 Catharina C. Moor
Catharina C. Moor Peter Heukels
Peter Heukels Mirjam Kool
Mirjam Kool Marlies S. Wijsenbeek
Marlies S. Wijsenbeek- Department of Respiratory Medicine, Erasmus Medical Center, University Hospital Rotterdam, Rotterdam, Netherlands
Idiopathic pulmonary fibrosis (IPF) is a progressive and ultimately fatal disease which has a major impact on patients’ quality of life (QOL). Except for lung transplantation, there is no curative treatment option. Fortunately, two disease-modifying drugs that slow down disease decline were recently approved. Though this is a major step forward, these drugs do not halt or reverse the disease, nor convincingly improve health-related QOL. In daily practice, disease behavior and response to therapy greatly vary among patients. It is assumed that this is related to the multiple biological pathways and complex interactions between genetic, molecular, and environmental factors that are involved in the pathogenesis of IPF. Recently, research in IPF has therefore started to focus on developing targeted therapy through identifying genetic risk factors and biomarkers. In this rapidly evolving field of personalized medicine, patient factors such as lifestyle, comorbidities, preferences, and experiences with medication should not be overlooked. This review describes recent insights and methods on how to integrate patient perspectives into personalized medicine. Furthermore, it provides an overview of the most used patient-reported outcome measures in IPF, to facilitate choices for both researchers and clinicians when incorporating the patient voice in their research and care. To enhance truly personalized treatment in IPF, biology should be combined with patient perspectives.
Introduction
Give different ones [therapeutic drinks] to different patients, for the sweet ones do not benefit everyone, nor do the astringent ones, nor are all patients able to drink the same things Hippocrates (1)
Idiopathic pulmonary fibrosis (IPF) is the most common idiopathic interstitial pneumonia (2). IPF is characterized by progressive decline of lung function, with a median survival of only 3–5 years (3). Common symptoms as breathlessness, cough, and fatigue have a major impact on the quality of life (QOL) of patients (4). IPF occurs more often in men than women and usually affects elderly patients, aged 50 years and above (3). There are two approved anti-fibrotic drugs that slow down disease decline, but these drugs do not halt or reverse the disease, and ultimately IPF remains a fatal disease (5, 6). The heterogeneity in disease behavior and response to therapy in IPF has (further) stimulated research to identify possible distinct underlying genetic, molecular, and environmental factors associated with IPF (7, 8).
The potential to enhance personalized treatment has prompted excitement also in the IPF field (7). Until now, the focus of personalized medicine has been on physiology and the use of this biological information to predict response to treatment and to develop targeted therapy (9). In this process, patient factors should not be overlooked. For real personalized treatment patient perceptions and preferences should also be taken into account. In this article, we focus on recent insights and methods on how to integrate patient perspectives into personalized medicine.
Impact of Disease
Idiopathic pulmonary fibrosis is a heterogeneous disease, with a highly variable disease course (10, 11). Additionally, different phenotypes of IPF exist. Most patients have a slow disease progression, while some patients display relative stable periods followed by acute exacerbations and a small group of patients experiences a rapid decline in lung function (12). Uncertainty about the disease course and prognosis can cause emotional distress and anxiety, and, as a result, IPF has a major impact on most patients’ health-related quality of life (HRQOL). HRQOL can be defined as a patient’s perceived well-being affected by disease and treatment of the disease (13). IPF affects patients in almost every domain of life; hence, the burden of the disease is high, not just for patients but also for their partners and families. Patients often struggle with loss of independence because of functional limitations and deteriorating symptoms. Not only can breathlessness, cough, and fatigue diminish QOL, but also other symptoms such as sleep disorders, loss of appetite, and psychological problems can (14–18).
Most clinical trials in IPF that have been performed so far, have shown no convincing improvement of patient HRQOL (5, 6, 19). To date, the main focus in research has been to stabilize or improve physiological outcomes rather than HRQOL. Physiological parameters, such as lung function, do not correlate well with HRQOL measurements (20, 21). To our knowledge for parameters as imaging and biomarkers, relationships with HRQOL have not yet been established. Thus, decline in lung function does not adequately reflect the perceived impact of the disease on patients’ lives.
Every person has a different lifestyle, personal circumstances, and coping strategies. These factors can play an important role in how a disease manifests itself; hence, the same disease affects each person in a different way (16, 22, 23). Medication may show promising results at group level in randomized controlled trials, but still in some individual patients, treatment may fail (22). For example, the side effects of medication may outweigh the positive effects of medication in daily practice, or the burden of treatment might be too high for patients. To improve and personalize treatment of IPF, we should also include patient perspectives and QOL.
Personomics
Personalized, stratified, or precision medicine is a broad term which can be referred to as “delivering the right treatment to the right patient at the right time” (24). Personalized medicine has gained increasing attention during the past decade (22, 25). However, the concept is not new; Hippocrates already mentioned the importance of a personalized approach to diagnosis and treatment in the fifth century BC, stating that “individuality of human beings affects predisposition to disease and response to treatment,” and also noting that “not all patients are able to drink the same therapeutic drinks” (1, 26). His concepts already include the notion that experiences with treatment differ among patients. This idea is also acknowledged by Britten et al., who suggest that because individuals are more than their genetic profile, the main concept of stratified medicine is too limited at the moment (22). Personalized treatment comprises not only “biology,” but should also focus on patient perspectives, needs, experiences, personality, environment, lifestyle, and other personal circumstances (Figure 1) (9, 22). Accordingly, the term “personomics” has been introduced to capture a patient’s life circumstances that may alter disease behavior and response to treatment (23). Below we will briefly touch on the role of biology and other aspects of personalized medicine as shown in Figure 1, but the focus will be on patient perspectives.
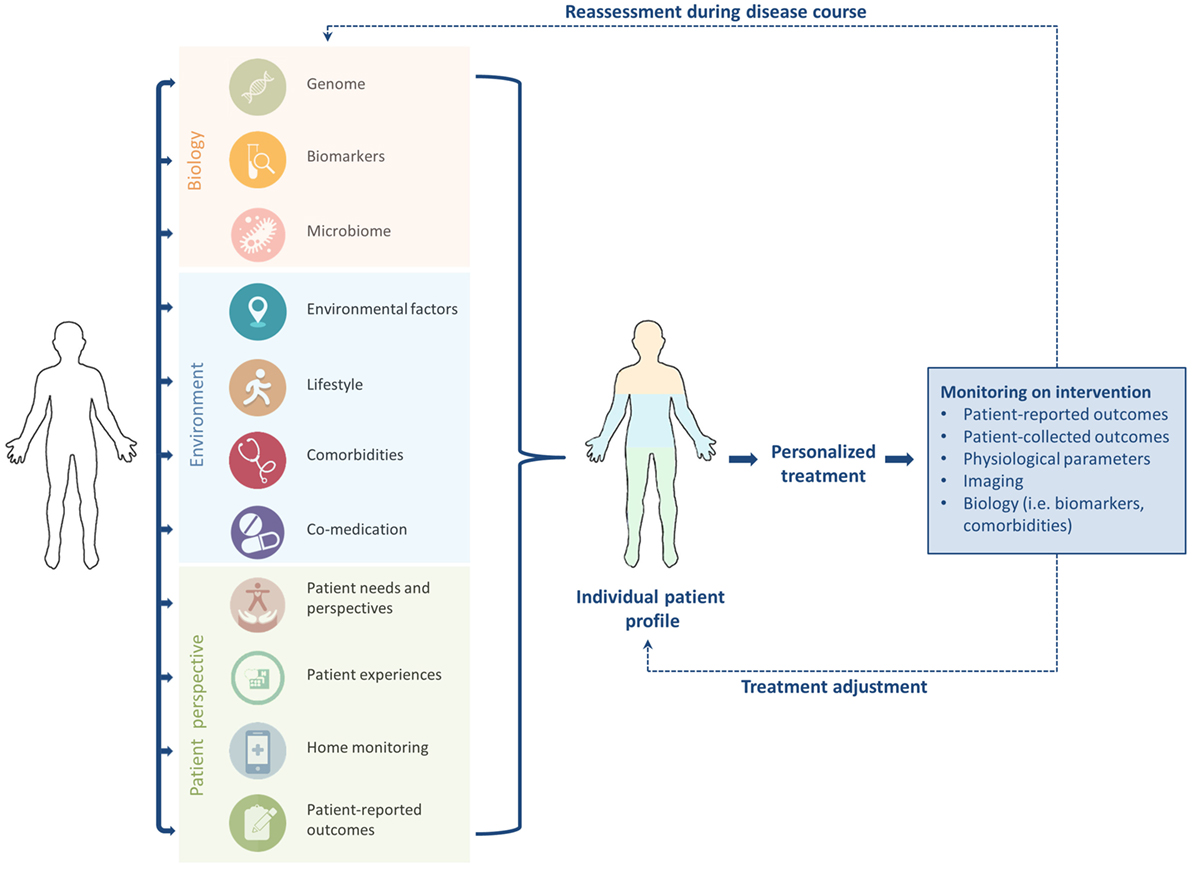
Figure 1. To enhance tailored treatment in idiopathic pulmonary fibrosis, “biology” should be combined with patient factors to generate an individual patient profile. Close monitoring, timely reassessment, and treatment adjustment during the disease course are required to optimize personalized care.
Current View of Personalized Medicine in IPF
In other fields, especially oncology, personalized medicine has dramatically changed clinical practice during the last few years. Biomarkers have been used to develop targeted therapy and allocate patients to individual treatment plans (27–29).
Currently, the diagnosis of IPF is based on clinical, radiological, and pathological findings (3). The exact etiology of IPF is, however, incompletely understood. One of the proposed hypotheses is the concept of dysfunctional wound healing: repeated epithelial injury and dysfunctional regeneration possibly in combination with a dysregulated immune system normally facilitating wound healing leads to fibrogenesis and, as a consequence, excessive scarring of the lung tissue (11, 30). Epithelial injury might be caused by risk factors such as cigarette smoking, micro-aspiration of gastric content, and lead to development of IPF in susceptible individuals (11). At present, it is assumed that multiple biological pathways and complex interactions between genetic, molecular, and environmental factors are involved in the pathogenesis of IPF. Improved understanding of the pathogenesis of IPF has led to the identification of potential molecular biomarkers (7, 11, 31–33). Genome-wide association studies found genetic mutations that correlate with disease risk and possibly also disease progression (34–37); subsequently, the first examples of drug–gene interactions in IPF were found (38). To date, the value of biomarkers in IPF has not been fully clarified, and, therefore biomarkers or genetic endotyping are not yet used in clinical practice (7, 33).
Novel studies in IPF suggest that the “respiratory microbiome” is also involved in IPF pathogenesis, disease progression, and mortality (39–41). Patients with IPF have a higher bacterial burden and abundance of specific pathogens in the lung microbiome than the normal population. Furthermore, interactions have been found between specific gene expression and an altered lung microbiome in IPF, which is the first evidence for host-environmental interactions in IPF (42, 43). The lung microbiome may serve as a prognostic factor in the future, and clinical trials aimed at altering the microbiome of patients with IPF have already started (44).
A detailed description of (molecular) biology and its current role and potential in the IPF field is beyond the scope of this review.
How to Integrate Personomics into Personalized Medicine
Patient Needs and Perspectives in IPF Care
The importance of engaging patients in IPF care has gained increasing attention during the last several years (45). Recent qualitative studies have reported a need for better education about IPF, information about specific treatment options and palliative care, and access to specialist centers and specialist nurses. Additionally, more support for caregivers is warranted (16, 17, 46–48). These recommendations underscore the idea that not only pharmacological treatment but also non-pharmacological treatment options such as oxygen therapy, pulmonary rehabilitation, psychological support, and palliative care, are an important part of personalized management. With regard to pharmacological treatment, it is important to assess the needs and perspectives of patients before starting treatment, thereby enhancing shared decision-making. For instance, some side effects of disease-modifying drugs might have a devastating impact on one patient, but be far less bothersome to other patients (22). At the moment, over-use and under-use of medication, compliance problems, and waste of medication are not unusual in IPF (22, 49, 50). Non-adherence to medication could therefore be prevented when patients’ preferences and lifestyle are taken into account (9). Since patient preferences and needs may change because of disease progression or personal circumstances, an important aspect of disease management is iterative evaluation of the situation of individual patients (16, 46, 51). Only in this way can “holistic” personalized care be given in IPF.
Comorbidities and Co-Medication
Holistic care also means looking further than the lungs. IPF is associated with a number of pulmonary and extra-pulmonary comorbidities, such as pulmonary hypertension, respiratory infection, cardiovascular disease, emphysema, lung cancer, diabetes mellitus, venous thromboembolism, and gastroesophageal reflux (52–56). Comorbidities are more prevalent in patients with IPF than in the normal population and have a negative influence on QOL and survival (54, 56–58). Hence, early identification and treatment of comorbid conditions have the potential to improve QOL, functional outcomes, and survival for patients with IPF (53). Kreuter et al. (54) proposed the “IPF comorbidome,” which visually displays prevalence of comorbidities and their strength of association with mortality in patients with IPF. This comorbidome could be used to predict prognosis for individual patients with IPF, and thus enhance personalized treatment.
Moreover, extra attention should be paid to the frail, elderly patients who have multiple comorbidities and functional impairment (55). As a consequence, these patients might have a higher risk of harmful side effects of disease-modifying medication and should be closely monitored during treatment. Besides, polypharmacy may play an important role in this group of patients. It is generally known that polypharmacy decreases medication compliance, increases risk of adverse drug events, and might lead to impaired functional status and cognitive impairment in elderly patients (59). Furthermore, co-medication can also interfere with disease-modifying medication, and subsequently increase side effects or reduce treatment efficacy (60). Accordingly, co-medication could play an important role in the choice of pharmacological treatment in IPF. Expected risk–benefit ratio, comorbidities, and co-medication should be taken into account before pharmacological treatment is started in individual patients.
Measuring QOL and Monitoring Treatment Response
It remains challenging how to measure patients’ disease burden, experiences, and response to treatment in IPF. For this purpose, it is important to receive structured patient input throughout the whole disease course, starting already when the diagnosis is established. At present, digital solutions can facilitate more collaboration with patients in monitoring disease behavior, their experiences, and response to therapy (Figure 2).
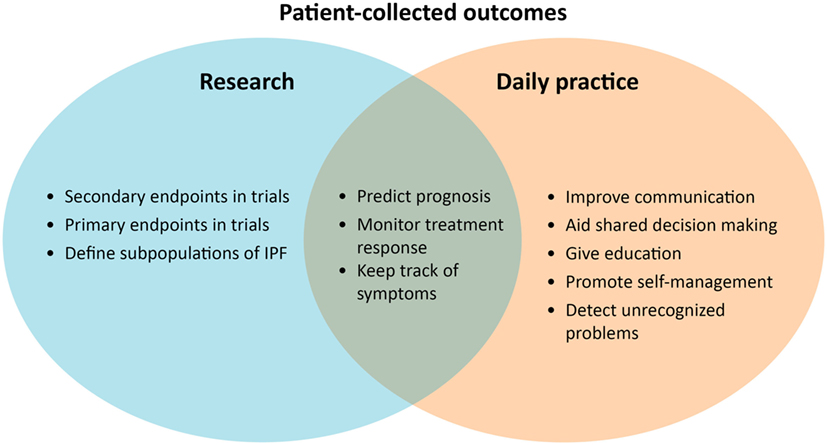
Figure 2. Patient-reported and recorded outcomes can be used to enhance personalized treatment.
Patient-Reported Outcome Measures (PROMs) in IPF
A PRO is defined as “any report of the status of a patient’s health condition that comes directly from the patient, without interpretation of the patient’s response by a clinician or anyone else” (61). Patient-reported outcome measures (PROMs) can be used to measure (HR)QOL, assess symptoms, and evaluate disease progression. There is a difference between generic and disease-specific PROMs. Disease-specific PROMs are developed to assess symptoms and (HR)QOL in a specific disease, whereas generic PROMs address more general questions and can be used in the whole population (62). One of the most commonly used generic PROMs in IPF trials are the short-form 36 and the Euroqol-5D, which is also a widely accepted instrument for economic evaluation in healthcare (63, 64). An overview of the most widely used PROMs in IPF is given in Table 1.
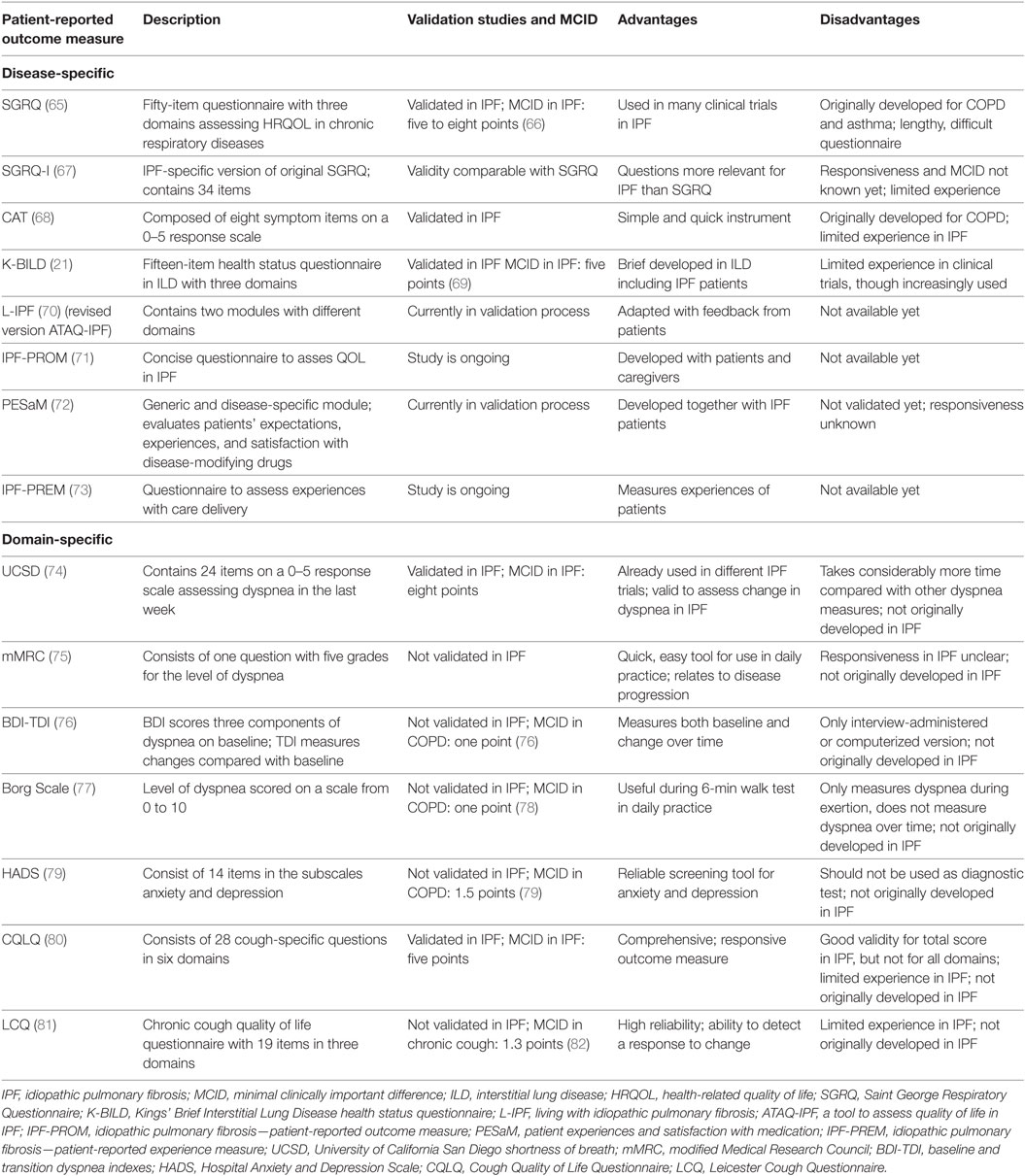
Table 1. Overview of most used patient-reported outcomes in IPF.
Disease-Specific PROMs
Although PROMs can play an important role to improve care for IPF, only a few well-validated, disease-specific questionnaires have been developed (19). Until a few years ago, most questionnaires used in clinical trials in IPF were originally intended for other chronic diseases (64, 65, 68). The validity of these questionnaires, such as the Saint George Respiratory Questionnaire (SGRQ) and COPD Assessment Test, has been confirmed in patients with IPF (66, 68). For the SGRQ, even an adapted version, the SGRQ-I, has been developed (67). This revised PROM consists of questions from the original SGRQ that were most relevant for patients with IPF. The reliability and validity of the SGRQ-I are comparable with the SGRQ. However, PROMs which are developed in a target population from the start, are thought to be more precise in capturing changes in HRQOL for this group of patients (58). One of the first questionnaires specifically developed in a population of patients with interstitial lung diseases (ILDs), among whom patients with IPF, is the Kings’ Brief Interstitial Lung Disease health status questionnaire (21). This is a brief, valid questionnaire that is increasingly used in IPF and other ILD clinical trials. One of the emerging PROMs in IPF is the “living with idiopathic pulmonary fibrosis” (L-IPF) questionnaire, which is a revised, electronic version of the ATAQ-IPF (a tool to assess quality of life in IPF). The L-IPF was adapted from the ATAQ-IPF following feedback from patients, and a validation study is underway at the moment (70). Another questionnaire which is currently being developed with the help of a multidisciplinary group of patients and carers is the IPF-PROM (71).
Domain-Specific PROMs
Additionally, domain-specific PROMs, which are questionnaires related to a specific symptom or organ, can be used to capture and objectify different aspects of disease. A few measures to evaluate breathlessness, such as the University of California San Diego Shortness of Breath Questionnaire, the modified Medical Research Council scale, the baseline and transition dyspnea indexes, and the Borg scale, are commonly used in IPF, although none were originally developed for IPF (74–77). Even though cough is a major problem in IPF, no specific cough questionnaires for IPF exist. However, the Leicester Cough Questionnaire and the Cough Quality of Life Questionnaire are currently used instead (80, 83). A widely known PROM to assess anxiety and depression is the Hospital Anxiety and Depression Scale, which is increasingly used in IPF (79). No specific fatigue questionnaires for IPF exist; however, the Fatigue Assessment Scale, originally developed for sarcoidosis, is used and might be adapted for IPF in the future (84).
PROs in Research and Daily Practice
PROs could be very helpful to enhance personalized treatment in IPF (Figure 2). Until now, PROMs have been mainly used for research purposes, as a secondary endpoint in clinical trials. The most used primary endpoint in IPF trials is forced vital capacity, which is accepted as a surrogate measure for mortality (85). One study showed that HRQOL, assessed with the SGRQ, is also an independent prognostic factor for mortality in IPF (86). PROMs probably reflect another dimension of disease compared with traditional physiological parameters (86). In the future, PROMs could possibly be used to predict treatment success in IPF.
PROM uses in daily practice can allow healthcare providers and patients to gain more insight into the individual disease and patient behavior. In a study of Sampson et al. (46), most patients were uncertain about their own disease course and progression and had difficulties interpreting objective hospital-based parameters. PROMs could allow both patients and healthcare providers to keep track of symptoms and disease progression easily. PRO results can even be used as a simple tool to communicate with patients, educate them, promote self-management, and aid shared decision making during the course of the disease (19, 87). A systematic review in oncology has shown strong evidence that routine collecting of PROs improved patient-centered care, patient satisfaction, and detection of unrecognized problems (88).
Patient-Reported Experience Measures in IPF
Optimal treatment requires close monitoring of the balance between the effects and side effects of disease-modifying drugs. Nonetheless, to our knowledge, a reliable measure to assess patient experiences with medication in IPF is not yet available in clinical practice. For this reason, a consortium of doctors, scientists, and patient representatives has joined forces to develop the patient experiences and satisfaction with medications (PESaM) questionnaire, which has a generic module and a disease-specific part for IPF (84). The PESaM questionnaire focuses on perceived effectiveness, side effects, and ease of use of medication and its impact on patients’ lives. This patient-reported experience measure (PREM) could not only be used in future clinical trials, but also in clinical practice to help with better detection of side effects and adjustment of medication. Moreover, Russell and colleagues, together with patients, are currently developing the “IPF-PREM.” This is a measure to assess patient experiences with healthcare and can possibly be used to improve the quality of care for patients (69).
Home Monitoring
Ideally, for a better tailored treatment, frequent monitoring with a low burden for the patient is needed. In the last decade, the use of e-health in chronic diseases has been growing, and shows mostly promising results (89–91). E-health involves the exchange of data between a patient and a healthcare provider using information and communication technologies (92). By using e-health tools, patients may better understand their health condition and become actively involved in the management of their own disease. It allows frequent monitoring in between regular visits and collection of PROs at home (93). Recently, a study showed that daily home spirometry in a population of patients with IPF was highly feasible and informative (94). Home-based spirometry predicts disease decline and mortality better than hospital-based measurements. Routine home spirometry could be very helpful to identify patients with rapid decline in lung function and to evaluate response to treatment. The authors suggest that daily home spirometry will allow for more individualized patient care. The feasibility of home-based spirometry in IPF was confirmed by Johannson et al. (95), who additionally showed that home spirometry might reduce sample size as well as the length of future clinical trials. Another promising example of home monitoring in IPF is the longitudinal follow-up of physical activity with activity trackers worn by patients at home (96). Decline in physical activity can provide reliable, objective data on disease progression and could be integrated into a home monitoring program. A comprehensive home monitoring program, consisting of an e-health tool combined with home spirometry and online collecting of PROs, has the potential to enhance trial design, stimulate self-management, allow for early treatment adaption to minimize side effects, prevent hospital admissions, and subsequently improve personalized management and QOL for patients with IPF.
Conclusion
The potential to enhance personalized treatment has prompted excitement also in the IPF field. In the future, patients’ genetic, biomarker, and microbiome profiles may guide clinical trial design and treatment decisions. In this process, patient perspectives should not be overlooked. Only by integrating biological information with patient-reported and patient-collected information, will we be able to realize truly personalized treatment.
Author Contributions
All authors conceptualized and designed the review, CM and MW wrote the paper, PH and MK provided critical feedback and input. All authors agree to be accountable for the content of the work and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Potter P. Hippocrates Vol VI. Diseases, Internal Affections. Cambridge, MA, London: Harvard University Press, William Heinemann LTD (1988).
2. American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med (2002) 165(2):277–304. doi:10.1164/ajrccm.165.2.ats01
3. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med (2011) 183(6):788–824. doi:10.1164/rccm.2009-040GL
4. The Voice of the Patient: A Series of Reports from the U.S. Food and Drug Administration’s (FDA’s) Patient-Focused Drug Development Initiative. Center for Drug Evaluation and Research (CDER) US Food and Drug Administration (FDA) (2015).
5. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med (2014) 370(22):2071–82. doi:10.1056/NEJMoa1402584
6. King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med (2014) 370(22):2083–92. doi:10.1056/NEJMoa1402582
7. Spagnolo P, Tzouvelekis A, Maher TM. Personalized medicine in idiopathic pulmonary fibrosis: facts and promises. Curr Opin Pulm Med (2015) 21(5):470–8. doi:10.1097/MCP.0000000000000187
8. Macagno F, Varone F, Leone PM, Mari PV, Panico L, Berardini L, et al. New treatment directions for IPF: current status of ongoing and upcoming clinical trials. Expert Rev Respir Med (2017) 11(7):533–48. doi:10.1080/17476348.2017.1335601
9. Di Paolo A, Sarkozy F, Ryll B, Siebert U. Personalized medicine in Europe: not yet personal enough? BMC Health Serv Res (2017) 17(1):289. doi:10.1186/s12913-017-2205-4
10. Clarke DL, Murray LA, Crestani B, Sleeman MA. Is personalised medicine the key to heterogeneity in idiopathic pulmonary fibrosis? Pharmacol Ther (2017) 169:35–46. doi:10.1016/j.pharmthera.2016.09.010
11. Spagnolo P, Cottin V. Genetics of idiopathic pulmonary fibrosis: from mechanistic pathways to personalised medicine. J Med Genet (2017) 54(2):93–9. doi:10.1136/jmedgenet-2016-103973
12. Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2011) 183(4):431–40. doi:10.1164/rccm.201006-0894CI
13. Ebrahim S. Clinical and public health perspectives and applications of health-related quality of life measurement. Soc Sci Med (1995) 41(10):1383–94. doi:10.1016/0277-9536(95)00116-O
14. Swigris JJ, Stewart AL, Gould MK, Wilson SR. Patients’ perspectives on how idiopathic pulmonary fibrosis affects the quality of their lives. Health Qual Life Outcomes (2005) 3:61. doi:10.1186/1477-7525-3-61
15. Duck A, Spencer LG, Bailey S, Leonard C, Ormes J, Caress AL. Perceptions, experiences and needs of patients with idiopathic pulmonary fibrosis. J Adv Nurs (2015) 71(5):1055–65. doi:10.1111/jan.12587
16. Overgaard D, Kaldan G, Marsaa K, Nielsen TL, Shaker SB, Egerod I. The lived experience with idiopathic pulmonary fibrosis: a qualitative study. Eur Respir J (2016) 47(5):1472–80. doi:10.1183/13993003.01566-2015
17. Russell AM, Ripamonti E, Vancheri C. Qualitative European survey of patients with idiopathic pulmonary fibrosis: patients’ perspectives of the disease and treatment. BMC Pulm Med (2016) 16:10. doi:10.1186/s12890-016-0171-y
18. Yount SE, Beaumont JL, Chen SY, Kaiser K, Wortman K, Van Brunt DL, et al. Health-related quality of life in patients with idiopathic pulmonary fibrosis. Lung (2016) 194(2):227–34. doi:10.1007/s00408-016-9850-y
19. Wijsenbeek M, van Manen M, Bonella F. New insights on patient-reported outcome measures in idiopathic pulmonary fibrosis: only PROMises? Curr Opin Pulm Med (2016) 22(5):434–41. doi:10.1097/MCP.0000000000000294
20. Swigris JJ, Esser D, Conoscenti CS, Brown KK. The psychometric properties of the St George’s Respiratory Questionnaire (SGRQ) in patients with idiopathic pulmonary fibrosis: a literature review. Health Qual Life Outcomes (2014) 12:124. doi:10.1186/s12955-014-0124-1
21. Patel AS, Siegert RJ, Brignall K, Gordon P, Steer S, Desai SR, et al. The development and validation of the King’s Brief Interstitial Lung Disease (K-BILD) health status questionnaire. Thorax (2012) 67(9):804–10. doi:10.1136/thoraxjnl-2012-201581
22. Britten N, Pope C, Halford S, Richeldi L. What if we made stratified medicine work for patients? Lancet Respir Med (2016) 4(1):8–10. doi:10.1016/S2213-2600(15)00499-3
23. Ziegelstein RC. Personomics. JAMA Intern Med (2015) 175(6):888–9. doi:10.1001/jamainternmed.2015.0861
24. MRC. (2017). Available from: https://www.mrc.ac.uk/research/initiatives/stratified-medicine/
25. Brownell R, Kaminski N, Woodruff PG, Bradford WZ, Richeldi L, Martinez FJ, et al. Precision medicine: the new frontier in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2016) 193(11):1213–8. doi:10.1164/rccm.201601-0169CI
26. Sykiotis GP, Kalliolias GD, Papavassiliou AG. Pharmacogenetic principles in the Hippocratic writings. J Clin Pharmacol (2005) 45(11):1218–20. doi:10.1177/0091270005281091
27. Daccord C, Maher TM. Recent advances in understanding idiopathic pulmonary fibrosis. F1000Res (2016) 5:F1000 Faculty Rev–1046. doi:10.12688/f1000research.8209.1
28. Dietel M. Molecular pathology: a requirement for precision medicine in cancer. Oncol Res Treat (2016) 39(12):804–10. doi:10.1159/000453085
29. Vargas AJ, Harris CC. Biomarker development in the precision medicine era: lung cancer as a case study. Nat Rev Cancer (2016) 16(8):525–37. doi:10.1038/nrc.2016.56
30. Hoyne GF, Elliott H, Mutsaers SE, Prele CM. Idiopathic pulmonary fibrosis and a role for autoimmunity. Immunol Cell Biol (2017) 95(7):577–83. doi:10.1038/icb.2017.22
31. Maher TM. Beyond the diagnosis of idiopathic pulmonary fibrosis; the growing role of systems biology and stratified medicine. Curr Opin Pulm Med (2013) 19(5):460–5. doi:10.1097/MCP.0b013e328363f4b7
32. Hambly N, Shimbori C, Kolb M. Molecular classification of idiopathic pulmonary fibrosis: personalized medicine, genetics and biomarkers. Respirology (2015) 20(7):1010–22. doi:10.1111/resp.12569
33. Ley B, Brown KK, Collard HR. Molecular biomarkers in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol (2014) 307(9):L681–91. doi:10.1152/ajplung.00014.2014
34. Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet (2013) 45(6):613–20. doi:10.1038/ng.2609
35. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med (2011) 364(16):1503–12. doi:10.1056/NEJMoa1013660
36. Peljto AL, Zhang Y, Fingerlin TE, Ma SF, Garcia JG, Richards TJ, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA (2013) 309(21):2232–9. doi:10.1001/jama.2013.5827
37. Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med (2013) 1(4):309–17. doi:10.1016/S2213-2600(13)70045-6
38. Oldham JM, Ma SF, Martinez FJ, Anstrom KJ, Raghu G, Schwartz DA, et al. TOLLIP, MUC5B, and the response to N-acetylcysteine among individuals with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2015) 192(12):1475–82. doi:10.1164/rccm.201505-1010OC
39. Molyneaux PL, Cox MJ, Willis-Owen SA, Mallia P, Russell KE, Russell AM, et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2014) 190(8):906–13. doi:10.1164/rccm.201403-0541OC
40. Han MK, Zhou Y, Murray S, Tayob N, Noth I, Lama VN, et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respir Med (2014) 2(7):548–56. doi:10.1016/S2213-2600(14)70069-4
41. Molyneaux PL, Cox MJ, Wells AU, Kim HC, Ji W, Cookson WO, et al. Changes in the respiratory microbiome during acute exacerbations of idiopathic pulmonary fibrosis. Respir Res (2017) 18(1):29. doi:10.1186/s12931-017-0511-3
42. Molyneaux PL, Willis-Owen SAG, Cox MJ, James P, Cowman S, Loebinger M, et al. Host-microbial interactions in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2017) 195(12):1640–50. doi:10.1164/rccm.201607-1408OC
43. Huang Y, Ma SF, Espindola MS, Vij R, Oldham JM, Huffnagle GB, et al. Microbes are associated with host innate immune response in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2017) 196(2):208–19. doi:10.1164/rccm.201607-1525OC
44. ClinicalTrials.gov. CleanUP IPF for the Pulmonary Trials Cooperative (CleanUp-IPF), NCT02759120. (2017). Available from: https://clinicaltrials.gov/ct2/show/NCT02759120
45. Russell AM, Swigris JJ. What’s it like to live with idiopathic pulmonary fibrosis? Ask the experts. Eur Respir J (2016) 47(5):1324–6. doi:10.1183/13993003.00109-2016
46. Sampson C, Gill BH, Harrison NK, Nelson A, Byrne A. The care needs of patients with idiopathic pulmonary fibrosis and their carers (CaNoPy): results of a qualitative study. BMC Pulm Med (2015) 15:155. doi:10.1186/s12890-015-0145-5
47. van Manen MJG, Kreuter M, van den Blink B, Oltmanns U, Palmowski K, Brunnemer E, et al. What patients with pulmonary fibrosis and their partners think: a live, educative survey in the Netherlands and Germany. ERJ Open Res (2017) 3(1):00065–2016. doi:10.1183/23120541.00065-2016
48. Bonella F, Wijsenbeek M, Molina-Molina M, Duck A, Mele R, Geissler K, et al. European IPF patient charter: unmet needs and a call to action for healthcare policymakers. Eur Respir J (2016) 47(2):597–606. doi:10.1183/13993003.01204-2015
49. Cottin V, Maher T. Long-term clinical and real-world experience with pirfenidone in the treatment of idiopathic pulmonary fibrosis. Eur Respir Rev (2015) 24(135):58–64. doi:10.1183/09059180.00011514
50. Koschel DCV, Skold M, Tomassetti S, Azuma A, Giot C, Yocum D, et al. Pirfenidone post-authorization safety registry (PASSPORT) – interim analysis of IPF treatment. Eur Respir J (2014) 44:1904.
51. van Manen MJ, Geelhoed JJ, Tak NC, Wijsenbeek MS. Optimizing quality of life in patients with idiopathic pulmonary fibrosis. Ther Adv Respir Dis (2017) 11(3):157–69. doi:10.1177/1753465816686743
52. Raghu G, Amatto VC, Behr J, Stowasser S. Comorbidities in idiopathic pulmonary fibrosis patients: a systematic literature review. Eur Respir J (2015) 46(4):1113–30. doi:10.1183/13993003.02316-2014
53. Suzuki A, Kondoh Y. The clinical impact of major comorbidities on idiopathic pulmonary fibrosis. Respir Investig (2017) 55(2):94–103. doi:10.1016/j.resinv.2016.11.004
54. Kreuter M, Ehlers-Tenenbaum S, Palmowski K, Bruhwyler J, Oltmanns U, Muley T, et al. Impact of comorbidities on mortality in patients with idiopathic pulmonary fibrosis. PLoS One (2016) 11(3):e0151425. doi:10.1371/journal.pone.0151425
55. Fulton BG, Ryerson CJ. Managing comorbidities in idiopathic pulmonary fibrosis. Int J Gen Med (2015) 8:309–18. doi:10.2147/IJGM.S74880
56. Hyldgaard C, Hilberg O, Bendstrup E. How does comorbidity influence survival in idiopathic pulmonary fibrosis? Respir Med (2014) 108(4):647–53. doi:10.1016/j.rmed.2014.01.008
57. Collard HR, Ward AJ, Lanes S, Cortney Hayflinger D, Rosenberg DM, Hunsche E. Burden of illness in idiopathic pulmonary fibrosis. J Med Econ (2012) 15(5):829–35. doi:10.3111/13696998.2012.680553
58. Kreuter M, Swigris J, Pittrow D, Geier S, Klotsche J, Prasse A, et al. Health related quality of life in patients with idiopathic pulmonary fibrosis in clinical practice: insights-IPF registry. Respir Res (2017) 18(1):139. doi:10.1186/s12931-017-0621-y
59. Maher RL, Hanlon J, Hajjar ER. Clinical consequences of polypharmacy in elderly. Expert Opin Drug Saf (2014) 13(1):57–65. doi:10.1517/14740338.2013.827660
60. Jo HE, Randhawa S, Corte TJ, Moodley Y. Idiopathic pulmonary fibrosis and the elderly: diagnosis and management considerations. Drugs Aging (2016) 33(5):321–34. doi:10.1007/s40266-016-0366-1
61. Guidance for Industry. Patient-Reported Outocome Measures: Use in Medical Product Development to Support Labeling Claims. US Department of Health and Human Services, Food and Drug Administration (2009). Available from: https://www.fda.gov/downloads/drugs/guidances/ucm193282.pdf
62. Black N. Patient reported outcome measures could help transform healthcare. BMJ (2013) 346:f167. doi:10.1136/bmj.f167
63. Brooks R. EuroQol: the current state of play. Health Policy (1996) 37(1):53–72. doi:10.1016/0168-8510(96)00822-6
64. Ware JE Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care (1992) 30(6):473–83. doi:10.1097/00005650-199206000-00002
65. Jones PW, Quirk FH, Baveystock CM. The St George’s respiratory questionnaire. Respir Med (1991) 85(Suppl B):25–31; discussion 33–7. doi:10.1016/S0954-6111(06)80166-6
66. Swigris JJ, Brown KK, Behr J, du Bois RM, King TE, Raghu G, et al. The SF-36 and SGRQ: validity and first look at minimum important differences in IPF. Respir Med (2010) 104(2):296–304. doi:10.1016/j.rmed.2009.09.006
67. Yorke J, Jones PW, Swigris JJ. Development and validity testing of an IPF-specific version of the St George’s respiratory questionnaire. Thorax (2010) 65(10):921–6. doi:10.1136/thx.2010.139121
68. Matsuda T, Taniguchi H, Ando M, Kondoh Y, Kimura T, Kataoka K, et al. COPD assessment test for measurement of health status in patients with idiopathic pulmonary fibrosis: a cross-sectional study. Respirology (2017) 22(4):721–7. doi:10.1111/resp.12936
69. Sinha APAS, Birring SS. The King’s Brief Interstitial Lung Disease (K-BILD) questionnaire; an updated minimal important difference. Eur Respir J (2016) 48(Suppl 60):PA808. doi:10.1183/13993003.congress-2016.PA808
70. Graney B, Johnson N, Evans CJ, Ryan A, Matsui A, Raimundo K, et al. Living with idiopathic pulmonary fibrosis (L-IPF): developing a patient-reported symptom and impact questionnaire to assess health-related quality of life in IPF. Am J Respir Crit Care Med (2017) 195:A5353.
71. Russell AM, Doyle AM, Fleming S, Burdett C, Ross D, Aden Z, et al. Development of a patient reported outcome measure (PROM) in idiopathic pulmonary fibrosis (IPF): incorporating a research partnership with patients. Am J Respir Crit Care Med (2015) 191:A5190.
72. Kimman ML, Rotteveel AH, Wijsenbeek M, Mostard R, Tak NC, van Jaarsveld X, et al. Development and pretesting of a questionnaire to assess patient experiences and satisfaction with medications (PESaM questionnaire). Patient (2017) 10(5):629–42. doi:10.1007/s40271-017-0234-z
73. Russell AM, Sonecha S, Datta A, Hewitt R, Howell I, Elliott A, Wickremasinghe M. Development of patient reported experience measure (PREM) for idiopathic pulmonary fibrosis (IPF). Thorax (2016) 71:A238. doi:10.1136/thoraxjnl-2016-209333.419
74. Swigris JJ, Han M, Vij R, Noth I, Eisenstein EL, Anstrom KJ, et al. The UCSD shortness of breath questionnaire has longitudinal construct validity in idiopathic pulmonary fibrosis. Respir Med (2012) 106(10):1447–55. doi:10.1016/j.rmed.2012.06.018
75. Nishiyama O, Taniguchi H, Kondoh Y, Kimura T, Kato K, Kataoka K, et al. A simple assessment of dyspnoea as a prognostic indicator in idiopathic pulmonary fibrosis. Eur Respir J (2010) 36(5):1067–72. doi:10.1183/09031936.00152609
76. Mahler DA, Witek TJ Jr. The MCID of the transition dyspnea index is a total score of one unit. COPD (2005) 2(1):99–103. doi:10.1081/COPD-200050666
77. Borg GA. Psychophysical bases of perceived exertion. Med Sci Sports Exerc (1982) 14(5):377–81. doi:10.1249/00005768-198205000-00012
78. Jones PW, Beeh KM, Chapman KR, Decramer M, Mahler DA, Wedzicha JA. Minimal clinically important differences in pharmacological trials. Am J Respir Crit Care Med (2014) 189(3):250–5. doi:10.1164/rccm.201310-1863PP
79. Matsuda T, Taniguchi H, Ando M, Kondoh Y, Kimura T, Kataoka K, et al. Depression is significantly associated with the health status in patients with idiopathic pulmonary fibrosis. Intern Med (2017) 56(13):1637–44. doi:10.2169/internalmedicine.56.7019
80. Lechtzin N, Hilliard ME, Horton MR. Validation of the cough quality-of-life questionnaire in patients with idiopathic pulmonary fibrosis. Chest (2013) 143(6):1745–9. doi:10.1378/chest.12-2870
81. Birring SS, Prudon B, Carr AJ, Singh SJ, Morgan MD, Pavord ID. Development of a symptom specific health status measure for patients with chronic cough: Leicester Cough Questionnaire (LCQ). Thorax (2003) 58(4):339–43. doi:10.1136/thorax.58.4.339
82. Raj AA, Pavord DI, Birring SS. Clinical cough IV: what is the minimal important difference for the Leicester Cough Questionnaire? Handb Exp Pharmacol (2009) 187:311–20. doi:10.1007/978-3-540-79842-2_16
83. Key AL, Holt K, Hamilton A, Smith JA, Earis JE. Objective cough frequency in idiopathic pulmonary fibrosis. Cough (2010) 6:4. doi:10.1186/1745-9974-6-4
84. Kotecha J, Atkins C, Wilson A. Patient confidence and quality of life in idiopathic pulmonary fibrosis and sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis (2016) 33(4):341–8.
85. Collard HR, Bradford WZ, Cottin V, Flaherty KR, King TE Jr, Koch GG, et al. A new era in idiopathic pulmonary fibrosis: considerations for future clinical trials. Eur Respir J (2015) 46(1):243–9. doi:10.1183/09031936.00200614
86. Furukawa T, Taniguchi H, Ando M, Kondoh Y, Kataoka K, Nishiyama O, et al. The St. George’s Respiratory Questionnaire as a prognostic factor in IPF. Respir Res (2017) 18(1):18. doi:10.1186/s12931-017-0503-3
87. Santana MJ, Feeny D. Framework to assess the effects of using patient-reported outcome measures in chronic care management. Qual Life Res (2014) 23(5):1505–13. doi:10.1007/s11136-013-0596-1
88. Chen J, Ou L, Hollis SJ. A systematic review of the impact of routine collection of patient reported outcome measures on patients, providers and health organisations in an oncologic setting. BMC Health Serv Res (2013) 13:211. doi:10.1186/1472-6963-13-211
89. Koff PB, Jones RH, Cashman JM, Voelkel NF, Vandivier RW. Proactive integrated care improves quality of life in patients with COPD. Eur Respir J (2009) 33(5):1031–8. doi:10.1183/09031936.00063108
90. Rasmussen LM, Phanareth K, Nolte H, Backer V. Internet-based monitoring of asthma: a long-term, randomized clinical study of 300 asthmatic subjects. J Allergy Clin Immunol (2005) 115(6):1137–42. doi:10.1016/j.jaci.2005.03.030
91. Denis F, Lethrosne C, Pourel N, Molinier O, Pointreau Y, Domont J, et al. Randomized trial comparing a web-mediated follow-up with routine surveillance in lung cancer patients. J Natl Cancer Inst (2017) 109:9. doi:10.1093/jnci/djx029
92. World Health Organization. eHealth. (2017). Available from: http://www.who.int/ehealth/en/
93. Wicks P, Stamford J, Grootenhuis MA, Haverman L, Ahmed S. Innovations in e-health. Qual Life Res (2014) 23(1):195–203. doi:10.1007/s11136-013-0458-x
94. Russell AM, Adamali H, Molyneaux PL, Lukey PT, Marshall RP, Renzoni EA, et al. Daily home spirometry: an effective tool for detecting progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2016) 194(8):989–97. doi:10.1164/rccm.201511-2152OC
95. Johannson KA, Vittinghoff E, Morisset J, Lee JS, Balmes JR, Collard HR. Home monitoring improves endpoint efficiency in idiopathic pulmonary fibrosis. Eur Respir J (2017) 50(1):1602406. doi:10.1183/13993003.02406-2016
Keywords: idiopathic pulmonary fibrosis, health-related quality of life, personalized medicine, patient-reported outcomes, personomics, patient experiences
Citation: Moor CC, Heukels P, Kool M and Wijsenbeek MS (2017) Integrating Patient Perspectives into Personalized Medicine in Idiopathic Pulmonary Fibrosis. Front. Med. 4:226. doi: 10.3389/fmed.2017.00226
Received: 30 August 2017; Accepted: 27 November 2017;
Published: 20 December 2017
Edited by:
Naftali Kaminski, Yale University, United StatesReviewed by:
Martin Petrek, Palacký University, Olomouc, CzechiaEsmaeil Mortaz, Utrecht Institute for Pharmaceutical Sciences, Netherlands
Copyright: © 2017 Moor, Heukels, Kool and Wijsenbeek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marlies S. Wijsenbeek, bS53aWpzZW5iZWVrLWxvdXJlbnNAZXJhc211c21jLm5s