 Andrea Angeletti
Andrea Angeletti Joselyn Reyes-Bahamonde1
Joselyn Reyes-Bahamonde1 Paolo Cravedi
Paolo Cravedi Kirk N. Campbell
Kirk N. Campbell- 1Department of Medicine, Division of Nephrology, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 2Department of Experimental Diagnostic and Specialty Medicine (DIMES), Nephrology, Dialysis and Renal Transplant Unit, St Orsola Hospital, University of Bologna, Bologna, Italy
The complement system is part of the innate immune response that plays important roles in protecting the host from foreign pathogens. The complement components and relative fragment deposition have long been recognized to be strongly involved also in the pathogenesis of autoantibody-related kidney glomerulopathies, leading to direct glomerular injury and recruitment of infiltrating inflammation pathways. More recently, unregulated complement activation has been shown to be associated with progression of non-antibody-mediated kidney diseases, including focal segmental glomerulosclerosis, C3 glomerular disease, thrombotic microangiopathies, or general fibrosis generation in progressive chronic kidney diseases. Some of the specific mechanisms associated with complement activation in these diseases were recently clarified, showing a dominant role of alternative activation pathway. Over the last decade, a growing number of anticomplement agents have been developed, and some of them are being approved for clinical use or already in use. Therefore, anticomplement therapies represent a realistic choice of therapeutic approaches for complement-related diseases. Herein, we review the complement system activation, regulatory mechanisms, their involvement in non-antibody-mediated glomerular diseases, and the recent advances in complement-targeting agents as potential therapeutic strategies.
Introduction
The complement cascade consists of 30 molecules that are activated as a proteolytic cascade regulated by three initiating pathways that function to protect the body from invading microorganisms (1, 2). Abnormal complement activation is also involved in many autoimmune inflammatory diseases. In particular, the pathogenesis of autoantibody-initiated kidney glomerulopathies suggests a role for complement-derived effector mechanisms leading to recruitment of infiltrating lymphocytes (3).
More recently, evidence has implicated a role for complement also in the pathogenesis of non-antibody-mediated kidney diseases that will be the topic of this review article. We will also discuss recent advances in complement-targeting strategies as potential therapeutic strategies for kidney disease.
The Complement Cascade
Activation and Amplification
The complement cascade is activated by the lectin pathway (LP), the classical pathway (CP), and the alternative pathway (AP) (Figure 1). These three pathways converge on C3 convertases, enzymatic multimeric protein complexes (2). C3 cleavage produces C3a and C3b, the latter triggering formation of C5 convertase. C5 cleavage results in formation of the membrane attack complex (MAC, C5b-9). Along with MAC, soluble and surface-bound split products, including C3a, C3b, iC3b, C3dg, and C5a, play a role in the inflammatory response (4).
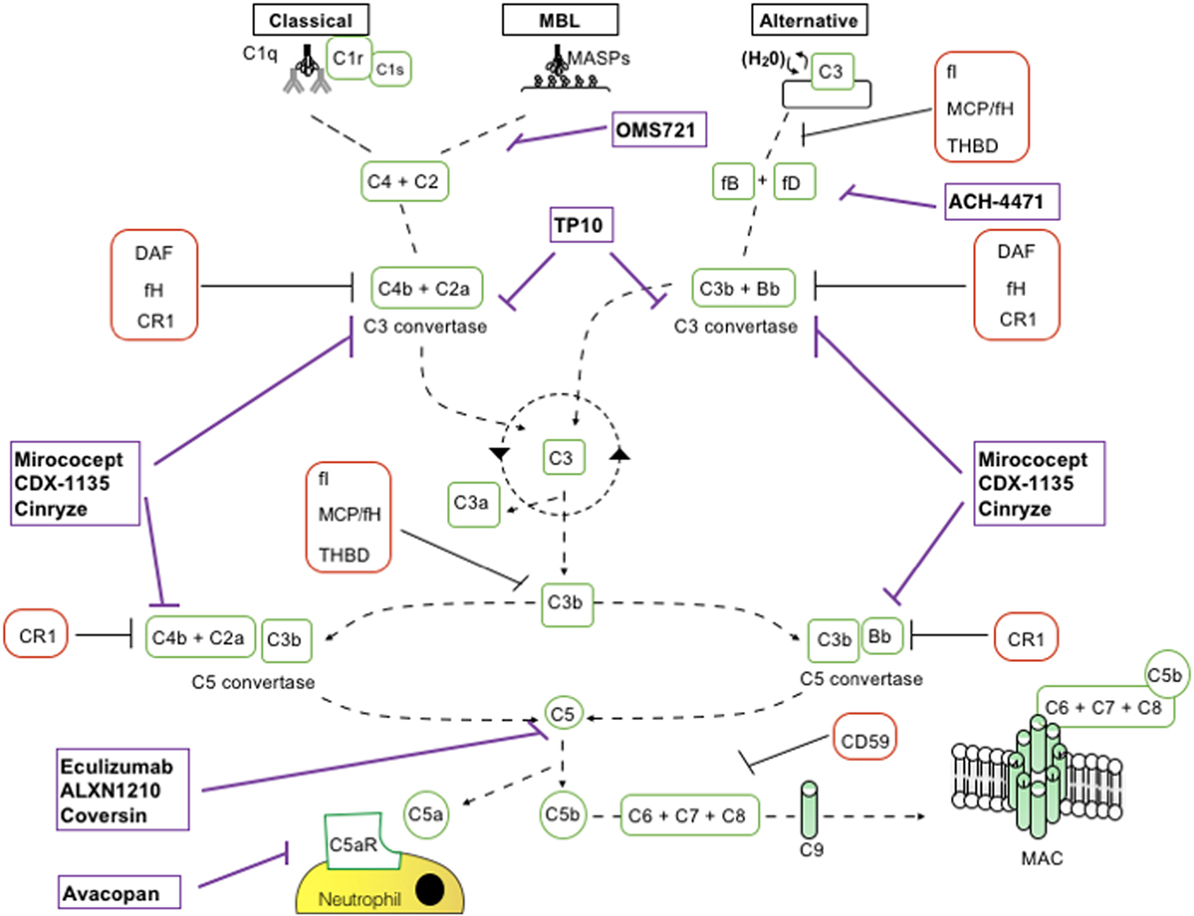
Figure 1. Schematic representation of complement activation pathways and complement-targeting agents. C1q,r,s cross-linking of antibodies activates the classical pathway. Mannose-associated serine proteases (MASPs) bind to mannose motifs expressed on bacteria to activate complement via the mannose-binding lectin (MBL) pathway. Subsequent cleavage and assembly of C2 and C4 proteins form the C3 convertase. The spontaneous hydrolysis of C3 on cell surfaces leads to the alternative pathway (AP): C3 convertase dependent on factor B (fB), factor D (fD), and properdin. The resultant C3 convertases can continuously cleave C3; however, after they are generated, the AP C3 convertase dominates in amplifying production of C3b (green looping arrow). C3 convertases cleave C3 into C3a and C3b. C3b permits the formation of C5 convertase. C3b has further roles in opsonization and immune complex clearance. C5b, in conjunction with C6–C9, allows formation of the membrane attack complex (MAC) and subsequent pathogen lysis. Decay accelerating factor (DAF) (CD55) and MCP (CD46) are cell surface-expressed complement regulators that accelerate the decay of all surface-assembled C3 convertases, thereby limiting amplification of the downstream cascade. MCP and factor H (fH) also have cofactor activity: in conjunction with soluble fI, they irreversibly cleave C3b to iC3b, thereby preventing reformation of the C3 convertase. CD59 inhibits formation of the MAC.
Regulation
It is essential to self-cell viability that complement activation is strictly controlled (4). Several molecules with discrete and synergistic roles regulate C3 convertase activity. Decay accelerating factor (DAF) encoded by the CD55 gene is a 70 kDa cell-surface regulator of the complement system. DAF inhibits C3 and C5 convertases thereby preventing downstream complement activation (5–8). Membrane cofactor protein encoded by CD46 is another inhibitory complement receptor with cofactor activity for C3b, C4b, and serum factor I inactivation (9). Crry is the murine homolog of human CD46 that also exhibits decay accelerating activity (10). Factor H (fH), a 155 kDa soluble glycoprotein exhibits both decay accelerating and cofactor activity to regulate the AP. Other complement cascade regulators include CD59 (protectin), the surface-expressed CR1 (11), and C1 inhibitor, a protease inhibitor of the serpin superfamily that inhibits the classical and LPs by binding and inactivating C1r, C1s, MASP-1, and MASP-2.
Complement Effector Mechanisms
Deposition of the MAC in the cell membranes of target cells results in the formation of transmembrane channels that promote cell lysis and death. In eukaryotic nucleated cells MAC insertion but can induce cellular activation (12) and/or promote tissue injury (13) but does not usually result in lysis.
Several complement cleavage products have distinct effector functions. For example, C3a and C5a promote vasodilation and chemokine release through their transmembrane-spanning G protein-coupled receptors. In addition, they regulate neutrophil and macrophage chemoattraction and contribute to T-cell and antigen-presenting cell (APC) activation, expansion, and survival (14–17).
Complement and Adaptive Immunity
The complement system’s role in innate immunity has been well established since the 1960s. Recently, complement has been found to act as a link between innate and adaptive immunity. Complement depletion decreases antibody production (18) through antigen-bound C3dg binding to CR2 (CD21). This facilitates antigen presentation to B cells and lowers the threshold for B-cell activation (19).
There is also evidence that locally produced complement acts as a regulator of T-cell immunity. During T cell and APC interaction, there is upregulation and secretion of C3, fB, and fD, C5 production, and upregulation of surface expression of C3aR and C5aR (20, 21). Locally generated C3a and C5a bind to their respective receptors to act as autocrine and paracrine stimulators of T cells and the APCs (20, 21). Subsequent signaling through these GPCRs in T cells activates phosphoinositide-3-kinase-γ and induces phosphorylation of phosphokinase B (AKT) (21, 22), upregulating the pro-survival protein Bcl-2 and downregulating the proapoptotic molecule Fas. Together, these complement-dependent mechanisms enhance T-cell proliferation and diminish T-cell apoptotic injury (22).
Regulatory T cells (Tregs) are essential for maintenance of self tolerance (23) with recent evidence showing that complement also regulates Treg induction, function, and stability (16). Peripheral, murine, natural regulatory T cells (nTregs) express C3aR and C5aR and signaling through these receptors inhibits Treg function (15). Genetic and pharmacologic blockade of C3aR/C5aR signal transduction in nTreg cells augments their in vitro and in vivo suppressive activity. Genetic deficiency or pharmacologic blockade of C3aR/C5aR signaling augments murine-induced regulatory T cell (iTreg) generation, stabilizes Foxp3 expression, and resists iTreg conversion to IFN-γ/TNF-α-producing effector T cells (16, 24). Pharmacologic antagonists to human C3aR and C5aR also augment in vitro generation and stability of human iTreg from naïve precursors (16, 24). These findings are an extension of previously published data that co-engagement of the T-cell receptor and the complement regulator CD46 promote regulatory IL-10 production (25). In summary, there is a crucial role for complement in modulating the balance between pathogenic and protective adaptive T-cell responses.
Source of Complement Components in Kidney Diseases
Complement deposition in the kidney in antibody-mediated glomerulonephritis was traditionally considered to derive from the circulating pool (mainly produced by the liver) (26). Subsequent studies have shown gene expression of complement in human kidneys (27) and the ability of resident cells (glomerular, tubular, epithelial, and mesangial cells) to synthesize several proteins of the complement cascade, such as C2, C3, C4, factor B (fB), and factor H (fH) (28–30). Sacks et al. (31) demonstrated in vivo the renal production of C3. Song et al. (32) described a higher expression of C2, C3, and C4 and C1q in the tubular cells of normal human kidneys than in glomerular cells. While the dominant role of renal versus systemic complement has been shown to mediate the ischemia–reperfusion injury, the role of circulating versus local complement in other physiological and pathological conditions has not yet been fully elucidated. Together, data support the conclusion that different inflammatory stimulation upregulate complement production in kidney tissue (11); the local effect of inflammatory stimuli on local complement regulators, as DAF, is still to clarify.
Complement in Glomerular Diseases
Focal Segmental Glomerulosclerosis (FSGS)
Focal segmental glomerulosclerosis is characterized by focal and segmental obliteration of glomerular capillary tufts with increased matrix (33). The incidence of FSGS has increased over the past decades and it is one of the leading causes of nephrotic syndrome in adults (34). Spontaneous remission is rare (<5%) and presence of persistent nephrotic syndrome portends a poor prognosis with 50% of patients progressing to end-stage renal disease (ESRD) 6–8 years after initial diagnosis (35).
While there are FSGS forms secondary to obesity, use of different drugs including lithium and anabolic steroid (36), primary cases historically have been attributed to a T cell disorder (possibly an imbalance between conventional and Tregs) resulting in the secretion of circulating factor(s) that increase glomerular permeability to plasma proteins (37). The identity of these permeability factor(s) is, however, still controversial (38). The origin of cells secreting the circulating factor(s) is also unclear, with new data pointing at neutrophils, monocyte/macrophages (39–41), and bone marrow immature myeloid cells (42). Efficacy of B cell depleting therapies in FSGS also implicates a role of B cell in disease pathogenesis (37).
Abnormal complement activation has also been implicated in the pathogenesis of the disease. Lenderink et al. (43) showed that fB-deficient mice have lower proteinuria than WT controls in the adriamycin-induced FSGS model, suggesting that activation of AP has a pathogenic role. Similarly, Turnberg et al. (44) reported lower proteinuria and less glomerular and tubulointerstitial injury, in fD-deficient mice compared to WT.
In a model of FSGS due to protein overload (45), fH-deficient mice display higher C3b glomerular deposition and more severe lesions than WT controls. In vitro experiments indicate that C3a activates podocytes to release glial cell line-derived neurotrophic factor that mediates the recruitment of parietal epithelial cell (PEC) and formation of sclerotic lesions. Signs of PEC activation were observed in renal biopsies from 10 patients with FSGS (45) (Table 1).
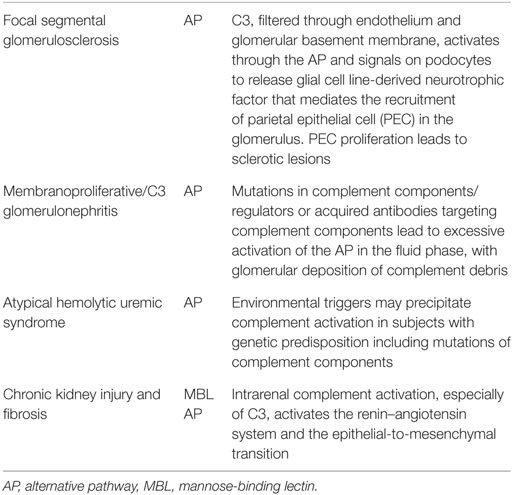
Table 1. Complement involvement in non-antibody renal disease.
Intriguingly, locally produced complement is implicated in abnormal T cell activation observed in the anti-podocyte model of FSGS. DAF-deficient mice develop more severe histological and ultrastructural features of FSGS than WT or CD59-deficient mice and severity of FSGS is reduced by depleting CD4+ T cells from DAF-deficient mice (46). Furthermore, WT kidneys transplanted into DAF-deficient recipients developed FSGS, suggesting that renal DAF is not implicated in mediating disease in this model (46). Altogether, these data indicate a major role for systemic and local complement dysregulation in murine models of FSGS.
In humans with FSGS, glomerular deposition of IgM and C3 deposits is frequently detected (47). In the urine and plasma of patients with FSGS, activated fragments of the complement cascade, such as C3a, C3b, Ba, Bb, C4a, and sC5b-9, are increased compared to patients with other renal diseases such as antineutrophilic cytoplasmic antibody (ANCA) vasculitis and lupus nephritis or in healthy controls (48). These findings could reflect complement activation within the glomeruli, mesangium, and areas of sclerosis, while activated complement fragments in urine could be due to activation of filtered proteins within the tubular lumen or urinary collecting system. The presence of high levels sC5b-9 is consistent with the hypothesis that there is an activation of the AP in FSGS (48). Importantly, fBa levels in the urine had an inverse relation with estimated glomerular filtration rate, further supporting a pathogenic role in disease progression (48). However, more studies are needed to better dissect the involvement of the AP of complement in the pathogenesis of FSGS as well as the implication of other complement pathway in the pathogenesis of the disease. Results of these studies could provide the background for clinical trials testing the hypothesis that complement blockade improves outcomes of FSGS patients.
Membranoproliferative/C3 Glomerulonephritis
C3 glomerular disease (C3GN) refers to a group of recently identified rare renal disorders characterized by the presence of C3 in the absence or in the presence of limited deposition of immunoglobulins in the renal tissue (49, 50). Clinical manifestations include proteinuria, hematuria, and approximately 50% of affected patients progress to kidney failure within 10 years from diagnosis (51). C3GN is subdivided into dense deposit disease (DDD) and C3 glomerulonephritis. DDD is characterized by mesangial and intramembranous highly electron-dense deposits, the composition of which has not been still completely clarify.
C3 glomerular disease shows isolated and less-dense deposits in the mesangial, subepithelial, subendothelial, and intramembranous areas of the glomeruli (52). Glomerular deposits of C3 alone, without immunoglobulin, are the hallmark of AP dysregulation via inherited or acquired defects. Reports from Servais et al. (53) showed frequently acquired or hereditary abnormalities mutation of the AP and MCP in about 24% of patients which included cases of C3GN, DDD, and membranoproliferative glomerulonephropathy type I (100% if only C3GN was included). This defect can be due to mutations in complement components or complement regulators (such as C3, fB, fH, and fI) or due to acquired autoantibodies that either stabilize the C3 convertase of the AP (e.g., C3 nephritic factors) or target the inhibitory complement factors (e.g., fH autoantibodies). In contrast to other diseases with AP involvement, these abnormalities promote excessive activation of the alternative complement pathway in the fluid phase, with deposition of complement debris, including breakdown products of C3b and components of the terminal complement cascade, in the glomerular capillary wall (54–56) (Table 1).
The optimal treatment for C3 glomerulopathy remains undefined. A recent KDIGO (Kidney Disease: Improving Global Outcomes) controversies conference recommended that all patients receive optimal blood pressure control and that patients with moderate disease (defined as urine protein of more than 500 mg/24 h despite supportive therapy or moderate inflammation on renal biopsy or recent rise in creatinine) receive prednisone or mycophenolate mofetil (57). Due to its pathogenesis, targeted therapies aimed at specific components of the alternative complement pathway may also be effective (58).
Complement in Thrombotic Microangiopathies
Thrombotic microangiopathy (TMA) is characterized by the presence of thrombi in small blood vessels, thrombocytopenia, non-immune hemolytic anemia, and peripheral blood schistocytes. The two main target organs of TMA are the kidney and the brain (59). The common denominator in each TMA forms is activation and dysfunction of the endothelium (60). Multiple etiologies can lead to development of TMA, including infection with Shiga-like toxin-producing bacteria causing typical HUS (STEC-HUS), genetically determined dysregulation of the AP, which predisposes to atypical hemolytic uremic syndrome (aHUS), or A Disintegrin and Metalloproteinase with ThromboSpondin motif repeats 13 (ADAMTS13) deficiency resulting in thrombotic thrombocytopenic purpura (TTP). TMA may also develop as a complication of various coexisting diseases or their treatments, such as malignant hypertension, systemic autoimmune disease such as systemic lupus erythematosus, cancer, drug treatment, hematopoietic stem cell transplantation, solid organ transplantation, open heart surgery, glomerulopathies or infections (61).
In STEC-HUS, bacterial exotoxins induce profound alterations in endothelial cells, upregulating expression of chemokines, chemokine receptors, and cell adhesion molecules that favor leukocyte recruitment and promote platelets activity (62). From the early 1970s, there have been reports of low levels of C3 in patients with STEC-HUS (63–67). Since 1980, increased levels of breakdown products of the components of the AP including C3 convertase, C3, fB, and MAC have been reported in the plasma of children with STEC-HUS (68–70). These findings suggest the activation of the AP of complement cascade, through to C5b–9 assembly, in STEC-HUS. Low levels of C4 have also occasionally been observed indicating activation of the CP and/or LP leading to C4 consumption (70). Evidence shows that exotoxin might directly contribute to AP activation (71), through endothelial complement deposition and loss of thromboresistance depended on exotoxin-induced upregulation of the membrane adhesion molecule P-selectin, which has been shown to bind C3b with high affinity (72).
HUS is defined as atypical when the disease occurs in the absence of a STEC infection, according to established criteria (57). The onset of aHUS ranges from the neonatal period to adulthood. Genetic aHUS accounts for an estimated 60%of all aHUS (73). It is likely that mutation of C3, CD46, CFB, CFH, CFI, and THBD confers a predisposition to developing aHUS, rather than directly causing the disease. Conditions that trigger complement activation may precipitate an acute event in those with the predisposing genetic background (74, 75) (Table 1). There are, however, non-complement inherited abnormalities such as mutations in DGKE, which can result in an aHUS phenotype. Until recently, the prognosis for aHUS was poor, with the majority of patients developing ESRD within 2 years of presentation. However, with the introduction of eculizumab, a humanized monoclonal antibody against C5, it is now possible to control the renal disease and prevent development of ESRD (57).
In TTP, systemic platelet thrombi are mainly composed by platelets and von-Willebrand Factor (VWF) (76). VWF is a high-molecular weight, multimeric plasma glycoprotein, produced by endothelial cells with highly thrombogenic property mediated by the availability of an array of docking sites for platelets on endothelial cells and extracellular matrix collagen. In normal conditions, the VWF thrombogenic potential is rapidly held in check through cleavage into smaller multimers by a plasma metalloprotease, ADAMTS13 (77), thus ADAMTS13 deficiency predisposes to microvascular thrombosis after a triggering event. ADAMST13 activity could also be helpful in predicting the disease clinical manifestation or, a relapsing course (78). Several studies investigated complement activation markers in TTP patients with documented severe ADAMTS13 deficiency, showing lower serum levels of C3 and MAC during the acute phase (79, 80), correlating also with disease activity. No significant differences in levels of CP or LP activation markers were observed (80). The above data point toward complement activation, through to the terminal C5b–9 complex, in TTP, but how complement is activated in TTP is still unclear.
Chronic Kidney Injury and Fibrosis
The pathogenesis of progressive renal fibrosis is complex and involves various cell types and molecular pathways. However, it is evident that the inflammatory microenvironment of the kidney, after sustained injury, plays a dominant role in the dynamic balance between tissue destruction (tubular atrophy and interstitial fibrosis) and repair (tubular cell growth and resolution of renal inflammation and fibrosis) (81). Recent evidence has implicated intrarenal complement activation in the progression of kidney injury of chronic renal failure (82). C3 plays a substantial role in the activation of the renin–angiotensin system and the epithelial-to-mesenchymal transition (83, 84). This is consistent with the concept that complement component generation by renal epithelial cells promotes tubular damage in proteinuria-associated renal disease. Further evidence is provided by the fact that absence/blockade of C5/C5aR1 (but not blocking MAC formation) limits kidney fibrosis in several animal models (85, 86). Taken together, these data suggest that kidney-derived complement participates in the progression of renal fibrosis.
In a murine model of ascending urinary tract infection C5aR1 deficiency or blockade not only reduces renal bacterial load at all stages of infection but also attenuates tissue inflammation and tubulointerstitial fibrosis, suggesting a pathogenic role for C5aR1 in experimental chronic kidney infection. Mechanistic studies suggest that C5aR1-mediated bacterial colonization of tubular epithelium, persistent local inflammatory responses, and impairment of the phagocytic function of monocytes/macrophages could contribute to the pathogenesis of chronic kidney infection (87) (Table 1).
Therapy
The last decade has witnessed a growing interest from pharmaceutical companies in the development of complement inhibitors for the most disparate indications, largely supported by the exceptional results obtained with anti-C5 blocking antibody eculizumab (Table 2; Figure 1).
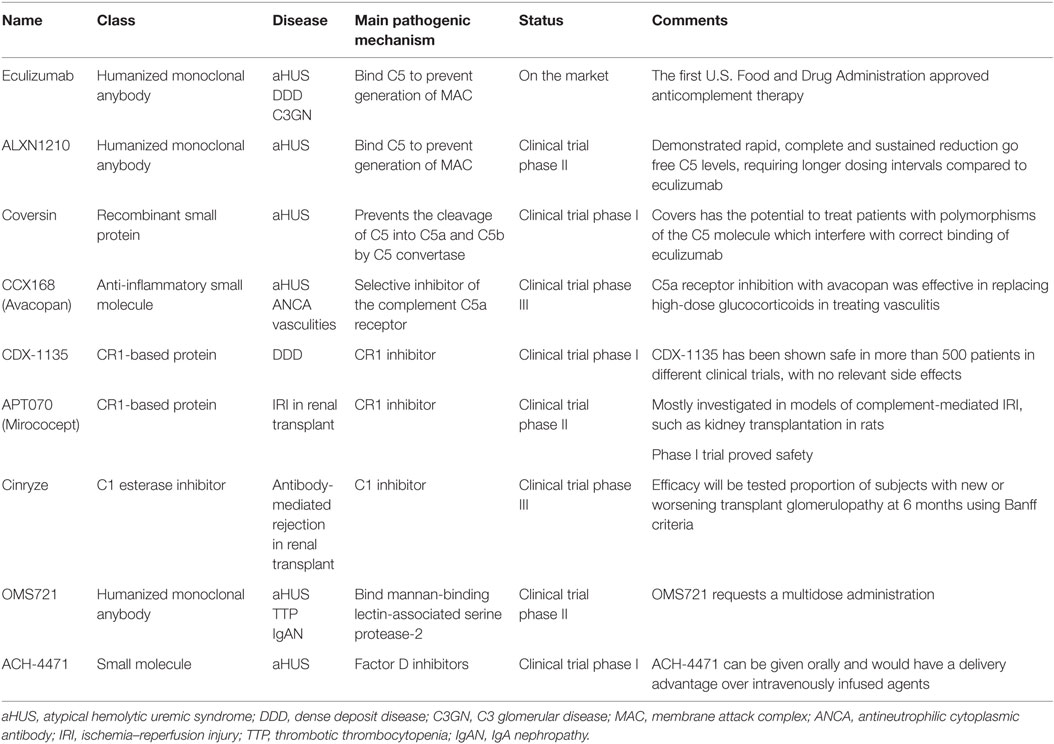
Table 2. Main complement-targeting therapies.
Eculizumab
Eculizumab is the first complement inhibitor approved for clinical use, initially for paroxysmal nocturnal hemoglobinuria (PNH) and subsequently for aHUS (88). It is an anti-C5 humanized mAb which prevents C5 cleavage by the C5 convertase, thereby inhibiting the terminal complement effector pathway. In absence of C5b the assembly of MAC is prevented (88).
Eculizumab was approved by the European Medicines Agency’s and the US Food and Drug Administration for the treatment of aHUS on the basis of results from two distinct prospective trials in 17 aHUS patients with thrombocytopenia and in 20 aHUS patients requiring persistent plasma exchange (PE), respectively (89). All patients achieved discontinuation of PE and 88% (33 of 37) reached normal hematological values after median of 63 weeks of eculizumab treatment. Eculizumab has clearly improved the renal outcome of aHUS patients with a dramatic decrease in the risk of ESRD. Nevertheless, the clinical use of eculizumab in aHUS still carries a number of unanswered questions, mostly concerning patient selection, timing and duration of the treatment: an ongoing multicenter single-arm trial is testing the safety of the discontinuation of eculizumab treatment in patients with aHUS (NCT02574403).
Efficacy of eculizumab in patients with DDD and C3GN, limited to case reports, suggested that eculizumab is a promising option in native disease, but not in C3GN recurrence after kidney transplantation (90–98). The only open-label study of eculizumab therapy in six patients (three C3GN and three DDD) reported complete to partial remission in four patients at one year of follow-up (99, 100).
Inhibitors of Terminal Effector Complement
Novel anti-C5 mAb antibodies are expected to reproduce the efficacy of eculizumab with longer half-lives and at lower costs. An ongoing single-arm study is testing the efficacy in controlling disease activity of a longer-acting anti-C5 humanized monoclonal antibody (ALXN1210) in patients with aHUS who have not previously used a complement inhibitor (NCT02949128).
Coversin is a recombinant small animal protein complement C5 inhibitor able to prevent the cleavage of C5 into C5a and C5b by C5 convertase (101). In vitro, it prevents hemolysis of PNH erythrocytes (102). In an open label, non-comparative clinical trial, Coversin reducing serum lactic dehydrogenase, will be tested in patients with PNH and proven resistance to eculizumab due to C5 polymorphisms (NCT02591862).
CCX168 (Avacopan) is a small molecule C5aR inhibitor. Jayne et al. recently conducted a randomized, double-blind, placebo-controlled trial in 67 adults with ANCA-associated vasculitis, treated with Avacopan with or without steroids. All patients received cyclophosphamide or rituximab. They achieved treatment responses in 86 and 81% of the avacopan with and without steroid groups, respectively, and in 70% in the control group, meeting non-inferiority criteria (103). One more randomized phase III trial Avacopan in patients with ANCA-associated vasculitis is in recruiting participants (NCT02994927), and an open-label phase II study to assess the effect of C5aR inhibitor in aHUS is in terminal status (NCT02464891).
TP10 is a C3 convertase inhibitor that acts as a soluble complement receptor. TP10 can be considered as a candidate for C3G and a phase I trial is currently underway (NCT02302755).
Inhibitors of Initial Complement Activation
Acting at the level of initial pathway-specific events that lead to C3 activation could represent an effective therapy to prevent C3 activation. Several preclinical trials are testing this strategy (102), and it is particularly useful in diseases where a specific complement pathway has a dominant pathogenic role. Examples would be the AP in PNH or the CP in antibody-mediated hemolytic anemias.
CR1 is a regulator of complement activation inhibiting C3/C5 convertases with effect on all the three complement pathway (104, 105). A soluble form of CR1, named CDX-1135 has been shown strongly effective in mouse model of DDD (106), and some clinical benefit has been reported in a short-term compassionate therapy in a child (106). A phase I clinical trial for testing CDX-1135 in DDD has been started and terminated (NCT01791686) with no results reported.
APT070 (Mirococept) (107) is an engineered molecule consisting of the first three short consensus domains of CR1and it is able to link it to cell membranes. Mirococept has been investigated in IRI preclinical studies achieving a significant increase in the number of surviving grafts, compared with control-treated grafts (63.6 versus 26.3%) (108). An ongoing phase III randomized, placebo-controlled trial is testing Mirococept as a protective agent to prevent functional impairment of transplanted kidneys (http://www.controlled-trials.com/ISRCTN49958194). Cinryze, a C1 esterase inhibitor currently used for hereditary angioedema, is being evaluated in a randomized double-blind study for the treatment of acute antibody-mediated rejection in donor-sensitized kidney transplants recipients (NCT02547220).
An antibody (OMS721) targeting mannan-binding lectin-associated serine protease-2 that cleaves C4 and C2, is currently being tested in clinical trials for use in TMA, aHUS (NCT02222545), and IgA nephropathy (NCT02682407).
Factor D inhibitors are agents that mitigate the complement-mediated amplification step of the AP. ACH-4471, a small molecule, demonstrated the ability to block the activation of the AP achieving decrease hemolysis and C3b deposition on red blood cells from patients with PNH (109) ACH-4471 is being tested in phase 1 clinical trials (110).
Conclusion
Increasing evidence has been accumulated showing that complement activation (mainly through the AP) is implicated in the pathogenesis of different non-antibody-mediated glomerular diseases and in the general progression of renal disease, regardless of the initial insult.
Eculizumab, an anti-C5 monoclonal antibody approved for the treatment of aHUS and PNH, revolutionized the treatment of TMA, with safe and effective inhibitors for different levels of complement cascade emerging. The advent of selective complement-targeting therapies has the potential provide new treatment options while enhancing our understanding of complement involvement in disease pathogenesis.
Author Contributions
All the authors participated in the drafting and critical appraisal of the manuscript, approved the final version, and agreed to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by NIH grant R01 DK103022 to KC.
References
1. Walport MJ. Complement. First of two parts. N Engl J Med (2001) 344:1058–66. doi:10.1056/NEJM200104053441406
2. Walport MJ. Complement. Second of two parts. N Engl J Med (2001) 344:1140–4. doi:10.1056/NEJM200104123441506
3. Farrar CA, Zhou W, Sacks SH. Role of the lectin complement pathway in kidney transplantation. Immunobiology (2016) 221:1068–72. doi:10.1016/j.imbio.2016.05.004
4. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11:785–97. doi:10.1038/ni.1923
5. Medof ME, Kinoshita T, Nussenzweig V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J Exp Med (1984) 160:1558–78. doi:10.1084/jem.160.5.1558
6. Fujita T, Inoue T, Ogawa K, Iida K, Tamura N. The mechanism of action of decay-accelerating factor (DAF). DAF inhibits the assembly of C3 convertases by dissociating C2a and Bb. J Exp Med (1987) 166:1221–8. doi:10.1084/jem.166.5.1221
7. Quigg RJ, Nicholson-Weller A, Cybulsky AV, Badalamenti J, Salant DJ. Decay accelerating factor regulates complement activation on glomerular epithelial cells. J Immunol (1989) 142:877–82.
8. Ichida S, Yuzawa Y, Okada H, Yoshioka K, Matsuo S. Localization of the complement regulatory proteins in the normal human kidney. Kidney Int (1994) 46:89–96. doi:10.1038/ki.1994.247
9. Cole JL, Housley GA Jr, Dykman TR, MacDermott RP, Atkinson JP. Identification of an additional class of C3-binding membrane proteins of human peripheral blood leukocytes and cell lines. Proc Natl Acad Sci U S A (1985) 82:859–63. doi:10.1073/pnas.82.3.859
10. Kim YU, Kinoshita T, Molina H, Hourcade D, Seya T, Wagner LM, et al. Mouse complement regulatory protein Crry/p65 uses the specific mechanisms of both human decay-accelerating factor and membrane cofactor protein. J Exp Med (1995) 181:151–9. doi:10.1084/jem.181.1.151
11. Cravedi P, Heeger PS. Complement as a multifaceted modulator of kidney transplant injury. J Clin Invest (2014) 124:2348–54. doi:10.1172/JCI72273
12. Jane-Wit D, Manes TD, Yi T, Qin L, Clark P, Kirkiles-Smith NC, et al. Alloantibody and complement promote T cell-mediated cardiac allograft vasculopathy through noncanonical nuclear factor-kappaB signaling in endothelial cells. Circulation (2013) 128:2504–16. doi:10.1161/CIRCULATIONAHA.113.002972
13. Adler S, Baker PJ, Johnson RJ, Ochi RF, Pritzl P, Couser WG. Complement membrane attack complex stimulates production of reactive oxygen metabolites by cultured rat mesangial cells. J Clin Invest (1986) 77:762–7. doi:10.1172/JCI112372
14. Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol (2005) 23:821–52. doi:10.1146/annurev.immunol.23.021704.115835
15. Kwan WH, van der Touw W, Paz-Artal E, Li MO, Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med (2013) 210:257–68. doi:10.1084/jem.20121525
16. van der Touw W, Cravedi P, Kwan WH, Paz-Artal E, Merad M, Heeger PS. Cutting edge: receptors for C3a and C5a modulate stability of alloantigen-reactive induced regulatory T cells. J Immunol (2013) 190:5921–5. doi:10.4049/jimmunol.1300847
17. Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J. The role of the anaphylatoxins in health and disease. Mol Immunol (2009) 46:2753–66. doi:10.1016/j.molimm.2009.04.027
18. Pepys MB. Role of complement in induction of antibody production in vivo. Effect of cobra factor and other C3-reactive agents on thymus-dependent and thymus-independent antibody responses. J Exp Med (1974) 140:126–45. doi:10.1084/jem.140.1.126
19. Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science (1996) 271:348–50. doi:10.1126/science.271.5247.348
20. Heeger PS, Lalli PN, Lin F, Valujskikh A, Liu J, Muqim N, et al. Decay-accelerating factor modulates induction of T cell immunity. J Exp Med (2005) 201:1523–30. doi:10.1084/jem.20041967
21. Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity (2008) 28:425–35. doi:10.1016/j.immuni.2008.02.001
22. Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood (2008) 112:1759–66. doi:10.1182/blood-2008-04-151068
23. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi:10.1016/j.cell.2008.05.009
24. Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4(+) cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3(+) regulatory T cells. Nat Immunol (2013) 14:162–71. doi:10.1038/ni.2499
25. Kemper C, Chan AC, Green JM, Brett KA, Murphy KM, Atkinson JP. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature (2003) 421:388–92. doi:10.1038/nature01315
26. Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol (2017) 188(2):183–94. doi:10.1111/cei.12952
27. Brooimans RA, Stegmann AP, van Dorp WT, van der Ark AA, van der Woude FJ, van Es LA, et al. Interleukin 2 mediates stimulation of complement C3 biosynthesis in human proximal tubular epithelial cells. J Clin Invest (1991) 88:379–84. doi:10.1172/JCI115314
28. Sacks S, Zhou W, Campbell RD, Martin J. C3 and C4 gene expression and interferon-gamma-mediated regulation in human glomerular mesangial cells. Clin Exp Immunol (1993) 93:411–7. doi:10.1111/j.1365-2249.1993.tb08193.x
29. Sacks SH, Zhou W, Pani A, Campbell RD, Martin J. Complement C3 gene expression and regulation in human glomerular epithelial cells. Immunology (1993) 79:348–54.
30. Zhou W, Campbell RD, Martin J, Sacks SH. Interferon-gamma regulation of C4 gene expression in cultured human glomerular epithelial cells. Eur J Immunol (1993) 23:2477–81. doi:10.1002/eji.1830231015
31. Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med (2002) 8:582–7. doi:10.1038/nm0602-582
32. Song D, Zhou W, Sheerin SH, Sacks SH. Compartmental localization of complement component transcripts in the normal human kidney. Nephron (1998) 78:15–22. doi:10.1159/000044876
33. D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis (2004) 43:368–82. doi:10.1053/j.ajkd.2003.10.024
34. Korbet SM. Treatment of primary FSGS in adults. J Am Soc Nephrol (2012) 23:1769–76. doi:10.1681/ASN.2012040389
35. Korbet SM. Clinical picture and outcome of primary focal segmental glomerulosclerosis. Nephrol Dial Transplant (1999) 14(Suppl 3):68–73. doi:10.1093/ndt/14.suppl_3.68
36. Doublier S, Lupia E, Catanuto P, Periera-Simon S, Xia X, Korach K, et al. Testosterone and 17beta-estradiol have opposite effects on podocyte apoptosis that precedes glomerulosclerosis in female estrogen receptor knockout mice. Kidney Int (2011) 79:404–13. doi:10.1038/ki.2010.398
37. Cravedi P, Kopp JB, Remuzzi G. Recent progress in the pathophysiology and treatment of FSGS recurrence. Am J Transplant (2013) 13:266–74. doi:10.1111/ajt.12045
38. Wada T, Nangaku M. A circulating permeability factor in focal segmental glomerulosclerosis: the hunt continues. Clin Kidney J (2015) 8:708–15. doi:10.1093/ckj/sfv090
39. Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardino L, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest (2004) 113:1390–7. doi:10.1172/JCI20402
40. Schlondorff D. Are serum suPAR determinations by current ELISA methodology reliable diagnostic biomarkers for FSGS? Kidney Int (2014) 85:499–501. doi:10.1038/ki.2013.549
41. Skorecki KL, Freedman BI. A suPAR biomarker for chronic kidney disease. N Engl J Med (2015) 373:1971–2. doi:10.1056/NEJMe1512997
42. Hahm E, Wei C, Fernandez I, Li J, Tardi NJ, Tracy M, et al. Bone marrow-derived immature myeloid cells are a main source of circulating suPAR contributing to proteinuric kidney disease. Nat Med (2017) 23:100–6. doi:10.1038/nm.4242
43. Lenderink AM, Liegel K, Ljubanovic D, Coleman KE, Gilkeson GS, Holers VM, et al. The alternative pathway of complement is activated in the glomeruli and tubulointerstitium of mice with adriamycin nephropathy. Am J Physiol Renal Physiol (2007) 293:F555–64. doi:10.1152/ajprenal.00403.2006
44. Turnberg D, Lewis M, Moss J, Xu Y, Botto M, Cook HT. Complement activation contributes to both glomerular and tubulointerstitial damage in adriamycin nephropathy in mice. J Immunol (2006) 177:4094–102. doi:10.4049/jimmunol.177.6.4094
45. Morigi M, Locatelli M, Rota C, Buelli S, Corna D, Rizzo P, et al. A previously unrecognized role of C3a in proteinuric progressive nephropathy. Sci Rep (2016) 6:28445. doi:10.1038/srep28445
46. Bao L, Haas M, Pippin J, Wang Y, Miwa T, Chang A, et al. Focal and segmental glomerulosclerosis induced in mice lacking decay-accelerating factor in T cells. J Clin Invest (2009) 119:1264–74. doi:10.1172/JCI36000
47. Strassheim D, Renner B, Panzer S, Fuquay R, Kulik L, Ljubanovic D, et al. IgM contributes to glomerular injury in FSGS. J Am Soc Nephrol (2013) 24:393–406. doi:10.1681/ASN.2012020187
48. Thurman JM, Wong M, Renner B, Frazer-Abel A, Giclas PC, Joy MS, et al. Complement activation in patients with focal segmental glomerulosclerosis. PLoS One (2015) 10:e0136558. doi:10.1371/journal.pone.0136558
49. Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, et al. C3 glomerulopathy: consensus report. Kidney Int (2013) 84:1079–89. doi:10.1038/ki.2013.377
50. Fakhouri F, Fremeaux-Bacchi V, Noel LH, Cook HT, Pickering MC. C3 glomerulopathy: a new classification. Nat Rev Nephrol (2010) 6:494–9. doi:10.1038/nrneph.2010.85
51. Medjeral-Thomas NR, O’Shaughnessy MM, O’Regan JA, Traynor C, Flanagan M, Wong L, et al. C3 glomerulopathy: clinicopathologic features and predictors of outcome. Clin J Am Soc Nephrol (2014) 9:46–53. doi:10.2215/CJN.04700513
52. Sethi S, Haas M, Markowitz GS, D’Agati VD, Rennke HG, Jennette JC, et al. Mayo clinic/renal pathology society consensus report on pathologic classification, diagnosis, and reporting of GN. J Am Soc Nephrol (2016) 27:1278–87. doi:10.1681/ASN.2015060612
53. Servais A, Noel LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int (2012) 82:454–64. doi:10.1038/ki.2012.63
54. Sethi S, Nester CM, Smith RJ. Membranoproliferative glomerulonephritis and C3 glomerulopathy: resolving the confusion. Kidney Int (2012) 81:434–41. doi:10.1038/ki.2011.399
55. Pickering M, Cook HT. Complement and glomerular disease: new insights. Curr Opin Nephrol Hypertens (2011) 20:271–7. doi:10.1097/MNH.0b013e328345848b
56. Smith RJ, Harris CL, Pickering MC. Dense deposit disease. Mol Immunol (2011) 48:1604–10. doi:10.1016/j.molimm.2011.04.005
57. Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Fremeaux-Bacchi V, Kavanagh D, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “kidney disease: improving global outcomes” (KDIGO) controversies conference. Kidney Int (2017) 91:539–51. doi:10.1016/j.kint.2016.10.005
58. Bomback AS. Eculizumab in the treatment of membranoproliferative glomerulonephritis. Nephron Clin Pract (2014) 128:270–6. doi:10.1159/000368592
59. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med (2014) 371:1847–8. doi:10.1056/NEJMra1312353
60. Goldberg RJ, Nakagawa T, Johnson RJ, Thurman JM. The role of endothelial cell injury in thrombotic microangiopathy. Am J Kidney Dis (2010) 56:1168–74. doi:10.1053/j.ajkd.2010.06.006
61. Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol (2012) 8:622–33. doi:10.1038/nrneph.2012.195
62. Nestoridi E, Tsukurov O, Kushak RI, Ingelfinger JR, Grabowski EF. Shiga toxin enhances functional tissue factor on human glomerular endothelial cells: implications for the pathophysiology of hemolytic uremic syndrome. J Thromb Haemost (2005) 3:752–62. doi:10.1111/j.1538-7836.2005.01205.x
63. Cameron JS, Vick R. Letter: plasma-C3 in haemolytic-uraemic syndrome and thrombotic thrombocytopenic purpura. Lancet (1973) 2:975. doi:10.1016/S0140-6736(73)92645-7
64. Kaplan BS, Thomson PD, MacNab GM. Letter: serum-complement levels in haemolytic-uraemic syndrome. Lancet (1973) 2:1505–6. doi:10.1016/S0140-6736(73)92782-7
65. Monnens L, Hendrickx G, van Wieringen P, van Munster P. Letter: serum-complement levels in haemolytic-uraemic syndrome. Lancet (1974) 2:294. doi:10.1016/S0140-6736(74)91463-9
66. Robson WL, Leung AK, Fick GH, McKenna AI. Hypocomplementemia and leukocytosis in diarrhea-associated hemolytic uremic syndrome. Nephron (1992) 62:296–9. doi:10.1159/000187063
67. Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, Oualha M, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med (2011) 364:2561–3. doi:10.1056/NEJMc1100859
68. Monnens L, Molenaar J, Lambert PH, Proesmans W, van Munster P. The complement system in hemolytic-uremic syndrome in childhood. Clin Nephrol (1980) 13:168–71.
69. Thurman JM, Marians R, Emlen W, Wood S, Smith C, Akana H, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol (2009) 4:1920–4. doi:10.2215/CJN.02730409
70. Stahl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood (2011) 117:5503–13. doi:10.1182/blood-2010-09-309161
71. Morigi M, Galbusera M, Gastoldi S, Locatelli M, Buelli S, Pezzotta A, et al. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol (2011) 187:172–80. doi:10.4049/jimmunol.1100491
72. Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med (2005) 201:871–9. doi:10.1084/jem.20041497
73. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al. Atypical aHUS: state of the art. Mol Immunol (2015) 67:31–42. doi:10.1016/j.molimm.2015.03.246
74. Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol (2010) 5:1844–59. doi:10.2215/CJN.02210310
75. Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood (2006) 108:1267–79. doi:10.1182/blood-2005-10-007252
76. Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood (2008) 112:11–8. doi:10.1182/blood-2008-02-078170
77. Plaimauer B, Zimmermann K, Volkel D, Antoine G, Kerschbaumer R, Jenab P, et al. Cloning, expression, and functional characterization of the von Willebrand factor-cleaving protease (ADAMTS13). Blood (2002) 100:3626–32. doi:10.1182/blood-2002-05-1397
78. Rurali E, Banterla F, Donadelli R, Bresin E, Galbusera M, Gastoldi S, et al. ADAMTS13 secretion and residual activity among patients with congenital thrombotic thrombocytopenic purpura with and without renal impairment. Clin J Am Soc Nephrol (2015) 10:2002–12. doi:10.2215/CJN.01700215
79. Ruiz-Torres MP, Casiraghi F, Galbusera M, Macconi D, Gastoldi S, Todeschini M, et al. Complement activation: the missing link between ADAMTS-13 deficiency and microvascular thrombosis of thrombotic microangiopathies. Thromb Haemost (2005) 93:443–52. doi:10.1160/TH04-07-0450
80. Reti M, Farkas P, Csuka D, Razso K, Schlammadinger A, Udvardy ML, et al. Complement activation in thrombotic thrombocytopenic purpura. J Thromb Haemost (2012) 10:791–8. doi:10.1111/j.1538-7836.2012.04674.x
81. Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol (2011) 7:684–96. doi:10.1038/nrneph.2011.149
82. Sheerin NS, Risley P, Abe K, Tang Z, Wong W, Lin T, et al. Synthesis of complement protein C3 in the kidney is an important mediator of local tissue injury. FASEB J (2008) 22:1065–72. doi:10.1096/fj.07-8719com
83. Tang Z, Lu B, Hatch E, Sacks SH, Sheerin NS. C3a mediates epithelial-to-mesenchymal transition in proteinuric nephropathy. J Am Soc Nephrol (2009) 20:593–603. doi:10.1681/ASN.2008040434
84. Zhou X, Fukuda N, Matsuda H, Endo M, Wang X, Saito K, et al. Complement 3 activates the renal renin-angiotensin system by induction of epithelial-to-mesenchymal transition of the nephrotubulus in mice. Am J Physiol Renal Physiol (2013) 305:F957–67. doi:10.1152/ajprenal.00344.2013
85. Boor P, Konieczny A, Villa L, Schult AL, Bucher E, Rong S, et al. Complement C5 mediates experimental tubulointerstitial fibrosis. J Am Soc Nephrol (2007) 18:1508–15. doi:10.1681/ASN.2006121343
86. Rangan GK, Pippin JW, Couser WG. C5b-9 regulates peritubular myofibroblast accumulation in experimental focal segmental glomerulosclerosis. Kidney Int (2004) 66:1838–48. doi:10.1111/j.1523-1755.2004.00957.x
87. Choudhry N, Li K, Zhang T, Wu KY, Song Y, Farrar CA, et al. The complement factor 5a receptor 1 has a pathogenic role in chronic inflammation and renal fibrosis in a murine model of chronic pyelonephritis. Kidney Int (2016) 90:540–54. doi:10.1016/j.kint.2016.04.023
88. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol (2007) 25:1256–64. doi:10.1038/nbt1207-1488c
89. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med (2013) 368:2169–81. doi:10.1056/NEJMoa1208981
90. Radhakrishnan S, Lunn A, Kirschfink M, Thorner P, Hebert D, Langlois V, et al. Eculizumab and refractory membranoproliferative glomerulonephritis. N Engl J Med (2012) 366:1165–6. doi:10.1056/NEJMc1106619
91. Vivarelli M, Pasini A, Emma F. Eculizumab for the treatment of dense-deposit disease. N Engl J Med (2012) 366:1163–5. doi:10.1056/NEJMc1111953
92. Daina E, Noris M, Remuzzi G. Eculizumab in a patient with dense-deposit disease. N Engl J Med (2012) 366:1161–3. doi:10.1056/NEJMc1112273
93. McCaughan JA, O’Rourke DM, Courtney AE. Recurrent dense deposit disease after renal transplantation: an emerging role for complementary therapies. Am J Transplant (2012) 12:1046–51. doi:10.1111/j.1600-6143.2011.03923.x
94. Kerns E, Rozansky D, Troxell ML. Evolution of immunoglobulin deposition in C3-dominant membranoproliferative glomerulopathy. Pediatr Nephrol (2013) 28:2227–31. doi:10.1007/s00467-013-2565-x
95. Gurkan S, Fyfe B, Weiss L, Xiao X, Zhang Y, Smith RJ. Eculizumab and recurrent C3 glomerulonephritis. Pediatr Nephrol (2013) 28:1975–81. doi:10.1007/s00467-013-2503-y
96. Rousset-Rouviere C, Cailliez M, Garaix F, Bruno D, Laurent D, Tsimaratos M. Rituximab fails where eculizumab restores renal function in C3nef-related DDD. Pediatr Nephrol (2014) 29:1107–11. doi:10.1007/s00467-013-2711-5
97. Ozkaya O, Nalcacioglu H, Tekcan D, Genc G, Meydan BC, Ozdemir BH, et al. Eculizumab therapy in a patient with dense-deposit disease associated with partial lipodystrophy. Pediatr Nephrol (2014) 29:1283–7. doi:10.1007/s00467-013-2748-5
98. Lebreton C, Bacchetta J, Dijoud F, Bessenay L, Fremeaux-Bacchi V, Sellier-Leclerc AL. C3 glomerulopathy and eculizumab: a report on four paediatric cases. Pediatr Nephrol (2017) 32(6):1023–8. doi:10.1007/s00467-017-3619-2
99. Herlitz LC, Bomback AS, Markowitz GS, Stokes MB, Smith RN, Colvin RB, et al. Pathology after eculizumab in dense deposit disease and C3 GN. J Am Soc Nephrol (2012) 23:1229–37. doi:10.1681/ASN.2011121186
100. Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, Herlitz L, et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol (2012) 7:748–56. doi:10.2215/CJN.12901211
101. Barratt-Due A, Thorgersen EB, Lindstad JK, Pharo A, Lissina O, Lambris JD, et al. Ornithodoros moubata complement inhibitor is an equally effective C5 inhibitor in pigs and humans. J Immunol (2011) 187:4913–9. doi:10.4049/jimmunol.1101000
102. Risitano AM, Marotta S. Therapeutic complement inhibition in complement-mediated hemolytic anemias: past, present and future. Semin Immunol (2016) 28:223–40. doi:10.1016/j.smim.2016.05.001
103. Jayne DR, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol (2017). doi:10.1681/ASN.2016111179
104. Khera R, Das N. Complement receptor 1: disease associations and therapeutic implications. Mol Immunol (2009) 46:761–72. doi:10.1016/j.molimm.2008.09.026
105. Medof ME, Nussenzweig V. Control of the function of substrate-bound C4b-C3b by the complement receptor Cr1. J Exp Med (1984) 159:1669–85. doi:10.1084/jem.159.6.1669
106. Zhang Y, Nester CM, Holanda DG, Marsh HC, Hammond RA, Thomas LJ, et al. Soluble CR1 therapy improves complement regulation in C3 glomerulopathy. J Am Soc Nephrol (2013) 24:1820–9. doi:10.1681/ASN.2013010045
107. Souza DG, Esser D, Bradford R, Vieira AT, Teixeira MM. APT070 (Mirococept), a membrane-localised complement inhibitor, inhibits inflammatory responses that follow intestinal ischaemia and reperfusion injury. Br J Pharmacol (2005) 145:1027–34. doi:10.1038/sj.bjp.0706286
108. Patel H, Smith RA, Sacks SH, Zhou W. Therapeutic strategy with a membrane-localizing complement regulator to increase the number of usable donor organs after prolonged cold storage. J Am Soc Nephrol (2006) 17:1102–11. doi:10.1681/ASN.2005101116
109. Yuan X, Gavriilaki E, Thanassi JA, Yang G, Baines AC, Podos SD, et al. Small-molecule factor D inhibitors selectively block the alternative pathway of complement in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Haematologica (2017) 102:466–75. doi:10.3324/haematol.2016.153312
Keywords: complement system, glomerular disease, thrombotic microangiopathy, fibrosis, focal segmental glomerulosclerosis
Citation: Angeletti A, Reyes-Bahamonde J, Cravedi P and Campbell KN (2017) Complement in Non-Antibody-Mediated Kidney Diseases. Front. Med. 4:99. doi: 10.3389/fmed.2017.00099
Received: 02 May 2017; Accepted: 21 June 2017;
Published: 12 July 2017
Edited by:
Michael L. Moritz, Children’s Hospital of Pittsburgh, United StatesReviewed by:
Wibke Bechtel-Walz, University Medical Center Freiburg, GermanyMinghui Zhao, Peking University First Hospital, China
Copyright: © 2017 Angeletti, Reyes-Bahamonde, Cravedi and Campbell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kirk N. Campbell, a2lyay5jYW1wYmVsbEBtc3NtLmVkdQ==