 Céline Louis1
Céline Louis1 Sandrine Vijgen2Kaveh Samii1Yves Chalandon1Louis Terriou3
Sandrine Vijgen2Kaveh Samii1Yves Chalandon1Louis Terriou3 David Launay3
David Launay3 David C. Fajgenbaum4
David C. Fajgenbaum4 Jörg D. Seebach5
Jörg D. Seebach5 Yannick D. Muller5*
Yannick D. Muller5*
- 1Division of Hematology, Geneva University Hospital, University of Geneva, Geneva, Switzerland
- 2Department of Pathology, Geneva University Hospital, University of Geneva, Geneva, Switzerland
- 3Department of Internal Medicine and Clinical Immunology CHU, University of Lille, U995, Lille Inflammation Research International Center, INSERM, Centre national de référence maladies systémiques et auto-immunes rares (sclérodermie systémique), Lille, France
- 4Division of Translational Medicine and Human Genetics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
- 5Division of Clinical Immunology and Allergy, Geneva University Hospital, University of Geneva, Geneva, Switzerland
Background: TAFRO syndrome has been reported in Japan among human herpesvirus 8 (HHV-8)-negative/idiopathic multicentric Castleman’s disease (iMCD) patients. To date, the majority of iMCD patients with TAFRO syndrome originate from Japan.
Case presentation: Herein, we report a 67-year-old HIV/HHV-8-negative Caucasian iMCD patient diagnosed with TAFRO. He presented with marked systemic inflammation, bicytopenia, terminal renal insufficiency, diffuse lymphadenopathies, and anasarca. Lymph node and bone marrow biopsies revealed atrophic germinal centers variably hyalinized and megakaryocytic hyperplasia with mild myelofibrosis. Several other biopsies performed in kidneys, liver, gastrointestinal tract, prostate, and lungs revealed unspecific chronic inflammation. The patient had a complete response to corticosteroids, tocilizumab, and rituximab. He relapsed twice following discontinuation of rituximab. When reviewing the literature, we found seven other Caucasian cases with TAFRO syndrome. There were no significant differences with those described by the Japanese cohort except for the higher frequency of kidney failure and auto-antibodies in Western patients.
Conclusion: This case illustrates that patients with TAFRO syndrome can develop non-specific inflammation in several tissue sites. Furthermore, this case and our review of the literature demonstrate that TAFRO syndrome can affect Caucasian and Japanese patients highlighting the importance of evaluating for this syndrome independently of ethnic background.
Background
Multicentric Castleman’s disease (MCD) is diagnosed clinicopathologically (1). Human herpesvirus 8 (HHV-8), a gamma herpesvirus first identified in Kaposi’s sarcoma, is the etiological cause of MCD in individuals who are HIV-positive or immunocompromised for another reason (2). In HHV-8-associated MCD, HHV-8 infects lymphocytes, macrophages, and endothelial and epithelial cells, lytically replicates in immunocompromised individuals, and signals for production of a viral homolog of human IL-6, which induces a cytokine storm and atypical lymphoproliferation (3). HHV-8-negative MCD patients in whom the etiology is not known are referred to as idiopathic multicentric Castleman’s disease (iMCD). A large study of Japanese MCD patients found that HHV-8-associated MCD occurs at a lower frequency than in European cohorts (4). None of the 79 Japanese HIV-negative MCD cases were HHV-8-positive whereas to 7/17 cases in a French cohort (2) and 6/14 cases in an Italian cohort (5) were HHV-8-positive (4). At the time, these controversial data were postulated to be due to the raciogeographical difference and the low prevalence of HHV-8 seropositivity in healthy Japanese individuals, although this hypothesis has never been confirmed/disproved (4).
In 2008, Kojima et al. proposed the first clinical sub-classification of HHV-8-negative/iMCD including idiopathic plasmacytic lymphadenopathy (IPL)-type and non-IPL type. IPL type exhibits marked hyperimmunoglobulinemia, severe inflammation, and thrombocytosis with follicular hyperplasia and interfollicular sheets of mature plasmatic cells. Non-IPL type exhibits anasarca, inflammation, and thrombocytopenia with atrophic lymphoid follicules and a hyaline vascular (HV)/mixed pattern of the germinal center (GC) (6). The clinicopathological description of non-IPL-iMCD corresponds very closely to the recently identified TAFRO syndrome or Castleman–Kojima disease described first by Takai et al. (7, 8). Since then, several cases have been published, and formal diagnostic criteria were established in 2015 based on 28 patients all originating from Japan (9). Major required criteria for TAFRO syndrome include (I) anasarca, (II) thrombocytopenia (<100 G/l), and (III) systemic inflammation. Two of the following four minor criteria are also required: (I) Castleman’s disease-like features on lymph node biopsy, (II) reticulin myelofibrosis and/or hyperplasia of megakaryocytes in the bone marrow, (III) mild organomegaly of the lymphoid organs and the liver, and (IV) progressive renal insufficiency. Malignancies (including POEMS), auto-immune disorders (including IgG4-related disease), infectious diseases, and auto-immune thrombocytopenia should be excluded before the diagnosis of TAFRO is made (9). Currently, it remains controversial as to whether TAFRO syndrome is a distinct entity from iMCD, a clinical subtype of iMCD, or a syndrome with multiple overlapping diseases (10).
We report the complex management of a Caucasian iMCD patient with TAFRO syndrome and identified seven other cases of Caucasian patients in the literature. The clinicopathological findings of this series of eight cases was systematically analyzed and compared with those described in Japan.
Case Presentation
Clinical Presentation
Herein, we report on a 67-year-old Caucasian originating from Portugal admitted to the hospital for fever of unknown origin and asthenia. His medical history was relevant for hypertension, insulin-dependent type two diabetes, and vitiligo and was on aspirin cardio, lisinopril, torasemid, omeprazole, and insulin therapy. On clinical examination, we noticed anasarca with pleural effusion, ascites, and edema of the lower limbs. Laboratory tests revealed microcytic anemia (hemoglobin 100 g/l, MCV 75 fl) with thrombocytopenia (73 G/l), marked elevation of C-reactive protein (204 mg/l), renal insufficiency (creatinine 463 µmol/l), and cholestasis (alkaline phosphatase 300 U/l, gamma-GT 56 U/l), with normal transaminases. Urinalysis showed severe proteinuria (5.29 g/l) and glomerular microhematuria. Immunological evaluation revealed normal IgG, IgM, IgA, and complement (C3, C4) levels, anti-nuclear antibodies (1/640 speckled), anti-SSA, anti-actine, and anti-parietal cell antibodies were positive. The infectious work-up was negative (HIV, hepatitis B/C, CMV, TB spot, and blood culture) except for EBV that was slightly positive by PCR (3,560 copies/ml). A thoraco-abdominal CT scan detected multiple mediastinal, axillary and retroperitoneal adenopathies, pleural and pericardial effusions, hepatosplenomegaly, and ascites. There was no sign of peripheral hemolysis (absence of schsitocytes). Finally, a cytokine profile showed elevation of IL-6, VEGF, soluble IL-2 receptor, and TNF-α, whereas IL-8 was normal.
Diagnostic Course and Biopsies
The etiology remained unclear and the patient underwent several supplementary investigations. An axillary lymph node was surgically removed and showed atreic secondary lymphoid follicles with hyalinized/vascular GCs. Interfollicular areas showed a marked vascular proliferation and contained a small number of CD138-positive plasma cells (Figures 1A–D). Immunohistochemistry for HHV-8 with latency-associated nuclear antigen-1 was negative. The histopathological findings were consistent with the newly defined diagnostic criteria for iMCD (11). The blood smear and the flow cytometry in the blood were normal without evidence for lymphoproliferative disease. A bone marrow trephine biopsy revealed megakaryocytic hyperplasia and mild reticulin myelofibrosis without abnormal lymphocytic infiltration (Figures 1E,F).
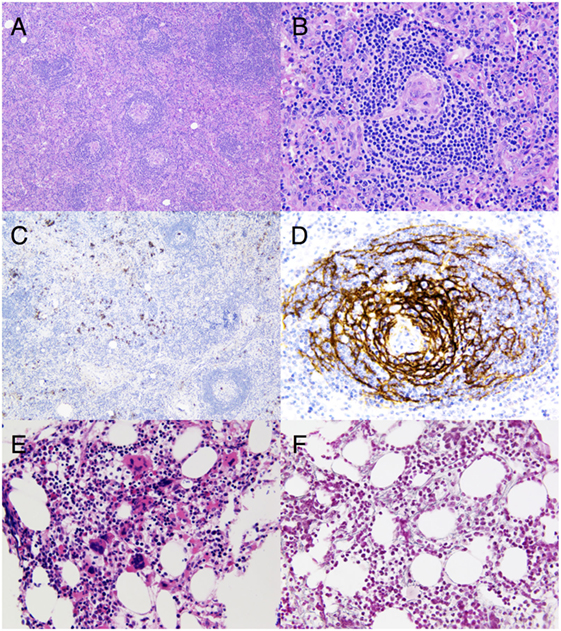
Figure 1. (A) Hematoxylin and eosin stain of a surgically removed axillary lymph node consistent with idiopathic multicentric Castleman’s disease (iMCD). Low magnification highlighting a prominent paracortex encircling atrophic secondary follicles (original magnification, ×10). (B) Hematoxylin and eosin stain focusing on an atrophic germinal center which is partially lymphocyte depleted and which is penetrated by a prominent, hyalinized blood vessel lined by plump endothelial cells with enlarged nuclei, giving the appearance of a lollipop. Note the marked vascular proliferation in the interfollicular areas (original magnification, ×40). (C) CD138 staining of sparsely scattered plasma cells in the interfollicular areas, less abundant compared to what is observed in the plasmacytic histopathological variant of iMCD (original magnification, ×10). (D) CD21 stain displaying a prominent network of follicular dendritic cells (FDCs) in an abnormal follicle with a concentric arrangement of the small lymphocytes of the mantle zone along with FDC nuclei (original magnification, ×40). (E) Hematoxylin and eosin stain of bone marrow showing megakaryocytic hyperplasia with a mixture of small hypolobated megakaryocytes and others having a multi-separated nucleus (original magnification, ×40). (F) Reticulin stain on the bone marrow trephine biopsy highlighting mild myelofibrosis (original magnification, ×40).
Renal biopsy showed (1) thickening of the peripheral basement membrane and the intern lamina rara, (2) mesangial proliferation without glomerular deposition of immunoglobulin, and (3) a band-like pattern of interstitial fibrosis associated with lymphohistiocytic inflammatory cell infiltrates. Malignancies were also actively searched for. Colon biopsies showed a collagenous colitis rich in eosinophils (Figure 2A). Gastric biopsies revealed chronic inflammation and glandular atrophia with intestinal metaplasia of the fundus suggestive of an atrophic gastritis (Figure 2B). Prostatic biopsies showed mild chronic inflammation without neoplasia (Figure 2C). Liver biopsy showed mild sinusoidal distension, unspecific portal and lobular inflammation, and rare epithelioid granulomas without necrosis (Figures 2D,E). Finally, the patient underwent a bronchial biopsy showing unspecific chronic inflammation without granuloma, which was not suspicious for sarcoidosis (Figure 2F).
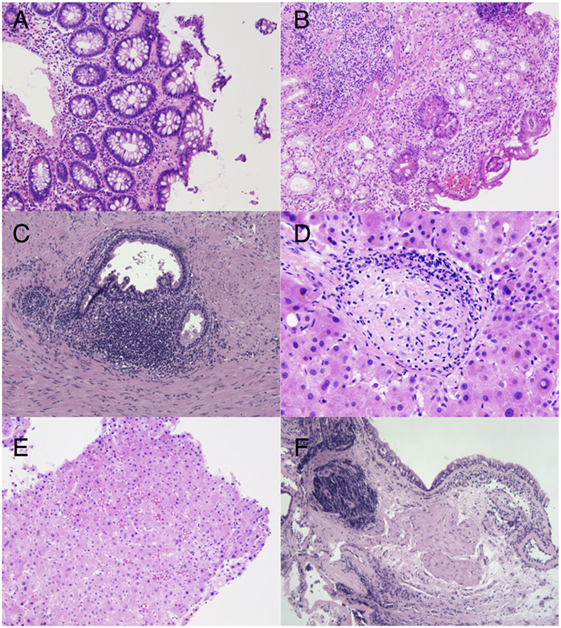
Figure 2. (A) Hematoxylin and eosin stain. Colon biopsy showing irregular thickening of the subepithelial basement membrane, encircling some capillaries, indicating a collagenous colitis (original magnification, ×20). (B) Hematoxylin and eosin stain. Glandular atrophy in the gastric body with foci of intestinal metaplasia and deep lymphoplasmacytic infiltrates, suggestive of an autoimune gastritis (original magnification, ×20). (C) Hematoxylin and eosin stain. Reactive periglandular chronic inflammation in the prostate (original magnification, ×10). (D) Hematoxylin and eosin stain. Rare lobular non-necrotizing epithelioid granulomas with peripheral lymphoid ring are found in addition to unspecific portal inflammation (original magnification, ×40). (E) Hematoxylin and eosin staining of a liver biopsy showing mild sinusoidal distension in the liver biopsy (original magnification, ×20). (F) Hematoxylin and eosin stain. Bronchial wall containing lymphoplasmocytic aggregates of variable densities with crush artifacts (original magnification, ×10).
Together, these clinicopathological findings are compatible with a diagnosis of iMCD (11) and TAFRO syndrome (9).
Treatment Course and Outcome
The best management of iMCD disease remains poorly defined with only one randomized clinical trial published in 2014 on siltuximab compared to best supportive care (12). Thus, this case required multidisciplinary discussion and consensus in 2011 when the diagnosed was made and judgment was based on available data with tocilizumab (13) and rituximab for the treatment of HHV-8-associated MCD (14). Thus, treatment was initiated with high dose methylprednisolone (1 g/day for 3 days) transitioned to prednisone 1 mg/kg for several weeks, one single infusion of tocilizumab (8 mg/kg), and 1 week later rituximab 375 mg/m2 (total 4 weekly doses) (Figure 3A). The rational to use methylprednisolone and tocilizumab was to rapidly stop IL-6-mediated inflammation as induction regimen, whereas rituximab was used as maintenance treatment. We observed resolution of the inflammatory syndrome and slow clinical improvement over the course of 3 weeks, thrombocyte values normalized (>100 G/l) after 5 weeks and cholestasis parameters several months later. Corticosteroids were rapidly weaned off because of poor control of the diabetes, and the patient was hemodialyzed for end-stage renal disease. In the following 5 years, the patient relapsed twice, each time about 1 year after rituximab was discontinued (Figure 3A). The first relapse was successfully treated with rituximab alone and the second with methylprednisolone/tocilizumab (2 doses)/rituximab (4 weekly doses 375 mg/m2). Five years after the diagnosis was made (before the second relapse), the PET-CT showed reduction of the lymphadenopathies (Figures 3B,C). Auto-antibodies disappeared over the years and hypogammaglobulinemia developed, probably due to repeated rituximab treatments. Follow-up bone marrow biopsies were not performed in this case.
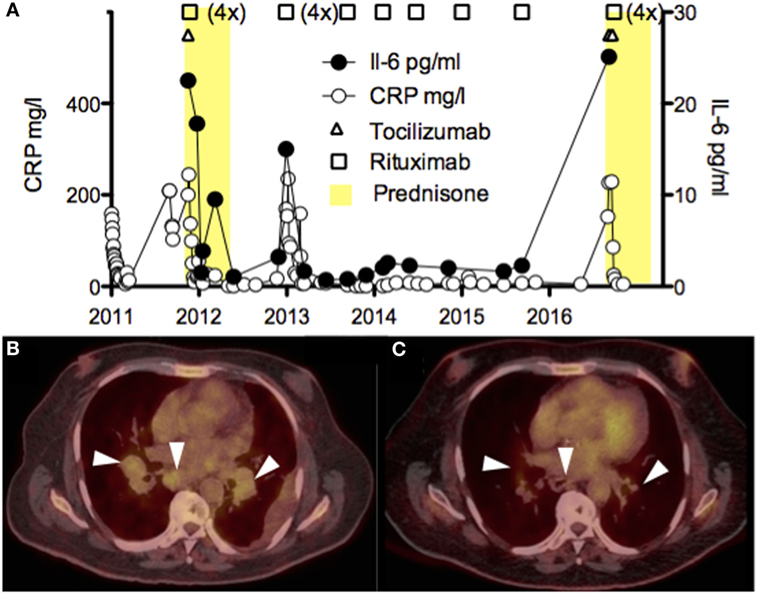
Figure 3. (A) Time course of the serum CRP (mg/l) and IL-6 (pg/ml) levels, and the different immunosuppressive drugs (corticosteroids, tocilizumab, and rituximab) administered to the patient. High values of IL-6 early after tocilizumab were excluded. (B,C) PET-CT scans showing pulmonary and mediastinal lymphadenopathies before (B) and after treatment with rituximab (C).
Discussion
TAFRO in Western Countries
TAFRO syndrome is a rare clinical syndrome of unknown origin that has been described in patients with iMCD. Most of the reported cases were Japanese patients. To analyze the role of ethnicity, we have reviewed the literature for all known Caucasian cases and have identified eight including the present one. When analyzing major and minor criteria recently established by Masaki et al., we confirmed that Caucasian cases were HIV/HHV-8-negative and meet both major and minor criteria (9, 15–20). In comparison with the Japanese cohort, of 18 patients we find Caucasian TAFRO cases presented more frequently with kidney failure and auto-immune antibodies, although these results need to be interpreted with caution due to the low number of patients (Table 1). Recently, a patient with TAFRO was also reported in Latin America (21) and the members of the Castleman Disease Collaborative Network report treating iMCD patients with TAFRO from diverse ethnic backgrounds throughout the world for decades (11). As to IgG4-related disease primary reported in Japan (22), TAFRO syndrome is a rare entity affecting patients with different ethnic background without major differences in the clinicopathological presentation.
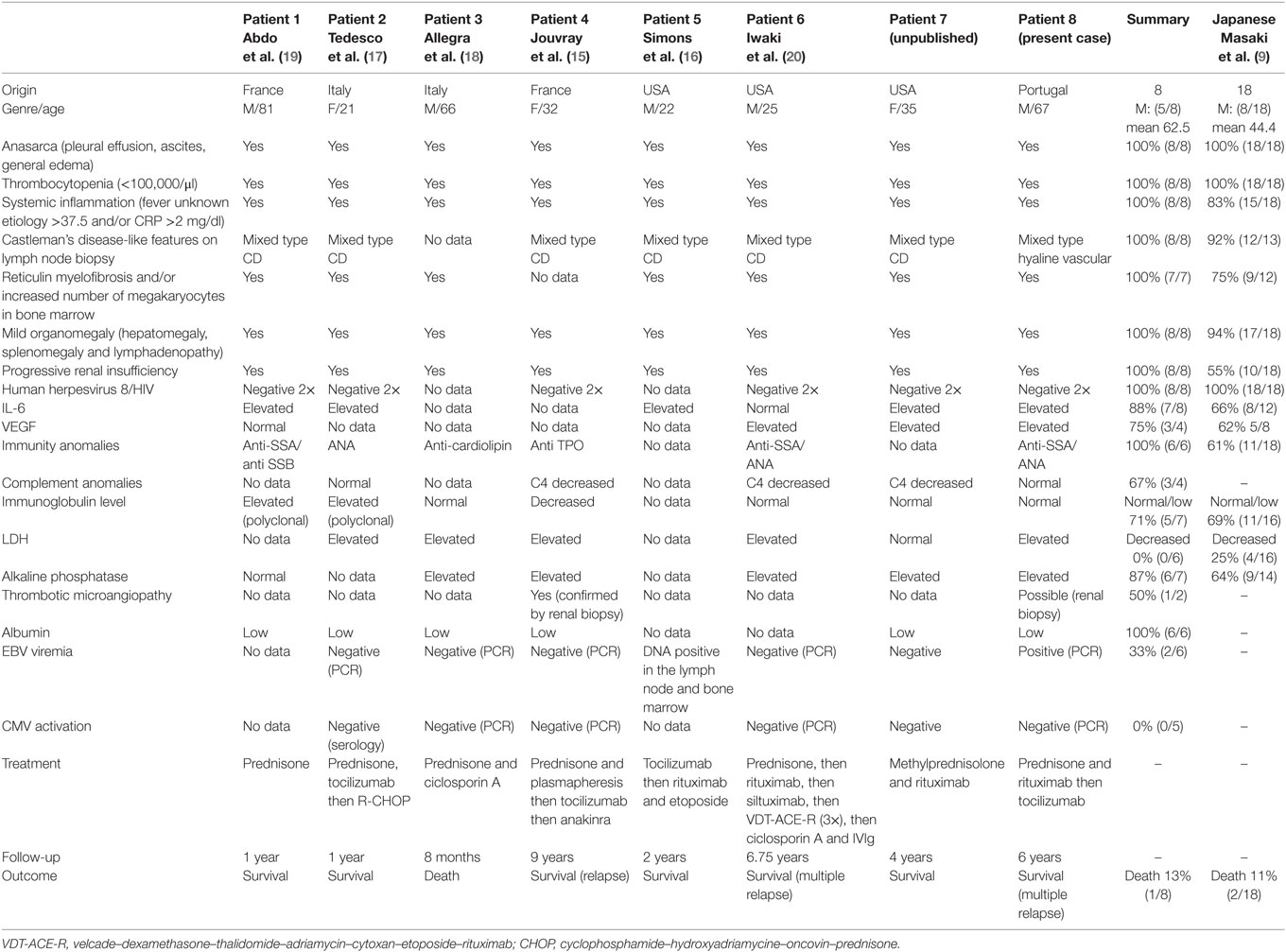
Table 1. Clinical characteristics and laboratory data of eight Caucasian patients with TAFRO compared to the Japanese cohort.
TAFRO: A Spectrum of Castleman’s Disease?
Recently, the international working group for the Castleman Disease Collaborative Network established the first-ever diagnostic criteria for HHV-8-negative-iMCD analyzing 244 patients with iMCD (128 patients from literature, 37 cases submitted by the working group members, and 79 from randomized control study) and more specifically the histopathological features of 85 lymph nodes with presumptive diagnosis of MCD. Importantly, this cohort included TAFRO and non-TAFRO-iMCD cases (11, 23). The authors argued that while there are clinical differences between iMCD patients with TAFRO syndrome and those who do not have TAFRO syndrome, there are a significant number of overlapping histopathological and clinical features. Furthermore, some patients with TAFRO clinical syndrome did not present with the more classical HV histopathology, and some patients with the more classical histopathology for TAFRO syndrome did not have TAFRO clinical features. There was consensus among the international working group that TAFRO syndrome is a clinical subtype of iMCD to be included in the diagnostic criteria, not a separate entity. Nevertheless, a consensus between both the international working group on iMCD and the Japanese working group on TAFRO is warranted to agree on criteria, prevalence, and incidence of TAFRO. We proposed a classification algorithm for MCD (Figure 4). An exhaustive list of major, minor, and exclusion criteria can been found elsewhere (11). More importantly, genomic, transcriptomic, and proteomic studies are needed to understand molecular differences that may exist. The clinical utility of subtyping the lymph node histological features into HV, plasmacytic, or mixed type also warrants further investigation.
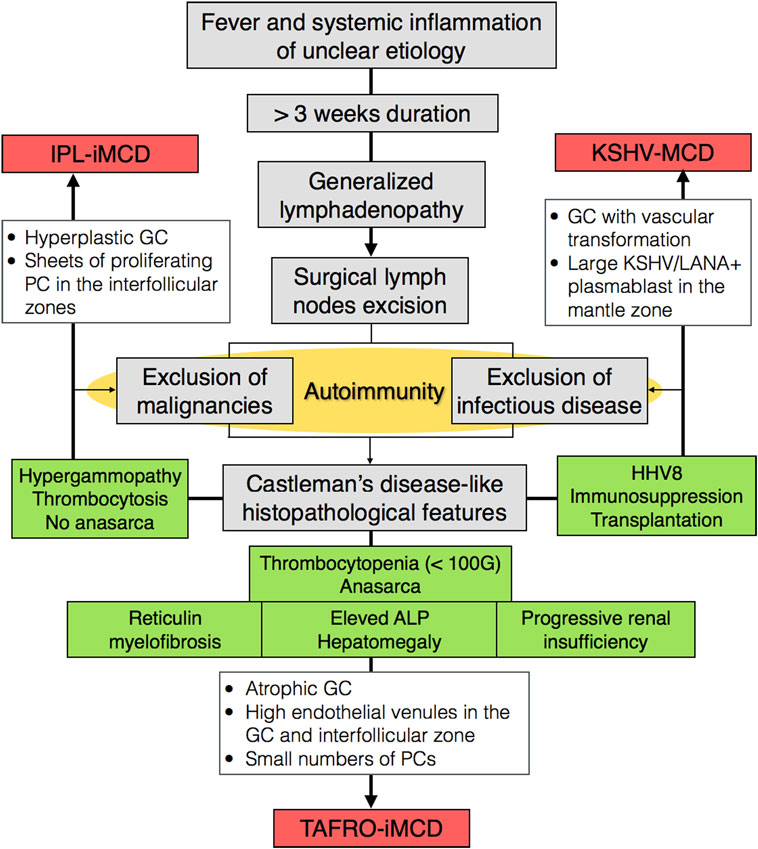
Figure 4. Classification algorithm for TAFRO-iMCD, IPL-iMCD, and KSVH-MCD. Abbreviations: iMCD, idiopathic multicentric Castleman disease; MCD, multicentric Castleman disease; HV, hyaline vascular; IPL, idiopathic plasmacytic lymphoadenpathy; KSHV, Kaposi sarcoma-associated herpesvirus; GC, germinal center; ALP, alkaline phosphatase; HHV-8, human herpesvirus 8; PC, plasma cell; LANA, latency-associated nuclear antigen.
TAFRO and Auto-Immunity
The relationship between TAFRO/iMCD and auto-immunity is poorly understood. In our study, auto-antibodies such as anti-nuclear or anti-SS-A (SS-A) were found in all six patients with available data (Table 1). Albeit auto-antibodies alone are insufficient to make a definite diagnosis of connective tissue disease such as systemic lupus erythematosus (SLE), Sjögren syndrome, systemic sclerosis, or dermatomyositis/polymyositis, the classification criteria to diagnose undifferentiated connective tissue disease can be met (24). Furthermore, generalized lymphadenopathies are a common finding in about a quarter of patients with SLE (25, 26). When analyzing the histopathological findings in lymph node biopsies from 19 patients with active SLE, iMCD-compatible lesions were present in 5; 3 of mixed type and 2 of HV type (27). As to anti-SS-A52 auto-antibodies, they seem to be more prevalent in patients with TAFRO as illustrated by this study and reported by others (19, 20, 28). Interestingly, undiagnosed anti-SS-A 52 positive patients do not frequently develop definitive connective tissue disease (29). Finally, the chemokine CXCL10 (IP-10) involved in the pathogenesis of many auto-immune disease including Sjögren syndrome (30) seems to be increased during the flares-up in TAFRO (31). In conclusion, these data suggest that auto-immunity is an important aspect of TAFRO.
TAFRO and Kidney Disease
Kidney involvement is frequently observed in patients with TAFRO syndrome, the underlying mechanisms, however, are poorly understood. Depending on the authors, the R in TAFRO may refer to renal dysfunction or reticulin myelofibrosis (9, 20). Iwaki et al.’s recently proposed diagnostic criteria for TAFRO that do not include renal failure as major or minor criteria (20). Importantly, all Caucasian TAFRO patients who we have found had renal failure (Table 1). The clinical presentation of iMCD patients with renal failure is frequently associated with arterial hypertension, glomerular hematuria, and proteinuria. The most frequent lesions were endotheliosis of the small-vessel and glomerular double contours with glomerular/arteriolar thrombi, a pattern similar to what can be found in thrombotic microangioapthy (TMA) (32, 33). Of interest, 2/7 patients with TAFRO had renal insufficiency and lesions compatible with MAT. Furthermore, complement, an important trigger for MAT (34), can also be frequently decreased in TAFRO (Table 1). Thus, these results stress the importance of regularly monitoring kidney function in patients with TAFRO syndrome particularly in the presence of other risk factors such as diabetes as was the case for our patient. Further studies into the link between TAFRO, complement dysregulation, and MAT are needed.
Conclusion
TAFRO syndrome is a rare subtype of iMCD resulting in generalized organ inflammation of unknown origin. Diagnosis remains very challenging and the exclusion of infectious, auto-immune, and neoplastic disorders is necessary. Patients undergo a series of clinical investigations and biopsies, which are often non-contributive. In this article, we demonstrate a Caucasian case of TAFRO syndrome and lend further support to the notion that TAFRO syndrome can be seen in iMCD patients around the world. Genomic, transcriptomic, and proteomic investigations into the etiology, pathogenesis, relationship between auto-immunity, complement dysregulation, and kidney failure, and molecular differences between TAFRO-iMCD and non-TAFRO-iMCD are warranted.
Consent
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Ethics Statement
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images as per our standard institutional rules. A copy of the written consent is available for review by the Editor of this journal.
Author Contributions
Conceived and designed the experiments: YM, CL, and SV. Analyzed the data: YM, CL, SV, DF, KS, JS, and YC. Contributed reagents/materials/analysis tools KS, YC, LT, DL, DF, JS, and SV. Critical discussion and reading: YM, JS, DF, KS, YC, LT, and DL. Wrote the paper: YM, CL, and DF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
iMCD, idiopathic multicentric Castleman’s disease; SLE, systemic lupus erythematosus; IPL, idiopathic plasmacytic lymphadenopathy; HHV-8, human herpesvirus 8.
References
1. Frizzera G, Peterson BA, Bayrd ED, Goldman A. A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease: clinical findings and clinicopathologic correlations in 15 patients. J Clin Oncol (1985) 3:1202–16. doi:10.1200/JCO.1985.3.9.1202
2. Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood (1995) 86:1276–80.
3. Neipel F, Albrecht JC, Ensser A, Huang YQ, Li JJ, Friedman-Kien AE, et al. Human herpesvirus 8 encodes a homolog of interleukin-6. J Virol (1997) 71:839–42.
4. Suda T, Katano H, Delsol G, Kakiuchi C, Nakamura T, Shiota M, et al. HHV-8 infection status of AIDS-unrelated and AIDS-associated multicentric Castleman’s disease. Pathol Int (2001) 51:671–9. doi:10.1046/j.1440-1827.2001.01266.x
5. Parravicini C, Corbellino M, Paulli M, Magrini U, Lazzarino M, Moore PS, et al. Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman’s disease. Am J Pathol (1997) 151:1517–22.
6. Kojima M, Nakamura N, Tsukamoto N, Otuski Y, Shimizu K, Itoh H, et al. Clinical implications of idiopathic multicentric Castleman disease among Japanese: a report of 28 cases. Int J Surg Pathol (2008) 16:391–8. doi:10.1177/1066896908315812
7. Takai K, Nikkuni K, Shibuya H, Hashidate H. [Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly]. Rinsho Ketsueki (2010) 51:320–5.
8. Kawabata H, Takai K, Kojima M, Nakamura N, Aoki S, Nakamura S, et al. Castleman-Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly: a status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J Clin Exp Hematop (2013) 53:57–61.
9. Masaki Y, Kawabata H, Takai K, Kojima M, Tsukamoto N, Ishigaki Y, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol (2016) 103:686–92. doi:10.1007/s12185-016-1979-1
10. Carbone A, Pantanowitz L. TAFRO syndrome: an atypical variant of KSHV-negative multicentric Castleman disease. Am J Hematol (2016) 91:171–2. doi:10.1002/ajh.24274
11. Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood (2017) 129:1646–57. doi:10.1182/blood-2016-10-746933
12. van Rhee F, Wong RS, Munshi N, Rossi JF, Ke XY, Fosså A, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol (2014) 15:966–74. doi:10.1016/S1470-2045(14)70319-5
13. Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood (2005) 106:2627–32. doi:10.1182/blood-2004-12-4602
14. Bower M. How I treat HIV-associated multicentric Castleman disease. Blood (2010) 116:4415–21. doi:10.1182/blood-2010-07-290213
15. Jouvray M, Terriou L, Meignin V, Bouchindhomme B, Jourdain M, Lambert M, et al. [Pseudo-adult Still’s disease, anasarca, thrombotic thrombocytopenic purpura and dysautonomia: an atypical presentation of multicentric Castleman’s disease. Discussion of TAFRO syndrome]. Rev Med Interne (2016) 37:53–7. doi:10.1016/j.revmed.2015.04.006
16. Simons M, Apor E, Butera JN, Treaba DO. TAFRO syndrome associated with EBV and successful triple therapy treatment: case report and review of the literature. Case Rep Hematol (2016) 2016:4703608. doi:10.1155/2016/4703608
17. Tedesco S, Postacchini L, Manfredi L, Goteri G, Luchetti MM, Festa A, et al. Successful treatment of a Caucasian case of multifocal Castleman’s disease with TAFRO syndrome with a pathophysiology targeted therapy – a case report. Exp Hematol Oncol (2015) 4:3. doi:10.1186/2162-3619-4-3
18. Allegra A, Rotondo F, Russo S, Calabrò L, Maisano V, Bacci F, et al. Castleman-Kojima disease (TAFRO syndrome) in a Caucasian patient: a rare case report and review of the literature. Blood Cells Mol Dis (2015) 55:206–7. doi:10.1016/j.bcmd.2015.06.013
19. Abdo LA, Morin CP, Collarino RP, Cabane JP, Gatfosse MA. First European case of TAFRO syndrome associated with Sjogren disease. Am J Intern Med (2014) 2:102–5. doi:10.11648/j.ajim.20140206.12
20. Iwaki N, Fajgenbaum DC, Nabel CS, Gion Y, Kondo E, Kawano M, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol (2016) 91:220–6. doi:10.1002/ajh.24242
21. José FF, Kerbauy LN, Perini GF, Blumenschein DI, Pasqualin DD, Malheiros DM, et al. A life-threatening case of TAFRO syndrome with dramatic response to tocilizumab, rituximab, and pulse steroids: the first case report in Latin America. Medicine (Baltimore) (2017) 96:e6271. doi:10.1097/MD.0000000000006271
22. Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med (2001) 344:732–8. doi:10.1056/NEJM200103083441005
23. Yu L, Tu M, Cortes J, Xu-Monette ZY, Miranda RN, Zhang J, et al. Clinical and pathological characteristics of HIV- and HHV-8-negative Castleman disease. Blood (2017) 129:1658–68. doi:10.1182/blood-2016-11-748855
24. Mosca M, Tani C, Carli L, Bombardieri S. Undifferentiated CTD: a wide spectrum of autoimmune diseases. Best Pract Res Clin Rheumatol (2012) 26:73–7. doi:10.1016/j.berh.2012.01.005
25. Shapira Y, Weinberger A, Wysenbeek AJ. Lymphadenopathy in systemic lupus erythematosus. Prevalence and relation to disease manifestations. Clin Rheumatol (1996) 15:335–8. doi:10.1007/BF02230354
26. Afzal W, Arab T, Ullah T, Teller K, Doshi KJ. Generalized lymphadenopathy as presenting feature of systemic lupus erythematosus: case report and review of the literature. J Clin Med Res (2016) 8:819–23. doi:10.14740/jocmr2717w
27. Kojima M, Nakamura S, Itoh H, Yoshida K, Asano S, Yamane N, et al. Systemic lupus erythematosus (SLE) lymphadenopathy presenting with histopathologic features of Castleman’s disease: a clinicopathologic study of five cases. Pathol Res Pract (1997) 193:565–71. doi:10.1016/S0344-0338(97)80015-5
28. Iwanaga N, Harada K, Tsuji Y, Kawahara C, Kurohama K, Izumi Y, et al. TAFRO syndrome with primary Sjogren’s syndrome. Nihon Rinsho Meneki Gakkai Kaishi (2016) 39:478–84. doi:10.2177/jsci.39.478
29. Alonso-Larruga A, Bustabad S, Navarro-Gonzálvez JA, Rodríguez-Lozano B, Franco A, Barrios Y. Isolated Ro52 antibodies as immunological marker of a mild phenotype of undifferentiated connective tissue diseases. Int J Rheumatol (2017) 2017:3076017. doi:10.1155/2017/3076017
30. Antonelli A, Ferrari SM, Giuggioli D, Ferrannini E, Ferri C, Fallahi P. Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun Rev (2014) 13:272–80. doi:10.1016/j.autrev.2013.10.010
31. Iwaki N, Gion Y, Kondo E, Kawano M, Masunari T, Moro H, et al. Elevated serum interferon γ-induced protein 10 kDa is associated with TAFRO syndrome. Sci Rep (2017) 7:42316. doi:10.1038/srep42316
32. El Karoui K, Vuiblet V, Dion D, Izzedine H, Guitard J, Frimat L, et al. Renal involvement in Castleman disease. Nephrol Dial Transplant (2011) 26:599–609. doi:10.1093/ndt/gfq427
33. Xu D, Lv J, Dong Y, Wang S, Su T, Zhou F, et al. Renal involvement in a large cohort of Chinese patients with Castleman disease. Nephrol Dial Transplant (2012) 27(Suppl 3):iii119–25. doi:10.1093/ndt/gfr245
Keywords: TAFRO, Caucasian, review of literature, Castleman–Kojima disease, multicentric Castleman’s disease
Citation: Louis C, Vijgen S, Samii K, Chalandon Y, Terriou L, Launay D, Fajgenbaum DC, Seebach JD and Muller YD (2017) TAFRO Syndrome in Caucasians: A Case Report and Review of the Literature. Front. Med. 4:149. doi: 10.3389/fmed.2017.00149
Received: 23 June 2017; Accepted: 31 August 2017;
Published: 22 September 2017
Edited by:
Meral Beksac, Ankara University, TurkeyReviewed by:
Michel Delforge, University Hospital Leuven, BelgiumBrian R. Branchford, University of Colorado Denver School of Medicine, United States
Copyright: © 2017 Louis, Vijgen, Samii, Chalandon, Terriou, Launay, Fajgenbaum, Seebach and Muller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yannick D. Muller, eWFubmljay5tdWxsZXJAdW5pZ2UuY2g=