 Maria Rosaria Galdiero1
Maria Rosaria Galdiero1 Gilda Varricchi1
Gilda Varricchi1 Mansour Seaf2
Mansour Seaf2 Giancarlo Marone3
Giancarlo Marone3 Francesca Levi-Schaffer2*
Francesca Levi-Schaffer2* Gianni Marone1,4*
Gianni Marone1,4*
- 1Department of Translational Medical Sciences (DiSMeT), Center for Basic and Clinical Immunology Research (CISI), University of Naples Federico II, Naples, Italy
- 2Pharmacology and Experimental Therapeutics Unit, Faculty of Medicine, School of Pharmacy, Institute for Drug Research, The Hebrew University of Jerusalem, Jerusalem, Israel
- 3Department of Public Health, University of Naples Federico II, Monaldi Hospital Pharmacy, Naples, Italy
- 4Institute of Experimental Endocrinology and Oncology “Gaetano Salvatore” (IEOS), National Research Council (CNR), Naples, Italy
Human mast cells (MCs) and eosinophils were first described and named by Paul Ehrlich. These cells have distinct myeloid progenitors and differ morphologically, ultrastructurally, immunologically, biochemically, and pharmacologically. However, MCs and eosinophils play a pivotal role in several allergic disorders. In addition, these cells are involved in autoimmune disorders, cardiovascular diseases, and cancer. MCs are distributed throughout all normal human tissues, whereas eosinophils are present only in gastrointestinal tract, secondary lymphoid tissues, and adipose tissue, thymus, mammary gland, and uterus. However, in allergic disorders, MCs and eosinophils can form the “allergic effector unit.” Moreover, in several tumors, MCs and eosinophils can be found in close proximity. Therefore, it is likely that MCs have the capacity to modulate eosinophil functions and vice versa. For example, interleukin 5, stem cell factor, histamine, platelet-activating factor (PAF), prostaglandin D2 (PGD2), cysteinyl leukotrienes, and vascular endothelial growth factors (VEGFs), produced by activated MCs, can modulate eosinophil functions through the engagement of specific receptors. In contrast, eosinophil cationic proteins such as eosinophil cationic protein and major basic protein (MBP), nerve growth factor, and VEGFs released by activated eosinophils can modulate MC functions. These bidirectional interactions between MCs and eosinophils might be relevant not only in allergic diseases but also in several inflammatory and neoplastic disorders.
Introduction
Mast cells (MCs) and eosinophils are important cells of the immune system with critical roles in allergic (1–3) and autoimmune disorders (4–7), cardiovascular diseases (8–15), and cancer (16–19). Human MCs and eosinophils were first described and named in 1878 and 1879, respectively, by Paul Ehrlich who discovered their property to be stained by specific dyes (20–22). Mature MCs and eosinophils differ morphologically, ultrastructurally, immunologically, biochemically, and pharmacologically (23, 24). Moreover, they synthesize a plethora of distinct mediators and display a constellation of different surface receptors (24, 25).
The recent assessment of the transcriptional profiles of MCs and eosinophils revealed the MC heterogeneity across different tissues and their different gene expression program compared to eosinophils (26). The latter findings are consistent with the identification of a distinct myeloid progenitor expressing the gene encoding the transcription factor GATA-1, which generates separately eosinophils and MCs (27). Human MCs derive from CD34+, CD117+ pluripotent hematopoietic stem cells in the bone marrow (28). MC progenitors enter the circulation and complete their maturation in different tissues such as skin, bronchi, tonsils, nasal and intestinal mucosa, conjunctiva, lymph nodes, and breast parenchyma (29). The main differentiation, maturation, survival, priming, and chemotactic factor for human MCs is stem cell factor (SCF), which acts by binding the tyrosine kinase receptor Kit (CD117) (30). CD34+ IL-5Rα+ eosinophil lineage-committed progenitors give rise to mature cells in the bone marrow under the control of critical transcription factors such as GATA-1, PU-1, and C/EBP (31). Eosinophil maturation in the bone marrow is driven by interleukin 5 (IL-5), IL-3, and granulocyte-macrophage colony-stimulating factor (GM-CSF) that share the common receptor β subunit (βc) (3, 32). Recent evidence indicates that IL-33 precedes IL-5 in regulating eosinophil commitment and is required for eosinophil homeostasis (33). Under the effect of chemotactic stimuli, together with IL-5, eosinophils migrate to the gastrointestinal tract, secondary lymphoid tissues, and adipose tissue, thymus, mammary gland, and uterus, where they reside under homeostatic conditions (see Marichal et al. in this issue) (34–36). In response to inflammatory stimuli (e.g., eotaxins), peripheral blood eosinophils migrate into inflamed tissues, where their survival is presumably prolonged (37, 38).
Bidirectional MC–Eosinophil Interactions
Although human MCs are distributed throughout, nearly all normal tissues (39, 40) their density is increased at sites of allergic reactions (1), autoimmune disorders (4, 5), and at the edge of several solid (41–55) and hematologic tumors (56–64). In several allergic disorders (e.g., bronchial asthma, allergic rhinitis, chronic urticaria, and eosinophilic esophagitis), MCs and eosinophils can be found in close proximity forming the “allergic effector unit” (AEU) (65). In addition, there is in vitro evidence that the physical interaction between MCs and eosinophils induces a hyperactivation state and release of soluble mediators (65–67). Therefore, MCs likely have the capacity to modulate eosinophil functions and vice versa. We discuss examples of such two-way interactions below.
MC Mediators
Histamine, released immunologically and non-immunologically from MCs, induces eosinophil chemotaxis through the engagement of the histamine 4 receptor (H4R) (68, 69). Similar to eosinophils, histamine-induced chemotaxis can be also observed in MCs (69).
Adenosine, an endogenous nucleoside released by activated MCs (70), acts in an autocrine and paracrine fashion via binding to four G protein-coupled receptors: the A1, A2a, A2b, and A3 receptors (71) and is involved in airway hyperresponsiveness in asthma (72). Adenosine and its stable analogs potentiate mediator release from human lung MCs (HLMCs) (73, 74) through the activation of adenosine receptors (75) and modulate eosinophil functions (76, 77). MC tryptase can stimulate eosinophil activation and degranulation by cleavage of protease-activated receptor 2 (78).
Eosinophil Mediators
On the other side, eosinophil granule proteins such as MBP and eosinophil cationic protein (ECP) act as complete secretagogues on MCs isolated from human heart (HHMC) (8, 9). ECP, and to a lesser extent MBP, induces the release of histamine and tryptase and the de novo synthesis of PGD2 from HHMC. This observation highlights a mechanism by which infiltrating eosinophils can cause myocardial damage in patients with eosinophilia (3, 79–84). ECP and MBP do not induce histamine release from isolated HLMCs (8, 9). Interestingly, Piliponsky et al. reported that HLMCs became responsive to MBP only in coculture with human lung fibroblasts (85). Recently, the Mas-related gene X2 (MRGPRX2) has been identified as a receptor for several basic peptides on human and rodent MCs (26, 86), and indeed ECP and MBP activate human MCs through the interaction of the MRGPRX2 receptor expressed on their surface (87). Eosinophil MBP-1 activates MCs through the interaction with integrin-β1 (88).
MC and Eosinophil Mediators
Stem cell factor (SCF) is a potent activator of human MCs (89, 90) and induces the release of eosinophil peroxidase (EPO) and cysteinyl leukotriene C4 (LTC4) from eosinophils (91). SCF, produced by both human MCs (90) and eosinophils (92), acts on Kit receptor (CD117) on MCs (30) and eosinophils (93).
Osteopontin (OPN) is a multifunctional glycoprotein implicated in allergic disorders and cancer. OPN can be released by IL-5-activated human eosinophils and induces their migration (94). OPN is also produced by MCs and modulates their IgE-mediated degranulation and migration (95).
Interleukin-5, produced by human MCs, activates the IL-5R, highly expressed on the surface of human eosinophils, basophils, and MCs (96). In addition to MCs, Th2 cells, group 2 innate lymphoid cells (ILC2), invariant NK T cells, and eosinophils themselves are major cellular sources of IL-5 (97). GM-CSF released by activated human MCs (98), and eosinophils binds its receptor expressed by both cell types (99). The cysteinyl leukotrienes (CysLTs LTC4 and LTD4), produced by activated MCs (18, 100), stimulate the proliferation of eosinophil progenitors in the presence of IL-5 and GM-CSF (101). In addition, CysLTs acting through CysLTR1/2 induce the release of IL-4 from human eosinophils (102). PGD2 is the major cyclooxygenase metabolite released by activated MCs (8) and a minor product of eosinophils (103). PGD2 is involved in asthma and allergic rhinitis (104, 105), mastocytosis, rheumatoid arthritis, and cardiac dysfunction (6, 106). PGD2 induces eosinophil and MC chemotaxis in a paracrine and autocrine fashion via binding to CRTH2 receptor on these cells (107, 108). Platelet-activating factor (PAF), synthesized by human MCs and eosinophils (109, 110), is involved in asthma (111) and exerts multiple effects on eosinophils (112, 113).
Nerve growth factor (NGF), produced by both MCs (114, 115) and eosinophils (116, 117), is increased in patients with asthma (118). NGF enhances MC survival (119) through the activation of TrkA receptor (115). NGF is preformed in and activates human eosinophils (116).
Human MCs produce several proangiogenic (VEGF-A, VEGF-B, and FGF-2) (120–125) and lymphangiogenic factors (VEGF-C and VEGF-D) (100, 124). Human eosinophils induce angiogenesis (126) through the production of VEGF-A (127, 128), MBP (129), and OPN (94). Interestingly, VEGF-A, produced by both MCs and eosinophils, is also chemotactic for MCs through the engagement of VEGFR-1/2 present on their surface (124).
The bidirectional interactions between MCs and eosinophils mediated by soluble mediators and the autocrine modulation of these cells are schematically illustrated in Figures 1A,B.
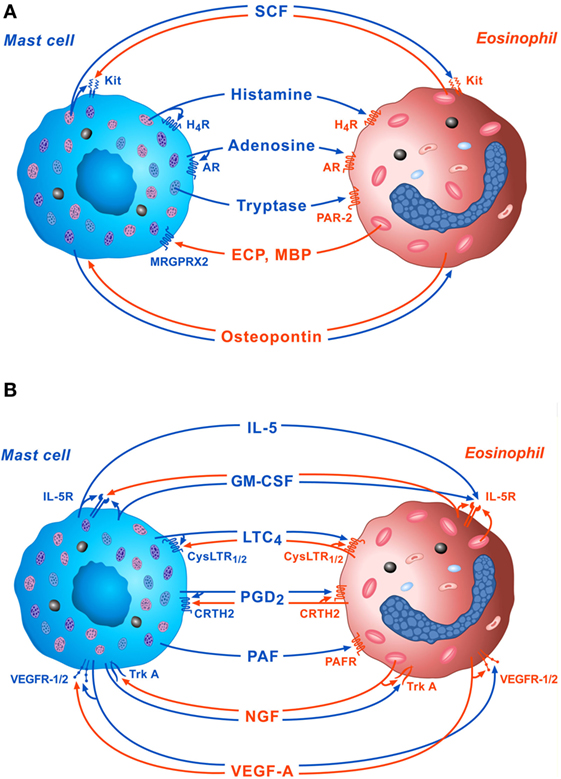
Figure 1. Schematic representation of some of the bidirectional interactions between MCs and eosinophils. (A) Several preformed mediators such as stem cell factor (SCF), histamine, adenosine, and tryptase, released by activated MCs can exert paracrine and/or autocrine functions through the engagement of Kit, H4R, adenosine receptors, and protease-activated receptor 2 (PAR-2), respectively. On the other side, cationic proteins [eosinophil cationic protein (ECP) and MBP], released by activated eosinophils modulate mast cell functions through the activation of MRGPRX2 on their surface. Osteopontin released by both activated eosinophils and MCs exert paracrine and autocrine effects. (B) Several de novo synthesized mediators such as IL-5, granulocyte-macrophage colony-stimulating factor (GM-CSF), LTC4, PGD2, platelet-activating factor (PFA), nerve growth factor (NGF), and VEGF-A, released by activated MCs, can modulate eosinophil functions via the activation of IL-5R, CysLTR1/2, CRTH2, platelet-activating factor receptor (PAFR), TrkA, and VEGF-R1/2, respectively, on their surface. IL-5, GM-CSF, LTC4, PGD2, NGF, and VEGF-A can also exert paracrine and/or autocrine effects.
Disorders in Which MCs and Eosinophils are Present and Likely Drive Disease Pathogenesis
Asthma
Asthma is a chronic inflammatory disorder of the airways in which cells of the innate and adaptive immune system act together with epithelial cells to cause bronchial hyperreactivity, mucus overproduction, airway wall remodeling, angiogenesis, and airway narrowing (123, 130, 131). MCs and their mediators display important roles in the pathogenesis of asthma (2, 39). Indeed, MC-derived histamine, proteases, chemotactic factors, cytokines, and metabolites of arachidonic acid act on vasculature, smooth muscle, connective tissue, goblet cells, and inflammatory cells in the airway inducing acute bronchoconstriction (1). MCs synthesize and release a vast array of pro-inflammatory chemokines and cytokines and recruit other immune cells, such as eosinophils, activated macrophages, and lymphocytes. Therefore, MCs are involved both in the early and the late phases of allergic responses in sensitized individuals (132). Compelling evidence suggests that in asthma MCs are constantly activated resulting in enhanced mediator release and the establishment of chronic airway inflammation. Moreover, MCs reside close to key structures of the bronchial wall, such as airway smooth muscle (ASM) epithelium and submucosal glands, contributing to ASM hypertrophy and other remodeling features (133).
Numerous stages of the MC life cycle have the potential for therapeutic intervention in allergic disorders (134). Targeting the progenitor recruitment offers an upstream checkpoint that could be used to limit tissue MC activity. However, since the mechanisms regulating MC progenitor recruitment to the human lung are not fully understood, no potential therapeutic targets at this level of MC biology have been defined so far. Once within tissue, MC survival, growth, differentiation, and maturation are driven by the local cytokine melieu, with a pivotal role played by SCF and its receptor Kit, which retains protein tyrosine kinase (TK) activity. MC eradication via TK inhibitors may also be a means to treat MC-driven diseases such as asthma. Indeed, the TK inhibitor imatinib decreased airway hyperresponsiveness, MC counts, and tryptase release in patients with severe asthma (135). In addition, masitinib, an inhibitor of Kit and the platelet-derived growth factor receptor, showed some benefit in a small phase II trial over 16 weeks in severe glucocorticoid-dependent asthma (136).
High-affinity receptor for the Fc region of IgE (FcεRI) is expressed on MCs and basophils as a tetrameric complex of three chains with the stoichiometry αβγ2. FcεRI is also expressed in either a trimeric form, αγ2, or the tetramer, on a range of other cell types [e.g., various antigen-presenting cells (APCs), dendritic cells, Langerhans cells, macrophages, eosinophils, and platelets] contributing to IgE-mediated allergic pathophysiology (137). The “low affinity” FcεRII, first discovered on B cells, is also expressed on several other cell types, including various APCs, and also airway and gut epithelial cells (137). FcεRI-dependent MC activation occurs following exposure to allergens, bacterial and viral superantigens, and IgE antibodies. This pathway has been targeted successfully with omalizumab, which prevents IgE binding to the FcεRI and has been approved for use in asthma and chronic urticaria (138, 139). Indeed, anti-IgE therapy with omalizumab, added to medium- or high-dose inhaled glucocorticoids, has proven effective in the treatment of patients with moderate-to-severe and severe allergic (IgE-mediated) asthma by reducing exacerbations and associated use of systemic glucocorticoids in addition to improving other clinical outcomes (140, 141). Since omalizumab reduces the expression of FcεRI on circulating basophils and MCs, it seems to lower the activity potentials of basophils and MCs, thereby reducing the potential reactivity of these cells. Concordantly, serum tryptase was reported to decrease under omalizumab therapy in two mastocytosis patients, but it remained unchanged in two other patients (142). A recent study performed on 18 non-atopic asthmatic patients showed improved lung function and reduced total bronchial mucosal IgE+ cells in bronchial biopsies, but not changed total MCs, plasma cells, B lymphocytes, eosinophils, and plasmablast (143). A pooled analysis of five randomized, double-blind, placebo-controlled trials demonstrated that the reduction of serum-free IgE by omalizumab was associated with a reduction in peripheral eosinophil counts in patients with moderate-to-severe asthma receiving moderate-to-high doses of glucocorticoids [see Stokes in this issue and Ref. (144)]. Smaller studies already reported an inhibitory effect of omalizumab on eosinophils, in the peripheral blood and in the sputum or in bronchial biopsies, but no significant results on tissue MC numbers (145–149). A decrease in blood eosinophilia during omalizumab therapy was proposed as predictor of less asthma exacerbations (150) as well as higher IL-13 levels in sputum predicted the response to omalizumab (151). However, despite these clinical evidences, the mechanisms whereby reductions in circulating IgE lead to a reduction in eosinophils remain unclear. It is possible that omalizumab leads to the inhibition of the release of pro-inflammatory mediators, cytokines, and chemokines from MCs/basophils or inhibition of the allergen-induced differentiation of T cells to Th2 cells by reducing the expression of FcεRI on APCs. Indeed, omalizumab was shown to reduce IL-4+ cells in the bronchial submucosa (145). A reduction in circulating IL-13 has also been reported in patients with moderate-to-severe allergic asthma treated with omalizumab (147). A decrease in eotaxin expression in exhaled breath condensate, exhaled NO, eosinophil blood count, serum ECP after 16 weeks of omalizumab treatment was observed (152). Increased eosinophil apoptosis and reduced numbers of GM-CSF+ lymphocytes have been observed in peripheral blood of omalizumab-treated patients with coexisting allergic asthma and rhinitis, which may also contribute to the inhibitory action of omalizumab on eosinophils (146). A direct effect of omalizumab on eosinophils may be possible via the FcεRI that have been detected on eosinophils, even though their functional significance has yet to be established (153).
Given the pivotal role played by eosinophils in the pathogenesis of severe eosinophilic asthma (3, 154), targeting IL-5 or IL-5Rα appears an interesting therapeutic approach (3, 131). Several randomized, double-blind, placebo-controlled studies demonstrated that mepolizumab (155, 156), reslizumab (157, 158), and benralizumab (159, 160) improved lung function and decreased asthma exacerbations in adult patients with severe eosinophilic asthma.
Eosinophilic Esophagitis
Eosinophils, normally present in the gastrointestinal tract, are absent in the esophagus of healthy subjects. Eosinophilic esophagitis (EoE) is a chronic, immune-mediated esophageal disease, characterized by dysphagia, abdominal pain, and presence of ≥15 eosinophils/field at 400× magnification in the proximal and distal esophagus (161). In EoE, eosinophils are present in all layers of the esophagus, but predominate in the lamina propria and submucosal regions, and are considered the main effector cells in this disorder (161). Activated MCs and their products (e.g., TGF-β) have been described in the esophageal biopsies of active EoE patients (162, 163). The relative contribution of MCs and eosinophils to disease pathogenesis is still under investigation. There is no evidence supporting MC-targeting therapies in EoE (164–166). However, an open label, single arm, unblinded small study showed a statistically significant reduction in MCs and eosinophils in endoscopic biopsies of EoE patients following omalizumab treatment, which correlated with clinical outcome (167). IL-5 targeting therapies resulted in a reduction of esophageal inflammation, but only in minimal symptom relief (168). Interestingly, mepolizumab did not deplete eosinophils nor MCs in the duodenal mucosa of patients (169). In contrast, a pediatric retrospective study showed a reduction in esophageal eosinophil numbers upon mepolizumab treatment, which was more pronounced in a subgroup of responders that also displayed a marked reduction of tryptase+ MCs after treatment. These esophageal MCs were found adjacent to eosinophils, and the frequency of these MC/eosinophil couplets in the esophagus of the responders was reduced after mepolizumab treatment. Moreover, activated MBP+ eosinophils and unidentified cells adjacent to tryptase+ MCs in the esophagus produced IL-9, a pleiotropic cytokine with a pivotal role in activation and maturation of MCs. Interestingly, the authors reported that the esophageal MC numbers correlated with the severity of EoE symptoms, but the reduction of eosinophil numbers did not correlate with symptoms severity. In the subgroup of patients with a greater than 70% decrease in MC density, numbers of MCs correlated with the severity of symptoms. By contrast, there was no correlation between eosinophil numbers and symptom severity. This study suggests an additional role for eosinophils in EoE, as providers of IL-9 that promotes esophageal mastocytosis and indicates that interactions between MCs and eosinophils can regulate the severity of EoE symptoms (170). Reslizumab reduced intraepithelial esophageal eosinophils without improvements in symptoms (171). Thus, although the involvement of eosinophils and presumably MCs in EoE is likely, their relative contribution to the pathogenesis and symptoms of EoE is not fully understood.
Eosinophilic Granulomatosis with Polyangiitis (EGPA)
Eosinophilic granulomatosis with polyangiitis, previously known as Churg–Strauss syndrome, is characterized by increased blood level of IL-5 and eosinophilia in peripheral blood and affected tissues (172). In EGPA, eosinophilic inflammation affects the upper (chronic rhinosinusitis) and lower airways (asthma) (173). Endocardial inflammation, coronary vasculitis, and pericarditis can be observed in patients with EGPA (79, 80). A preliminary study in a small group of EGPA patients demonstrated the efficacy of mepolizumab in reducing blood eosinophils, but not in improving the pulmonary functions (174). A recent multicenter, double-blind, parallel-group, phase 3 trial demonstrated that in patients with EGPA mepolizumab (300 mg s.c. every 4 weeks) was associated with more accrued time in remission than was placebo, which allowed for reductions in the glucocorticoid dose over a period of 52 weeks (175). We have found that omalizumab resulted in clinical improvement of asthma, reduction of peripheral blood eosinophils, and prednisone administration in EGPA patients (173, 176). However, the role of MC in the pathogenesis of EGPA is not fully understood.
Eosinophilic Endomyocarditis and Atherosclerosis
The association between endomyocardial disease and eosinophilia was first described by Löffler in 1936 (177). Cardiac involvement is the most common cause of morbidity and mortality in patients with hypereosinophilia (3, 81–84). Eosinophils and their granule proteins have been found in cardiac biopsies from patients with eosinophilic endomyocardial disease (178, 179). Recently, an association of EoE and cardiomyopathy has been reported (180).
Eosinophil cationic protein and, to a lesser extent, MBP stimulate the release of preformed (histamine and tryptase) and the de novo synthesis of PGD2 and LTC4 from human HHMC (8, 181). Activated HHMCs release histamine and CysLTs, which exert profound cardiovascular and metabolic effects (182, 183). In addition, MBP and eosinophil peroxidase induce platelet aggregation (184). These observations suggest that infiltrating eosinophils and their mediators contribute to cardiac dysfunction in patients with eosinophilia.
Activated MCs are increased at site of atheromatous rupture in myocardial infarction (10). MCs in human coronary plaques release angiogenic factors, such as FGF-β (11), which enhance atherosclerotic plaque progression. Cardiac MC-derived renin promotes local angiotensin formation leading to cardiac dysfunction (12). Activated MCs may also promote abdominal aortic aneurysms (13, 14) presumably through the release of chymase (185, 186) and CysLTs (15).
Skin Disorders
Bullous pemphigoid (BP) is the most frequent autoimmune blistering dermatosis, characterized by autoantibodies directed against the dermal–epidermal junction proteins BP180/BP230 typically causing pruritic bullous eruptions. The immune response leading to blister formation in BP involves different inflammatory cells and molecules, including CD4 T cells, B cells, complement factors, neutrophils, as well as MCs and eosinophils (187). Serum levels of ECP were elevated in patients with active BP compared with healthy controls. Moreover, MC tryptase serum levels were associated with circulating anti-BP180 autoantibodies and decreased at the time of clinical remission (188). In a murine model of BP, blistering was dependent on C5a–C5aR interaction on MCs, which led to the activation of the p38 MAPK pathway in MC and their degranulation (189). Moreover, blood, skin, and blister-derived eosinophils were activated in patients with BP compared to controls. Activated eosinophils produced CCL26, IL-6, IL-8, and IL-1α in BP skin and blister fluid and displayed apoptosis features (190). Interestingly, IL-5-activated eosinophils were shown ex vivo to directly contribute to BP blister formation in the presence of BP autoantibodies. Indeed, IL-5-activated eosinophils induced dermal–epidermal separation, which was dependent on eosinophil adhesion, FcγR activation, ROS production, degranulation, and eosinophil extracellular trap formation (191).
Psoriasis is a frequent, chronic recurrent inflammatory skin disease which results from dysregulation between environmental factors, epithelial cells and immune cells (100). MCs infiltrate skin lesions of psoriatic patients and were identified as high producers of IL-17A and IL-22, both cytokines involved in psoriasis pathogenesis (192, 193). MCs and keratinocytes also induced angiogenesis by producing IL-8 and VEGF-A (194). In contrast, eosinophils were not increased in the skin or peripheral blood of psoriatic patients.
Atopic dermatitis (AD) is a common chronic inflammatory skin disease driven by specific genetic and immunological mechanisms (100). MC-derived histamine, tryptase, chymase, and other inflammatory mediators contribute to itching and inflammation in patients with AD (195). However, MCs were not required for the development of disease in a murine model of AD (196). AD is characterized by an increased number of circulating eosinophils and dermal and epidermal infiltrates of eosinophils. Tissue and blood eosinophilia and increased circulating levels of ECP, MBP, and eosinophil-derived neurotoxin have been correlated with disease activity. Serum levels of IL-5 were increased in AD patients and correlated with disease activity. However, although eosinophils might have important roles in AD pathogenesis, their exact mechanisms are not fully understood (197).
Tumor-Associated MCs (TAMCs) and Tumor-Associated Eosinophils in Cancer
Tumor-associated eosinophilia was first described in 1893 (198). Eosinophilia is frequently observed in patients with solid tumors (199–201) and Hodgkin’s lymphoma (202). Eosinophils are recruited to tumors by chemoattractant CCL11 (eotaxin-1), which binds to CCR3 (203) and damage-associated molecular patterns, notably the alarmin high-mobility group box 1, released by necrotic tumor cells (204, 205).
Clinical studies addressing the role of eosinophils in tumors provided conflicting results. Tumor-associated eosinophilia was related to good prognosis in colorectal, head and neck, bladder and prostate cancers (206–208). By contrast, in Hodgkin’s lymphoma, oral squamous cell carcinoma, and cervical carcinoma, eosinophils have been linked to poor prognosis (206, 207, 209, 210).
Experimental studies also provided inconclusive results (211). Indeed, human eosinophils exert tumoricidal activity toward cancer cells through the release of TNF-α and granzyme A, contained in their secondary granules (17, 212). On the other hand, tumor-recruited eosinophils influence tumor angiogenesis, through distinct mechanisms. Human eosinophils and their supernatants induce endothelial cell proliferation in vitro and angiogenesis in vivo (213). Eosinophils contain VEGF in their secretory granules that can be secreted upon activation by IL-5 (127). In addition, eosinophils can contribute to tumor angiogenesis through the release of other proangiogenic molecules such as OPN (94) and MBP (129). Recently, activated eosinophils were shown to be essential for tumor rejection (16). Indeed, tumor-homing eosinophils secreted chemoattractants such as CCL5, CXCL9, and CXCL10, which recruited CD8+ T cells to the tumor (16).
Tumor-associated MCs are present in several human solid (41–55) and hematologic tumors (56–64). Peritumoral and/or intratumoral MC density is increased in different types of human cancer (18). Although the role of MCs and their mediators in experimental and human tumors is still controversial (19, 214), the bidirectional interaction between MCs and eosinophils can influence tumor angiogenesis and lymphangiogenesis.
Tumor immunologists have just scratched the surface of the complexity of the multidirectional interactions between eosinophils, MCs, and their neighboring tumor cells in tumor microenvironment.
MCs and Eosinophils in Myeloproliferative Disorders
Mast cells and eosinophils can also found to be increased in primary myeloproliferative disorders of the bone marrow. The mechanism of the increased numbers of MCs and eosinophils in myeloproliferative disorders involves a primary defect in a tyrosine kinase gene resulting in uncontrolled proliferation and dysregulated apoptosis. Two such disorders are particularly associated with increased numbers of both cell types: systemic mastocytosis (SM) and chronic eosinophilic leukemia (CEL).
Mastocytosis is an abnormal clonal MC expansion and accumulation in several tissues including the bone marrow and the skin (215, 216). Cutaneous mastocytosis is associated with gain-of-function Kit mutations in approximately 8% of cases (217). Almost all patients with SM present a somatic mutation in codon 816 (D816V) of the gene encoding the receptor Kit, which leads to the substitution of a valine for an aspartate in the protein. Because of the D816V mutation, Kit is constitutively active, resulting in autophosphorylation and enhancement of MC differentiation and survival. A variable percentage (15–28%) of patients with SM also presents peripheral blood eosinophilia, which predicted poorer prognosis in some studies (218–220). In patients with cutaneous or SM, a correlation between the levels of soluble IL-5Rα (sIL-5Rα) and eosinophils in peripheral blood was also found (219).
In 2003, the FIP1L1–PDGFRA fusion was identified in patients with idiopathic hypereosinophilic syndrome and its presence redefined such patients having a neoplasm instead of idiopathic hypereosinophilic syndrome. Before the discovery of this cytogenetic rearrangement, the patients carried a poor prognosis due to early cardiac death in the absence of effective treatment. The identification of this fusion rearrangement as therapeutic target of imatinib dramatically changed the perspectives of these patients, due to a prompt hematologic and clinical remission. Patients with FIP1L1–PDGFRA+ CEL exhibit features of myeloproliferative syndromes such as splenomegaly, hypercellular bone marrow, and clinicopathological aspects that overlap with systemic MC diseases, such as increased number of abnormal MCs, elevated circulating tryptase levels, and bone marrow fibrosis (221). These similarities raised the doubt that FIP1L1–PDGFRA+ CEL could be considered a subtype of SM, rather than a primary eosinophil disease (222). Indeed, even though less dense clusters of MCs compared to the typical multifocal aggregates of D816V Kit+ SM, in some cases MCs exhibited spindle-shaped morphology and aberrant surface expression of CD25, both minor criteria for SM according to the WHO criteria (223). In the revised 2008 WHO semi-molecular classification of myeloid neoplasms, FIP1L1–PDGFRA+ disease is not considered a subtype of SM. To date, FIP1L1–PDGFRA and D816V Kit mutations appear to be mutually exclusive. In the D816V Kit+ patients, gastrointestinal symptoms, urticaria pigmentosa, thrombocytosis, median serum tryptase value, and the presence of MC dense infiltrates in the bone marrow were increased compared to patients with FIP1L1–PDGFRA mutation. By contrast, cardiac and pulmonary symptoms, median eosinophil count, eosinophil to tryptase ratio, and serum B12 levels were higher in the FIP1L1–PDGFRA+ patients. Whether a patient with peripheral eosinophilia and increased bone marrow MC infiltration carries a D816V Kit or FIP1L1–PDGFRA mutation is important for guiding therapeutic decisions. Indeed, FIP1L1–PDGFRA mutation is highly sensitive to imatinib treatment, which induces clinical remission as early as 4 weeks. By contrast, the vast majority of SM carrying D816V Kit mutation are imatinib resistant and candidate to second-line tyrosine kinase inhibitors or cytoreductive therapy (224).
Conclusion
Mast cells and eosinophils were identified and named by Paul Ehrlich based on their capacity to be stained by specific dyes. These cells and their mediators have been classically associated with the pathogenesis of allergic disorders. However, there is now evidence that MCs and eosinophils are involved in autoimmune disorders, vasculitis, cardiovascular diseases, as well as solid and hematologic tumors. MCs and eosinophils play complex, sometimes complementary, but also distinct roles in these conditions. The latter findings are not surprising given the observations that these cells have distinct myeloid progenitors, are activated by different agonists, and differ morphologically, ultrastructurally, immunologically, biochemically, and pharmacologically.
In allergic disorders (e.g., asthma, allergic rhinitis, chronic urticaria) and certain solid (e.g., gastric and prostate cancers) and hematologic tumors (e.g., Hodgkin’s lymphoma), MCs and eosinophils can be found in close proximity. In particular, in allergic diseases, these cells can form the AEU (65). It is now clear that MCs modulate several eosinophil functions through the release of a plethora of preformed (e.g., SCF, histamine, and adenosine) and de novo synthesized mediators (e.g., IL-5, LTC4, SCF, PGD2, and PAF). On the other side, eosinophils modulate MC functions through the production of several mediators (e.g., IL-5, PAF, ECP, MBP, and NGF). These bidirectional interactions between MCs and eosinophils might be important not only in allergic diseases but also in several inflammatory and neoplastic disorders.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and have approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Funding
This work was supported in part by grants from Regione Campania CISI-Lab Project, CRèME Project, and TIMING Project (GM), United States – Israel Binational Science Foundation (BSF) (Grant No. 2015045); Israel Science Foundation (Grant No. 472/15); Israel Cancer Association (Grant Nos. 20140118 and 20161131) (FL-S).
Abbreviations
SCF, stem cell factor; MCs, mast cells; H4R, histamine 4 receptor; PAR-2, protease-activated receptor 2; ECP, eosinophil cationic protein; MBP; major basic protein; MRGPRX2, Mas-related G protein-coupled receptor member X2; OPN, osteopontin; IL-5, interleukin 5; GM-CSF, granulocyte-macrophage colony-stimulating factor; LTC4, leukotriene C4; PGD2, prostaglandin D2; PAF, platelet-activating factor; NGF, nerve growth factor; VEGF-A, vascular endothelial growth factor A; IL-5R, interleukin 5 receptor; CysLTR1/2, cysteinyl leukotriene receptor 1/2; CRTH2, chemoattractant receptor-homologous molecule expressed on Th2 cells; PAFR, platelet-activating factor receptor; TrkA, tyrosine kinase receptor A; VEGF-R1/2, vascular endothelial growth factor receptor 1/2.
References
1. Bradding P, Arthur G. Mast cells in asthma – state of the art. Clin Exp Allergy (2016) 46(2):194–263. doi:10.1111/cea.12675
2. Marone G, Triggiani M, de Paulis A. Mast cells and basophils: friends as well as foes in bronchial asthma? Trends Immunol (2005) 26(1):25–31. doi:10.1016/j.it.2004.10.010
3. Varricchi G, Bagnasco D, Borriello F, Heffler E, Canonica GW. Interleukin-5 pathway inhibition in the treatment of eosinophilic respiratory disorders: evidence and unmet needs. Curr Opin Allergy Clin Immunol (2016) 16(2):186–200. doi:10.1097/ACI.0000000000000251
4. Rivellese F, Suurmond J, Habets K, Dorjee AL, Ramamoorthi N, Townsend MJ, et al. Ability of interleukin-33- and immune complex-triggered activation of human mast cells to down-regulate monocyte-mediated immune responses. Arthritis Rheumatol (2015) 67(9):2343–53. doi:10.1002/art.39192
5. Suurmond J, Rivellese F, Dorjee AL, Bakker AM, Rombouts YJ, Rispens T, et al. Toll-like receptor triggering augments activation of human mast cells by anti-citrullinated protein antibodies. Ann Rheum Dis (2015) 74(10):1915–23. doi:10.1136/annrheumdis-2014-205562
6. Cho C, Nguyen A, Bryant KJ, O’Neill SG, McNeil HP. Prostaglandin D2 metabolites as a biomarker of in vivo mast cell activation in systemic mastocytosis and rheumatoid arthritis. Immun Inflamm Dis (2016) 4(1):64–9. doi:10.1002/iid3.94
7. Henault J, Riggs JM, Karnell JL, Liarski VM, Li J, Shirinian L, et al. Self-reactive IgE exacerbates interferon responses associated with autoimmunity. Nat Immunol (2016) 17(2):196–203. doi:10.1038/ni.3326
8. Patella V, de Crescenzo G, Marino I, Genovese A, Adt M, Gleich GJ, et al. Eosinophil granule proteins activate human heart mast cells. J Immunol (1996) 157(3):1219–25.
9. Patella V, de Crescenzo G, Marino I, Genovese A, Adt M, Gleich GJ, et al. Eosinophil granule proteins are selective activators of human heart mast cells. Int Arch Allergy Immunol (1997) 113(1–3):200–2. doi:10.1159/000237546
10. Kovanen PT, Kaartinen M, Paavonen T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation (1995) 92(5):1084–8. doi:10.1161/01.CIR.92.5.1084
11. Lappalainen H, Laine P, Pentikainen MO, Sajantila A, Kovanen PT. Mast cells in neovascularized human coronary plaques store and secrete basic fibroblast growth factor, a potent angiogenic mediator. Arterioscler Thromb Vasc Biol (2004) 24(10):1880–5. doi:10.1161/01.ATV.0000140820.51174.8d
12. Mackins CJ, Kano S, Seyedi N, Schafer U, Reid AC, Machida T, et al. Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J Clin Invest (2006) 116(4):1063–70. doi:10.1172/JCI25713
13. Tsuruda T, Kato J, Hatakeyama K, Kojima K, Yano M, Yano Y, et al. Adventitial mast cells contribute to pathogenesis in the progression of abdominal aortic aneurysm. Circ Res (2008) 102(11):1368–77. doi:10.1161/CIRCRESAHA.108.173682
14. Sun J, Sukhova GK, Yang M, Wolters PJ, MacFarlane LA, Libby P, et al. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J Clin Invest (2007) 117(11):3359–68. doi:10.1172/JCI31311
15. Di Gennaro A, Wagsater D, Mayranpaa MI, Gabrielsen A, Swedenborg J, Hamsten A, et al. Increased expression of leukotriene C4 synthase and predominant formation of cysteinyl-leukotrienes in human abdominal aortic aneurysm. Proc Natl Acad Sci U S A (2010) 107(49):21093–7. doi:10.1073/pnas.1015166107
16. Carretero R, Sektioglu IM, Garbi N, Salgado OC, Beckhove P, Hammerling GJ. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8(+) T cells. Nat Immunol (2015) 16(6):609–17. doi:10.1038/ni.3159
17. Legrand F, Driss V, Delbeke M, Loiseau S, Hermann E, Dombrowicz D, et al. Human eosinophils exert TNF-alpha and granzyme A-mediated tumoricidal activity toward colon carcinoma cells. J Immunol (2010) 185(12):7443–51. doi:10.4049/jimmunol.1000446
18. Marone G, Varricchi G, Loffredo S, Granata F. Mast cells and basophils in inflammatory and tumor angiogenesis and lymphangiogenesis. Eur J Pharmacol (2016) 778:146–51. doi:10.1016/j.ejphar.2015.03.088
19. Galdiero MR, Varricchi G, Marone G. The immune network in thyroid cancer. Oncoimmunology (2016) 5(6):e1168556. doi:10.1080/2162402X.2016.1168556
21. Ehrlich P. Beitrage zur Kenntnis der granulierten Bindegewbszellen und der eosinophilen Leukocyten. Arch Anat Physiol (Leipzig) (1879):166–9.
22. Ehrlich P. Uber die specifischen Granulationen des Blutes. Arch Anat Physiol (Leipzig) (1879):571–9.
23. Borriello F, Granata F, Varricchi G, Genovese A, Triggiani M, Marone G. Immunopharmacological modulation of mast cells. Curr Opin Pharmacol (2014) 17:45–57. doi:10.1016/j.coph.2014.07.002
24. Gangwar RS, Landolina N, Arpinati L, Levi-Schaffer F. Mast cell and eosinophil surface receptors as targets for anti-allergic therapy. Pharmacol Ther (2017) 170:37–63. doi:10.1016/j.pharmthera.2016.10.010
25. Borriello F, Iannone R, Marone G. Histamine release from mast cells and basophils. Handb Exp Pharmacol (2017) 241:121–39. doi:10.1007/164_2017_18
26. Dwyer DF, Barrett NA, Austen KF. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat Immunol (2016) 17(7):878–87. doi:10.1038/ni.3445
27. Drissen R, Buza-Vidas N, Woll P, Thongjuea S, Gambardella A, Giustacchini A, et al. Distinct myeloid progenitor-differentiation pathways identified through single-cell RNA sequencing. Nat Immunol (2016) 17(6):666–76. doi:10.1038/ni.3412
28. Kirshenbaum AS, Goff JP, Semere T, Foster B, Scott LM, Metcalfe DD. Demonstration that human mast cells arise from a progenitor cell population that is CD34(+), c-kit(+), and expresses aminopeptidase N (CD13). Blood (1999) 94(7):2333–42.
29. Frossi B, Mion F, Tripodo C, Colombo MP, Pucillo CE. Rheostatic functions of mast cells in the control of innate and adaptive immune responses. Trends Immunol (2017). doi:10.1016/j.it.2017.04.001
30. Meininger CJ, Yano H, Rottapel R, Bernstein A, Zsebo KM, Zetter BR. The c-kit receptor ligand functions as a mast cell chemoattractant. Blood (1992) 79(4):958–63.
31. Mori Y, Iwasaki H, Kohno K, Yoshimoto G, Kikushige Y, Okeda A, et al. Identification of the human eosinophil lineage-committed progenitor: revision of phenotypic definition of the human common myeloid progenitor. J Exp Med (2009) 206(1):183–93. doi:10.1084/jem.20081756
32. Molfino NA, Gossage D, Kolbeck R, Parker JM, Geba GP. Molecular and clinical rationale for therapeutic targeting of interleukin-5 and its receptor. Clin Exp Allergy (2012) 42(5):712–37. doi:10.1111/j.1365-2222.2011.03854.x
33. Johnston LK, Hsu CL, Krier-Burris RA, Chhiba KD, Chien KB, McKenzie A, et al. IL-33 precedes IL-5 in regulating eosinophil commitment and is required for eosinophil homeostasis. J Immunol (2016) 197(9):3445–53. doi:10.4049/jimmunol.1600611
34. Lamouse-Smith ES, Furuta GT. Eosinophils in the gastrointestinal tract. Curr Gastroenterol Rep (2006) 8(5):390–5. doi:10.1007/s11894-006-0024-6
35. Jung Y, Rothenberg ME. Roles and regulation of gastrointestinal eosinophils in immunity and disease. J Immunol (2014) 193(3):999–1005. doi:10.4049/jimmunol.1400413
36. Lee JJ, Jacobsen EA, Ochkur SI, McGarry MP, Condjella RM, Doyle AD, et al. Human versus mouse eosinophils: “that which we call an eosinophil, by any other name would stain as red”. J Allergy Clin Immunol (2012) 130(3):572–84. doi:10.1016/j.jaci.2012.07.025
37. Blanchard C, Rothenberg ME. Biology of the eosinophil. Adv Immunol (2009) 101:81–121. doi:10.1016/S0065-2776(08)01003-1
38. Foster PS, Mould AW, Yang M, Mackenzie J, Mattes J, Hogan SP, et al. Elemental signals regulating eosinophil accumulation in the lung. Immunol Rev (2001) 179:173–81. doi:10.1034/j.1600-065X.2001.790117.x
39. Marone G, Galli SJ, Kitamura Y. Probing the roles of mast cells and basophils in natural and acquired immunity, physiology and disease. Trends Immunol (2002) 23(9):425–7. doi:10.1016/S1471-4906(02)02274-3
40. Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med (2012) 18(5):693–704. doi:10.1038/nm.2755
41. Aoki M, Pawankar R, Niimi Y, Kawana S. Mast cells in basal cell carcinoma express VEGF, IL-8 and RANTES. Int Arch Allergy Immunol (2003) 130(3):216–23. doi:10.1159/000069515
42. Beer TW, Ng LB, Murray K. Mast cells have prognostic value in Merkel cell carcinoma. Am J Dermatopathol (2008) 30(1):27–30. doi:10.1097/DAD.0b013e31815c932a
43. Dvorak AM, Mihm MC Jr, Osage JE, Dvorak HF. Melanoma. An ultrastructural study of the host inflammatory and vascular responses. J Invest Dermatol (1980) 75(5):388–93. doi:10.1111/1523-1747.ep12523627
44. Giannou AD, Marazioti A, Spella M, Kanellakis NI, Apostolopoulou H, Psallidas I, et al. Mast cells mediate malignant pleural effusion formation. J Clin Invest (2015) 125(6):2317–34. doi:10.1172/JCI79840
45. Johansson A, Rudolfsson S, Hammarsten P, Halin S, Pietras K, Jones J, et al. Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. Am J Pathol (2010) 177(2):1031–41. doi:10.2353/ajpath.2010.100070
46. Ma Y, Hwang RF, Logsdon CD, Ullrich SE. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res (2013) 73(13):3927–37. doi:10.1158/0008-5472.CAN-12-4479
47. Melillo RM, Guarino V, Avilla E, Galdiero MR, Liotti F, Prevete N, et al. Mast cells have a protumorigenic role in human thyroid cancer. Oncogene (2010) 29(47):6203–15. doi:10.1038/onc.2010.348
48. Ng L, Beer TW, Murray K. Vascular density has prognostic value in Merkel cell carcinoma. Am J Dermatopathol (2008) 30(5):442–5. doi:10.1097/DAD.0b013e318172364d
49. Pittoni P, Tripodo C, Piconese S, Mauri G, Parenza M, Rigoni A, et al. Mast cell targeting hampers prostate adenocarcinoma development but promotes the occurrence of highly malignant neuroendocrine cancers. Cancer Res (2011) 71(18):5987–97. doi:10.1158/0008-5472.CAN-11-1637
50. Ribatti D, Vacca A, Ria R, Marzullo A, Nico B, Filotico R, et al. Neovascularisation, expression of fibroblast growth factor-2, and mast cells with tryptase activity increase simultaneously with pathological progression in human malignant melanoma. Eur J Cancer (2003) 39(5):666–74. doi:10.1016/S0959-8049(02)00150-8
51. Ribatti D, Ennas MG, Vacca A, Ferreli F, Nico B, Orru S, et al. Tumor vascularity and tryptase-positive mast cells correlate with a poor prognosis in melanoma. Eur J Clin Invest (2003) 33(5):420–5. doi:10.1046/j.1365-2362.2003.01152.x
52. Siiskonen H, Poukka M, Bykachev A, Tyynela-Korhonen K, Sironen R, Pasonen-Seppanen S, et al. Low numbers of tryptase+ and chymase+ mast cells associated with reduced survival and advanced tumor stage in melanoma. Melanoma Res (2015) 25(6):479–85. doi:10.1097/CMR.0000000000000192
53. Takahashi K, Mulliken JB, Kozakewich HP, Rogers RA, Folkman J, Ezekowitz RA. Cellular markers that distinguish the phases of hemangioma during infancy and childhood. J Clin Invest (1994) 93(6):2357–64. doi:10.1172/JCI117241
54. Toth-Jakatics R, Jimi S, Takebayashi S, Kawamoto N. Cutaneous malignant melanoma: correlation between neovascularization and peritumor accumulation of mast cells overexpressing vascular endothelial growth factor. Hum Pathol (2000) 31(8):955–60. doi:10.1053/hupa.2000.16658
55. Johnson C, Huynh V, Hargrove L, Kennedy L, Graf-Eaton A, Owens J, et al. Inhibition of mast cell-derived histamine decreases human cholangiocarcinoma growth and differentiation via c-kit/stem cell factor-dependent signaling. Am J Pathol (2016) 186(1):123–33. doi:10.1016/j.ajpath.2015.09.016
56. Ribatti D, Vacca A, Nico B, Quondamatteo F, Ria R, Minischetti M, et al. Bone marrow angiogenesis and mast cell density increase simultaneously with progression of human multiple myeloma. Br J Cancer (1999) 79(3–4):451–5. doi:10.1038/sj.bjc.6690070
57. Rabenhorst A, Schlaak M, Heukamp LC, Forster A, Theurich S, von Bergwelt-Baildon M, et al. Mast cells play a protumorigenic role in primary cutaneous lymphoma. Blood (2012) 120(10):2042–54. doi:10.1182/blood-2012-03-415638
58. Vyzoukaki R, Tsirakis G, Pappa CA, Devetzoglou M, Tzardi M, Alexandrakis MG. The impact of mast cell density on the progression of bone disease in multiple myeloma patients. Int Arch Allergy Immunol (2015) 168(4):263–8. doi:10.1159/000443275
59. Tripodo C, Gri G, Piccaluga PP, Frossi B, Guarnotta C, Piconese S, et al. Mast cells and Th17 cells contribute to the lymphoma-associated pro-inflammatory microenvironment of angioimmunoblastic T-cell lymphoma. Am J Pathol (2010) 177(2):792–802. doi:10.2353/ajpath.2010.091286
60. Taskinen M, Karjalainen-Lindsberg ML, Leppa S. Prognostic influence of tumor-infiltrating mast cells in patients with follicular lymphoma treated with rituximab and CHOP. Blood (2008) 111(9):4664–7. doi:10.1182/blood-2007-11-125823
61. Franco G, Guarnotta C, Frossi B, Piccaluga PP, Boveri E, Gulino A, et al. Bone marrow stroma CD40 expression correlates with inflammatory mast cell infiltration and disease progression in splenic marginal zone lymphoma. Blood (2014) 123(12):1836–49. doi:10.1182/blood-2013-04-497271
62. Molin D, Edstrom A, Glimelius I, Glimelius B, Nilsson G, Sundstrom C, et al. Mast cell infiltration correlates with poor prognosis in Hodgkin’s lymphoma. Br J Haematol (2002) 119(1):122–4. doi:10.1046/j.1365-2141.2002.03768.x
63. Andersen MD, Kamper P, Nielsen PS, Bendix K, Riber-Hansen R, Steiniche T, et al. Tumour-associated mast cells in classical Hodgkin’s lymphoma: correlation with histological subtype, other tumour-infiltrating inflammatory cell subsets and outcome. Eur J Haematol (2016) 96(3):252–9. doi:10.1111/ejh.12583
64. Englund A, Molin D, Enblad G, Karlen J, Glimelius I, Ljungman G, et al. The role of tumour-infiltrating eosinophils, mast cells and macrophages in classical and nodular lymphocyte predominant Hodgkin lymphoma in children. Eur J Haematol (2016) 97(5):430–8. doi:10.1111/ejh.12747
65. Minai-Fleminger Y, Elishmereni M, Vita F, Soranzo MR, Mankuta D, Zabucchi G, et al. Ultrastructural evidence for human mast cell-eosinophil interactions in vitro. Cell Tissue Res (2010) 341(3):405–15. doi:10.1007/s00441-010-1010-8
66. Elishmereni M, Alenius HT, Bradding P, Mizrahi S, Shikotra A, Minai-Fleminger Y, et al. Physical interactions between mast cells and eosinophils: a novel mechanism enhancing eosinophil survival in vitro. Allergy (2011) 66(3):376–85. doi:10.1111/j.1398-9995.2010.02494.x
67. Elishmereni M, Bachelet I, Nissim Ben-Efraim AH, Mankuta D, Levi-Schaffer F. Interacting mast cells and eosinophils acquire an enhanced activation state in vitro. Allergy (2013) 68(2):171–9. doi:10.1111/all.12059
68. Hofstra CL, Desai PJ, Thurmond RL, Fung-Leung WP. Histamine H4 receptor mediates chemotaxis and calcium mobilization of mast cells. J Pharmacol Exp Ther (2003) 305(3):1212–21. doi:10.1124/jpet.102.046581
69. Jemima EA, Prema A, Thangam EB. Functional characterization of histamine H4 receptor on human mast cells. Mol Immunol (2014) 62(1):19–28. doi:10.1016/j.molimm.2014.05.007
70. Marquardt DL, Gruber HE, Wasserman SI. Adenosine release from stimulated mast cells. Proc Natl Acad Sci U S A (1984) 81(19):6192–6. doi:10.1073/pnas.81.19.6192
71. Rudich N, Ravid K, Sagi-Eisenberg R. Mast cell adenosine receptors function: a focus on the a3 adenosine receptor and inflammation. Front Immunol (2012) 3:134. doi:10.3389/fimmu.2012.00134
72. Cushley MJ, Tattersfield AE, Holgate ST. Inhaled adenosine and guanosine on airway resistance in normal and asthmatic subjects. Br J Clin Pharmacol (1983) 15(2):161–5. doi:10.1111/j.1365-2125.1983.tb01481.x
73. Peachell PT, Columbo M, Kagey-Sobotka A, Lichtenstein LM, Marone G. Adenosine potentiates mediator release from human lung mast cells. Am Rev Respir Dis (1988) 138(5):1143–51. doi:10.1164/ajrccm/138.5.1143
74. Sereda MJ, Bradding P, Vial C. Adenosine potentiates human lung mast cell tissue plasminogen activator activity. J Immunol (2011) 186(2):1209–17. doi:10.4049/jimmunol.1001563
75. Yip KH, Lau HY, Wise H. Reciprocal modulation of anti-IgE induced histamine release from human mast cells by A(1) and A(2B) adenosine receptors. Br J Pharmacol (2011) 164(2b):807–19. doi:10.1111/j.1476-5381.2011.01446.x
76. Reeves JJ, Harris CA, Hayes BP, Butchers PR, Sheehan MJ. Studies on the effects of adenosine A3 receptor stimulation on human eosinophils isolated from non-asthmatic or asthmatic donors. Inflamm Res (2000) 49(12):666–72. doi:10.1007/s000110050644
77. Ezeamuzie CI, Philips E. Positive coupling of atypical adenosine A3 receptors on human eosinophils to adenylyl cyclase. Biochem Biophys Res Commun (2003) 300(3):712–8. doi:10.1016/S0006-291X(02)02910-8
78. Temkin V, Kantor B, Weg V, Hartman ML, Levi-Schaffer F. Tryptase activates the mitogen-activated protein kinase/activator protein-1 pathway in human peripheral blood eosinophils, causing cytokine production and release. J Immunol (2002) 169(5):2662–9. doi:10.4049/jimmunol.169.5.2662
79. Neumann T, Manger B, Schmid M, Kroegel C, Hansch A, Kaiser WA, et al. Cardiac involvement in Churg-Strauss syndrome: impact of endomyocarditis. Medicine (Baltimore) (2009) 88(4):236–43. doi:10.1097/MD.0b013e3181af35a5
80. Dennert RM, van Paassen P, Schalla S, Kuznetsova T, Alzand BS, Staessen JA, et al. Cardiac involvement in Churg-Strauss syndrome. Arthritis Rheum (2010) 62(2):627–34. doi:10.1002/art.27263
81. Chusid MJ, Dale DC, West BC, Wolff SM. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore) (1975) 54(1):1–27. doi:10.1097/00005792-197501000-00001
82. Parrillo JE, Borer JS, Henry WL, Wolff SM, Fauci AS. The cardiovascular manifestations of the hypereosinophilic syndrome. Prospective study of 26 patients, with review of the literature. Am J Med (1979) 67(4):572–82. doi:10.1016/0002-9343(79)90227-4
83. Harley JB, Fauci AS, Gralnick HR. Noncardiovascular findings associated with heart disease in the idiopathic hypereosinophilic syndrome. Am J Cardiol (1983) 52(3):321–4. doi:10.1016/0002-9149(83)90131-5
84. Gottdiener JS, Maron BJ, Schooley RT, Harley JB, Roberts WC, Fauci AS. Two-dimensional echocardiographic assessment of the idiopathic hypereosinophilic syndrome. Anatomic basis of mitral regurgitation and peripheral embolization. Circulation (1983) 67(3):572–8. doi:10.1161/01.CIR.67.3.572
85. Piliponsky AM, Gleich GJ, Nagler A, Bar I, Levi-Schaffer F. Non-IgE-dependent activation of human lung- and cord blood-derived mast cells is induced by eosinophil major basic protein and modulated by the membrane form of stem cell factor. Blood (2003) 101(5):1898–904. doi:10.1182/blood-2002-05-1488
86. Fujisawa D, Kashiwakura J, Kita H, Kikukawa Y, Fujitani Y, Sasaki-Sakamoto T, et al. Expression of Mas-related gene X2 on mast cells is upregulated in the skin of patients with severe chronic urticaria. J Allergy Clin Immunol (2014) 134(3):622–33.e9. doi:10.1016/j.jaci.2014.05.004
87. Subramanian H, Gupta K, Ali H. Roles of Mas-related G protein-coupled receptor X2 on mast cell-mediated host defense, pseudoallergic drug reactions, and chronic inflammatory diseases. J Allergy Clin Immunol (2016) 138(3):700–10. doi:10.1016/j.jaci.2016.04.051
88. Ben-Zimra M, Bachelet I, Seaf M, Gleich GJ, Levi-Schaffer F. Eosinophil major basic protein activates human cord blood mast cells primed with fibroblast membranes by integrin-beta1. Allergy (2013) 68(10):1259–68. doi:10.1111/all.12232
89. Columbo M, Horowitz EM, Botana LM, MacGlashan DW Jr, Bochner BS, Gillis S, et al. The human recombinant c-kit receptor ligand, rhSCF, induces mediator release from human cutaneous mast cells and enhances IgE-dependent mediator release from both skin mast cells and peripheral blood basophils. J Immunol (1992) 149(2):599–608.
90. de Paulis A, Minopoli G, Arbustini E, de Crescenzo G, Dal Piaz F, Pucci P, et al. Stem cell factor is localized in, released from, and cleaved by human mast cells. J Immunol (1999) 163(5):2799–808.
91. Oliveira SH, Taub DD, Nagel J, Smith R, Hogaboam CM, Berlin A, et al. Stem cell factor induces eosinophil activation and degranulation: mediator release and gene array analysis. Blood (2002) 100(13):4291–7. doi:10.1182/blood.V100.13.4291
92. Hartman M, Piliponsky AM, Temkin V, Levi-Schaffer F. Human peripheral blood eosinophils express stem cell factor. Blood (2001) 97(4):1086–91. doi:10.1182/blood.V97.4.1086
93. Yuan Q, Austen KF, Friend DS, Heidtman M, Boyce JA. Human peripheral blood eosinophils express a functional c-kit receptor for stem cell factor that stimulates very late antigen 4 (VLA-4)-mediated cell adhesion to fibronectin and vascular cell adhesion molecule 1 (VCAM-1). J Exp Med (1997) 186(2):313–23. doi:10.1084/jem.186.2.313
94. Puxeddu I, Berkman N, Ribatti D, Bader R, Haitchi HM, Davies DE, et al. Osteopontin is expressed and functional in human eosinophils. Allergy (2010) 65(2):168–74. doi:10.1111/j.1398-9995.2009.02148.x
95. Nagasaka A, Matsue H, Matsushima H, Aoki R, Nakamura Y, Kambe N, et al. Osteopontin is produced by mast cells and affects IgE-mediated degranulation and migration of mast cells. Eur J Immunol (2008) 38(2):489–99. doi:10.1002/eji.200737057
96. Dahl C, Hoffmann HJ, Saito H, Schiotz PO. Human mast cells express receptors for IL-3, IL-5 and GM-CSF; a partial map of receptors on human mast cells cultured in vitro. Allergy (2004) 59(10):1087–96. doi:10.1111/j.1398-9995.2004.00606.x
97. Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature (2013) 502(7470):245–8. doi:10.1038/nature12526
98. Suurmond J, Dorjee AL, Knol EF, Huizinga TW, Toes RE. Differential TLR-induced cytokine production by human mast cells is amplified by FcvarepsilonRI triggering. Clin Exp Allergy (2015) 45(4):788–96. doi:10.1111/cea.12509
99. Broughton SE, Nero TL, Dhagat U, Kan WL, Hercus TR, Tvorogov D, et al. The betac receptor family – structural insights and their functional implications. Cytokine (2015) 74(2):247–58. doi:10.1016/j.cyto.2015.02.005
100. Varricchi G, Granata F, Loffredo S, Genovese A, Marone G. Angiogenesis and lymphangiogenesis in inflammatory skin disorders. J Am Acad Dermatol (2015) 73(1):144–53. doi:10.1016/j.jaad.2015.03.041
101. Braccioni F, Dorman SC, O’Byrne PM, Inman MD, Denburg JA, Parameswaran K, et al. The effect of cysteinyl leukotrienes on growth of eosinophil progenitors from peripheral blood and bone marrow of atopic subjects. J Allergy Clin Immunol (2002) 110(1):96–101. doi:10.1067/mai.2002.125000
102. Bandeira-Melo C, Hall JC, Penrose JF, Weller PF. Cysteinyl leukotrienes induce IL-4 release from cord blood-derived human eosinophils. J Allergy Clin Immunol (2002) 109(6):975–9. doi:10.1067/mai.2002.124269
103. Luna-Gomes T, Magalhaes KG, Mesquita-Santos FP, Bakker-Abreu I, Samico RF, Molinaro R, et al. Eosinophils as a novel cell source of prostaglandin D2: autocrine role in allergic inflammation. J Immunol (2011) 187(12):6518–26. doi:10.4049/jimmunol.1101806
104. Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, et al. Prostaglandin D2 as a mediator of allergic asthma. Science (2000) 287(5460):2013–7. doi:10.1126/science.287.5460.2013
105. Shiraishi Y, Takeda K, Domenico J, Gelfand EW. Role of prostaglandin D2 and CRTH2 blockade in early- and late-phase nasal responses. Clin Exp Allergy (2014) 44(8):1076–82. doi:10.1111/cea.12280
106. Hattori Y, Levi R. Effect of PGD2 on cardiac contractility: a negative inotropism secondary to coronary vasoconstriction conceals a primary positive inotropic action. J Pharmacol Exp Ther (1986) 237(3):719–24.
107. Gervais FG, Cruz RP, Chateauneuf A, Gale S, Sawyer N, Nantel F, et al. Selective modulation of chemokinesis, degranulation, and apoptosis in eosinophils through the PGD2 receptors CRTH2 and DP. J Allergy Clin Immunol (2001) 108(6):982–8. doi:10.1067/mai.2001.119919
108. Schratl P, Royer JF, Kostenis E, Ulven T, Sturm EM, Waldhoer M, et al. The role of the prostaglandin D2 receptor, DP, in eosinophil trafficking. J Immunol (2007) 179(7):4792–9. doi:10.4049/jimmunol.179.7.4792
109. Bartemes KR, McKinney S, Gleich GJ, Kita H. Endogenous platelet-activating factor is critically involved in effector functions of eosinophils stimulated with IL-5 or IgG. J Immunol (1999) 162(5):2982–9.
110. Triggiani M, Hubbard WC, Chilton FH. Synthesis of 1-acyl-2-acetyl-sn-glycero-3-phosphocholine by an enriched preparation of the human lung mast cell. J Immunol (1990) 144(12):4773–80.
111. Triggiani M, De Marino V, Sofia M, Faraone S, Ambrosio G, Carratu L, et al. Characterization of platelet-activating factor acetylhydrolase in human bronchoalveolar lavage. Am J Respir Crit Care Med (1997) 156(1):94–100. doi:10.1164/ajrccm.156.1.9608084
112. Kato M, Kimura H, Motegi Y, Tachibana A, Minakami H, Morikawa A, et al. Platelet-activating factor activates two distinct effector pathways in human eosinophils. J Immunol (2002) 169(9):5252–9. doi:10.4049/jimmunol.169.9.5252
113. Liu L, Zuurbier AE, Mul FP, Verhoeven AJ, Lutter R, Knol EF, et al. Triple role of platelet-activating factor in eosinophil migration across monolayers of lung epithelial cells: eosinophil chemoattractant and priming agent and epithelial cell activator. J Immunol (1998) 161(6):3064–70.
114. Leon A, Buriani A, Dal Toso R, Fabris M, Romanello S, Aloe L, et al. Mast cells synthesize, store, and release nerve growth factor. Proc Natl Acad Sci U S A (1994) 91(9):3739–43. doi:10.1073/pnas.91.9.3739
115. Nilsson G, Forsberg-Nilsson K, Xiang Z, Hallbook F, Nilsson K, Metcalfe DD. Human mast cells express functional TrkA and are a source of nerve growth factor. Eur J Immunol (1997) 27(9):2295–301. doi:10.1002/eji.1830270925
116. Solomon A, Aloe L, Pe’er J, Frucht-Pery J, Bonini S, Levi-Schaffer F. Nerve growth factor is preformed in and activates human peripheral blood eosinophils. J Allergy Clin Immunol (1998) 102(3):454–60. doi:10.1016/S0091-6749(98)70135-6
117. Kobayashi H, Gleich GJ, Butterfield JH, Kita H. Human eosinophils produce neurotrophins and secrete nerve growth factor on immunologic stimuli. Blood (2002) 99(6):2214–20. doi:10.1182/blood.V99.6.2214
118. Bonini S, Lambiase A, Angelucci F, Magrini L, Manni L, Aloe L. Circulating nerve growth factor levels are increased in humans with allergic diseases and asthma. Proc Natl Acad Sci U S A (1996) 93(20):10955–60. doi:10.1073/pnas.93.20.10955
119. Munitz A, Levi-Schaffer F. Eosinophils: ’new’ roles for ’old’ cells. Allergy (2004) 59(3):268–75. doi:10.1111/j.1398-9995.2003.00442.x
120. Abdel-Majid RM, Marshall JS. Prostaglandin E2 induces degranulation-independent production of vascular endothelial growth factor by human mast cells. J Immunol (2004) 172(2):1227–36. doi:10.4049/jimmunol.172.2.1227
121. Theoharides TC, Zhang B, Kempuraj D, Tagen M, Vasiadi M, Angelidou A, et al. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc Natl Acad Sci U S A (2010) 107(9):4448–53. doi:10.1073/pnas.1000803107
122. Boesiger J, Tsai M, Maurer M, Yamaguchi M, Brown LF, Claffey KP, et al. Mast cells can secrete vascular permeability factor/vascular endothelial cell growth factor and exhibit enhanced release after immunoglobulin E-dependent upregulation of fc epsilon receptor I expression. J Exp Med (1998) 188(6):1135–45. doi:10.1084/jem.188.6.1135
123. Detoraki A, Granata F, Staibano S, Rossi FW, Marone G, Genovese A. Angiogenesis and lymphangiogenesis in bronchial asthma. Allergy (2010) 65(8):946–58. doi:10.1111/j.1398-9995.2010.02372.x
124. Detoraki A, Staiano RI, Granata F, Giannattasio G, Prevete N, de Paulis A, et al. Vascular endothelial growth factors synthesized by human lung mast cells exert angiogenic effects. J Allergy Clin Immunol (2009) 123(5):1142–9, 1149.e1–5. doi:10.1016/j.jaci.2009.01.044
125. Grutzkau A, Kruger-Krasagakes S, Baumeister H, Schwarz C, Kogel H, Welker P, et al. Synthesis, storage, and release of vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) by human mast cells: implications for the biological significance of VEGF206. Mol Biol Cell (1998) 9(4):875–84. doi:10.1091/mbc.9.4.875
126. Puxeddu I, Alian A, Piliponsky AM, Ribatti D, Panet A, Levi-Schaffer F. Human peripheral blood eosinophils induce angiogenesis. Int J Biochem Cell Biol (2005) 37(3):628–36. doi:10.1016/j.biocel.2004.09.001
127. Horiuchi T, Weller PF. Expression of vascular endothelial growth factor by human eosinophils: upregulation by granulocyte macrophage colony-stimulating factor and interleukin-5. Am J Respir Cell Mol Biol (1997) 17(1):70–7. doi:10.1165/ajrcmb.17.1.2796
128. Nissim Ben-Efraim AH, Eliashar R, Levi-Schaffer F. Hypoxia modulates human eosinophil function. Clin Mol Allergy (2010) 8:1–8. doi:10.1186/1476-7961-8-10
129. Puxeddu I, Berkman N, Nissim Ben-Efraim AH, Davies DE, Ribatti D, Gleich GJ, et al. The role of eosinophil major basic protein in angiogenesis. Allergy (2009) 64(3):368–74. doi:10.1111/j.1398-9995.2008.01822.x
130. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol (2015) 16(1):45–56. doi:10.1038/ni.3049
131. Varricchi G, Senna G, Loffredo S, Bagnasco D, Ferrando M, Canonica GW. Reslizumab and eosinophilic asthma: one step closer to precision medicine? Front Immunol (2017) 8:242. doi:10.3389/fimmu.2017.00242
132. Thiriou D, Morianos I, Xanthou G, Samitas K. Innate immunity as the orchestrator of allergic airway inflammation and resolution in asthma. Int Immunopharmacol (2017) 48:43–54. doi:10.1016/j.intimp.2017.04.027
133. Carter RJ, Bradding P. The role of mast cells in the structural alterations of the airways as a potential mechanism in the pathogenesis of severe asthma. Curr Pharm Des (2011) 17(7):685–98. doi:10.2174/138161211795428975
134. Arthur G, Bradding P. New developments in mast cell biology: clinical implications. Chest (2016) 150(3):680–93. doi:10.1016/j.chest.2016.06.009
135. Cahill KN, Katz HR, Cui J, Lai J, Kazani S, Crosby-Thompson A, et al. KIT inhibition by imatinib in patients with severe refractory asthma. N Engl J Med (2017) 376(20):1911–20. doi:10.1056/NEJMoa1613125
136. Humbert M, de Blay F, Garcia G, Prud’homme A, Leroyer C, Magnan A, et al. Masitinib, a c-kit/PDGF receptor tyrosine kinase inhibitor, improves disease control in severe corticosteroid-dependent asthmatics. Allergy (2009) 64(8):1194–201. doi:10.1111/j.1398-9995.2009.02122.x
137. Sutton BJ, Davies AM. Structure and dynamics of IgE-receptor interactions: FcepsilonRI and CD23/FcepsilonRII. Immunol Rev (2015) 268(1):222–35. doi:10.1111/imr.12340
138. Normansell R, Walker S, Milan SJ, Walters EH, Nair P. Omalizumab for asthma in adults and children. Cochrane Database Syst Rev (2014) 1:CD003559. doi:10.1002/14651858.CD003559.pub4
139. Kaplan A, Ledford D, Ashby M, Canvin J, Zazzali JL, Conner E, et al. Omalizumab in patients with symptomatic chronic idiopathic/spontaneous urticaria despite standard combination therapy. J Allergy Clin Immunol (2013) 132(1):101–9. doi:10.1016/j.jaci.2013.05.013
140. Bousquet J, Cabrera P, Berkman N, Buhl R, Holgate S, Wenzel S, et al. The effect of treatment with omalizumab, an anti-IgE antibody, on asthma exacerbations and emergency medical visits in patients with severe persistent asthma. Allergy (2005) 60(3):302–8. doi:10.1111/j.1398-9995.2004.00770.x
141. Humbert M, Beasley R, Ayres J, Slavin R, Hebert J, Bousquet J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy (2005) 60(3):309–16. doi:10.1111/j.1398-9995.2004.00772.x
142. Molderings GJ, Raithel M, Kratz F, Azemar M, Haenisch B, Harzer S, et al. Omalizumab treatment of systemic mast cell activation disease: experiences from four cases. Intern Med (2011) 50(6):611–5. doi:10.2169/internalmedicine.50.4640
143. Pillai P, Chan YC, Wu SY, Ohm-Laursen L, Thomas C, Durham SR, et al. Omalizumab reduces bronchial mucosal IgE and improves lung function in non-atopic asthma. Eur Respir J (2016) 48(6):1593–601. doi:10.1183/13993003.01501-2015
144. Massanari M, Holgate ST, Busse WW, Jimenez P, Kianifard F, Zeldin R. Effect of omalizumab on peripheral blood eosinophilia in allergic asthma. Respir Med (2010) 104(2):188–96. doi:10.1016/j.rmed.2009.09.011
145. Djukanovic R, Wilson SJ, Kraft M, Jarjour NN, Steel M, Chung KF, et al. Effects of treatment with anti-immunoglobulin E antibody omalizumab on airway inflammation in allergic asthma. Am J Respir Crit Care Med (2004) 170(6):583–93. doi:10.1164/rccm.200312-1651OC
146. Noga O, Hanf G, Brachmann I, Klucken AC, Kleine-Tebbe J, Rosseau S, et al. Effect of omalizumab treatment on peripheral eosinophil and T-lymphocyte function in patients with allergic asthma. J Allergy Clin Immunol (2006) 117(6):1493–9. doi:10.1016/j.jaci.2006.02.028
147. Noga O, Hanf G, Kunkel G. Immunological and clinical changes in allergic asthmatics following treatment with omalizumab. Int Arch Allergy Immunol (2003) 131(1):46–52. doi:10.1159/000070434
148. van Rensen EL, Evertse CE, van Schadewijk WA, van Wijngaarden S, Ayre G, Mauad T, et al. Eosinophils in bronchial mucosa of asthmatics after allergen challenge: effect of anti-IgE treatment. Allergy (2009) 64(1):72–80. doi:10.1111/j.1398-9995.2008.01881.x
149. Riccio AM, Dal Negro RW, Micheletto C, De Ferrari L, Folli C, Chiappori A, et al. Omalizumab modulates bronchial reticular basement membrane thickness and eosinophil infiltration in severe persistent allergic asthma patients. Int J Immunopathol Pharmacol (2012) 25(2):475–84. doi:10.1177/039463201202500217
150. Skiepko R, Zietkowski Z, Lukaszyk M, Budny W, Skiepko U, Milewski R, et al. Changes in blood eosinophilia during omalizumab therapy as a predictor of asthma exacerbation. Postepy dermatologii i alergologii (2014) 31(5):305–9. doi:10.5114/pdia.2014.40973
151. Kallieri M, Papaioannou AI, Papathanasiou E, Ntontsi P, Papiris S, Loukides S. Predictors of response to therapy with omalizumab in patients with severe allergic asthma – a real life study. Postgrad Med (2017):1–7. doi:10.1080/00325481.2017.1321945
152. Zietkowski Z, Skiepko R, Tomasiak-Lozowska MM, Bodzenta-Lukaszyk A. Airway inflammation and eotaxin in exhaled breath condensate of patients with severe persistent allergic asthma during omalizumab therapy. Adv Med Sci (2011) 56(2):318–22. doi:10.2478/v10039-011-0024-0
153. Seminario MC, Saini SS, MacGlashan DW Jr, Bochner BS. Intracellular expression and release of Fc epsilon RI alpha by human eosinophils. J Immunol (1999) 162(11):6893–900.
154. Fahy JV. Type 2 inflammation in asthma – present in most, absent in many. Nat Rev Immunol (2015) 15(1):57–65. doi:10.1038/nri3807
155. Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med (2014) 371(13):1198–207. doi:10.1056/NEJMoa1403290
156. Bel EH, Ortega HG, Pavord ID. Glucocorticoids and mepolizumab in eosinophilic asthma. N Engl J Med (2014) 371(25):2434. doi:10.1056/NEJMoa1403291
157. Bjermer L, Lemiere C, Maspero J, Weiss S, Zangrilli J, Germinaro M. Reslizumab for inadequately controlled asthma with elevated blood eosinophil levels: a randomized phase 3 study. Chest (2016) 150(4):789–98. doi:10.1016/j.chest.2016.03.032
158. Corren J, Weinstein S, Janka L, Zangrilli J, Garin M. Phase 3 study of reslizumab in patients with poorly controlled asthma: effects across a broad range of eosinophil counts. Chest (2016) 150(4):799–810. doi:10.1016/j.chest.2016.03.018
159. Bleecker ER, FitzGerald JM, Chanez P, Papi A, Weinstein SF, Barker P, et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting beta2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. Lancet (2016) 388(10056):2115–27. doi:10.1016/S0140-6736(16)31324-1
160. FitzGerald JM, Bleecker ER, Nair P, Korn S, Ohta K, Lommatzsch M, et al. Benralizumab, an anti-interleukin-5 receptor alpha monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet (2016) 388(10056):2128–41. doi:10.1016/S0140-6736(16)31322-8
161. Liacouras CA, Furuta GT, Hirano I, Atkins D, Attwood SE, Bonis PA, et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. J Allergy Clin Immunol (2011) 128(1):3–20.e6; quiz 21–2. doi:10.1016/j.jaci.2011.02.040
162. Dellon ES, Chen X, Miller CR, Fritchie KJ, Rubinas TC, Woosley JT, et al. Tryptase staining of mast cells may differentiate eosinophilic esophagitis from gastroesophageal reflux disease. Am J Gastroenterol (2011) 106(2):264–71. doi:10.1038/ajg.2010.412
163. Aceves SS, Chen D, Newbury RO, Dohil R, Bastian JF, Broide DH. Mast cells infiltrate the esophageal smooth muscle in patients with eosinophilic esophagitis, express TGF-beta1, and increase esophageal smooth muscle contraction. J Allergy Clin Immunol (2010) 126(6):1198–204.e4. doi:10.1016/j.jaci.2010.08.050
164. Lucendo AJ, Bellon T, Lucendo B. The role of mast cells in eosinophilic esophagitis. Pediatr Allergy Immunol (2009) 20(6):512–8. doi:10.1111/j.1399-3038.2008.00798.x
165. Konikoff MR, Noel RJ, Blanchard C, Kirby C, Jameson SC, Buckmeier BK, et al. A randomized, double-blind, placebo-controlled trial of fluticasone propionate for pediatric eosinophilic esophagitis. Gastroenterology (2006) 131(5):1381–91. doi:10.1053/j.gastro.2006.08.033
166. Liacouras CA, Spergel JM, Ruchelli E, Verma R, Mascarenhas M, Semeao E, et al. Eosinophilic esophagitis: a 10-year experience in 381 children. Clin Gastroenterol Hepatol (2005) 3(12):1198–206. doi:10.1016/S1542-3565(05)00885-2
167. Loizou D, Enav B, Komlodi-Pasztor E, Hider P, Kim-Chang J, Noonan L, et al. A pilot study of omalizumab in eosinophilic esophagitis. PLoS One (2015) 10(3):e0113483. doi:10.1371/journal.pone.0113483
168. Straumann A, Conus S, Grzonka P, Kita H, Kephart G, Bussmann C, et al. Anti-interleukin-5 antibody treatment (mepolizumab) in active eosinophilic oesophagitis: a randomised, placebo-controlled, double-blind trial. Gut (2010) 59(1):21–30. doi:10.1136/gut.2009.178558
169. Conus S, Straumann A, Bettler E, Simon HU. Mepolizumab does not alter levels of eosinophils, T cells, and mast cells in the duodenal mucosa in eosinophilic esophagitis. J Allergy Clin Immunol (2010) 126(1):175–7. doi:10.1016/j.jaci.2010.04.029
170. Otani IM, Anilkumar AA, Newbury RO, Bhagat M, Beppu LY, Dohil R, et al. Anti-IL-5 therapy reduces mast cell and IL-9 cell numbers in pediatric patients with eosinophilic esophagitis. J Allergy Clin Immunol (2013) 131(6):1576–82. doi:10.1016/j.jaci.2013.02.042
171. Spergel JM, Rothenberg ME, Collins MH, Furuta GT, Markowitz JE, Fuchs G III, et al. Reslizumab in children and adolescents with eosinophilic esophagitis: results of a double-blind, randomized, placebo-controlled trial. J Allergy Clin Immunol (2012) 129(2):456–63, 463.e1–3. doi:10.1016/j.jaci.2011.11.044
172. Jakiela B, Szczeklik W, Plutecka H, Sokolowska B, Mastalerz L, Sanak M, et al. Increased production of IL-5 and dominant Th2-type response in airways of Churg-Strauss syndrome patients. Rheumatology (Oxford) (2012) 51(10):1887–93. doi:10.1093/rheumatology/kes171
173. Detoraki A, Di Capua L, Varricchi G, Genovese A, Marone G, Spadaro G. Omalizumab in patients with eosinophilic granulomatosis with polyangiitis: a 36-month follow-up study. J Asthma (2016) 53(2):201–6. doi:10.3109/02770903.2015.1081700
174. Kahn JE, Grandpeix-Guyodo C, Marroun I, Catherinot E, Mellot F, Roufosse F, et al. Sustained response to mepolizumab in refractory Churg-Strauss syndrome. J Allergy Clin Immunol (2010) 125(1):267–70. doi:10.1016/j.jaci.2009.10.014
175. Wechsler ME, Akuthota P, Jayne D, Khoury P, Klion A, Langford CA, et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med (2017) 376(20):1921–32. doi:10.1056/NEJMoa1702079
176. Detoraki A, Varricchi G, Genovese A, Marone G, Spadaro G. The role of omalizumab in patients with eosinophilic granulomatosis with polyangiitis: comment on the article by Jachiet et al. Arthritis Rheumatol (2016) 69(4):868–70. doi:10.1002/art.40012
177. Loeffler W. Endocarditis parietalis fibroplastica mit Blut-eosinophilie, ein eigenartiges Krankheitsbild. Schweiz Med Wochen (1936) 66:817.
178. Desreumaux P, Janin A, Dubucquoi S, Copin MC, Torpier G, Capron A, et al. Synthesis of interleukin-5 by activated eosinophils in patients with eosinophilic heart diseases. Blood (1993) 82(5):1553–60.
179. Tai PC, Ackerman SJ, Spry CJ, Dunnette S, Olsen EG, Gleich GJ. Deposits of eosinophil granule proteins in cardiac tissues of patients with eosinophilic endomyocardial disease. Lancet (1987) 1(8534):643–7. doi:10.1016/S0140-6736(87)90412-0
180. Davis BP, Epstein T, Kottyan L, Amin P, Martin LJ, Maddox A, et al. Association of eosinophilic esophagitis and hypertrophic cardiomyopathy. J Allergy Clin Immunol (2016) 137(3):934–6.e5. doi:10.1016/j.jaci.2015.08.026
181. Marone G, de Crescenzo G, Adt M, Patella V, Arbustini E, Genovese A. Immunological characterization and functional importance of human heart mast cells. Immunopharmacology (1995) 31(1):1–18. doi:10.1016/0162-3109(95)00037-3
182. Marone G, Giordano A, Cirillo R, Triggiani M, Vigorito C. Cardiovascular and metabolic effects of peptide leukotrienes in man. Ann N Y Acad Sci (1988) 524:321–33. doi:10.1111/j.1749-6632.1988.tb38555.x
183. Vigorito C, Poto S, Picotti GB, Triggiani M, Marone G. Effect of activation of the H1 receptor on coronary hemodynamics in man. Circulation (1986) 73(6):1175–82. doi:10.1161/01.CIR.73.6.1175
184. Rohrbach MS, Wheatley CL, Slifman NR, Gleich GJ. Activation of platelets by eosinophil granule proteins. J Exp Med (1990) 172(4):1271–4. doi:10.1084/jem.172.4.1271
185. Tsunemi K, Takai S, Nishimoto M, Jin D, Sakaguchi M, Muramatsu M, et al. A specific chymase inhibitor, 2-(5-formylamino-6-oxo-2-phenyl-1,6-dihydropyrimidine-1-yl)-N-[[3,4-dioxo-1-phenyl-7-(2-pyridyloxy)]-2-heptyl]acetamide (NK3201), suppresses development of abdominal aortic aneurysm in hamsters. J Pharmacol Exp Ther (2004) 309(3):879–83. doi:10.1124/jpet.103.063974
186. Sun J, Zhang J, Lindholt JS, Sukhova GK, Liu J, He A, et al. Critical role of mast cell chymase in mouse abdominal aortic aneurysm formation. Circulation (2009) 120(11):973–82. doi:10.1161/CIRCULATIONAHA.109.849679
187. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (2013) 381(9863):320–32. doi:10.1016/S0140-6736(12)61140-4
188. Bieber K, Ernst AL, Tukaj S, Holtsche MM, Schmidt E, Zillikens D, et al. Analysis of serum markers of cellular immune activation in patients with bullous pemphigoid. Exp Dermatol (2017). doi:10.1111/exd.13382
189. Heimbach L, Li Z, Berkowitz P, Zhao M, Li N, Rubenstein DS, et al. The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem (2011) 286(17):15003–9. doi:10.1074/jbc.M111.221036
190. Engmann J, Rudrich U, Behrens G, Papakonstantinou E, Gehring M, Kapp A, et al. Increased activity and apoptosis of eosinophils in blister fluids, skin and peripheral blood of patients with bullous pemphigoid. Acta Derm Venereol (2017) 97(4):464–71. doi:10.2340/00015555-2581
191. de Graauw E, Sitaru C, Horn M, Borradori L, Yousefi S, Simon HU, et al. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy (2017) 72(7):1105–13. doi:10.1111/all.13131
192. Brembilla NC, Stalder R, Senra L, Bohencke WH. IL-17A localizes in the exocytic compartment of mast cells in psoriatic skin. Br J Dermatol (2017). doi:10.1111/bjd.15358
193. Mashiko S, Bouguermouh S, Rubio M, Baba N, Bissonnette R, Sarfati M. Human mast cells are major IL-22 producers in patients with psoriasis and atopic dermatitis. J Allergy Clin Immunol (2015) 136(2):351–9.e1. doi:10.1016/j.jaci.2015.01.033
194. Heidenreich R, Rocken M, Ghoreschi K. Angiogenesis drives psoriasis pathogenesis. Int J Exp Pathol (2009) 90(3):232–48. doi:10.1111/j.1365-2613.2009.00669.x
195. Oyoshi MK, He R, Kumar L, Yoon J, Geha RS. Cellular and molecular mechanisms in atopic dermatitis. Adv Immunol (2009) 102:135–226. doi:10.1016/S0065-2776(09)01203-6
196. Alenius H, Laouini D, Woodward A, Mizoguchi E, Bhan AK, Castigli E, et al. Mast cells regulate IFN-gamma expression in the skin and circulating IgE levels in allergen-induced skin inflammation. J Allergy Clin Immunol (2002) 109(1):106–13. doi:10.1067/mai.2002.120553
197. Guttman-Yassky E, Nograles KE, Krueger JG. Contrasting pathogenesis of atopic dermatitis and psoriasis – part II: immune cell subsets and therapeutic concepts. J Allergy Clin Immunol (2011) 127(6):1420–32. doi:10.1016/j.jaci.2011.01.054
198. Rheinbach G. Ueber das Verhalten der Leucocyten bei malignen Tumoren. Arch Klin Chir (1893) 46:486–562.
199. Looi LM. Tumor-associated tissue eosinophilia in nasopharyngeal carcinoma. A pathologic study of 422 primary and 138 metastatic tumors. Cancer (1987) 59(3):466–70. doi:10.1002/1097-0142(19870201)59:3<466::AID-CNCR2820590319>3.0.CO;2-P
200. Nielsen HJ, Hansen U, Christensen IJ, Reimert CM, Brunner N, Moesgaard F. Independent prognostic value of eosinophil and mast cell infiltration in colorectal cancer tissue. J Pathol (1999) 189(4):487–95. doi:10.1002/(SICI)1096-9896(199912)189:4<487::AID-PATH484>3.0.CO;2-I
201. Dorta RG, Landman G, Kowalski LP, Lauris JR, Latorre MR, Oliveira DT. Tumour-associated tissue eosinophilia as a prognostic factor in oral squamous cell carcinomas. Histopathology (2002) 41(2):152–7. doi:10.1046/j.1365-2559.2002.01437.x
202. Teruya-Feldstein J, Jaffe ES, Burd PR, Kingma DW, Setsuda JE, Tosato G. Differential chemokine expression in tissues involved by Hodgkin’s disease: direct correlation of eotaxin expression and tissue eosinophilia. Blood (1999) 93(8):2463–70.
203. Kampen GT, Stafford S, Adachi T, Jinquan T, Quan S, Grant JA, et al. Eotaxin induces degranulation and chemotaxis of eosinophils through the activation of ERK2 and p38 mitogen-activated protein kinases. Blood (2000) 95(6):1911–7.
204. Lotfi R, Herzog GI, DeMarco RA, Beer-Stolz D, Lee JJ, Rubartelli A, et al. Eosinophils oxidize damage-associated molecular pattern molecules derived from stressed cells. J Immunol (2009) 183(8):5023–31. doi:10.4049/jimmunol.0900504
205. Cormier SA, Taranova AG, Bedient C, Nguyen T, Protheroe C, Pero R, et al. Pivotal advance: eosinophil infiltration of solid tumors is an early and persistent inflammatory host response. J Leukoc Biol (2006) 79(6):1131–9. doi:10.1189/jlb.0106027
206. Davis BP, Rothenberg ME. Eosinophils and cancer. Cancer Immunol Res (2014) 2(1):1–8. doi:10.1158/2326-6066.CIR-13-0196
207. Gatault S, Legrand F, Delbeke M, Loiseau S, Capron M. Involvement of eosinophils in the anti-tumor response. Cancer Immunol Immunother (2012) 61(9):1527–34. doi:10.1007/s00262-012-1288-3
208. Lotfi R, Lee JJ, Lotze MT. Eosinophilic granulocytes and damage-associated molecular pattern molecules (DAMPs): role in the inflammatory response within tumors. J Immunother (2007) 30(1):16–28. doi:10.1097/01.cji.0000211324.53396.f6
209. von Wasielewski R, Seth S, Franklin J, Fischer R, Hubner K, Hansmann ML, et al. Tissue eosinophilia correlates strongly with poor prognosis in nodular sclerosing Hodgkin’s disease, allowing for known prognostic factors. Blood (2000) 95(4):1207–13.
210. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ’Immunoscore’ in the classification of malignant tumours. J Pathol (2014) 232(2):199–209. doi:10.1002path.4287
211. Sakkal S, Miller S, Apostolopoulos V, Nurgali K. Eosinophils in cancer: favourable or unfavourable? Curr Med Chem (2016) 23(7):650–66. doi:10.2174/0929867323666160119094313
212. Beil WJ, Weller PF, Tzizik DM, Galli SJ, Dvorak AM. Ultrastructural immunogold localization of tumor necrosis factor-alpha to the matrix compartment of eosinophil secondary granules in patients with idiopathic hypereosinophilic syndrome. J Histochem Cytochem (1993) 41(11):1611–5. doi:10.1177/41.11.8409368
213. Nissim Ben-Efraim AH, Levi-Schaffer F. Roles of eosinophils in the modulation of angiogenesis. Chem Immunol Allergy (2014) 99:138–54. doi:10.1159/000353251
214. Varricchi G, Galdiero MR, Marone G, Granata F, Borriello F, Marone G. Controversial role of mast cells in skin cancers. Exp Dermatol (2017) 26(1):11–7. doi:10.1111/exd.13107
215. Valent P, Horny HP, Escribano L, Longley BJ, Li CY, Schwartz LB, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res (2001) 25(7):603–25. doi:10.1016/S0145-2126(01)00038-8
216. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med (2015) 373(2):163–72. doi:10.1056/NEJMra1409760
217. Bodemer C, Hermine O, Palmerini F, Yang Y, Grandpeix-Guyodo C, Leventhal PS, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol (2010) 130(3):804–15. doi:10.1038/jid.2009.281
218. Bohm A, Fodinger M, Wimazal F, Haas OA, Mayerhofer M, Sperr WR, et al. Eosinophilia in systemic mastocytosis: clinical and molecular correlates and prognostic significance. J Allergy Clin Immunol (2007) 120(1):192–9. doi:10.1016/j.jaci.2007.03.015
219. Wilson TM, Maric I, Shukla J, Brown M, Santos C, Simakova O, et al. IL-5 receptor alpha levels in patients with marked eosinophilia or mastocytosis. J Allergy Clin Immunol (2011) 128(5):1086–92.e1–3. doi:10.1016/j.jaci.2011.05.032
220. Wimazal F, Germing U, Kundi M, Noesslinger T, Blum S, Geissler P, et al. Evaluation of the prognostic significance of eosinophilia and basophilia in a larger cohort of patients with myelodysplastic syndromes. Cancer (2010) 116(10):2372–81. doi:10.1002/cncr.25036
221. Klion AD, Noel P, Akin C, Law MA, Gilliland DG, Cools J, et al. Elevated serum tryptase levels identify a subset of patients with a myeloproliferative variant of idiopathic hypereosinophilic syndrome associated with tissue fibrosis, poor prognosis, and imatinib responsiveness. Blood (2003) 101(12):4660–6. doi:10.1182/blood-2003-01-0006
222. Pardanani A, Brockman SR, Paternoster SF, Flynn HC, Ketterling RP, Lasho TL, et al. FIP1L1-PDGFRA fusion: prevalence and clinicopathologic correlates in 89 consecutive patients with moderate to severe eosinophilia. Blood (2004) 104(10):3038–45. doi:10.1182/blood-2004-03-0787
223. Valent P, Akin C, Hartmann K, Nilsson G, Reiter A, Hermine O, et al. Advances in the classification and treatment of mastocytosis: current status and outlook toward the future. Cancer Res (2017) 77(6):1261–70. doi:10.1158/0008-5472.CAN-16-2234
Keywords: allergy, asthma, cancer, eosinophils, inflammation, mast cells, mastocytosis
Citation: Galdiero MR, Varricchi G, Seaf M, Marone G, Levi-Schaffer F and Marone G (2017) Bidirectional Mast Cell–Eosinophil Interactions in Inflammatory Disorders and Cancer. Front. Med. 4:103. doi: 10.3389/fmed.2017.00103
Received: 17 March 2017; Accepted: 26 June 2017;
Published: 24 July 2017
Edited by:
Florence Emmanuelle Roufosse, Free University of Brussels, BelgiumReviewed by:
Josiane Sabbadini Neves, Federal University of Rio de Janeiro, BrazilWei Li, Marshall University, United States
Copyright: © 2017 Galdiero, Varricchi, Seaf, Marone, Levi-Schaffer and Marone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Levi-Schaffer, ZnJhbmNlc2NhbEBla21kLmh1amkuYWMuaWw=;
Gianni Marone, bWFyb25lQHVuaW5hLml0