 Miriam Peinhaupt
Miriam Peinhaupt Eva M. Sturm
Eva M. Sturm Akos Heinemann*
Akos Heinemann*
- Institute of Experimental and Clinical Pharmacology, Medical University of Graz, Graz, Austria
Of the known prostanoid receptors, human eosinophils express the prostaglandin D2 (PGD2) receptors DP1 [also D-type prostanoid (DP)] and DP2 (also chemoattractant receptor homologous molecule, expressed on Th2 cells), the prostaglandin E2 receptors EP2 and EP4, and the prostacyclin (PGI2) receptor IP. Prostanoids can bind to either one or multiple receptors, characteristically have a short half-life in vivo, and are quickly degraded into metabolites with altered affinity and specificity for a given receptor subtype. Prostanoid receptors signal mainly through G proteins and naturally activate signal transduction pathways according to the G protein subtype that they preferentially interact with. This can lead to the activation of sometimes opposing signaling pathways. In addition, prostanoid signaling is often cell-type specific and also the combination of expressed receptors can influence the outcome of the prostanoid impulse. Accordingly, it is assumed that eosinophils and their (patho-)physiological functions are governed by a sensitive prostanoid signaling network. In this review, we specifically focus on the functions of PGD2, PGE2, and PGI2 and their receptors on eosinophils. We discuss their significance in allergic and non-allergic diseases and summarize potential targets for drug intervention.
The Prostanoid—Eosinophil Axis in Allergic Diseases
Atopy is a genetically determined disorder, which results in characteristic inflammatory responses to per se innocuous antigens. Atopic diseases can manifest in different tissues as allergic rhinitis, conjunctivitis, bronchial asthma, dermatitis, or food allergies, and are associated with a major reduction in quality of life and life expectancy. In addition, some diseases, such as intrinsic asthma, aspirin sensitivity, nasal polyposis, adenoid hyperplasia, or chronic idiopathic urticaria, share several clinical and pathophysiological aspects of allergy, but with less clear ties to allergens. The basic concept of atopic reactions is grounded in an inadequate activation of immune cells by both specific and non-specific stimuli, with a shift toward the type-2 spectrum of inflammatory mediators, such as interleukin (IL)-4, -5, -9, and -13 (1). In allergen-specific IgE-mediated hypersensitivity reactions mast cells release preformed and newly synthesized mediators [histamine, leukotriene C4, prostaglandin (PG) D2, TNFα, and many others] (2). This is the pivotal step in the inflammatory cascade as it initiates the early phase of an allergic reaction. On the one hand, these mediators provoke symptoms such as sneezing, nasal congestion, rhinorrhea, wheezing, skin rash, etc., on the other hand, they trigger the infiltration of innate and adaptive immune cells, which favors the development of the late phase response that is characterized by symptoms such as bronchoconstriction, mucus hypersecretion, edema, pain, heat, and erythema.
Eosinophils are regarded as crucial effector cells in chronic allergic inflammation. Activated eosinophils release an array of cytotoxic and pro-inflammatory mediators promoting mucosal damage in chronic asthma and allergic inflammation. The tissue damage repeatedly initiates repair mechanisms that can lead to imbalance of epithelial-to-mesenchymal transition (3, 4). Consequently, eosinophils also play a role in airway remodeling and angiogenesis in chronically inflamed tissue, and hence contribute to the progression of the disease (5, 6). Consequently, eosinophil-deficient mice are protected against allergen-induced pulmonary inflammation and airway hyperresponsiveness (7, 8). The pathogenic role of eosinophils was eventually highlighted in a pivotal study showing that patients whose treatment is adjusted according to sputum eosinophil counts have significantly fewer severe asthma exacerbations than patients on standard management therapy (9). Therefore, eosinophils are currently considered a major therapeutic target in allergic diseases, such as conjunctivitis, rhinosinusitis, asthma, and atopic dermatitis, but they might also play pathogenic roles in several other diseases, such as eosinophilic esophagitis and gastroenteritis, pancreatitis, colitis ulcerosa, hypereosinophilic syndrome, renal disease, and cancer (10–19).
Importantly, the role of eosinophils in murine models of allergic airway inflammation is discussed controversially. IL-5 transgenic mice show pronounced eosinophilia and intrinsic airway hyperreactivity whereas the latter is abolished when CD4+ cells are depleted in these mice (20). However, it has also been observed that IL-5 transgenic mice are protected from airway hyperreactivity, and eosinophils isolated from BAL of OVA-challenged IL-5 transgenic mice do not release superoxides when activated with physiological stimuli (eotaxin, IL-5, PAF, or IgG) (21), which is in sharp contrast to human eosinophils. Therefore, the role of mouse vs. human eosinophils might differ in the pathophysiology of allergic diseases.
Human eosinophils express a distinct pattern of prostanoid receptors, comprising the receptors for PGD2, DP1 [also D-type prostanoid (DP)] (22) and DP2 [also chemoattractant receptor homologous molecule expressed on Th2 cells (CRTH2)] (23), the prostaglandin E2 receptors EP2 and EP4 (24), and the PGI2 (prostacyclin) receptor IP (25). When activated, these seven-transmembrane receptors couple to G proteins, which initiate further intracellular signaling events and are eventually eliciting a cellular response. Depending on the G protein subtypes involved, this can lead to the activation of opposing signaling pathways (26–29). For instance, the DP2 receptor couples to Gαi and Gαq causing eosinophil shape change and migration, while the IP receptor inhibits these eosinophil responses, likely through Gαs. In the mouse, eosinophils express DP1 and DP2 (30). EP2 is expressed on murine eosinophils since the EP2 agonist butaprost inhibits eosinophil trafficking, and in OVA-sensitized mice, the infiltrating leukocytes after allergen challenge were immunohistologically stained EP2 positive (31). The expression of EP1, EP3, EP4, and IP remains elusive; however, IP-deficient OVA-sesitized mice show less eosinophils in the brochoalveolar lavage and airway inflammation after allergen challenge as compared to wild type mice (32, 33).
Prostaglandin D2 (PGD2)
Prostaglandin D2 is the principal ligand for two receptors, DP1 and DP2 (34), of which both are expressed on the surface of eosinophils (35). At micromolar concentrations, PGD2 is also an agonist of the thromboxane receptor, TP, which mediates the direct bronchoconstrictor effect of PGD2 (36). Moreover, a major metabolite of PGD2, 15-deoxy-Δ12,14-PGJ2 is a potent agonist of peroxisome proliferator-activated receptor (PPAR)-γ, which is also expressed by eosinophils (37). PGD2 had been known to stimulate eosinophil locomotion for some time (38, 39), but it was only in 2001 that the DP2 receptor was found to mediate this effect (22, 40, 41). Also, DP2 activation by PGD2 or DP2-selective ligands triggers Ca2+ flux, CD11b upregulation, respiratory burst, and release of eosinophil cationic protein (22, 40–42). Eosinophil responses to DP2 activation seem to depend on Gαq proteins, exemplified by the lack of effect of pertussis toxin on PGD2-induced eosinophil shape change, which—however—is abrogated by phospholipase C inhibition (43). However, PGD2-induced chemotaxis was abrogated by pretreatment of eosinophils with pertussis toxin (unpublished observation). In addition to directly stimulating eosinophil migration, we also observed that PGD2 is capable of priming eosinophils for other chemoattractants like eotaxin, 5-oxo-6,8,11,14-eicosatetraenoic acid (5-oxo-ETE), or complement factor C5a, an effect that is likewise mediated by the DP2 receptor (42, 44). Conversely, eosinophil migration toward PGD2 is impaired by eotaxin or 5-oxo-ETE in a pathway depending on phosphoinositide 3-kinase as well as p38 mitogen-activated protein kinase (44). The subcellular signaling cascades that mediate the priming effect of PGD2 are not yet understood, while the priming effect of the PGD2 metabolite 15-deoxy-Δ12,14-PGJ2 seems to involve PPAR-γ (45). Thus, it appears that a hierarchy exists among eosinophil chemoattractants: PGD2 might be regarded as an initial chemoattractant, since its potency is sustained also in whole blood and primes eosinophils for other chemoattractants; however, eotaxin seems to be an end-point chemoattractant, as it has reduced efficacy in blood as compared to isolated eosinophils, and effectively downmodulates eosinophil migration toward other chemoattractants (44).
Besides PGD2, DP2 is also activated by the PGD2 metabolites 13,14-dihydro-15-keto- (DK-) PGD2, PGJ2, Δ12-PGJ2 and 15-deoxy-Δ12,14-PGJ2 (42, 46, 47). Considering that PGD2 is as short-lived molecule and rapidly degraded into metabolites (48), it is interesting that the PGD2 actions on eosinophils are maintained through metabolites binding to DP2. Moreover, one of the major metabolites of the thromboxane pathway, 11-dehydro-TXB2, and even the common precursor of all prostanoids, PGH2, are also potent DP2 agonists (49, 50). Similarly, PGF2α has been found to activate eosinophils through DP2 (51).
In human disease, DP2 on peripheral blood eosinophils is upregulated in allergic dermatitis and rhinitis patients (52, 53), but it is diminished in active ulcerative colitis (26).
Although PGD2 binds to DP1 with similar affinity as to DP2 (34), the exact function of this receptor in immune cells has not been fully elucidated yet, and both pro- and anti-inflammatory effects have been reported (29). For instance, DP1 mediates the PGD2-induced expression of the airway mucin MUC5B in human nasal epithelial cells (54) and stimulates mucus production in vitro (55) but inhibits the functions of platelet, neutrophils, basophils, and dendritic cells (56–62). Unlike DP2, which is preferentially expressed on immune cells, such as eosinophils, basophils, macrophages, mast cells, a subset of Th2 lymphocytes and group 2 innate lymphoid cells (23, 40, 63–66), DP1 is more widely expressed, including the vasculature, the central nervous system, the retina, and the lungs (55, 67–69).
DP1-deficient mice were shown to be protected from development of allergic lung inflammation in terms of airway hyperresponsiveness, reduced numbers of BAL eosinophils, and BAL levels of IL-4, IL-5, and IL-13 (70). In contrast, intratracheal administration of DP1 agonist BW245c protected mice from airway hyperresponsiveness and lung eosinophilia in a OVA models of experimental asthma, thereby counteracting DP2-mediated proinflammatory responses (30, 71). DP1 activation has also been linked to inhibition of dendritic cell function (60) and to reduce inflammation in an IL-10-dependent mechanism (71). DP1, but not DP2, expression in lung tissue (mRNA) is upregulated upon OVA challenge (72). More recently, in guinea pigs, PGD2 aerosols were shown to induce the activation of sensory nerves and cough via DP1 receptor activation. Interestingly, DK-PGD2 modulated the sensory nerve activity by inhibiting the response to capsaicin (73).
In eosinophils, the DP1 receptor transmits antiapoptotic signals by PGD2 (22), but has been found to limit DP2-mediated CD11b upregulation (41). At micromolar concentrations, however, PGD2 and 15-deoxy-Δ12,14-PGJ2 drive eosinophils into apoptosis in a nuclear factor κB-dependent manner (74). Regarding other eosinophil responses, there is growing literature reporting cooperative signaling of DP1 and DP2 receptors. In guinea pigs, both DP1 and DP2 activation can stimulate the mobilization of eosinophils from the bone marrow (75). Moreover, DP1-dependent eosinophil responses such as migration and production of reactive oxygen species are—to some extent—co-mediated by DP1 (75, 76). On the molecular level, we have shown that DP1 activation is substantially involved in DP2-triggered Ca2+ signaling in a heterologous expression system and in human peripheral blood eosinophils and, therefore, might be an important regulator of DP2-mediated pro-inflammatory signaling (35). Cooperative signaling of the two receptors also converges in the PGD2-induced synthesis of leukotriene C4 synthesis in eosinophils. Only a simultaneous activation of DP1 and DP2 led to a sufficient response while the activation of either one or the other receptor did not equal the full PGD2 response (77). This finding does not only substantiate the significance of PGD2 in stimulating the synthesis of LTC4 but also highlights the cooperative function of the two PGD2 receptors (Figure 1).
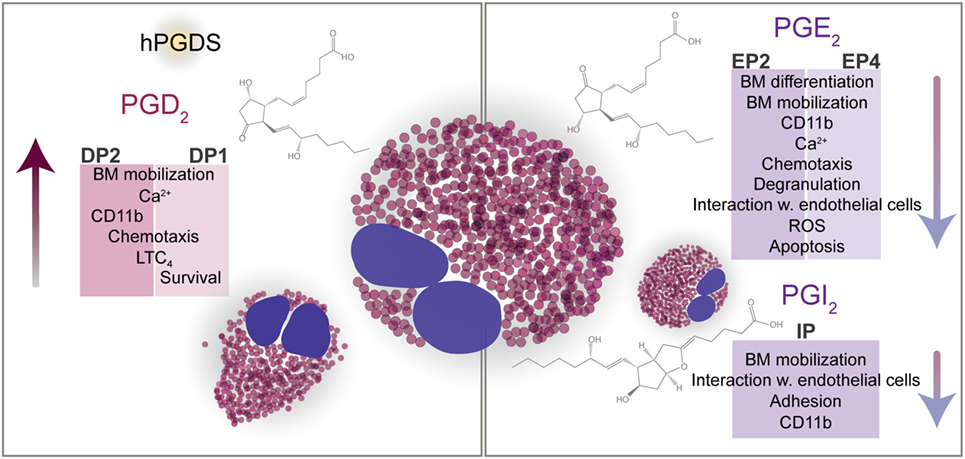
Figure 1. PGD2, PGE2, and PGI2 direct the functions of eosinophils. Eosinophils express receptors for PGD2 (DP1, DP2), PGE2 (EP2, EP4), and PGI2 (IP). Via DP2, PGD2 attracts eosinophils to the site of inflammation, enhances eosinophil mobilization from the bone marrow, and upregulates CD11b expression. In line with the chemotactic response, PGD2-mediated activation of eosinophils results in increased size and altered cell shape. DP1 and DP2 cooperatively regulate the synthesis of LTC4. DP1 has been shown to enhance the DP2-mediated Ca2+ response and to prolong the survival of eosinophils in vitro. Counteracting pro-inflammatory mechanisms PGE2 and PGI2 suppress the activation of eosinophils and hence dampen pro-inflammatory signals. Despite the negative regulation of eosinophil effector and chemotactic functions by PGE2 and PGI2, PGE2 was shown to decrease eosinophil apoptosis in vitro.
Targeting PGD2 Signaling in Eosinophilic Diseases
DP2 Receptor Antagonists
Blood and tissue eosinophilia is a key feature of allergy and asthma. It correlates with the severity of the disease on the one hand, and levels of PGD2 on the other hand (78). Exogenously applied PGD2 and DP2 agonists provoke peripheral blood eosinophilia and infiltration of eosinophils into the conjunctiva, lung, nose, and skin in animal models (30, 38, 79–82), whereas pharmacological blockade of DP2 can ameliorate models of atopic dermatitis, asthma, rhinitis, and conjunctivitis (83–88). Interestingly, DP2-deficient mice develop a normal chronic allergic inflammatory response to allergen challenge after sensitization and challenge, while the acute inflammatory response and eosinophil infiltration in the skin are abrogated (89).
The effects of the DP2 antagonist timapiprant (OC-459) was studied in a large patient cohort (n = 482) of mild-to-moderate persistent asthma. In this randomized, double-blind placebo-controlled study, the DP2 antagonist was given over 12 weeks with overall beneficial effects on lung function. A post hoc analysis revealed that the greatest improvement of lung function by timapiprant was observed in patients with active eosinophilia (≥250/μl peripheral blood) and—even more pronounced—in younger patients (90). This applies also for the humanized murine IL-5 antibody mepolizumab, which is most effective and only given in asthma patients with severe eosinophilic airway inflammation (91). In eosinophil esophagitis, timapiprant significantly reduced the esophageal eosinophil load and induced some clinical improvement (92). Timapiprant also successfully reduced nasal and ocular symptoms in allergic subjects exposed to grass pollen (93).
Fevipiprant (QAW039) is another DP2 antagonist, but as compared to timapiprant, it has slower dissociating properties and is, therefore, a candidate compound with potentially improved efficacy (94). In 170 patients with uncontrolled asthma, however, fevipiprant administered once daily did not meet the overall expected primary clinical end point (increase in FEV1), but led to an improvement of clinical symptoms in a sub-cohort with severe asthma (FEV1 < 70%), leading to a significant improvement in FEV1 and the asthma control questionnaire score, in addition to being well tolerated by the patients (95). It has to be considered, however, that post hoc analyses like these performed with fevipirant and timapiprant (90) need to be interpreted with caution. Importantly, fevipirant reduced eosinophilic airway inflammation in a separate, small trial comprising 61 patients with persistent moderate-to-severe asthma, uncontrolled by inhaled corticosteroids and elevated sputum eosinophil counts (96).
Several other DP2 antagonists have been subject to clinical trials in asthma or even COPD, but showed little efficacy and are discussed elsewhere (97).
DP1 Receptor Antagonists
Based on in situ hybridization and immunohistochemistry, DP1 mRNA and DP2 protein expression were detectable in eosinophils in nasal polyp tissue of allergic rhinitis patients; in contrast, only DP1 but not DP2 was observed in nasal tissue of healthy subjects (67).
A pivotal study using DP1-knockout mice suggested that DP1 plays an important role in the OVA-induced asthma model. DP1-deficient mice not only showed markedly reduced eosinophils in BAL fluid but also did not develop airway hyperresponsiveness (70). In a rat model of OVA-induced pulmonary inflammation, DP1 expression was upregulated in the lungs while bronchial hyperresponsiveness and immune cell infiltration was diminished by the DP1 antagonist S-5751 (98). In an OVA-induced allergic rhinitis model in guinea pigs, S-5751 inhibited late phase responses such as infiltration of eosinophils and mucosal plasma exudation (99). A newly developed DP1 antagonist (S-555739, asapiprant) showed improved affinity and bioavailability, and reversed antigen- and PGD2-induced nasal congestion and airway hyperresponsiveness in guinea pigs and sheep, respectively, along with significantly decreased eosinophils and other inflammatory cells in nasal lavage fluid (100). A phase II clinical trial in the USA (NCT01651871) and a phase III clinical trial (JapicCTI-132046) in Japan are underway testing asapiprant in seasonal allergic rhinitis. The results are yet to be announced. Previously, another DP1 antagonist, laropiprant (MK-0524), was shown to prevent nasal congestion induced by PGD2 in healthy subjects (101) but failed in phase II trials in allergic rhinitis and asthma (102). Similarly, the dual DP1/DP2 antagonist vidupiprant (AMG 853) provided no benefit as an add-on to inhaled corticosteroid therapy in moderate-to-severe asthma (103).
Inhibition of PGD2 Synthases—HPGDS and Lipocaline Prostaglandin D2 Synthase (LPGDS)
In mammals, two isoforms of PGD2 synthases are expressed: the lipocaline type (LPGDS), which is highly abundant in the central nervous system and the hematopoietic type (HPGDS), which is mainly expressed in mast cells, but also can be found in macrophages and Th2 lymphocytes (Table 1). Additionally, resident eosinophils themselves might be a late source of PGD2 at the site of allergic inflammation acting in an autocrine manner to attract and activate further eosinophils (104, 105). An interesting novel link between PGD2 and eosinophils is the recent discovery of pro-eosinophilic, so-called pathogenic effector (pe)Th2 lymphocytes, which highly express IL-5 and IL-13, and can be found at elevated levels in eosinophilic patients suffering from atopic dermatitis and eosinophilic gastrointestinal disease. These cells express not only DP2 but also HPGDS (106).
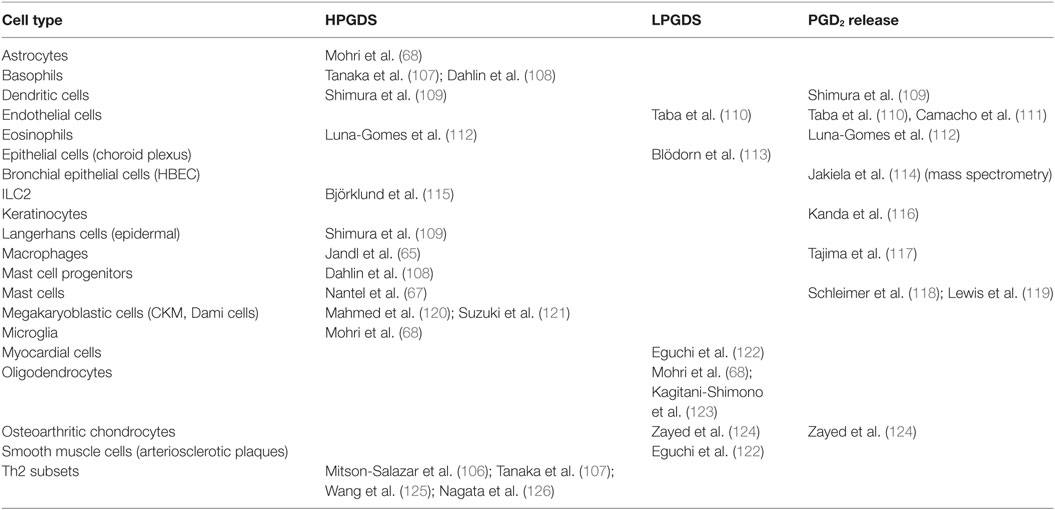
Table 1. PGD2 release and expression of hematopoietic prostaglandin D2 synthase (HPGDS) and lipocaline prostaglandin D2 synthase (LPGDS) in human cells.
Both PGD synthases are regarded as promising drug targets in a variety of diseases, such as allergic inflammation, mastocytosis, asthma and chronic obstructive pulmonary disease, metabolic disorders, muscular dystrophy, Alzheimer’s disease, or spinal cord injury (127), stimulating the development of several selective inhibitors (128–136). Transgenic mice overexpressing LPGDS show exaggerated eosinophilic pulmonary inflammation (72), which was reversed by AT-56, a LPGDS inhibitor (129). In contrast, eosinophil numbers in OVA-induced pulmonary inflammation are not significantly increased in transgenic mice overexpressing HPGDS, but the HPGDS inhibitor HQL-79 abrogated eosinophilic pulmonary inflammation in OVA-challenged mice (128). HPGDS in healthy nasal mucosa is expressed only in mast cells, but in allergic rhinitis and nasal polyps also in infiltrating inflammatory cells including eosinophils (67, 137). In a guinea-pig model of allergic inflammation, the HPGDS inhibitor TAS-204 prevented OVA-induced nasal obstruction and eosinophil infiltration (132).
Activation of PPAR-γ
In an OVA-induced allergic model, 15-deoxy-Δ12,14-PGJ2 and the PPAR-γ agonist rosiglitazone abrogated peritoneal accumulation of eosinophils and eosinophil proliferation in bone marrow (138). Similarly, several studies have shown that synthetic PPAR-γ agonists are beneficial in mouse models of allergic pulmonary inflammation and rhinitis (139, 140). Pioglitazone was tested in patients with mild asthma but did not reproduce the results from animal studies (141).
Prostaglandin E2
Infiltration of eosinophils along with other proinflammatory parameters in OVA-induced asthma model was found to be markedly enhanced in COX-1 and COX-2 knockout mice (142) and after pharmacological blockade of these enzymes (143). Conversely, inhaled PGE2 reduced airway inflammation, hyperresponsiveness, and eosinophil counts in BAL fluid of asthmatic patients (144). These findings suggested a possible inhibitory effect of PGs on eosinophils.
In airways, PGE2 is released by epithelial-, endothelial-, and smooth muscle cells, macrophages, and fibroblasts, and potently counteracts the pro-inflammatory actions of PGD2. PGE2 has bronchodilator functions and reduces airway hyperresponsiveness via activation of EP2 receptors (145). Recently, we found that PGE2 promotes the endothelial barrier by EP4 receptors expressed on the endothelium and protects against thrombin-induced junctional disruption (146).
Early studies indicated that PGE2 inhibits the release of eosinophil cationic protein (39) and homotypic aggregation of eosinophils (147) that is mediated by the β2-integrin CD18 (148). Of the known PGE2 receptors (EP1, EP2, EP3, and EP4), eosinophils express mRNA for EP2 and EP4 (24). Accordingly, we found both EP2 and EP4 protein in eosinophils using flow cytometry and Western blot, respectively (27, 31). By directly addressing the significance of PGE2 in eosinophil function, we could show that PGE2 acts to suppress eosinophil responses such as chemotaxis and degranulation, which seemed to be mediated by both EP2 and EP4 receptors (27, 31). On the subcellular level, EP4 receptor activation resulted in blockade of intracellular Ca2+ release, cytoskeletal reorganization, and production of reactive oxygen species (27). EP4 agonist treatment inhibited CD11b upregulation, activation, and clustering of β2 integrins, and L-selectin shedding of eosinophils, which were all abolished using an EP4 antagonist (149). We could delineate the underlying signaling pathways to involve phosphoinositide 3-kinase, phosphoinositide-dependent kinase 1, and protein kinase C but not the cyclic AMP/protein kinase A pathway (27, 150). Likewise, the PGE2—EP4 axis acted inhibitory on the interaction of eosinophils with endothelial cells, including adhesion and transmigration (149). In contrast, mobilization of eosinophils from guinea pig bone marrow was mediated by the EP2 receptor (31). Previously, in vitro eosinophilopoiesis stimulated by IL-5 was also observed to be under negative control of PGE2 in normal and OVA-sensitized mice by selectively inducing apoptosis in developing eosinophils (151, 152). Unexpectedly, PGE2 has been found to be antiapoptotic for peripheral blood eosinophils (153, 154), which might be linked to elevated PGE2 levels in airways of asthmatic patients (155), and even more in non-asthmatic eosinophilic bronchitis (156). Another study, however, found an inverse relationship between sputum eosinophil counts and PGE2 levels (157). Nevertheless, activation of the EP2 receptor inhibited the allergen-induced increase of eosinophils in the bronchoalveolar lavage fluid of OVA-sensitized mice (31).
Hence, the activation of EP2/EP4 receptors can be protective against the accumulation and activation of eosinophils in the affected tissue, and is therefore considered as a potential treatment strategy in allergy (Figure 1).
Prostaglandin I2
Parts of the immune-suppressive effects of PGE2 are shared by PGI2 (prostacyclin). In contrast to EP2/EP4 signaling, the activation of PGI2 receptors (IP) is mediated by intracellular cAMP, thereby inhibiting eosinophil functions. PGI2 and the stable PGI2 mimetic iloprost negatively regulate the trafficking of guinea pig bone marrow eosinophils via IP receptor activation (158). In experimental asthma in mice, iloprost attenuates dendritic cell function and the concomitant allergen-specific Th2 response and inhibits eosinophilia in lung tissue (159). After repeated allergen challenge, endogenous PGI2 abrogates airway remodeling (32).
In an in vitro study using human eosinophils and endothelial cells, we found that endothelium-derived PGI2 is an important modulator of eosinophil–endothelial interaction and might have a bearing on eosinophil accumulation at sites of allergic reaction. Moreover, PGI2 promotes the barrier function of lung endothelial cells and limits eosinophil adhesion and transendothelial migration (25). Our data might hence explain previous findings that deletion of IP receptors in mice augments the eosinophilic infiltrate in allergic responses of the lung and skin and enhances airway remodeling (32, 33).
The Prostanoid—Eosinophil Axis in Non-Allergic Diseases
Aspirin-Exacerbated Respiratory Disease (AERD)
Also referred to as aspirin intolerance or Samter’s triad, AERD is a chronic inflammatory state of the airways resulting in rhinosinusitis, nasal polyps, and asthma. In some patients, these symptoms are accompanied by skin rash such as urticaria or angioedema, while in others the skin manifestations are prevailing. These symptoms are aggravated after intake of aspirin (acetylsalicylic acid) or any other non-selective COX inhibitor, occasionally culminating in massive anaphylactoid reactions or even death. In contrast, selective COX-2 inhibitors are mostly tolerated. A comprehensive overview on clinical presentations and pathobiologic mechanisms is provided elsewhere (160–162). In brief, an imbalance of anti-inflammatory PGE2 and proinflammatory LTC4 exists in these patients at baseline, which is further enhanced after intake of COX inhibitors, which alludes into activation of mast cells, eosinophils, and several other immune cells. In addition to mast cells, LTC4 biosynthesis in eosinophils is upregulated in AERD patients. Similarly, both cell types express more HPGDS and release excessive levels of PGD2 in this condition (163). Urinary levels of a stable PGD2 metabolite were found to be twofold higher in patients with AERD relative to those in control subjects and—most remarkably—increased further upon aspirin exposure. This correlated with reductions in blood eosinophil counts and lung function, and clinical symptoms such as nasal congestion (164). Aspirin-induced secretion of PGD2 was abrogated after successful aspirin desensitization therapy (165). Aspirin by itself was found to activate blood eosinophils in terms of Ca2+ flux, degranulation, and CD11b upregulation, the latter being more pronounced in AERD patients (166, 167). These effects were reversed by PGE2. We observed that the expression of the EP4 receptor in blood eosinophils tended to be reduced in AERD patients, and inhibition of eosinophil chemotaxis by PGE2 or an EP4 agonist was less pronounced in AERD patients as compared to healthy controls (168). Single nucleotide polymorphisms of the ptger2 and ptger4 were detected in aspirin-intolerant Korean patients, predicting lower EP2 and EP4 receptor expression levels (169, 170). A single nucleotide polymorphism in the DP2 gene crth2 was also observed to correlate with increased levels of the eosinophil chemoattractant, eotaxin-2 in Korean AERD patients (171). Similarly, the prevalence of a crth2 single nucleotide polymorphism was found to be increased in a female Japanese AERD patient cohort (172). These findings suggest that targeting PGE2 and PGD2 receptors might provide potential novel treatment options for AERD. Whether these genetic alterations specifically contribute to AERDS pathophysiology is still unclear, as similar finding have also been made for allergic disease and asthma (173).
Miscellaneous
Eosinophil infiltration into tumor-surrounding areas is observed in various types of cancer (174). The presence of tumor-associated tissue eosinophils (TATEs) seems to beneficially influence the prognosis of oral squamous cell carcinoma and other types of cancer. Davoine et al. have shown that eosinophil lysates inhibit the growth of the oral squamous carcinoma cells line (SCC-9) in vitro and correlates with the amount of released eosinophil peroxidase. Inhibition of HPGDS by HQL-79 in oral squamous cell carcinoma abrogated the migration of eosinophils toward the tumor cells. These results suggest an antitumor activity of PGD2 via the activation of release of eosinophil peroxidase from, or by cytolysis of, eosinophils (175). By using HPGDS-deficient mice, Murata et al. have shown that mast cell-derived PGD2 is an antiangiogenic factor in lung carcinoma (176). Therefore, stimulating the HPGDS/PGD2 axis could be a beneficial strategy in cancer, with TATEs serving as an additional biomarker.
Eosinophils have been shown to play a significant role in inflammatory bowel disease, ulcerative colitis, and Crohn’s disease (13, 177, 178). We have shown in experimental Crohn’s disease that eosinophils contribute to intestinal inflammation via activation of DP2. Timapiprant inhibited the recruitment of eosinophils into the colon, reduced intestinal inflammation, and decreases cytokine levels (TNFα, IL-1β, IL-6) in mice. In Crohn’s patients, PGD2 and Δ12-PGJ2 levels were increased as compared to control individuals (179). In a subsequent study, increased expression of LPGDS in myenteric and submucosal neurons, and enhanced PGD2 release, was observed in tissue samples from colon of patients with active Crohn’s disease (180). In ulcerative colitis, we observed opposing effects of DP1 and DP2 as blockade of DP2 improved, whereas a DP1 antagonist worsened, inflammation in a mouse model of colitis (26). In ulcerative colitis patients, DP2 expression was downregulated on peripheral blood eosinophils, while DP1 was upregulated, and both findings correlated with disease activity. Biopsies of colitis patients revealed an increase of DP2-positive cells in the colonic mucosa and high DP2 protein content. Both PGD2 and PGE2 levels were elevated in serum of colitis patients (26). Eosinophils and macrophages were suggested to be the main source of PGE2 in colitis (181). Current literature suggests that, like in allergy, PGE2 through its EP4 receptor opposes the pro-inflammatory action of PGD2 in inflammatory bowel disease and plays a protective role in mouse models of colitis (182–184). In contrast, a large body of evidence supports EP4 receptors to predominantly mediate the overall pro-tumorigenic action of PGE2 (185). Whether inhibition of eosinophil function is involved in the anti-inflammatory and pro-tumorigenic roles of the EP4 receptor in the gut has not been investigated yet.
Concluding Remarks
Accumulating data suggest that the DP2 receptor is an important activator of eosinophils, as it does not only respond to its cognate ligand, PGD2, but also to most of its metabolites, and even unrelated prostanoid species. PGD2 is generated by a large variety of immune cells under different conditions. Among other leukocytes, eosinophils are probably the most important DP2-bearing cells. Thus, it is believed that DP2, and to some extent also DP1, crucially contribute to various pathologies that involve eosinophils, and provide novel therapeutic approaches to conditions such as asthma, allergic rhinitis, conjunctivitis, esophagitis and skin disease, nasal polyposis, aspirin-intolerance, Crohn’s disease, and certain types of cancer. In contrast, PGE2 transmits inhibitory signals onto eosinophils through EP2 and EP4 receptors, and is thus a natural antipode to its isomer, PGD2. For instance, HPGDS expression is enhanced, while microsomal PGE2 synthase is decreased in chronic rhinosinusitis that results in eosinophilic inflammation favoring polyp formation (186). In asthma patients, a decrease of PGE2 as compared to other prostanoids including PGD2 correlates with airway obstruction (187). Similar findings are typical for AERD. An imbalance of PGD2/PGE2 secretion might hence potentially underlie and/or sustain the abovementioned, eosinophilic pathologies, and might constitute novel therapeutic targets.
Author Contributions
MP, ES, and AH wrote and edited the review.
Conflict of Interest Statement
MP and ES declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. AH received consulting fees from AstraZeneca and Bayer.
Funding
This work was supported by the Austrian Science Fund FWF (DK MOLIN-W1241), the Medical University of Graz, and BioTechMed Graz.
References
1. Barnes PJ. Pathophysiology of allergic inflammation. Immunol Rev (2011) 242:31–50. doi:10.1111/j.1600-065X.2011.01020.x
2. Modena BD, Dazy K, White AA. Emerging concepts: mast cell involvement in allergic diseases. Transl Res (2016) 174:98–121. doi:10.1016/j.trsl.2016.02.011
3. Kagalwalla AF, Akhtar N, Woodruff SA, Rea BA, Masterson JC, Mukkada V, et al. Eosinophilic esophagitis: epithelial mesenchymal transition contributes to esophageal remodeling and reverses with treatment. J Allergy Clin Immunol (2012) 129:1387–96.e7. doi:10.1016/j.jaci.2012.03.005
4. Yasukawa A, Hosoki K, Toda M, Miyake Y, Matsushima Y, Matsumoto T, et al. Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS One (2013) 8:e64281. doi:10.1371/journal.pone.0064281
5. Aceves SS, Broide DH. Airway fibrosis and angiogenesis due to eosinophil trafficking in chronic asthma. Curr Mol Med (2008) 8:350–8. doi:10.2174/156652408785161023
6. Wilson SJ, Rigden HM, Ward JA, Laviolette M, Jarjour NN, Djukanović R. The relationship between eosinophilia and airway remodelling in mild asthma. Clin Exp Allergy (2013) 43:1342–50. doi:10.1111/cea.12156
7. Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, et al. A critical role for eosinophils in allergic airways remodeling. Science (2004) 305:1776–9. doi:10.1126/science.1100283
8. Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, O’Neill KR, et al. Defining a link with asthma in mice congenitally deficient in eosinophils. Science (2004) 305:1773–6. doi:10.1126/science.1099472
9. Green RH, Brightling CE, McKenna S, Hargadon B, Parker D, Bradding P, et al. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet (2002) 360:1715–21. doi:10.1016/S0140-6736(02)11679-5
10. Cottin V. Eosinophilic lung diseases. Clin Chest Med (2016) 37:535–56. doi:10.1016/j.ccm.2016.04.015
11. Kovalszki A, Weller PF, Israel B, Medical D, Israel B, Medical D. Eosiniophilia. Prim Care (2017) 43:607–17. doi:10.1016/j.pop.2016.07.010
12. Manohar M, Verma AK, Venkateshaiah SU, Sanders NL, Mishra A. Pathogenic mechanisms of pancreatitis. World J Gastrointest Pharmacol Ther (2017) 8:10. doi:10.4292/wjgpt.v8.i1.10
13. Park S, Abdi T, Gentry M, Laine L. Histological disease activity as a predictor of clinical relapse among patients with ulcerative colitis: systematic review and meta-analysis. Am J Gastroenterol (2016) 111:1692–701. doi:10.1038/ajg.2016.418
14. Praga M, González E. Acute interstitial nephritis. Kidney Int (2010) 77:956–61. doi:10.1038/ki.2010.89
15. Shah SA, Ishinaga H, Takeuchi K. Pathogenesis of eosinophilic chronic rhinosinusitis. J Inflamm (Lond) (2016) 13:11. doi:10.1186/s12950-016-0121-8
16. Sakkal S, Miller S, Apostolopoulos V, Nurgali K. Eosinophils in cancer: favourable or unfavourable? Curr Med Chem (2016) 23:650–66. doi:10.2174/0929867323666160119094313
17. Schoepfer A, Safroneeva E, Straumann A. Eosinophilic esophagitis: impact of latest insights into pathophysiology on therapeutic strategies. Dig Dis (2016) 34:462–8. doi:10.1159/000445201
18. Shih H-M, Bair M-J, Chen H-L, Lin I-T. Eosinophilic gastroenteritis: brief review. Acta Gastroenterol Belg (2016) 79:239–44.
19. Werfel T, Allam JP, Biedermann T, Eyerich K, Gilles S, Guttman-Yassky E, et al. Cellular and molecular immunologic mechanisms in patients with atopic dermatitis. J Allergy Clin Immunol (2016) 138:336–49. doi:10.1016/j.jaci.2016.06.010
20. Borchers MT, Crosby J, Justice P, Farmer S, Hines E, Lee JJ, et al. Intrinsic AHR in IL-5 transgenic mice is dependent on CD4+ cells and CD49d-mediated signaling. Am J Physiol Lung Cell Mol Physiol (2001) 281:L653–9.
21. Kobayashi T, Iijima K, Kita H. Marked airway eosinophilia prevents development of airway hyper-responsiveness during an allergic response in IL-5 transgenic mice. J Immunol (2003) 170:5756–63. doi:10.4049/jimmunol.170.11.5756
22. Gervais FG, Cruz RP, Chateauneuf A, Gale S, Sawyer N, Nantel F, et al. Selective modulation of chemokinesis, degranulation, and apoptosis in eosinophils through the PGD2 receptors CRTH2 and DP. J Allergy Clin Immunol (2001) 108:982–8. doi:10.1067/mai.2001.119919
23. Nagata K, Hirai H, Tanaka K, Ogawa K, Aso T, Sugamura K, et al. CRTH2, an orphan receptor of T-helper-2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factor(s). FEBS Lett (1999) 459:195–9. doi:10.1016/S0014-5793(99)01251-X
24. Mita H, Hasegawa M, Higashi N, Akiyama K. Characterization of PGE2 receptor subtypes in human eosinophils. J Allergy Clin Immunol (2002) 110:457–9. doi:10.1067/mai.2002.127001
25. Konya V, Sturm EM, Schratl P, Beubler E, Marsche G, Schuligoi R, et al. Endothelium-derived prostaglandin I(2) controls the migration of eosinophils. J Allergy Clin Immunol (2010) 125:1105–13. doi:10.1016/j.jaci.2009.12.002
26. Sturm EM, Radnai B, Jandl K, Stančić A, Parzmair GP, Högenauer C, et al. Opposing roles of prostaglandin d 2 receptors in ulcerative colitis. J Immunol (2014) 15:827–39. doi:10.4049/jimmunol.1303484
27. Luschnig-Schratl P, Sturm EM, Konya V, Philipose S, Marsche G, Fröhlich E, et al. EP4 receptor stimulation down-regulates human eosinophil function. Cell Mol Life Sci (2011) 68:3573–87. doi:10.1007/s00018-011-0642-5
28. Schuligoi R, Sturm E, Luschnig P, Konya V, Philipose S, Sedej M, et al. CRTH2 and D-type prostanoid receptor antagonists as novel therapeutic agents for inflammatory diseases. Pharmacology (2010) 85:372–82. doi:10.1159/000313836
29. Kostenis E, Ulven T. Emerging roles of DP and CRTH2 in allergic inflammation. Trends Mol Med (2006) 12:148–58. doi:10.1016/j.molmed.2006.02.005
30. Spik I, Brénuchon C, Angéli V, Staumont D, Fleury S, Capron M, et al. Activation of the prostaglandin D2 receptor DP2/CRTH2 increases allergic inflammation in mouse. J Immunol (2005) 174:3703–8. doi:10.4049/jimmunol.174.6.3703
31. Sturm EM, Schratl P, Schuligoi R, Konya V, Sturm GJ, Lippe IT, et al. Prostaglandin E2 inhibits eosinophil trafficking through E-prostanoid 2 receptors. J Immunol (2008) 181:7273–83. doi:10.4049/jimmunol.181.10.7273
32. Nagao K, Tanaka H, Komai M, Masuda T, Narumiya S, Nagai H. Role of prostaglandin I2 in airway remodeling induced by repeated allergen challenge in mice. Am J Respir Cell Mol Biol (2003) 29:314–20. doi:10.1165/rcmb.2003-0035OC
33. Takahashi Y, Tokuoka S, Masuda T, Hirano Y, Nagao M, Tanaka H, et al. Augmentation of allergic inflammation in prostanoid IP receptor deficient mice. Br J Pharmacol (2002) 137:315–22. doi:10.1038/sj.bjp.0704872
34. Sawyer N, Cauchon E, Chateauneuf A, Cruz RPG, Nicholson DW, Metters KM, et al. Molecular pharmacology of the human prostaglandin D2 receptor, CRTH2. Br J Pharmacol (2002) 137:1163–72. doi:10.1038/sj.bjp.0704973
35. Sedej M, Schröder R, Bell K, Platzer W, Vukoja A, Kostenis E, et al. D-type prostanoid receptor enhances the signaling of chemoattractant receptor-homologous molecule expressed on T(H)2 cells. J Allergy Clin Immunol (2012) 129:492–500,500–509. doi:10.1016/j.jaci.2011.08.015
36. Hamid-Bloomfield S, Payne AN, Petrovic AA, Whittle BJR. The role of prostanoid TP- and DP-receptors in the bronchoconstrictor effect of inhaled PGD2 in anaesthetized guinea-pigs: effect of the DP-antagonist BW A868C. Br J Pharmacol (1990) 100:761–6. doi:10.1111/j.1476-5381.1990.tb14089.x
37. Ueki S, Adachi T, Bourdeaux J, Oyamada H, Yamada Y, Hamada K, et al. Expression of PPARgamma in eosinophils and its functional role in survival and chemotaxis. Immunol Lett (2003) 86:183–9. doi:10.1016/S0165-2478(03)00003-8
38. Woodward DF, Hawley SB, Williams LS, Ralston TR, Protzman CE, Spada CS, et al. Studies on the ocular pharmacology of prostaglandin D2. Investig Ophthalmol Vis Sci (1990) 31:138–46.
39. Butchers PR, Vardey CJ. The effect of prostanoids on the function of human eosinophils. Agents Actions Suppl (1990) 31:103–12.
40. Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med (2001) 193:255–61. doi:10.1084/jem.193.2.255
41. Monneret G, Gravel S, Diamond M, Rokach J, Powell WS. Prostaglandin D 2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood (2001) 98(6):1942–8. doi:10.1182/blood.V98.6.1942
42. Heinemann A, Schuligoi R, Sabroe I, Hartnell A, Peskar BA. Delta 12-prostaglandin J2, a plasma metabolite of prostaglandin D2, causes eosinophil mobilization from the bone marrow and primes eosinophils for chemotaxis. J Immunol (2003) 170:4752–8. doi:10.4049/jimmunol.170.9.4752
43. Stubbs VEL, Schratl P, Hartnell A, Williams TJ, Peskar BA, Heinemann A, et al. Indomethacin causes prostaglandin D2-like and eotaxin-like selective responses in eosinophils and basophils. J Biol Chem (2002) 277:26012–20. doi:10.1074/jbc.M201803200
44. Schratl P, Sturm EM, Royer JF, Sturm GJ, Lippe IT, Peskar BA, et al. Hierarchy of eosinophil chemoattractants: role of p38 mitogen-activated protein kinase. Eur J Immunol (2006) 36:2401–9. doi:10.1002/eji.200535672
45. Kobayashi Y, Ueki S, Mahemuti G, Chiba T, Oyamada H, Saito N, et al. Physiological levels of 15-deoxy-Δ12,14-prostaglandin J2 prime eotaxin-induced chemotaxis on human eosinophils through peroxisome proliferator-activated receptor-γ ligation. J Immunol (2005) 175:5744–50. doi:10.4049/jimmunol.175.9.5744
47. Fitzpatrickz FA, Wynalda MA. Albumin-catalyzed metabolism of prostaglandin D2. J Biol Chem (1983) 258:11713–11718.
48. Schuligoi R, Schmidt R, Geisslinger G, Kollroser M, Peskar BA, Heinemann A. PGD2 metabolism in plasma: kinetics and relationship with bioactivity on DP1 and CRTH2 receptors. Biochem Pharmacol (2007) 74:107–17. doi:10.1016/j.bcp.2007.03.023
49. Böhm E, Sturm GJ, Weiglhofer I, Sandig H, Shichijo M, McNamee A, et al. 11-dehydro-thromboxane B2, a stable thromboxane metabolite, is a full agonist of chemoattractant receptor-homologous molecule expressed on TH2 cells (CRTH2) in human eosinophils and basophils. J Biol Chem (2004) 279:7663–70. doi:10.1074/jbc.M310270200
50. Schuligoi R, Sedej M, Waldhoer M, Vukoja A, Sturm EM, Lippe IT, et al. Prostaglandin H2 induces the migration of human eosinophils through the chemoattractant receptor homologous molecule of Th2 cells, CRTH2. J Leukoc Biol (2009) 85:136–45. doi:10.1189/jlb.0608387
51. Sandig H, Andrew D, Barnes AA, Sabroe I, Pease J. 9a,11b-PGF2 and its stereoisomer PGF2a are novel agonists of the chemoattractant receptor, CRTH2. FEBS Lett (2006) 580:373–9. doi:10.1016/j.febslet.2005.11.052
52. Yahara H, Satoh T, Miyagishi C, Yokozeki H. Increased expression of CRTH2 on eosinophils in allergic skin diseases. J Eur Acad Dermatol Venereol (2010) 24:75–6. doi:10.1111/j.1468-3083.2009.03267.x
53. El-Shazly AE, Moonen V, Mawet M, Begon D, Henket M, Arafa M, et al. IFN-γ and TNF-α potentiate prostaglandin D2-induced human eosinophil chemotaxis through up-regulation of CRTH2 surface receptor. Int Immunopharmacol (2011) 11:1864–70. doi:10.1016/j.intimp.2011.07.017
54. Choi YH, Lee SN, Aoyagi H, Yamasaki Y, Yoo JY, Park B, et al. The extracellular signal-regulated kinase mitogen-activated protein kinase/ribosomal S6 protein kinase 1 cascade phosphorylates cAMP response element-binding protein to induce MUC5B gene expression via D-prostanoid receptor signaling. J Biol Chem (2011) 286:34199–214. doi:10.1074/jbc.M111.247684
55. Wright DH, Ford-Hutchinson AW, Chadee K, Metters KM. The human prostanoid DP receptor stimulates mucin secretion in LS174T cells. Br J Pharmacol (2000) 131:1537–45. doi:10.1038/sj.bjp.0703688
56. Song W-L, Stubbe J, Ricciotti E, Alamuddin N, Ibrahim S, Crichton I, et al. Niacin and biosynthesis of PGD2 by platelet COX-1 in mice and humans. J Clin Invest (2012) 122:1459–68. doi:10.1172/JCI59262
57. Miller OV, Gorman RR. Evidence for distinct prostaglandin I2 and D2 receptors in human platelets. J Pharmacol Exp Ther (1979) 210:134–40.
58. Giles H, Leff P, Bolofo ML, Kelly MG, Robertson AD. The classification of prostaglandin DP-receptors in platelets and vasculature using BW A868C, a novel, selective and potent competitive antagonist. Br J Pharmacol (1989) 96:291–300. doi:10.1111/j.1476-5381.1989.tb11816.x
59. Boie Y, Sawyer N, Slipetz DM, Metters KM, Abramovitz M. Molecular cloning and characterization of the human prostanoid DP receptor. J Biol Chem (1995) 270:18910–6. doi:10.1074/jbc.270.32.18910
60. Hammad H, de Heer HJ, Soullie T, Hoogsteden HC, Trottein F, Lambrecht BN. Prostaglandin D2 inhibits airway dendritic cell migration and function in steady state conditions by selective activation of the D prostanoid receptor 1. J Immunol (2003) 171:3936–40. doi:10.4049/jimmunol.171.8.3936
61. Wheeldon A, Vardey CJ. Characterization of the inhibitory prostanoid receptors on human neutrophils. Br J Pharmacol (1993) 108:1051–4. doi:10.1111/j.1476-5381.1993.tb13504.x
62. Yoshimura-Uchiyama C, Iikura M, Yamaguchi M, Nagase H, Ishii A, Matsushima K, et al. Differential modulation of human basophil functions through prostaglandin D2 receptors DP and chemoattractant receptor-homologous molecule expressed on Th2 cells/DP2. Clin Exp Allergy (2004) 34:1283–90. doi:10.1111/j.1365-2222.2004.02027.x
63. Xue L, Fergusson J, Salimi M, Panse I, Ussher JE, Hegazy AN, et al. Prostaglandin D2 and leukotriene E4 synergize to stimulate diverse TH2 functions and TH2 cell/neutrophil crosstalk. J Allergy Clin Immunol (2015) 135:.1358–66.e1–11. doi:10.1016/j.jaci.2014.09.006
64. Mjösberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol (2011) 12:1055–62. doi:10.1038/ni.2104
65. Jandl K, Stacher E, Bálint Z, Sturm EM, Maric J, Peinhaupt M, et al. Activated prostaglandin D2 receptors on macrophages enhance neutrophil recruitment into the lung. J Allergy Clin Immunol (2016) 137:833–43. doi:10.1016/j.jaci.2015.11.012
66. Moon TC, Campos-Alberto E, Yoshimura T, Bredo G, Rieger AM, Puttagunta L, et al. Expression of DP2 (CRTh2), a prostaglandin D2 receptor, in human mast cells. PLoS One (2014) 9:e108595. doi:10.1371/journal.pone.0108595
67. Nantel F, Fong C, Lamontagne S, Wright DH, Giaid A, Desrosiers M, et al. Expression of prostaglandin D synthase and the prostaglandin D2 receptors DP and CRTH2 in human nasal mucosa. Prostaglandins Other Lipid Mediat (2004) 73:87–101. doi:10.1016/j.prostaglandins.2003.12.002
68. Mohri I, Kadoyama K, Kanekiyo T, Sato Y, Kagitani-Shimono K, Saito Y, et al. Hematopoietic prostaglandin D synthase and DP1 receptor are selectively upregulated in microglia and astrocytes within senile plaques from human patients and in a mouse model of Alzheimer disease. J Neuropathol Exp Neurol (2007) 66:469–80. doi:10.1097/01.jnen.0000240472.43038.27
69. Gerashchenko D, Beuckmann CT, Kanaoka Y, Eguchi N, Gordon WC, Urade Y, et al. Dominant expression of rat prostanoid DP receptor mRNA in leptomeninges, inner segments of photoreceptor cells, iris epithelium, and ciliary processes. J Neurochem (1998) 71:937–45. doi:10.1046/j.1471-4159.1998.71030937.x
70. Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, et al. Prostaglandin D2 as a mediator of allergic asthma. Science (2000) 287:2013–7. doi:10.1126/science.287.5460.2013
71. Hammad H, Kool M, Soullié T, Narumiya S, Trottein F, Hoogsteden HC, et al. Activation of the D prostanoid 1 receptor suppresses asthma by modulation of lung dendritic cell function and induction of regulatory T cells. J Exp Med (2007) 204:357–67. doi:10.1084/jem.20061196
72. Fujitani Y, Kanaoka Y, Aritake K, Uodome N, Okazaki-Hatake K, Urade Y. Pronounced eosinophilic lung inflammation and Th2 cytokine release in human lipocalin-type prostaglandin D synthase transgenic mice. J Immunol (2002) 168:443–9. doi:10.4049/jimmunol.168.1.443
73. Maher SA, Birrell MA, Adcock JJ, Wortley MA, Dubuis ED, Bonvini SJ, et al. Prostaglandin D2 and the role of the DP1, DP2 and TP receptors in the control of airway reflex events. Eur Respir J (2015) 45(4):1108–18. doi:10.1183/09031936.00061614
74. Ward C, Dransfield I, Murray J, Farrow SN, Haslett C, Rossi AG. Prostaglandin D2 and its metabolites induce caspase-dependent granulocyte apoptosis that is mediated via inhibition of IκBα degradation using a peroxisome proliferator-activated receptor-γ-independent mechanism. J Immunol (2002) 168:6232–43. doi:10.4049/jimmunol.168.12.6232
75. Schratl P, Royer JF, Kostenis E, Ulven T, Sturm EM, Waldhoer M, et al. The role of the prostaglandin D2 receptor, DP, in eosinophil trafficking. J Immunol (2007) 179:4792–9. doi:10.4049/jimmunol.179.7.4792
76. Royer JF, Schratl P, Lorenz S, Kostenis E, Ulven T, Schuligoi R, et al. A novel antagonist of CRTH2 blocks eosinophil release from bone marrow, chemotaxis and respiratory burst. Allergy (2007) 62:1401–9. doi:10.1111/j.1398-9995.2007.01452.x
77. Mesquita-Santos FP, Bakker-Abreu I, Luna-Gomes T, Bozza PT, Diaz BL, Bandeira-Melo C. Co-operative signalling through DP(1) and DP(2) prostanoid receptors is required to enhance leukotriene C(4) synthesis induced by prostaglandin D(2) in eosinophils. Br J Pharmacol (2011) 162:1674–85. doi:10.1111/j.1476-5381.2010.01086.x
78. Fajt ML, Gelhaus SL, Freeman B, Uvalle CE, Trudeau JB, Holguin F, et al. Prostaglandin D2 pathway upregulation: relation to asthma severity, control, and TH2 inflammation. J Allergy Clin Immunol (2013) 131:1504–12. doi:10.1016/j.jaci.2013.01.035
79. Shichijo M, Sugimoto H, Nagao K, Inbe H, Encinas JA, Takeshita K, et al. Chemoattractant receptor-homologous molecule expressed on Th2 cells activation in vivo increases blood leukocyte counts and its blockade abrogates 13,14-dihydro-15-keto-prostaglandin d2-induced eosinophilia in rats. J Pharmacol Exp Ther (2003) 307:518–25. doi:10.1124/jpet.103.055442
80. Shiraishi Y, Takeda K, Domenico J, Gelfand EW. Role of prostaglandin D2 and CRTH2 blockade in early- and late-phase nasal responses. Clin Exp Allergy (2014) 44:1076–82. doi:10.1111/cea.12280
81. Almishri W, Cossette C, Rokach J, Martin JG, Hamid Q, Powell WS. Effects of prostaglandin D2, 15-deoxy-Delta12,14-prostaglandin J2, and selective DP1 and DP2 receptor agonists on pulmonary infiltration of eosinophils in Brown Norway rats. J Pharmacol Exp Ther (2005) 313:64–9.
82. Shiraishi Y, Asano K, Nakajima T, Oguma T, Suzuki Y, Shiomi T, et al. Prostaglandin D2-induced eosinophilic airway inflammation is mediated by CRTH2 receptor. J Pharmacol Exp Ther (2005) 312:954–60. doi:10.1124/jpet.104.078212
83. Kagawa S, Fukunaga K, Oguma T, Suzuki Y, Shiomi T, Sayama K, et al. Role of prostaglandin D2 receptor CRTH2 in sustained eosinophil accumulation in the airways of mice with chronic asthma. Int Arch Allergy Immunol (2011) 155:6–11. doi:10.1159/000327257
84. He R, Oyoshi MK, Wang JY, Hodge MR, Jin H, Geha RS. The prostaglandin D2 receptor CRTH2 is important for allergic skin inflammation after epicutaneous antigen challenge. J Allergy Clin Immunol (2010) 126:784–90. doi:10.1016/j.jaci.2010.07.006
85. Nomiya R, Okano M, Fujiwara T, Maeda M, Kimura Y, Kino K, et al. CRTH2 plays an essential role in the pathophysiology of Cry j 1-induced pollinosis in mice. J Immunol (2008) 180:5680–8. doi:10.4049/jimmunol.180.8.5680
86. Satoh T, Moroi R, Aritake K, Urade Y, Kanai Y, Sumi K, et al. Prostaglandin D2 plays an essential role in chronic allergic inflammation of the skin via CRTH2 receptor. J Immunol (2006) 177:2621–9. doi:10.4049/jimmunol.177.4.2621
87. Uller L, Mathiesen JM, Alenmyr L, Korsgren M, Ulven T, Högberg T, et al. Antagonism of the prostaglandin D2 receptor CRTH2 attenuates asthma pathology in mouse eosinophilic airway inflammation. Respir Res (2007) 8:16. doi:10.1186/1465-9921-8-16
88. Stebbins KJ, Broadhead AR, Musiyenko A, Barik S, Scott JM, Truong YP, et al. DP2 (CRTh2) antagonism reduces ocular inflammation induced by allergen challenge and respiratory syncytial virus. Int Arch Allergy Immunol (2012) 157:259–68. doi:10.1159/000328769
89. He R, Oyoshi MK, Wang JYT, Hodge MR, Jin H, Geha RS. The prostaglandin D2 receptor CRTH2 is important for allergic skin inflammation after epicutaneous antigen challenge. J Allergy Clin Immunol (2010) 126:784–90. doi:10.1016/j.jaci.2010.07.006
90. Pettipher R, Hunter MG, Perkins CM, Collins LP, Lewis T, Baillet M, et al. Heightened response of eosinophilic asthmatic patients to the CRTH2 antagonist OC000459. Allergy (2014) 69:1223–32. doi:10.1111/all.12451
91. Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med (2009) 360:973–84. doi:10.1056/NEJMoa0808991
92. Straumann A, Hoesli S, Bussmann C, Stuck M, Perkins M, Collins LP, et al. Anti-eosinophil activity and clinical efficacy of the CRTH2 antagonist OC000459 in eosinophilic esophagitis. Allergy (2013) 68:375–85. doi:10.1111/all.12096
93. Horak F, Zieglmayer P, Zieglmayer R, Lemell P, Collins LP, Hunter MG, et al. The CRTH2 antagonist OC000459 reduces nasal and ocular symptoms in allergic subjects exposed to grass pollen, a randomised, placebo-controlled, double-blind trial. Allergy (2012) 67:1572–9. doi:10.1111/all.12042
94. Sykes DA, Bradley ME, Riddy DM, Willard E, Reilly J, Miah A, et al. Fevipiprant (QAW039), a slowly dissociating CRTh2 antagonist with the potential for improved clinical efficacy. Mol Pharmacol (2016) 89:593–605. doi:10.1124/mol.115.101832
95. Erpenbeck VJ, Popov TA, Miller D, Weinstein SF, Spector S, Magnusson B, et al. The oral CRTh2 antagonist QAW039 (fevipiprant): a phase II study in uncontrolled allergic asthma. Pulm Pharmacol Ther (2016) 39:54–63. doi:10.1016/j.pupt.2016.06.005
96. Gonem S, Berair R, Singapuri A, Hartley R, Laurencin MFM, Bacher G, et al. Fevipiprant, a prostaglandin D2 receptor 2 antagonist, in patients with persistent eosinophilic asthma: a single-centre, randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir Med (2016) 4:699–707. doi:10.1016/S2213-2600(16)30179-5
97. Santus P, Radovanovic D. Prostaglandin D2 receptor antagonists in early development as potential therapeutic options for asthma. Expert Opin Investig Drugs (2016) 25:1083–92. doi:10.1080/13543784.2016.1212838
98. Hirano Y, Shichijo M, Ikeda M, Kitaura M, Tsuchida J, Asanuma F, et al. Prostanoid DP receptor antagonists suppress symptomatic asthma-like manifestation by distinct actions from a glucocorticoid in rats. Eur J Pharmacol (2011) 666:233–41. doi:10.1016/j.ejphar.2011.05.003
99. Arimura A, Yasui K, Kishino J, Asanuma F, Hasegawa H, Kakudo S, et al. Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S-5751. J Pharmacol Exp Ther (2001) 298:411–9.
100. Takahashi G, Asanuma F, Suzuki N, Hattori M, Sakamoto S, Kugimiya A, et al. Effect of the potent and selective DP1 receptor antagonist, asapiprant (S-555739), in animal models of allergic rhinitis and allergic asthma. Eur J Pharmacol (2015) 765:15–23. doi:10.1016/j.ejphar.2015.08.003
101. Van Hecken A, Depré M, De Lepeleire I, Thach C, Oeyen M, Van Effen J, et al. The effect of MK-0524, a prostaglandin D2 receptor antagonist, on prostaglandin D2-induced nasal airway obstruction in healthy volunteers. Eur J Clin Pharmacol (2007) 63:135–41. doi:10.1007/s00228-006-0211-2
102. Philip G, van Adelsberg J, Loeys T, Liu N, Wong P, Lai E, et al. Clinical studies of the DP1 antagonist laropiprant in asthma and allergic rhinitis. J Allergy Clin Immunol (2009) 124:942–8.e1–9. doi:10.1016/j.jaci.2009.07.006
103. Busse WW, Wenzel SE, Meltzer EO, Kerwin EM, Liu MC, Zhang N, et al. Safety and efficacy of the prostaglandin D2 receptor antagonist AMG 853 in asthmatic patients. J Allergy Clin Immunol (2013) 131:339–45. doi:10.1016/j.jaci.2012.10.013
104. Luna-Gomes T, Bozza PT, Bandeira-Melo C. Eosinophil recruitment and activation: the role of lipid mediators. Front Pharmacol (2013) 4:27. doi:10.3389/fphar.2013.00027
105. Hyo S, Kawata R, Kadoyama K, Eguchi N, Kubota T, Takenaka H, et al. Expression of prostaglandin D2 synthase in activated eosinophils in nasal polyps. Arch Otolaryngol Neck Surg (2007) 133:693. doi:10.1001/archotol.133.7.693
106. Mitson-Salazar A, Yin Y, Wansley DL, Young M, Bolan H, Arceo S, et al. Hematopoietic prostaglandin D synthase defines a proeosinophilic pathogenic effector human TH2 cell subpopulation with enhanced function. J Allergy Clin Immunol (2016) 137:907–18.e9. doi:10.1016/j.jaci.2015.08.007
107. Tanaka K, Ogawa K, Sugamura K, Nakamura M, Takano S, Nagata K. Cutting edge: differential production of prostaglandin D2 by human helper T cell subsets. J Immunol (2000) 164:2277–80. doi:10.4049/jimmunol.164.5.2277
108. Dahlin JS, Malinovschi A, Ohrvik H, Sandelin M, Janson C, Alving K, et al. Lin- CD34hi CD117int/hi FceRI+ cells in human blood constitute a rare population of mast cell progenitors. Blood (2016) 127:383–92. doi:10.1182/blood-2015-06-650648
109. Shimura C, Satoh T, Igawa K, Aritake K, Urade Y, Nakamura M, et al. Dendritic cells express hematopoietic prostaglandin D synthase and function as a source of prostaglandin D2 in the skin. Am J Pathol (2010) 176:227–37. doi:10.2353/ajpath.2010.090111
110. Taba Y, Sasaguri T, Miyagi M, Abumiya T, Miwa Y, Ikeda T, et al. Fluid shear stress induces lipocalin-type prostaglandin D(2) synthase expression in vascular endothelial cells. Circ Res (2000) 86:967–73. doi:10.1161/01.RES.86.9.967
111. Camacho M, López-Belmonte J, Vila L. Rate of vasoconstrictor prostanoids released by endothelial cells depends on cyclooxygenase-2 expression and prostaglandin I synthase activity. Circ Res (1998) 83(4):353–65. doi:10.1161/01.RES.83.4.353
112. Luna-Gomes T, Magalhães KG, Mesquita-Santos FP, Bakker-Abreu I, Samico RF, Molinaro R, et al. Eosinophils as a novel cell source of prostaglandin D2: autocrine role in allergic inflammation. J Immunol (2011) 187:6518–26. doi:10.4049/jimmunol.1101806
113. Blödorn B, Mäder M, Urade Y, Hayaishi O, Felgenhauer K, Brück W. Choroid plexus: the major site of mRNA expression for the β-trace protein (prostaglandin D synthase) in human brain. Neurosci Lett (1996) 209:117–20. doi:10.1016/0304-3940(96)12614-8
114. Jakiela B, Gielicz A, Plutecka H, Hubalewska M, Mastalerz L, Bochenek G, et al. Eicosanoid biosynthesis during mucociliary and mucous metaplastic differentiation of bronchial epithelial cells. Prostaglandins Other Lipid Mediat (2013) 106:116–23. doi:10.1016/j.prostaglandins.2013.05.001
115. Björklund ÅK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, et al. The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol (2016) 17:451–60. doi:10.1038/ni.3368
116. Kanda N, Kano R, Ishikawa T, Watanabe S. The antimycotic drugs itraconazole and terbinafine hydrochloride induce the production of human β-defensin-3 in human keratinocytes. Immunobiology (2011) 216:497–504. doi:10.1016/j.imbio.2010.08.008
117. Tajima T, Murata T, Aritake K, Urade Y, Hirai H, Nakamura M, et al. Lipopolysaccharide induces macrophage migration via prostaglandin D2 and prostaglandin E2. J Pharmacol Exp Ther (2008) 326:493–501. doi:10.1124/jpet.108.137992
118. Schleimer RP, Fox CC, Naclerio RM, Plaut M, Creticos PS, Togias AG, et al. Role of human basophils and mast cells in the pathogenesis of allergic diseases. J Allergy Clin Immunol (1985) 76:369–74. doi:10.1016/0091-6749(85)90656-6
119. Lewis RA, Soter NA, Diamond PT, Austen KF, Oates JA, Roberts LJ. Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE. J Immunol (1982) 129:1627–31.
120. Mahmud I, Ueda N, Yamaguchi H, Yamashita R, Yamamoto S, Kanaoka Y, et al. Prostaglandin D synthase in human megakaryoblastic cells. J Biol Chem (1997) 272:28263–6. doi:10.1074/jbc.272.45.28263
121. Suzuki T, Watanabe K, Kanaoka Y, Sato T, Hayaishi O. Induction of hematopoietic prostaglandin D synthase in human megakaryocytic cells by phorbol ester. Biochem Biophys Res Commun (1997) 241:288–93.
122. Eguchi Y, Eguchi N, Oda H, Seiki K, Kijima Y, Matsu-ura Y, et al. Expression of lipocalin-type prostaglandin D synthase (beta-trace) in human heart and its accumulation in the coronary circulation of angina patients. Proc Natl Acad Sci U S A (1997) 94:14689–94. doi:10.1073/pnas.94.26.14689
123. Kagitani-Shimono K, Mohri I, Oda H, Ozono K, Suzuki K, Urade Y, et al. Lipocalin-type prostaglandin D synthase (beta-trace) is upregulated in the alphaB-crystallin-positive oligodendrocytes and astrocytes in the chronic multiple sclerosis. Neuropathol Appl Neurobiol (2006) 32:64–73. doi:10.1111/j.1365-2990.2005.00690.x
124. Zayed N, Li X, Chabane N, Benderdour M, Martel-Pelletier J, Pelletier J-P, et al. Increased expression of lipocalin-type prostaglandin D2 synthase in osteoarthritic cartilage. Arthritis Res Ther (2008) 10:R146. doi:10.1186/ar2581
125. Wang YH, Ito T, Wang YH, Homey B, Watanabe N, Martin R, et al. Maintenance and polarization of human TH2 central memory T cells by thymic stromal lymphopoietin-activated dendritic cells. Immunity (2006) 24:827–38. doi:10.1016/j.immuni.2006.03.019
126. Nagata K, Tanaka K, Ogawa K, Kemmotsu K, Imai T, Yoshie O, et al. Selective expression of a novel surface molecule by human Th2 cells in vivo. J Immunol (1999) 162:1278–86.
127. Thurairatnam S. Hematopoietic prostaglandin D synthase inhibitors. Prog Med Chem (2012) 51:97–133. doi:10.1016/B978-0-12-396493-9.00004-2
128. Aritake K, Kado Y, Inoue T, Miyano M, Urade Y. Structural and functional characterization of HQL-79, an orally selective inhibitor of human hematopoietic prostaglandin D synthase. J Biol Chem (2006) 281:15277–86. doi:10.1074/jbc.M506431200
129. Irikura D, Aritake K, Nagata N, Maruyama T, Shimamoto S, Urade Y. Biochemical, functional, and pharmacological characterization of AT-56, an orally active and selective inhibitor of lipocalin-type prostaglandin D synthase. J Biol Chem (2009) 284:7623–30. doi:10.1074/jbc.M808593200
130. Carron CP, Trujillo JI, Olson KL, Huang W, Hamper BC, Dice T, et al. Discovery of an oral potent selective inhibitor of hematopoietic prostaglandin D synthase (HPGDS). ACS Med Chem Lett (2010) 1:59–63. doi:10.1021/ml900025z
131. Christ AN, Labzin L, Bourne GT, Fukunishi H, Weber JE, Sweet MJ, et al. Development and characterization of new inhibitors of the human and mouse hematopoietic prostaglandin D2 synthases. J Med Chem (2010) 53:5536–48. doi:10.1021/jm100194a
132. Kajiwara D, Aoyagi H, Shigeno K, Togawa M, Tanaka K, Inagaki N, et al. Role of hematopoietic prostaglandin D synthase in biphasic nasal obstruction in guinea pig model of experimental allergic rhinitis. Eur J Pharmacol (2011) 667:389–95. doi:10.1016/j.ejphar.2011.05.041
133. Nabe T, Kuriyama Y, Mizutani N, Shibayama S, Hiromoto A, Fujii M, et al. Inhibition of hematopoietic prostaglandin D synthase improves allergic nasal blockage in guinea pigs. Prostaglandins Other Lipid Mediat (2011) 95:27–34. doi:10.1016/j.prostaglandins.2011.05.001
134. Moyo R, Chimponda T, Mukanganyama S. Inhibition of hematopoietic prostaglandin D2 synthase (H-PGDS) by an alkaloid extract from Combretum molle. BMC Complement Altern Med (2014) 14:221. doi:10.1186/1472-6882-14-221
135. Mazari AMA, Hegazy UM, Mannervik B. Identification of new inhibitors for human hematopoietic prostaglandin D2 synthase among FDA-approved drugs and other compounds. Chem Biol Interact (2015) 229:91–9. doi:10.1016/j.cbi.2015.01.014
136. Edfeldt F, Evenäs J, Lepistö M, Ward A, Petersen J, Wissler L, et al. Identification of indole inhibitors of human hematopoietic prostaglandin D2 synthase (hH-PGDS). Bioorg Med Chem Lett (2015) 25:2496–500. doi:10.1016/j.bmcl.2015.04.065
137. Asaka C, Honda K, Ito E, Fukui N, Chihara J, Ishikawa K. Peroxisome proliferator-activated receptor-γ is expressed in eosinophils in nasal polyps. Int Arch Allergy Immunol (2011) 155:57–63. doi:10.1159/000327294
138. Farnesi-de-Assunção TS, Alves CF, Carregaro V, de Oliveira JR, da Silva CAT, Cheraim AB, et al. PPAR-γ agonists, mainly 15d-PGJ(2), reduce eosinophil recruitment following allergen challenge. Cell Immunol (2012) 273:23–9. doi:10.1016/j.cellimm.2011.11.010
139. Woerly G, Honda K, Loyens M, Papin J-P, Auwerx J, Staels B, et al. Peroxisome proliferator-activated receptors alpha and gamma down-regulate allergic inflammation and eosinophil activation. J Exp Med (2003) 198:411–21. doi:10.1084/jem.20021384
140. Fukui N, Honda K, Ito E, Ishikawa K. Peroxisome proliferator-activated receptor gamma negatively regulates allergic rhinitis in mice. Allergol Int (2009) 58:247–53. doi:10.2332/allergolint.08-OA-0047
141. Anderson JR, Mortimer K, Pang L, Smith KM, Bailey H, Hodgson DB, et al. Evaluation of the PPAR-γ agonist pioglitazone in mild asthma: a double-blind randomized controlled trial. PLoS One (2016) 11:e0160257. doi:10.1371/journal.pone.0160257
142. Gavett SH, Madison SL, Chulada PC, Scarborough PE, Qu W, Boyle JE, et al. Allergic lung responses are increased in prostaglandin H synthase-deficient mice. J Clin Invest (1999) 104:721–32. doi:10.1172/JCI6890
143. Peebles RS, Hashimoto K, Morrow JD, Dworski R, Collins RD, Hashimoto Y, et al. Selective cyclooxygenase-1 and -2 inhibitors each increase allergic inflammation and airway hyperresponsiveness in mice. Am J Respir Crit Care Med (2002) 165:1154–60. doi:10.1164/ajrccm.165.8.2106025
144. Gauvreau GM, Watson RM, O’Byrne PM. Protective effects of inhaled PGE2 on allergen-induced airway responses and airway inflammation. Am J Respir Crit Care Med (1999) 159:31–6. doi:10.1164/ajrccm.159.1.9804030
145. Vancheri C, Mastruzzo C, Sortino MA, Crimi N. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol (2004) 25:40–6. doi:10.1016/j.it.2003.11.001
146. Konya V, Üllen A, Kampitsch N, Theiler A, Philipose S, Parzmair GP, et al. Endothelial E-type prostanoid 4 receptors promote barrier function and inhibit neutrophil trafficking. J Allergy Clin Immunol (2013) 131:532–40.e1–2. doi:10.1016/j.jaci.2012.05.008
147. Teixeira MM, al-Rashed S, Rossi AG, Hellewell PG. Characterization of the prostanoid receptors mediating inhibition of PAF-induced aggregation of guinea-pig eosinophils. Br J Pharmacol (1997) 121:77–82. doi:10.1038/sj.bjp.0701107
148. Teixeira M, Rossi G, Hellewell PG. Adhesion mechanisms involved homotypic aggregation in C5a-induced eosinophil. J Leukoc Biol (1996) 59:389–96.
149. Konya V, Philipose S, Bálint Z, Olschewski A, Marsche G, Sturm EM, et al. Interaction of eosinophils with endothelial cells is modulated by prostaglandin EP4 receptors. Eur J Immunol (2011) 41:2379–89. doi:10.1002/eji.201141460
150. Sturm EM, Parzmair GP, Radnai B, Frei RB, Sturm GJ, Hammer A, et al. Phosphoinositide-dependent protein kinase 1 (PDK1) mediates potent inhibitory effects on eosinophils. Eur J Immunol (2015) 45:1548–59. doi:10.1002/eji.201445196
151. Gaspar Elsas MIC, Joseph D, Lintomen L, Maximiano ES, Bodstein M, Elsas PX, et al. Murine myeloid progenitor responses to GM-CSF and eosinophil precursor responses to IL-5 represent distinct targets for downmodulation by prostaglandin E2. Br J Pharmacol (2000) 130:1362–8. doi:10.1038/sj.bjp.0703403
152. Jones CP, Paula Neto HA, Assreuy J, Vargaftig BB, Gaspar Elsas MI, Elsas PX. Prostaglandin E2 and dexamethasone regulate eosinophil differentiation and survival through a nitric oxide- and CD95-dependent pathway. Nitric Oxide (2004) 11:184–93. doi:10.1016/j.niox.2004.08.001
153. Peacock CD, Misso NLA, Watkins DN, Thompson PJ. PGE2 and dibutyryl cyclic adenosine monophosphate prolong eosinophil survival in vitro. J Allergy Clin Immunol (1999) 104:153–62. doi:10.1016/S0091-6749(99)70127-2
154. Daffern PJ, Jagels MA, Saad JJ, Fischer W, Hugli TE. Upper airway epithelial cells support eosinophil survival in vitro through production of GM-CSF and prostaglandin E2: regulation by glucocorticoids and TNF-alpha. Allergy Asthma Proc (1999) 20:243–53. doi:10.2500/108854199778339008
155. Profita M, Sala A, Bonanno A, Riccobono L, Siena L, Melis MR, et al. Increased prostaglandin E2 concentrations and cyclooxygenase-2 expression in asthmatic subjects with sputum eosinophilia. J Allergy Clin Immunol (2003) 112:709–16. doi:10.1016/S0091-6749(03)01889-X
156. Sastre B, Fernández-Nieto M, Mollá R, López E, Lahoz C, Sastre J, et al. Increased prostaglandin E2 levels in the airway of patients with eosinophilic bronchitis. Allergy (2008) 63:58–66. doi:10.1111/j.1398-9995.2007.01515.x
157. Aggarwal S, Moodley YP, Thompson PJ, Misso NL. Prostaglandin E2 and cysteinyl leukotriene concentrations in sputum: association with asthma severity and eosinophilic inflammation. Clin Exp Allergy (2010) 40:85–93. doi:10.1111/j.1365-2222.2009.03386.x
158. Sturm EM, Schuligoi R, Konya V, Sturm GJ, Heinemann A. Inhibitory effect of prostaglandin I2 on bone marrow kinetics of eosinophils in the guinea pig. J Leukoc Biol (2011) 90:285–91. doi:10.1189/jlb.0211087
159. Idzko M, Hammad H, Van Nimwegen M, Kool M, Vos N, Hoogsteden HC, et al. Inhaled iloprost suppresses the cardinal features of asthma via inhibition of airway dendritic cell function. J Clin Invest (2007) 117:464–72. doi:10.1172/JCI28949
160. Woessner KM. Update on aspirin-exacerbated respiratory disease. Curr Allergy Asthma Rep (2017) 17:2. doi:10.1007/s11882-017-0673-6
161. Le Pham D, Lee J-H, Park H-S. Aspirin-exacerbated respiratory disease. Curr Opin Pulm Med (2017) 23:89–96. doi:10.1097/MCP.0000000000000328
162. Steinke JW, Payne SC, Borish L. Eosinophils and mast cells in aspirin-exacerbated respiratory disease. Immunol Allergy Clin North Am (2016) 36:719–34. doi:10.1016/j.iac.2016.06.008
163. Feng X, Ramsden MK, Negri J, Baker MG, Payne SC, Borish L, et al. Eosinophil production of prostaglandin D2 in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol (2016) 138:1089–97.e3. doi:10.1016/j.jaci.2016.04.042
164. Buchheit KM, Cahill KN, Katz HR, Murphy KC, Feng C, Lee-Sarwar K, et al. Thymic stromal lymphopoietin controls prostaglandin D2 generation in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol (2016) 137:1566–76.e5. doi:10.1016/j.jaci.2015.10.020
165. Cahill KN, Bensko JC, Boyce JA, Laidlaw TM. Prostaglandin D2: a dominant mediator of aspirin-exacerbated respiratory disease. J Allergy Clin Immunol (2015) 135:245–52. doi:10.1016/j.jaci.2014.07.031
166. Isogai S, Hayashi M, Yamamoto N, Morishita M, Minezawa T, Okamura T, et al. Upregulation of CD11b on eosinophils in aspirin induced asthma. Allergol Int (2013) 62:367–73. doi:10.2332/allergolint.12-OA-0499
167. Steinke JW, Negri J, Liu L, Payne SC, Borish L. Aspirin activation of eosinophils and mast cells: implications in the pathogenesis of aspirin-exacerbated respiratory disease. J Immunol (2014) 193:41–7. doi:10.4049/jimmunol.1301753
168. Luschnig P, Frei R, Lang-Loidolt D, Rozsasi A, Tomazic PV, Lippe IT, et al. Altered inhibitory function of the E-type prostanoid receptor 4 in eosinophils and monocytes from aspirin-intolerant patients. Pharmacology (2014) 94:280–6. doi:10.1159/000369827
169. Jinnai N, Sakagami T, Sekigawa T, Kakihara M, Nakajima T, Yoshida K, et al. Polymorphisms in the prostaglandin E2 receptor subtype 2 gene confer susceptibility to aspirin-intolerant asthma: a candidate gene approach. Hum Mol Genet (2004) 13:3203–17. doi:10.1093/hmg/ddh332
170. Palikhe NS, Sin HJ, Kim SH, Sin HJ, Hwang EK, Ye YM, et al. Genetic variability of prostaglandin E2 receptor subtype EP4 gene in aspirin-intolerant chronic urticaria. J Hum Genet (2012) 57:494–9. doi:10.1038/jhg.2012.55
171. Palikhe NS, Kim S-H, Cho B-Y, Ye Y-M, Choi G-S, Park H-S. Genetic variability in CRTH2 polymorphism increases eotaxin-2 levels in patients with aspirin exacerbated respiratory disease. Allergy (2010) 65:338–46. doi:10.1111/j.1398-9995.2009.02158.x
172. Kohyama K, Hashimoto M, Abe S, Kodaira K, Yukawa T, Hozawa S, et al. Thromboxane A2 receptor +795T>C and chemoattractant receptor-homologous molecule expressed on Th2 cells -466T>C gene polymorphisms in patients with aspirin-exacerbated respiratory disease. Mol Med Rep (2012) 5:477–82. doi:10.3892/mmr.2011.680
173. Cornejo-García JA, Perkins JR, Jurado-Escobar R, García-Martín E, Agúndez JA, Viguera E, et al. Pharmacogenomics of prostaglandin and leukotriene receptors. Front Pharmacol (2016) 7:316. doi:10.3389/fphar.2016.00316
174. Davis BP, Rothenberg ME. Eosinophils and cancer. Cancer Immunol Res (2014) 2:1–8. doi:10.1158/2326-6066.CIR-13-0196
175. Davoine F, Sim A, Tang C, Fisher S, Ethier C, Puttagunta L, et al. Eosinophils in human oral squamous carcinoma; role of prostaglandin D2. J Inflamm (2013) 10:1. doi:10.1186/1476-9255-10-4
176. Murata T, Aritake K, Matsumoto S, Kamauchi S, Nakagawa T, Hori M, et al. Prostagladin D2 is a mast cell-derived antiangiogenic factor in lung carcinoma. Proc Natl Acad Sci U S A (2011) 108:19802–7. doi:10.1073/pnas.1110011108
177. Hogan SP, Waddell A, Fulkerson PC. Eosinophils in infection and intestinal immunity. Curr Opin Gastroenterol (2013) 29:7–14. doi:10.1097/MOG.0b013e32835ab29a
178. Wedemeyer J, Vosskuhl K. Role of gastrointestinal eosinophils in inflammatory bowel disease and intestinal tumours. Best Pract Res Clin Gastroenterol (2008) 22:537–49. doi:10.1016/j.bpg.2007.12.001
179. Radnai B, Sturm EM, Stančić A, Jandl K, Labocha S, Ferreirós N, et al. Eosinophils contribute to intestinal inflammation via chemoattractant receptor-homologous molecule expressed on Th2 cells, CRTH2, in experimental Crohn’s disease. J Crohns Colitis (2016) 10:1087–95. doi:10.1093/ecco-jcc/jjw061
180. Le Loupp A-G, Bach-Ngohou K, Bourreille A, Boudin H, Rolli-Derkinderen M, Denis MG, et al. Activation of the prostaglandin D2 metabolic pathway in Crohn’s disease: involvement of the enteric nervous system. BMC Gastroenterol (2015) 15:112. doi:10.1186/s12876-015-0338-7
181. Raab Y, Sundberg C, Hällgren R, Knutson L, Gerdin B. Mucosal synthesis and release of prostaglandin E2 from activated eosinophils and macrophages in ulcerative colitis. Am J Gastroenterol (1995) 90:614–20.
182. Kabashima K, Saji T, Murata T, Nagamachi M, Matsuoka T, Segi E, et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest (2002) 109:883–93. doi:10.1172/JCI0214459
183. Nitta M, Hirata I, Toshina K, Murano M, Maemura K, Hamamoto N, et al. Expression of the EP4 prostaglandin E2 receptor subtype with rat dextran sodium sulphate colitis: colitis suppression by a selective agonist, ONO-AE1-329. Scand J Immunol (2002) 56:66–75. doi:10.1046/j.1365-3083.2002.01096.x
184. Jiang G-L, Nieves A, Im WB, Old DW, Dinh DT, Wheeler L. The prevention of colitis by E prostanoid receptor 4 agonist through enhancement of epithelium survival and regeneration. J Pharmacol Exp Ther (2006) 320:22–8. doi:10.1124/jpet.106.111146
185. Konya V, Marsche G, Schuligoi R, Heinemann A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol Ther (2013) 138:485–502. doi:10.1016/j.pharmthera.2013.03.006
186. Okano M, Fujiwara T, Yamamoto M, Sugata Y, Matsumoto R, Fukushima K, et al. Role of prostaglandin D2 and E2 terminal synthases in chronic rhinosinusitis. Clin Exp Allergy (2006) 36:1028–38. doi:10.1111/j.1365-2222.2006.02528.x
Keywords: allergy, inflammation, respiratory and gastrointestinal tract, bone marrow, chemotaxis, endothelium
Citation: Peinhaupt M, Sturm EM and Heinemann A (2017) Prostaglandins and Their Receptors in Eosinophil Function and As Therapeutic Targets. Front. Med. 4:104. doi: 10.3389/fmed.2017.00104
Received: 25 April 2017; Accepted: 27 June 2017;
Published: 19 July 2017
Edited by:
Mats W. Johansson, University of Wisconsin-Madison, United StatesReviewed by:
Pedro Xavier-Elsas, Federal University of Rio de Janeiro, BrazilLena Uller, Lund University, Sweden
Copyright: © 2017 Peinhaupt, Sturm and Heinemann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Akos Heinemann, YWtvcy5oZWluZW1hbm5AbWVkdW5pZ3Jhei5hdA==