 Dhanunjay Mukhi
Dhanunjay Mukhi Rajkishor Nishad
Rajkishor Nishad Ram K. Menon
Ram K. Menon Anil Kumar Pasupulati
Anil Kumar Pasupulati- 1Department of Biochemistry, School of Life Sciences, University of Hyderabad, Hyderabad, India
- 2Department of Pediatric Endocrinology and Physiology, University of Michigan, Ann Arbor, MI, United States
The kidney regulates water, electrolyte, and acid-base balance and thus maintains body homeostasis. The kidney’s potential to ensure ultrafiltered and almost protein-free urine is compromised in various metabolic and hormonal disorders such as diabetes mellitus (DM). Diabetic nephropathy (DN) accounts for ~20–40% of mortality in DM. Proteinuria, a hallmark of renal glomerular diseases, indicates injury to the glomerular filtration barrier (GFB). The GFB is composed of glomerular endothelium, basement membrane, and podocytes. Podocytes are terminally differentiated epithelial cells with limited ability to replicate. Podocyte shape and number are both critical for the integrity and function of the GFB. Podocytes are vulnerable to various noxious stimuli prevalent in a diabetic milieu that could provoke podocytes to undergo changes to their unique architecture and function. Effacement of podocyte foot process is a typical morphological alteration associated with proteinuria. The dedifferentiation of podocytes from epithelial-to-mesenchymal phenotype and consequential loss results in proteinuria. Poorly controlled type 1 DM is associated with elevated levels of circulating growth hormone (GH), which is implicated in the pathophysiology of various diabetic complications including DN. Recent studies demonstrate that functional GH receptors are expressed in podocytes and that GH may exert detrimental effects on the podocyte. In this review, we summarize recent advances that shed light on actions of GH on the podocyte that could play a role in the pathogenesis of DN.
Introduction
The vertebrate kidney plays an essential role in filtration of blood, regulation of water, electrolyte, and acid-base balance, and thereby maintenance of overall body homeostasis. The function of the kidney to ensure almost protein-free ultrafiltered urine depends on the collective action of millions of nephrons (1). A nephron comprises two highly coordinated units: glomerulus and renal tubule. The glomerulus filters plasma to prevent protein loss into the glomerular filtrate. The renal tubule reabsorbs water and electrolytes in addition to contributing selective salts and Tamm–Horsfall proteins to the glomerular filtrate. The contribution of renal tubular absorption and secretion notwithstanding, the final composition of urine is largely determined by the integrity of glomerular filtration barrier (GFB, Figure 1A). The GFB consists of three critical components—endothelium, glomerular basement membrane (GBM), and podocytes (1, 2). The endothelium lines the inner surface of glomerular capillaries and is fenestrated (70–100 nm), which allows for small molecules and a limited amount of protein to pass through. The GBM (250–300 nm) is an extracellular matrix (ECM) material made up of structural proteins, the most abundant being collagen, fibronectin, and heparan sulfate proteoglycans. Podocytes are terminally differentiated visceral cells that adhere firmly to GBM and offer epithelial coverage to the surface of glomerular capillaries (1, 2). The slit pore between neighboring podocytes is 30–50 nm wide (3). This three-layered GFB with a distinct architecture serves as a size-selective and charge-dependent molecular sieve that facilitate the filtration of water, electrolytes, and small solutes while restricting the passage of negatively charged macromolecules including proteins and polypeptides. Proteins with a molecular weight of up to 20 kDa filter easily across the GFB (4). However, the smaller proteins are largely reabsorbed by proximal convoluted tubule, and only small amounts of protein are excreted. When the nephron function is disturbed, varying amounts of plasma proteins, particularly albumin (~67 kDa), are excreted in urine (Figure 1B) and albuminuria is a well-known index of adverse renal function (5).
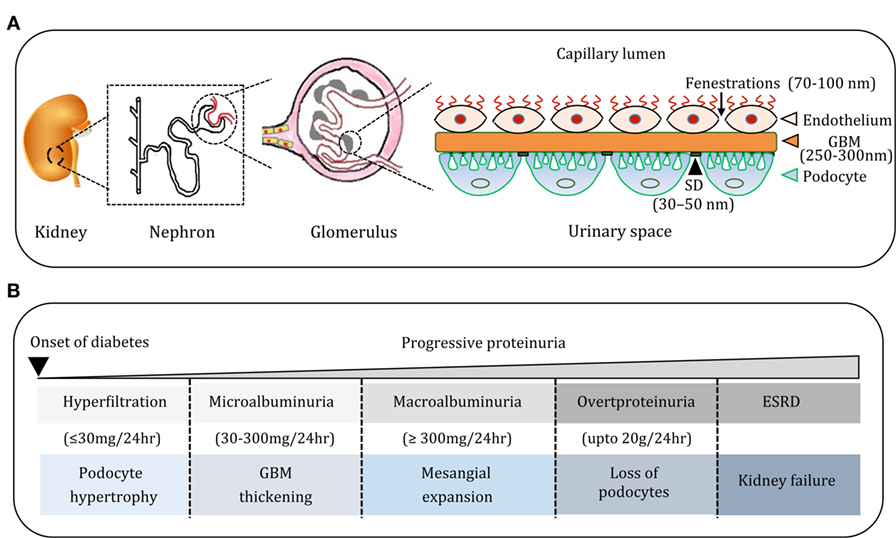
Figure 1. (A) Cartoon depicting the organization of nephron, glomerulus, and glomerular filtration barrier. The glomerular filtration barrier (GFB) comprises endothelial cells (ECs) of glomerular capillaries, basement membrane (BM), and podocytes (PC). Podocytes are specialized glomerular epithelial cells and slit diaphragm (SD) forms a contact between them and contributes to the glomerular permselectivity. (B) The natural course of diabetic nephropathy (DN). Progression of DN is associated with progressive proteinuria from micro albuminuria to overt proteinuria. The course of DN is also associated with histological manifestations in the glomerulus such as hypertrophy of podocytes, widening of glomerular BM, glomerulosclerosis, and depletion of podocytes, and these events culminate in end-stage renal disease.
Proteinuria can be classified as glomerular, tubular, and overflow (6). Glomerular disease is the most common cause of pathologic proteinuria that presents with large protein losses (>4 g per 24 h). Glomerular diseases that elicit proteinuria include minimal change disease (MCD), focal segmental glomerulonephritis (FSGS), membranous proliferative glomerulonephritis, IgA nephropathy, DN, collagen vascular disorders, amyloidosis, preeclampsia, and certain infections. Since GFB is the main constituent of the renal filtration apparatus, the appearance of protein in urine can be explained by damage to GFB, particularly to the podocytes as they play a key role in ensuring permselectivity of GFB. Tubular proteinuria is caused by tubulointerstitial disease results from decreased tubular reabsorption of proteins from the glomerular ultrafiltrate. In the case of overflow proteinuria, low-molecular-weight proteins overwhelm the ability of the proximal tubules to reabsorb proteins from the glomerular ultrafiltrate. Most often, this is a result of the immunoglobulin overproduction that occurs in multiple myeloma. In general, in tubular and overflow proteinuria less than 2 g of protein is excreted in 24 h.
Podocytes have a unique architecture consisting of a cell body with major and minor processes that extend outward from the cell body and interdigitated foot processes (FPs) that enwrap the glomerular capillaries (7, 8). Adjacent FPs are connected with slit diaphragm (SD) that forms the sole contact between podocytes. The extracellular domains of several transmembrane proteins such as nephrin, Neph-1, podocin, P-cadherin, and CD2AP constitute the SD. The unique structure and localization of podocytes enable them to oppose hydrostatic pressure within the glomerular capillaries and ensure glomerular filtration. Since podocytes remain attached to the GBM by only their FPs, they are always in danger of becoming detached and excreted in the urine (9). Furthermore, podocytes are continually exposed to metabolic waste products, drugs, and toxins that are routed to the kidneys for elimination. Podocyte injury is observed in both diabetic and non-diabetic renal diseases. The spectrum of podocyte diseases includes MCD, FSGS, collapsing glomerulonephropathy, inflammatory glomerulonephropathy, and DN, to name a few (10, 11).
Podocyte Injury During Diabetes
It is generally considered that podocytes are terminally differentiated visceral epithelial cells of the glomerulus with no or limited proliferating capacity. Hence, the loss of a podocyte cannot be compensated by regeneration of a podocyte from neighboring healthy podocytes. Morphologically podocyte injury is manifested by distortion and fusion of FPs, which leaves the GBM uncovered and such injury is termed as effacement (12). Either injury or loss of podocytes is an early event in the pathology of DN in both humans and animal models. Podocytes are vulnerable to various insults during diabetes that result in apoptosis or dedifferentiation. Another noticeable feature of podocyte injury is hypertrophy by which podocytes could compensate for the loss of neighboring podocytes. Adaptive hypertrophy of podocytes in response to the loss of neighboring podocytes has limited effectiveness since these hypertrophic podocytes have to cover increased area of filtration surface that could decrease adhesiveness to the GBM (13). Also, it is hypothesized that hypertrophied podocytes are more vulnerable to prevailing stress conditions (13–15). Progressive loss of podocytes is one of the hallmarks of glomerulosclerosis (16).
Aberrations in various cellular and molecular events mediate podocytopathy during diabetes including hyperglycemia-induced increased ROS production, advanced glycation end products formation, aberrant Notch signaling, elevated vascular endothelial growth factor and transforming growth factor type β (TGF-β) signaling, and inappropriate activation of the renal renin–angiotensin system (1). Increased concentrations of angiotensin II in diabetes mellitus (DM) induces podocyte apoptosis by enhancing the expression of transient receptor potential cation channel 6 and this effect is reduced by inhibiting the nuclear translocation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) (17, 18). TGF-β transgenic mice develop progressive mesangial expansion, which is associated with podocyte apoptosis through activation of mitogen-activated protein kinase (MAPK) dependent caspase-3 pathway (19). Growth factors elicit glomerular hypertrophy by acting directly on podocytes and/or on the other glomerular cells such as endothelial and mesangial cells. In addition to these signaling molecules and cytokines, endocrine mediators can also exert an adverse effect on podocytes. Hence, elevated levels of growth hormone (GH) are implicated in the early renal hypertrophy and proteinuria in type 1 DM (T1DM) (20). Excess GH secretion is a feature of poorly controlled T1DM and is posited to be a causative factor in the development of microangiopathic complications of diabetes including retinopathy and nephropathy (Table 1). The focus of this review is to summarize the novel actions of GH in the pathogenesis of podocyte injury in T1DM.

Table 1. Significant findings in growth hormone (GH)-mediated podocytopathy and diabetic nephropathy (DN).
Altered GH/GH Receptor (GHR) Axis and Renal Dysfunction
Human GH is synthesized by somatotrophs of the anterior pituitary gland and stimulates postnatal growth, cell proliferation, and regeneration (21). Secretion of GH from the pituitary is primarily controlled by GH-releasing hormone and somatostatin, where former induces GH secretion and later inhibits GH secretion. GH elicits its actions at the cellular, molecular, and organ levels via interacting with GHR. Binding of GH dimerizes GHR and induces a conformational change in its cytoplasmic portion, which allows activation of Janus kinase 2 (JAK2) (22). GH-mediated cell signaling events are summarized in Figure 2. Autophosphorylation of JAK2 by GHR facilitates the activation of downstream signaling proteins including signal transducers and activators of transcription (STATs), MAPK, insulin receptor substrate (IRS-1&2), and phosphatidylinositol 3-kinase (22–25). Upon activation (phosphorylation), STAT proteins translocate to the nucleus and stimulate transcription of an array of GH-regulated genes. The JAK2-STAT pathway is regulated by suppressor of cytokine signaling proteins. The association of growth factor receptor-bound 2-son of sevenless complex with JAK2 is critical for the activation of MAPK pathway by GH (26, 27). GH potentially exerts its actions directly on target cells and indirectly by stimulating the production of insulin-like growth factor (IGF-1) (28). An intact JAK2–STAT5b signaling pathway is essential for stimulating the production of IGF-1. Circulating levels of IGF-1 regulate GH secretion by the anterior pituitary via a negative feedback loop and is used in clinical medicine as a surrogate marker of GH action (29). Although GH induces IGF-1 synthesis and secretion by hepatocytes, the rate of IGF-1 clearance and levels of IGF-binding proteins (IGFBPs) in blood also determine the concentrations of free IGF-1 at the tissue level. Several components of GH/GHR axis, including GHR, IGF-1, IGF-1 receptor, and IGFBPs are expressed in the kidney with precise spatial distribution across various segments of the nephron (30, 31). GH and IGF-1 have significant effects on renal function by regulating cellular hyperplasia and, hypertrophy, intrarenal blood flow, and tubular reabsorption (2, 32, 33). GH signaling regulates the intrarenal hemodynamics and glomerular arteriolar vasodilation by inducing cyclooxygenase activity and generation of nitric oxide (2, 34).
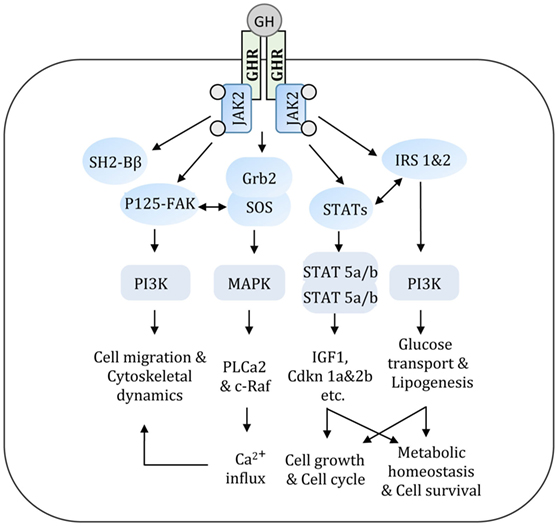
Figure 2. GH acts via a variety of signal transduction pathways. Multiple GH signaling pathways can contribute to specific GH responses. GH binds to GHR and activates JAK2 that in turn triggers an array of signaling cascade. These interconnected signal transduction pathways regulate various metabolic and cellular events. GH, growth hormone; GHR, growth hormone receptor; JAK2, Janus kinase 2; IRS, insulin receptor substrate; PI3K, phosphatidyl inositol 3-kinase; STAT, signal transducer and activator of transcription; Grb2-SOS, growth factor receptor-bound 2-son of sevenless complex; FAK, focal adhesion kinase; SH2-Bβ, src-homology 2 domain Bβ; MAPK, mitogen-activated protein kinase.
Alterations in the GH/GHR axis have been described in T1DM and diabetic kidney diseases (2). The mean 24 h concentration of circulating GH is elevated in poorly controlled T1DM. Elevation of GH levels in diabetes can be explained by two interrelated mechanisms (Figure 3). In poorly controlled T1DM, decreased hepatic GHR expression results in GH resistance and consequent attenuation of hepatic IGF-1 production (35). The resulting low levels of circulating IGF-1 stimulate GH secretion by feedback mechanism (36). Additionally, hypoinsulinemia in T1DM results in increased hepatic production of IGFBPs (37). The increase in serum IGFBPs, in particular IGFBP-1, dampens IGF-1 action at the cellular level and thus feedbacks on the pituitary somatotroph to stimulate GH secretion (38, 39). However, in contrast to the liver, GHR expression in the kidney is unaltered or even increased in poorly controlled DM, thus exposing the kidney to the effects of elevated levels of GH (40). Administration of GH for a week to healthy volunteers resulted in increased glomerular filtration rate (GFR) (41, 42). On the other hand, administration of somatostatin analogs: octreotide (SMS 201-995) and somatulin, which serves as inhibitors of GH, attenuated GFR, renal hyperfiltration, and kidney size in subjects with T1DM (43, 44). A study of a cohort of patients with acromegaly (a condition resulting from excessive GH levels secondary to a pituitary adenoma) demonstrated adverse effects of elevated GH levels on renal structure and function (45). Microalbuminuria is significantly increased in patients with acromegaly compared to healthy volunteers. Surgical ablation of pituitary adenoma results in decreased GH levels and kidney size (46). An increase in the GFR and kidney size was observed in both humans and rats injected with GH (47, 48). A 25–50% elevation in the GFR was observed in early course of T1DM (49). It was reported that glomerular hypertrophy and increased kidney size typically accompany the rise in GFR (50). Hyperfiltration, increased renal plasma flow, and hypertrophy that are induced by GH administration may accelerate the renal injury (51, 52). However, the clinical relevance of increase in GFR with GH administration is not precisely known. Elevated levels of GH in transgenic mice have been linked to diabetic kidney disease and other renal complications (53, 54). On the other hand, mice with a disrupted GHR gene or those expressing GH antagonist are protected from glomerular hypertrophy (55). Although these studies highlight the importance of GH/GHR axis in renal biology and the association of overactivity of GH signaling with the adverse renal outcome, the precise molecular mechanism(s) of GH action on podocytes remained uncertain until the recent identification of GHR in glomerular podocytes (56).
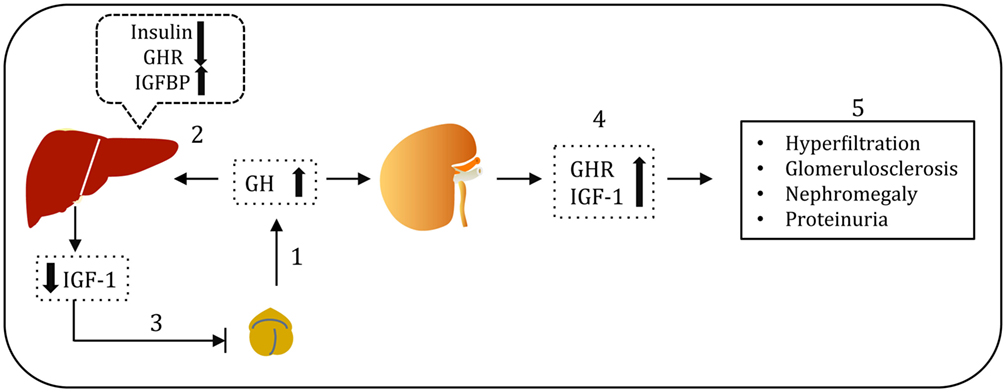
Figure 3. Growth hormone (GH)–GH receptor (GHR)–IGF-1 axis in type 1 diabetes. Pituitary GH acts via GHR and induces hepatic insulin-like growth factor (IGF-1) production, which serves as a surrogate marker for GH action (1). However, in type 1 diabetes, reduced portal insulin levels results in decreased expression of hepatic GHR, impaired IGF-1 production and elevated IGFBP-1 levels (2). Low bioavailability of IGF-1 leads to compensatory GH hypersecretion via negative feedback loop mechanism (3). Except in liver, GHR expression in other tissues including kidney is not compromised. Elevated GH levels in poorly controlled type 1 diabetes associated with elevated GHR and IGF-1 in the kidney (4). Elevated GH levels are implicated in the renal hyperfiltration, glomerulosclerosis, nephromegaly, and proteinuria (5).
Identification of GHR in Podocytes
It had been known for some time that increased levels of circulating GH was associated with glomerular hypertrophy, hyperfiltration, and proteinuria (57, 58). However, it was not clear whether these effects of GH were due to a direct action of GH on various cell types of the glomerulus or an indirect effect secondary to GH’s effects on hemodynamic parameters such as blood pressure and vascular tone (59). A significant advancement in this field was the identification of GHR in mouse and human podocytes with activation of canonical JAK–STAT signaling cascade and MAP kinase pathways in a GH-dependent manner (56). Exposure of podocytes to GH resulted in intracellular redistribution of the JAK2 adapter protein SH2-Bβ, activation of focal adhesion kinase, production of reactive oxygen species, and GH-dependent remodeling of actin cytoskeleton (56). These findings provide evidence for a direct effect of GH on podocytes. Besides podocytes, the GHR is also expressed in mesangial cells, proximal tubule, ascending limb of Henle’s loop, and collecting ducts (60–62). The expression of GHR exhibits a decreasing gradient from the renal cortex to the medulla. GHR levels are approximately 10–20 times higher in the proximal tubule than in downstream segments of the renal tubule.
GH Induces Dedifferentiation of Podocytes
Podocyte loss has been reported in IgA nephropathy, HIV-associated nephropathy, and DN. Several studies have reported the presence of podocytes in urine sediments of patients with glomerular diseases supporting the argument that podocytes detach from GBM (63, 64). It was reported that patients with glomerular diseases shed viable podocytes in urine (65). Interestingly, these podocytes isolated from urine of the patients with the glomerular disease can be cultured under suitable conditions (65). The presence of viable podocytes in urinary sediments suggests that these podocytes could detach from basement membrane (BM) into urinary space (Figure 4).
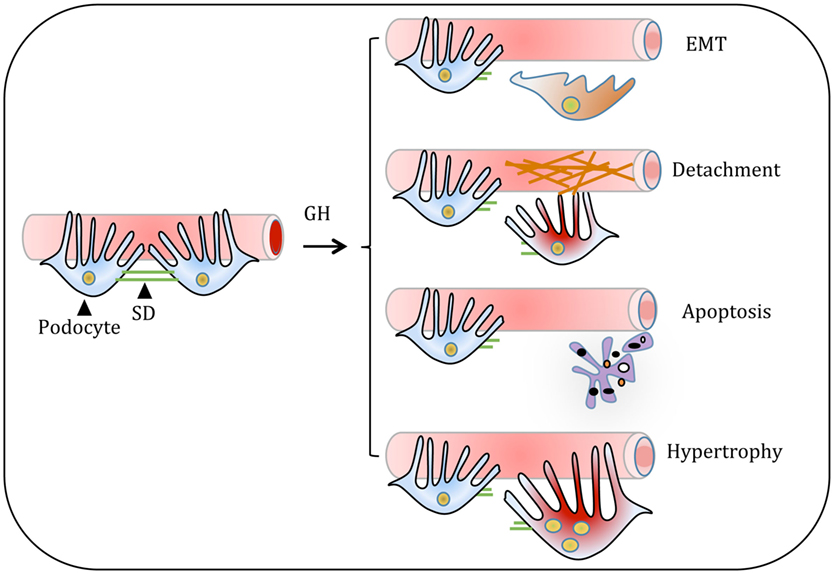
Figure 4. Proposed model for the growth hormone (GH) action on glomerular podocyte. GH-induced cellular events in podocyte include dedifferentiation of podocytes or thickening and/or cross-linking of the basement membrane. Both these events result in the shedding of podocytes. Alternatively, GH induces podocyte apoptosis and hypertrophy. All these changes in podocytes manifest in decreased podocyte count and impair glomerular function in pathological states of overactivity of GH/GH receptor axis such as acromegaly and type 1 diabetes.
Podocytes originate from columnar epithelial cells, which in turn arise from the metanephric mesenchyme via a process termed as mesenchymal–epithelial transition (MET). Columnar epithelial cells possess apical tight junctions and adherent junctions, which migrate basally and transform into the SD during podocyte morphogenesis (66). It is reasonable to posit that in glomerular diseases mature podocytes undergo dedifferentiation that mimics the reversal of MET. Epithelial cells are fully differentiated and immobile having cell–cell junctions and cell–matrix interactions. Epithelial cells express several adhesion molecules such as E-cadherin, epithelial cell adhesion molecule, and tight junction proteins including occludins and junction adhesion molecules. Unlike epithelial cells, mesenchymal cells are motile and invasive and express a distinct set of markers such as vimentin, snail, slug, and N-cadherin (67). Epithelial cells upon undergoing dedifferentiation detach from the substratum by disrupting cell–cell contacts and cytoskeletal rearrangements and attain migratory properties (68) Podocyte dedifferentiation during injury is evidenced by loss of epithelial polarity, loss of cell–cell junctions, and increase in mesenchymal characteristics (69). The process of podocyte dedifferentiation recapitulates some of the features with a fundamental cell remodeling mechanism known as epithelial–mesenchymal transition (EMT) that occurs during embryonic development and wound healing. EMT has been categorized into three types based on their origin: type 1, associated with embryogenesis and organ development; type 2, associated with wound healing and fibrosis; and type 3, associated with cancer cell invasion, migration, and metastasis. The mechanism of podocyte dedifferentiation resembles type 2 EMT (69, 70).
Recent studies shed light on the mechanisms of GH-mediated dedifferentiation in podocytes. Studies from our group revealed that in both animal models and cultured podocytes, GH induces the expression of transcription factor zinc finger E-box binding homeobox2 (ZEB2) (71). ZEB2 orchestrates dedifferentiation of epithelial cells by promoting a cadherin switch—decreasing expression of E- and P-cadherin with a concomitant increase in expression of N-cadherin (71). It was further identified that GH induces the expression of a natural antisense transcript specific to ZEB2 (ZEB2-NAT). ZEB2-NAT, synthesized from 5′UTR of ZEB2 gene in an antisense manner, specifically binds to a complementary region located in the internal ribosomal entry site (IRES) of ZEB2 mRNA (72). The complimentary base pairing of ZEB2-NAT to the ZEB2 mRNA prevents splicing of an IRES element, which in turn facilitates increased translation of ZEB2 mRNA (71). Thus, GH induces dedifferentiation of podocytes by regulating both ZEB2 and ZEB2-NAT. The dedifferentiation has significant consequences on the function of the podocyte, particularly on the epithelial cell–cell junctions: expression of SD is lost and FPs are effaced in the diseased podocytes. It is noteworthy that a recent study has elucidated the role of the microRNA 96-182-183 cluster in GH-dependent cell transition and invasion in breast cancer (73).
GH Promotes Accumulation of ECM Components
Extracellular matrix offers mechanical strength and provides signals for differentiation and tissue morphogenesis. The components of ECM include matrix polysaccharides, structural proteins, and interlinking proteins. The most abundant matrix polysaccharides of ECM are glycosaminoglycans, hyaluronan, and heparan sulfates, which aggregate with linker proteins to form larger complex proteoglycans. Proteoglycans such as agrin and perlecan offer negative charge throughout the GBM (74). Negative charges on the GBM offer electrostatic repulsion to anionic molecules and impede the flow of these molecules into the glomerular filtrate (75). Collagen type IV, type XVII, and nidogen are most prevalent matrix structural proteins of the GBM. Laminin and fibronectin are matrix interlinking proteins known to interact with proteoglycans and collagen. Matrix metalloproteases (MMPs), serine, and cysteine proteases are classes of enzymes that degrade ECM proteins. Tissue inhibitors of metalloproteases (TIPMs) are the known inhibitors of MMPs that are upregulated in the early pathogenesis of DN (76). Together TIPMs and MMPs maintain ECM homeostasis. Excess synthesis and deposition with decreased clearance of ECM proteins in the glomerulus result in glomerulosclerosis.
During DN, accumulation of ECM proteins predominantly occurs in the glomerular mesangium, tubulointerstitium, and GBM. Glomerular endothelial cells (ECs) and podocytes mainly dictate the composition of GBM (77). Mass spectrometry-based proteomics approach revealed that both glomerular ECs and podocytes synthesize more than 50% of ECM proteins that comprises the core of the ECM (77). However, the mechanism(s) for increased expression of ECM proteins during DN is not well understood. Increased deposition of ECM proteins, including collagen type IV, laminin B2, and heparan sulfate proteoglycans, in GH transgenic mice resulted in progressive glomerulosclerosis (53, 78). Studies with GH transgenic mice, experiments with cultured podocytes, and rats injected with GH have provided evidence that GH promotes synthesis and accumulation of ECM components (79). Thickening of GBM and mesangial sclerosis was observed in glomerular sections of rats administered with GH (79, 80). GH administration to diabetic rats showed further increase (30%) in glomerulosclerosis and tubulointerstitial fibrosis (80). A significant decrease in the expression of MMP2 and MMP9 was observed with GH treatment in streptozotocin-induced diabetic male rats (80). MMP2 and MMP9 are the major matrix proteases associated with ECM turnover. In our earlier study, we employed ECM and adhesion molecules profiler PCR array and analyzed the effect of GH on the expression of 84 genes that are important for cell–cell and cell–matrix interactions (81). This array contains ECM proteins including BM constituents, collagens, and other genes defining ECM structure. GH induced the expression of collagen type II α1, integrin E, laminin subunit α3, and laminin subunit β3 (81). The data suggest that GH promotes increased synthesis of ECM components. Coincident with the synthesis of ECM components, GH treatment resulted in reduced expression of MMPs, such as MMP7 and MMP13-15, which degrade extracellular molecules (81). We observed elevated expression of transforming growth factor beta-induced protein (TGFBIp) in podocytes treated with GH (81). TGFBIp is also induced by TGF-β and inhibit cell adhesion and interacts with collagen and other matrix structural proteins including integrins. Overall, these studies indicate that GH increases the synthesis of ECM components and contributes to their cross-linking and stiffness. These effects of GH impede the adhesion of podocytes to the GBM resulting in loss of podocytes (Figure 4).
GH Induces Podocyte Apoptosis
Unlike other cell types of the glomerulus, podocytes are postmitotic, terminally differentiated cells, which can replicate DNA but do not undergo proliferation because they arrest in the G0/G1 phase of cell cycle (82). Podocyte depletion is well documented in DN and apoptosis of podocyte is considered as one of the mechanisms for decreased podocyte count (83). Podocytes undergo apoptosis at early stages in the course of progressive glomerulosclerosis and podocyte loss precedes mesangial expansion. Several nutrients, cytokines, and growth factors differentially contribute to podocyte viability. Insulin and IGF-1 are critical for podocyte survival (84, 85), whereas high concentrations of glucose and TGF-β are known to induce podocyte apoptosis (19, 86).
Studies from our group demonstrated that mouse podocytes exposed to GH undergo apoptosis (81). A time-dependent increase in the rate of GH-induced apoptosis of mouse podocytes was observed (81). GH-induced podocyte apoptosis can be explained in two possible ways. GH induces reactive oxygen species (ROS) in podocytes and these ROS are implicated in cell death (56, 61). Quenching of GH-induced ROS by N-acetyl cysteine prevented GH-mediated podocyte cell death (56). Alternatively, GH-induced TGFBIp is also implicated in podocyte cell death (81). Podocytes exposed to exogenous TGFBIp underwent apoptosis (81). Furthermore, TGFBIp-induced apoptosis has been reported in other renal cell types including renal tubular epithelial cells and retinal ECs (87, 88). Anti-TGFBIp antibody inhibited apoptosis that was induced by exogenous TGFBIp (88). These studies suggest that GH induces podocyte apoptosis that could contribute to decreased podocyte density in conditions, such as poorly controlled diabetes, with elevated concentrations of GH (81).
The Role of GH/GHR Axis in Podocyte Hypertrophy
Renal hypertrophy and enlarged glomeruli are prominent features during the early course of T1DM. This glomerular enlargement is associated with podocyte hypertrophy rather than hyperplasia (Figure 4) (89). It is postulated that hypertrophy of podocytes is the compensatory cellular response for the loss of neighboring podocytes. In addition to compensating for the loss of podocyte function, the podocyte enlargement may also help to cover the areas of on glomerular capillaries that have been exposed because of detachment and loss of podocytes. These advantages notwithstanding the hypertrophic response could be a temporary adaptive response, which could ultimately result in detachment and shedding of podocytes in the urine. Recovery of viable and cultivable podocytes from the urine of subjects with DN supports such a scenario (90). It is noteworthy that 10–50% of viable podocytes in the urinary sediments are multinucleated and can be maintained for several days in in vitro conditions (65). The presence of multinucleated podocytes suggests impaired coordination between karyokinesis and cytokinesis.
Elevated GH levels are associated with renal hypertrophy (91). We have observed an increase in kidney size in rats injected with GH for 7 days. Increase in kidney size in streptozotocin-induced diabetes is ameliorated in mice where GHR in podocytes is knocked out. However, the cellular mechanism(s) regulating podocyte hypertrophy remains unclear. Mammalian target of rapamycin complex1 (mTORC1) regulates cell size. mTORC1 is a major cell survival kinase activated by Akt and inhibited by tuberous sclerosis complex (TSC1/2) (92, 93). Constitutive activation of mTORC1 recapitulated several DN features including podocyte hypertrophy, proteinuria, and glomerular sclerosis (92). Inhibition of podocyte-specific mTORC1 prevents podocyte hypertrophy and progressive glomerulosclerosis during diabetes (93, 94). GH potentially stimulates protein synthesis by activation of mTOR kinase in human cell lines (95). Mice transgenic for GH and null to IGF1 showed substantial podocyte hypertrophy, mesangial hypertrophy, proliferation, and overt proteinuria (91). Although GH actions are partially mediated through IGF-1, studies from GH transgenic mice demonstrated that alterations in the kidney and liver development are independent of IGF-1 (96). Furthermore, GH transgenic mice exhibit higher degrees of glomerular hypertrophy and podocyte damage compared to IGF-1 transgenic mice (53, 54). Conversely, GHR null diabetic mice display a substantial reduction in glomerular hypertrophy and are protected from development of DN (55). Overall, these findings indicate that elevated GH can induce glomerular hypertrophy.
Effects of Blunting GH/GHR Axis in the Diabetic Kidney on Renal Function
On the basis of the association between elevated levels of GH and DN (2), it was hypothesized that blockade of GH action by somatostatin analogs and/or GHR antagonists might reduce the risk of diabetic renal disease. Streptozotocin-induced diabetic rats treated with octreotide, a long-acting somatostatin analog, showed reduced kidney weight and urinary albumin excretion (97). Lanreotide, another somatostatin analog, prevented diabetic renal and glomerular growth suggesting that blunting GH action could prevent experimental diabetic kidney disease (98). Administration of SMS 201-995 (a somatostatin analog) attenuated the kidney weight and ensured a significant decrease in urinary albumin excretion in experimental diabetic rats (99). GH antagonism by PTR-3173 (a somatostatin analog) elicited a blunting effect on renal hypertrophy, albuminuria, and GFR in non-obese diabetic mice (100). GHR/GH binding protein gene-disrupted mice were protected against streptozotocin-induced DN (55). Similar to somatostatin analogs, administration of GHR antagonists also elicited renoprotective effect in diabetic rodent models. GHR antagonist (G120K-PEG) ameliorated renal enlargement, glomerular hypertrophy, and urinary albumin excretion in diabetic mice (101). Furthermore, GHR antagonism offered salutary effects in non-obese diabetic mice as evidenced by normalization of kidney weight, glomerular volume, and attenuation of albuminuria (102). Therefore, modulation of GH effects may have beneficial therapeutic effects in DN.
Summary
Podocytes are the most vulnerable component of the glomerular filtration apparatus. Podocytes have a limited capacity to divide and do not regenerate in response to injury and loss. Therefore, maintaining podocyte integrity and number is critical for normal renal function. Accumulated evidence suggests a role for the GH/GHR axis in the kidney in DN in T1DM. Studies from our group and others demonstrate that elevated levels of GH results in podocyte injury via multiple mechanisms including altering podocyte permselectivity, podocyte phenotypic switch, podocyte detachment, and apoptosis. In the setting of T1DM wherein elevated GH levels are associated with podocyte injury and diminished renal filtration, targeting the GH/GHR axis in podocytes could be a potential approach to combat podocytopathy. Whereas significant progress has been made in our understanding of the role of podocyte injury in DN, several questions related to podocyte repair mechanisms and regulatory pathways remain unanswered and warrant further investigation.
Author Contributions
DM, RN, RM, and AP contributed to the review article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Science and Engineering Research Board, India (EMR/2015/002076) supports research work in AKP laboratory. DM acknowledges the University Grants Commission, India, for providing research fellowship.
References
1. Anil Kumar P, Welsh GI, Saleem MA, Menon RK. Molecular and cellular events mediating glomerular podocyte dysfunction and depletion in diabetes mellitus. Front Endocrinol (2014) 5:151. doi:10.3389/fendo.2014.00151
2. Kumar PA, Brosius FC III, Menon RK. The glomerular podocyte as a target of growth hormone action: implications for the pathogenesis of diabetic nephropathy. Curr Diabetes Rev (2011) 7:50–5. doi:10.2174/157339911794273900
3. Khurana S, Bruggeman LA, Kao HY. Nuclear hormone receptors in podocytes. Cell Biosci (2012) 2:33. doi:10.1186/2045-3701-2-33
4. Larson TS. Evaluation of proteinuria. Mayo Clin Proc (1994) 69:1154–8. doi:10.1016/S0025-6196(12)65767-X
5. Kumar PA, Chitra PS, Reddy GB. Metabolic syndrome and associated chronic kidney diseases: nutritional interventions. Rev Endocr Metab Disord (2013) 14:273–86. doi:10.1007/s11154-013-9268-2
6. Carroll MF, Temte JL. Proteinuria in adults: a diagnostic approach. Am Fam Physician (2000) 62:1333–40.
7. Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev (2003) 83:253–307. doi:10.1152/physrev.00020.2002
8. Ichimura K, Miyazaki N, Sadayama S, Murata K, Koike M, Nakamura K-I, et al. Three-dimensional architecture of podocytes revealed by block-face scanning electron microscopy. Sci Rep (2015) 5:8993. doi:10.1038/srep08993
9. Kriz W. Glomerular diseases: podocyte hypertrophy mismatch and glomerular disease. Nat Rev Nephrol (2012) 8:618–9. doi:10.1038/nrneph.2012.198
10. Nagata M. Podocyte injury and its consequences. Kidney Int (2016) 89:1221–30. doi:10.1016/j.kint.2016.01.012
12. Mundel P, Reiser J. Proteinuria: an enzymatic disease of the podocyte? Kidney Int (2010) 77:571–80. doi:10.1038/ki.2009.424
13. Kriz W, Lemley KV. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol (2015) 26:258–69. doi:10.1681/ASN.2014030278
14. Liapis H, Romagnani P, Anders H-J. New insights into the pathology of podocyte loss: mitotic catastrophe. Am J Pathol (2013) 183:1364–74. doi:10.1016/j.ajpath.2013.06.033
15. Lasagni L, Lazzeri E, Shankland SJ, Anders HJ, Romagnani P. Podocyte mitosis – a catastrophe. Curr Mol Med (2013) 13:13–23. doi:10.2174/156652413804486250
16. Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol (2005) 16:2941–52. doi:10.1681/ASN.2005010055
17. Ilatovskaya DV, Levchenko V, Lowing A, Shuyskiy LS, Palygin O, Staruschenko A. Podocyte injury in diabetic nephropathy: implications of angiotensin II-dependent activation of TRPC channels. Sci Rep (2015) 5:17637. doi:10.1038/srep17637
18. Zhang H, Ding J, Fan Q, Liu S. TRPC6 up-regulation in Ang II-induced podocyte apoptosis might result from ERK activation and NF-kappaB translocation. Exp Biol Med (2009) 234:1029–36. doi:10.3181/0901-RM-11
19. Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, et al. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest (2001) 108:807–16. doi:10.1172/JCI200112367
20. Landau D, Israel E, Rivkis I, Kachko L, Schrijvers BF, Flyvbjerg A, et al. The effect of growth hormone on the development of diabetic kidney disease in rats. Nephrol Dial Transplant (2003) 18:694–702. doi:10.1093/ndt/gfg142
21. Baumann G. Growth hormone heterogeneity: genes, isohormones, variants, and binding proteins. Endocr Rev (1991) 12:424–49. doi:10.1210/edrv-12-4-424
22. Lanning NJ, Carter-Su C. Recent advances in growth hormone signaling. Rev Endocr Metab Disord (2006) 7:225–35. doi:10.1007/s11154-007-9025-5
23. Schlaepfer DD, Hunter T. Focal adhesion kinase overexpression enhances ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interactions with and activation of c-Src. J Biol Chem (1997) 272:13189–95. doi:10.1074/jbc.272.20.13189
24. Smit LS, Meyer DJ, Billestrup N, Norstedt G, Schwartz J, Carter-Su C. The role of the growth hormone (GH) receptor and JAK1 and JAK2 kinases in the activation of Stats 1, 3, and 5 by GH. Mol Endocrinol (1996) 10:519–33. doi:10.1210/me.10.5.519
25. Herrington J, Carter-Su C. Signaling pathways activated by the growth hormone receptor. Trends Endocrinol Metab (2001) 12:252–7. doi:10.1016/S1043-2760(01)00423-4
26. VanderKuur J, Allevato G, Billestrup N, Norstedt G, Carter-Su C. Growth hormone-promoted tyrosyl phosphorylation of SHC proteins and SHC association with Grb2. J Biol Chem (1995) 270:7587–93. doi:10.1074/jbc.270.13.7587
27. Vanderkuur JA, Butch ER, Waters SB, Pessin JE, Guan KL, Carter-Su C. Signaling molecules involved in coupling growth hormone receptor to mitogen-activated protein kinase activation. Endocrinology (1997) 138:4301–7. doi:10.1210/endo.138.10.5453
28. Velloso CP. Regulation of muscle mass by growth hormone and IGF-I. Br J Pharmacol (2008) 154:557–68. doi:10.1038/bjp.2008.153
29. Clemmons DR. The relative roles of growth hormone and IGF-1 in controlling insulin sensitivity. J Clin Invest (2004) 113:25–7. doi:10.1172/JCI20660
30. Lindenbergh-Kortleve DJ, Rosato RR, van Neck JW, Nauta J, van Kleffens M, Groffen C, et al. Gene expression of the insulin-like growth factor system during mouse kidney development. Mol Cell Endocrinol (1997) 132:81–91. doi:10.1016/S0303-7207(97)00123-8
31. Oh Y. The insulin-like growth factor system in chronic kidney disease: pathophysiology and therapeutic opportunities. Kidney Res Clin Pract (2012) 31:26–37. doi:10.1016/j.krcp.2011.12.005
32. Feld S, Hirschberg R. Growth hormone, the insulin-like growth factor system, and the kidney. Endocr Rev (1996) 17:423–80. doi:10.1210/edrv-17-5-423
33. Johannsson G, Sverrisdottir YB, Ellegard L, Lundberg PA, Herlitz H. GH increases extracellular volume by stimulating sodium reabsorption in the distal nephron and preventing pressure natriuresis. J Clin Endocrinol Metab (2002) 87:1743–9. doi:10.1210/jcem.87.4.8394
34. Boger RH, Skamira C, Bode-Boger SM, Brabant G, von zur Muhlen A, Frolich JC. Nitric oxide may mediate the hemodynamic effects of recombinant growth hormone in patients with acquired growth hormone deficiency. A double-blind, placebo-controlled study. J Clin Invest (1996) 98:2706–13. doi:10.1172/JCI119095
35. Menon RK, Stephan DA, Rao RH, Shen-Orr Z, Downs LS Jr, Roberts CT Jr, et al. Tissue-specific regulation of the growth hormone receptor gene in streptozocin-induced diabetes in the rat. J Endocrinol (1994) 142:453–62. doi:10.1677/joe.0.1420453
36. Yakar S, Setser J, Zhao H, Stannard B, Haluzik M, Glatt V, et al. Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1-deficient mice. J Clin Invest (2004) 113:96–105. doi:10.1172/JCI200417763
37. Brismar K, Gutniak M, Povoa G, Werner S, Hall K. Insulin regulates the 35 kDa IGF binding protein in patients with diabetes mellitus. J Endocrinol Invest (1988) 11:599–602. doi:10.1007/BF03350189
38. Salgado LR, Semer M, Nery M, Knoepfelmacher M, Lerario AC, Povoa G, et al. Effect of glycemic control on growth hormone and IGFBP-1 secretion in patients with type I diabetes mellitus. J Endocrinol Invest (1996) 19:433–40. doi:10.1007/BF03349888
39. Munoz MT, Barrios V, Pozo J, Argente J. Insulin-like growth factor I, its binding proteins 1 and 3, and growth hormone-binding protein in children and adolescents with insulin-dependent diabetes mellitus: clinical implications 1. Pediatr Res (1996) 39:992–8. doi:10.1203/00006450-199606000-00011
40. Landau D, Domene H, Flyvbjerg A, Grønbaek H, Roberts CT, Argov S, et al. Differential expression of renal growth hormone receptor and its binding protein in experimental diabetes mellitus. Growth Horm IGF Res (1998) 8:39–45. doi:10.1016/S1096-6374(98)80320-1
41. Hirschberg R, Kopple JD. Effects of growth hormone on GFR and renal plasma flow in man. Kidney Int Suppl (1987) 22:S21–4.
42. Ogle GD, Rosenberg AR, Kainer G. Renal effects of growth hormone. I. Renal function and kidney growth. Pediatr Nephrol (1992) 6:394–8. doi:10.1007/BF00869745
43. Jacobs ML, Derkx FH, Stijnen T, Lamberts SW, Weber RF. Effect of long-acting somatostatin analog (Somatulin) on renal hyperfiltration in patients with IDDM. Diabetes Care (1997) 20:632–6. doi:10.2337/diacare.20.4.632
44. Bak M, Thomsen K, Flyvbjerg A. Effects of the somatostatin analogue octreotide on renal function in conscious diabetic rats. Nephrol Dial Transplant (2001) 16:2002–7. doi:10.1093/ndt/16.10.2002
45. Auriemma RS, Galdiero M, De Martino MC, De Leo M, Grasso LF, Vitale P, et al. The kidney in acromegaly: renal structure and function in patients with acromegaly during active disease and 1 year after disease remission. Eur J Endocrinol (2010) 162:1035–42. doi:10.1530/EJE-10-0007
46. Sonksen PH, Russell-Jones D, Jones RH. Growth hormone and diabetes mellitus. A review of sixty-three years of medical research and a glimpse into the future? Horm Res (1993) 40:68–79.
47. Hirschberg R, Kopple JD. Effects of growth hormone and IGF-I on renal function. Kidney Int Suppl (1989) 27:S20–6.
48. Tonshoff B, Nowack R, Kurilenko S, Blum WF, Seyberth HW, Mehls O, et al. Growth hormone-induced glomerular hyperfiltration is dependent on vasodilating prostanoids. Am J Kidney Dis (1993) 21:145–51. doi:10.1016/S0272-6386(12)81085-7
49. Bank N. Mechanisms of diabetic hyperfiltration. Kidney Int (1991) 40:792–807. doi:10.1038/ki.1991.277
50. Tuttle KR, Bruton JL, Perusek MC, Lancaster JL, Kopp DT, DeFronzo RA. Effect of strict glycemic control on renal hemodynamic response to amino acids and renal enlargement in insulin-dependent diabetes mellitus. N Engl J Med (1991) 324:1626–32. doi:10.1056/NEJM199106063242304
51. O’Shea MH, Layish DT. Growth hormone and the kidney: a case presentation and review of the literature. J Am Soc Nephrol (1992) 3:157–61.
52. Ritz E, Tonshoff B, Worgall S, Kovacs G, Mehls O. Influence of growth hormone and insulin-like growth factor-I on kidney function and kidney growth. Pediatr Nephrol (1991) 5:509–12. doi:10.1007/BF01453692
53. Doi T, Striker LJ, Quaife C, Conti FG, Palmiter R, Behringer R, et al. Progressive glomerulosclerosis develops in transgenic mice chronically expressing growth hormone and growth hormone releasing factor but not in those expressing insulinlike growth factor-1. Am J Pathol (1988) 131:398–403.
54. Doi T, Striker LJ, Gibson CC, Agodoa LY, Brinster RL, Striker GE. Glomerular lesions in mice transgenic for growth hormone and insulinlike growth factor-I. I. Relationship between increased glomerular size and mesangial sclerosis. Am J Pathol (1990) 137:541–52.
55. Bellush LL, Doublier S, Holland AN, Striker LJ, Striker GE, Kopchick JJ. Protection against diabetes-induced nephropathy in growth hormone receptor/binding protein gene-disrupted mice. Endocrinology (2000) 141:163–8. doi:10.1210/endo.141.1.7284
56. Reddy GR, Pushpanathan MJ, Ransom RF, Holzman LB, Brosius FC III, Diakonova M, et al. Identification of the glomerular podocyte as a target for growth hormone action. Endocrinology (2007) 148:2045–55. doi:10.1210/en.2006-1285
57. Bak M, Thomsen K, Christiansen T, Flyvbjerg A. Renal enlargement precedes renal hyperfiltration in early experimental diabetes in rats. J Am Soc Nephrol (2000) 11:1287–92.
58. Thirone AC, Scarlett JA, Gasparetti AL, Araujo EP, Lima MH, Carvalho CR, et al. Modulation of growth hormone signal transduction in kidneys of streptozotocin-induced diabetic animals: effect of a growth hormone receptor antagonist. Diabetes (2002) 51:2270–81. doi:10.2337/diabetes.51.7.2270
59. Haffner D, Ritz E, Mehls O, Rosman J, Blum W, Heinrich U, et al. Growth hormone induced rise in glomerular filtration rate is not obliterated by angiotensin-converting enzyme inhibitors. Nephron (1990) 55:63–8. doi:10.1159/000185920
60. Kamenicky P, Mazziotti G, Lombes M, Giustina A, Chanson P. Growth hormone, insulin-like growth factor-1, and the kidney: pathophysiological and clinical implications. Endocr Rev (2014) 35:234–81. doi:10.1210/er.2013-1071
61. Doi SQ, Jacot TA, Sellitti DF, Hirszel P, Hirata MH, Striker GE, et al. Growth hormone increases inducible nitric oxide synthase expression in mesangial cells. J Am Soc Nephrol (2000) 11:1419–25.
62. Kamenicky P, Viengchareun S, Blanchard A, Meduri G, Zizzari P, Imbert-Teboul M, et al. Epithelial sodium channel is a key mediator of growth hormone-induced sodium retention in acromegaly. Endocrinology (2008) 149:3294–305. doi:10.1210/en.2008-0143
63. Nakamura T, Ushiyama C, Suzuki S, Hara M, Shimada N, Ebihara I, et al. Urinary excretion of podocytes in patients with diabetic nephropathy. Nephrol Dial Transplant (2000) 15:1379–83. doi:10.1093/ndt/15.9.1379
64. Mundel P. Urinary podocytes: lost and found alive. Kidney Int (2003) 64:1529–30. doi:10.1046/j.1523-1755.2003.00339.x
65. Vogelmann SU, Nelson WJ, Myers BD, Lemley KV. Urinary excretion of viable podocytes in health and renal disease. Am J Physiol Renal Physiol (2003) 285:F40–8. doi:10.1152/ajprenal.00404.2002
66. Reidy K, Susztak K. Epithelial-mesenchymal transition and podocyte loss in diabetic kidney disease. Am J Kidney Dis (2009) 54:590–3. doi:10.1053/j.ajkd.2009.07.003
67. Diepenbruck M, Christofori G. Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr Opin Cell Biol (2016) 43:7–13. doi:10.1016/j.ceb.2016.06.002
68. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15:178–96. doi:10.1038/nrm3758
69. May CJ, Saleem M, Welsh GI. Podocyte dedifferentiation: a specialized process for a specialized cell. Front Endocrinol (2014) 5:148. doi:10.3389/fendo.2014.00148
70. Nishad R, Nakuluri K, Motrapu M, Pasupulati AK. Epithelial-mesenchymal transition of glomerular podocytes: implications in proteinuria. MGM J Med Sci (2017) 4:1–9. doi:10.5005/jp-journals-10036-1133
71. Kumar PA, Kotlyarevska K, Dejkhmaron P, Reddy GR, Lu C, Bhojani MS, et al. Growth hormone (GH)-dependent expression of a natural antisense transcript induces zinc finger E-box-binding homeobox 2 (ZEB2) in the glomerular podocyte: a novel action of gh with implications for the pathogenesis of diabetic nephropathy. J Biol Chem (2010) 285:31148–56. doi:10.1074/jbc.M110.132332
72. Beltran M, Puig I, Pena C, Garcia JM, Alvarez AB, Pena R, et al. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev (2008) 22:756–69. doi:10.1101/gad.455708
73. Zhang W, Qian P, Zhang X, Zhang M, Wang H, Wu M, et al. Autocrine/paracrine human growth hormone-stimulated microRNA 96-182–183 cluster promotes epithelial-mesenchymal transition and invasion in breast cancer. J Biol Chem (2015) 290:13812–29. doi:10.1074/jbc.M115.653261
74. Miner JH. The glomerular basement membrane. Exp Cell Res (2012) 318:973–8. doi:10.1016/j.yexcr.2012.02.031
75. Menzel S, Moeller MJ. Role of the podocyte in proteinuria. Pediatr Nephrol (2011) 26:1775–80. doi:10.1007/s00467-010-1725-5
76. Genovese F, Manresa AA, Leeming DJ, Karsdal MA, Boor P. The extracellular matrix in the kidney: a source of novel non-invasive biomarkers of kidney fibrosis? Fibrogenesis Tissue Repair (2014) 7:4. doi:10.1186/1755-1536-7-4
77. Byron A, Randles MJ, Humphries JD, Mironov A, Hamidi H, Harris S, et al. Glomerular cell cross-talk influences composition and assembly of extracellular matrix. J Am Soc Nephrol (2014) 25:953–66. doi:10.1681/ASN.2013070795
78. Machado MO, Hirata RD, Sellitti DF, Iotti R, Iotti A, Cusumano AM, et al. Growth hormone promotes glomerular lipid accumulation in bGH mice. Kidney Int (2005) 68:2019–28. doi:10.1111/j.1523-1755.2005.00656.x
79. Østerby R, Seyer-Hansen K, Gundersen HJG, Lundbaek K. Growth hormone enhances basement membrane thickening in experimental diabetes. Diabetologia (1978) 15:487–9. doi:10.1007/BF02342875
80. Whitney JL, Bilkan CM, Sandberg K, Myers AK, Mulroney SE. Growth hormone exacerbates diabetic renal damage in male but not female rats. Biol Sex Differ (2013) 4:12. doi:10.1186/2042-6410-4-12
81. Chitra PS, Swathi T, Sahay R, Reddy GB, Menon RK, Kumar PA. Growth hormone induces transforming growth factor-beta-induced protein in podocytes: implications for podocyte depletion and proteinuria. J Cell Biochem (2015) 116:1947–56. doi:10.1002/jcb.25150
82. Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney Int (2006) 69:2131–47. doi:10.1038/sj.ki.5000410
83. Eid AA, Gorin Y, Fagg BM, Maalouf R, Barnes JL, Block K, et al. Mechanisms of podocyte injury in diabetes. Diabetes (2009) 58:1201. doi:10.2337/db08-1536
84. Bridgewater DJ, Ho J, Sauro V, Matsell DG. Insulin-like growth factors inhibit podocyte apoptosis through the PI3 kinase pathway. Kidney Int (2005) 67:1308–14. doi:10.1111/j.1523-1755.2005.00208.x
85. Welsh GI, Hale LJ, Eremina V, Jeansson M, Maezawa Y, Lennon R, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab (2010) 12:329–40. doi:10.1016/j.cmet.2010.08.015
86. Lee SH, Moon SJ, Paeng J, Kang HY, Nam BY, Kim S, et al. Podocyte hypertrophy precedes apoptosis under experimental diabetic conditions. Apoptosis (2015) 20:1056–71. doi:10.1007/s10495-015-1134-0
87. Mondragon AA, Betts-Obregon BS, Moritz RJ, Parvathaneni K, Navarro MM, Kim HS, et al. BIGH3 protein and macrophages in retinal endothelial cell apoptosis. Apoptosis (2015) 20:29–37. doi:10.1007/s10495-014-1052-6
88. Moritz RJ, LeBaron RG, Phelix CF, Rupaimoole R, Kim HS, Tsin A, et al. Macrophage TGF-beta1 and the proapoptotic extracellular matrix protein BIGH3 induce renal cell apoptosis in prediabetic and diabetic conditions. Int J Clin Med (2016) 7:496–510. doi:10.4236/ijcm.2016.77055
89. Wiggins JE, Goyal M, Sanden SK, Wharram BL, Shedden KA, Misek DE, et al. Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: prevention by calorie restriction. J Am Soc Nephrol (2005) 16:2953–66. doi:10.1681/ASN.2005050488
90. Petermann AT, Pippin J, Krofft R, Blonski M, Griffin S, Durvasula R, et al. Viable podocytes detach in experimental diabetic nephropathy: potential mechanism underlying glomerulosclerosis. Nephron Exp Nephrol (2004) 98:e114–23. doi:10.1159/000081555
91. Blutke A, Schneider MR, Wolf E, Wanke R. Growth hormone (GH)-transgenic insulin-like growth factor 1 (IGF1)-deficient mice allow dissociation of excess GH and IGF1 effects on glomerular and tubular growth. Physiol Rep (2016) 4:e12709. doi:10.14814/phy2.12709
92. Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest (2011) 121:2181–96. doi:10.1172/JCI44771
93. Godel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest (2011) 121:2197–209. doi:10.1172/JCI44774
94. Mori H, Inoki K, Masutani K, Wakabayashi Y, Komai K, Nakagawa R, et al. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem Biophys Res Commun (2009) 384:471–5. doi:10.1016/j.bbrc.2009.04.136
95. Hayashi AA, Proud CG. The rapid activation of protein synthesis by growth hormone requires signaling through mTOR. Am J Physiol Endocrinol Metab (2007) 292:E1647–55. doi:10.1152/ajpendo.00674.2006
96. Blutke A, Schneider MR, Renner-Muller I, Herbach N, Wanke R, Wolf E. Genetic dissection of IGF1-dependent and -independent effects of permanent GH excess on postnatal growth and organ pathology of mice. Mol Cell Endocrinol (2014) 394:88–98. doi:10.1016/j.mce.2014.07.002
97. Flyvbjerg A, Marshall SM, Frystyk J, Hansen KW, Harris AG, Orskov H. Octreotide administration in diabetic rats: effects on renal hypertrophy and urinary albumin excretion. Kidney Int (1992) 41:805–12. doi:10.1038/ki.1992.124
98. Gronbaek H, Nielsen B, Frystyk J, Flyvbjerg A, Orskov H. Effect of lanreotide on local kidney IGF-I and renal growth in experimental diabetes in the rat. Exp Nephrol (1996) 4:295–303.
99. Igarashi K, Nakazawa A, Tani N, Yamazaki M, Ito S, Shibata A. Effect of a somatostatin analogue (SMS 201–995) on renal function and urinary protein excretion in diabetic rats. J Diabet Complications (1991) 5:181–3. doi:10.1016/0891-6632(91)90066-X
100. Landau D, Segev Y, Afargan M, Silbergeld A, Katchko L, Podshyvalov A, et al. A novel somatostatin analogue prevents early renal complications in the nonobese diabetic mouse. Kidney Int (2001) 60:505–12. doi:10.1046/j.1523-1755.2001.060002505.x
101. Flyvbjerg A, Bennett WF, Rasch R, Kopchick JJ, Scarlett JA. Inhibitory effect of a growth hormone receptor antagonist (G120K-PEG) on renal enlargement, glomerular hypertrophy, and urinary albumin excretion in experimental diabetes in mice. Diabetes (1999) 48:377–82. doi:10.2337/diabetes.48.2.377
Keywords: growth hormone, podocytes, diabetic nephropathy, zinc finger E-box binding homeobox2, dedifferentiation, hypertrophy
Citation: Mukhi D, Nishad R, Menon RK and Pasupulati AK (2017) Novel Actions of Growth Hormone in Podocytes: Implications for Diabetic Nephropathy. Front. Med. 4:102. doi: 10.3389/fmed.2017.00102
Received: 25 March 2017; Accepted: 26 June 2017;
Published: 12 July 2017
Edited by:
Maik Gollasch, Charité Universitätsmedizin Berlin, GermanyReviewed by:
Marcela Abbott Galvão Ururahy, Federal University of Rio Grande do Norte, BrazilDeepak Nihalani, Medical University of South Carolina, United States
Copyright: © 2017 Mukhi, Nishad, Menon and Pasupulati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anil Kumar Pasupulati, cGFzdXB1bGF0aS5hbmlsa3VtYXJAZ21haWwuY29t