 Gabriela Leuschner
Gabriela Leuschner Jürgen Behr
Jürgen Behr- 1Department of Internal Medicine V, Ludwig Maximilians University, Comprehensive Pneumology Center (CPC-M), German Center for Lung Research (DZL), Munich, Germany
- 2Asklepios Fachkliniken München-Gauting, Gauting, Germany
Acute exacerbation of idiopathic pulmonary fibrosis (AE-IPF) has been defined as an acute, clinically significant deterioration that develops within less than 1 month without obvious clinical cause like fluid overload, left heart failure, or pulmonary embolism. Pathophysiologically, damage of the alveoli is the predominant feature of AE-IPF which manifests histopathologically as diffuse alveolar damage and radiologically as diffuse, bilateral ground-glass opacification on high-resolution computed tomography. A growing body of literature now focuses on acute exacerbations of interstitial lung disease (AE-ILD) other than idiopathic pulmonary fibrosis. Based on a shared pathophysiology it is generally accepted that AE-ILD can affect all patients with interstitial lung disease (ILD) but apparently occurs more frequently in patients with an underlying usual interstitial pneumonia pattern. The etiology of AE-ILD is not fully understood, but there are distinct risk factors and triggers like infection, mechanical stress, and microaspiration. In general, AE-ILD has a poor prognosis and is associated with a high mortality within 6–12 months. Although there is a lack of evidence based data, in clinical practice, AE-ILD is often treated with a high dose corticosteroid therapy and antibiotics. This article aims to provide a summary of the clinical features, diagnosis, management, and prognosis of AE-ILD as well as an update on the current developments in the field.
Introduction
Interstitial lung diseases (ILD) are a heterogeneous group of diseases. Despite various types of clinical presentation, disease progression, and prognosis, the common feature in most ILDs is a fibrotic destruction of the lung parenchyma. Within the clinical course of ILD, an acute exacerbation [acute exacerbations of interstitial lung disease (AE-ILD)] can occur at any time and is associated with significant morbidity and mortality (1–5). Initially, AE-ILD was described in idiopathic pulmonary fibrosis (IPF), and according to the official American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Society IPF guideline, an acute exacerbation of idiopathic pulmonary fibrosis (AE-IPF) has been defined as an acute clinical worsening of dyspnea which develops within less than 1 month without an alternative etiology (6).
Pathophysiologically, AE-ILD resembles an acute lung injury (ALI), which presents histopathologically as diffuse alveolar damage (DAD) in most cases (7). However, DAD is not only found in autopsy studies of patients with IPF but also in patients with connective tissue-related ILD (CTD-ILD), idiopathic fibrotic non-specific interstitial pneumonia (NSIP), and chronic hypersensitivity pneumonitis (HP) (8, 9). Besides the histopathological DAD, AE-ILD and ALI have more clinical features in common, such as an increased oxygen requirement and new bilateral infiltrates on high-resolution computed tomography (HRCT) (e.g., ground-glass opacification/consolidation) (10–12).
While AE-IPF is increasingly recognized better and is perceived as a severe event with high mortality, there is only a limited amount of clinical data on AE-ILD in non-IPF ILD. The aim of this review is to provide a summary of the definition, clinical features, diagnosis, prognosis, and management of AE-ILD. Furthermore, this review will update the current developments in the field of AE-ILD not only in IPF but also in non-IPF ILD.
Definition
Especially in IPF, great efforts have been made to establish a clear definition and diagnosis criteria for AE-IPF (6, 10, 13). In 2007, the IPF Clinical Trials Network (IPFnet) described the clinical presentation, radiological and histopathological findings of AE-IPF and developed diagnostic criteria based on the published literature (13). Just recently, an international working group revised an update on the definition of AE-IPF (Table 1) (10). In this document, AE-IPF is defined by a clinically significant respiratory deterioration developing within typically less than 1 month, accompanied by new radiologic abnormalities on HRCT such as diffuse, bilateral ground-glass opacification, and the absence of other obvious clinical causes like fluid overload, left heart failure, or pulmonary embolism (10). In contrast to the previous definition, the authors promote discrimination between a triggered AE-IPF (e.g., infection, post-procedural/postoperative or drug toxicity) and an idiopathic AE-IPF, where no trigger is identified (10). The revised definition aims to be broader and thus allow more inclusion possibilities. In the event of a clinical deterioration with unknown cause, where the criteria for AE-IPF are not met, the term “suspected AE-IPF” can be used (10). This might be the case if there are only unilateral ground glass abnormalities on HRCT or if HRCT data are even missing (13).
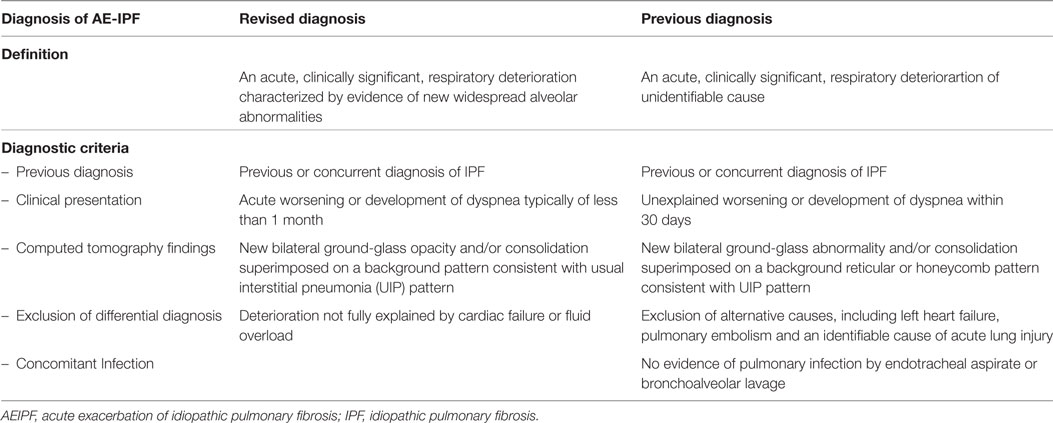
Table 1. Revised and previous definitions and diagnostic criteria for AE-IPF.
Clincal Features and Diagnostic Evaluation
Unfortunately, so far, there is no existing official definition of AE-ILD in non-IPF ILD. Since an AE-ILD in non-IPF patients resembles AE-IPF (14–16), in the clinical setting it might be reasonable to apply the definition of AE-IPF to all AE-ILD. Still, it should be pointed out, that the current definition of AE-IPF refers exclusively to IPF and that the authors of the working group report decided against a definition including other ILD (10).
The clinical presentation of AE-ILD is usually a rapid worsening of respiratory symptoms with increased dyspnea within less than 1 month (10, 13). Additional findings can be cough, increased sputum production, fever, and flu-like symptoms (1, 14, 15, 17). Since many patients present with a severe hypoxemia in the arterial blood gas analysis and respiratory failure, admission to the intensive care unit and assisted ventilation is often required (13). Established criteria for a presenting abnormal gas exchange is a PaO2/FiO2 ratio <225 or a decrease in PaO2 of ≥10 mmHg over time (13).
Still, establishing the diagnosis of AE-ILD often comprises a challenge. In order to get to this diagnosis, various diagnostic tests should be performed and differential diagnosis like myocardial infarction, pulmonary embolism, or fluid overload need to be excluded (Figure 1). An elementary part of the diagnosis is the HRCT, which should be carried out in all patients who are clinically stable. The important finding in AE-ILD is newly developed, bilateral alveolar infiltrates like ground-glass opacification with or without consolidation on HRCT (Figure 2) (6, 14, 16). Three suggested HRCT abnormality patterns are peripheral, multifocal and diffuse ground glass, with the latter two being associated with histologically DAD (13, 18). Several studies have shown that the extent of disease on HRCT seems to be related with the clinical outcome (1, 19–21). If there is no previous HRCT scan available, bilateral ground-glass opacity and/or consolidation on a background of usual interstitial pneumonia (UIP) pattern is sufficient to confirm the radiographic diagnostic criteria of AE-IPF (10). The term “suspected AE-IPF” should be used if there are only unilateral ground glass abnormalities (13).
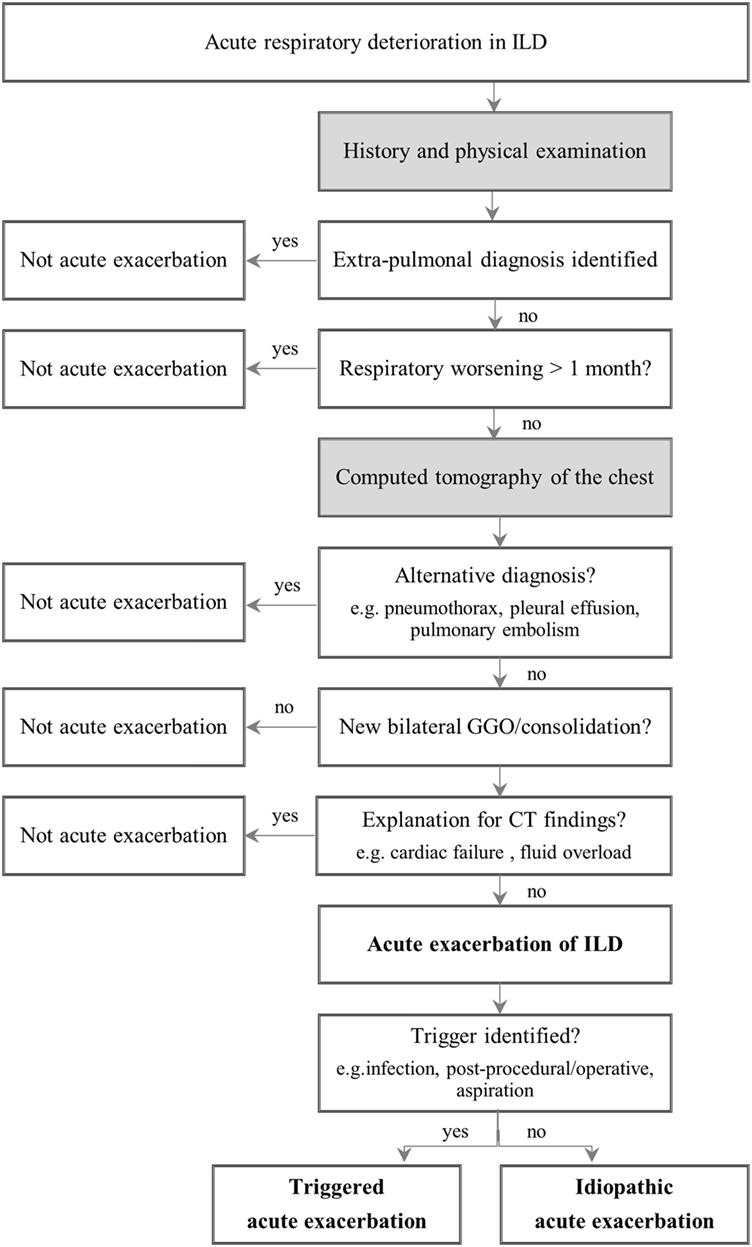
Figure 1. Diagnostic approach to acute exacerbation in interstitial lung disease. Adapted from Ref. (10). Abbreviation: ILD, interstitial lung disease; GGO, ground glass opacity; CT, computed tomography.
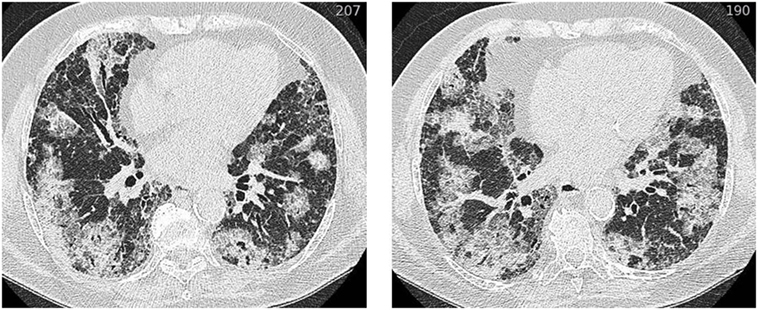
Figure 2. HRCT of an acute exacerbation in IPF. Axial HRCT of a patient with IPF at the time of an acute exacerbation shows extensive bilateral ground-glass opacification. Abbreviation: HRCT, high resolution computed tomography; IPF, idiopathic pulmonary fibrosis.
Histologically, most AE-ILDs are characterized by a DAD, while alternative histological appearances comprise organizing pneumonia, alveolar hemorrhage, and unspecific inflammatory changes (1, 13, 15, 22, 23). In early stages, the acute injury of the lung is characterized by an interstitial edema and hyaline membranes (10). Furthermore, type II pneumocyte hyperplasia and fibroblast foci have been reported in biopsies/autopsies as well as squamous metaplasia and honeycombing with and without hyaline membranes (24).
It has been reported, that patients with AE-ILD present with elevated inflammatory laboratory values such as increased white blood cell count, elevated values of erythrocyte sedimentation rate, and C-reactive protein and increased lactate dehydrogenase (14, 23–25). Although bronchoalveolar lavage (BAL) is not done routinely in AE-ILD, it has also been shown that AE-IPF and AE-HP are associated with an increase in neutrophils in BAL (1, 9, 17, 24–27). Rarely, lymphocytosis has been observed (23, 28), and reactive type II cells hyperplasia has been dectected on BAL (24). Furthermore, BAL is still the subject of research in terms of investigating the pathogenesis of AE and identifying possible prognostic factors.
Epidemiology
Acute exacerbations of interstitial lung disease can occur at any time during the disease, and in some cases it can be the presenting manifestation of an ILD (1, 13, 17). The exact frequency is unknown and the reported incidence rates of AE-ILD broadly vary, most likely due to differences in definition, ILD-entity and disease severity (10, 29). Furthermore, due to incomplete clinical information, definite AE-ILD cannot be confirmed in some cases, although AE-ILD is the suspected and the most probable diagnosis (10). The impact of this relevant difference was investigated in a post hoc analysis of the STEP-IPF trail, where a definite AE-IPF occurred in 40 per 1,000 patient-years but combining definite and suspected AE-IPF raised the number to 200 per 1,000 patient-years (30). In a recently published central adjudication on three randomized controlled trials, only 33.2% of the investigator-reported AE-IPF met the criteria (31). A meta-analysis of six randomized-controlled clinical trials identified a weighted average of 41 AE-IPF per 1,000 patient-years (32). In the INPULSIS I and II trial, the 1-year incidence of AE-IPF in the placebo-arm was 7.6% (33).
Compared to clinical trials, retrospective studies report even higher 1-year incidences of AE-IPF, ranging from 7 to 19.1% with highest risk in advanced IPF (1, 2, 34–36). Retrospective analyses of studies from the US and Japan identified the incidences of AE-IPF in approximately 52 per 1,000 patient-years (37, 38). In a registry-based US study, the annual rate of AE in IPF was 133 per 1,000 patient-years (39).
There is much less data on the frequency of AE-ILD in non-IPF ILD compared to AE-IPF. However, the majority of studies indicate that patients with IPF are at a higher risk for developing AE compared to non-IPF ILD (40–43). The estimated 1-year incidence of AE-NSIP is reported to be 4.2%, and the estimated 1-year incidence of AE-CTD ranges from 1.25 to 3.3% (14, 16). Within CTD-ILD, AE seems to be most common in patients with rheumatoid arthritis ILD (RA-ILD) (16). Since the frequency of a UIP pattern is higher in RA-ILD compared to other ILDs, the higher number of AE-RA may be explained by the observation that a UIP pattern per se is associated with a higher risk of AE-ILD. Thus, in patients with CTD-ILD and RA-ILD with UIP pattern, a 1-year incidence of 5.6 and 11.1% was found, respectively (14). Furthermore, the 2-year incidence of AE-HP was 11.5% among patients with chronic HP and UIP-like lesions on surgical biopsies (9).
Moreover, ethnicity may play a role, since AE-ILD were initially observed and reported in Japan and Korea, and the literature still is dominated by reports from Asian countries (1, 14, 19, 35). However, two randomized, controlled studies did not support this observation (33, 44, 45).
Pathogenesis and Etiology
The onset and development of an AE-ILD is unpredictable and until now, it is uncertain, whether an AE-ILD is triggered by an intrinsic factor causing a progression of the underlying disease or a response to an external factor (e.g., infection, aspiration, pulmonary emboli, mechanical stretch) or both (6, 10). Most likely, environmental and genetic factors interact individually leading to AE-IPF in only a subset of patients (13). Concerning the parallels between AE-IPF and acute respiratory distress syndrome, the IPF lung may be generally more vulnerable to intrinsic and extrinsic triggers (10). Still, further research is needed to identify the underlying causes and potential biomarkers for AE-ILD.
Epithelial Injury
During AE-IPF, alveolar injury and loss of epithelial cell integrity may be involved leading to an increased fibrin production and remodeling (13, 46). Morphologically, this leads to neutrophilia in BAL and histopathological DAD (8, 17, 24). Neutrophilic processes are potentially transmitted via α-defensins, as they have been shown to be upregulated in patients with AE-IPF (47, 48). α-defensins belong to a family of antimicrobial and cytotoxic peptides contained in mammalian neutrophils (49, 50). Supporting the hypothesis of epithelial injury and proliferation during AE-IPF, a gene expression study of lung tissue detected an increased expression of cyclin A2 and α-defensins together with widespread apoptosis in lungs of patients suffering from AE-IPF in comparison to stable IPF and healthy controls (47). Furthermore, α-defensins were increased in the peripheral blood of patients with AE-IPF, suggesting a potential role as biomarker (47). In a later study including patients with idiopathic interstitial pneumonia (IIP), elevated plasma levels of α-defensins in AE-IIP compared to stable IIP were also seen, but they were not useful as biomarkers due to a lack of specificity (51). Therefore, further studies are needed to clarify the role of α-defensins as biomarker.
Moreover, it could be shown that fibrocytes, which are increased in stable IPF, are even more elevated during AE-IPF (52). Fibrocytes, CD45 and collagen-1 positive cells, are mesenchymal derived progenitor cells which can migrate into injured tissue and can differentiate to fibroblast-like cells playing a role in wound repair, tissue regeneration and pulmonary fibrosis (53, 54). Patients with fibrocytes >5% of total blood leukocytes had a significantly worse survival compared to patients with fibrocytes <5% (52).
Another theory includes the involvement of alternative, so called M2, activation of macrophages in AE-IPF. M2 macrophages play an important role in tumor progression and wound healing (55), and seem to be associated with ILD (56–58). It could be shown that the pro-inflammatory chemokines CXCL1 and Interleukin 8 (produced by classically activated macrophages) and the anti-inflammatory chemokines CC-chemokine ligand (CCL)2, CCL17, CCL18, CCL22, and Interleukin 1ra (produced by alternatively/M2 activated macrophages) were elevated in BAL of patients with AE-IPF in comparison to stable IPF (26). High CCL18 levels in BAL at baseline were further highly predictive for a future AE-IPF (26).
Further markers have been studied, including Krebs von den Lungen-6 (KL-6) and surfactant protein D, which were both identified to be elevated in AE-IPF compared to stable IPF (59) and KL-6 was also increased during the event of AE-HP (60). Furthermore, an elevated serum level of KL-6 at baseline was identified as a predictor for developing AE-ILD in both, IPF and combined pulmonary fibrosis and emphysema (34, 61). KL-6 is a mucin-like glycoprotein, which is mainly expressed in type II penumocytes and bronchial epithelial cells (62). In ILD, a high expression of KL-6 has been detected in regenerating type II cells likely being the primary source of serum KL-6 (63).
Increased levels of Interleukin 6 and Interleukin 8 were also detected in patients with AE-IPF and an increase in either of them was identified to be associated with worse outcome (59, 64). Total protein C, thrombomodulin, plasminogen activator inhibitor 1 (59), and leptin (65) have also been shown to be elevated in the serum of patients with AE-IPF in comparison to stable IPF. In chronic HP, patients with elevated levels of KL-6 and surfactant protein D as well as increased neutrophils in the BAL fluid also have a higher risk for developing AE-HP.
Autoimmunity
Heat shock protein (HSP) 47 is a human collagen-specific molecular chaperone, which is involved in the early stages of biosynthesis and secretion of collagen molecules (66). In AE-IPF, it has been shown that HSP47 serum levels were significantly higher in comparison to stable IPF (67). Furthermore, immunohistochemical analysis detected more HSP47 expression in DAD than in UIP tissues (67). Interestingly, another study identified anti-HSP70 IgG autoantibodies in 25% of patients with IPF and anti-HSP70 positivity was associated with a higher mortality and risk for AE-IPF (68). Mortality among patients with positive anti-HSP70 antibodies was significantly higher compared to patients with negative antibodies (68).
Supporting autoimmune involvement in AE-IPF, one study identified annexin 1 as an autoantigen which increased antibody production and T cell response in AE-IPF with the N-terminus of annexin 1 potentially playing a role in the pathogensis of AE-IPF (69).
Infection
There is an increasing number of findings indicating that infection, both viral and bacterial, might be involved in some cases of AE-ILD. First, in a minority of patients with IIP suffering from AE-IIP, viral ribonucleic acid or a rise in specific immunoglobulins was detected by polymerase chain reaction or pan-viral microarray (70–74). Moreover, changes in the respiratory microbiome were recently identified showing an increased bacterial burden in BAL during an AE-IPF (27). Patients with AE-IPF experienced a markable change in the respiratory microbiome with an increase in Campylobacter sp. and Stenotrophomonas sp., as well as a significant decrease in Veillonella sp. compared to stable IPF (27). The hypothesis of an underlying infection is supported by the fact, that AE-IPF occurs more often between December and May (30, 75) and in the majority of studies an immunosuppressive therapy increases the risk for developing AE-IPF (30, 37, 76).
Microaspiration
Microaspiration might also have a connection to the development of AE-ILD (10). In a post hoc analysis of the placebo-treated IPF patients in three clinical trials, none of the patients developing AE-IPF was on an anti-acid therapy (77). Furthermore, patients with AE-IPF had significantly higher levels of pepsin in BAL compared to stable controls, suggesting an involvement of occult aspiration (78).
Risk Factors
Several clinical risk factors are discussed playing a potentially crucial role in developing an AE-IPF. First of all, a functionally and clinically advanced stage of disease appears to be an important risk factor. In this context, a low forced vital capacity (FVC) seemed to be the most stable risk factor (2, 26, 30, 34, 35). Other clinical risk factors include a recent decline in FVC (35, 38, 79), a low diffusing capacity of the lung for carbon monoxide (DLCO) (9, 26, 30), a low total lung capacity (9), a low 6-min walking distance (30), an impaired baseline oxygenation (30, 38), an increased dyspnea (30, 35), and a previous AE-IPF (37, 79). However, it should also be considered, that the apparent association between advanced IPF and risk of AE-IPF may be biased by the fact that in advanced disease an AE may have more obvious clinical consequences, while it may even be overlooked in less advanced disease.
In 2007, Selman et al. discriminated IPF patients as rapid or slow progressors based on the duration of symptoms before first presentation (80). Although this study did not analyze AE-IPF, the authors stated that the rapid progression of AE-IPF does not correspond to an AE-IPF (80). Still, some links might be present as the rapid progressors showed a higher rate of fibroblast migration than slow progressors and survival was significantly reduced (80). Until now there is no proof for the theory that rapid progressors and AE-IPF could have a connection but obviously, there is not enough data in this field. Given the association between a recent decline of FVC and an increased risk of AE-IPF (35, 38, 79), it would be further interesting to acknowledge daily variability of FVC. In this context, daily home spirometry is a promising clinical tool to follow the clinical course of IPF patients more closely and potentially detect AE-IPF earlier (81).
Although AE-ILD can occur in different histological forms of ILD, UIP-like lesions were identified to be associated with a higher risk for AE-ILD in patients with chronic HP and CTD-ILD (9, 14). Additional risk factors including male gender (15), a co-existing pulmonary hypertension (36), coronary artery disease (30), a higher body-mass-index (35), and exposure to increased ozone and nitrogen dioxide levels (37) have been reported. Some studies observed a higher risk in former smokers (9, 34), but this finding is inconsistent (2, 26, 82). Similarly, there are different findings concerning age as a potential risk factor: in IPF, younger patients seem to be at a higher risk for AE-IPF (26), whereas a study on CTD-ILD identified higher age as a risk factor for developing AE-CTD (16).
In a retrospective study on patients with ILD and lung cancer undergoing chemotherapy, 21.9% of the patients experienced AE-ILD during the time from diagnosis to the end of the chemotherapy treatment period (83). The authors suggested tegafur–gimeracil–oteracil potassium (S-1) and etoposide as relatively safe options in these patients. Moreover, there is one case-report about a patient with primary lung cancer and subclinical IPF, developing an AE-IPF after hypofractionated stereotactic radiotherapy (84).
A surgical biopsy is another important risk factor triggering AE-ILD. Whereas the incidence rates of developing an AE-ILD after a surgical biopsy for ILD-diagnosis finding are reported to be less than 2.5% (40, 41, 43, 85), AE-ILD after pulmonary resection due to lung cancer can occur in 3–32% (40, 86–88). A decreased FVC and DLCO seemed to be additional risk factors for patients with ILD developing a respiratory deterioration after lung surgery (43, 89). Pulmonary surgery itself seems to be a risk factor, but AE-ILD has also been reported in non-pulmonary surgery and throughout major surgeries the incidences was 3.3% in a study from Korea (90, 91). In IPF, there might be an association between AE-IPF and BAL, as few, individual cases of AE-IPF following BAL exist (92, 93). Furthermore, as this will become increasingly important in the future, the risk of AE-ILD after cryobiopsy needs to be investigated. However, data in this field is still limited and so far, only single cases of AE-ILD following cyrobiopsy have been reported (94, 95).
Prognosis
Acute exacerbations of interstitial lung disease is a life-threatening event and the mortality rate is high. It is assumed that between 35 and 46% of deaths in IPF are caused by AE-IPF (35, 96, 97). In a large number of studies, the in-hospital mortality in AE-IPF is estimated over 50% (1–4, 17, 19, 20, 36, 70, 98) and the median survival after AE-IPF is between 1 and 4 months (2, 36, 75, 82). In IPF, the 1-month mortality ranges between 37 and 53% (37, 99), and the 3-month mortality rate ranges from 63.8 to 73.7% (5, 19, 99). The existing data suggest that patients with IPF have a worse survival compared to ILD patients other than IPF; nonetheless, AE-ILD is also fatal in non-IPF ILD (5, 74). In a study including IPF and non-IPF patients, the overall survival after admission for AE-ILD was 67% at 1 month and 40% at 3 months (4). Similarly to IPF, the highest overall mortality rate of AE-ILD is seen in AE-HP (75–100% mortality) (9, 15). Mortality of AE-ILD in other ILDs ranges from 34 to 83% (5, 16).
Some potential prognostic factors have been identified. First of all, lower baseline pulmonary function parameters (FVC and DLCO) as well as a more impaired oxygenation are associated with a worse outcome in AE-IPF (2, 35, 75). Furthermore, a higher fibrosis score or more extensive disease on HRCT seems to be of prognostic relevance (1, 19–21). A lymphocytosis >15% in the BAL might be another prognostic factor for a favorable outcome in patients with AE-IIP (28). Several markers in the blood could also be potential prognostic markers including lactate dehydrogenase (9, 19, 75), C-reactive protein (2), KL-6 (19, 21), circulating fibrocytes (52), and anti-HSP70 autoantibodies (68). Just recently, Kishaba et al. developed a staging system for AE-IPF, which includes some of these prognostic factors (19).
Treatment
So far, there is a lack of evidence based data on effective therapies in AE-ILD. In clinical practice, AE-ILD is often treated with high-dose systemic corticosteroid therapy and antibiotics (9, 14, 16). In AE-IPF, the current international guidelines give a weak recommendation on the treatment with corticosteroids emphasizing that this recommendation is based on anecdotal reports of benefit and the high overall mortality in AE-IPF (6). The authors further point out, that there was a consensus to promote supportive care as an important therapy strategy (6). This includes palliation of symptoms, e.g., with opioids, and supply of oxygen in hypoxemia. Still, there are different opinions on the length of supportive care, and regarding the use of mechanical ventilation (10). Based on an estimated 90% in-hospital mortality, the international guidelines on the management of IPF make a weak recommendation against the use of mechanical ventilation in the case of respiratory failure due to the underlying lung disease (6). The authors point out that this decision has to be made case-by-case together with the physician, the patient, and the family and in accordance with the individual goals of care (6). As a bridge to lung transplantation, mechanical ventilation, or extra-corporal membrane oxygenation may be appropriate and successful in selected patients (6, 100). In a recent retrospective cohort study, the mortality rate in IPF patients undergoing mechanical ventilation significantly decreased from 58.4% in 2006 to 49.3% in 2012 (101). This reminds us to carefully analyze every single patient before a decision for or against mechanical ventilation is taken.
Several studies on different therapy regimens in AE-IPF, other than high-dose intravenous corticosteroids mono, have been published (Table 2). In smaller, observational studies, it could be shown that the combination of a steroid-pulse therapy with oral tacrolimus (82) or cyclosporine (102–104) was superior to the corticosteroid mono therapy in terms of prognosis in IPF. Other studies identified a positive effect of a treatment with rituximab with plasma exchange and intravenous immunoglobulin (105), polymyxin B-immoblilized fiber column perfusion (99, 106–109), and intra-venous thrombomodulin (110–114). Still, the benefits seen in these studies have to be critically assessed since these were all observational studies with either a historical control or a parallel, untreated control arm, potentially excluding very ill patients from the experimental arm (10). One randomized trial investigated the benefit of a procalcitonin-guided antibiotic therapy compared to a clinical-driven antibiotic therapy but no difference in mechanical ventilation and mortality was seen (115). Several studies reported about a combination therapy of corticosteroids with other immunosuppressant drugs like cyclophosphamide, but it remains unclear whether this is beneficial (20, 21, 116).
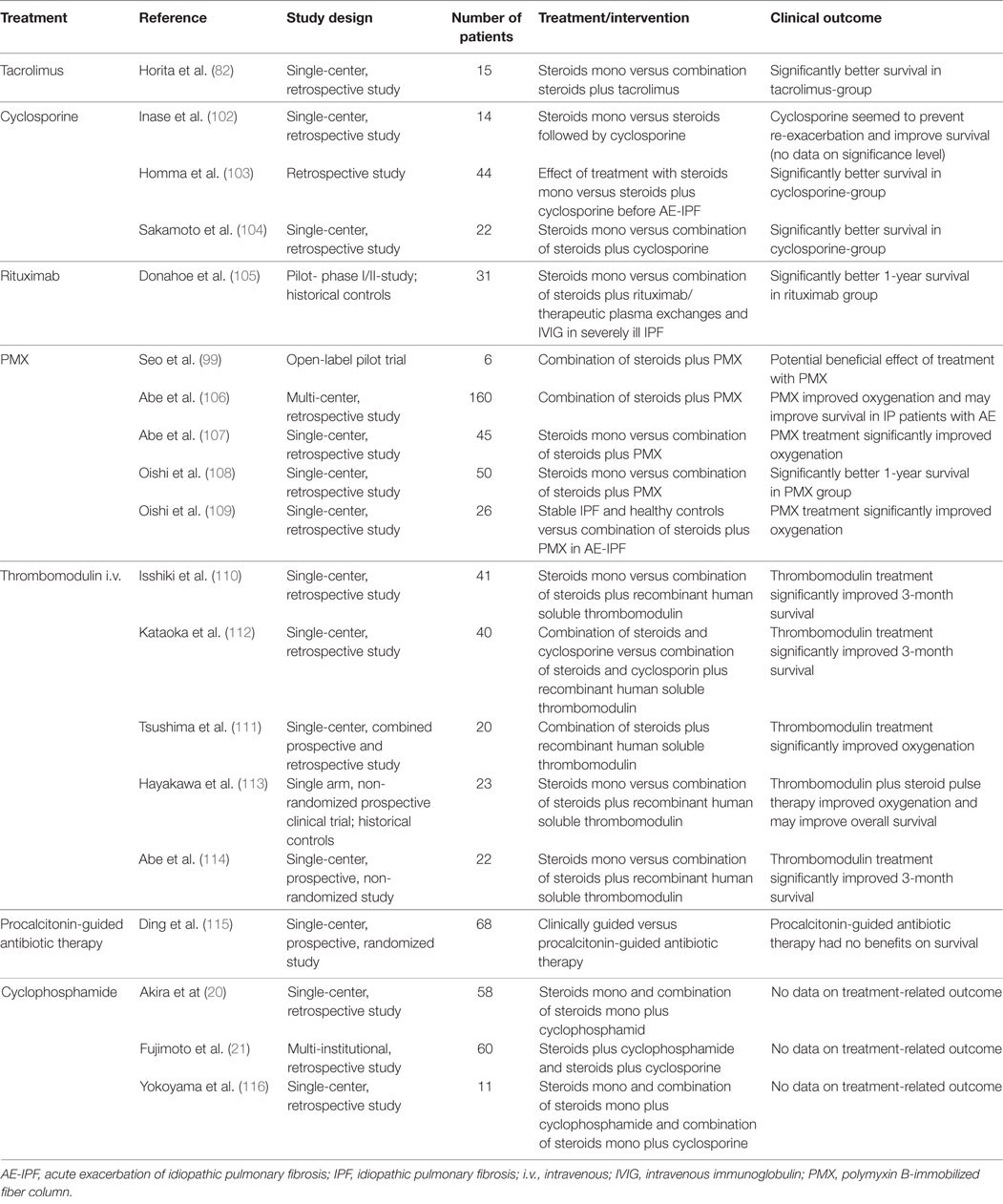
Table 2. Medical treatment of AE-IPF other than high-dose intravenous corticosteroids mono therapy.
Currently, there are indications that an anti-acid therapy could have a protective effect against AE-IPF, as in an analysis of patients from the placebo arm of three large clinical trials, an anti-acid therapy was reported to have a potentially preventive effect on the development of AE-IPF (77). The same potentially preventive effect applies for antifibrotic drugs, although, so far, there is no sufficient data on whether an antifibrotic therapy with nintedanib or pirfenidone should be paused or continued in the event of an AE-IPF. In a phase II trial, patients receiving pirfenidone had a significant reduction in AE-IPF compared to placebo (117). However, a subsequent phase II trial could not reproduce this finding (44). After AE-IPF was not included as an endpoint in the three phase III trials ASCEND and CAPACITY (118, 119), a pooled analysis recently showed that patients receiving pirfenidone had a lower risk for respiratory-related hospitalization compared to healthy controls (120). Interestingly, there is data that the perioperative use of pirfenidone might prevent postoperative AE-IPF (121). In contrast to pirfenidone, AE-IPF was consequently included in the nintedanib clinical study program as a key secondary endpoint; however, the role of nintedanib on AE-IPF needs to be fully understood. Whereas the phase II trial of nintedanib identified a delay in time to the first investigator-reported AE in the nintedanib arm (122), only one of the two INPULSIS phase III twin-trials showed a significant effect of nintedanib on AE-IPF (33). Pooled analysis of the data showed a highly significant prolongation of the time to first AE in IPF due to treatment with nintedanib, thus confirming its preventive effect (45, 123). Still, at this point, more data is needed to validate the effect of pirfenidone and nintedanib in AE-ILD.
Apart from these potentially effective therapeutic approaches, there are a number of drugs that seem to have no preventive effect on AE-IPF, including acetylcystein mono therapy (124), sildenafil (125), bosentan (126), interferon-gamma 1b (127), warfarin (128), ambrisentan (129), and imatinib (130). A combination “triple” therapy (prednisone, azathioprine and acetylcysteine), might even increase the risk for developing AE-IPF (131).
As in AE-IPF, AE-ILD in non-IPF ILD is often treated with a high dose, systemic corticosteroid therapy together with broad-spectrum antibiotics (9, 14, 16). There are some studies additionally using cyclosporine A or cyclophosphamide, but there are no reports on whether there was any benefit in these therapies (9, 16). Similar to IPF, treatment with intra-venous thrombomodulin significantly improved 3-month survival in AE-NSIP (114).
In order to address all therapeutic approaches in this context, one study should be mentioned focusing on a non-steroid approach in AE-IPF: in the event of AE-IPF, prior immunosuppression was immediately stopped and patients were only treated with best supportive care and broad-spectrum antibiotics (132). The median survival of all patients was 1.73 months. Analyzing the single event of AE-IPF, 50% of AE-IPF episodes were survived. Overall, 35.3% of the patients survived AE-IPF and the 1-year survival of the survivors was 83%. Interestingly, patients who had never been treated with immunosuppressant drugs before had a significantly better survival. The 1-year survival in the “never treated” group was 65%, whereas the patients, who had a history of immunosuppression, had a 1-year survival of 17%. Unfortunately, no comparison with a high-dose steroid therapy during AE-IPF was investigated in the study. Nevertheless, this underlines again the lack of evidence based data on therapy strategies in AE-ILD and the necessity for further studies in this field.
Future
Acute exacerbations of interstitial lung diseases are severe events with a high mortality rate. Therefore, it is important to gain further knowledge in this field. As it has been shown that in IPF, an early referral to a specialized center is crucial for survival in general (133), in AE-ILD an early diagnosis and referral might also be important for the patients’ prognosis. Therefore, effort should be made to detect early signs of AE-ILD and identify patients who are at a higher risk for developing AE-ILD.
Potential treatment options should be studied in randomized, controlled trials. The currently revised definition of AE-IPF will hopefully allow a more uniform diagnosis, which will help to conduct well designed clinical trials in IPF (10). However, it should be emphasized that, even if it largely follows the framework of AE-IPF guidelines, an official, uniform definition for AE-ILD is needed in the future.
Biomarkers, which can be obtained in an easy and harmless way, are needed to identify patients at a higher risk for developing AE-ILD before symptoms and HRCT features are present. Since biomarkers in BAL might be difficult to obtain in a severely ill patient, serum markers are particularly interesting because of their easier accessibility. Furthermore, daily home spirometry might be a potential tool to understand the clinical course of AE-PF better and even possibly detecting AE-IPF in an early stage (81). There is evidence that home spirometry can potentially improve endpoint efficacy in clinical trials of IPF-therapeutics (134). Therefore, an effort should be made to design studies in that field analyzing the benefit of daily home spirometry in patients with ILD and help establishing home spirometry in clinical, daily routine.
Conclusion
Acute exacerbations of interstitial lung disease is a life-threatening event with a high in-hospital mortality rate. The clinical presentation of AE-ILD is similar in non-IPF and IPF, but AE-ILD in non-IPF ILD is less common and the clinical course is less fatal compared to IPF. The new working group report on AE-IPF supports that there are both, idiopathic and triggered AE (e.g., triggered by an infection) (10). So far, there is no evidence as to whether a triggered AE-ILD has a worse prognosis than an idiopathic AE-ILD. Due to the lack of evidence-based therapy options, more studies in this field are urgently needed.
Author Contributions
GL and JB wrote the manuscript and have approved the final version of the manuscript for submission.
Conflict of Interest Statement
GL received travel funding from Intermune and Novartis. JB received personal fees from Actelion, grants from Actelion, personal fees from Bayer, personal fees from Boehringer-Ingelheim, personal fees from Roche; JB is member of national and international IPF guideline committees.
Abbreviations
AE-CTD, acute exacerbation of connective tissue disease related interstitial lung disease; AE-HP, acute exacerbation of chronic hypersensitivity pneumonitis; AE-IIP, acute exacerbation of idiopathic interstitial pneumonia; AE-ILD, acute exacerbations of interstitial lung disease; AE-IPF, acute exacerbation of idiopathic pulmonary fibrosis; AE-NSIP, acute exacerbation of non-specific interstitial pneumonia; AE-RA, acute exacerbation of rheumatoid arthritis and interstitial lung disease; ALI, acute lung injury; BAL, bronchoalveolar lavage; CCL, CC-chemokine ligand; CTD-ILD, connective tissue-related interstitial lung disease; DAD, diffuse alveolar damage; DLCO, diffusing capacity of the lung for carbon monoxide; FVC, forced vital capacity; HP, hypersensitivity pneumonitis; HRCT, high-resolution computed tomography; HSP, heat shock protein; IIP, idiopathic interstitial pneumonia; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; KL-6, Krebs von den Lungen-6; NSIP, non-specific interstitial pneumonia; RA-ILD, interstitial lung disease in patients with rheumatoid arthritis; UIP, usual interstitial pneumonia.
References
1. Kim DS, Park JH, Park BK, Lee JS, Nicholson AG, Colby T. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J (2006) 27:143–50. doi:10.1183/09031936.06.00114004
2. Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J (2011) 37:356–63. doi:10.1183/09031936.00159709
3. Al-Hameed FM, Sharma S. Outcome of patients admitted to the intensive care unit for acute exacerbation of idiopathic pulmonary fibrosis. Can Respir J (2004) 11:117–22. doi:10.1155/2004/379723
4. Usui Y, Kaga A, Sakai F, Shiono A, Komiyama K, Hagiwara K, et al. A cohort study of mortality predictors in patients with acute exacerbation of chronic fibrosing interstitial pneumonia. BMJ Open (2013) 3:e002971. doi:10.1136/bmjopen-2013-002971
5. Tachikawa R, Tomii K, Ueda H, Nagata K, Nanjo S, Sakurai A, et al. Clinical features and outcome of acute exacerbation of interstitial pneumonia: collagen vascular diseases-related versus idiopathic. Respiration (2012) 83:20–7. doi:10.1159/000329893
6. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med (2011) 183:788–824. doi:10.1164/rccm.2009-040GL
7. Oda K, Ishimoto H, Yamada S, Kushima H, Ishii H, Imanaga T, et al. Autopsy analyses in acute exacerbation of idiopathic pulmonary fibrosis. Respir Res (2014) 15:109. doi:10.1186/s12931-014-0109-y
8. Rice AJ, Wells AU, Bouros D, du Bois RM, Hansell DM, Polychronopoulos V, et al. Terminal diffuse alveolar damage in relation to interstitial pneumonias. An autopsy study. Am J Clin Pathol (2003) 119:709–14. doi:10.1309/UVARMDY8FE9FJDKU
9. Miyazaki Y, Tateishi T, Akashi T, Ohtani Y, Inase N, Yoshizawa Y. Clinical predictors and histologic appearance of acute exacerbations in chronic hypersensitivity pneumonitis. Chest (2008) 134:1265–70. doi:10.1378/chest.08-0866
10. Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med (2016) 194:265–75. doi:10.1164/rccm.201604-0801CI
11. de Hemptinne Q, Remmelink M, Brimioulle S, Salmon I, Vincent JL. ARDS: a clinicopathological confrontation. Chest (2009) 135:944–9. doi:10.1378/chest.08-1741
12. Ferguson ND, Frutos-Vivar F, Esteban A, Fernandez-Segoviano P, Aramburu JA, Najera L, et al. Acute respiratory distress syndrome: underrecognition by clinicians and diagnostic accuracy of three clinical definitions. Crit Care Med (2005) 33:2228–34. doi:10.1097/01.CCM.0000181529.08630.49
13. Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE Jr, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2007) 176:636–43. doi:10.1164/rccm.200703-463PP
14. Park IN, Kim DS, Shim TS, Lim CM, Lee SD, Koh Y, et al. Acute exacerbation of interstitial pneumonia other than idiopathic pulmonary fibrosis. Chest (2007) 132:214–20. doi:10.1378/chest.07-0323
15. Olson AL, Huie TJ, Groshong SD, Cosgrove GP, Janssen WJ, Schwarz MI, et al. Acute exacerbations of fibrotic hypersensitivity pneumonitis: a case series. Chest (2008) 134:844–50. doi:10.1378/chest.08-0428
16. Suda T, Kaida Y, Nakamura Y, Enomoto N, Fujisawa T, Imokawa S, et al. Acute exacerbation of interstitial pneumonia associated with collagen vascular diseases. Respir Med (2009) 103:846–53. doi:10.1016/j.rmed.2008.12.019
17. Parambil JG, Myers JL, Ryu JH. Histopathologic features and outcome of patients with acute exacerbation of idiopathic pulmonary fibrosis undergoing surgical lung biopsy. Chest (2005) 128:3310–5. doi:10.1378/chest.128.5.3310
18. Akira M, Hamada H, Sakatani M, Kobayashi C, Nishioka M, Yamamoto S. CT findings during phase of accelerated deterioration in patients with idiopathic pulmonary fibrosis. AJR Am J Roentgenol (1997) 168:79–83. doi:10.2214/ajr.168.1.8976924
19. Kishaba T, Tamaki H, Shimaoka Y, Fukuyama H, Yamashiro S. Staging of acute exacerbation in patients with idiopathic pulmonary fibrosis. Lung (2014) 192:141–9. doi:10.1007/s00408-013-9530-0
20. Akira M, Kozuka T, Yamamoto S, Sakatani M. Computed tomography findings in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2008) 178:372–8. doi:10.1164/rccm.200709-1365OC
21. Fujimoto K, Taniguchi H, Johkoh T, Kondoh Y, Ichikado K, Sumikawa H, et al. Acute exacerbation of idiopathic pulmonary fibrosis: high-resolution CT scores predict mortality. Eur Radiol (2012) 22:83–92. doi:10.1007/s00330-011-2211-6
22. Churg A, Muller NL, Silva CI, Wright JL. Acute exacerbation (acute lung injury of unknown cause) in UIP and other forms of fibrotic interstitial pneumonias. Am J Surg Pathol (2007) 31:277–84. doi:10.1097/01.pas.0000213341.70852.9d
23. Dallari R, Foglia M, Paci M, Cavazza A. Acute exacerbation of idiopathic pulmonary fibrosis. Eur Respir J (2004) 23:792. doi:10.1183/09031936.04.00004404
24. Ambrosini V, Cancellieri A, Chilosi M, Zompatori M, Trisolini R, Saragoni L, et al. Acute exacerbation of idiopathic pulmonary fibrosis: report of a series. Eur Respir J (2003) 22:821–6. doi:10.1183/09031936.03.00022703
25. Kondoh Y, Taniguchi H, Kawabata Y, Yokoi T, Suzuki K, Takagi K. Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest (1993) 103:1808–12. doi:10.1378/chest.103.6.1808
26. Schupp JC, Binder H, Jager B, Cillis G, Zissel G, Muller-Quernheim J, et al. Macrophage activation in acute exacerbation of idiopathic pulmonary fibrosis. PLoS One (2015) 10:e0116775. doi:10.1371/journal.pone.0116775
27. Molyneaux PL, Cox MJ, Wells AU, Kim HC, Ji W, Cookson WO, et al. Changes in the respiratory microbiome during acute exacerbations of idiopathic pulmonary fibrosis. Respir Res (2017) 18:29. doi:10.1186/s12931-017-0511-3
28. Takei R, Arita M, Kumagai S, Ito Y, Noyama M, Tokioka F, et al. Impact of lymphocyte differential count > 15% in BALF on the mortality of patients with acute exacerbation of chronic fibrosing idiopathic interstitial pneumonia. BMC Pulm Med (2017) 17:67. doi:10.1186/s12890-017-0412-8
29. Ryerson CJ, Cottin V, Brown KK, Collard HR. Acute exacerbation of idiopathic pulmonary fibrosis: shifting the paradigm. Eur Respir J (2015) 46:512–20. doi:10.1183/13993003.00419-2015
30. Collard HR, Yow E, Richeldi L, Anstrom KJ, Glazer C, IPFnet Investigators. Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir Res (2013) 14:73. doi:10.1186/1465-9921-14-73
31. de Andrade J, Schwarz M, Collard HR, Gentry-Bumpass T, Colby T, Lynch D, et al. The idiopathic pulmonary fibrosis clinical research network (IPFnet): diagnostic and adjudication processes. Chest (2015) 148:1034–42. doi:10.1378/chest.14-2889
32. Atkins CP, Loke YK, Wilson AM. Outcomes in idiopathic pulmonary fibrosis: a meta-analysis from placebo controlled trials. Respir Med (2014) 108:376–87. doi:10.1016/j.rmed.2013.11.007
33. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med (2014) 370:2071–82. doi:10.1056/NEJMoa1402584
34. Ohshimo S, Ishikawa N, Horimasu Y, Hattori N, Hirohashi N, Tanigawa K, et al. Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir Med (2014) 108:1031–9. doi:10.1016/j.rmed.2014.04.009
35. Kondoh Y, Taniguchi H, Katsuta T, Kataoka K, Kimura T, Nishiyama O, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis (2010) 27:103–10.
36. Judge EP, Fabre A, Adamali HI, Egan JJ. Acute exacerbations and pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Eur Respir J (2012) 40:93–100. doi:10.1183/09031936.00115511
37. Johannson KA, Vittinghoff E, Lee K, Balmes JR, Ji W, Kaplan GG, et al. Acute exacerbation of idiopathic pulmonary fibrosis associated with air pollution exposure. Eur Respir J (2014) 43:1124–31. doi:10.1183/09031936.00122213
38. Kondoh Y, Taniguchi H, Ebina M, Azuma A, Ogura T, Taguchi Y, et al. Risk factors for acute exacerbation of idiopathic pulmonary fibrosis – extended analysis of pirfenidone trial in Japan. Respir Investig (2015) 53:271–8. doi:10.1016/j.resinv.2015.04.005
39. Fernandez Perez ER, Daniels CE, Schroeder DR, St Sauver J, Hartman TE, Bartholmai BJ, et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest (2010) 137:129–37. doi:10.1378/chest.09-1002
40. Sakamoto S, Homma S, Mun M, Fujii T, Kurosaki A, Yoshimura K. Acute exacerbation of idiopathic interstitial pneumonia following lung surgery in 3 of 68 consecutive patients: a retrospective study. Intern Med (2011) 50:77–85. doi:10.2169/internalmedicine.50.3390
41. Samejima J, Tajiri M, Ogura T, Baba T, Omori T, Tsuboi M, et al. Thoracoscopic lung biopsy in 285 patients with diffuse pulmonary disease. Asian Cardiovasc Thorac Ann (2015) 23:191–7. doi:10.1177/0218492314550724
42. Rotolo N, Imperatori A, Dominioni L, Facchini A, Conti V, Castiglioni M, et al. Efficacy and safety of surgical lung biopsy for interstitial disease. Experience of 161 consecutive patients from a single institution in Italy. Sarcoidosis Vasc Diffuse Lung Dis (2015) 32:251–8.
43. Bando M, Ohno S, Hosono T, Yanase K, Sato Y, Sohara Y, et al. Risk of acute exacerbation after video-assisted thoracoscopic lung biopsy for interstitial lung disease. J Bronchology Interv Pulmonol (2009) 16:229–35. doi:10.1097/LBR.0b013e3181b767cc
44. Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J (2010) 35:821–9. doi:10.1183/09031936.00005209
45. Costabel U, Inoue Y, Richeldi L, Collard HR, Tschoepe I, Stowasser S, et al. Efficacy of nintedanib in idiopathic pulmonary fibrosis across prespecified subgroups in INPULSIS. Am J Respir Crit Care Med (2016) 193:178–85. doi:10.1164/rccm.201503-0562OC
46. Kuhn C III, Boldt J, King TE Jr, Crouch E, Vartio T, McDonald JA. An immunohistochemical study of architectural remodeling and connective tissue synthesis in pulmonary fibrosis. Am Rev Respir Dis (1989) 140:1693–703. doi:10.1164/ajrccm/140.6.1693
47. Konishi K, Gibson KF, Lindell KO, Richards TJ, Zhang Y, Dhir R, et al. Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2009) 180:167–75. doi:10.1164/rccm.200810-1596OC
48. Mukae H, Iiboshi H, Nakazato M, Hiratsuka T, Tokojima M, Abe K, et al. Raised plasma concentrations of alpha-defensins in patients with idiopathic pulmonary fibrosis. Thorax (2002) 57:623–8. doi:10.1136/thorax.57.7.623
49. van Wetering S, Sterk PJ, Rabe KF, Hiemstra PS. Defensins: key players or bystanders in infection, injury, and repair in the lung? J Allergy Clin Immunol (1999) 104:1131–8. doi:10.1016/S0091-6749(99)70004-7
50. Ganz T, Lehrer RI. Defensins. Curr Opin Immunol (1994) 6:584–9. doi:10.1016/0952-7915(94)90145-7
51. Sakamoto N, Ishimatsu Y, Kakugawa T, Yura H, Tomonaga M, Harada T, et al. Elevated plasma alpha-defensins in patients with acute exacerbation of fibrotic interstitial pneumonia. Respir Med (2015) 109:265–71. doi:10.1016/j.rmed.2014.12.015
52. Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2009) 179:588–94. doi:10.1164/rccm.200810-1534OC
53. Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med (1994) 1:71–81.
54. Gomperts BN, Strieter RM. Fibrocytes in lung disease. J Leukoc Biol (2007) 82:449–56. doi:10.1189/jlb.0906587
55. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25:677–86. doi:10.1016/j.it.2004.09.015
56. Mora AL, Torres-Gonzalez E, Rojas M, Corredor C, Ritzenthaler J, Xu J, et al. Activation of alveolar macrophages via the alternative pathway in herpesvirus-induced lung fibrosis. Am J Respir Cell Mol Biol (2006) 35:466–73. doi:10.1165/rcmb.2006-0121OC
57. Willems S, Verleden SE, Vanaudenaerde BM, Wynants M, Dooms C, Yserbyt J, et al. Multiplex protein profiling of bronchoalveolar lavage in idiopathic pulmonary fibrosis and hypersensitivity pneumonitis. Ann Thorac Med (2013) 8:38–45. doi:10.4103/1817-1737.105718
58. Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, et al. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med (2006) 173:781–92. doi:10.1164/rccm.200509-1518OC
59. Collard HR, Calfee CS, Wolters PJ, Song JW, Hong SB, Brady S, et al. Plasma biomarker profiles in acute exacerbation of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol (2010) 299:L3–7. doi:10.1152/ajplung.90637.2008
60. Okamoto T, Fujii M, Furusawa H, Tsuchiya K, Miyazaki Y, Inase N. The usefulness of KL-6 and SP-D for the diagnosis and management of chronic hypersensitivity pneumonitis. Respir Med (2015) 109:1576–81. doi:10.1016/j.rmed.2015.10.005
61. Kishaba T, Shimaoka Y, Fukuyama H, Yoshida K, Tanaka M, Yamashiro S, et al. A cohort study of mortality predictors and characteristics of patients with combined pulmonary fibrosis and emphysema. BMJ Open (2012) 2:e000988. doi:10.1136/bmjopen-2012-000988
62. Kohno N, Akiyama M, Kyoizumi S, Hakoda M, Kobuke K, Yamakido M. Detection of soluble tumor-associated antigens in sera and effusions using novel monoclonal antibodies, KL-3 and KL-6, against lung adenocarcinoma. Jpn J Clin Oncol (1988) 18:203–16.
63. Kohno N, Kyoizumi S, Awaya Y, Fukuhara H, Yamakido M, Akiyama M. New serum indicator of interstitial pneumonitis activity. Sialylated carbohydrate antigen KL-6. Chest (1989) 96:68–73. doi:10.1378/chest.96.1.68
64. Papiris SA, Tomos IP, Karakatsani A, Spathis A, Korbila I, Analitis A, et al. High levels of IL-6 and IL-8 characterize early-on idiopathic pulmonary fibrosis acute exacerbations. Cytokine (2017). doi:10.1016/j.cyto.2017.08.019
65. Cao M, Swigris JJ, Wang X, Cao M, Qiu Y, Huang M, et al. Plasma leptin is elevated in acute exacerbation of idiopathic pulmonary fibrosis. Mediators Inflamm (2016) 2016:6940480. doi:10.1155/2016/6940480
66. Dafforn TR, Della M, Miller AD. The molecular interactions of heat shock protein 47 (Hsp47) and their implications for collagen biosynthesis. J Biol Chem (2001) 276:49310–9. doi:10.1074/jbc.M108896200
67. Kakugawa T, Yokota S, Ishimatsu Y, Hayashi T, Nakashima S, Hara S, et al. Serum heat shock protein 47 levels are elevated in acute exacerbation of idiopathic pulmonary fibrosis. Cell Stress Chaperones (2013) 18:581–90. doi:10.1007/s12192-013-0411-5
68. Kahloon RA, Xue J, Bhargava A, Csizmadia E, Otterbein L, Kass DJ, et al. Patients with idiopathic pulmonary fibrosis with antibodies to heat shock protein 70 have poor prognoses. Am J Respir Crit Care Med (2013) 187:768–75. doi:10.1164/rccm.201203-0506OC
69. Kurosu K, Takiguchi Y, Okada O, Yumoto N, Sakao S, Tada Y, et al. Identification of annexin 1 as a novel autoantigen in acute exacerbation of idiopathic pulmonary fibrosis. J Immunol (2008) 181:756–67. doi:10.4049/jimmunol.181.1.756
70. Tomioka H, Sakurai T, Hashimoto K, Iwasaki H. Acute exacerbation of idiopathic pulmonary fibrosis: role of Chlamydophila pneumoniae infection. Respirology (2007) 12:700–6. doi:10.1111/j.1440-1843.2007.01119.x
71. Ushiki A, Yamazaki Y, Hama M, Yasuo M, Hanaoka M, Kubo K. Viral infections in patients with an acute exacerbation of idiopathic interstitial pneumonia. Respir Investig (2014) 52:65–70. doi:10.1016/j.resinv.2013.07.005
72. Wootton SC, Kim DS, Kondoh Y, Chen E, Lee JS, Song JW, et al. Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2011) 183:1698–702. doi:10.1164/rccm.201010-1752OC
73. Bando M, Ohno S, Oshikawa K, Takahashi M, Okamoto H, Sugiyama Y. Infection of TT virus in patients with idiopathic pulmonary fibrosis. Respir Med (2001) 95:935–42. doi:10.1053/rmed.2001.1151
74. Huie TJ, Olson AL, Cosgrove GP, Janssen WJ, Lara AR, Lynch DA, et al. A detailed evaluation of acute respiratory decline in patients with fibrotic lung disease: aetiology and outcomes. Respirology (2010) 15:909–17. doi:10.1111/j.1440-1843.2010.01774.x
75. Simon-Blancal V, Freynet O, Nunes H, Bouvry D, Naggara N, Brillet PY, et al. Acute exacerbation of idiopathic pulmonary fibrosis: outcome and prognostic factors. Respiration (2012) 83:28–35. doi:10.1159/000329891
76. Petrosyan F, Culver DA, Reddy AJ. Role of bronchoalveolar lavage in the diagnosis of acute exacerbations of idiopathic pulmonary fibrosis: a retrospective study. BMC Pulm Med (2015) 15:70. doi:10.1186/s12890-015-0066-3
77. Lee JS, Collard HR, Anstrom KJ, Martinez FJ, Noth I, Roberts RS, et al. Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomised controlled trials. Lancet Respir Med (2013) 1:369–76. doi:10.1016/S2213-2600(13)70105-X
78. Lee JS, Song JW, Wolters PJ, Elicker BM, King TE Jr, Kim DS, et al. Bronchoalveolar lavage pepsin in acute exacerbation of idiopathic pulmonary fibrosis. Eur Respir J (2012) 39:352–8. doi:10.1183/09031936.00050911
79. Reichmann WM, Yu YF, Macaulay D, Wu EQ, Nathan SD. Change in forced vital capacity and associated subsequent outcomes in patients with newly diagnosed idiopathic pulmonary fibrosis. BMC Pulm Med (2015) 15:167. doi:10.1186/s12890-015-0161-5
80. Selman M, Carrillo G, Estrada A, Mejia M, Becerril C, Cisneros J, et al. Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS One (2007) 2:e482. doi:10.1371/journal.pone.0000482
81. Russell AM, Adamali H, Molyneaux PL, Lukey PT, Marshall RP, Renzoni EA, et al. Daily home spirometry: an effective tool for detecting progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2016) 194:989–97. doi:10.1164/rccm.201511-2152OC
82. Horita N, Akahane M, Okada Y, Kobayashi Y, Arai T, Amano I, et al. Tacrolimus and steroid treatment for acute exacerbation of idiopathic pulmonary fibrosis. Intern Med (2011) 50:189–95. doi:10.2169/internalmedicine.50.4327
83. Kakiuchi S, Hanibuchi M, Tezuka T, Saijo A, Otsuka K, Sakaguchi S, et al. Analysis of acute exacerbation of interstitial lung disease associated with chemotherapy in patients with lung cancer: a feasibility of S-1. Respir Investig (2017) 55:145–52. doi:10.1016/j.resinv.2016.10.008
84. Takeda A, Enomoto T, Sanuki N, Nakajima T, Takeda T, Sayama K, et al. Acute exacerbation of subclinical idiopathic pulmonary fibrosis triggered by hypofractionated stereotactic body radiotherapy in a patient with primary lung cancer and slightly focal honeycombing. Radiat Med (2008) 26:504–7. doi:10.1007/s11604-008-0261-8
85. Kondoh Y, Taniguchi H, Kitaichi M, Yokoi T, Johkoh T, Oishi T, et al. Acute exacerbation of interstitial pneumonia following surgical lung biopsy. Respir Med (2006) 100:1753–9. doi:10.1016/j.rmed.2006.02.002
86. Mizuno Y, Iwata H, Shirahashi K, Takamochi K, Oh S, Suzuki K, et al. The importance of intraoperative fluid balance for the prevention of postoperative acute exacerbation of idiopathic pulmonary fibrosis after pulmonary resection for primary lung cancer. Eur J Cardiothorac Surg (2012) 41:e161–5. doi:10.1093/ejcts/ezs147
87. Suzuki H, Sekine Y, Yoshida S, Suzuki M, Shibuya K, Yonemori Y, et al. Risk of acute exacerbation of interstitial pneumonia after pulmonary resection for lung cancer in patients with idiopathic pulmonary fibrosis based on preoperative high-resolution computed tomography. Surg Today (2011) 41:914–21. doi:10.1007/s00595-010-4384-z
88. Watanabe A, Higami T, Ohori S, Koyanagi T, Nakashima S, Mawatari T. Is lung cancer resection indicated in patients with idiopathic pulmonary fibrosis? J Thorac Cardiovasc Surg (2008) 136:1357–63, 1363.e1–2. doi:10.1016/j.jtcvs.2008.07.016
89. Park JS, Kim HK, Kim K, Kim J, Shim YM, Choi YS. Prediction of acute pulmonary complications after resection of lung cancer in patients with preexisting interstitial lung disease. Thorac Cardiovasc Surg (2011) 59:148–52. doi:10.1055/s-0030-1250644
90. Ghatol A, Ruhl AP, Danoff SK. Exacerbations in idiopathic pulmonary fibrosis triggered by pulmonary and nonpulmonary surgery: a case series and comprehensive review of the literature. Lung (2012) 190:373–80. doi:10.1007/s00408-012-9389-5
91. Choi SM, Lee J, Park YS, Cho YJ, Lee CH, Lee SM, et al. Postoperative pulmonary complications after surgery in patients with interstitial lung disease. Respiration (2014) 87:287–93. doi:10.1159/000357046
92. Hiwatari N, Shimura S, Takishima T, Shirato K. Bronchoalveolar lavage as a possible cause of acute exacerbation in idiopathic pulmonary fibrosis patients. Tohoku J Exp Med (1994) 174:379–86. doi:10.1620/tjem.174.379
93. Sakamoto K, Taniguchi H, Kondoh Y, Wakai K, Kimura T, Kataoka K, et al. Acute exacerbation of IPF following diagnostic bronchoalveolar lavage procedures. Respir Med (2012) 106:436–42. doi:10.1016/j.rmed.2011.11.006
94. Tomic R, Cortes-Puentes GA, Murugan P, Joo Kim H, Amin K, Dincer HE. Acute exacerbation of interstitial lung disease after cryobiopsy. J Bronchology Interv Pulmonol (2017) 24:319–22. doi:10.1097/LBR.0000000000000369
95. Casoni GL, Tomassetti S, Cavazza A, Colby TV, Dubini A, Ryu JH, et al. Transbronchial lung cryobiopsy in the diagnosis of fibrotic interstitial lung diseases. PLoS One (2014) 9:e86716. doi:10.1371/journal.pone.0086716
96. Jeon K, Chung MP, Lee KS, Chung MJ, Han J, Koh WJ, et al. Prognostic factors and causes of death in Korean patients with idiopathic pulmonary fibrosis. Respir Med (2006) 100:451–7. doi:10.1016/j.rmed.2005.06.013
97. Natsuizaka M, Chiba H, Kuronuma K, Otsuka M, Kudo K, Mori M, et al. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med (2014) 190:773–9. doi:10.1164/rccm.201403-0566OC
98. Parambil JG, Myers JL, Aubry MC, Ryu JH. Causes and prognosis of diffuse alveolar damage diagnosed on surgical lung biopsy. Chest (2007) 132:50–7. doi:10.1378/chest.07-0104
99. Seo Y, Abe S, Kurahara M, Okada D, Saito Y, Usuki J, et al. Beneficial effect of polymyxin B-immobilized fiber column (PMX) hemoperfusion treatment on acute exacerbation of idiopathic pulmonary fibrosis. Intern Med (2006) 45:1033–8. doi:10.2169/internalmedicine.45.6018
100. Bozso S, Sidhu S, Garg M, Freed DH, Nagendran J. Canada’s longest experience with extracorporeal membrane oxygenation as a bridge to lung transplantation: a case report. Transplant Proc (2015) 47:186–9. doi:10.1016/j.transproceed.2014.10.039
101. Rush B, Wiskar K, Berger L, Griesdale D. The use of mechanical ventilation in patients with idiopathic pulmonary fibrosis in the United States: a nationwide retrospective cohort analysis. Respir Med (2016) 111:72–6. doi:10.1016/j.rmed.2015.12.005
102. Inase N, Sawada M, Ohtani Y, Miyake S, Isogai S, Sakashita H, et al. Cyclosporin A followed by the treatment of acute exacerbation of idiopathic pulmonary fibrosis with corticosteroid. Intern Med (2003) 42:565–70. doi:10.2169/internalmedicine.42.565
103. Homma S, Sakamoto S, Kawabata M, Kishi K, Tsuboi E, Motoi N, et al. Cyclosporin treatment in steroid-resistant and acutely exacerbated interstitial pneumonia. Intern Med (2005) 44:1144–50. doi:10.2169/internalmedicine.44.1144
104. Sakamoto S, Homma S, Miyamoto A, Kurosaki A, Fujii T, Yoshimura K. Cyclosporin A in the treatment of acute exacerbation of idiopathic pulmonary fibrosis. Intern Med (2010) 49:109–15. doi:10.2169/internalmedicine.49.2359
105. Donahoe M, Valentine VG, Chien N, Gibson KF, Raval JS, Saul M, et al. Autoantibody-targeted treatments for acute exacerbations of idiopathic pulmonary fibrosis. PLoS One (2015) 10:e0127771. doi:10.1371/journal.pone.0127771
106. Abe S, Azuma A, Mukae H, Ogura T, Taniguchi H, Bando M, et al. Polymyxin B-immobilized fiber column (PMX) treatment for idiopathic pulmonary fibrosis with acute exacerbation: a multicenter retrospective analysis. Intern Med (2012) 51:1487–91. doi:10.2169/internalmedicine.51.6965
107. Abe S, Hayashi H, Seo Y, Matsuda K, Kamio K, Saito Y, et al. Reduction in serum high mobility group box-1 level by polymyxin B-immobilized fiber column in patients with idiopathic pulmonary fibrosis with acute exacerbation. Blood Purif (2011) 32:310–6. doi:10.1159/000330325
108. Oishi K, Aoe K, Mimura Y, Murata Y, Sakamoto K, Koutoku W, et al. Survival from an acute exacerbation of idiopathic pulmonary fibrosis with or without direct hemoperfusion with a polymyxin B-immobilized fiber column: a retrospective analysis. Intern Med (2016) 55:3551–9. doi:10.2169/internalmedicine.55.6056
109. Oishi K, Mimura-Kimura Y, Miyasho T, Aoe K, Ogata Y, Katayama H, et al. Association between cytokine removal by polymyxin B hemoperfusion and improved pulmonary oxygenation in patients with acute exacerbation of idiopathic pulmonary fibrosis. Cytokine (2013) 61:84–9. doi:10.1016/j.cyto.2012.08.032
110. Isshiki T, Sakamoto S, Kinoshita A, Sugino K, Kurosaki A, Homma S. Recombinant human soluble thrombomodulin treatment for acute exacerbation of idiopathic pulmonary fibrosis: a retrospective study. Respiration (2015) 89:201–7. doi:10.1159/000369828
111. Tsushima K, Yamaguchi K, Kono Y, Yokoyama T, Kubo K, Matsumura T, et al. Thrombomodulin for acute exacerbations of idiopathic pulmonary fibrosis: a proof of concept study. Pulm Pharmacol Ther (2014) 29:233–40. doi:10.1016/j.pupt.2014.04.008
112. Kataoka K, Taniguchi H, Kondoh Y, Nishiyama O, Kimura T, Matsuda T, et al. Recombinant human thrombomodulin in acute exacerbation of idiopathic pulmonary fibrosis. Chest (2015) 148:436–43. doi:10.1378/chest.14-2746
113. Hayakawa S, Matsuzawa Y, Irie T, Rikitake H, Okada N, Suzuki Y. Efficacy of recombinant human soluble thrombomodulin for the treatment of acute exacerbation of idiopathic pulmonary fibrosis: a single arm, non-randomized prospective clinical trial. Multidiscip Respir Med (2016) 11:38. doi:10.1186/s40248-016-0074-z
114. Abe M, Tsushima K, Matsumura T, Ishiwata T, Ichimura Y, Ikari J, et al. Efficacy of thrombomodulin for acute exacerbation of idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia: a nonrandomized prospective study. Drug Des Devel Ther (2015) 9:5755–62. doi:10.2147/DDDT.S90739
115. Ding J, Chen Z, Feng K. Procalcitonin-guided antibiotic use in acute exacerbations of idiopathic pulmonary fibrosis. Int J Med Sci (2013) 10:903–7. doi:10.7150/ijms.4972
116. Yokoyama T, Kondoh Y, Taniguchi H, Kataoka K, Kato K, Nishiyama O, et al. Noninvasive ventilation in acute exacerbation of idiopathic pulmonary fibrosis. Intern Med (2010) 49:1509–14. doi:10.2169/internalmedicine.49.3222
117. Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2005) 171:1040–7. doi:10.1164/rccm.200404-571OC
118. King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med (2014) 370:2083–92. doi:10.1056/NEJMoa1402582
119. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet (2011) 377:1760–9. doi:10.1016/S0140-6736(11)60405-4
120. Ley B, Swigris J, Day BM, Stauffer JL, Raimundo K, Chou W, et al. Pirfenidone reduces respiratory-related hospitalizations in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2017) 196(6):756–61. doi:10.1164/rccm.201701-0091OC
121. Iwata T, Yoshida S, Nagato K, Nakajima T, Suzuki H, Tagawa T, et al. Experience with perioperative pirfenidone for lung cancer surgery in patients with idiopathic pulmonary fibrosis. Surg Today (2015) 45:1263–70. doi:10.1007/s00595-014-1071-5
122. Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med (2011) 365:1079–87. doi:10.1056/NEJMoa1103690
123. Kreuter M, Koegler H, Trampisch M, Geier S, Richeldi L. Efficacy of nintedanib on acute exacerbations reported as serious adverse events in the INPULSIS® trials in idiopathic pulmonary fibrosis (IPF). American Thoracic Society 2016 International Conference. (Vol. 193), San Francisco: Am J Respir Crit Care Med (2016).
124. Idiopathic Pulmonary Fibrosis Clinical Research Network Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr, Raghu G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med (2014) 370:2093–101. doi:10.1056/NEJMoa1401739
125. Idiopathic Pulmonary Fibrosis Clinical Research Network Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, et al. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med (2010) 363:620–8. doi:10.1056/NEJMoa1002110
126. King TE Jr, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2011) 184:92–9. doi:10.1164/rccm.201011-1874OC
127. King TE Jr, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet (2009) 374:222–8. doi:10.1016/S0140-6736(09)60551-1
128. Noth I, Anstrom KJ, Calvert SB, de Andrade J, Flaherty KR, Glazer C, et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2012) 186:88–95. doi:10.1164/rccm.201202-0314OC
129. Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med (2013) 158:641–9. doi:10.7326/0003-4819-158-9-201305070-00003
130. Daniels CE, Lasky JA, Limper AH, Mieras K, Gabor E, Schroeder DR, et al. Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo-controlled trial results. Am J Respir Crit Care Med (2010) 181:604–10. doi:10.1164/rccm.200906-0964OC
131. Idiopathic Pulmonary Fibrosis Clinical Research Network Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med (2012) 366:1968–77. doi:10.1056/NEJMoa1113354
132. Papiris SA, Kagouridis K, Kolilekas L, Papaioannou AI, Roussou A, Triantafillidou C, et al. Survival in idiopathic pulmonary fibrosis acute exacerbations: the non-steroid approach. BMC Pulm Med (2015) 15:162. doi:10.1186/s12890-015-0146-4
133. Lamas DJ, Kawut SM, Bagiella E, Philip N, Arcasoy SM, Lederer DJ. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med (2011) 184:842–7. doi:10.1164/rccm.201104-0668OC
Keywords: acute exacerbation, interstitial lung disease, idiopathic pulmonary fibrosis, definition, diagnosis, management
Citation: Leuschner G and Behr J (2017) Acute Exacerbation in Interstitial Lung Disease. Front. Med. 4:176. doi: 10.3389/fmed.2017.00176
Received: 27 July 2017; Accepted: 02 October 2017;
Published: 23 October 2017
Edited by:
Argyrios Tzouvelekis, Alexander Fleming Biomedical Sciences Research Center, GreeceReviewed by:
Venerino Poletti, Aarhus University Hospital, DenmarkJustin M. Oldham, University of California, Davis, United States
Copyright: © 2017 Leuschner and Behr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriela Leuschner, Z2FicmllbGEubGV1c2NobmVyQG1lZC51bmktbXVlbmNoZW4uZGU=