–60. doi:10.1007/s12192-012-0364-0
169. Chow AM, Tang DW, Hanif A, Brown IR. Localization of heat shock proteins in cerebral cortical cultures following induction by celastrol. Cell Stress Chaperones (2014) 19(6):845–51. doi:10.1007/s12192-014-0508-5
170. Zhao Y, Zhao H, Lobo N, Guo X, Gentleman SM, Ma D. Celastrol enhances cell viability and inhibits amyloid-beta production induced by lipopolysaccharide in vitro. J Alzheimers Dis (2014) 41(3):835–44. doi:10.3233/JAD-131799
171. Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res (1992) 587(2):250–6. doi:10.1016/0006-8993(92)91004-X
172. Kim YS, Joh TH. Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s disease. Exp Mol Med (2006) 38(4):333–47. doi:10.1038/emm.2006.40
173. Jung HW, Chung YS, Kim YS, Park YK. Celastrol inhibits production of nitric oxide and proinflammatory cytokines through MAPK signal transduction and NF-kappaB in LPS-stimulated BV-2 microglial cells. Exp Mol Med (2007) 39(6):715–21. doi:10.1038/emm.2007.78
174. Hafizi Abu Bakar M, Kian Kai C, Wan Hassan WN, Sarmidi MR, Yaakob H, Zaman Huri H. Mitochondrial dysfunction as a central event for mechanisms underlying insulin resistance: the roles of long chain fatty acids. Diabetes Metab Res Rev (2015) 31(5):453–75. doi:10.1002/dmrr.2601
175. Abu Bakar MH, Cheng KK, Sarmidi MR, Yaakob H, Huri HZ. Celastrol protects against antimycin A-induced insulin resistance in human skeletal muscle cells. Molecules (2015) 20(5):8242–69. doi:10.3390/molecules20058242
176. Kim JE, Lee MH, Nam DH, Song HK, Kang YS, Lee JE, et al. Celastrol, an NF-kappaB inhibitor, improves insulin resistance and attenuates renal injury in db/db mice. PLoS One (2013) 8(4):e62068. doi:10.1371/journal.pone.0062068
177. Han LP, Li CJ, Sun B, Xie Y, Guan Y, Ma ZJ, et al. Protective effects of celastrol on diabetic liver injury via TLR4/MyD88/NF-kappaB signaling pathway in type 2 diabetic rats. J Diabetes Res (2016) 2016:2641248. doi:10.1155/2016/2641248
178. Guan Y, Cui ZJ, Sun B, Han LP, Li CJ, Chen LM. Celastrol attenuates oxidative stress in the skeletal muscle of diabetic rats by regulating the AMPK-PGC1alpha-SIRT3 signaling pathway. Int J Mol Med (2016) 37(5):1229–38. doi:10.3892/ijmm.2016.2549
179. Grant CW, Moran-Paul CM, Duclos SK, Guberski DL, Arreaza-Rubin G, Spain LM. Testing agents for prevention or reversal of type 1 diabetes in rodents. PLoS One (2013) 8(8):e72989. doi:10.1371/journal.pone.0072989
180. Zhang J, Li CY, Xu MJ, Wu T, Chu JH, Liu SJ, et al. Oral bioavailability and gender-related pharmacokinetics of celastrol following administration of pure celastrol and its related tablets in rats. J Ethnopharmacol (2012) 144(1):195–200. doi:10.1016/j.jep.2012.09.005
181. Ju SM, Youn GS, Cho YS, Choi SY, Park J. Celastrol ameliorates cytokine toxicity and pro-inflammatory immune responses by suppressing NF-kappaB activation in RINm5F beta cells. BMB Rep (2015) 48(3):172–7. doi:10.5483/BMBRep.2015.48.3.147
182. Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, Ozcan U. Treatment of obesity with celastrol. Cell (2015) 161(5):999–1011. doi:10.1016/j.cell.2015.05.011
183. Aragones G, Ardid-Ruiz A, Ibars M, Suarez M, Blade C. Modulation of leptin resistance by food compounds. Mol Nutr Food Res (2016) 60(8):1789–803. doi:10.1002/mnfr.201500964
184. Ma X, Xu L, Alberobello AT, Gavrilova O, Bagattin A, Skarulis M, et al. Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1-PGC1alpha transcriptional axis. Cell Metab (2015) 22(4):695–708. doi:10.1016/j.cmet.2015.08.005
185. Zhu F, Li C, Jin XP, Weng SX, Fan LL, Zheng Z, et al. Celastrol may have an anti-atherosclerosis effect in a rabbit experimental carotid atherosclerosis model. Int J Clin Exp Med (2014) 7(7):1684–91.
186. Gu L, Bai W, Li S, Zhang Y, Han Y, Gu Y, et al. Celastrol prevents atherosclerosis via inhibiting LOX-1 and oxidative stress. PLoS One (2013) 8(6):e65477. doi:10.1371/journal.pone.0065477
187. Lu C, Yu X, Zuo K, Zhang X, Cao C, Xu J, et al. Tripterine treatment improves endothelial progenitor cell function via integrin-linked kinase. Cell Physiol Biochem (2015) 37(3):1089–103. doi:10.1159/000430234
188. Lu C, Zhang X, Zhang D, Pei E, Xu J, Tang T, et al. Short time tripterine treatment enhances endothelial progenitor cell function via heat shock protein 32. J Cell Physiol (2015) 230(5):1139–47. doi:10.1002/jcp.24849
189. Selimoglu E. Aminoglycoside-induced ototoxicity. Curr Pharm Des (2007) 13(1):119–26. doi:10.2174/138161207779313731
191. Francis SP, Kramarenko II, Brandon CS, Lee FS, Baker TG, Cunningham LL. Celastrol inhibits aminoglycoside-induced ototoxicity via heat shock protein 32. Cell Death Dis (2011) 2:e195. doi:10.1038/cddis.2011.76
192. Kim HJ, So HS, Lee JH, Lee JH, Park C, Park SY, et al. Heme oxygenase-1 attenuates the cisplatin-induced apoptosis of auditory cells via down-regulation of reactive oxygen species generation. Free Radic Biol Med (2006) 40(10):1810–9. doi:10.1016/j.freeradbiomed.2006.01.018
193. Qi X, Qin J, Ma N, Chou X, Wu Z. Solid self-microemulsifying dispersible tablets of celastrol: formulation development, characterization and bioavailability evaluation. Int J Pharm (2014) 472(1–2):40–7. doi:10.1016/j.ijpharm.2014.06.019
194. Li Z, Yao L, Li J, Zhang W, Wu X, Liu Y, et al. Celastrol nanoparticles inhibit corneal neovascularization induced by suturing in rats. Int J Nanomedicine (2012) 7:1163–73. doi:10.2147/IJN.S27860
195. Rita Cascão TC, Goncalves J, Moita L, Fonseca J. Efficacy and Safety of Oral Administration of Pure Celastrol in AIA Rats. Madrid: EULAR (2017). Abstract Book: Annals of Rheumatic Diseases ARD.
196. Bai JP, Shi YL, Fang X, Shi QX. Effects of demethylzeylasteral and celastrol on spermatogenic cell Ca2+ channels and progesterone-induced sperm acrosome reaction. Eur J Pharmacol (2003) 464(1):9–15. doi:10.1016/S0014-2999(03)01351-7
197. Sun H, Liu X, Xiong Q, Shikano S, Li M. Chronic inhibition of cardiac Kir2.1 and HERG potassium channels by celastrol with dual effects on both ion conductivity and protein trafficking. J Biol Chem (2006) 281(9):5877–84. doi:10.1074/jbc.M600072200
198. Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry (2001) 25(7):1341–57. doi:10.1016/S0278-5846(01)00192-0
199. Yu X, Tao W, Jiang F, Li C, Lin J, Liu C. Celastrol attenuates hypertension-induced inflammation and oxidative stress in vascular smooth muscle cells via induction of heme oxygenase-1. Am J Hypertens (2010) 23(8):895–903. doi:10.1038/ajh.2010.75
200. Aqil F, Kausar H, Agrawal AK, Jeyabalan J, Kyakulaga AH, Munagala R, et al. Exosomal formulation enhances therapeutic response of celastrol against lung cancer. Exp Mol Pathol (2016) 101(1):12–21. doi:10.1016/j.yexmp.2016.05.013
201. Zhang X, Zhang T, Zhou X, Liu H, Sun H, Ma Z, et al. Enhancement of oral bioavailability of tripterine through lipid nanospheres: preparation, characterization, and absorption evaluation. J Pharm Sci (2014) 103(6):1711–9. doi:10.1002/jps.23967
202. Sanna V, Chamcheu JC, Pala N, Mukhtar H, Sechi M, Siddiqui IA. Nanoencapsulation of natural triterpenoid celastrol for prostate cancer treatment. Int J Nanomedicine (2015) 10:6835–46. doi:10.2147/IJN.S93752
203. Boridy S, Soliman GM, Maysinger D. Modulation of inflammatory signaling and cytokine release from microglia by celastrol incorporated into dendrimer nanocarriers. Nanomedicine (Lond) (2012) 7(8):1149–65. doi:10.2217/nnm.12.16
204. Wolfram J, Suri K, Huang Y, Molinaro R, Borsoi C, Scott B, et al. Evaluation of anticancer activity of celastrol liposomes in prostate cancer cells. J Microencapsul (2014) 31(5):501–7. doi:10.3109/02652048.2013.879932
205. Huang Y, Zhou D, Hang T, Wu Z, Liu J, Xu Q, et al. Preparation, characterization, and assessment of the antiglioma effects of liposomal celastrol. Anticancer Drugs (2012) 23(5):515–24. doi:10.1097/CAD.0b013e3283514b68
206. Song J, Shi F, Zhang Z, Zhu F, Xue J, Tan X, et al. Formulation and evaluation of celastrol-loaded liposomes. Molecules (2011) 16(9):7880–92. doi:10.3390/molecules16097880
207. Peng X, Wang J, Song H, Cui D, Li L, Li J, et al. Optimized preparation of celastrol-loaded polymeric nanomicelles using rotatable central composite design and response surface methodology. J Biomed Nanotechnol (2012) 8(3):491–9. doi:10.1166/jbn.2012.1398
208. Li Z, Wu X, Li J, Yao L, Sun L, Shi Y, et al. Antitumor activity of celastrol nanoparticles in a xenograft retinoblastoma tumor model. Int J Nanomedicine (2012) 7:2389–98. doi:10.2147/IJN.S29945
209. Yuan L, Liu C, Chen Y, Zhang Z, Zhou L, Qu D. Antitumor activity of tripterine via cell-penetrating peptide-coated nanostructured lipid carriers in a prostate cancer model. Int J Nanomedicine (2013) 8:4339–50. doi:10.2147/IJN.S51621
210. Chen Y, Zhou L, Yuan L, Zhang ZH, Liu X, Wu Q. Formulation, characterization, and evaluation of in vitro skin permeation and in vivo pharmacodynamics of surface-charged tripterine-loaded nanostructured lipid carriers. Int J Nanomedicine (2012) 7:3023–32. doi:10.2147/IJN.S32476
211. Zhou L, Chen Y, Zhang Z, He J, Du M, Wu Q. Preparation of tripterine nanostructured lipid carriers and their absorption in rat intestine. Pharmazie (2012) 67(4):304–10.
212. Niemela E, Desai D, Nkizinkiko Y, Eriksson JE, Rosenholm JM. Sugar-decorated mesoporous silica nanoparticles as delivery vehicles for the poorly soluble drug celastrol enables targeted induction of apoptosis in cancer cells. Eur J Pharm Biopharm (2015) 96:11–21. doi:10.1016/j.ejpb.2015.07.009
213. Shanmugam MK, Nguyen AH, Kumar AP, Tan BK, Sethi G. Targeted inhibition of tumor proliferation, survival, and metastasis by pentacyclic triterpenoids: potential role in prevention and therapy of cancer. Cancer Lett (2012) 320(2):158–70. doi:10.1016/j.canlet.2012.02.037
214. Wang S, Wu X, Tan M, Gong J, Tan W, Bian B, et al. Fighting fire with fire: poisonous Chinese herbal medicine for cancer therapy. J Ethnopharmacol (2012) 140(1):33–45. doi:10.1016/j.jep.2011.12.041
215. Abbas S, Bhoumik A, Dahl R, Vasile S, Krajewski S, Cosford ND, et al. Preclinical studies of celastrol and acetyl isogambogic acid in melanoma. Clin Cancer Res (2007) 13(22 Pt 1):6769–78. doi:10.1158/1078-0432.CCR-07-1536
216. Sun H, Xu L, Yu P, Jiang J, Zhang G, Wang Y. Synthesis and preliminary evaluation of neuroprotection of celastrol analogues in PC12 cells. Bioorg Med Chem Lett (2010) 20(13):3844–7. doi:10.1016/j.bmcl.2010.05.066
217. Wei W, Wu S, Wang X, Sun CK, Yang X, Yan X, et al. Novel celastrol derivatives inhibit the growth of hepatocellular carcinoma patient-derived xenografts. Oncotarget (2014) 5(14):5819–31. doi:10.18632/oncotarget.2171
218. Wu J, Zhou Y, Wang L, Zuo J, Zhao W. Terpenoids from root bark of Celastrus orbiculatus. Phytochemistry (2012) 75:159–68. doi:10.1016/j.phytochem.2011.11.023
219. Tang K, Huang Q, Zeng J, Wu G, Huang J, Pan J, et al. Design, synthesis and biological evaluation of C6-modified celastrol derivatives as potential antitumor agents. Molecules (2014) 19(7):10177–88. doi:10.3390/molecules190710177
Keywords: celastrol, inflammation, cancer, neurodegenerative diseases, toxicity, treatment, clinical trials
Citation: Cascão R, Fonseca JE and Moita LF (2017) Celastrol: A Spectrum of Treatment Opportunities in Chronic Diseases. Front. Med. 4:69. doi: 10.3389/fmed.2017.00069
Received: 13 March 2017; Accepted: 19 May 2017;
Published: 15 June 2017
Edited by:
Peter S. Steyger, Oregon Health & Science University, United StatesReviewed by:
Imtiaz Ahmad Siddiqui, University of Wisconsin-Madison, United StatesCarole Guillonneau, Centre national de la recherche scientifique (CNRS), France
Copyright: © 2017 Cascão, Fonseca and Moita. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: João E. Fonseca, amVjZm9uc2VjYUBnbWFpbC5jb20=