 András Tamás Mészáros
András Tamás Mészáros Ágnes Lilla Szilágyi
Ágnes Lilla Szilágyi László Juhász
László Juhász Eszter Tuboly
Eszter Tuboly Dániel Érces
Dániel Érces Gabriella Varga
Gabriella Varga Petra Hartmann*
Petra Hartmann*
- Institute of Surgical Research, University of Szeged, Szeged, Hungary
This review summarizes the current knowledge on the role of mitochondria in the context of hypoxic cell biology, while providing evidence of how these mechanisms are modulated by methane (CH4). Recent studies have unambiguously confirmed CH4 bioactivity in various in vitro and in vivo experimental models and established the possibility that CH4 can affect many aspects of mitochondrial physiology. To date, no specific binding of CH4 to any enzymes or receptors have been reported, and it is probable that many of its effects are related to physico-chemical properties of the non-polar molecule. (i) Mitochondria themselves can be sources of endogenous CH4 generation under oxido-reductive stress conditions; chemical inhibition of the mitochondrial electron transport chain with site-specific inhibitors leads to increased formation of CH4 in eukaryote cells, in plants, and in animals. (ii) Conventionally believed as physiologically inert, studies cited in this review demonstrate that exogenous CH4 modulates key events of inflammation. The anti-apoptotic effects of exogenously administered CH4 are also recognized, and these properties also suggest that CH4-mediated intracellular signaling is closely associated with mitochondria. (iii) Mitochondrial substrate oxidation is coupled with the reduction of molecular oxygen, thus providing energy for cellular metabolism. Interestingly, recent in vivo studies have shown improved basal respiration and modulated mitochondrial oxidative phosphorylation by exogenous CH4. Overall, these data suggest that CH4 liberation and effectiveness in eukaryotes are both linked to hypoxic events and redox regulation and support the notion that CH4 has therapeutic roles in mammalian pathophysiologies.
Introduction
Methane (CH4) is a small omnipresent molecule, the simplest alkane, and the most abundant organic gas in the atmosphere (1). It can help control the amount of hydroxyl radicals and neutralizes ozone in the troposphere (2) and also plays a role in global warming. More importantly, CH4 can be synthesized biologically and several recent studies have revealed its bioactivity. In a pioneering study, radioactive 14C-labeled CH4 was administered to the systemic circulation of sheep, and radioactive carbon dioxide (CO2) was detected in the breath of animals. Since apart from burning, non-biological decomposition of CH4 would need high temperature (above 1,000°C) or catalysts, recovery of 14CO2 in the exhaled air suggest involvement of CH4 in the cellular metabolism. Unfortunately, the underlying mechanisms were never resolved (3). In line with the data of Dougherty and coworkers, in a pilot study both 3H- and 14C-labeled CH4 was administered to rats. The subsequent analyses revealed organ-dependent rates of retention and decomposition CH4 (4, 5), which was interpreted as involvement in the carbon metabolism.
In the past decades, several signaling cascades of small gaseous molecules, including nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S), have been recognized. These gases play vital roles in biological systems, and they are in the focus of research interest (5). Due to its characteristics, availability and effectiveness, CH4 became also a candidate gasotransmitter (6). The first reports about the protective effect of CH4 against oxidative stress and inflammation caused by ischemia and reperfusion (IR) (7, 8) was followed by several studies (9–12). Because of its wide-ranging protective effects in many diverse disease models, it was proposed that CH4 could be a new medical gas (13). Indeed, CH4 is intrinsically non-toxic, without any known side effects. However, before the use in human clinical settings, the specific mechanism of action needs to be elucidated.
The mitochondrion is feasible target. Mitochondria are specialized subcellular structures that power various physiological roles, such as energy production, reactive oxygen species (ROS) formation, calcium homeostasis, and intrinsic apoptosis, all of which may be targets of CH4 administration (3, 4). A previous review summarized the available findings on the biological role of CH4, and it was proposed that CH4 liberation is related to hypoxic events resulting in, or associated with mitochondrial dysfunction (6). Indeed, several studies have demonstrated that the effects of exogenously administered CH4 in IR injuries can be grouped around a typical triad, namely anti-inflammatory, anti-oxidative, and anti-apoptotic properties. Notably, all these changes are also associated with mitochondrial functions, probably via non-specific physico-chemical alterations of membranes.
Importantly, concentrations of exogenously applied CH4 are orders of magnitude higher than those reported for endogenous production (reviewed in Section “Endogenous CH4 Formation Is Associated with Mitochondrial Dysfunction”). Due to the low solubility of CH4 in the watery phase, the majority of the gas is exhaled while CH4 is enriched at biological membrane interfaces, leading to higher local concentrations. Therefore, no direct conclusion can be made about the role of CH4 as a messenger only based on studies with CH4 treatment.
Overall, this review summarizes the effects of CH4 on mitochondria along with the current knowledge and the best available evidences on the possible mode of action. First, mitochondria are discussed as sources of endogenous CH4 generation under oxido-reductive stress conditions. Next, the consequences of exogenous CH4 supplementation are outlined: how it modulates key events of inflammation that are associated with mitochondrial functions. Thereafter, CH4-mediated intracellular signaling events are overviewed that are likely involved in cellular protection. Finally, the impact on the mitochondrial substrate oxidation in relationship with the anti-apoptotic effects is discussed.
Endogenous CH4 Formation is Associated with Mitochondrial Dysfunction
Mammalian methanogenesis has been considered an exclusive attribute of methanogenic Archaea, a group well distinguished from bacteria and eukaryotes. Nevertheless, to date, a number of studies have demonstrated the generation of non-bacterial CH4 also in aerobic living systems, and it has also been proposed that the CH4-producing phenomenon can be linked to the loss of the redox homeostasis.
It was shown in 2003 that hypoxia could lead to the generation of measurable amounts of non-bacterial CH4 in isolated liver mitochondria (14). Increasingly, high amounts of CH4 (between 0 and 2.3 nmol/mg protein) were generated after the addition of ascorbic acid and 1–100 mM hydrogen peroxide (H2O2), and the formation of CH4 was related linearly to the quantity of mitochondria incubated. A breakthrough came when Keppler and colleagues (15) provided direct evidence of CH4 generation in multi-cellular organisms under aerobic conditions. This key paper was followed by many studies that either supported or disagreed with the initial findings (16–19) where the common denominator was likely to be mitochondrial dysfunction. More importantly, it has been shown that oxido-reductive stress elicits aerobic CH4 emission in plants (20).
Interestingly, in 2008, a study by Ghyczy et al. (21) demonstrated aerobic CH4 emission in cultured endothelial cells exposed to hypoxia and metabolic distress. Mitochondrial dysfunction in this setting led to significant CH4 generation (~2–23 nmol/mg mitochondrial protein), depending on the nature and intensity of the metabolic distress, and a similarly high and dose-dependent CH4 generation was detected after ROS attack in the Udenfriend reaction.
Furthermore, it has been shown that the CH4-producing phenomenon can be mimicked by the administration of sodium azide (NaN3), a compound known to disrupt mitochondrial electron transport flow by specifically binding to cytochrome c (Cyt c) oxidase. In this in vivo study, the whole-body CH4 production profile was determined in unrestrained animals after chronic NaN3 administration (22). In this scenario, the stress-related methanogenic capacity of the rats was revealed in animals treated with antibiotics to eradicate the CH4-producing intestinal flora (22). In a model of transient mitochondrial distress, the CH4 generation of rats and healthy human volunteers was evaluated before and after excessive ethanol intake, and significant CH4 production was demonstrated in both species (23). The phenomenon was again independent from the activity of methanogenic prokaryotes (23).
The CH4-generating capacity of NaN3 administration may also be associated with the generation of ROS (24, 25). It has been hypothesized that electrophylic methyl groups of biomolecules such as the phosphatidylcholine molecule might be carbon precursors (14, 18) and a potential source of CH4 liberation. Nevertheless, the underlying mechanism is complicated by the various oxido-reductive stress answers of the mitochondria, e.g., throughout DNA methylation patterns and therefore gene expression changes. Such conditions can both affect gene expressions and activity of S-adenosylhomocysteine hydrolase (26) and may change the methionine metabolism, where CH4 may be liberated as an intermediate compound. Recently, Althoff et al. (27) presented a novel chemical reaction that readily forms CH4 from organosulphur compounds such as methionine, under highly oxidative conditions, ambient atmospheric pressure, and temperature. In this reaction, methyl sulfides are oxidized to the corresponding sulphoxides by a ferryl species, then, in the next phase, demethylation of the sulfoxide via homolytic bond cleavage leads to CH4 formation (27).
Novel Signaling Pathways Involved in CH4-Mediated Nuclear and Mitochondrial Effects
CH4 might alter the pattern of the activation of various signal transduction pathways and vice versa, the well-described triple effect (i.e., anti-inflammatory, antioxidant, and anti-apoptotic) of CH4 may influence the upregulation and downregulation of cellular signaling cascades. A direct link between exogenously administered CH4 and signaling targets have been reported recently (28, 29); however, a crosstalk with other bioactive gases and pathways cannot be excluded. Furthermore, no specific binding of CH4 to any enzymes or receptors have been reported to date, and it is highly probable that many (if not all) of its effects are related to physico-chemical properties of the non-polar molecule.
In a recent study by Wang et al. (29, 30), a supersaturated (~1.5 mmol/l) CH4-enriched saline solution was administered in a rat model of IR injury. High tissue concentrations of CH4 (between 90 and 145 µmol/g) were achieved, which lead to increased expression of Nrf2 (also known as nuclear factor erythroid 2-related factor 2). Nrf2 undoubtedly plays a central role in the activation of antioxidant defense system in most living organisms. It is now well characterized that Nrf2 translocates and binds to the antioxidant response element (ARE) forming a complex in the nucleus and induces the expression genes antioxidant and detoxifying enzymes (Figure 1) (31, 32). A negative regulator of Nrf2–ARE pathway is the kelch-like ECH associating protein 1 (Keap1), which forms a cytoplasmic complex with Nrf2 and inhibits its translocation to the nucleus under basal conditions (33).
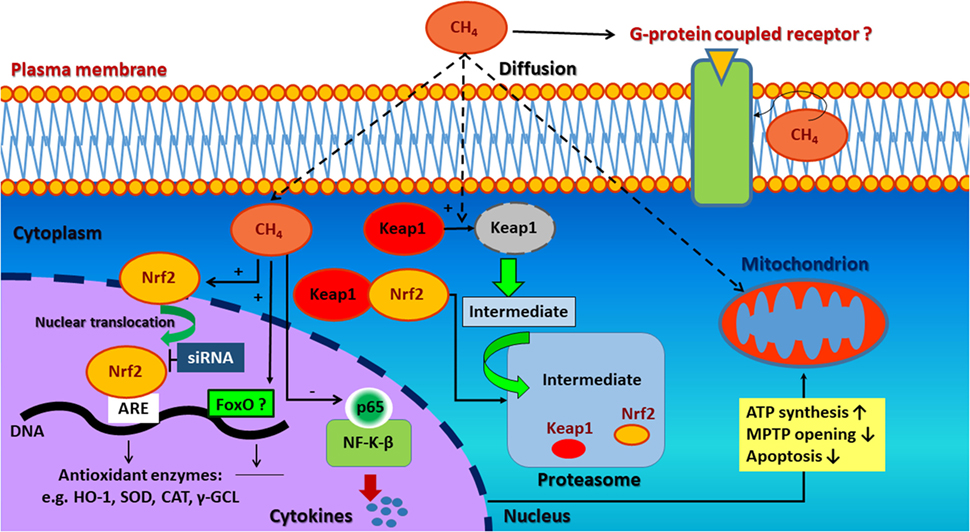
Figure 1. Possible signaling pathways involved in the antioxidant, anti-apoptotic, and anti-inflammatory effect of methane. CH4 may induce Nrf2/ARE-mediated activation of antioxidant and detoxifying enzymes. These attenuate the excessive production of reactive oxygen species (ROS) resulting preserved mitochondrial function as well as anti-inflammatory and anti-apoptotic effects. Second, complementary antioxidant pathways (e.g., FoxO) are also hypothesized to be activated. The non-polar nature of CH4 may influence cell membrane permeability and ion channel function-related signal transductionas well. Nrf2, nuclear factor erythroid 2-related factor 2; ARE, antioxidant response element; HO-1, hem oxygenase-1; SOD, superoxide dismutase; CAT, catalase; γ-GCL, γ-glutamyl cysteine ligase; siRNA, small-interfering RNA; FoxO, forkhead box transcription factor class O; CH4, methane; Keap1, kelch-like ECH associating protein 1; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; p65, transcription factor p65.
Wang and coworkers (29, 30) have found that CH4 administration enhances Nrf2 expression at both the mRNA and protein levels. In this study, marked elevations in Nrf2 mRNA and protein levels were found while the regulatory Keap1 protein was degraded in a time dependent manner. Besides, activation of phosphatidylinositol 3-kinase–Akt pathway, an indirect mechanism involved in the synergistic activation of Nrf2–ARE oxidative stress response, may also play a role in CH4 action (28, 34). As a result of CH4-upregulated Nrf2/ARE signaling, activated downstream enzymes [e.g., hem oxygenase-1, superoxide dismutase (SOD), catalase, and γ-glutamyl cysteine ligase] attenuate the excessive production of ROS and result in preserved mitochondrial function (35) as well as anti-inflammatory (36–38) and anti-apoptotic effects (39, 40).
Among several transduction pathways, the forkhead box transcription factor class O (FoxO)-related antioxidant enzyme induction seems to be a novel candidate in the protective effects of CH4 (41). Although FoxO in cooperation with tumor suppressor p53 (42) regulates cell cycle arrest, it is also responsible for ROS elimination by promoting the expression of numerous antioxidant genes and detoxifying enzymes (43); such as SOD, catalase, glutathione peroxidase 2, glutathione-S-transferase, and a sulfiredoxin (42). Moreover, it may influence mitochondrial homeostasis (44) through the modulation a serine/threonine-protein kinase, PTEN-induced putative kinase, thereby contributing to cell survival. Nevertheless, in the absence of data confirming that CH4 acts on FoxO pathway (45) in animal disease models, this remains only a hypothesis.
CH4-Mediated Actions on the Mitochondrial Electron Transport System (ETS)
CH4 has favorable distribution characteristics by penetrating membranes and diffusing into organelles including mitochondria (46); therefore, a potential effect of CH4 on the mitochondrial respiration has also been emerged. Upon exogenous administration, CH4 gets into the bloodstream through the alveoli of the lung and dissolves in the plasma. The cytoplasm/plasma solubility is near uniform for CH4; however, this ratio is much higher in hydrophobic substances such as the phospholipid biomembranes of mitochondria (47, 48). Of interest, the protein complexes of the mitochondrial respiratory chain are partially embedded in the inner mitochondrial membrane, exposing parts of them to the hydrophobic lipid bilayer, which makes them potential targets of the CH4.
The modulator effects of small gaseous molecules on mitochondrial respiration have been demonstrated in animal models of IR injury both in vivo and in hypoxic assays in vitro (11, 30, 49–51). Specifically, NO exerted protection through the activation of the mitochondrial KATP channel opening (50) and induced a sustained mitochondrial depolarization (49). H2S preserved mitochondrial membrane integrity and the complex I- and II-linked oxygen consumption rate (51). CH4 restored the ADP-dependent mitochondrial respiration, i.e., the oxydative phosphorylation (11, 30). During oxygen deprivation, the mitochondrial ETS is manifested lower rates of non-phosphorylating basal respiration. In addition, the ADP-dependent oxygen consumption, or in other words oxidative phosphorylation, is significantly depressed. In contrast, reperfusion conditions induce leakage of electrons from the ETS into the intermembranous space (52) that leads to increased ROS formation. These results suggest the sensitivity of both the resting state of ETS and the mitochondrial bioenergetic function to IR injury.
General effects of CH4 on IR-related mitochondrial dysfunction involve the restoration of the electron transport machinery of the inner mitochondrial membrane when oxygen concentration rises. In a study by Strifler et al. (11), mitochondria incubated in a medium with a gas phase containing 2.2% CH4–air mixture displayed a significantly improved leak respiration and increased recovery of oxidative phosphorylation. These effects of CH4 on the inner mitochondrial membrane can be explained with a hypothesis which presumes that CH4 dissolves in biological membranes thereby changing its oxidative stress-related rigidity (7).
In parallel with the general effects on the mitochondrial ETS, CH4 seems to exert site-specific action on protein complexes. Among the protein complexes of the mitochondrial ETS, complex IV (cytochrome c oxidase), which catalyzes the reduction of oxygen by ferrycytochrome to H2O, is target of the CH4 action. Indeed, endogenous CH4 generation occurs in plant mitochondria (53) and in mammalian cells after inhibition of complex IV by NaN3 (22). Meanwhile, exogenous CH4 administration in IR injury conditions resulted in reduced Cyt c release from the inner mitochondrial membrane and lower Cyt c oxidase activity in liver mitochondria (11).
Nonetheless, the above observations are all related to the in vivo effects of exogenous CH4 supplementation on mitochondria in oxido-reductive stress conditions. Interestingly, direct mitochondrial effects could not be shown when a 2.2% CH4–air mixture was administered isolated mitochondria in vitro (11). In other words, direct effect of CH4 on oxidative phosphorylation capacity and leak respiration in intact liver mitochondria cannot be shown in vitro. CH4 has relatively low solubility in watery phase, ranging from 37.2 to 19.1 mg/l between 0 and 35°C at 1 standard atmosphere. Therefore, high concentrations should be applied exogenously to overcome the limitation of low gas solubility. This means that approximately 1 mmol/l/min CH4 was usually applied as gas therapy (7) and 1.6 mmol/l in CH4-enriched saline (29). Consequently, CH4 concentrations in tissues upon CH4 treatment are certainly higher than the average endogenous levels. Still, due to the high affinity of Cyt c oxidase for oxygen (54), CH4 does not dysproportionate oxygen and thus does not limit mitochondrial respiration.
CH4-Mediated Actions on Apoptosis
The anti-apoptotic effect is one of the most studied properties of CH4. Several studies demonstrated that CH4 modulates the intrinsic pathway of apoptosis (9–11, 29, 30, 55–58). The intrinsic pathway of apoptosis, also called the mitochondrial pathway owing to the essential involvement of mitochondria (59), which are not only the site where anti- and pro-apoptotic proteins interact but also the origins of high range of signal pathways that initiate the activation of caspases through various mechanisms (60). The large family of Bcl-2 homologs involves key proteins of intrinsic apoptosis described to organize the process (61). They can be divided into two classes: anti-apoptotic Bcl-2 family proteins (such as Bcl-XL, Bcl-w, Mcl-1, A1, Bcl-Rambo, Bcl-L10, and Bcl-G) and pro-apoptotic proteins [such as Bcl-2 associated X protein (Bax), Bak, and Bok] (62). The primary anti-apoptotic function of Bcl-2 is to block the release of Cyt c. In contrast, upon stress, pro-apoptotic members of the Bcl-2 family are activated (Bak or Bax) which leads to the mitochondrial outer membrane permeabilization and subsequent release of intermembrane space proteins such as Cyt c. Cyt c is attached to the inner mitochondrial membrane and shuttles electrons between complex III and complex IV. In response to membrane damage, it releases to the cytosol and activate the initiator procaspase-9 within the apoptotic protease-activating factor-1 (Apaf-1) apoptosome complex (63). Once activated, caspase-9 activates effector procaspase-3 which, in turn, can cleave various protein substrates, leading to the morphological and biochemical features of apoptosis (64).
In IR, CH4 supplementation may improve the level of key regulators of apoptosis, such as Bcl-2, Apaf-1, and caspases indirectly through an unknown pathway (10). Two recent studies of experimental retinal IR provided evidence for the effectiveness of CH4 treatment on upregulation of anti-apoptotic Bcl-2 proteins, while reduced cleaved caspase 3- and 9 quantities (10, 57). Likewise, in an experimental spinal cord IR scenario, CH4 was proven to significantly reduce the number of apoptotic cells (confirmed by TUNEL staining) and also the amount of cleaved caspase-3 and caspase-9 proteins and to attenuate cytosolic Cyt c release (29). In addition, CH4-rich fluids significantly reduced the expression of cleaved caspase-3 and also decreased the apoptotic cell number in rodents exposed to LPS-induced acute lung injury (58). In a hepatic IR model, CH4 inhalation could effectively attenuate the apoptosis-linked morphological changes in the liver and the TUNEL positivity of hepatocytes (11). Similarly, caspase-3 expression was attenuated with CH4-rich saline treatment which substantiated this finding (56). Moreover, CH4-rich saline has pronounced neuroprotective effect in diabetic retinopathy in streptozotocin-induced diabetes, possibly by upregulating those cell cycle related microRNAs, which contribute to post-transcriptional regulation (65). A possible indirect way how CH4 supplementation modulates apoptosis is to reduce the level of mitochondrial ROS formation. ROS and their by-products can oxidize the reduced thioredoxin-apoptosis signal-regulating kinase 1 complex, then activate apoptosis signal-regulating kinase and its downstream stress signaling targets, such as c-Jun NH(2)-terminal kinase (66). Another plausible explanation for the anti-apoptotic effect of CH4 is reducing Cyt c release from the inner membrane, which has already been demonstrated in hepatic, myocardial, and spinal cord IR as well (9, 11, 29) (Figure 2).
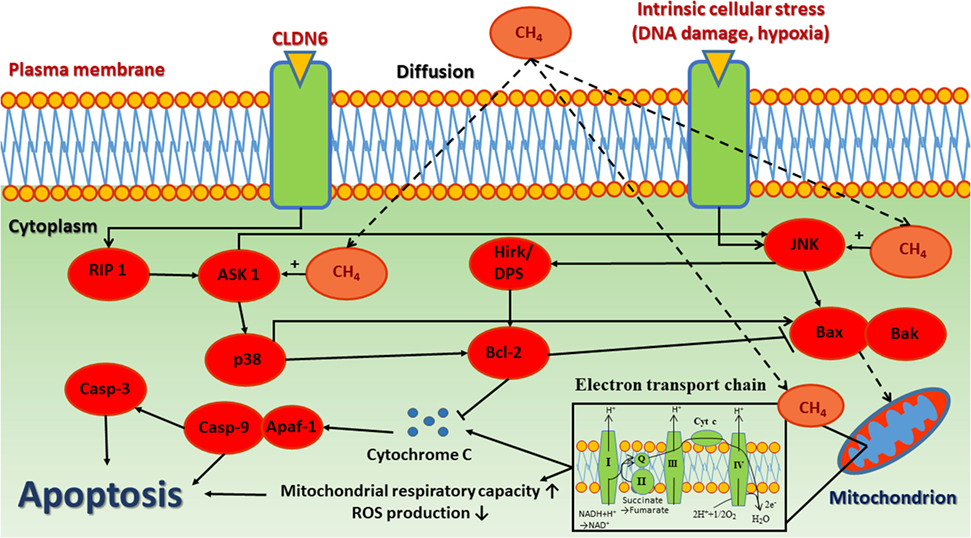
Figure 2. The anti-apoptotic effect of CH4. The release of cytochrome c and other inner mitochondrial membrane proteins are regulated by Bcl-2 family proteins through interplay between pro-apoptotic and anti-apoptotic proteins, which converge to Bax/Bak activation, thereby inducing mitochondrial outer membrane permeability. CH4, methane; CLDN6, claudin 6; RIP 1, receptor-interacting kinase 1; ASK-1, apoptosis signal-regulating kinase 1; p-38, mitogen-activated protein kinase; Casp-3, caspase 3; Casp-9, caspase 9; Apaf-1, apoptotic protease-activating factor 1; Hrk/DP5, harakiri gene; Bcl-2, B-cell lymphoma 2 regulation protein; JNK, c-Jun N-terminal kinase; BAX, Bcl-2 associated X protein; BAK, Bcl-2 homologous antagonist/killer.
Perspectives and Concluding Statements
In the human body, many gases are biologically active. Signaling roles were demonstrated for NO, CO, and H2S, and it has become clear that gaseous mediators form complex intracellular pathways and regulate numerous physiological processes, separately, or more often, in antagonistic or synergistic ways. CH4 is a small, less reactive gas molecule, having a close symbiosis with bioactive gases in the intracellular spaces. The effects of exogenous CH4 were clearly illustrated in detail in various tissues under different conditions. Of particular interest is that the recognized biological effects of CH4 are not cell- or tissue specific, and an increased input may result in anti-inflammatory changes in cells and tissues. In this regard, it is tempting to speculate on a much broader, controller role for CH4 in acute and chronic oxido-reductive stress conditions.
Author Contributions
AM and PH designed and developed the concept of the manuscript. AS, LJ, ET, DÉ, and GV wrote the manuscript. ÁS prepared Figure 2. LJ prepared Figure 1. PH supervised and edited the manuscript. All authors discussed and commented on the manuscript at all stages.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer UA and handling editor declared their shared affiliation.
Funding
This work was supported by Hungarian National Research, Development and Innovation Office (NKFIH) grant K120232 and K116861, EFOP-3.6.2-16-2017-00006, GINOP 2.3.2-15-2016-00015 and New National Excellence Program of the Ministry of Human Capacities UNKP-17-4. Nemzeti Kutatási és Technológiai Hivatal 10.13039/501100003827.
References
1. Wuebbles DJ, Hayhoe K. Atmospheric methane and global change. Earth Sci Rev (2002) 57:177–210. doi:10.1016/S0012-8252(01)00062-9
2. Fiore AM, West JJ, Horowitz LW, Naik V, Schwarzkopf MD. Characterizing the tropospheric ozone response to methane emission controls and the benefits to climate and air quality. J Geophys Res (2008) 113:D08307. doi:10.1029/2007JD009162
3. Dougherty RW, O’Toole JJ, Allison MJ. Oxidation of intra-arterially administered carbon 14-labelled methane in sheep. Proc Soc Exp Biol Med (1967) 124(4):1155–7. doi:10.3181/00379727-124-31949
4. Carlisle SM, Burchart PA, McCauley C, Surette RA. Biokinetics of inhaled radioactive methane in rats: a pilot study. Appl Radiat Isot (2005) 62:847–60. doi:10.1016/j.apradiso.2005.01.010
5. Smith RA, Murphy MP. Mitochondria-targeted antioxidants as therapies Discov Med (2011) 11:106–14.
6. Boros M, Tuboly E, Mészáros A, Amann A. The role of methane in mammalian physiology-is it a gasotransmitter? J Breath Res (2015) 9:014001. doi:10.1088/1752-7155/9/1/014001
7. Boros M, Ghyczy M, Erces D, Varga G, Tokes T, Kupai K, et al. The anti-inflammatory effects of methane. Crit Care Med (2012) 40:1269–78. doi:10.1097/CCM.0b013e31823dae05
8. Varga G, Erces D, Tuboly E, Kaszaki J, Ghyczy M, Boros M. Characterization of the antiinflammatory properties of methane inhalation during ischaemia-reperfusion. Magy Seb (2012) 65:205–11. doi:10.1556/MaSeb.65.2012.4.6
9. Chen O, Ye Z, Cao Z, Manaenko A, Ning K, Zhai X, et al. Methane attenuates myocardial ischemia injury in rats through anti-oxidative, anti-apoptotic and anti-inflammatory actions. Free Radic Biol Med (2016) 90:1–11. doi:10.1016/j.freeradbiomed.2015.11.017
10. Song K, Zhang M, Hu J, Liu Y, Liu Y, Wang Y, et al. Methane-rich saline attenuates ischemia/reperfusion injury of abdominal skin flaps in rats via regulating apoptosis level. BMC Surg (2015) 15:92. doi:10.1186/s12893-015-0075-4
11. Strifler G, Tuboly E, Szél E, Kaszonyi E, Cao C, Kaszaki J, et al. Inhaled methane limits the mitochondrial electron transport chain dysfunction during experimental liver ischemia-reperfusion injury. PLoS One (2016) 11:e0146363. doi:10.1371/journal.pone.0146363
12. Mészáros AT, Buki T, Fazekas B, Tuboly E, Horvath K, Poles MZ, et al. Inhalation of methane preserves the epithelial barrier during ischemia and reperfusion in the rat small intestine. Surgery (2017) 161:1696–709. doi:10.1016/j.surg.2016.12.040
13. Liu W, Wang D, Tao H, Sun X. Is methane a new therapeutic gas? Med Gas Res (2012) 2:25. doi:10.1186/2045-9912-2-25
14. Ghyczy M, Torday C, Boros M. Simultaneous generation of methane, carbon dioxide, and carbon monoxide from choline and ascorbic acid: a defensive mechanism against reductive stress? FASEB J (2003) 17:1124–6. doi:10.1096/fj.02-0918fje
15. Keppler F, Hamilton JT, Brass M, Röckmann T. Methane emissions from terrestrial plants under aerobic conditions. Nature (2006) 439:187–91. doi:10.1038/nature04420
16. McLeod AR, Fry SC, Loake GJ, Messenger DJ, Reay DS, Smith KA, et al. Ultraviolet radiation drives methane emissions from terrestrial plant pectins. New Phytol (2008) 180:124–32. doi:10.1111/j.1469-8137.2008.02571.x
17. Vigano I, van Weelden H, Holzinger R, Keppler F, McLeod A, Röckmann T. Effect of UV radiation and temperature on the emission of methane from plant biomass and structural components. Biogeosciences (2008) 5:937–47. doi:10.5194/bg-5-937-2008
18. Bruggemann N, Meier R, Steigner D, Zimmer I, Louis S, Schnitzler JP. Nonmicrobial aerobic methane emission from poplar shoot cultures under low-light conditions. New Phytol (2009) 182:912–8. doi:10.1111/j.1469-8137.2009.02797.x
19. Messenger DJ, McLeod AR, Fry SC. The role of ultraviolet radiation, photosensitizers, reactive oxygen species and ester groups in mechanisms of methane formation from pectin. Plant Cell Environ (2009) 32:1–9. doi:10.1111/j.1365-3040.2008.01892.x
20. Wang R. Gasotransmitters: growing pains and joys. Trends Biochem Sci (2014) 39:227–32. doi:10.1016/j.tibs.2014.03.003
21. Ghyczy M, Torday C, Kaszaki J, Szabo A, Czóbel M, Boros M. Hypoxia-induced generation of methane in mitochondria and eukaryotic cells: an alternative approach to methanogenesis. Cell Physiol Biochem (2008) 21:251–8. doi:10.1159/000113766
22. Tuboly E, Szabo A, Garab D, Bartha G, Janovszky A, Eros G, et al. Methane biogenesis during sodium azide-induced chemical hypoxia in rats. Am J Physiol Cell Physiol (2013) 304:C207–14. doi:10.1152/ajpcell.00300.2012
23. Tuboly E, Molnár R, Tőkés T, Turányi RN, Hartmann P, Mészáros A, et al. Excessive alcohol consumption induces methane production in humans and rats. Sci Rep (2017) 7(1):7329. doi:10.1038/s41598-017-07637-3
24. Smith TS, Bennett JP. Mitochondrial toxins in models of neurodegenerative diseases. I: in vivo brain hydroxyl radical production during systemic MPTP treatment or following microdialysis infusion of methylpyridinium or azide ions. Brain Res (1997) 765:183–8. doi:10.1016/S0006-8993(97)00429-0
25. Duranteau J, Chandel NS, Kulisz A, Shao Z, Schumacker PT. Intracellular signalling by reactive oxygen species during hypoxia in cardiomyocytes. J Biol Chem (1998) 273:11619–24. doi:10.1074/jbc.273.19.11619
26. Watson WH, Song Z, Kirpich IA, Deaciuc IV, Chen T, McClain CJ. Ethanol exposure modulates hepatic S-adenosylmethionine and S-adenosylhomocysteine levels in the isolated perfused rat liver through changes in the redox state of the NADH/NAD(+) system. Biochim Biophys Acta (2011) 1812:613–8. doi:10.1016/j.bbadis.2011.01.016
27. Althoff F, Benzing K, Comba P, McRoberts C, Boyd DR, Greiner S, et al. Abiotic methanogenesis from organosulphur compounds under ambient conditions. Nat Commun (2014) 24:4205. doi:10.1038/ncomms5205
28. Zhang X, Li N, Shao H, Meng Y, Wang L, Wu Q, et al. Methane limit LPS-induced NF-KB/MAPKs signal in macrophages and suppress immune response in mice by enhancing PI3K/AKT/GSK3β-mediated IL-10 expression. Sci Rep (2016) 6:29359. doi:10.1038/srep29359
29. Wang L, Yao Y, He R, Meng Y, Li N, Zhang D, et al. Methane ameliorates spinal cord ischaemia-reperfusion injury in rats: antioxidant, anti-inflammatory and anti-apoptotic activity mediated by Nrf2 activation. Free Rad Biol Med (2017) 103:69–89. doi:10.1016/j.freeradbiomed.2016.12.014
30. Wang R, Sun Q, Xia F, Chen Z, Wu J, Zhang Y, et al. Methane rescues retinal ganglion cells and limits retinal mitochondrial dysfunction following optic nerve crush. Exp Eye Res (2017) 159:49–57. doi:10.1016/j.exer.2017.03.008
31. Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem (1991) 266:11632–9.
32. Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun (1997) 236:313–22. doi:10.1006/bbrc.1997.6943
33. Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells (2003) 8:379–91. doi:10.1046/j.1365-2443.2003.00640.x
34. Lee JM, Hanson JM, Chu WA, Johnson JA. Phosphatidylinositol 3-kinase, not extracellular signal-regulated kinase, regulates activation of the antioxidant-responsive element in IMR-32 human neuroblastoma cells. J Biol Chem (2001) 276:20011–6. doi:10.1074/jbc.M100734200
35. Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med (2015) 88:179–88. doi:10.1016/j.freeradbiomed.2015.04.036
36. Pan H, Wang H, Wang X, Zhu L, Mao L. The absence of Nrf2 enhances NF-KB-dependent inflammation following scratch injury in mouse primary cultured astrocytes. Mediators Inflamm (2012) 2012:217580. doi:10.1155/2012/217580
37. Qin SY, Du RH, Yin SS, Liu XF, Xu GL, Cao W. Nrf2 is essential for the anti-inflammatory effect of carbon monoxide in LPS-induced inflammation. Inflamm Res (2015) 64:537–48. doi:10.1007/s00011-015-0834-9
38. Onasanwo SA, Velagapudi R, El-Bakoush A, Olajide OA. Inhibition of neuroinflammation in BV2 microglia by the biflavonoid kolaviron is dependent on the Nrf2/ARE antioxidant protective mechanism. Mol Cell Biochem (2016) 414:23–6. doi:10.1007/s11010-016-2655-8
39. Xu J, Huang G, Zhang K, Sun J, Xu T, Li R, et al. Nrf2 activation in astrocytes contributes to spinal cord ischemic tolerance induced by hyperbaric oxygen preconditioning. J Neurotrauma (2014) 31:1343–53. doi:10.1089/neu.2013.3222
40. Dwivedi S, Rajasekar N, Hanif K, Nath C, Shukla R. Sulforaphane ameliorates okadaic acid-induced memory impairment in rats by activating the Nrf2/HO-1 antioxidant pathway. Mol Neurobiol (2016) 53:5310–23. doi:10.1007/s12035-015-9451-4
41. Li H, Wang Y, Wang S, Chen O. Comments and hypotheses on the mechanism of methane against ischemia/reperfusion injury. Med Gas Res (2017) 7(2):120–3. doi:10.4103/2045-9912.208518
42. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov (2013) 12:931–47. doi:10.1038/nrd4002
43. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11:85–95. doi:10.1038/nrc2981
44. Mei Y, Zhang Y, Yamamoto K, Xie W, Mak TW, You H. FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signalling in response to cytokine deprivation. Proc Natl Acad Sci U S A (2009) 106:5153–8. doi:10.1073/pnas.0901104106
45. Li H, Wang J, Wang S, Chen O. Comments and hypotheses on the mechanism of methane against ischemia/reperfusion injury. Med Gas Res (2017) 7:120–3. doi:10.4103/2045-9912.208518
46. Pimentel M, Lin HC, Enayati P, van den Burg B, Lee HR, Chen JH, et al. Methane, a gas produced by enteric bacteria, slows intestinal transit and augments small intestinal contractile activity. Am J Physiol Gastrointest Liver Physiol (2006) 290:G1089–95. doi:10.1152/ajpgi.00574.2004
47. Miller KW, Hammond L, Porter EG. The solubility of hydrocarbon gases in lipid bilayers. Chem Phys Lipids (1977) 20:229–41. doi:10.1016/0009-3084(77)90039-1
48. Meyer M, Tebbe U, Piiper J. Solubility of inert gases in dog blood and skeletal muscle. Pflugers Arch (1980) 384:131–4. doi:10.1007/BF00584428
49. Rakhit R, Mojet M, Marber M, Duchen M. Mitochondria as targets for nitric oxide-induced protection during simulated ischemia and reoxygenation in isolated neonatal cardiomyocytes. Ciruculation (2001) 103:2617–23. doi:10.1161/01.CIR.103.21.2617
50. Bolli R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J Mol Cell Cardiol (2001) 33:1897–918. doi:10.1006/jmcc.2001.1462
51. Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A (2007) 104:15560–5. doi:10.1073/pnas.0705891104
52. Rose S, Frye RE, Slattery J, Wynne R, Tippett M, Melnyk S, et al. Oxidative stress induces mitochondrial dysfunction in a subset of autistic lymphoblastoid cell lines. Transl Psychiatry (2014) 4:e377. doi:10.1038/tp.2014.15
53. Wishkerman A, Greiner S, Ghyczy M, Boros M, Rausch T, Lenhart K, et al. Enhanced formation of methane in plant cell cultures by inhibition of cytochrome c oxidase. Plant Cell Environ (2011) 34:457–64. doi:10.1111/j.1365-3040.2010.02255.x
54. Krab K, Kempe H, Wikstrom M. Explaining the enigmatic K(M) for oxygen in cytochrome c oxidase: a kinetic model. Biochim Biophys Acta (2011) 3:348–58. doi:10.1016/j.bbabio.2010.12.015
55. Fan DF, Hu HJ, Sun Q, Lv Y, Ye ZH, Sun XJ, et al. Neuroprotective effects of exogenous methane in a rat model of acute carbon monoxide poisoning. Brain Res (2016) 1633:62–72. doi:10.1016/j.brainres.2015.12.019
56. Ye Z, Chen O, Zhang R, Nakao A, Fan D, Zhang T, et al. Methane attenuates hepatic ischemia/reperfusion injury in rats through antiapoptotic, anti-inflammatory, and antioxidative actions. Shock (2015) 44:181–7. doi:10.1097/SHK.0000000000000385
57. Liu L, Sun Q, Wang R, Chen Z, Wu J, Xia F, et al. Methane attenuates retinal ischemia/reperfusion injury via anti-oxidative and anti-apoptotic pathways. Brain Res (2016) 1646:327–33. doi:10.1016/j.brainres.2016.05.037
58. Sun A, Wang W, Ye X, Wang Y, Yang X, Ye Z, et al. Protective effects of methane-rich saline on rats with lipopolysaccharide-induced acute lung injury. Oxid Med Cell Longev (2017) 2017:7430193. doi:10.1155/2017/7430193
59. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol (2007) 35:495–516. doi:10.1080/01926230701320337
60. Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet (2009) 43:95–118. doi:10.1146/annurev-genet-102108-134850
61. Lindsay J, Esposti MD, Gilmore AP. Bcl-2 proteins and mitochondria – specificity in membrane targeting for death. Biochim Biophys Acta (2011) 1813:532–9. doi:10.1016/j.bbamcr.2010.10.017
62. Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer (2002) 2:647–56. doi:10.1038/nrc883
64. Shawgo ME, Shelton SN, Robertson JD. Caspase-mediated Bak activation and cytochrome c release during intrinsic apoptotic cell death in Jurkat cells. J Biol Chem (2008) 283:35532–8. doi:10.1074/jbc.M807656200
65. Wu J, Wang R, Ye Z, Sun X, Chen Z, Xia F, et al. Protective effects of methane-rich saline on diabetic retinopathy via anti-inflammation in a streptozotocin-induced diabetic rat model. Biochem Biophys Res Commun (2015) 466:155–61. doi:10.1016/j.bbrc.2015.08.121
66. Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, et al. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem (2005) 280:33917–25. doi:10.1074/jbc.M505818200
Keywords: methane, review, mitochondrion, apoptosis, gasotransmitter
Citation: Mészáros AT, Szilágyi ÁL, Juhász L, Tuboly E, Érces D, Varga G and Hartmann P (2017) Mitochondria As Sources and Targets of Methane. Front. Med. 4:195. doi: 10.3389/fmed.2017.00195
Received: 01 August 2017; Accepted: 25 October 2017;
Published: 13 November 2017
Edited by:
Mert Şentürk, Istanbul University, TurkeyReviewed by:
Victoria Bunik, Moscow State University, RussiaInge Bauer, University Hospital Duesseldorf, Germany
Ugur Aksu, Istanbul University, Turkey
Copyright: © 2017 Mészáros, Szilágyi, Juhász, Tuboly, Érces, Varga and Hartmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petra Hartmann, aGFydG1hbm4ucGV0cmFAbWVkLnUtc3plZ2VkLmh1