 Irina Khamaganova
Irina Khamaganova- Pirogov Russian National Research Medical University, Moscow, Russia
Similar clinical and histhopathological features in progressive hemifacial atrophy and linear scleroderma en coup de sabre are well known. Trauma may predispose to the development of both diseases. The lack of association with anti-Borrelia antibodies was shown in both cases as well. The otolaryngological and endocrine disorders may be associated findings in both diseases. However, there are certain differences in neurological and ophthalmological changes in the diseases.
Introduction
The Parry–Romberg syndrome has an old history. Appenzeller and colleagues scrutinized about 200 mummy colored portraits painted at the beginning of the first millennium. The Parry–Romberg syndrome was diagnosed in two persons (1).
Charlier and colleagues retrospectively described the disease in a major French revolution leader Mirabeau (2).
The “Collections from the Unpublished Medical Writings of the Late Caleb Hillier Parry” (1825) presented the first description of Parry–Romberg syndrome (3). Romberg described the disorder in 1846 (4). Eulenburg proposed the title “progressive hemifacial atrophy” (PHA) in 1871 (5).
Tolkachjov and colleagues (6) named the synonyms of Parry–Romberg syndrome: idiopathic hemifacial atrophy, PHA, and Romberg syndrome. Otherwise, Nomura and colleagues pointed out that PHA could progress in lupus profundus, lipodystrophy, morphea, and Parry–Romberg syndrome (7). The term PHA is often used as a synonym of Parry–Romberg syndrome.
In spite of the long history, the Parry–Romberg syndrome has unknown etiology and pathogenesis. Earlier it was highlighted that PHA and linear scleroderma “en coup de sabre” (ECDS) often coexisted (6, 8), but some authors described PHA as a unique disease (9–11).
The problem of differential diagnosis of PHA and ECDS is being discussed (12, 13). The goal of the review is to analyze the data on these opinions.
Material
The original articles, reviews, and cases from Medline PubMed were the main source of information. The words “progressive hemifacial atrophy,” “Parry–Romberg syndrome,” “localized scleroderma en coup de sabre,” “linear scleroderma en coup de sabre,” and “linear morphoea” were used for the search. The inclusion criteria contained:
– publication no later than 2001.
– publication presenting the problems named in the search words.
The exclusion criteria contained:
– publications later than 2001.
– articles on another subject 5,479 papers published from March 1897 to August 2017 were analyzed.
We excluded 5,100 papers published before 2001 as far as they were earlier discussed in some reviews (6, 8, 10). We selected 125 publications that presented clinical and histopathological characteristics of PHA and ECDS, predisposing and provocative factors for their progressing. 254 articles that were written on other topics were excluded.
Results
Clinical and Histopathological Features of PHA and ECDS
Our results are provided in Table 1. So, Tolkachjov and colleagues (6) summarized the published data on the relationship of ECDS and PHA. The average age of the beginning in ECDS was 10 years, in PHA 13.6, respectively. Both diseases have female predilection (6, 14, 15) and started in childhood previously (16).
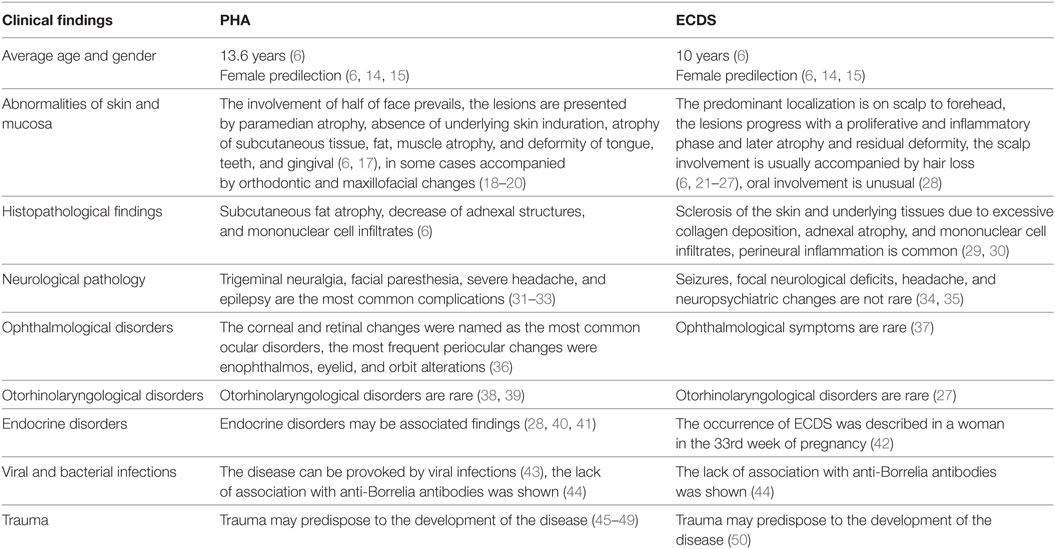
Table 1. Clinical and histopathological findings in progressive hemifacial atrophy (PHA) and en coup de saber (ECDS) (1).
The PHA usually presents in the first 20 years of life, late onset is rare (6). The disorder is usually slowly progressive. The defect becomes more pronounced in long duration, leading to esthetic and functional deficits (51). Patients may experience halting of the facial atrophy. But in fact, it may be difficult to differentiate disease acceleration from normal evolution. The PHA main clinical features included paramedian atrophy, absence of underlying skin induration and atrophy which might extend down entire face (6). Typical cases demonstrate the involvement of one or more branches of the fifth cranial nerve. The atrophy of such structures as subcutaneous tissue, fat, muscle and osteocartilaginous was revealed in PHA patients. The changes create a sunken hemiface appearance (6, 17). As far as it is “hemifacial” the involvement of half of face is usual, but Pathi and colleagues reported a case of involving one-half of the body (52).
En coup de sabre is usually a linear unilateral depression on the frontoparietal scalp or paramedian forehead. ECDS develops through the stages of proliferation, inflammation phase, atrophy, and residual deformity. The predominant localization is on scalp to forehead (6, 21–23). The abnormalities of the skin, subcutaneous tissues, and fascia were shown in localized scleroderma (24, 25). Besides, the atrophic shiny plaque may be associated with madarosis (6).
The scalp involvement was usually accompanied by hair loss (26, 27). The lesion really resembles “stroke from a sword” (“en coup de sabre”) (6). Gradual progress over 60 years was described (53). The outcome of the disease is characterized by certain variety from cosmetic problem to invalidism (6, 23, 53).
The atrophy and deformity detected in PHA were noted in the tongue, teeth and gingiva (6). Verma and colleagues reported a PHA patient with marked left-sided facial atrophy and wasting of the tongue (49). Mucosa can be involved in PHA (54) and in ECDS (28). In some cases mucosal involvement may be accompanied by orthodontic and maxillofacial changes (18, 20). ECDS and PHA can be deforming and irreversibly disabling (55, 56). Segna and colleagues emphasized the gravity of the associated deformity and its impact on facial function and appearance (57).
As for scleroderma, xerostomia, microstomia, idiopathic resorption of tooth and mucosal erosions are diagnosed previously in diffuse scleroderma (58). Oral involvement in ECDS is rare, and it may be presented by white linear scar-like fibrotic areas, atrophical tongue papillae, gingival recession, and alveolar bone resorption (28). Hørberg and colleagues presented a case of ECDS in 6-year and 10-month-old Turkish girl. The dental clinical and radiographic examination was performed. The malformed left maxillary incisors with short roots and lack of eruption were revealed (59). A progressive recession on teeth 11 and 12 was described in 19-year-old patient with ECDS (60). A progressive recession on gingiva was shown in 13-year-old girl with ECDS as well (28).
Bilateral involvement seems to be more common in PHA and in ECDS than previously reported (8).
The histopathological examination revealed dermal sclerosis in both diseases. Besides, PHA was characterized by subcutaneous fat atrophy, decrease of adnexal structures, and mononuclear cell infiltrates (6). In ECDS, sclerotic changes of the skin and hypoderma due to excessive collagen deposition, adnexal atrophy, and mononuclear cell infiltrates were revealed as well (6, 61). Walker and colleagues underlined that severe inflammation might lead to pain and functional limitation in morphea (62). Perineural inflammation is common in morphea (29). Goh and colleagues described a substantial perineural lymphocytic and plasmacytic infiltrate, extending into deep layers in ECDS patient (30). Pierre-Louis and colleagues presented ECDS patient with a 1.5-year history of linear morphea and alopecia with morphological changes presented by atrophic follicular remnants (63).
Neurological Pathology
The extracutaneous changes in PHA were noted as well. The neurological complications are the most frequent (6, 64). Chokar and colleagues presented a PHA case with alien-hand syndrome (65).
Okumura and colleagues (66) performed a detailed neuroimaging study in a PHA patient. Parents noticed asymmetry of the face at 4 months of patient’s age. PHA was diagnosed at 18 months of age. At 22 months of age, patient had widespread white matter lesions in the left hemisphere which presented diminished signal intensities on T1-weighted images and enhanced signal intensities on T2-weighted images and fluid-attenuated inversion recovery (FLAIR) images. Examining of the occipital areas in the right hemisphere showed a similar white matter lesion. No changes were noted while neurologic and electroencephalographic examination. MR angiography was performed at 33 and 44 months. It did not reveal any vascular abnormalities. The single-photon emission (SPECT) was performed at 31 months of age. It demonstrated the blood flow within the white matter in the left hemisphere being 55% of the corresponding region of the right hemisphere. Both conventional MR imaging and diffusion tension imaging showed the widespread white matter abnormalities (66).
So, the absence of pathologic neurologic changes in some cases (67, 68) does not prove the intactness of neural system.
The advanced magnetic resonance imaging (MRI) detected the white matter hypersignal on T2-weighted and FLAIR sequences, leptomeningeal enhancement, intracranial calcifications, and brain atrophy (31).
Vix and colleagues (69) showed that central nervous system involvement was frequent among PHA patients. The seizures, headaches, movement disturbances, neuropsychological changes, and focal symptoms were noted.
The trigeminal neuralgia (32, 33, 70, 71), facial paresthesia, severe headache, and epilepsy are the most common complications of PHA (31–33). Variable neurological pathology was described in PHA patients. So, Verma and colleagues reported mirror movements in a hand of a teenager with PHA and epilepsy (72). Epilepsy (epilepsia partialis continua) can arise from the hemisphere opposite to the side of facial atrophy (73). Asai and colleagues (74) presented a case of PHA syndrome associated with contralateral cerebral atrophy. The facial myokymias were also noted in PHA (75). Seifert and colleagues reported two cases PHA with chronic focal encephalitis (76). Gupta and Patil described PHA with multiple intracranial cysts (77). Yanagishita and colleagues presented a case of dyschromatosis symmetrica hereditaria complicated by intracranial hemangiomas and PHA (78). PHA combined with hemifacial spasm was described (79). Zahlane and colleagues reported a case of PHA associated with complete agenesis of the corpus callosum (80). The abnormalities of brain glucose metabolism in PHA may be observed before anatomical changes and therefore facilitate early diagnosis (71). De Lange and colleagues reported a case of PHA with a giant intracranial aneurysm (81).
Neurological changes were noted in ECDS as well, they are more often described in children than in adults (37, 82). Such neurological manifestations as seizures, focal neurological deficits, headache, and neuropsychiatric changes are not rare in ECDS (34, 35, 83). Recurrent myelitis was described in ECDS (84). Takahashi and colleagues (85) presented an ECDS case in a 39-year-old man. The brain hemorrhage was revealed, but any signs of cerebral vascular abnormalities were not noted. The implication of developmental abnormalities was proposed. Lis-Święty and colleagues examined 20 patients with localized scleroderma affecting the face and head. The symptoms and/or abnormalities in the central nervous system in high-resolution computed tomography and/or MRI were observed in 12 patients (60%). It was concluded that the involvement of central nervous system was not correlated with the clinical course of the facial and head localized scleroderma (86). Brain cavernomas associated with ECDS were described in children (26). Gambichler and colleagues presented an ECDS associated with epileptic seizures, as well oculomotor and facial nerve palsy (87). Holland and colleagues emphasized the underrecognized relationship between neurologic complications and ECDS and highlighted thorough skin examination in patients with unexplained neurologic disease (88). Besides, ECDS can be accompanied by focal neurologic deficits, movement disorders, trigeminal neuralgia, and mimics of hemiplegic migraines (89).
The neurological disorders may precede the appearance of skin lesions (40, 90). Verhelst and colleagues presented hippocampal atrophy and developmental regression as initial clinical feature of ECDS (90).
Bergler-Czop and colleagues presented two cases: a case of a 49-year-old woman with clinical signs of PHA and minor neurological symptoms and a case of a 33-year-old patient with ECDS without any neurological symptoms when only special neurological methods had revealed the presence of CNS tumor (91).
Cranial neuropathies, seizure disorder, and vision loss were diagnosed both in PHA and ECDS (6). The neurological symptoms in both diseases may resemble those in Rasmussen encephalitis (13, 92, 93).
Ophthalmological and Otorhinolaryngological Disorders
Bucher and colleagues (36) summarized the ocular, periocular, and neuro-ophthalmological findings in PHA The most frequent periocular changes were enophthalmos, eyelid, and orbit alterations. The corneal and retinal changes were named as the most common ocular disorders. The optic nerve, ocular motor, and pupillary dysfunction are the most frequent neuro-ophthalmological disorders (36).
Ayyildiz and colleagues presented a patient with PHA at right facial side who developed granulomatous uveitis and periferic retinal vasculitis in his left eye (94). Fea and colleagues described enophthalmos, uveitis, and iris atrophy in 23-year-old female Caucasian patient with PHA (95). The total atrophy of iris and ciliary body with associated ocular hypotony was describes in PHA patient (8). Kaya and colleagues described severe atrophy and loss of one eye in another patient (96).
According to Careta and Romiti, ECDS is rarely associated with ocular abnormalities, the most part of them noted in children (37). On the other side, a multicenter study showed that ophthalmological symptoms were not unusual in patients with juvenile localized scleroderma, particularly in the ECDS subtype (97). Generally in juvenile localized scleroderma ocular abnormalities include episcleritis, uveitis, xerophthalmia, glaucoma, and papilledema (98).
Otorhinolaryngological disorders are rare in PHA. The association of PHA with dysphonia was described (38, 39).
Otorhinolaryngological disorders are not common in ECDS as well. Pathologic changes may extend into nose rarely (27).
Predisposing and Triggering Factors
The only one case of familial PHA was described by Anderson and colleagues (99).
The family cases of variable localized scleroderma are presented (100–103), but no cases of ECDS in family.
The involvement of X chromosome monosomy (104), the influence of HLA haplotypes (105), the specific HLA classes are associated with localized scleroderma (106). The overlapping gene expression profiles (107), downregulation of microRNA-196a (108), and role of the CAV1 gene (109) are being examined.
However, the genetic predisposition may be realized due to certain triggers, such as endocrine changes, infections, traumas.
Panda and colleagues presented a case of the development of intermittent unilateral painful spasms of jaw during the fifth month of pregnancy in previously healthy woman. Besides, the patient developed hemifacial and hemitongue atrophy. PHA might be correlated either with some hormonal imbalance or some unknown mechanisms (110).
Unterberger and colleagues reported the neurological disorders such as right sided hemiparesis in association with the occurrence of ECDS and preexisting plaque-morphea in a woman in the 33rd week of pregnancy (42).
Endocrine disorders may be associated findings in PHA (28, 40, 41).
The examination of 17 patients with different types of morphea revealed normal thyroid functions in two patients with linear morphoea (111), but another types of localized scleroderma were accompanied in some cases by thyroid diseases (43, 112).
Progressive hemifacial atrophy can be provoked by viral infections (43). Zhang and colleagues reported a 41-year-old woman with PHA who showed an uncharacteristic “relapsing-remitting” evolution of brain lesions and chronic HBV infection may have triggered the relapse in this case (113).
The role of Borrelia infection is being discussed in the development of PHA and ECDS (114, 115). Prinz and colleagues concluded that Borrelia burgdorferi might be relevant for the induction of severe morphoea (116). Salpietro and colleagues proposed a possibility of molecular mimicry in PHA patient seropositive for B. burgdorferi (117).
The lack of association in PHA and ECDS with anti-Borrelia antibodies was shown (44).
Trauma may predispose to the development of PHA (45–49, 118).
The same is noted in ECDS (50). The development of ECDS was presented by Arif and colleagues in a 26-year-old woman. The lesion located on the frontal and forehead regions. Six years before the examination patient had had a trauma at the same site 6 years back (119). Lipson and colleagues reported a case of congenital ECDS, misdiagnosed since birth as birth trauma. The ECDS was diagnosed in adult (120).
Coexistence of PHA and ECDS
28–42% of patients have been reported to have coexistence of these diseases. Some findings might overlap in the same patient (6). Blaszczyk and colleagues investigated the relationship of PHA and ECDS by approving the presence and type of central nervous system involvement in both diseases (121). The investigation indicated a close relationship between PHA and ECDS. Dixit and colleagues presented a case of mucosal involvement in morphea, associated with PHA (122). The development or transition of ECDS into PHA in the same physical location was described as well (6, 123).
Conclusion
The genetic predisposition to ECDS may be realized due to certain triggers, such as endocrine changes, infections, and traumas. The only one case of familial PHA was described. The lack of association both in PHA and ECDS with anti-Borrelia antibodies was shown. Both PHA and ECDS may be correlated with hormonal imbalance. PHA and ECDS have certain similar clinical and histopathological features that may indicate to the same spectrum of diseases. The PHA patients suffer from esthetic and functional deficits which may be seriously complicated by the systemic involvement. The outcome of ECDS may vary from cosmetic problem to invalidism. Some differences can be noted in the diseases belonging to the same group. Mucosal involvement and ophthalmological disorders are more frequent in PHA than in ECDS. Variable neurological disorders were described both in PHA and ECDS. The presented data indicate the need for further neuroimaging studies in PHA and ECDS.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AP and the handling Editor declared their shared affiliation.
References
1. Appenzeller O, Stevens JM, Kruszynski R, Walker S. Neurology in ancient faces. J Neurol Neurosurg Psychiatry (2001) 70:524–9. doi:10.1136/jnnp.70.4.524
2. Charlier P, Froesch P, Tollefson M. Parry-Romberg syndrome on a major French revolution leader: Mirabeau, 1791. J Craniofac Surg (2017) 28(2):582. doi:10.1097/SCS.0000000000003091
3. Parry CH, editor. Facial hemiatrophy. Collections from the Unpublished Medical Writings of the Late Caleb Hillier Parry. (Vol. 1), London: Underwoods, Fleet Street (1825). p. 478–80.
5. Eulenburg A. Lehrbuch der functionellen Nervenkrankheiten auf Physiologischer Basis. Berlin: A. Hirschwald (1871). 712 p.
6. Tolkachjov SN, Patel NG, Tollefson MM. Progressive hemifacial atrophy: a review. Orphanet J Rare Dis (2015) 10:39. doi:10.1186/s13023-015-0250-9
7. Nomura H, Egami S, Yokoyama T, Sugiura M. Case of rapid progression of hemiatrophy on the face: a new clinical entity? Case Rep Dermatol Med (2015) 2015:478640. doi:10.1155/2015/478640
8. Tollefson MM, Witman PM. En coup de sabre morphea and Parry-Romberg syndrome: a retrospective review of 54 patients. J Am Acad Dermatol (2007) 56(2):257–63. doi:10.1016/j.jaad.2006.10.959
9. Kini TA, Prakash VS, Puthalath S, Bhandari PL. Progressive hemifacial atrophy with ciliary body atrophy and ocular hypotony. Indian J Ophthalmol (2015) 63(1):61–3. doi:10.4103/0301-4738.151474
10. Wong M, Phillips CD, Hagiwara M, Shatzkes DR. Parry Romberg syndrome: 7 cases and literature review. AJNR Am J Neuroradiol (2015) 36(7):1355–61. doi:10.3174/ajnr.A4297
11. Rodby KA, Kaptein YE, Roring J, Jacobs RJ, Kang V, Quinn KP, et al. Evaluating autologous lipofilling for Parry-Romberg syndrome-associated defects: a systematic literature review and case report. Cleft Palate Craniofac J (2016) 53(3):339–50. doi:10.1597/14-232
12. Wakhlu A, Agarwal V, Aggarwal A, Misra R. Parry Romberg syndrome: a close differential diagnosis of linear scleroderma en coup de sabre. J Assoc Physicians India (2003) 51:980.
13. Paprocka J, Jamroz E, Adamek D, Marszal E, Mandera M. Difficulties in differentiation of Parry-Romberg syndrome, unilateral facial sclerodermia, and Rasmussen syndrome. Childs Nerv Syst (2006) 22(4):409–15. doi:10.1007/s00381-005-1262-x
14. Lis-Święty A, Skrzypek-Salamon A, Ranosz-Janicka I, Brzezińska-Wcisło L. Localized scleroderma: clinical and epidemiological features with emphasis on adulthood- versus childhood-onset disease differences. J Eur Acad Dermatol Venereol (2017) 31(10):1595–603. doi:10.1111/jdv.14197
15. Knobler R, Moinzadeh P, Hunzelmann N, Kreuter A, Cozzio A, Mouthon L, et al. European Dermatology Forum S1-guideline on the diagnosis and treatment of sclerosing diseases of the skin, Part 1: localized scleroderma, systemic sclerosis and overlap syndromes. J Eur Acad Dermatol Venereol (2017) 31(9):1401–24. doi:10.1111/jdv.14458
16. Marzano AV, Menni S, Parodi A, Borghi A, Fuligni A, Fabbri P, et al. Localized scleroderma in adults and children. Clinical and laboratory investigations on 239 cases. Eur J Dermatol (2003) 13(2):171–6.
17. Balaji SM. Subdermal fat grafting for Parry-Romberg syndrome. Ann Maxillofac Surg (2014) 4(1):55–9. doi:10.4103/2231-0746.133081
18. El-Kehdy J, Abbas O, Rubeiz N. A review of Parry-Romberg syndrome. J Am Acad Dermatol (2012) 67(4):769–84. doi:10.1016/j.jaad.2012.01.019
19. Qiao J, Gui L, Fu X, Niu F, Liu J, Chen Y, et al. A novel method of mild to moderate Parry-Romberg syndrome reconstruction: computer-assisted surgery with mandibular outer cortex and fat grafting. J Craniofac Surg (2017) 28(2):359–65. doi:10.1097/SCS.0000000000003293
20. Al-Aizari NA, Azzeghaiby SN, Al-Shamiri HM, Darwish S, Tarakji B. Oral manifestations of Parry-Romberg syndrome: a review of literature. Avicenna J Med (2015) 5(2):25–8. doi:10.4103/2231-0770.154193
22. Pekiner FN, Yücelten D, Gümrü B, Sinanoğlu EA. Frontal linear scieroderma (en Coup de Sabre): a case report. J Dent Child (Chic) (2006) 73(3):175–8.
23. Franco JP, Serra MS, Lima RB, D’Acri AM, Martins CJ. Scleroderma en coup de sabre treated with polymethylmethacrylate – case report. An Bras Dermatol (2016) 91(2):209–11. doi:10.1590/abd1806-4841.20163867
24. Eutsler EP, Horton DB, Epelman M, Finkel T, Averill LW. Musculoskeletal MRI findings of juvenile localized scleroderma. Pediatr Radiol (2017) 47(4):442–9. doi:10.1007/s00247-016-3765-x
25. Gutwein F, Tratenberg M, Rao V, Sperber K, Wasserrman A, Ash J. Localized scleroderma: a clinical review. Curr Rheumatol Rev (2017) 13(2):86–92. doi:10.2174/1573397112666160907105434
26. Fain ET, Mannion M, Pope E, Young DW, Laxer RM, Cron RQ. Brain cavernomas associated with en coup de sabre linear scleroderma: two case reports. Pediatr Rheumatol (2011) 9:18. doi:10.1186/1546-0096-9-18
27. Allmendinger AM, Ricci JA, Desai NS, Viswanadhan N, Rogriguez D. Atypical neuroimaging manifestations of linear scleroderma “en coup de sabre”. Iran J Child Neurol (2015) 9(3):62–8.
28. Niklander S, Marín C, Martínez R, Esguep A. Morphea “en coup de sabre”: an unusual oral presentation. J Clin Exp Dent (2017) 9(2):e315–8. doi:10.4317/jced.53151
29. Dhaliwal CA, MacKenzie AI, Biswas A. Perineural inflammation in morphea (localized scleroderma): systematic characterization of a poorly recognized but potentially useful histopathological feature. J Cutan Pathol (2014) 41(1):28–35. doi:10.1111/cup.12242
30. Goh C, Biswas A, Goldberg LJ. Alopecia with perineural lymphocytes: a clue to linear scleroderma en coup de sabre. J Cutan Pathol (2012) 39(5):518–20. doi:10.1111/j.1600-0560.2012.01889.x
31. Alfenas R, Niemeyer B, Bahia PTV, Niemeyer R, Balbi L. Parry-Romberg syndrome: findings in advanced magnetic resonance imaging sequences – case report. Radiol Bras (2014) 47(3):186–8. doi:10.1590/0100-3984.2013.1699
32. Sande A, Risbud M, Kshar A, Paranjpe AO. Progressive hemifacial atrophy. Dent Res J (Isfahan) (2013) 10(1):108–11. doi:10.4103/1735-3327.111810
33. Bucher F, Fricke J, Cursiefen C, Heindl LM. Trigeminal involvement in parry-romberg syndrome: an in vivo confocal microscopy study of the cornea. Cornea (2015) 34(4):e10–1. doi:10.1097/ICO.0000000000000390
34. Pinho J, Rocha J, Sousa F, Macedo C, Soares-Fernandes J, Cerqueira J, et al. Localized scleroderma en coup de sabre in the Neurology Clinic. Mult Scler Relat Disord (2016) 8:96–8. doi:10.1016/j.msard.2016.05.013
35. Polcari I, Moon A, Mathes EF, Gilmore ES, Paller AS. Headaches as a presenting symptom of linear morphea en coup de sabre. Pediatrics (2014) 134(6):e1715–9. doi:10.1542/peds.2014-0019
36. Bucher F, Fricke J, Neugebauer A, Cursiefen C, Heindl LM. Ophthalmological manifestations of Parry-Romberg syndrome. Surv Ophthalmol (2016) 61(6):693–701. doi:10.1016/j.survophthal.2016.03.009
37. Careta MF, Romiti R. Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol (2015) 90(1):62–73. doi:10.1590/abd1806-4841.20152890
38. Rafai MA, Boulaajaj FZ, El Moutawakil B, Bourezgui M, Sibai M, Mahtar M, et al. Parry-Romberg syndrome with dysphonia. Rev Neurol (Paris) (2007) 163(12):1246–8. doi:10.1016/S0035-3787(07)78412-6
39. Mugundhan K, Selvakumar CJ, Gunasekaran K, Thiruvarutchelvan K, Sivakumar S, Anguraj M, et al. Parry-Romberg syndrome (progressive hemifacial atrophy) with spasmodic dysphonia – a rare association. J Assoc Physicians India (2014) 62(4):340–2.
40. Corbally CM, Breckenridge A, Jampana R. Imaging and clinical findings in a case of linear scleroderma en coup de sabre. BJR Case Rep (2016) 2:20150203. doi:10.1259/bjrcr20150203
41. Pinheiro TP, Silva CC, Silveira CS, Botelho PC, Pinheiro MD, Pinheiro Jde J. Progressive hemifacial atrophy – case report. Med Oral Patol Oral Cir Bucal (2006) 11(2):E112–4.
42. Unterberger I, Trinka E, Engelhardt K, Muigg A, Eller P, Wagner M, et al. Linear scleroderma “en coup de sabre” coexisting with plaque-morphea: neuroradiological manifestation and response to corticosteroids. J Neurol Neurosurg Psychiatry (2003) 74(5):661–4. doi:10.1136/jnnp.74.5.661
43. Patel H, Thakkar C, Patel K. Parry-Romberg syndrome: a rare entity. J Maxillofac Oral Surg (2010) 9(3):247–50. doi:10.1007/s12663-010-0103-y
44. Gutiérrez-Gómez C, Godínez-Hana AL, García-Hernández M, Suárez-Roa Mde L, Toussaint-Caire S, Vega-Memije E, et al. Lack of IgG antibody seropositivity to Borrelia burgdorferi in patients with Parry-Romberg syndrome and linear morphea en coup de sabre in Mexico. Int J Dermatol (2014) 53(8):947–51. doi:10.1111/ijd.12105
45. Buonaccorsi S, Leonardi A, Covelli E, Indrizzi E, Perdicchi A, Fini G. Parry-Romberg syndrome. J Craniofac Surg (2005) 16(6):1132–5. doi:10.1097/01.scs.0000183466.08332.be
46. Sommer A, Gambichler T, Bacharach-Buhles M, von Rothenburg T, Altmeyer P, Kreuter A. Clinical and serological characteristics of progressive facial hemiatrophy: a case series of 12 patients. J Am Acad Dermatol (2006) 54(2):227–33. doi:10.1016/j.jaad.2005.10.020
47. Clauser LC, Tieghi R, Consorti G. Parry-Romberg syndrome: volumetric regeneration by structural fat grafting technique. J Craniomaxillofac Surg (2010) 38(8):605–9. doi:10.1016/j.jcms.2010.02.005
48. Deshingkar SA, Barpande SR, Bhavthankar JD, Humbe JG. Progressive hemifacial atrophy (Parry-Romberg syndrome). Contemp Clin Dent (2012) 3(Suppl 1):S78–81. doi:10.4103/0976-237X.95111
49. Verma R, Ram H, Gupta M, Vidhate MR. A case of extensive left-sided facial atrophy of Romberg. Natl J Maxillofac Surg (2013) 4(1):77–80. doi:10.4103/0975-5950.117881
50. Mohan SV, Nittur V, Stevens KJ. Late-onset en coup de sabre of the skull. Skeletal Radiol (2013) 42(10):1447–50. doi:10.1007/s00256-013-1617-4
51. Lóderer Z, Janovszky Á, Lázár P, Piffkó J. Surgical management of progressive hemifacial atrophy with de-epithelialized profunda artery perforator flap: a case report. J Oral Maxillofac Surg (2017) 75(3):596–602. doi:10.1016/j.joms.2016.10.020
52. Pathi J, Mishra P, Kumar H, Panda A. Parry-Romberg syndrome affecting one half of the body. J Int Soc Prev Community Dent (2016) 6(4):387–90. doi:10.4103/2231-0762.186792
53. Saigusa R, Asano Y, Sumida H, Sato S. A case of scleroderma en coup de sabre progressing gradually over 60 years. Scand J Rheumatol (2017) 46(1):83–4. doi:10.3109/03009742.2016.1158315
54. Koutlas IG. Syndromes affecting skin and mucosa. Atlas Oral Maxillofac Surg Clin North Am (2014) 22(2):135–51. doi:10.1016/j.cxom.2014.06.001
55. Pope E, Laxer RM. Diagnosis and management of morphea and lichen sclerosus and atrophicus in children. Pediatr Clin North Am (2014) 61(2):309–19. doi:10.1016/j.pcl.2013.11.006
56. Agostini T, Spinelli G, Marino G, Perello R. Esthetic restoration in progressive hemifacial atrophy (Romberg disease): structural fat grafting versus local/free flaps. J Craniofac Surg (2014) 25(3):783–7. doi:10.1097/SCS.0000000000000831
57. Segna E, Pucciarelli V, Beltramini GA, Sforza C, Silvestre FJ, Giannì AB, et al. Parry Romberg syndrome and linear facial scleroderma: management in pediatric population. J Biol Regul Homeost Agents (2017) 31(2 Suppl 1):131–8.
58. Leader DM. Scleroderma and dentistry: every dentist is a scleroderma specialist. J Mass Dent Soc (2007) 56(2):16–9.
59. Hørberg M, Lauesen SR, Daugaard-Jensen J, Kjaer I. Linear scleroderma en coup de sabre including abnormal dental development. Eur Arch Paediatr Dent (2015) 16(2):227–31. doi:10.1007/s40368-014-0148-6
60. Van der Veken D, De Haes P, Hauben E, Teughels W, Lambrechts P. A rare cause of gingival recession: morphea with intra-oral involvement. Oral Surg Oral Med Oral Pathol Oral Radiol (2015) 119(5):e257–64. doi:10.1016/j.oooo.2015.02.002
61. Miller K, Lehrhoff S, Fischer M, Meehan S, Latkowski JA. Linear morphea of the forehead (en coup de sabre). Dermatol Online J (2012) 18(12):22.
62. Walker D, Susa JS, Currimbhoy S, Jacobe H. Histopathological changes in morphea and their clinical correlates: results from the Morphea in adults and children cohort V. J Am Acad Dermatol (2017) 76(6):1124–30. doi:10.1016/j.jaad.2016.12.020
63. Pierre-Louis M, Sperling LC, Wilke MS, Hordinsky MK. Distinctive histopathologic findings in linear morphea (en coup de sabre) alopecia. J Cutan Pathol (2013) 40(6):580–4. doi:10.1111/cup.12124
64. Aydın H, Yologlu Z, Sargın H, Metin MR. Parry-Romberg syndrome. Physical, clinical, and imaging features. Neurosciences (Riyadh) (2015) 20(4):368–71. doi:10.17712/nsj.2015.4.20150142
65. Chokar G, Cerase A, Gough A, Hasan S, Scullion D, El-Sayeh H, et al. A case of Parry-Romberg syndrome and alien hand. J Neurol Sci (2014) 341(1–2):153–7. doi:10.1016/j.jns.2014.04.004
66. Okumura A, Ikuta T, Tsuji T, Kato T, Fukatsu H, Naganawa S, et al. Parry-Romberg syndrome with a clinically silent white matter lesion. AJNR Am J Neuroradiol (2006) 27(8):1729–31.
67. Kumar S, Kumar D, Kumar R, Rajkumar, Prabhakar R. Parry-Romberg syndrome: a case of late onset with rapid progression. J Clin Diagn Res (2014) 8(9):ZD27–8. doi:10.7860/JCDR/2014/10292.4849
68. Trisal D, Kumar N, Dembla G, Sundriyal D. Parry-Romberg syndrome: uncommon but interesting. BMJ Case Rep (2014) 2014:bcr2013201969. doi:10.1136/bcr-2013-201969
69. Vix J, Mathis S, Lacoste M, Guillevin R, Neau JP. Neurological manifestations in Parry-Romberg syndrome: 2 case reports. Medicine (Baltimore) (2015) 94(28):e1147. doi:10.1097/MD.0000000000001147
70. Dibaj P, Herrendorf G, Bahn E, Obermann M. Late progression of neurological symptoms and MRI T2 hyperintensities in Parry-Romberg syndrome. J Neurol Neurosurg Psychiatry (2016) 87(11):1254–5. doi:10.1136/jnnp-2016-313091
71. Uhrhan K, Rabenstein M, Kobe C, Drzezga A, Fink GR, Burghaus L. Brain glucose metabolism in Parry-Romberg syndrome. Clin Nucl Med (2017) 42(5):e251–2. doi:10.1097/RLU.0000000000001558
72. Verma R, Dixit PK, Lalla R, Singh B. Mirror movements in progressive hemifacial atrophy. Ann Indian Acad Neurol (2015) 18(2):246–8. doi:10.4103/0972-2327.150606
73. Kakisaka Y, So NK, Jones SE, Wang ZI, Mosher JC, Alexopoulos AV, et al. Intractable focal epilepsy contralateral to the side of facial atrophy in Parry-Romberg syndrome. Neurol Sci (2012) 33(1):165–8. doi:10.1007/s10072-011-0643-z
74. Asai M, Sago T, Utani A. Parry-Romberg syndrome associated with contralateral cerebral atrophy. Eur J Dermatol (2015) 25(6):624–5. doi:10.1684/ejd.2015.2651
75. Pistoia F, Carolei A, Pistoia ML, Sacco S. Facial myokymias in Parry-Romberg syndrome. Neurohospitalist (2017) 7(2):N1–2. doi:10.1177/1941874416656731
76. Seifert F, Bien CG, Schellinger PD, Saake M, Blümcke I, Weigel D, et al. Parry-Romberg syndrome with chronic focal encephalitis: two cases. Clin Neurol Neurosurg (2017) 113(2):170–2. doi:10.1016/j.clineuro.2010.10.009
77. Gupta R, Patil H. Parry-Romberg syndrome with multiple intracranial cysts: a rare case report. J Pediatr Neurosci (2016) 11(2):145–9. doi:10.4103/1817-1745.187645
78. Yanagishita S, Fukai K, Tsuruta D, Seto T, Shimono T, Okamura K, et al. Dyschromatosis symmetrica hereditaria complicated by intracranial hemangiomas and Parry-Romberg syndrome. J Dermatol (2016) 43(9):1106–8. doi:10.1111/1346-8138.13353
79. Wei-Feng Z, Yi-Bo Z, Jun Z, Wei-Guo Z. Parry-Romberg syndrome combined with hemifacial spasm: association or chance event? J Craniofac Surg (2011) 22(6):2397–9. doi:10.1097/SCS.0b013e318231fe86
80. Zahlane S, Adali N, Kissani N. Parry-Romberg syndrome associated with complete agenesis of the corpus callosum: an uncommon case report. Rev Neurol (Paris) (2015) 171(10):737–9. doi:10.1016/j.neurol.2015.04.011
81. De Lange IH, van Oploo AM, Halbertsma FJ, Roos FG, Bok LA. Parry Romberg syndrome presenting with a giant intracranial aneurysm: a case report. Oxford Med Case Reports (2017) 2017(5):omx017. doi:10.1093/omcr/omx017
82. Chang GY, Park SH, Youn YC, Kwon OS. Neuroimaging findings in scleroderma en coup de sabre. Neurology (2004) 63(11):2197–8. doi:10.1212/WNL.63.11.2197-b author reply 2197-8,
83. Khan MA, Shaw L, Eleftheriou D, Prabhakar P, Chong WK, Glover M. Radiologic improvement after early medical intervention in localised facial morphea. Pediatr Dermatol (2016) 33(2):e95–8. doi:10.1111/pde.12799
84. Miyamoto M, Kinoshita M, Tanaka K, Tanaka M. Recurrent myelitis in localized scleroderma. Clin Neurol Neurosurg (2014) 127:140–2. doi:10.1016/j.clineuro.2014.10.005
85. Takahashi T, Asano Y, Oka T, Miyagaki T, Tamaki Z, Nonaka S, et al. Scleroderma en coup de sabre with recurrent episodes of brain hemorrhage. J Dermatol (2016) 43(2):203–6. doi:10.1111/1346-8138.13023
86. Lis-Święty A, Brzezińska-Wcisło L, Arasiewicz H. Neurological abnormalities in localized scleroderma of the face and head: a case series study for evaluation of imaging findings and clinical course. Int J Neurosci (2017) 127(9):835–9. doi:10.1080/00207454.2016.1244823
87. Gambichler T, Kreuter A, Hoffmann K, Bechara FG, Altmeyer P, Jansen T. Bilateral linear scleroderma “en coup de sabre” associated with facial atrophy and neurological complications. BMC Dermatol (2001) 1:9. doi:10.1186/1471-5945-1-9
88. Holland KE, Steffes B, Nocton JJ, Schwabe MJ, Jacobson RD, Drolet BA. Linear scleroderma en coup de sabre with associated neurologic abnormalities. Pediatrics (2006) 117(1):e132–6. doi:10.1542/peds.2005-0470
89. Amaral TN, Neto JFM, Lapa AT, Peres FA, Guirao CR, Appenzeller S. Neurologic involvement in scleroderma en coup de sabre. Autoimmune Dis (2012):6. doi:10.1155/719685
90. Verhelst HE, Beele H, Joos R, Vanneuville B, Van Coster RN. Hippocampal atrophy and developmental regression as first sign of linear scleroderma “en coup de sabre”. Eur J Paediatr Neurol (2008) 12(6):508–11. doi:10.1016/j.ejpn.2007.12.001
91. Bergler-Czop B, Lis-Swiety A, Brzezińska-Wcisło L. Scleroderma linearis: hemiatrophia faciei progressiva (Parry-Romberg syndrom) without any changes in CNS and linear scleroderma “en coup de sabre” with CNS tumor. BMC Neurol (2009) 9:39. doi:10.1186/1471-2377-9-39
92. Carreño M, Donaire A, Barceló MI, Rumià J, Falip M, Agudo R, et al. Parry Romberg syndrome and linear scleroderma in coup de sabre mimicking Rasmussen encephalitis. Neurology (2007) 68(16):1308–10. doi:10.1212/01.wnl.0000259523.09001.7a
93. Echenne B, Sébire G. Parry Romberg syndrome and linear scleroderma en coup de sabre mimicking Rasmussen encephalitis. Neurology (2007) 69(24):2274. doi:10.1212/01.wnl.0000295701.56822.6f author reply 2274,
94. Ayyildiz O, Ayyildiz S, Durukan AH, Sobaci G. Progressive hemifacial atrophy with contralateral uveitis: a case report. Iran Red Crescent Med J (2015) 17(9):e16168. doi:10.5812/ircmj.16168
95. Fea AM, Aragno V, Briamonte C, Franzone M, Putignano D, Grignolo FM. Parry Romberg syndrome with a wide range of ocular manifestations: a case report. BMC Ophthalmol (2015) 15:119. doi:10.1186/s12886-015-0093-0
96. Kaya M, Sel Yilmaz C, Kurtaran H, Gunduz M. Chronologic presentation of a severe case of progressive hemifacial atrophy (parry-romberg syndrome) with the loss of an eye. Case Rep Otolaryngol (2014) 2014:703017. doi:10.1155/2014/703017
97. Zannin ME, Martini G, Athreya BH, Russo R, Higgins G, Vittadello F, et al. Ocular involvement in children with localised scleroderma: a multi-centre study. Br J Ophthalmol (2007) 91(10):1311–4. doi:10.1136/bjo.2007.116038 Epub 2007 May 2.,
98. Zulian F, Vallongo C, Woo P, Russo R, Ruperto N, Harper J, et al. Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum (2005) 52(9):2873–81. doi:10.1002/art.21264
99. Anderson PJ, Molony D, Haan E, David DJ. Familial Parry-Romberg disease. Int J Pediatr Otorhinolaryngol (2005) 69(5):705–8. doi:10.1016/j.ijporl.2004.12.004
100. Iranzo P, Lopez I, Palou J, Herrero C, Lecha M. Morphoea in three siblings. J Eur Acad Dermatol Venereol (2001) 15(1):46–7. doi:10.1046/j.1468-3083.2001.00153.x
101. Englert H, Roberts-Thomson PJ, Byth K, Manolios N. Familial scleroderma: nature, nurture or both? Intern Med J (2008) 38(4):235–42. doi:10.1111/j.1445-5994.2007.01525.x
102. Pham CM, Browning JC. Morphoea affecting a father and son. Pediatr Dermatol (2010) 27(5):536–7. doi:10.1111/j.1525-1470.2010.01277.x
103. Lis-Świety A, Mierzwińska K, Wodok-Wieczorek K, Widuchpwska M, Brzezińska-Wcisło L. Co-existence oif lichen sclerosus and localized scleroderma in female monozygotic twins. J Pediatr Adolesc Gynecol (2014) 27(6):e133–6. doi:10.1016/j.jpag.2013.11.010
104. Karaca NE, Aksu G, Karaca E, Tuzun F, Gunes AT, Ozkinay F, et al. Progressive morphea of early childhood tracing Blaschko’s lines on the face: involvement of X chromosome monosomy in pathogenesis and clinical prognosis. Int J Dermatol (2011) 50(11):1406–10. doi:10.1111/j.1365-4632.2011.04900
105. Canady J, Karrer S, Fleck M, Bosserhoff AK. Fibrosing connective tissue disorders of the skin: molecular similarities and distinctions. J Dermatol Sci (2013) 70(3):151–8. doi:10.1016/j.jdermsci.2013.03.005
106. Jacobe H, Ahn C, Arnett FC, Reveille JD. Major histocompatibility complex class Ι and class ΙΙ alleles may confer susceptibility to or protection against morphoea: findings from the morphoea in adults and children cohort. Arthritis Rheumatol (2014) 66(11):3170–7. doi:10.1002/art.38814
107. Limpers A, van Royen-Kerkhof A, van Roon JA, Radstake TR, Broen JC. Overlapping gene expression profiles indicative of antigen processing and the interferon pathway characterize inflammatory fibrotic skin diseases. Expert Rev Clin Immunol (2014) 10(2):231–41. doi:10.1586/1744666X.2014.872561
108. Makino T, Jinnin M, Etoh M, Yamane K, Kajihara I, Makino K, et al. Down-regulation of microRNA-196a in the sera and involved skin of localized scleroderma patients. Eur J Dermatol (2014) 24(4):470–6. doi:10.1684/ejd.2014.2384
109. Wortsman X, Ma L, Chung WK, Wortsman J. Evaluation of the CAV1 gene in clinically, sonographically and histologically proven morphea patients. Exp Dermatol (2015) 24(9):718–20. doi:10.1111/exd.12743
110. Panda AK, Gopinath G, Singh S. Parry-Romberg syndrome with hemimasticatory spasm in pregnancy; A dystonia mimic. J Neurosci Rural Pract (2014) 5(2):184–6. doi:10.4103/0976-3147.131675
111. Hassan I, Arif T, Anwar P. Thyroid dysfunctions in morphoea: a preliminary report. Indian J Dermatol Venereol Leprol (2014) 80(6):579. doi:10.4103/0378-6323.144230
112. Bonilla-Abadía F, Muñoz-Buitrón E, Ochoa CD, Carrascal E, Cañas CA. A rare association of localized scleroderma type morphoea, vitiligo, autoimmune hypothyroidism, pneumonotis, autoimmune thrombocytopenic purpura and central nervous system vasculitis. Case report. BMC Res Notes (2012) 5:689. doi:10.1186/1756-0500-5-689
113. Zhang Y, Zhu M, Li X, Miao J, Duan C, Cui L, et al. Relapsing-remitting lesions in a woman with progressive hemifacial atrophy and chronic hepatitis B virus infection: a case report. Medicine (Baltimore) (2016) 95(47):e5390. doi:10.1097/MD.0000000000005390
114. Sahin MT, Bariş S, Karaman A. Parry-Romberg syndrome: a possible association with borreliosis. J Eur Acad Dermatol Venereol (2004) 18(2):204–7. doi:10.1111/j.1468-3083.2004.00862.x
115. di Meo N, Stinco G, Nan K, Pinzani C, Trevisan G. Parry-Romberg syndrome: a case with a possible association with Lyme disease. Acta Dermatovenerol Alp Pannonica Adriat (2015) 24(4):77–9. doi:10.15570/actaapa.2015.20
116. Prinz JC, Kutasi Z, Weisenseel P, Pótó L, Battyáni Z, Ruzicka T. “Borrelia-associated early-onset morphea”: a particular type of scleroderma in childhood and adolescence with high titer antinuclear antibodies? Results of a cohort analysis and presentation of three cases. J Am Acad Dermatol (2009) 60(2):248–55. doi:10.1016/j.jaad.2008.09.023
117. Salpietro DC, Merlino MV, Bruglia S, Guarneri F, Vaccaro M. Linear scleroderma ‘en coup de sabre’ associated with facial atrophy in a patient seropositive for Borrelia burgdorferi: a true case of molecular mimicry? Pediatr Allergy Immunol (2004) 15(6):570–2. doi:10.1111/j.1399-3038.2004.00189.x
118. Kurian K, Shanmugam S, Elongavan MB. Facial hemiatrophy – a report of 5 cases. Indian J Dent Res (2003) 14(4):238–45.
119. Arif T, Majid I, Ishtiyaq Haji ML. Late onset ‘en coup de sabre’ following trauma: rare presentation of a rare disease. Our Dermatol Online (2015) 6(1):49–51. doi:10.7241/ourd.20151.12
120. Lipson J, O’Toole A, Kapur S. Delay in diagnosis of congenital linear scleroderma until adulthood. J Cutan Med Surg (2015) 19(2):156–8. doi:10.2310/7750.2014.14028
121. Blaszczyk M, Królicki L, Krasu M, Glinska O, Jablonska S. Progressive facial hemiatrophy: central nervous system involvement and relationship with scleroderma en coup de sabre. J Rheumatol (2003) 30(9):1997–2004.
122. Dixit S, Kalkur C, Sattur AP, Bornstein MM, Melton F. Scleroderma and dentistry: two case reports. J Med Case Rep (2016) 10(1):297. doi:10.1186/s13256-016-1086-1
Keywords: progressive hemifacial atrophy, linear scleroderma en coup de sabre, clinical features, histopathological findings, predisposing factors
Citation: Khamaganova I (2018) Progressive Hemifacial Atrophy and Linear Scleroderma En Coup de Sabre: A Spectrum of the Same Disease? Front. Med. 4:258. doi: 10.3389/fmed.2017.00258
Received: 16 October 2017; Accepted: 26 December 2017;
Published: 31 January 2018
Edited by:
Ivan V. Litvinov, McGill University, CanadaReviewed by:
Oleg E. Akilov, University of Pittsburgh, United StatesFeras M. Ghazawi, University of Ottawa, Canada
Anie Philip, McGill University, Canada
Copyright: © 2018 Khamaganova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irina Khamaganova, c3VwZXJiaXJpbmFAeWFuZGV4LnJ1