 Vítor Teixeira
Vítor Teixeira Lai-Shan Tam
Lai-Shan Tam- 1Rheumatology Department, Centro Hospitalar de Lisboa Norte, EPE, Hospital de Santa Maria, Lisbon, Portugal
- 2Department of Medicine and Therapeutics, The Prince of Wales Hospital, The Chinese University of Hong Kong, Sha Tin, Hong Kong
Introduction: The systemic inflammatory nature of systemic lupus erythematosus (SLE) is well patent not only in the diverse clinical manifestations of the disease but also in the increased risk of premature atherosclerosis and cardiovascular events (CVE), making SLE one of the most complex diseases to study and manage in clinical practice.
Aim: To travel from old aspects to modern insights on the physiopathology, new molecular biomarkers, imaging methods of atherosclerosis assessment, and the potential treatments of atherosclerosis in SLE.
Methods: We conducted a literature search using PubMed database and performed a critical review.
Conclusion/discussion: Several developments have taken place in the understanding of the relationship between SLE and premature atherosclerosis. Nevertheless, cardiovascular diseases are still the major cause of reduced life expectancy in SLE and the main cause of death. The lack of standardization methods for the imaging assessment of atherosclerosis in SLE and the multifactorial nature of the disease are well patriated in the difficulty of achieving consistent and reproducible results among studies that focus in cardiovascular risk assessment and prediction. A raising number of molecular biomarkers of atherosclerosis have been proposed, but the combination of several biomarkers and risk factors may better estimate cardiovascular disease risk. Moreover, the development of effective therapies to prevent progression of atherosclerosis and CVE shall address systemic inflammation.
Introduction
Atherosclerotic lesions were initially thought to result mainly from an abnormal accumulation of lipoproteins associated with a disorganization of the intima and deformation of the arterial wall (1). However, since inflammation was implicated in the pathogenesis of atherosclerosis, several studies have focused on the immunologic aspects of atherosclerosis. Alterations of specific immune functions play a pivotal role in all stages of atherosclerotic plaque development from its initiation to progression (2). Diseases characterized by systemic inflammation, like systemic lupus erythematosus (SLE), have been strongly linked to accelerated atherosclerosis and increased cardiovascular morbidity and mortality. This association is only partially explained by the presence of traditional Framingham cardiovascular risk factors (3).
About half a century ago, the reported mortality of patients with SLE was 50% in the first 5 years after diagnosis and was mainly related to disease activity (4). In Europe, the 5-year survival of SLE is currently at 95% (5) and the 10-year survival is around 90% (6). Despite this improvement of survival in the first years of disease, the pattern of mortality of SLE is for a long time known to be bimodal, with a late peak of mortality largely due to cardiovascular disease (7). This peak has remained almost unchanged in recent years, contributing to an important reduction in the average life expectancy of 20 years in SLE (8). In fact, community-based studies have found that cardiovascular diseases are currently the main cause of death in SLE (9).
Review Methodology
We conducted a selected sampling of the literature, using PubMed as a database, and we performed a critical review on the relationship of lupus and atherosclerosis. We focused on cardiovascular events (CVE), atherosclerosis physiopathology, molecular and imaging biomarkers, and treatment strategies for atherosclerosis prevention in SLE.
Results
Cardiovascular Events
Patients with SLE have several comorbidities that are related not only to the disease itself but also to treatments and associated autoimmune diseases, such as antiphospholipid syndrome (APS), making it difficult to stablish the exact etiology of CVE in many patients. Nevertheless, data suggests that SLE is per se a strong independent risk factor for the development of CVE, comparable even to type I diabetes mellitus (DM) (10). A large study that included 1874 SLE patients estimated a risk of 2.7-fold in acute CVE (stroke, myocardial infarction, angina, coronary intervention, and peripheral vascular disease) relative to the risk that would be expected based on the Framingham risk score (11). This risk was remarkably higher in the sub-group of younger women aged 35–44 years, in whom it reached a 50-fold risk (12). In the Toronto lupus cohort, the mean age of myocardial infarction was 49 years compared with the peak years of the general population of 65–74 years (13). Coronary artery disease is responsible for 30% of deaths in SLE (14). Also of concern, SLE patients have striking poorer outcomes after percutaneous coronary intervention (PCI) than non-SLE patients, being more likely to suffer a new myocardial infarction (15.6 versus 4.8%, p = 0.01) and repeat PCI at 1 year after the first PCI (31.3 versus 11.8%, p = 0.009) (15). Moreover, SLE patients have lower 3-year survival and higher need of re-intervention after coronary artery bypass grafting (16). A recent report from the USA Nationwide Inpatient Sample highlighted that from 1996 to 2012 there was an increase in the rates of hospitalization due to acute myocardial infarction and ischemic stroke in SLE patients, despite a reduction in hospitalization due to unstable angina (17).
The relative risk of cerebrovascular events in SLE is lower than for coronary events, but twice higher the risk in the general population. It is also particularly higher in the first year after diagnosis and in women younger than 50 years (18). Despite that and after exclusion of APS, the absolute risk is considered low before the age of 60, and the highest rate is found in those individuals ≥60 years old (18). SLE patients without clinical neurological involvement also have more visible perivascular spaces and white-matter hyperintensities on magnetic resonance imaging (MRI) than controls, suggesting a subclinical compromise of the cerebral small vessel integrity (19).
The risk of peripheral artery occlusive disease (PAOD) is also high and has been reported to be ninefold higher than the general population, and higher in the first year after diagnosis; afterward it tends to decline (20). As in myocardial infarction and ischemic stroke, the relative risk was particularly higher in younger women, principally those less than 34 years (HR = 47.6, 95% CI = 26.8–84.4, p < 0.001) (20). In a Spanish study, age was found to be the only independent variable of increased PAOD risk among the traditional risk factors (21). Apart from age, Hassan and colleagues, additionally found a positive correlation with DM, dyslipidaemia, smoking, extended duration of steroid use, the Systemic Lupus International Collaborating Clinics Damage Index, use of azathioprine or warfarin, and plasma levels of thrombotic variables (22). No association was found with age in a study by Bhatt and colleagues, while dyslipidaemia was the only traditional risk factor in this study associated with increased risk of PAOD (23).
Mechanisms of Atherosclerosis
Systemic lupus erythematosus may affect the integrity and repair mechanisms of endothelial cells through direct binding of antibodies to endothelial cells or deposition of circulating immune complexes (24). The consequent endothelial damage promotes atherogenesis, which has been divided into three stages (Figure 1) (2). The inflammatory process triggers the first stage, which consists of expression of surface molecules in the vascular wall that are responsible for adhesion of leukocytes [such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1)], their rolling (selectins) and attachment (integrins) (2, 25). In the second stage, the adherent leukocytes migrate across the intima layer and penetrate the media layer (2). The transmigration process is mediated by monocyte chemotactic protein-1 (MCP-1). In mice models of accelerated atherosclerosis, the knockout of MCP-1 is associated with a reduction of atherosclerosis (26), while in humans, elevated circulating levels of MCP-1 correlate to increased carotid intima-media thickness (IMT) (27). l-Homocysteine is another promoter of the leukocyte recruitment by inducing expression of MCP-1 and interleukin-8 (28). In SLE, the elevation of plasma homocysteine concentrations after oral methionine load is associated with elevated markers of endothelial damage and platelet activation, inducing a prothrombotic tendency (29).
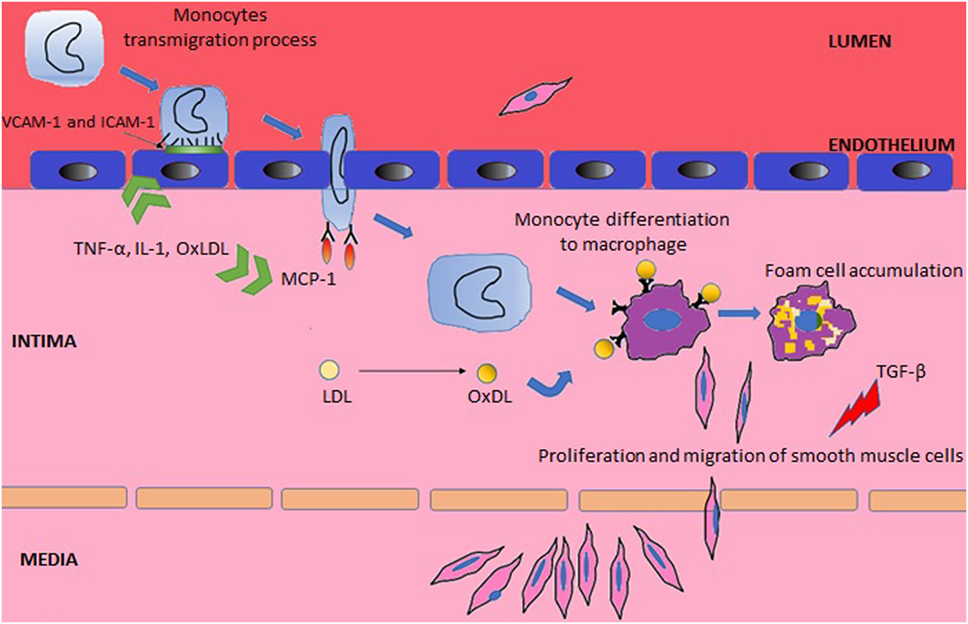
Figure 1. Main steps in the pathogenesis of atherosclerosis.
Tumor necrosis factor (TNF)-α, interleukin (IL)-1, and oxidized low-density lipoprotein (OxLDL) have an important role in the two first stages of atherosclerosis, as they upregulate adhesion molecules and MCP-1 (26, 30). Conversely, the transforming growth factor (TGF) supresses smooth muscle and endothelial cell proliferation (31). Decreased levels of TGF-β was associated with premature atherosclerosis in lupus-prone mice (32), supporting the hypothesis that downregulation of this cytokine can lead to arterial wall dysfunction with subsequent atherosclerosis development.
Finally, in the last stage, there is formation of macrophages foam cells and replication of smooth muscle cells leading to plaque propagation and rupture (2). This complex stage starts with the entrapment of low-density lipoprotein (LDL) in the subendothelial space where it is exposed to reactive oxygen species and then converted to OxLDL (33). The exposure of endothelial cells to OxLDL results in production of more MCP-1 and macrophage colony-stimulating factor, which further contribute to monocyte binding, chemotaxis, and differentiation to macrophages (34). In addition, when OxLDL is being exposed to macrophages, it contributes toward the inhibition of the phagocytosis of apoptotic cells and promotes the expression of the scavenger receptor CD36, which further increases the phagocytosis of OxLDL (35), leading to foam cells formation (36).
Oxidized low-density lipoprotein, unlike native LDL, can also form complexes with β2-GPI, which are significantly elevated in SLE with or without APS (37). The presence of IgG β2-GP1 increases OxLDL uptake by macrophages (38). Both, elevated levels of circulating OxLDL and of antibodies against OxLDL, are more common in SLE patients with CVD, than patients without CVD (39). Elevation of antibodies against epitopes of OxLDL has also been associated with maximum IMT and progression of atherosclerosis (40). Nevertheless, the heterogeneity among Ig subclasses and epitopes specificity and affinity might explain diverse properties for oxLDL antibodies (41). In fact, only IgG antibodies are thought to promote atherogenesis, while IgM antibodies seem to protect atherosclerosis prone mice from inflammatory oxidized moieties (42).
Tumor necrosis factor-α also plays a role in the last stage as it suppresses lipoprotein lipase synthesis and thus inhibits the metabolism of triglycerides and very LDL (43). The protective mechanism of efflux of cellular cholesterol is thought to be impaired in SLE, as the responsible enzyme for this process, cholesterol 27-hydroxylase, has been showed to be decreased in incubated human monocytes and aortic endothelial cells of SLE patients (44).
Notably, one of the most important overexpressed cytokine in SLE, interferon (IFN)-γ, upregulates several pro-atherogenic processes such as the production of lipid mediators, platelet-activating factors and eicosanoids, antigen presentation, and synthesis of TNF-α and IL-1 (45). The activation of toll-like receptors 7 and 9 is responsible for the upregulation of IFN-α expression (46). Mice models exposed to IFN-α have increased apoptosis, dysfunctional endothelial progenitor cells (EPCs), and reduced number of endothelial and smooth muscle cells (47). The pro-atherogenic effect of IFN is substantiated in other mice experiments, where a correlation was found between the depletion and dysfunction of EPCs and excessive type I IFN levels (48). The levels of these EPCs correlate inversely with cardiovascular risk in the general population (49). Furthermore, IFN is also responsible for plaque instability, as it inhibits the growth of smooth muscle cells, endothelial cells, and the production of collagen trough mechanisms that are not fully understood (50).
Accelerated atherosclerosis in SLE may be also related with the presence of antiphospholipid antibodies (aPL), known to increase the risk of thrombosis in SLE through several mechanisms. APL interact with endothelial cells and monocytes inducing a pro-inflammatory and pro-coagulant phenotype (51, 52) and activate the complement, that generates C5a which then activates neutrophils and expression of tissue factor (responsible for the initiating of the extrinsic coagulation cascade) (53). Specific autoantibodies against phospholipids also induce the expression of adhesion molecules, such as ICAM-1, VCAM-1, and E-selectin (54). Other mechanisms of atherogenesis include augmentation of the production of intracellular ROS (55) and promotion of lipid peroxidation of lipoproteins by reducing the activity of paraxonase 1 activity (56).
A new input for the understanding of the relationship between lupus and atherosclerosis came with the identification of NETosis as a key pathophysiological element. NETosis is a type of cell death pathway that results from externalization of chromatin fibers decorated with granule-derived antimibrobial peptides and is one of the defense mechanisms of neutrophils against pathogens (57). Neutrophil extracellular traps (NETs) contain many proinflammatory antimicrobial molecules, such as neutrophil elastase, IL17, human cathelicidin (LL 37), myeloperoxidase, histones, and MMP-9 (58–60). Many of these molecules induce endothelial cell death and vascular disfunction.
In SLE, there is a deficient clearing of these NETs, which increases production and release of type I IFN and further enhances NETosis (59). Impaired degradation of NETs is presumably due to the presence of antibodies against DNase (deoxyribonuclease) I, the main degrader of NETs, or due to the presence of anti-NET autoantibodies that protect NETs from degradation (61). NETs also lead to increased inflammasome activation, increasing the synthesis of activated IL1β and IL-18, which induce a positive loop of NET formation (62).
Low-density granulocytes (LDGs), a subtype of neutrophils prevalent in SLE, are particular prone to predispose NETosis (63). LDG NETs are deleterious for the endothelium and thus thought to contribute for accelerated atherosclerosis in SLE, as they synthesize increased levels of pro-inflammatory cytokines, mainly IFN-α and disrupt the differentiation of EPC to mature endothelial cells (59, 60, 63). LDGs also display significant increases in mitochondrial ROS production (63). The oxidation of mitochondrial DNA (mtDNA), mediated by ROS, allows migration of mtDNA to the cellular surface, thereby triggering potent pro-inflammatory and interferogenic responses (63). In fact, mitochondrial ROS are required for maximal NET stimulation in animal models. There is additional evidence favoring a role of NETosis in atherosclerosis progression, coming from studies that demonstrated the presence of NETs in atherosclerotic plaques (64) and that the inhibition of NET formation protected mice from atherosclerosis and mitigated type I IFN response (65).
Molecular Biomarkers
The progressive knowledge on the physiopathology of atherosclerosis has led to the search for potential biomarkers of cardiovascular disease and atherosclerosis progression, derived mainly from plasma measurements. In general, most molecular biomarkers studied so far have low to modest prediction value of atherosclerosis progression and CVE (66). Moreover, strong epidemiological studies that make direct comparisons of the value of different biomarkers in patients with SLE are lacking. Examining the individual role of each biomarker in the pathophysiology of atherosclerosis in SLE and the predictive value for atherosclerosis and CVE is not the objective of this review, and some of them have been previously discussed in Section “Mechanisms of Atherosclerosis.” Table 1 resumes some of the available literature regarding several molecular biomarkers and associations found with CVE and mortality and correlation with imaging markers of atherosclerosis in patients with SLE. We will focus on three newly identified atherosclerosis biomarkers in SLE: pentraxin-3 (PTX3), pro-inflammatory HDL (piHDL), and endocan. PTX3 is produced from mononuclear phagocytes, myeloid-derived dendritic cells, and endothelium cells in response to local inflammation, being considered a biomarker of local vascular inflammation (67). Levels of PTX3 are increased in SLE and correlate with disease activity (68). Furthermore, levels of PTX3 correlate with other indicators of endothelial dysfunction such as the soluble VCAM-1 and vWf (69). These data suggest that PTX3 could be a novel biomarker for premature atherosclerosis in SLE.
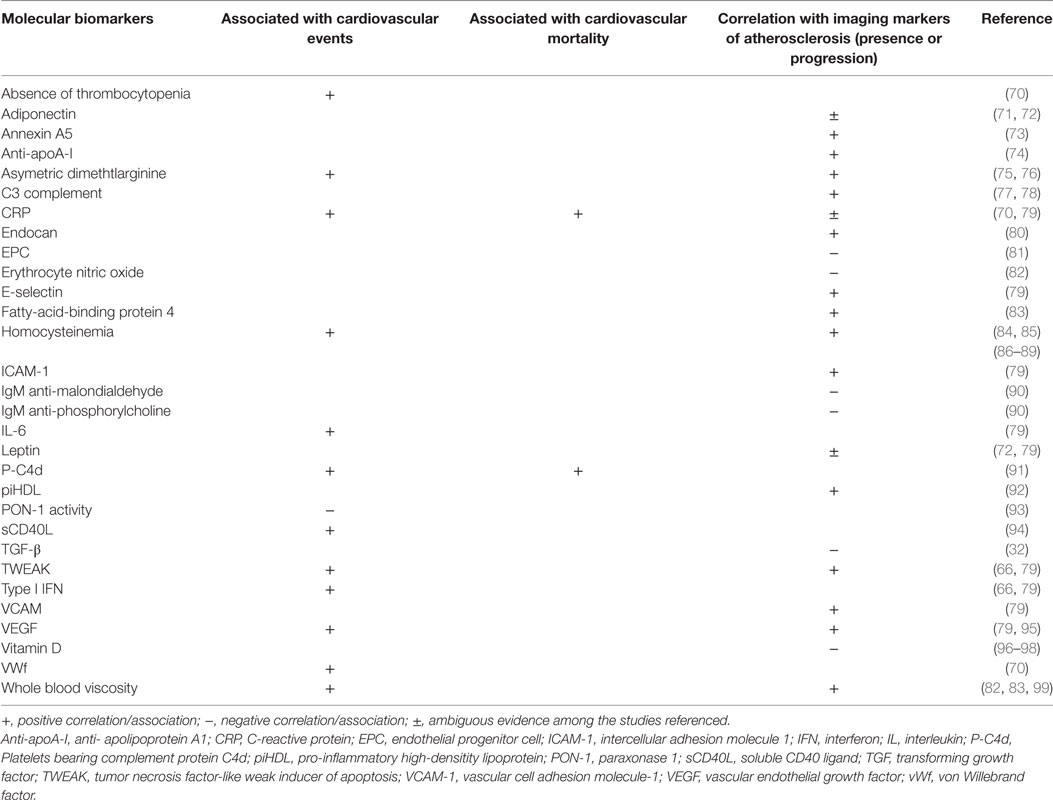
Table 1. Molecular biomarkers and associations with cardiovascular events and mortality and atherosclerosis imaging surrogates.
Cholesterol HDL is generally considered atheroprotective, but can lose its protective effect against the oxidation of LDL. This happens when it is converted from the usual anti-inflammatory form to the piHDL form, which usually occurs in the advent of chronic inflammatory states, like in SLE (92). The piHDL form is associated with progression of carotid plaques (odds ratio 16.1, p < 0.001) and IMT (odds ratio 2.5, p = 0.02) in patients with SLE (66, 100).
Endocan is an indicator of angiogenesis and endothelial cell activation (101) and participates in the recruitment, adhesion, and migration of leukocytes across the endothelium (102). In SLE, one study addressed its effects in atherosclerosis and concluded that serum levels were greater in these patients than in controls; in addition, endocan levels correlated positively with cIMT (r = 0.469, p < 0.01) (80).
Combinations of biomarkers may predict more accurately the atherosclerosis risk and CVE risk than isolated biomarkers (66). For SLE, it has been developed the Predictors of Risk for Elevated Flares, Damage Progression, and Increased Cardiovascular Disease in SLE (PREDICTS) model, which includes four inflammatory biomarkers (homocysteine, piHDL, TWEAK, and leptin) and two risk factors (age and diabetes) (66). In the population studied, none of the individual biomarkers had a good balance of strong positive predictive value and negative predictive value plus high specificity and sensitivity. On the contrary, the complete panel had a better predictive capacity for the longitudinal presence of plaque in SLE patients than did the individual parameters. Remarkably, a high-risk score conferred a 28-fold increased odds ratio of carotid plaque and an 8-fold increased odds ratio for cIMT progression, both with statistical significance.
Currently, however, combining several biomarkers has limited use in daily clinical practice, is probably not cost effective and needs to be validated in other SLE cohorts. Moreover, evidence-based treatment decisions according to a determinate risk score will be needed before wide use.
Imaging Assessment of Atherosclerosis
Subclinical atherosclerosis is an early finding in SLE patients and an important predictor of cardiovascular risk and morbidity (84). Subclinical atherosclerosis should not be overlooked, because it contributes to peripheral embolism, pre-hypertension, or hypertension and increased left ventricular afterload that can result in left ventricular hypertrophy and diastolic dysfunction (103, 104).
To access subclinical atherosclerosis, several non-invasive imaging surrogate indexes have been used for evaluation of morphological or functional arterial alterations: IMT, detection of atherosclerotic plaques, flow-mediated dilation (FMD), pulse wave velocity (PWV), and myocardial perfusion studies using single photon emission computed tomography (SPECT) dual isotope or MRI (Table 2). For large-scale populations, cIMT has been the most widely used, being able to predict stroke and myocardial infarction in the general population (105, 106). The detection of atherosclerotic plaques is, however, a better predictor of cardiovascular risk than IMT alone (107, 108). For early stages of atherosclerosis, IMT may be a more sensitive marker (109).
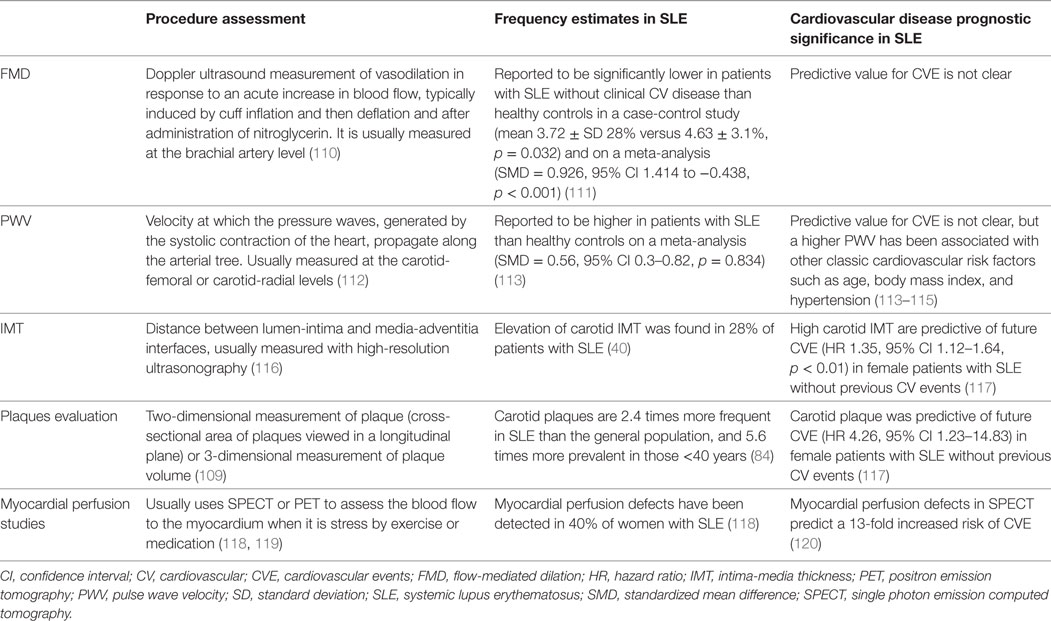
Table 2. Summary of atherosclerosis imaging surrogates in SLE.
Flow-Mediated Dilatation of the Brachial Artery
Vascular stiffness is usually estimated from FMD or PWV using vascular ultrasonography. A higher pulse pressure to achieve a certain degree of vessel distension suggests a lower vessel distensibility and thus a higher stiffness (121). Impaired FMD is thought to represent an early stage in the process of atherogenesis. SLE patients have significantly lower FMD, not fully explained by classic cardiovascular risk factors (111). Lupus nephritis, higher disease activity measured with SLE Disease Activity Index (SLEDAI), and higher levels of CRP have been correlated with impaired brachial endothelial function (122). Aortic stiffness, evaluated directly with transoesophageal echocardiography, is higher in SLE patients with hypertension, but normotensive SLE patients also have higher aortic stiffness than normotensive controls (123).
Pulse Wave Velocity
Systemic lupus erythematosus patients also have higher PWV (carotid-femoral or carotid-radial) than controls, but the predictive value for CVE is not clear (113). Despite that, PWV velocity has been associated with other classic cardiovascular risk factors such as age, body mass index, and hypertension (113–115, 124).
Intima-Media Thickness
Most studies determine IMT at the carotid level due to the better accessibility, but the prevalence, characteristics, risk factors, or predictors of aortic atherosclerosis are probably similar to carotid atherosclerosis (125–127). There is considerable heterogeneity regarding studies protocols in the method of measuring IMT in terms of the carotid segment investigated, unilateral/bilateral measurements, far wall, near wall, or both walls measurements and expression and mean or a maximal IMT. Nevertheless, there is clear evidence for a significant increase of cIMT in SLE patients, as showed in a systematic review and meta-analysis (116). In women with SLE without previous CVE, the cIMT at baseline was predictive of future CVE (117). This was especially true in the presence of concomitant carotid plaques. As for aortic IMT, values are also higher in SLE patients than age- and gender-matched controls (117).
Imaging Evaluation of Atherosclerotic Plaques
In SLE, carotid plaques are reported to be 2.4 times higher than the general population, with a peak of 5.6 times higher among patients <40 years (84). Patients at higher risk also include those with longer disease duration, higher damage index score, and less aggressive immunosuppressive therapies (84). Likewise, coronary artery calcifications evaluated through computed tomography appear to be more pronounced in younger SLE patients when compared to age-matched controls, but also in patients with increased disease activity (128). When assessing the risk of CVE related to atherosclerotic plaques, it has been suggested that quantification of plaques may reflect better the extent of atherosclerosis, rather than only assessing the presence or absence of plaques (109).
Myocardial Perfusion Studies
Using SPECT, myocardial perfusion defects were detected in 40% of women with SLE (118). A 13-fold increased risk of CVE was found in patients with SLE who had perfusion abnormalities on SPECT after a mean follow-up of 8.7 years (120). In other study using positron emission tomography, myocardial blood flow was measured in the resting phase and after administration of adenosine (hyperemic phase) (119). Inclusion criteria were patients suffering from either SLE or rheumatoid arthritis, with normal or minimally diseased coronary arteries (<20% luminal diameter reduction) at angiography and without cardiovascular risk factors. Overall, the study found that myocardial blood flow during adenosine hyperemia and coronary flow reserve were significantly blunted in patients compared with controls. Despite the small sample size, the study alerted for the fact that a reduced coronary flow reserve can be found in the absence of significant coronary artery disease and might be the result of prolonged inflammation that precedes premature coronary artery disease.
Myocardial perfusion studies using MRI have, additionally, led to conclude that SLE patients tend to have a distinct and more diffuse pattern of coronary artery wall contrast enhancement than coronary artery patients without SLE or healthy controls. These differences were explained by a more diffuse vascular inflammation in SLE (129).
Future Perspectives
Carotid ultrasound will remain probably the most used imaging method to assess atherosclerosis as it is relatively non-inexpensive and non-invasive and already validated. Still, there is great variation in IMT acquisition and analysis among studies. Although for SLE no specific recommendations exist for assessment of IMT, compliance with other published guidelines, such as from the American Society of Echocardiography cIMT task force (2012) or the Advisory Board for the “watching the risk symposia” at the European Stroke Conferences (2011), can help to standardize methods of assessment, improve the quality of future studies, and facilitate the comparison of results among them (130, 131). Similarly, the International Brachial Artery Reactivity Task Force guidelines should be applied when assessing FMD of the brachial artery (132). A combined approach, for example, assessment of IMT and plaque, may improve sensibility and risk prediction.
Predictive Factors of Premature Atherosclerosis
Traditional Risk Factors
As described elsewhere in this article, traditional risk factors could not account for the total burden of CVE in SLE; however, they are still very important and frequent in this population. In the Hopkins Lupus Cohort, several traditional risk factors like age, hypertension, obesity, and cholesterol >200 mg/day were showed to be the main predictors of coronary artery disease (109). Despite being a relatively young SLE cohort (average 38.3 ± 12.1 years), half the patients had three or more coronary artery disease risk factors, largely exceeding that of other same-nationality populations matched for age, race, and sex. Other cohorts have also shown, compared to healthy controls, a higher prevalence of sedentary lifestyle and obesity (85), older age (12, 14, 133), hypertension (14), dyslipidemia (12), and DM (85).
Tobacco use has also been linked to coronary events in SLE (7, 134), although other studies failed to prove this (135). Differences among the cardiovascular risk associated with smoking have been hypothesized to be due to race specific effects, as one study showed that history of smoking in SLE was associated with higher mean IMT measures in black women, but not white woman (136).
Disability caused by SLE can limit the ability to exercise. A sedentary life style has been identified as one of the most common risk factors for coronary artery disease, being present in 70% of patients (135). This was attributed to disease and treatment-related variables such as fatigue, anemia, arthritis, and avascular necrosis of bone.
Lupus Associated Risk Factors
Autoantibodies
Patients with primary aPL have not only an increased thrombophilic profile but also thicker cIMT than controls (137). In SLE, experimental models of atherosclerosis also support the role of aPL in the development of atherosclerotic plaques and a correlation with cIMT or carotid plaques has been demonstrated (138–140). Nonetheless, these associations are not reported in other large cohorts (141–143) and some studies even found a protective role for aPL in atherosclerosis in LDL receptor-deficient mice (144). These conflicting evidence warrants further studies to clarify the influence of aPL in atherosclerosis.
Antibodies against OxLDL, although not usually determined in clinical practice, have also been correlated with lupus activity and cIMT (145).
Whether anti-double-stranded DNA (anti-dsDNA) antibodies may contribute to premature atherosclerosis remained controversial as there are studies reporting positive association (140, 146), while other studies cannot find any association between cIMT or presence of carotid plaques (133, 147). The levels of complement were not found to correlate with cIMT (147).
Disease Activity and Clinical Manifestations
Differences among studies regarding disease activity indices are remarkable. In a Japanese SLE population, the SLEDAI was associated with cIMT in a dose-dependent manner (148). One Italian study found that active disease at baseline according to the European Consensus Lupus Activity Measurement index was predictive of carotid plaque and thickened mean IMT measured after 5 years of follow-up (40). Another Italian study did not show a correlation of cIMT with the SELENA-SLEDAI index measured in the preceding 3 months after cIMT assessment (147). In a study by Manzi and colleagues, the use of the Systemic Lupus Activity Measure (SLAM) index at the time of ultrasound examination was even inversely related to the presence of atherosclerotic plaque (109). These variances can be due to differences among the used activity indices and the time of assessment of disease activity and ultrasound. It has been proposed that studies should consider average activity scores over a long period of time, rather than single measures, as they may reflect better lupus activity and its relationship to atherosclerosis (20).
Neurolupus was found to be a strong predictor of CVE, with a 2.21 hazard ratio for psychosis (95% CI 1.10–4.44, p < 0.001) and 1.85 for seizures (95% CI 1.00–3.24, p = 0.007), suggesting it could also be a risk factor for accelerated atherosclerosis (149).
As for lupus nephritis, one study showed that cIMT, unlike FMD, wasn’t significantly different between SLE patients with and without nephritis (122). In a different study, women with nephrotic syndrome followed prospectively for 3 years were more likely to have progression of atherosclerosis, defined as increase in the cIMT >0.15 mm and/or an increase of the plaque score (relative risk = 4.22, 95% CI = 2.18–8.15, p = 0.22) (150).
Therapeutic Strategies
Large-scale randomized trials focusing on the prevention of atherosclerosis and CVE in SLE are lacking. Additionally, there are no current guidelines for the treatment and prevention of atherosclerosis in SLE. The available studies have mainly focused on the use of immunosuppressants and treatments for traditional cardiovascular risk factors. The results from studies of the effect of several drugs on atherosclerosis and CVE in SLE are summarized on Table 3.
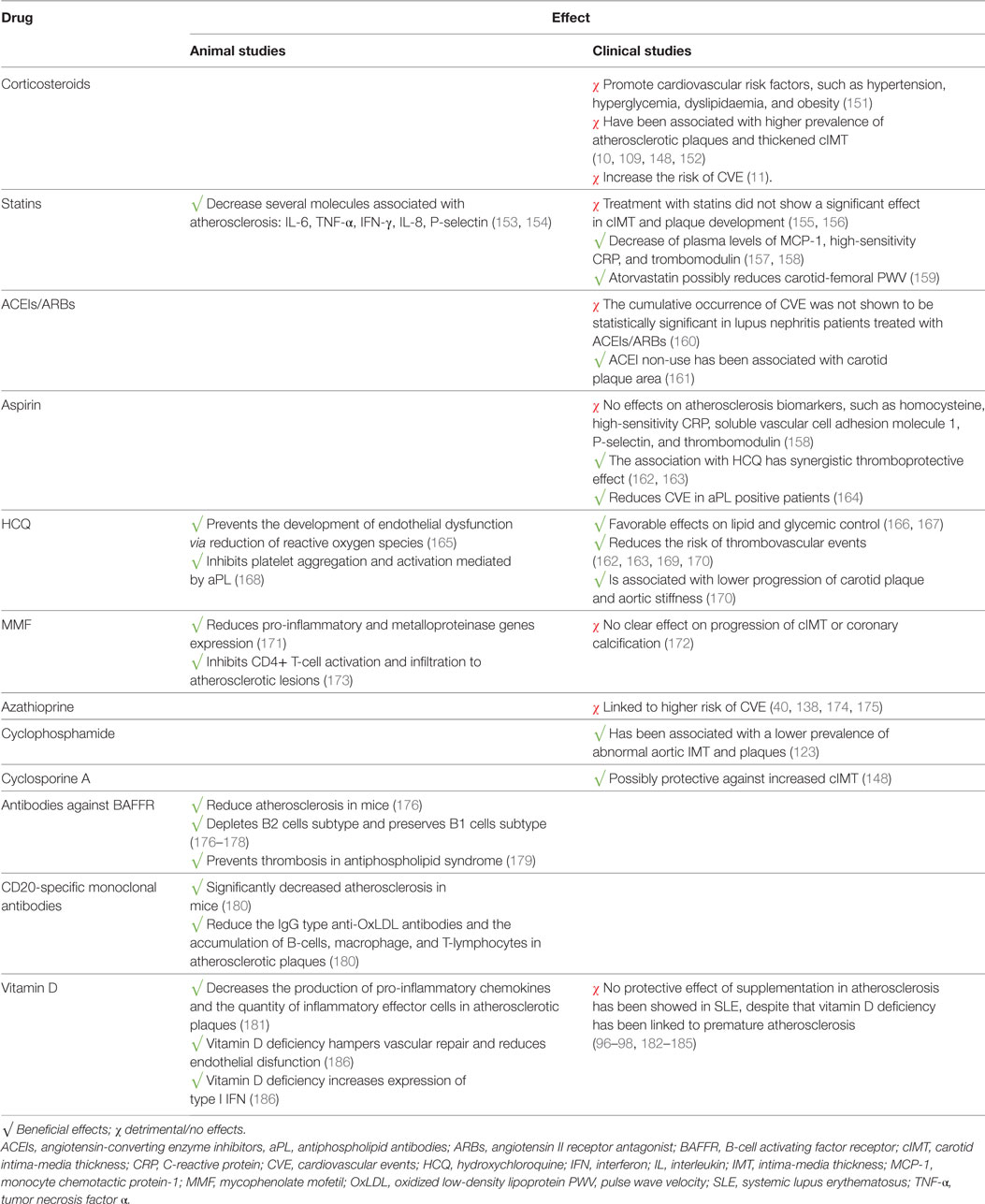
Table 3. Summary of the effects of several drugs on atherosclerosis and CVE in SLE.
Corticosteroids
There has been a long debate on the theoretical dual action of corticosteroids in atherosclerosis. Since inflammation plays a significant role in atherogenesis, one might think that the anti-inflammatory action of corticosteroids would reduce this risk. On the other hand, corticosteroids enhance classical cardiovascular risk factors such as hypertension, hyperglycemia, dyslipidaemia, and obesity (151). Different variables such as duration of treatment, cumulative, average (all time, only few years) or maximum doses have been evaluated in different studies, which can in part explain the mixed results. A positive correlation has been found with both duration of treatment (152) and cumulative dose (10, 148, 187).
When addressing the causative role of corticosteroids, one shall also consider that cumulative disease activity and some clinical phenotypes, such as nephritis and neuropsychiatric lupus, often warrant high doses of corticosteroids and have also been associated with more atherosclerosis in SLE and thus could be confounding factors. After adjusting for all these confounding factors, Manzi and colleagues demonstrated that a longer treatment duration with prednisolone (but not current use, maximum dose, or cumulative dose) was an independent determinant of carotid plaque, while disease activity (SLAM) determined at the time of the detection of the plaque was found to be inversely related to the presence of plaque (109). Completely different results were obtained by another study that reported current disease activity (SELENA-SLEDAI) and current dose of corticosteroids (>20 mg/day) were associated with a higher cardiovascular risk (11). Finally, it has been purposed that the action of corticosteroids may be dose-dependent, as one study in pediatric SLE showed an association between the highest and lowest cumulative doses of corticosteroids and a higher cIMT, while moderate cumulative doses were associated with a decreased cIMT (188).
Statins
Statins theoretical benefits are related to their effect in decreasing proinflammatory cytokines and chemokines such as IL-6, IL-8, TNF-α, and MCP-1 (153, 154, 157, 189). In a Chinese randomized, double-blind, placebo-controlled trial, low-dose rosuvastatin (10 mg/day) was associated with reduction of LDL, CRP, P-selectin, and thrombomodulin (158). Moreover, atorvastatin (20 mg/day) therapy for 8 weeks has shown to reduce arterial stiffness of SLE female patients who had baseline pathological carotid-femoral PWV, although this was only significant for middle-aged patients (36–59 years) (159). Atorvastatin at a dose of 40 mg/day may stabilize the coronary artery calcium score, although it did not ameliorate perfusion defects in myocardium SPECT (190). Results from the 2-year double-blinded Lupus Atherosclerosis Prevention Study were, unfortunately, more disappointing (155). In this study, 200 SLE patients without previous cardiovascular disease were randomized to receive atorvastatin (40 mg/day) or placebo. Helical-CT scanning for coronary artery calcium measurement and cIMT and plaque detection were performed at baseline and after 2 years, but no significant difference was found between the groups in terms of progression of these variables. Similar negative results have also been obtained in SLE pediatric populations (156).
ACEIs and Angiotensin II Receptor Antagonists (ARBs)
The renin–angiotensin–aldosterone system (RAAS) has been implicated in atherogenesis (191). In a SLE mice model, an enhance vasoconstriction response to the RAAS which promotes vascular changes during the SLE course was noticed (192). Despite this possible contribution for atherosclerosis development, only few studies have focused on the potentially protective role of ACEIs and ARBs in SLE. A non-randomized prospective trial, comprising 144 patients with lupus nephritis treated with ACEIs/ARBs and 301 non-treated patients, showed no significant differences in the cumulative occurrence of CVE between the groups (160). One cross-sectional study comprising 51 SLE patients, mainly African-American, of whom only 12 were being treated with ACEIs, showed a strong association between total plaque area and 25(OH)-vitamin D insufficiency or ACEI non-use (161).
Aspirin
In healthy women, aspirin had no significant effect on the risk of myocardial infarction or death from cardiovascular causes, with the exception of women 65 years of age or older (relative risk 0.66, 95% CI 0.44–0.97, p = 0.04) (193). In SLE, aspirin did not alter the level of several atherosclerosis biomarkers (homocysteine, high-sensitivity CRP, soluble VCAM-1, P-selectin, and thrombomodulin) (158). Moreover, in the SLE cohort of the SOLVABLE study, baseline aspirin was even correlated with progression of coronary artery calcifications (194). Nevertheless, the authors hypothesized that patients under aspirin were already identified as those who may benefit from aspirin use. The role of aspirin in patients with APS positivity should not be forgotten, as in these patients, aspirin reduces the cardiovascular risk and thrombotic events (164).
Hydroxychloroquine
Hydroxychloroquine (HCQ) has well-known benefits for lipid (166) and glycemic control (166, 167) and even reduces the risk of thrombovascular events (169, 170). In an animal model of SLE, early treatment with HCQ prevented the development of endothelial dysfunction via reduction of reactive oxygen species (165). Furthermore, HCQ has been shown to reduce the carotid plaque burden and aortic stiffness in SLE patients (170). One recent paper from Fasano and colleagues supported the association of HCQ and aspirin in patients with Lupus for primary prevention of CVE (162). The authors performed an observational study and multivariate analysis that lead to the conclusion that both aspirin and HCQ reduced the risk of the first CVE (hazard ratio 0.24 and 0.027, respectively), and they found a time-dependent effect of HCQ, as HCQ protective effect was only significant after 5 years of treatment. The same author found in a similar designed study that the association of aspirin to HCQ had a synergistic thromboprotective effect (163).
Mycophenolate Mofetil (MMF)
In mice, MMF slows down the progression of atherosclerosis by inhibiting CD4+ T-cell activation and infiltration to the atherosclerotic lesion (173). One interesting clinical study enrolled 22 SLE patients who were undergoing carotid endarterectomy and randomized them in two groups (171). One group received 1,000 mg of MMF for 2 weeks prior to the surgery, while the other group received placebo. When compared to the placebo group, the carotid plaques of the MMF group had reduced number of activated T-cells and increased number of regulatory T-cells and had also a reduced pro-inflammatory and metalloproteinase genes expression. Contrary to this trial, no beneficial effect of MMF in the progression of cIMT or coronary calcification was noted in a 2-year longitudinal cohort study, despite the fact that only 25 patients of the study received the drug and at variable doses (172).
Azathioprine
Several data point out that azathioprine is linked to a higher risk of cardiovascular disease (40, 138, 174, 175), but as for corticosteroids, azathioprine use is also associated with higher disease activity which may cofound the results.
Cyclophosphamide and Cyclosporine
Studies on other less commonly used immunosuppressive drugs are scarce. The prevalence of abnormal aortic IMT and plaques was found to be negatively correlated with cyclophosphamide therapy (123). Similarly, current use of cyclosporine A was found to be protective against increased cIMT (148).
Monoclonal Antibodies
CD20-specific monoclonal antibodies administered to apoE−/− and LDLr−/− mice were shown to significantly decrease atherosclerosis, probably through reduction of the IgG type anti-oxLDL antibodies and through reduction in the accumulation of B-cells, macrophage, and T-lymphocytes in atherosclerotic plaques (180).
Mice treated with antibodies against the B-cell activating factor receptor (BAFFR) or that lacked the BAFFR exhibited reduced atherosclerosis (176). This was postulated to be related with the consequent depletion of the atherogenic B2 cells subtype and the preservation of B1 cells subtype. This last subtype is considered atheroprotective, as it produces IgM antibodies against oxLDL and apoptotic cells (177, 178). BAFF inhibition also prevents APS in lupus-prone mice, suggesting it could have a potential benefit in preventing thrombosis in patients with SLE (179).
Vitamin D
Vitamin D regulates several important immune functions and its deficiency has been linked to premature atherosclerosis (182–185). In apolipoprotein E knockout mice, calcitriol treatment changes the function or differentiation of dendritic cells and regulatory T-cells, decreases the production of pro-inflammatory chemokines, and reduces the quantity of inflammatory effector cells in atherosclerotic plaques (181). For patients with SLE, however, evidence supporting the supplementation with vitamin D to reduce atherosclerosis progression are lacking. In fact, several studies failed to prove a protective effect in atherosclerosis (96–98).
Lifestyle Modifications
Smoking cessation should be recommended for all SLE patients, because smoking is a strong predictor of cardiovascular diseases with an odds ratio of 3.731 (CI 1.39–10.0) (195).
To our knowledge, only one non-randomized small-scale study analyzed the effect of supervised physical exercise on the endothelial function and ergospirometric test variables in SLE. Improved FMD, exercise tolerance, and threshold velocity after training was noted in the group of physical exercise (196).
Future Perspectives
No strong conclusion can be drawn from the currently available literature concerning the effects in atherosclerosis of most of the currently used drugs in SLE. Despite the lack of recommendations, it is prudent for clinicians to treat modifiable risk factors whenever possible.
Understanding that atherogenesis is an inflammatory process by nature is important for the future development of drugs aimed at stopping or reversing plaque formation. A meta-analysis concluded that regression/progression of cIMT induced by several drugs did not predict changes in the occurrence of major CVE (197), therefore, addressing both the reduction of atherosclerosis surrogate markers and CVE will be crucial for treatment recommendations.
Celastrol is a potential future candidate for slowing down atherosclerosis in SLE. Although not used currently for the treatment of SLE, the effect of this drug was tested in active chromatin-induced SLE BALB/c mice, where it was shown to improve proteinuria, lower anti-nuclear, and anti-dsDNA antibodies, reduce renal histological changes, and increase survival rate (198). In addition, celastrol inhibits several important atherosclerosis pathways such as the production of LDL, expression of VEGF, formation of lectin-like oxidized LDL receptor-1, and reactive oxygen species (199, 200). Plaque ratio reduction was achieved with this drug in a rabbit experimental model (199).
The 4-F peptide, an apoA-I fragment, was shown to convert HDL from pro-inflammatory to anti-inflammatory, prevent the inflammation induced by oxidized lipids and to dramatically reduce atherosclerosis in mice, suggesting that it could also be used in the future for the prevention of atherosclerosis (201, 202).
We have showed before that IL-1 is involved in atherosclerosis development, thus targeting the IL-1 pathway could be a potential therapeutic approach against atherosclerosis in patients with SLE. The Canakinumab Anti-Inflammatory Thrombosis Outcomes Study trial provided evidence that targeting IL-1β with canakinumab (150 mg every 3 months) was associated with significant lower incidence of nonfatal myocardial infarction, nonfatal stroke, or cardiovascular death (hazard ratio versus placebo, 0.85, p = 0.02) (203). The higher incidence of fatal infections noted in the canakinumab group, the lack of an established role in the treatment of SLE and their expensive cost will, however, limit the use of IL-1 blockers for preventing atherosclerosis in SLE.
If promising results are obtained in these trials, subsequent studies in SLE may be warranted.
Conclusion
We presented the current state of the research in premature atherosclerosis in SLE.
In SLE, cardiovascular diseases are the main cause of death, with risk ratios for several CVE (myocardial infarction, stroke, and peripheral artery disease) overcoming that of the general population. In our review, systemic inflammation was shown to be the cornerstone of the pathophysiological process of atherogenesis in lupus. New contributions for atherosclerosis development have been highlighted in recent years, namely, the role of EPCs, OxLDL and OxLDL autoantibodies, NETosis, and piHDL. Additionally, a raising number of molecular biomarkers have been proposed, but a direct comparison of these markers in terms of atherosclerotic events prediction risk has not been done so far. Biomarkers and risk factors integrated in panels are a promising strategy for enhancing CVD risk prediction, but they need to be validated in other cohorts.
We have also shown several conflicting results regarding lupus-related risk factors, namely, disease activity scores and the protective/harmful effect of several drugs, with most of the existing studies being observational. Different variable definitions may contribute to these differences. For example, many studies only evaluated the presence/absence of a drug as a CVD risk factor, others considered the highest dose or the cumulative dose; another example is the significant heterogeneity in IMT measurements among studies. In the future, standardization of variables definitions and of the imaging methods for evaluation of atherosclerosis should be sought according to their greatest clinical utility.
There is still a great need for controlled prospective studies for evaluating the efficacy of drugs in reducing not only atherosclerosis surrogates, but also CVE. So far, conventional treatment strategies for CVD, such as statins and ACEIs/ARBs, have not proved to be effective in preventing to reduce atherosclerosis progression in SLE. On the other hand, some immunomodulating agents, such as HCQ have demonstrated several favorable effects in CVE prevention, suggesting that disease-related factors may be more important in lupus atherogenesis than conventional risk factors and that future treatment strategies shall address systemic inflammation.
In conclusion, SLE and atherosclerosis have a multifactorial nature and an intricate relationship that has been considerably studied in the literature. Nevertheless, the current knowledge remains insufficient, as from a clinical point of view, it has not yet translated into effective approaches for prevention of both atherosclerosis progression and CVE and related mortality.
Author Contributions
All authors wrote the main manuscript text and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ACEI, angiotensin-converting enzyme inhibitor; Apo-AI, apolipoprotein A1; aPL, antiphospholipid antibodies; APS, antiphospholipid syndrome; ARB, angiotensin II receptor antagonist; BAFFR, B-cell activating factor receptor; cIMT, carotid intima-media thickness; CRP, C-reactive protein; CVE, cardiovascular events; DM, diabetes mellitus; dsDNA, double-stranded DNA; EPC, endothelial progenitor cell; FABP4, fatty-acid-binding protein 4; FMD, flow-mediated dilation; HCQ, hydroxychloroquine; ICAM-1, intercellular adhesion molecule 1; IFN, interferon; IL, interleukin; IMT, intima-media thickness; LDL, low-density lipoprotein; MCP-1, monocyte chemotactic protein-1; MMF, mycophenolate mofetil; MRI, magnetic resonance imaging; oxLDL, oxidized low-density lipoprotein; PAOD, peripheral arterial occlusive disease; PCI, percutaneous coronary intervention; piHDL, pro-inflammatory high-density lipoprotein; PON-1, paraxonase 1; PTX3, pentraxin-3; PWV, pulse wave velocity; RAAS, renin–angiotensin–aldosterone system; sCD40L, soluble CD40 ligand; SLE, systemic lupus erythematosus; SPECT, single photon emission computed tomography dual isotope; TGF, transforming growth factor; TNF-α, tumor necrosis factor α; TWEAK, tumor necrosis factor-like weak inducer of apoptosis; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor; vWf, von Willebrand factor.
References
1. Stary HC, Chandler AB, Glagov S, Guyton JR, Insull WJ, Rosenfeld ME, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb (1994) 14(5):840–56. doi:10.1161/01.ATV.14.5.840
2. Libby P. Inflammation in atherosclerosis. Nature (2002) 420(6917):868–74. doi:10.1038/nature01323
3. Esdaile JM, Abrahamowicz M, Grodzicky T, Li Y, Panaritis C, Berger RD, et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum (2001) 44(10):2331–7. doi:10.1002/1529-0131(200110)44:10<2331::AID-ART395>3.0.CO;2-I
4. Merrell M, Shulman LE. Determination of prognosis in chronic disease, illustrated by systemic lupus erythematosus. J Chronic Dis (1955) 1(1):12–32. doi:10.1016/0021-9681(55)90018-7
5. Cervera R, Khamashta MA, Font J, Sebastiani GD, Gil A, Lavilla P, et al. Morbidity and mortality in systemic lupus erythematosus during a 5-year period: a multicenter prospective study of 1,000 patients. Medicine (1999) 78(3):167–75. doi:10.1097/00005792-199905000-00003
6. Doria A, Iaccarino L, Ghirardello A, Zampieri S, Arienti S, Sarzi-Puttini P, et al. Long-term prognosis and causes of death in systemic lupus erythematosus. Am J Med (2006) 119(8):700–6. doi:10.1016/j.amjmed.2005.11.034
7. Urowitz MB, Bookman AA, Koehler BE, Gordon DA, Smythe HA, Ogryzlo MA. The bimodal mortality pattern of systemic lupus erythematosus. Am J Med (1976) 60(2):221–5. doi:10.1016/0002-9343(76)90431-9
8. Mok CC, Kwok CL, Ho LY, Chan PT, Yip SF. Life expectancy, standardized mortality ratios, and causes of death in six rheumatic diseases in Hong Kong, China. Arthritis Rheum (2011) 63(5):1182–9. doi:10.1002/art.30277
9. Stahl-Hallengren C, Jonsen A, Nived O, Sturfelt G. Incidence studies of systemic lupus erythematosus in Southern Sweden: increasing age, decreasing frequency of renal manifestations and good prognosis. J Rheumatol (2000) 27(3):685–91.
10. Tektonidou MG, Kravvariti E, Konstantonis G, Tentolouris N, Sfikakis PP, Protogerou A. Subclinical atherosclerosis in systemic lupus erythematosus: comparable risk with diabetes mellitus and rheumatoid arthritis. Autoimmun Rev (2017) 16(3):308–12. doi:10.1016/j.autrev.2017.01.009
11. Magder LS, Petri M. Incidence of and risk factors for adverse cardiovascular events among patients with systemic lupus erythematosus. Am J Epidemiol (2012) 176(8):708–19. doi:10.1093/aje/kws130
12. Manzi S, Meilahn EN, Rairie JE, Conte CG, Medsger TA Jr, Jansen-McWilliams L, et al. Age-specific incidence rates of myocardial infarction and angina in women with SLE: comparison with the Framingham Study. Am J Epidemiol (1997) 145(5):408–15. doi:10.1093/oxfordjournals.aje.a009122
13. Nikpour M, Urowitz MB, Gladman DD. Premature atherosclerosis in systemic lupus erythematosus. Rheum Dis Clin North Am (2005) 31(2):329–54. doi:10.1016/j.rdc.2005.01.001
14. Aranow C, Ginzler EM. Epidemiology of cardiovascular disease in systemic lupus erythematosus. Lupus (2000) 9(3):166–9. doi:10.1191/096120300678828208
15. Maksimowicz-Mckinnon K, Selzer F, Manzi S, Kip KE, Mulukutla SR, Marroquin OC, et al. Poor 1-year outcomes after percutaneous coronary interventions in systemic lupus erythematosus: report from the national heart, lung, and blood institute dynamic registry. Circ Cardiovasc Interv (2008) 1(3):201–8. doi:10.1161/CIRCINTERVENTIONS.108.788745
16. Birdas TJ, Landis JT, Haybron D, Evers D, Papasavas PK, Caushaj PF. Outcomes of coronary artery bypass grafting in patients with connective tissue diseases. Ann Thorac Surg (2005) 79(5):1610–4. doi:10.1016/j.athoracsur.2004.10.052
17. Tektonidou MG, Wang Z, Ward MM. Brief report: trends in hospitalizations due to acute coronary syndromes and stroke in patients with systemic lupus erythematosus, 1996 to 2012. Arthritis Rheumatol (2016) 68(11):2680–5. doi:10.1002/art.39758
18. Arkema EV, Svenungsson E, Von Euler M, Sjöwall C, Simard JF. Stroke in systemic lupus erythematosus: a Swedish population-based cohort study. Ann Rheum Dis (2017) 76(9):1544–9. doi:10.1136/annrheumdis-2016-210973
19. Wiseman SJ, Bastin ME, Jardine CL, Barclay G, Hamilton IF, Sandeman E, et al. Cerebral small vessel disease burden is increased in systemic lupus erythematosus. Stroke (2016) 47(11):2722–8. doi:10.1161/STROKEAHA.116.014330
20. Chuang YW, Yu MC, Lin CL, Yu TM, Shu KH, Kao CH. Risk of peripheral arterial occlusive disease in patients with systemic lupus erythematosus: a nationwide population-based cohort study. Medicine (2015) 94(46):e21211. doi:10.1097/MD.0000000000002121
21. Erdozain JG, Villar I, Nieto J, Ruiz-Irastorza G. Peripheral arterial disease in systemic lupus erythematosus: prevalence and risk factors. J Rheumatol (2014) 41(2):310–7. doi:10.3899/jrheum.130817
22. Hassan AA, Habib HM, Eissa AA. Peripheral arterial disease in patients with systemic lupus erythematosus: a prospective controlled study. Int J Rheum Dis (2013) 16(3):319–24. doi:10.1111/1756-185x.12025
23. Bhatt S, Handa R, Gulati G, Sharma S, Pandey R, Aggarwal P, et al. Peripheral vascular disease in systemic lupus erythematosus. Lupus (2007) 16(9):720–3. doi:10.1177/0961203307081123
24. Prechl JCB, Czirják L. The endothelial deprotection hypothesis for lupus pathogenesis: the dual role of C1q as a mediator of clearance and regulator of endothelial permeability. F1000Research (2015) 4:24. doi:10.12688/f1000research.6075.2
25. Hansson GK. Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol (2001) 21(12):1876–90. doi:10.1161/hq1201.100220
26. Wang JM, Sica A, Peri G, Walter S, Padura IM, Libby P, et al. Expression of monocyte chemotactic protein and interleukin-8 by cytokine-activated human vascular smooth muscle cells. Arterioscler Thromb (1991) 11(5):1166–74. doi:10.1161/01.ATV.11.5.1166
27. Larsson PT, Hallerstam S, Rosfors S, Wallén NH. Circulating markers of inflammation are related to carotid artery atherosclerosis. Int Angiol (2005) 24(1):43–51.
28. Poddar R, Sivasubramanian N, Dibello PM, Robinson K, Jacobsen DW. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: implications for vascular disease. Circulation (2001) 103(22):2717–23. doi:10.1161/01.CIR.103.22.2717
29. Tam LS, Fan B, Li EK, Thomas GN, Yim SF, Haines CJ, et al. Patients with systemic lupus erythematosus show increased platelet activation and endothelial dysfunction induced by acute hyperhomocysteinemia. J Rheumatol (2003) 30(7):1479–84.
30. Torzewski J, Oldroyd R, Lachmann P, Fitzsimmons C, Proudfoot D, Bowyer D. Complement-induced release of monocyte chemotactic protein-1 from human smooth muscle cells: a possible initiating event in atherosclerotic lesion formation. Arterioscler Thromb Vasc Biol (1996) 16(5):673–7. doi:10.1161/01.ATV.16.5.673
31. Grainger DJ, Kemp PR, Liu AC, Lawn RM, Metcalfe JC. Activation of transforming growth factor-β is inhibited in transgenic apolipoprotein(a) mice. Nature (1994) 370(6489):460–2. doi:10.1038/370460a0
32. Jackson M, Ahmad Y, Bruce IN, Coupes B, Brenchley PE. Activation of transforming growth factor-β1 and early atherosclerosis in systemic lupus erythematosus. Arthritis Res Ther (2006) 8(3):R81. doi:10.1186/ar1951
33. Camejo G, Olofsson SO, Lopez F, Carlsson P, Bondjers G. Identification of Apo B-100 segments mediating the interaction of low density lipoproteins with arterial proteoglycans. Arteriosclerosis (1988) 8(4):368–77. doi:10.1161/01.ATV.8.4.368
34. Navab M, Hama SY, Cooke CJ, Anantharamaiah GM, Chaddha M, Jin L, et al. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: step 1. J Lipid Res (2000) 41(9):1481–94.
35. Miller YI, Viriyakosol S, Binder CJ, Feramisco JR, Kirkland TN, Witztum JL. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J Biol Chem (2003) 278(3):1561–8. doi:10.1074/jbc.M209634200
36. Navab M, Berliner JA, Watson AD, Hama SY, Territo MC, Lusis AJ, et al. The Yin and Yang of oxidation in the development of the fatty streak: a review based on the 1994 George Lyman Duff memorial lecture. Arterioscler Thromb Vasc Biol (1996) 16(7):831–42. doi:10.1161/01.ATV.16.7.831
37. Lopez D, Garcia-Valladares I, Palafox-Sanchez CA, De La Torre IG, Kobayashi K, Matsuura E, et al. Oxidized low-density lipoprotein/b2-glycoprotein I complexes and autoantibodies to oxLig-1/b2-glycoprotein I in patients with systemic lupus erythematosus and antiphospholipid syndrome. Am J Clin Pathol (2004) 121:426–36. doi:10.1309/2AUE-6HD4-W6TL-EUU5
38. Kobayashi K, Matsuura E, Liu Q, Furukawa J, Atsumi T, Sakairi N, et al. A specific ligand for beta(2)-glycoprotein I mediates autoantibody-dependent uptake of oxidized low density lipoprotein by macrophages. J Lipid Res (2001) 42:697–709.
39. Svenungsson E, Jensen-Urstad K, Heimbürger M, Silveira A, Hamsten A, de Faire U, et al. Risk factors for cardiovascular disease in systemic lupus erythematosus. Circulation (2001) 104(16):1887–93. doi:10.1161/hc4101.097518
40. Doria A, Shoenfeld Y, Wu R, Gambari PF, Puato M, Ghirardello A, et al. Risk factors for subclinical atherosclerosis in a prospective cohort of patients with systemic lupus erythematosus. Ann Rheum Dis (2003) 62(11):1071–7. doi:10.1136/ard.62.11.1071
41. Shoenfeld Y, Wu R, Dearing LD, Matsuura E. Are anti-oxidized lowdensity lipoprotein antibodies pathogenic or protective? Circulation (2004) 110:2552–8. doi:10.1161/01.CIR.0000143225.07377.EA
42. Matsuura E, Kobayashi K, Tabuchi M, Lopez LR. Oxidative modification of low-density lipoprotein and immune regulation of atherosclerosis. Prog Lipid Res (2006) 45:466–86. doi:10.1016/j.plipres.2006.05.001
43. Semb H, Peterson J, Tavernier J, Olivecrona T. Multiple effects of tumor necrosis factor on lipoprotein lipase in vivo. J Biol Chem (1987) 262(17):8390–4.
44. Reiss AB, Anwar K, Merrill JT, Chan ESL, Awadallah NW, Cronstein BN, et al. Plasma from systemic lupus patients compromises cholesterol homeostasis: a potential mechanism linking autoimmunity to atherosclerotic cardiovascular disease. Rheumatol Int (2010) 30(5):591–8. doi:10.1007/s00296-009-1020-6
45. Peilot H, Rosengren B, Bondjers G, Hurt-Camejo E. Interferon-gamma induces secretory group IIA phospholipase A2in human arterial smooth muscle cells. Involvement of cell differentiation, STAT-3 activation, and modulation by other cytokines. J Biol Chem (2000) 275(30):22895–904. doi:10.1074/jbc.M002783200
46. Avalos AM, Busconi L, Marshak-Rothstein A. Regulation of autoreactive B cell responses to endogenous TLR ligands. Autoimmunity (2010) 43(1):76–83.1. doi:10.3109/08916930903374618
47. Thacker SG, Zhao W, Smith CK, Luo W, Wang H, Vivekanandan-Giri A, et al. Type I interferons modulate vascular function, repair, thrombosis, and plaque progression in murine models of lupus and atherosclerosis. Arthritis Rheum (2012) 64(9):2975–85. doi:10.1002/art.34504
48. Thacker S, Duquaine D, Park J, Kaplan M. Lupus-prone New Zealand Black/New Zealand White F1 mice display endothelial dysfunction and abnormal phenotype and function of endothelial progenitor cells. Lupus (2010) 19(3):288–99. doi:10.1177/0961203309353773
49. Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med (2005) 353(10):999–1007. doi:10.1056/NEJMoa043814
50. Hansson GK, Jonasson L, Holm J, Clowes MM, Clowes AW. Gamma-interferon regulates vascular smooth muscle proliferation and Ia antigen expression in vivo and in vitro. Circ Res (1988) 63(4):712–9. doi:10.1161/01.RES.63.4.712
51. Meroni P, Raschi E, Testoni C, Borghi M. Endothelial cell activation by antiphospholipid antibodies. Clin Immunol (2004) 112(2):169–74. doi:10.1016/j.clim.2004.02.015
52. Harris E, Pierangeli S. Primary, secondary, and catastrophic antiphospholipid syndrome: what’s in a name? Semin Thromb Hemost (2008) 34(03):219–26. doi:10.1055/s-0028-1082265
53. Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol (2006) 177(7):4794–802. doi:10.4049/jimmunol.177.7.4794
54. Sallai KK, Nagy E, Bodó I, Mohl A, Gergely P. Thrombosis risk in systemic lupus erythematosus: the role of thrombophilic risk factors. Scand J Rheumatol (2007) 36(3):198–205. doi:10.1080/03009740601089283
55. Perez-Sanchez C, Ruiz-Limon P, Aguirre MA, Bertolaccini ML, Khamashta MA, Rodriguez-Ariza A, et al. Mitochondrial dysfunction in antiphospholipid syndrome: implications in the pathogenesis of the disease and effects of coenzyme Q(10) treatment. Blood (2012) 119(24):5859–70. doi:10.1182/blood-2011-12-400986
56. Lopez LR, Salazar-Paramo M, Palafox-Sanchez C, Hurley BL, Matsuura E, Garcia-De La Torre I. Oxidized low-density lipoprotein and beta2-glycoprotein I in patients with systemic lupus erythematosus and increased carotid intima-media thickness: implications in autoimmune-mediated atherosclerosis. Lupus (2006) 15(2):80–6. doi:10.1191/0961203306lu2267oa
57. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi:10.1126/science.1092385
58. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol (2011) 187(1):538–52. doi:10.4049/jimmunol.1100450
59. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra20. doi:10.1126/scitranslmed.3001201
60. Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis (2015) 74(7):1417–24. doi:10.1136/annrheumdis-2013-204837
61. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A (2010) 107(21):9813–8. doi:10.1073/pnas.0909927107
62. Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol (2013) 190(3):1217–26. doi:10.4049/jimmunol.1202388
63. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22(2):146–53. doi:10.1038/nm.4027
64. Megens RT, Vijayan S, Lievens D, Döring Y, van Zandvoort MA, Grommes J, et al. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb Haemost (2012) 107(3):597–8. doi:10.1160/TH11-09-0650
65. Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao W, Smith CK, et al. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis (2015) 74(12):2199–206. doi:10.1136/annrheumdis-2014-205365
66. McMahon M, Skaggs BJ, Grossman JM, Sahakian L, Fitzgerald J, Wong WK, et al. A panel of biomarkers is associated with increased risk of the presence and progression of atherosclerosis in women with systemic lupus erythematosus. Arthritis Rheumatol (2014) 66(1):130–9. doi:10.1002/art.38204
67. Mantovani A, Garlanda C, Doni A, Bottazzi B. Pentraxins in innate immunity: from C-reactive protein to the long pentraxin PTX3. J Clin Immunol (2008) 28(1):1–13. doi:10.1007/s10875-007-9126-7
68. Pang Y, Tan Y, Li Y, Zhang J, Guo Y, Guo Z, et al. Pentraxin 3 is closely associated with tubulointerstitial injury in lupus nephritis: a large multicenter cross-sectional study. Medicine (2016) 95(3):e2520. doi:10.1097/MD.0000000000002520
69. Cieślik PC, Hrycek A. Pentraxin 3 as a biomarker of local inflammatory response to vascular injury in systemic lupus erythematosus. Autoimmunity (2015) 48(4):242–50. doi:10.3109/08916934.2014.983264
70. Gustafsson JT, Simard JF, Gunnarsson I, Elvin K, Lundberg IE, Hansson LO, et al. Risk factors for cardiovascular mortality in patients with systemic lupus erythematosus, a prospective cohort study. Arthritis Res Ther (2012) 14(2):R46. doi:10.1186/ar3759
71. Reynolds HR, Buyon J, Kim M, Rivera TL, Izmirly P, Tunick P, et al. Association of plasma soluble E-selectin and adiponectin with carotid plaque in patients with systemic lupus erythematosus. Atherosclerosis (2010) 210(2):569–74. doi:10.1016/j.atherosclerosis.2009.12.007
72. Chung C, Long A, Solus J, Rho Y, Oeser A, Raggi P, et al. Adipocytokines in systemic lupus erythematosus: relationship to inflammation, insulin resistance and coronary atherosclerosis. Lupus (2009) 18(9):799–806. doi:10.1177/0961203309103582
73. Valer P, Paul B, Eugenia B, Camelia B. Annexin A5 as independent predictive biomarker for subclinical atherosclerosis and endothelial dysfunction in systemic lupus erythematosus patients. Clin Lab (2013) 59(3–4):359–67. doi:10.7754/Clin.Lab.2012.120521
74. Kiani AN, Fang H, Akhter E, Quiroga C, Simpson N, Alaupovic P, et al. Apolipoprotein-containing lipoprotein subclasses and subclinical atherosclerosis in systemic lupus erythematosus. Arthritis Care Res (2015) 67(3):442–6. doi:10.1002/acr.22430
75. Perna M, Roman MJ, Alpert DR, Crow MK, Lockshin MD, Sammaritano L, et al. Relationship of asymmetric dimethylarginine and homocysteine to vascular aging in systemic lupus erythematosus patients. Arthritis Rheum (2010) 62(6):1718–22. doi:10.1002/art.27392
76. Elmageed AM, Ahmed IK, Saleh BI, Ali SR. Exploring disease activity and cardiovascular events by raised serum asymmetric dimethyl arginine among systemic lupus erythematosus patients. Egypt J Immunol (2011) 18(1):43–9.
77. Rua-Figueroa I, Arencibia-Mireles O, Elvira M, Erausquin C, Ojeda S, Francisco F, et al. Factors involved in the progress of preclinical atherosclerosis associated with systemic lupus erythematosus: a 2-year longitudinal study. Ann Rheum Dis (2010) 69(6):1136–9. doi:10.1136/ard.2008.104349
78. Maksimowicz-McKinnon K, Magder LS, Petri M. Predictors of carotid atherosclerosis in systemic lupus erythematosus. J Rheumatol (2006) 33:2458–63.
79. Tselios K, Sheane BJ, Gladman DD, Urowitz MB. Optimal monitoring for coronary heart disease risk in patients with systemic lupus erythematosus: a systematic review. J Rheumatol (2016) 43(1):54–65. doi:10.3899/jrheum.150460
80. Icli A, Cure E, Cure MC, Uslu AU, Balta S, Mikhailidis DP, et al. Endocan levels and subclinical atherosclerosis in patients with systemic lupus erythematosus. Angiology (2016) 67(8):749–55. doi:10.1177/0003319715616240
81. Castejon R, Jimenez-Ortiz C, Valero-Gonzalez S, Rosado S, Mellor S, Yebra-Bango M. Decreased circulating endothelial progenitor cells as an early risk factor of subclinical atherosclerosis in systemic lupus erythematosus. Rheumatology (2014) 53(4):631–8. doi:10.1093/rheumatology/ket367
82. Santos MJCA, Pedro LM, Canhão H, Fernandes JFE, Silva JCD, Fonseca JE, et al. Hemorheological parameters are related to subclinical atherosclerosis in systemic lupus erythematosus and rheumatoid arthritis patients. Atherosclerosis (2011) 219(2):821–6. doi:10.1016/j.atherosclerosis.2011.08.026
83. Parra S, Cabré A, Marimon F, Ferré R, Ribalta J, Gonzàlez M, et al. Circulating FABP4 is a marker of metabolic and cardiovascular risk in SLE patients. Lupus (2014) 23(3):245–54. doi:10.1177/0961203313517405
84. Roman MJ, Shanker B-A, Davis A, Lockshin MD, Sammaritano L, Simantov R, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med (2003) 349(25):2399–406. doi:10.1056/NEJMoa035471
85. Bruce IN, Urowitz MB, Gladman DD, Ibañez D, Steiner G. Risk factors for coronary heart disease in women with systemic lupus erythematosus: the Toronto Risk Factor Study. Arthritis Rheum (2003) 48(11):3159–67. doi:10.1002/art.11296
86. Petri M. Thrombosis and systemic lupus erythematosus: the Hopkins lupus cohort perspective. Scand J of Rheumatol (1996) 25(4):191–3. doi:10.3109/03009749609069986
87. Petri M. Detection of coronary artery disease and the role of traditional risk factors in the Hopkins lupus cohort. Lupus (2000) 9(3):170–5. doi:10.1191/096120300678828226
88. Feldt JMV, Scalzi LV, Cucchiara AJ, Morthala S, Kealey C, Flagg SD, et al. Homocysteine levels and disease duration independently correlate with coronary artery calcification in patients with systemic lupus erythematosus. Arthritis Rheum (2006) 54(7):2220–7. doi:10.1002/art.21967
89. Asanuma Y, Oeser A, Shintani AK, Turner E, Olsen N, Fazio S, et al. Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N Engl J Med (2003) 349(25):2407–15. doi:10.1056/NEJMoa035611
90. Rahman M, Sing S, Golabkesh Z, Fiskesund R, Gustafsson T, Jogestrand T, et al. IgM antibodies against malondialdehyde and phosphorylcholine are together strong protection markers for atherosclerosis in systemic lupus erythematosus: regulation and underlying mechanisms. Clin Immunol (2016) 16(6–167):27–37. doi:10.1016/j.clim.2016.04.007
91. Kao AH, McBurney CA, Sattar A, Lertratanakul A, Wilson NL, Rutman S, et al. Relation of platelet C4d with all-cause mortality and ischemic stroke in patients with systemic lupus erythematosus. Transl Stroke Res (2014) 5(4):510–8. doi:10.1007/s12975-013-0295-9
92. Lenten BJV, Hama SY, Beer FCD, Stafforini DM, Mcintyre TM, Prescott SM, et al. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J Clin Invest (1995) 96(6):2758–67. doi:10.1172/JCI118345
93. Kiss E, Seres I, Tarr T, Kocsis Z, Szegedi G, Paragh G. Reduced paraoxonase1 activity is a risk for atherosclerosis in patients with systemic lupus erythematosus. Ann N Y Acad Sci (2007) 1108:83–91. doi:10.1196/annals.1422.009
94. Schonbeck U, Varo N, Libby P, Buring J, Ridker PM. Soluble CD40L and cardiovascular risk in women. Circulation (2001) 104(19):2266–8. doi:10.1161/hc4401.099447
95. Colombo BM, Cacciapaglia F, Puntoni M, Murdaca G, Rossi E, Rodriguez G, et al. Traditional and non traditional risk factors in accelerated atherosclerosis in systemic lupus erythematosus: role of vascular endothelial growth factor (VEGATS Study). Autoimmun Rev (2009) 8(4):309–15. doi:10.1016/j.autrev.2008.10.002
96. Kiani AN, Fang H, Magder LS, Petri M. Vitamin D deficiency does not predict progression of coronary artery calcium, carotid intima-media thickness or high-sensitivity C-reactive protein in systemic lupus erythematosus. Rheumatol (2013) 52(11):2071–6. doi:10.1093/rheumatology/ket271
97. Mok CC, Birmingham DJ, Leung HW, Hebert LA, Song H, Rovin BH. Vitamin D levels in Chinese patients with systemic lupus erythematosus: relationship with disease activity, vascular risk factors and atherosclerosis. Rheumatology (2012) 51(4):644–52. doi:10.1093/rheumatology/ker212
98. Jung JY, Koh BR, Bae CB, Kim HA, Suh CH. Carotid subclinical atherosclerosis is associated with disease activity but not vitamin D in Korean systemic lupus erythematosus. Lupus (2014) 23(14):1517–22. doi:10.1177/0961203314544185
99. Booth S, Chohan S, Curran JC, Karrison T, Schmitz A, Utset TO. Whole blood viscosity and arterial thrombotic events in patients with systemic lupus erythematosus. Arthritis Rheum (2007) 57(5):845–50. doi:10.1002/art.22766
100. Mcmahon M, Grossman J, Skaggs B, Fitzgerald J, Sahakian L, Ragavendra N, et al. Dysfunctional proinflammatory high-density lipoproteins confer increased risk of atherosclerosis in women with systemic lupus erythematosus. Arthritis Rheum (2009) 60(8):2428–37. doi:10.1002/art.24677
101. Sarrazin S, Adam E, Lyon M, Depontieu F, Motte V, Landolfi C, et al. Endocan or endothelial cell specific molecule-1 (ESM-1): a potential novel endothelial cell marker and a new target for cancer therapy. Biochim Biophys Acta (2006) 1765(1):25–37. doi:10.1016/j.bbcan.2005.08.004
102. Lee HG, Choi HY, Bae JS. Endocan as a potential diagnostic or prognostic biomarker for chronic kidney disease. Kidney Int (2014) 86(6):1079–81. doi:10.1038/ki.2014.292
103. Ryan MJ. The pathophysiology of hypertension in systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol (2009) 296(4):R1258–67. doi:10.1152/ajpregu.90864.2008
104. Roldan CA, Alomari IB, Awad K, Boyer NM, Qualls CR, Greene ER, et al. Aortic stiffness is associated with left ventricular diastolic dysfunction in systemic lupus erythematosus: a controlled transesophageal echocardiographic study. Clin Cardiol (2014) 37:83–90. doi:10.1002/clc.22218
105. O’Leary DH, Bots ML. Imaging of atherosclerosis: carotid intima-media thickness. Eur Heart J (2010) 31(14):1682–9. doi:10.1093/eurheartj/ehq185
106. Lorenz MW, Markus HS, Bots ML, Rosvall M, Sitzer M. Prediction of clinical cardiovascular events with carotid intima media thickness. A systematic review and meta-analysis. Circulation (2007) 115(4):459–67. doi:10.1161/CIRCULATIONAHA.106.628875
107. Inaba Y, Chen JA, Bergmann SR. Carotid plaque, compared with carotid intima-media thickness, more accurately predicts coronary artery disease events: a meta-analysis. Atherosclerosis (2012) 220(1):128–33. doi:10.1016/j.atherosclerosis.2011.06.044
108. Frerix M, Stegbauer J, Kreuter A, Weiner S. Atherosclerotic plaques occur in absence of intima-media thickening in both systemic sclerosis and systemic lupus erythematosus: a duplexsonography study of carotid and femoral arteries and follow-up for cardiovascular events. Arthritis Res Ther (2014) 16(1):R54. doi:10.1186/ar4489
109. Manzi S, Selzer F, Sutton-Tyrrell K, Fitzgerald SG, Rairie JE, Tracy RP, et al. Prevalence and risk factors of carotid plaque in women with systemic lupus erythematosus. Arthritis Rheum (1999) 42(1):51–60. doi:10.1002/1529-0131(199901)42:1<51::AID-ANR7>3.0.CO;2-D
110. Harris RA, Nishiyama SK, Wray DW, Richardson RS. Ultrasound assessment of flow-mediated dilation. Hypertension (2010) 55(5):1075–85. doi:10.1161/HYPERTENSIONAHA.110.150821
111. El-Magadmi M, Bodill H, Ahmad Y, Durrington PN, Mackness M, Walker M, et al. Systemic lupus erythematosus: an independent risk factor for endothelial dysfunction in women. Circulation (2004) 110(4):399–404. doi:10.1161/01.CIR.0000136807.78534.50
112. Pereira T, Correia C, Cardoso J. Novel methods for pulse wave velocity measurement. J Med Biol Eng (2015) 35(5):555–65. doi:10.1007/s40846-015-0086-8
113. Sacre K, Escoubet B, Pasquet B, Chauveheid MP, Zennaro MC, Tubach F, et al. Increased arterial stiffness in systemic lupus erythematosus (SLE) patients at low risk for cardiovascular disease: a cross-sectional controlled study. PLoS One (2014) 9(4):e94511. doi:10.1371/journal.pone.0094511
114. Cypiene A, Kovaite M, Venalis A, Dadoniene J, Rugiene R, Petrulioniene Z, et al. Arterial wall dysfunction in systemic lupus erythematosus. Lupus (2009) 18(6):522–9. doi:10.1177/0961203308099625
115. Sabio JM, Vargas-Hitos J, Zamora-Pasadas M, Mediavilla JD, Navarrete N, Ramirez A, et al. Metabolic syndrome is associated with increased arterial stiffness and biomarkers of subclinical atherosclerosis in patients with systemic lupus erythematosus. J Rheumatol (2009) 36(10):2204–11. doi:10.3899/jrheum.081253
116. Tyrrell PN, Beyene J, Feldman BM, Mccrindle BW, Silverman ED, Bradley TJ. Rheumatic disease and carotid intima-media thickness: a systematic review and meta-analysis. Arterioscler Thromb Vasc Biol (2010) 30(5):1014–26. doi:10.1161/ATVBAHA.109.198424
117. Kao AH, Lertratanakul A, Elliott JR, Sattar A, Santelices L, Shaw P, et al. Relation of carotid intima-media thickness and plaque with incident cardiovascular events in women with systemic lupus erythematosus. Am J Cardiol (2013) 112(7):1025–32. doi:10.1016/j.amjcard.2013.05.040
118. Bruce IN, Burns RJ, Gladman DD, Urowitz MB. Single photon emission computed tomography dual isotope myocardial perfusion imaging in women with systemic lupus erythematosus. I. Prevalence and distribution of abnormalities. J Rheumatol (2000) 27:2372–7.
119. Recio-Mayoral A, Mason JC, Kaski JC, Rubens MB, Harari OA, Camici PG. Chronic inflammation and coronary microvascular dysfunction in patients without risk factors for coronary artery disease. Eur Heart J (2009) 30(15):1837–43. doi:10.1093/eurheartj/ehp205
120. Nikpour M, Gladman DD, Ibanez D, Bruce IN, Burns RJ, Urowitz MB. Myocardial perfusion imaging in assessing risk of coronary events in patients with systemic lupus erythematosus. J Rheumatol (2009) 36(2):288–94. doi:10.3899/jrheum.080776
121. Oliver JJ, Webb DJ. Noninvasive assessment of arterial stiffness and risk of atherosclerotic events. Arterioscler Thromb Vasc Biol (2003) 23(4):554–66. doi:10.1161/01.ATV.0000060460.52916.D6
122. Sharma SK, Rathi M, Sahoo S, Prakash M, Dhir V, Singh S. Assessment of premature atherosclerosis in systemic lupus erythematosus patients with and without nephritis. Lupus (2016) 25(5):525–31. doi:10.1177/0961203315622822
123. Roldan C, Joson J, Qualls C, Sharrar J, Sibbitt W. Premature aortic stiffness in systemic lupus erythematosus by transesophageal echocardiography. Lupus (2010) 19(14):1599–605. doi:10.1177/0961203310377088
124. Selzer F, Sutton-Tyrrell K, Fitzgerald SG, Pratt JE, Tracy RP, Kuller LH, et al. Comparison of risk factors for vascular disease in the carotid artery and aorta in women with systemic lupus erythematosus. Arthritis Rheum (2004) 50(1):151–9. doi:10.1002/art.11418
125. Roldan PC, Ratliff M, Snider R, Macias L, Rodriguez R, Sibbitt W, et al. Aortic atherosclerosis in systemic lupus erythematosus. Rheumatology (2014) (Suppl 5). doi:10.4172/2161-1149.S5-006
126. Roldan CA, Joson J, Sharrar J, Qualls CR, Sibbitt WL. Premature aortic atherosclerosis in systemic lupus erythematosus: a controlled transesophageal echocardiographic study. J Rheumatol (2010) 37(1):71–8. doi:10.3899/jrheum.090665
127. Sen S, Hinderliter A, Sen PK, Simmons J, Beck J, Offenbacher S, et al. Aortic arch atheroma progression and recurrent vascular events in patients with stroke or transient ischemic attack. Circulation (2007) 116(8):928–35. doi:10.1161/CIRCULATIONAHA.106.671727
128. Sangiorgi G, Rumberger JA, Severson A, Edwards WD, Gregoire J, Fitzpatrick LA, et al. Arterial calcification and not lumen stenosis is highly correlated with atherosclerotic plaque burden in humans: a histologic study of 723 coronary artery segments using nondecalcifying methodology. J Am Coll Cardiol (1998) 31(1):126–33. doi:10.1016/S0735-1097(97)00443-9
129. Varma N, Hinojar R, D’Cruz D, Ucar EA, Indermuehle A, Peel S, et al. Coronary vessel wall contrast enhancement imaging as a potential direct marker of coronary involvement. JACC Cardiovasc Imaging (2014) 7(8):762–70. doi:10.1016/j.jcmg.2014.03.012
130. Stein JH, Korcarz CE, Hurst RT, Lonn E, Kendall CB, Mohler ER, et al. Use of carotid ultrasound to identify subclinical vascular disease and evaluate cardiovascular disease risk: a consensus statement from the American Society of Echocardiography Intima-Media Thickness Task Force. J Am Soc of Echocardiogr (2008) 21(2):93–111. doi:10.1016/j.echo.2007
131. Touboul PJ, Hennerici MG, Meairs S, Adams H, Amarenco P, Bornstein N, et al. Mannheim carotid intima-media thickness and plaque consensus (2004–2006–2011). An update on behalf of the advisory board of the 3rd, 4th and 5th watching the risk symposia, at the 13th, 15th and 20th European Stroke Conferences, Mannheim, Germany, 2004, Brussels, Belgium, 2006, and Hamburg, Germany, 2011. Cerebrovasc Dis (2012) 34(4):290–6. doi:10.1159/000343145
132. Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery. J Am Coll Cardiol (2002) 39(2):257–65. doi:10.1016/S0735-1097(01)01746-6
133. Wu GC, Liu HR, Leng RX, Li XP, Li XM, Pan HF, et al. Subclinical atherosclerosis in patients with systemic lupus erythematosus: a systemic review and meta-analysis. Autoimmun Rev (2016) 15(1):22–37. doi:10.1016/j.autrev.2015.10.002
134. Calvo-Alén J, Toloza SMA, Fernández M, Bastian HM, Fessler BJ, Roseman JM, et al. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA): XXV. Smoking, older age, disease activity, lupus anticoagulant, and glucocorticoid dose as risk factors for the occurrence of venous thrombosis in lupus patients. Arthritis Rheum (2005) 52(7):2060–8. doi:10.1002/art.21149
135. Petri M, Perez-Gutthann S, Spence D, Hochberg MC. Risk factors for coronary artery disease in patients with systemic lupus erythematosus. Am J Med (1992) 93(5):513–9. doi:10.1016/0002-9343(92)90578-Y
136. Scalzi L, Bhatt S, Gilkeson R, Shaffer M. The relationship between race, cigarette smoking and carotid intimal medial thickness in systemic lupus erythematosus. Lupus (2009) 18(14):1289–97. doi:10.1177/0961203309345781
137. Ames PRJ, Margarita A, Sokoll KB, Weston M, Brancaccio V. Premature atherosclerosis in primary antiphospholipid syndrome: preliminary data. Ann Rheum Dis (2005) 64(2):315–7. doi:10.1136/ard.2004.023952
138. Ahmad Y, Shelmerdine J, Bodill H, Lunt M, Pattrick MG, Teh LS, et al. Subclinical atherosclerosis in systemic lupus erythematosus (SLE): the relative contribution of classic risk factors and the lupus phenotype. Rheumatology (2007) 46(6):983–8. doi:10.1093/rheumatology/kem002
139. Gallelli B, Burdick L, Quaglini L, Banfi G, Novembrino C, Bamonti F, et al. Carotid plaques in patients with long-term lupus nephritis. Clin Exp Rheumatol (2010) 28(3):386–92.
140. Kiani AN, Vogel-Claussen J, Arbab-Zadeh A, Magder LS, Lima J, Petri M. Semiquantified noncalcified coronary plaque in systemic lupus erythematosus. J Rheumatol (2012) 39(12):2286–93. doi:10.3899/jrheum.120197
141. Leeuw KD, Freire B, Smit AJ, Bootsma H, Kallenberg CG, Bijl M. Traditional and non-traditional risk factors contribute to the development of accelerated atherosclerosis in patients with systemic lupus erythematosus. Lupus (2006) 15(10):675–82. doi:10.1177/0961203306069972
142. Vlachoyiannopoulos PG, Kanellopoulos PG, Ioannidis JP, Tektonidou MG, Mastorakou I, Moutsopoulos HM. Atherosclerosis in premenopausal women with antiphospholipid syndrome and systemic lupus erythematosus: a controlled study. Rheumatology (2003) 42(5):645–51. doi:10.1093/rheumatology/keg182
143. Farzaneh-Far A, Roman MJ, Lockshin MD, Devereux RB, Paget SA, Crow MK, et al. Relationship of antiphospholipid antibodies to cardiovascular manifestations of systemic lupus erythematosus. Arthritis Rheum (2006) 54(12):3918–25. doi:10.1002/art.22265
144. Nicolo D, Goldman BI, Monestier M. Reduction of atherosclerosis in low-density lipoprotein receptor-deficient mice by passive administration of antiphospholipid antibody. Arthritis Rheum (2003) 48(10):2974–8. doi:10.1002/art.11255
145. Fesmire J, Wolfson-Reichlin M, Reichlin M. Effects of autoimmune antibodies anti-lipoprotein lipase, anti-low density lipoprotein, and anti-oxidized low density lipoprotein on lipid metabolism and atherosclerosis in systemic lupus erythematosus. Rev Bras Reumatol (2010) 50:539–51. doi:10.1590/S0482-50042010000500007
146. Skaggs BJ, Hahn BH, Mcmahon M. Accelerated atherosclerosis in patients with SLE—mechanisms and management. Nat Rev Rheumatol (2012) 8(4):214–23. doi:10.1038/nrrheum.2012.14
147. Ammirati E, Bozzolo E, Contri R, Baragetti A, Palini A, Cianflone D, et al. Cardiometabolic and immune factors associated with increased common carotid artery intima-media thickness and cardiovascular disease in patients with systemic lupus erythematosus. Nutr Metab Cardiovasc Dis (2014) 24(7):751–9. doi:10.1016/j.numecd.2014.01.006
148. Oryoji K, Kiyohara C, Horiuchi T, Tsukamoto H, Niiro H, Shimoda T, et al. Reduced carotid intima-media thickness in systemic lupus erythematosus patients treated with cyclosporine A. Mod Rheumatol (2014) 24(1):86–92.1. doi:10.3109/14397595.2013.852838
149. Bertoli A, Vilá L, Alarcón G, Mcgwin G, Edberg J, Petri M, et al. Factors associated with arterial vascular events in PROFILE: a multiethnic lupus cohort. Lupus (2009) 18(11):958–65. doi:10.1177/0961203309104862
150. Telles RW, Lanna CC, Sousa AJ, Navarro TP, Souza FL, Rodrigues LA, et al. Progression of carotid atherosclerosis in patients with systemic lupus erythematosus. Clin Rheumatol (2013) 32(9):1293–300. doi:10.1007/s10067-013-2264-9
151. Sarnes E, Crofford L, Watson M, Dennis G, Kan H, Bass D. Incidence and US costs of corticosteroid-associated adverse events: a systematic literature review. Clin Ther (2011) 357:1413–32. doi:10.1016/j.clinthera.2011.09.009
152. Thompson T, Sutton-Tyrrell K, Wildman RP, Kao A, Fitzgerald SG, Shook B, et al. Progression of carotid intima-media thickness and plaque in women with systemic lupus erythematosus. Arthritis Rheum (2008) 58(3):835–42. doi:10.1002/art.23196
153. Aktas O, Waiczies S, Smorodchenko A, Dorr J, Seeger B, Prozorovski T, et al. Treatment of relapsing paralysis in experimental encephalomyelitis by targeting Th1 cells through atorvastatin. J Exp Med (2003) 197(6):725–33. doi:10.1084/jem.20021425
154. Leung BP, Sattar N, Crilly A, Prach M, Mccarey DW, Payne H, et al. A novel anti-inflammatory role for simvastatin in inflammatory arthritis. J Immunol (2003) 170(3):1524–30. doi:10.4049/jimmunol.170.3.1524
155. Petri MA, Kiani AN, Post W, Christopher-Stine L, Magder LS. Lupus atherosclerosis prevention study (LAPS). Ann Rheum Dis (2011) 70(5):760–5. doi:10.1136/ard.2010.136762
156. Schanberg LE, Sandborg C, Barnhart HX, Ardoin SP, Yow E, Evans GW, et al. Use of atorvastatin in systemic lupus erythematosus in children and adolescents. Arthritis Rheum (2012) 64(1):285–96. doi:10.1002/art.30645
157. Xu ZM, Zhao SP, Li QZ, Nie S, Zhou HN. Atorvastatin reduces plasma MCP-1 in patients with acute coronary syndrome. Clin Chim Acta (2003) 338(1–2):17–24. doi:10.1016/S0009-8981(03)00321-8
158. Mok CC, Wong CK, To CH, Lai JPS, Lam CS. Effects of rosuvastatin on vascular biomarkers and carotid atherosclerosis in lupus: a randomized, double-blind, placebo-controlled trial. Arthritis Care Res (2011) 63(6):875–83. doi:10.1002/acr.20440
159. Castejon R, Castañeda A, Sollet A, Mellor-Pita S, Tutor-Ureta P, Jimenez-Ortiz C, et al. Short-term atorvastatin therapy improves arterial stiffness of middle-aged systemic lupus erythematosus patients with pathological pulse wave velocity. Lupus (2016) 26(4):355–64. doi:10.1177/0961203316662719
160. Tselios K, Gladman DD, Su J, Urowitz MB. Does renin-angiotensin system blockade protect lupus nephritis patients from atherosclerotic cardiovascular events? A case-control study. Arthritis Care Res (2016) 68(10):1497–504. doi:10.1002/acr.22857
161. Ravenell RL, Kamen DL, Fleury TJ, Spence JD, Hollis BW, Janech MG, et al. Premature atherosclerosis is associated with hypovitaminosis D and angiotensin-converting enzyme inhibitor non-use in lupus patients. Am J Med Sci (2012) 344(4):268–73. doi:10.1097/MAJ.0b013e31823fa7d9
162. Fasano S, Margiotta DP, Navarini L, Pierro L, Pantano I, Riccardi A, et al. Primary prevention of cardiovascular disease in patients with systemic lupus erythematosus: case series and literature review. Lupus (2017) 26(14):1463–72. doi:10.1177/0961203317722847
163. Fasano S, Pierro L, Pantano I, Iudici M, Valentini G. Longterm hydroxychloroquine therapy and low-dose aspirin may have an additive effectiveness in the primary prevention of cardiovascular events in patients with systemic lupus erythematosus. J Rheumatol (2017) 44(7):1032–8. doi:10.3899/jrheum.161351
164. Tektonidou MG, Laskari K, Panagiotakos DB, Moutsopoulos HM. Risk factors for thrombosis and primary thrombosis prevention in patients with systemic lupus erythematosus with or without antiphospholipid antibodies. Arthritis Rheum (2009) 61(1):29–36. doi:10.1002/art.24232
165. Virdis A, Tani C, Duranti E, Vagnani S, Carli L, Kühl AA, et al. Early treatment with hydroxychloroquine prevents the development of endothelial dysfunction in a murine model of systemic lupus erythematosus. Arthritis Res Ther (2015) 6(17):277. doi:10.1186/s13075-015-0790-3
166. Petri M. Hydroxychloroquine use in the Baltimore lupus cohort: effects on lipids, glucose and thrombosis. Lupus (1996) 5(1_suppl):16–22. doi:10.1177/096120339600500105
167. Penn SK, Kao AH, Schott LL, Elliott JR, Toledo FGS, Kuller L, et al. Hydroxychloroquine and glycemia in women with rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol (2010) 37(6):1136–42. doi:10.3899/jrheum.090994
168. Espinola RG, Pierangeli SS, Gharavi AE, Harris EN. Hydroxychloroquine reverses platelet activation induced by human IgG antiphospholipid antibodies. Thromb Haemost (2002) 87(3):518–22.
169. Kaiser R, Cleveland CM, Criswell LA. Risk and protective factors for thrombosis in systemic lupus erythematosus: results from a large, multi-ethnic cohort. Ann Rheum Dis (2009) 68(2):238–41. doi:10.1136/ard.2008.093013
170. Selzer F, Sutton-Tyrrell K, Fitzgerald S, Tracy R, Kuller L, Manzi S. Vascular stiffness in women with systemic lupus erythematosus. Hypertension (2001) 37(4):1075–82. doi:10.1161/01.HYP.37.4.1075
171. van Leuven SI, van Wijk DF, Volger OL, de Vries JP, van der Loos CM, de Kleijn DV, et al. Mycophenolate mofetil attenuates plaque inflammation in patients with symptomatic carotid artery stenosis. Atherosclerosis (2010) 211(1):231–6. doi:10.1016/j.atherosclerosis.2010.01.043
172. Kiani AN, Magder LS, Petri M. Mycophenolate mofetil (MMF) does not slow the progression of subclinical atherosclerosis in SLE over 2 years. Rheumatol Int (2012) 32(9):2701–5. doi:10.1007/s00296-011-2048-y
173. van Leuven SI, Mendez-Fernandez YV, Wilhelm AJ, Wade NS, Gabriel CL, Kastelein JJ, et al. Mycophenolate mofetil but not atorvastatin attenuates atherosclerosis in lupus-prone LDLr(-/-)mice. Ann Rheum Dis (2011) 71(3):408–14. doi:10.1136/annrheumdis-2011-200071
174. Ho KT, Ahn CW, Alarcon GS, Baethge BA, Tan FK, Roseman J, et al. Systemic lupus erythematosus in a multiethnic cohort (LUMINA): XXVIII. Factors predictive of thrombotic events. Rheumatology (2005) 44(10):1303–7. doi:10.1093/rheumatology/kei014
175. Haque S, Gordon C, Isenberg D, Rahman A, Lanyon P, Bell A, et al. Risk factors for clinical coronary heart disease in systemic lupus erythematosus: the lupus and atherosclerosis evaluation of risk (LASER) study. J Rheumatol (2010) 37(2):322–9. doi:10.3899/j090306
176. Kyaw T, Cui P, Tay C, Kanellakis P, Hosseini H, Liu E, et al. BAFF receptor mAb treatment ameliorates development and progression of atherosclerosis in hyperlipidemic ApoE−/− mice. PLoS One (2013) 8(4):e60430. doi:10.1371/journal.pone.0060430
177. Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K, et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. The J Immunol (2010) 185(7):4410–9. doi:10.4049/jimmunol.1000033
178. Kyaw T, Tipping P, Toh BH, Bobik A. Current understanding of the role of B cell subsets and intimal and adventitial B cells in atherosclerosis. Curr Opin Lipidol (2011) 22(5):373–9. doi:10.1097/MOL.0b013e32834adaf3
179. Kahn P, Ramanujam M, Bethunaickan R, Huang W, Tao H, Madaio MP, et al. Prevention of murine antiphospholipid syndrome by BAFF blockade. Arthritis Rheum (2008) 58(9):2824–34. doi:10.1002/art.23764
180. Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med (2010) 207(8):1579–87. doi:10.1084/jem.20100155
181. Takeda M, Yamashita T, Sasaki N, Nakajima K, Kita T, Shinohara M, et al. Oral administration of an active form of vitamin D3 (Calcitriol) decreases atherosclerosis in mice by inducing regulatory T cells and immature dendritic cells with tolerogenic functions. Arterioscler Thromb Vasc Biol (2010) 30(12):2495–503. doi:10.1161/ATVBAHA.110.215459
182. Menezes AR, Lamb MC, Lavie CJ, Dinicolantonio JJ. Vitamin D and atherosclerosis. Curr Opin Cardiol (2014) 29(6):571–7. doi:10.1097/HCO.0000000000000108
183. Reis JP, Mühlen DV, Michos ED, Miller ER, Appel LJ, Araneta MR, et al. Serum vitamin D, parathyroid hormone levels, and carotid atherosclerosis. Atherosclerosis (2009) 207(2):585–90. doi:10.1016/j.atherosclerosis.2009.05.030
184. Carrelli AL, Walker MD, Lowe H, Mcmahon DJ, Rundek T, Sacco RL, et al. Vitamin D deficiency is associated with subclinical carotid atherosclerosis: the Northern Manhattan Study. Stroke (2011) 42(8):2240–5. doi:10.1161/STROKEAHA.110.608539
185. Lupoli R, Vaccaro A, Ambrosino P, Poggio P, Amato M, Minno MN. Impact of vitamin D deficiency on subclinical carotid atherosclerosis: a pooled analysis of cohort studies. J Clin Endocrinol Metab (2017). doi:10.1210/jc.2017-00342
186. Reynolds JA, Rosenberg AZ, Smith CK, Sergeant JC, Rice GI, Briggs TA, et al. Brief report: vitamin D deficiency is associated with endothelial dysfunction and increases type I interferon gene expression in a murine model of systemic lupus erythematosus. Arthritis Rheumatol (2016) 68(12):2929–35. doi:10.1002/art.39803
187. Nikpour M, Urowitz MB, Ibanez D, Harvey PJ, Gladman DD. Importance of cumulative exposure to elevated cholesterol and blood pressure in development of atherosclerotic coronary artery disease in systemic lupus erythematosus: a prospective proof-of-concept cohort study. Arthritis Res Ther (2011) 13(5):R156. doi:10.1186/ar3473
188. Schanberg LE, Sandborg C, Barnhart HX, Ardoin SP, Yow E, Evans GW, et al. Premature atherosclerosis in pediatric systemic lupus erythematosus: risk factors for increased carotid intima-media thickness in the atherosclerosis prevention in pediatric lupus erythematosus cohort. Arthritis Rheum (2009) 60(5):1496–507. doi:10.1002/art.24469
189. Chen CH, Jiang W, Via DP, Luo S, Li TR, Lee YT, et al. Oxidized low-density lipoproteins inhibit endothelial cell proliferation by suppressing basic fibroblast growth factor expression. Circulation (2000) 101(2):171–7. doi:10.1161/01.CIR.101.2.171
190. Plazak W, Gryga K, Dziedzic H, Tomkiewicz-Pajak L, Konieczynska M, Podolec P, et al. Influence of atorvastatin on coronary calcifications and myocardial perfusion defects in systemic lupus erythematosus patients: a prospective, randomized, double-masked, placebo-controlled study. Arthritis Res Ther (2011) 13(4):R117. doi:10.1186/ar3402
191. Nehme A, Zibara K. Cellular distribution and interaction between extended renin-angiotensin-aldosterone system pathways in atheroma. Atherosclerosis (2017) 263:334–42. doi:10.1016/j.atherosclerosis.2017.05.029
192. Bagi Z, Hamar P, Kardos M, Koller A. Lack of flow mediated dilation and enhanced angiotensin II-induced constriction in skeletal muscle arterioles of lupus-prone autoimmune mice. Lupus (2006) 15(6):326–34. doi:10.1191/0961203306lu2297oa
193. Ridker P, Cook N, Lee IM, Gordon D, Gaziano JM, Manson JE, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Eng J Med (2005) 352(13):1293–304. doi:10.1056/NEJMoa050613
194. Lertratanakul A, Wu P, Dyer AR, Kondos G, Edmundowicz D, Carr J, et al. Risk factors in the progression of subclinical atherosclerosis in women with systemic lupus erythematosus. Arthritis Care Res (2014) 66(8):1177–85. doi:10.1002/acr.22271
195. Toloza SMA, Uribe AG, Mcgwin G, Alarcón GS, Fessler BJ, Bastian HM, et al. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA): XXIII. Baseline predictors of vascular events. Arthritis Rheum (2004) 50(12):3947–57. doi:10.1002/art.20622
196. Reis-Neto ETD, Silva AED, Monteiro CMDC, Camargo LMD, Sato EI. Supervised physical exercise improves endothelial function in patients with systemic lupus erythematosus. Rheumatology (2013) 52(12):2187–95. doi:10.1093/rheumatology/ket283
197. Costanzo P, Perrone-Filardi P, Vassallo E, Paolillo S, Cesarano P, Brevetti G, et al. Does carotid intima-media thickness regression predict reduction of cardiovascular events? A meta-analysis of 41 randomized trials. J Am Coll Cardiol (2010) 56(24):2006–20. doi:10.1016/j.jacc.2010.05.059
198. Li H, Zhang YY, Huang XY, Sun YN, Jia YF, Li D. Beneficial effect of tripterine on systemic lupus erythematosus induced by active chromatin in BALB/c mice. Eur J Pharmacol (2005) 512(2–3):231–7. doi:10.1016/j.ejphar.2005.02.030
199. Zhu F, Li C, Jin XP, Weng SX, Fan LL, Zheng Z, et al. Celastrol may have an anti-atherosclerosis effect in a rabbit experimental carotid atherosclerosis model. Int J Clin Exp Med (2014) 7(7):1684–91.
200. Gu L, Bai W, Li S, Zhang Y, Han Y, Gu Y, et al. Celastrol prevents atherosclerosis via inhibiting LOX-1 and oxidative stress. PLoS One (2013) 8(6):e65477. doi:10.1371/journal.pone.0065477
201. Navab M, Anantharamaiah GM, Hama S, Garber DW, Chaddha M, Hough G, et al. Oral administration of an Apo A-I mimetic peptide synthesized from d-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation (2002) 105(3):290–2. doi:10.1161/hc0302.103711
202. Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, et al. Oral D-4F causes formation of pre-high-density lipoprotein and improves high-density lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from macrophages in apolipoprotein E-null mice. Circulation (2004) 109(25):3215–20. doi:10.1161/01.CIR.0000134275.90823.87
Keywords: systemic lupus erythematosus, atherosclerosis, cardiovascular events, physiopathology, biomarkers
Citation: Teixeira V and Tam L-S (2018) Novel Insights in Systemic Lupus Erythematosus and Atherosclerosis. Front. Med. 4:262. doi: 10.3389/fmed.2017.00262
Received: 29 August 2017; Accepted: 28 December 2017;
Published: 29 January 2018
Edited by:
Dimitrios Vassilopoulos, National and Kapodistrian University of Athens Medical School, GreeceReviewed by:
George Bertsias, University of Crete, GreeceCristina Pamfil, Iuliu Hatieganu University of Medicine and Pharmacy, Romania
Theodoros Dimitroulas, Aristotle University of Thessaloniki, Greece
Teresa Padro, Centre d’Investigació Cardiovascular (CSIC), Spain
Copyright: © 2018 Teixeira and Tam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vítor Teixeira, dml0b3IuYXMudGVpeGVpcmFAZ21haWwuY29t