 Ivana Bratić Hench
Ivana Bratić Hench Jürgen Hench
Jürgen Hench Markus Tolnay
Markus Tolnay
- Institute for Medical Genetics and Pathology, University Hospital Basel, Basel, Switzerland
Examination of tumor molecular characteristics by liquid biopsy is likely to greatly influence personalized cancer patient management. Analysis of circulating tumor DNA (ctDNA), circulating tumor cells (CTCs), and tumor-derived exosomes, all collectively referred to as “liquid biopsies,” are not only a modality to monitor treatment efficacy, disease progression, and emerging therapy resistance mechanisms, but they also assess tumor heterogeneity and evolution in real time. We review the literature concerning the examination of ctDNA and CTC in a diagnostic setting, evaluating their prognostic, predictive, and monitoring capabilities. We discuss the advantages and limitations of various leading ctDNA/CTC analysis technologies. Finally, guided by the results of clinical trials, we discuss the readiness of cell-free DNA and CTC as routine biomarkers in the context of various common types of neoplastic disease. At this moment, one cannot conclude whether or not liquid biopsy will become a mainstay in oncology practice.
Introduction
Testing of bodily fluids in medical diagnostics has a long history with Greek humorism and the Indian Ayurveda system as prominent examples. These antique ideas remain applicable since analysis of bodily fluids can reveal diseases. “Liquid biopsies” are commonly blood and urine samples and the term usually refers to neoplasia diagnostics in analogy to classical biopsies. The most prominent example of a “liquid biopsy” is testing for prostate-specific antigen (PSA) in blood samples, which, if obtained lege artis, is a robust predictor of prostate cancer. However, its specificity can be hampered by non-neoplastic prostate damage since PSA is present in both cancer and normal cells. Many cells and tissues release some of their constituents to the bloodstream, including fragmented, cell-free DNA (cfDNA) which can also arise from tumor cells, i.e., circulating tumor DNA (ctDNA). As opposed to PSA and other proteins, ctDNA sequences are tumor specific. Such changes have been associated with a variety of neoplastic, but also as hereditary disorders. The BRAF(p.V600E) point mutation, e.g., is found in a wide range of malignant and benign neoplastic diseases in many organ systems. Other mutations, such as MYD88(p.L265P), have a narrower occurrence spectrum restricted to hematological disorders. Lastly, some mutations, such as in the VHL gene, occur both in sporadic but also hereditary hemangioblastomas.
In contrast to PSA and alike, the diagnostic value of ctDNA dramatically increases with prior knowledge about the mutational landscape of a tumor based on a conventional biopsy. Targeted NGS reveals traceable mutations. With these patient-specific data at hand, liquid biopsies, designed to determine the exact amount of ctDNA fragments in blood, may become an invaluable personalized surveillance tool. The ultimate goal of precision medicine is to deliver the most suitable, personalized cancer treatment at the most appropriate dose and time point to maximize quality of life and OS. Liquid biopsies might pave the road toward this aim, offering a way to quantify treatment response and detect emerging resistance in real time without the need of serial conventional biopsies.
Herein, we will discuss the use of cfDNA/ctDNA as diagnostic, prognostic, and predictive biomarkers and their applicability in clinical settings. In addition, the modalities by which circulating tumor cells (CTCs) can be detected in peripheral blood and to which extend this principle has been adopted in clinical practice will be reviewed. Due to the overwhelming number of studies, we will, for the sake of data comparability, focus on the three most frequently investigated diseases: breast, lung, and colorectal cancer.
cfDNA and ctDNA
Biology and Origin of cfDNA/ctDNA
Cell-free DNA gained increased attention upon the discovery that part of it originates from tumor cells and can be isolated from peripheral blood (here: ctDNA), urine, and other bodily fluids (1). The amount of ctDNA likely depends on tumor burden. It was estimated in CRC that a tumor load of 100 g, corresponding to 3 × 1010 neoplastic cells, would release 3.3% of tumor (tissue) DNA into the blood daily (2). Mutations found in cfDNA are likely to represent a mixture of alterations in primary tumor and/or metastatic sites (3). The discovery that blood-derived cfDNA contains tumor-specific genetic and epigenetic alterations has provided a solid ground to clinical usage of ctDNA as a biomarker. Most of the respective studies showed high concordance between individual mutations found in tDNA and cfDNA samples (4). There is evidence that ctDNA analysis could inform about clonal heterogeneity and subclonal changes in real time (5–8). In healthy subjects, 70–90% of the cfDNA pool originates from white blood cells (mostly neutrophils and lymphocytes). Neoplastic cells, tumor-infiltrating T-lymphocytes, and degenerating endothelial cells found in the vicinity of expanding carcinoma tissue, likely contribute to cfDNA (9). However, the exact mechanisms of how cfDNA is released remains elusive. Three major hypotheses of ctDNA origin exist: (i) from dying tumor cells; (ii) from CTCs, and (iii) via active release. The majority of ctDNA fragments is likely derived from disintegrating cells (i.e., apoptosis, oncosis, and necrosis). Apoptosis is suggested to generate DNA fragments of about 180 bp and multiples of this length, appearing as ladder pattern in electrophoresis. Necrosis should result in longer fragments (>10.000 bp). Apoptosis, however, is likely impaired in most neoplasms. NGS to characterize plasma cfDNA profiles at single base resolution revealed that most cfDNA fragments in hepatocellular carcinoma (HCC) patients, healthy subjects as well as individuals with hepatitis B virus infections with and without cirrhosis, had a peak size near 166 bp. This length corresponds to DNA wrapped around a nucleosome and, thus, may be due to caspase-dependent endonucleases (10), supporting the idea that apoptosis is indeed a major source of cfDNA release. Atomic force microscopy of plasma cfDNA in stage IV CRC and healthy controls (11) indicated that over 80% of cfDNA fragments are shorter than 145 bp in CRC with no cfDNA fragment being larger than 300 bp. Likewise, controls had 65% fragments <145 bp but also 10% >300 bp. Blood-borne nuclease activities are likely responsible for this cfDNA size range. The discrepancies strongly argue against the diagnostic use of fragment length patterns. Many studies found larger sized DNA fragments and increased amounts of cfDNA in the plasma during advanced cancer stages and cytotoxic treatment, suggesting necrosis as release mechanism (2, 12–15). It was found that ctDNA fraction within cfDNA are enriched in short fragments (100–300 bp) while the same tumor-specific ctDNA mutations were not present in longer (1,000 bp) fragments (2, 15). Since most studies focused on frequent tumor types and were entity specific, the major contributor of ctDNA release remains elusive, might depend on cancer type, and could even also vary between patients (16). Further discrepancies among studies stem from different preanalytical conditions (serum vs. plasma, cfDNA extraction protocols), patient selection (varying tumor load and cancer stages) and the diversity of analytical methods. cfDNA fragments are likely protected from nuclease cleavage due to their association with nucleosomes (16). Determination of the nucleosome occupancy profile in cfDNA could potentially determine its tissue origin which might be beneficial in localizing “cancers of unknown primary” (17). ctDNA could originate from CTCs too. However, ctDNA levels are typically too high considering the low CTC counts in blood samples, and ctDNA is also present in absence of CTCs, making this hypothesis rather doubtable (4). Lastly, spontaneous, active release of ctDNA by tumors is the least investigated possibility. Such released ctDNA might have the role of an intercellular messenger and could either integrate into the genome of a host cell leading to genetic instability or it would bind to receptors leading to transformation of target recipient cells at distant locations. This effect gave rise to the theory of “genometastasis” (18–21). An in vitro study on breast cancer cell lines showed that active cfDNA release, at least partially, occurs via exosomes which would further regulate proliferation of the neoplastic cells (22). Nevertheless, further work is needed to identify the underlying mechanisms and, more importantly, the biological significance of active cfDNA release.
While cancer patients generally have much higher cfDNA levels than healthy subjects, the total amount varies considerably even among patients with comparable cancer type and stage (23, 24). Nevertheless, direct correlations between ctDNA level and tumor burden, stage, vascularity, cellular turnover, and therapy response were reported for various neoplasms (4, 15, 23, 25). Opposedly, increased cfDNA levels are not specific to cancer, as they are equally found in various pathologic (e.g., chronic inflammation, autoimmune disease) (26) and physiologic conditions (e.g., extensive exercise) (27), making it difficult to use cfDNA levels as a cancer-specific biomarker.
Knowledge about cfDNA clearance mechanisms comes from prenatal diagnostics. Clearance of fetal DNA from maternal blood occurs in two phases: a first rapid phase with a mean half-life of ~1 h (28) and a second slow phase of ~13 h (29). It has been speculated that the liver, spleen, and kidney might be responsible for cfDNA elimination (30). Since ctDNA is much less defined than circulating free fetal DNA, further investigation is required to understand its removal from the blood.
Methodology in cfDNA/ctDNA Analysis
Preanalytics
Comparison of cfDNA concentrations between different malignancies and estimation of the prognostic potential of cfDNA levels is currently impossible due to disparities among sample preparation techniques (plasma vs. serum, containers used for blood withdraw, storage conditions), cfDNA isolation (amount of blood, centrifugal speed, cfDNA isolation kits), and DNA concentration measurement (colorimetric/fluorometric assays, real-time PCR, Picogreen or SYBR Green I dsDNA quantification assays, PCR assays targeting different genes, etc.) (31–34). Moreover, the lack of generally accepted units for cfDNA quantification largely impairs comparative retrospective, and even prospective studies. The result from the first large-scale quality control external quality assessment (EQA) scheme that investigated the impact of preanalytical conditions on cfDNA quality, quantity, and integrity showed that different extraction kits produce a wide range of cfDNA yields ranging from 2.87 to 224 pg/μl (35). Moreover, results from the first EQA scheme for isolation and analysis of ctDNA that involved 42 laboratories from 10 European countries reported high variability in multiple phases of cfDNA processing and in choice of genotyping technologies with regard to cfDNA analysis and overall error rate of 6.09% (36).
Increased cfDNA concentrations have been observed more frequently in serum (3- to 24-fold higher) than in plasma from both healthy control subjects and cancer patients (37–40). These higher values are likely the result of in vitro hemolysis during clotting (37, 39). Higher cfDNA levels have been observed in advanced tumor stages than in patients with non-metastatic disease (23, 40). However, the increase in serum, but not plasma, cfDNA concentration in advanced tumor stages strongly correlated with leukocyte counts (40).
The current preanalytical recommendations for cfDNA analysis are as follows: (1) blood processing within 3 h if an EDTA Monovette® (Sarstedt, Germany) or similar instrument is used. Otherwise, vacuum containers (e.g., Vacutainer®, Becton-Dickinson, USA) with stabilizing reagents should be used; (2) plasma should be isolated by at least two sequential centrifugation steps; (3) plasma should be stored at −80°C and frequent freeze–thaw cycles should be avoided; and (4) use of dedicated cfDNA extraction kits (41).
Analytics
The possibility to identify oncogenic driver mutations offers advantage of ctDNA testing over solid biopsies and conventional biomarkers (e.g. serum PSA, CA15, CEA, and so on) the latter of which are not causally involved in tumorigenesis and, hence, not specific for neoplasia. The technical challenge in ctDNA detection stems from its low (e.g., 0.01%) fraction of cfDNA (2), demanding high sensitivity and specificity techniques, such as qPCR (ARMS, see below), dPCR [BEAMing; Droplet Digital™ PCR (ddPCR™)], and targeted parallel sequencing (next-generation sequencing, NGS). PCR-based methods, such as Cobas (FDA, CE-IVD) and Therascreen (CE-IVD) assays, based on the Scorpion Amplification-Refractory Mutation System (ARMS) reaction, are able to detect single base changes or small deletions by use of allele-specific primers. qPCR readout is cut-off based distinction of presence or absence of the mutation in question, detecting AFs down to 1% (42–45). Their sensitivity is only moderate compared to dPCR and targeted NGS with detection thresholds around 0.1% or lower (46, 47).
Droplet Digital™ PCR (ddPCR™, Bio-Rad or RainDrop™, RainDance Technologies) and BEAMing (beads, emulsion, amplification, magnetics; Sysmex) are two PCR-based techniques based on a similar principle, where DNA is diluted down to single DNA molecules that get physically separated into individual reaction compartments. Before quantification, DNA templates are amplified separately either on beads (BEAMing) or in water droplets engulfed by oil (ddPCR). Readout—analogous to flow cytometry—detects AFs down to 0.005% (4, 48, 49). Despite a sensitivity of BEAMing comparable to ddPCR™, the more complex protocol limits its clinical use. While the concentration of mutant fragments detected by qPCR is calculated relatively to a standard curve, ddPCR™ allows an end point analysis in a cell counter-like manner. The advantage of digital approaches is their high specificity, high sensitivity, speed, independence of qPCR equipment, ease of use, and combined with comparatively low cost. A specialized droplet/bead reader is required for ddPCR™ but not for BEAMing which works on flow cytometers. The major disadvantage of qPCR and dPCR is their restriction to predefined genetic alterations unlike NGS that is able to detect novel changes without modification of the protocol. Yet, given current clinical consequences, detection of previously unreported alterations is neither required nor helpful for therapeutic decisions. To monitor the disease after determining distinctive mutations, specific probes can be individually designed as a personalized assay for peripheral blood samples, potentially beneficial in early recurrence detection, disease progression monitoring, and identification of resistance mechanisms prior to conventional clinical signs (43, 49, 50).
NGS detects multiple somatic mutations in plasma cfDNA and informs about intratumoral heterogeneity, potentially useful in metastatic disease. However, the sequencing error rate of conventional NGS approaches is too high for detection of rare cfDNA variants. The Illumina® platform has error rates from 0.29 to 1% depending on read length, library preparation, base misincorporation, base calling algorithms, and variant type (6, 51, 52). Therefore, adaptations were made for ultrasensitive detection of low mutation AFs, in particular, an alternative library preparation, read depth, and coverage. Targeted gene panels infer high coverage per base at moderate cost. CAncer Personalized Profiling by deep Sequencing (CAPP-Seq) is a capture-based NGS ctDNA detection method for SNVs, indels, rearrangements, and CNVs (47). This method enriches for recurrently mutated genomic regions chosen for specific cancer types prior to sequencing by hybridization to a pool of antisense biotinylated oligonucleotides. High coverage (e.g., 10,000×) sequencing of the captured DNA detected ctDNA in 100% of stage II–IV and 50% of stage I non-small cell lung cancer (NSCLC) patients, with 96% specificity for mutant AFs down to 0.02% (47). Even though no patient-specific customization is needed, the multi-phase bioinformatics framework for CAPP-Seq data analysis is rather unattractive for clinical use. Recently, the detection limit of CAPP-Seq was decreased to 0.004% AF by incorporating molecular barcoding and iDES (53). The alternative Safe-Sequencing System (Safe-SeqS) detects variants down to 9/106 by assignment of UIDs to each DNA molecule prior to PCR amplification. The number of molecules in the sample is estimated based on UIDs and precisely quantified. A variant is called only if contained in >95% of PCR fragments within the same UID family (52). Polymerase and oligonucleotide synthesis errors can, hence, be excluded, making it possible to detect a mutant AF of 0.0001%. Safe-SeqS furthermore corrects for the PCR amplification bias. Its disadvantages are the relatively long turnaround time and the possibility of false-positives due to imperfect amplification (52). targeted error correction sequencing (TEC-Seq) is an alternative approach based on targeted capture of multiple regions of the genome labeled with dual-index barcode adapters and subsequent deep sequencing (30,000×) with an analytical specificity of >99.9999% and sensitivity of 97.4%. Lower false-positive rates (< 3 × 10−7) comparable to iDES were reported (54). Evaluation of 200 plasma samples with TEC-Seq detected somatic mutations in 71, 59, 59, and 68% of patients with early-stage breast, colorectal, lung, and ovarian cancer (54). Importantly, the detection limit depends on the number of free DNA molecules present in a sample. A typical plasma sample of 1 ml contains approximately 3,000 copies of any given gene leading to a sensitivity limit for detection of only 1 in 15,000 copies in 5 ml (55). Despite the low detection limits observed in CAPP-Seq, iDES, Safe-SaqS, and TEC-Seq they have, to our knowledge, only been systematically tested in research settings. Besides the Illumina platform, semiconductor-based targeted Ion Torrent™ sequencing (Thermo Fisher) has been successfully applied in analysis of cfDNA, too (56–58).
Only few laboratories have analyzed genome-wide copy number alterations, rearrangements, and mutations in cfDNA. Shotgun massively parallel sequencing method was applied to detect CNVs and point mutations in plasma of a four-patient case study with HCC and two patients with synchronous breast and ovarian cancers. Changes in ctDNA level in pre- and post-surgery blood samples were tracked to monitor disease burden during tumor evolution (59). The largest tumor had the highest ctDNA level and most CNVs (59). Plasma ctDNA WGS performed on 10 CRC and BC patients (60) demonstrated the feasibility of detecting chromosomal aberrations. WES of cfDNA in advanced cancers was able to objectify tumor evolution in response to therapy. Plasma cfDNA collected at the beginning of treatment and at relapse in 4/6 patients was analyzed (3). Since the mutations were present at high AFs due to metastatic state, the study’s clinical significance is limited. WES of six NSCLC stage III patients identified a median of 17.19% of tumor variants in serum (61). Interestingly, a median of 1,218 additional variants were present in serum only and of unknown origin. The average 68.5× WES sequencing depth would be unable to detect commonly low ctDNA AFs. The key advantage of WGS and WES is their general applicability without personalization.
Only small amounts (200 μl–2 ml) of plasma were used for cfDNA extraction in the abovementioned studies, making it unlikely to detect low AFs (51): (i) typically low ctDNA counts prohibit high throughput approaches and could lead to artifacts, in particular false-positives. (ii) The fraction of false-negatives correlates with the sensitivity of the method. (iii) In order to achieve high sensitivity and specificity, normal reference samples (skin, lymphocytes, etc.) need to be analyzed concomitantly. However, imperfect sequencing of normal tissue would induce false-positives (4). Besides determination of extensive genetic profiles of the tumors in individual patients, WGS and WES remain unable to identify the origin of ctDNA and currently remain confined to translational research. Here, they offer the potential of discovering novel pathogenic variants to be tested in clinical trials. Their long turnaround time, need for extensive bioinformatic support, and lack of clinical associations for the majority of genomic alterations preclude their routine use.
DNA methylation signatures differ between tissues, allowing to determine their relative contributions to cfDNA by genome-wide bisulfite sequencing (8). Tissue of origin of genomic aberrations identified in cfDNA from 29 HCCs was elucidated by deconvolution of plasma bisulfite sequencing data into tissue contribution percentages. HCC patients had a higher liver contribution than healthy controls (8). This approach might, thus, be diagnostically helpful in identifying the origin of elevated cfDNA levels.
The multitude and diversity of techniques aimed at obtaining similar molecular information has led to discordant and even conflicting data. The majority of authors measured artificially spiked healthy donor blood samples to define sensitivity and specificity of their methods. Being a good starting point, it remains problematic that patient samples are hard to preserve in their native state which makes testing of multiple methods on the same specimen almost impossible. Furthermore, the ctDNA fraction of cfDNA is low, requiring relatively large blood volumes per test. SNP-based results from different platforms vary markedly (62). Physicians started to request confidence levels for clinical use of cancer genetic results and only fraction of them routinely requests genomic tumor profiling (63). Targeted approaches, currently featuring highest sensitivity and specificity, are likely the first to find acceptance among oncologists for disease monitoring.
cfDNA/ctDNA in Cancer Patient Management
Circulating tumor DNA is present in over 75% of patients with advanced pancreatic, ovarian, colorectal, bladder, gastroesophageal, breast, melanoma, hepatocellular, and head, and neck cancers while rates below 50% were found in primary brain tumors, in renal, prostate, and thyroid cancers as well as in patients with localized tumors (4). Herein, we discuss the potential use of ctDNA as biomarker in a (i) diagnostic, (ii) prognostic, and (iii) predictive setting. The regimes comprise (a) total cfDNA level, (b) identification of tumor-specific genetic alterations, and (c) quantification of mutant alleles. Depending on cancer type, different combinations of these strategies might prove beneficial. The largest and—in our opinion—most relevant studies are summarized in Tables 1–3.
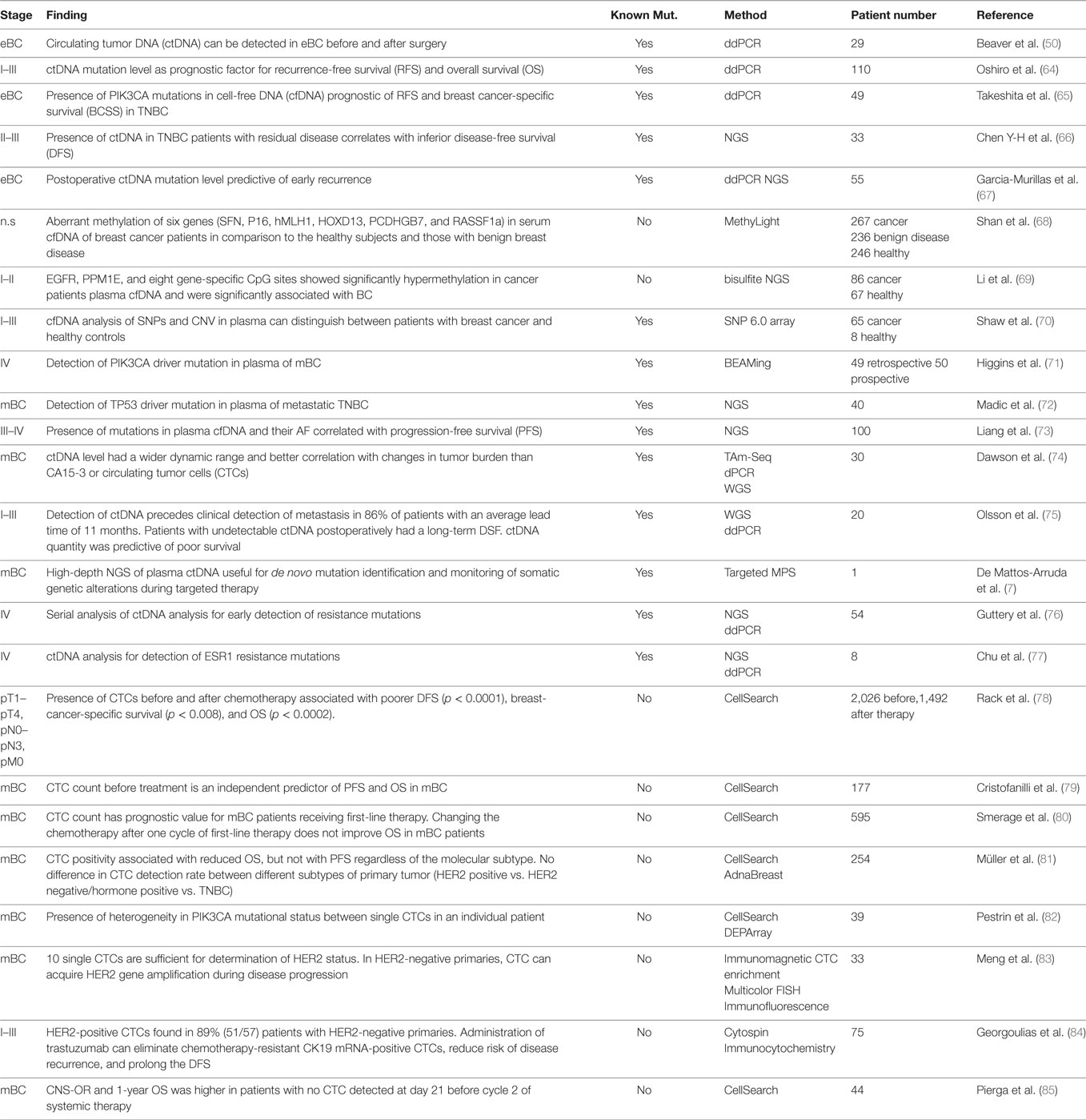
Table 1. Breast cancer.
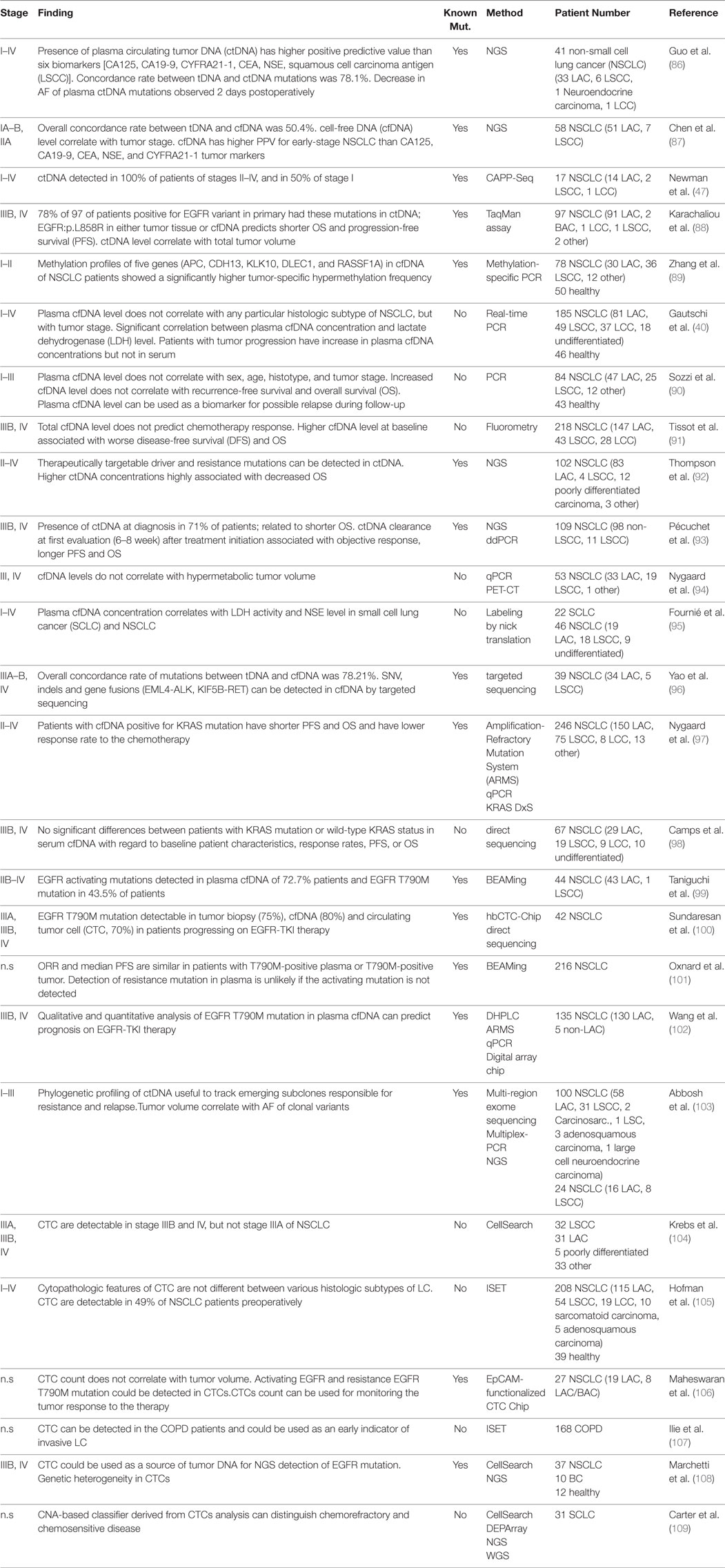
Table 2. Lung cancer.
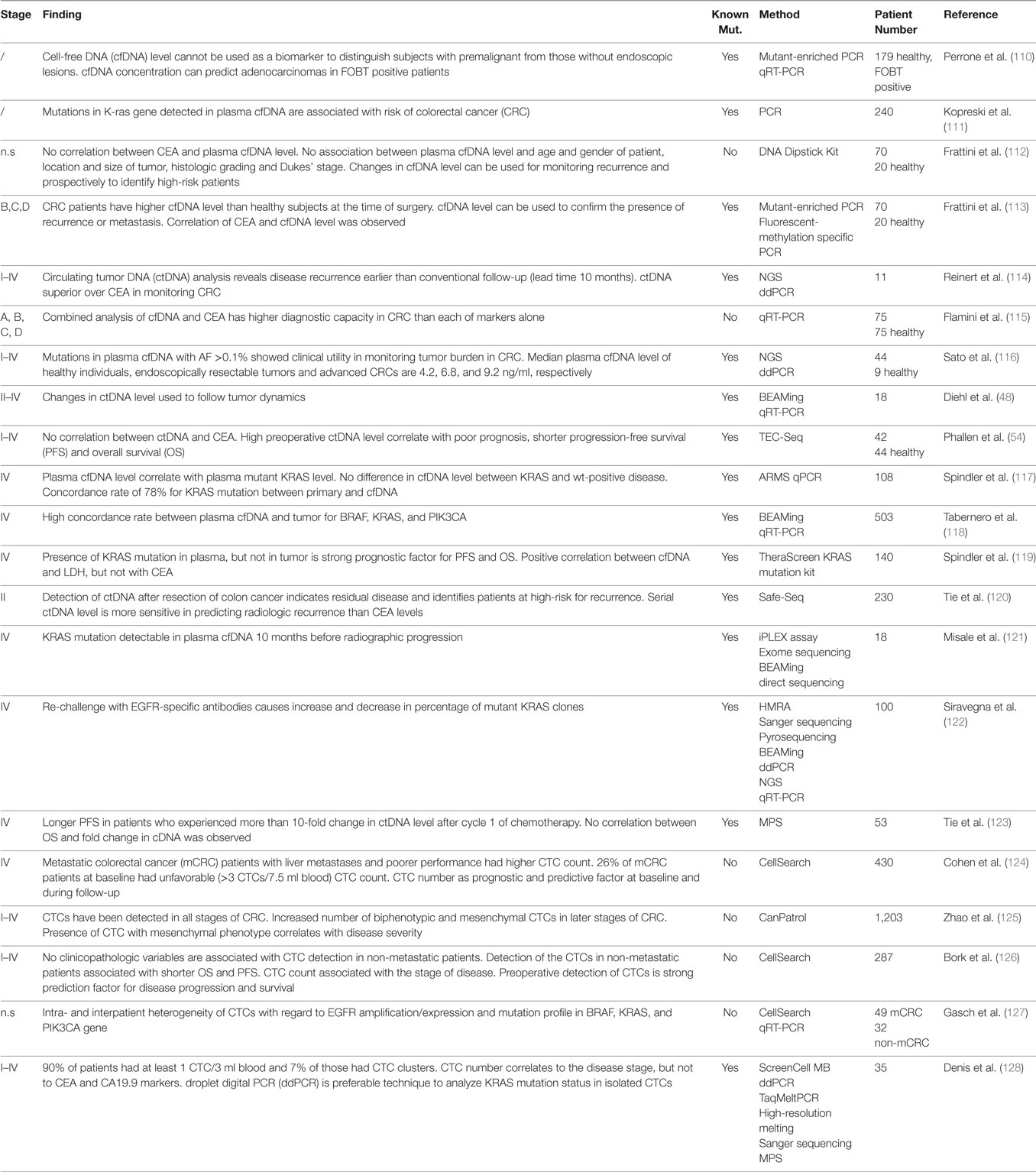
Table 3. Colorectal cancer.
Early Detection of Cancer in Clinically Healthy Subjects
The potential of ctDNA to detect presymptomatic early-stage cancer or even precursor lesions would be clinically advantageous but was infrequently studied in clinically healthy subjects. In a longitudinal study, TP53 and KRAS mutations in cfDNA of non-smokers and ex-smokers were correlated with occurrence of various neoplasms potentially caused by tobacco smoke and air pollution (129). Plasma cfDNA from 550 healthy subjects was tested for mutations in two genes (TP53/n = 550; KRAS/n = 1,098). Such events were detected at an average 20.8 (TP53) and 14.3 months (KRAS) before cancer diagnosis (129). Mutant detection did not correlate with total cfDNA amounts. cfDNA mutations in healthy subjects were good predictors of bladder but not lung, upper-digestive tract, and blood cancers (129). Since tumor tissue was not tested, it remains unknown whether the mutations were present in the tumor cells. Adversely, some of the TP53/KRAS mutations were also detected in some healthy subjects not developing cancer during follow-up (129). A mutation in codon 249 of TP53 [NM_000546.5 (TP53):c.747G>T (p.Arg249Ser)], common after dietary exposure to the high levels of aflatoxin B1, was detected in plasma of 4/8 patients at least 1 year before initial diagnosis of HCC (130). To determine the specificity of such ab initio approaches, blood from 134 healthy subjects was ultra-deep sequenced in 50 cancer-associated genes with an average 30,000× coverage and a detection limit for variant AFs of 0.001% (131). Somatic mutations in blood cells significantly contribute to the mutational load in cfDNA, suggesting to rather sequence both cfDNA and blood cells in parallel for background removal (131). This finding was independently replicated in 2,728 patients with various tumors (132). Somatic alterations accumulate in solid tissues and the hematopoietic system as a function of age (133). The Circulating Cell-free Genome Atlas (CCGA, Clinical Trial NCT02889978, sponsored by Illumina®) is a 5-year prospective, multicenter, observational study aimed at developing ctDNA blood tests for early cancer detection (134). Ultra-broad, ultra-deep NGS combined with machine learning may found a database on mutations in the blood of subjects with and without cancer (134). This unbiased strategy might later provide risk assessment based on blood screening.
Breast Cancer
Detection of oncogenic driver mutations in early-stage presurgical breast cancer might tremendously impact on clinical management. Oncogenic driver mutations were screened for in 29 patients with early-stage BC (I-III) positive for 1/3 PIK3CA mutations (p.H1047R, p.E545K, p.E542K) before and after surgery. Mutant AFs in blood before surgery were low (0.01–0.07%) with the exception of one case with 2.99% who relapsed 26 months later (50). The same cfDNA mutations were found in 22% of 110 stage I–III BC patients (64), indicating a prognostic value for ctDNA AFs: higher values correlated with shorter RFS and OS (64) which holds true for TNBC, too (65). ctDNA detection in stage II–III TNBC patients with residual disease after neoadjuvant chemotherapy predicts recurrence with high specificity, but moderate sensitivity (66), potentially due to low plasma volume (1 ml) and a non-optimized NGS approach. ctDNA content shows significant correlation with prognosis at early cancer stages, as opposed to conventional protein tumor markers (64). Postoperative plasma ctDNA abundance after neoadjuvant chemotherapy and surgery, but not baseline levels predicted early recurrence (67). 50% of relapsing patients were ctDNA-positive postsurgically which increased to 80% during follow-up. None of the relapse-free patients had a ctDNA-positive blood sample. Importantly, ctDNA detection had a median lead time of 7.9 months over clinical recurrence diagnosis (67).
Serum cfDNA was analyzed for promoter methylation of six genes (SFN, P16, hMLH1, HOXD13, PCDHGB7, and RASSF1a) by the qPCR-based MethyLight test in 749 patients with BC, benign breast lesions, and healthy subjects for early disease detection (68). The method is able to discriminate between cancer and health with 79.6% sensitivity and 72.4% specificity. Malignant and benign lesions were distinguished with 82.4% sensitivity and 78.1% specificity (68). Likewise, the methylation profiles of EGFR, PPM1E, and eight more CpG islands in plasma cfDNA were used as biomarker for early BC detection (69). Alternatively, genome-wide CNV screening in cfDNA could distinguish cancer from health during routine follow-up (70). Even though the aforementioned studies suggest cfDNA analysis as a screening tool in early-stage BC, larger prospective trials are required to determine its reliability.
Lung Cancer
Unfortunately, despite WHO-defined (135) LC entities, several recent studies on liquid biopsies use the old, outdated SCLC and NSCLC categorization and lack precise distinction between LAC and LSCC, which in terms of tumor biology and targeted treatment options would be more informative. We tried to comply with the WHO classification (135) wherever possible.
Lung cancer commonly presents at advanced stage due to the lack of screening. Therefore, many studies assessed cfDNA testing for early LC detection and for recurrence monitoring. Concordance rate between tumor DNA and ctDNA mutations in the pretreatment plasma at early-stage (I, II) NSCLC patients was 78.1% (positive predictive value 94.7%), making it an indicator of early-stage LC (86). 89.7% of 58 early-stage (I–II) NSCLC patients had increased cfDNA out of whom 60.3% were ctDNA-positive with tumor-specific mutations (87). Others detected ctDNA in 100% of stage II–IV and 50% of stage I NSCLC patients (47). Reasons for this discordance are differences in detection technologies, small tumor size and molecular analysis of potentially non-representative tumor sections (87). 78% of 97 advanced-stage (IIIB, IV) NSCLCs featuring an EGFR variant in the primary had the same mutations in ctDNA; EGFR(p.L858R) in either tumor tissue or cfDNA predicted shorter OS and PFS (88). Changes in ctDNA AFs were observed when comparing pre- and postoperative cfDNA: AFs drop 11.52% in stage Ia and 14.63% in stage Ib, but only 0.57% in stage IIa, and 0.13% in stage IIIa. This drop already occurs 2 days postoperatively (86). cfDNA may have a higher positive predictive value compared to serum protein tumor biomarkers for early-stage LC (86, 87).
Methylation profiling in early-stage NSCLC might become a diagnostic and prognostic biomarker in analogy to breast cancer: Aberrant methylation profiles of five genes (APC, CDH13, KLK10, DLEC1, RASSF1A) in cfDNA from 110 early-stage mixed entity NSCLC cases showed a significantly higher tumor-specific hypermethylation frequency when compared to healthy controls, reaching 83.64% sensitivity and 74% specificity to diagnose LC (89).
Colorectal Cancer
Approximately 50% of localized CRC patients will develop metastases (136). Although there has been dramatic decline in the number of cases due to screening, CRC incidence remains high. Therefore, cfDNA testing might further improve screening efficiency. Traditional serum protein biomarkers (e.g., CEA, CA19-9) lack high specificity and sensitivity. There is only a limited number of studies investigating cfDNA testing for early CRC detection. A study on 170 subjectively healthy patients positive for occult fecal blood assessed the predictive power of plasma cfDNA levels and ctDNA KRAS mutations (110). Adenocarcinoma, but not intraepithelial precursor lesions, including HGIN, could be detected based on these values alone. Yet, the KRAS mutant AF was low (3%) compared to the AF in the tumor itself (45% in AC and HGIN), leaving the positive predictive value of the test questionable (110). Prospectively collected plasma cfDNA of 232 patients subjected to colonoscopy was analyzed for KRAS mutations which had previously been identified in the tumor tissues of 35 patients. These mutations were detectable in cfDNA in 29 patients (81%). 39% of patients positive for a KRAS mutation in cfDNA had a KRAS mutant colorectal neoplasia (111), suggesting to add cfDNA testing for frequent CRC mutations in screening programmes despite restriction to well-known CRC genetic aberrations.
CEA is the only routine tumor marker for estimation of tumor burden and progression monitoring despite low sensitivity and specificity, being elevated in 40% of CRC cases only (137). Several studies found that cfDNA performed better than CEA (138). No correlation between plasma cfDNA and CEA level was found (112). 151 plasma samples from six relapsing and five non-relapsing CRC patients were analyzed by NGS and ddPCR. Detection of ctDNA in cfDNA may provide an average of 10 months lead time over detection of metastatic recurrence by CEA (114). Combining CEA and cfDNA testing further improved diagnostic power (115).
cfDNA/ctDNA Level as a Prognostic Biomarker
Clinical use of cfDNA levels alone as cancer biomarker is currently not recommended due to its highly variable amount; a broad spectrum of cfDNA levels in healthy controls has been observed. In a large multicenter study on 776 healthy individuals, the mean plasma cfDNA concentration was 67 ng/ml with an exorbitant standard deviation of 405 ng/ml (139). Meta analysis of 39 studies revealed cfDNA concentrations as low as 2.5 to 27 ng/ml (24), prohibiting to make cancer diagnoses based on a cut-off value: Other phenomena including inflammatory processes or tissue decay unrelated to cancer are impossible to distinguish. If still considering cfDNA levels as diagnostic modality, blood withdraw and cfDNA concentration measurement would require stringent standardization (140). Therefore, most studies summarized below analyzed ctDNA rather than cfDNA concentrations alone.
Breast Cancer
Numerous studies assessed the prognostic value of cfDNA levels in BC (141). A concentration range from ~60 to 550 ng/ml plasma or serum was found, only partially overlapping with healthy controls in whom values ranged from 3 to 63 ng/ml (142, 143). Many studies agree that cfDNA levels increase in patients with malignant lesions and correlated with tumor size, lymph node metastasis, histopathological grade, and clinical stage (141, 144–147). Two laboratories could not confirm the association between baseline total cfDNA level, pathologic complete response to neoadjuvant chemotherapy, and OS (145, 148). These conflicting results could be due to the variety of preanalytical and analytical methods as well as differences in patient cohorts. While the prognostic usefulness of ctDNA levels at baseline in a cohort of 30 metastatic luminal type BC patients was documented, another cohort of 36 patients with metastatic TNBC failed to verify this (72, 74), suggesting cancer type and burden dependency. Several studies addressed the prognostic value of cfDNA integrity reflected by the ratio of longer to shorter fragments. Analysis of Alu DNA repeats by qPCR in the serum of 51 healthy women and 83 with stage I–IV BC before surgery showed that mean DNA integrity was higher in BC patients with stage II, III, and IV, but no significant difference between stage I patients and healthy controls could be observed (149). A second comparable study confirmed this (150) while a third showed the opposite with lowest cfDNA integrity in metastatic BC (151). In summary, cfDNA levels alone are currently unlikely to become a clinically useful prognostic biomarker for BC, mainly due to their overall inability to stratify malignancy, benign lesions, and health (142, 152).
Analysis of genetic alterations seems more promising in cfDNA research. PIK3CA mutations were screened for in cfDNA of mBC patients in retrospective (n = 49) and prospective (n = 60) cohorts and were detected in approximately 30% of subjects with 100% concordance between ctDNA and FFPE specimens (71). A high mutation concordance of 81% between ctDNA and FFPE tumor samples was observed in TNBC too (72). An increased PIK3CA mutant AF predicted a short PFS in a retrospective cohort of 100 advanced-stage (III–IV) BC cases (73).
Lung Cancer
A broad spectrum of cfDNA concentrations, ranging, from ~3.7 to 318 ng/ml plasma/serum in LC have been reported (142). cfDNA concentration in stage II NSCLC (51 LAC, 7 LSCC) was significantly higher than in stage I, where no association was found between cfDNA level and age, sex, smoking history, tumor histology, differentiation level, and vascular invasion (87). While some authors found the highest plasma cfDNA concentrations in stage IV and, thus, a correlation between cfDNA level and tumor stage (40, 95), other studies could not confirm this (90). cfDNA levels in LC patients are significantly lower during follow-up than before surgery, suggesting a way to objectify surgical effectiveness (23, 90). High plasma cfDNA concentrations at baseline were significantly associated with decreased OS and increased tumor progression rates (40, 91, 92). 105 NSCLC patients (98 non-LSCC, 11 SCC) were categorized according to ctDNA concentration at baseline into the tertiles “high” (>0.50 ng/ml), “intermediate” (0.027–0.50 ng/ml), and “low” (<0.027 ng/ml) (93). “High” ctDNA levels were associated with high tumor burden, liver metastases, and high proliferative indices. The median OS was 13, 13.4, and 21.5 months; the median PFS was 4.1, 5.7, and 10.4 months for the ‘high,” “intermediate,” and “low” groups (93). Meta analysis of 22 studies concerning stages III and IV treated with chemotherapy suggests that higher levels of cfDNA are significantly associated with a shorter PFS and OS (153). Other studies could not recapitulate this correlation in different tumor types (47 LAC, 25 LSCC, 12 other unspecified types) (90). It remains unclear whether total tumor volume correlates with ctDNA level (47), or not (94).
Several studies addressed the relation between cfDNA level and other prognostic follow-up markers. A highly significant correlation was observed between increased plasma cfDNA level, elevated LDH level, advanced tumor stage, and poor OS (40). The association between OS and cfDNA concentration, NSE levels, and LDH activity has been reported earlier for SCLCs and NSCLCs, respectively (95).
Taken together, studies on LC share the major conceptual shortcoming of mixing biologically and clinically distinct NSCLC entities which, combined with the diversity in preanalytical and analytical methods, make data rather inconsistent concerning cfDNA levels as a standalone prognostic marker and suggest rather limited benefits for clinical practice.
Many studies assessed qualitative cfDNA analysis to identify ctDNA. A large meta-analysis reviewing 25 studies (2,605 NSCLCs) found a high concordance rate in EGFR mutation profiles between blood and tumor tissue. The authors concluded that EGFR mutation positivity in blood could be used to guide treatment decisions for EGFR TKIs in advanced NSCLC (154). 39 advanced NSCLC patients (34 LAC, 5 LSCC, stages III–IV) had 78.21% overall concordance between tumor tissue and cfDNA (96); point mutations, indels, and gene rearrangements (EML4-ALK, KIF5B-RET) were considered. AFs of concordant point mutations and indels in EGFR, KRAS, and PIK3CA ranged from 0.3 to 52% in tumor tissue DNA and in plasma cfDNA from 0.2 11.6% (96). The feasibility to detect NSCLC-related driver mutations in EGFR, KRAS, BRAF, and PIK3CA in cfDNA was demonstrated (99, 155). Mutant EGFR ctDNA levels correlate well with clinical response stage III and IV LACs (156). There are conflicting data concerning ctDNA KRAS mutations: Some studies found KRAS mutant ctDNA to be associated with recurrence and shorter OS (97, 157) while others did not (98). Two large meta-studies suggest that the presence but not type of ctDNA KRAS mutations (variants unspecified) was associated with shorter OS in NSCLC (153, 158). In summary, qualitative assessment of plasma ctDNA seems more appealing for LC diagnostics than cfDNA levels, particularly in cases without molecular access to tumor tissue.
Colorectal Cancer
Colonoscopy remains the gold standard for early detection of CRC with sensitivity rates of >80% (159) which can be complemented by testing fecal occult blood for mutations, currently having a higher sensitivity than plasma cfDNA analysis. cfDNA levels are elevated in metastatic CRC and have prognostic value (138, 160). Median plasma cfDNA levels of healthy individuals, endoscopically resectable tumors, and advanced CRC differ (116); a broad spectrum has been reported (142). cfDNA quantification may be useful for monitoring early-stage CRC postoperatively (112). cfDNA levels progressively decreased in patients who became tumor-free after surgery, while it increased in those developing recurrence and metastasis (112). CRC patients with a postoperatively detectable ctDNA relapsed within 1 year whereas those negative for ctDNA had no recurrence (48). 31 CRC patients across stages I–IV were preoperatively tested for ctDNA mutation rates. Patients with mutant AFs higher than 2% had significantly shorter PFS and OS compared to those <2% mutant AF (54). KRAS mutant ctDNA increase correlates with decreased OS (4). Meta-analysis of 1,076 patients with mCRC treated with chemotherapy confirmed that baseline total cfDNA levels correlate with OS (160).
Circulating tumor DNA in CRC was qualitatively examined in numerous studies which found a high agreement of mutations between tumor tissue and ctDNA (117, 138). Data divergence stems not only from different methodologies used for mutation detection but also from different samples sizes, clinical settings, and intervals between tumor biopsies and blood withdraw. Analysis of 108 chemotherapy-refractory mCRCs demonstrated that cfDNA and KRAS mutant ctDNA levels at baseline show a clear correlation. Patients with plasma mutant KRAS levels above 75% had a disease control rate of 0%, whereas those with lower levels had a rate of 42% (p = 0.048), indicating that ctDNA KRAS mutation AF predicts disease behavior (117). The applicability of cfDNA as prognostic parameter and alternative modality for mutation detection before treatment in mCRC was demonstrated in the randomized CORRECT phase III trial (118): 166 mCRC patients received placebo and 337 were treated with the TKI regorafenib. Plasma cfDNA was collected before treatment and tumor DNA from archival tissue. Presence of KRAS, PIK3CA, and BRAF mutations was tested in cfDNA and tumor tissue with an overall concordance rate of 76% for KRAS, 88% for PIK3CA, and 97% for BRAF mutations. Mean mutant AF detected in plasma was 11.05% for KRAS and 8.23% for PIK3CA. Despite high interpatient variability, high plasma cfDNA concentrations were associated with shorter median OS and PFS in both placebo and regorafenib groups (118). Shorter PFS during regorafenib treatment in patients with ctDNA KRAS mutations compared to those without was independently reported (161). The prognostic value of KRAS mutations in ctDNA but not tumor tissue was repeatedly confirmed (117, 119).
The utility of postoperative ctDNA as an indicator of MRD was demonstrated in a prospective cohort of 230 patients with resected stage II CRC (120). In subjects without adjuvant chemotherapy, tumor-specific mutations in cfDNA were detected postoperatively in 7.9% (14/178) patients of whom 79% (11/14) relapsed after a median follow-up of 27 months. In 164 patients without ctDNA, only 9.8% (16/164) experienced recurrence (120). Postoperative ctDNA positivity correlated with reduced RFS: 0% for ctDNA-positives and 90% for ctDNA-negatives after 3 years. Postoperative ctDNA status had a higher impact on RFS than any individual clinicopathological risk factor or any combination thereof (120). Presence of ctDNA immediately after chemotherapy completion was associated with a high risk of radiological relapse (120). ctDNA positivity precedes radiological recurrence up to several months, leaving more time to adapt the next line of treatment. In addition, detection of tumor-specific mutations in cfDNA has a higher specificity than CT scans (120). A strong association between ctDNA-positivity, RFS, and OS in patients with CRC irrespective of tumor stage, study size, tumor markers, detection methods, and sample type (plasma vs. serum) was revealed in a meta-analysis of 9 studies (1,022 CRC patients) (162).
In summary, total cfDNA levels and detection of ctDNA feature a strong prognostic value in metastatic CRC and are directly related to disease burden. cfDNA level and presence of ctDNA are prognostic markers of RFS, PFS, and OS. Disease progression is reflected by increasing cfDNA and ctDNA levels. Rapid decline in cfDNA level and absence ctDNA in plasma after initiation of therapy reflect treatment response.
cfDNA/ctDNA for Monitoring Treatment Response and Resistance Mechanisms
As discussed above, studies in colorectal, breast, and lung cancer have shown that ctDNA, rather than cfDNA, levels correlate with the clinical disease course and can precede clinical progression detected by imaging by weeks or months. Spatial and temporal genomic heterogeneity in primary tumors makes it nearly impossible to capture their complete genomic profile in a single biopsy. Analysis of tumor heterogeneity would require sequencing of multiple regions from the primary and metastases, which to date is difficult in clinical settings. ctDNA allows longitudinal evolution and heterogeneity tracking, relapse prediction, quantifying therapy response, and resistance identification as reviewed in the cancer type-specific sections below.
Breast Cancer
Matched tumor and serial plasma samples from a prospective cohort of 30 mBC patients receiving systemic therapy were tested for somatic genetic alterations in TP53 and PIK3CA; structural variants were identified by deep targeted amplicon sequencing and WGS (74). ctDNA levels in serial samples at intervals ≥3 weeks during follow-up were analyzed by personalized dPCR and tagged amplicon deep sequencing. ctDNA was detected in ≥1 samples in 29/30 patients while CA15-3 was positive in only 21/27 cases. Plasma levels of ctDNA mutations and structural variants but not total cfDNA showed dynamic patterns correlating with CT-morphological treatment responses. In 10/19 patients ctDNA increased in average 5 months before radiographic disease progression. In some cases, multiple ctDNA mutations exhibited similar AF dynamics, while in others, a ctDNA TP53 mutation not found in the initial tumor biopsy showed elevated levels after paclitaxel chemotherapy despite decrease of a PIK3CA mutation AF during stable disease. This suggests clonally divergent treatment responses. ctDNA testing achieved higher sensitivity and better correlation with tumor burden than CA15-3 or CTC count (74). Major limitations were the small sample size and that somatic mutations and structural genetic variants could be identified in only 60% of primaries. The limited clinical utility of CA15-3 was repeatedly reported (4, 163, 164). Chromosomal rearrangements were retrospectively investigated by WGS in resection specimens of 20 patients with surgically treated, otherwise therapy-naive primary BC (stages I–III) (75). These rearrangements were targeted by ddPCR assays for 4–6 patient-specific changes. Plasma samples were taken at surgery and three to six times during follow-up. Detection of ctDNA in ≥1 samples indicated relapse. Again, ctDNA positivity preceded clinical detection of metastatic growth in 86% (12/14) of patients with an average lead time of 11 months, while ctDNA remained undetectable after surgery in long-term disease-free patients (93% sensitivity, 100% specificity), making ctDNA a significant predictor of short RFS (75). Somatic alterations of archival primary tumor tissue and synchronous liver metastases collected at the time of diagnosis, as well as serial plasma samples collected during fourth line treatment with an AKT inhibitor (ipatasertib) were analyzed in a single patient with estrogen receptor (ER) positive, HER2-negative invasive mixed ductal–lobular adenocarcinoma of the breast. Sequencing was targeted on the high-depth massively parallel sequencing platform Integrated Mutation Profiling of Actionable Cancer Targets that comprises 300 cancer genes with actionable mutations (7). Parallel ctDNA analysis revealed mutations from both, primary and metastatic sites. Changes in ctDNA mutant AF mirrored the pharmacodynamic response to targeted monotherapy (7). This case study points out that ctDNA can deliver a holistic view of the tumoral genetic landscape. Genetic aberrations occurring under sequential targeted treatment with tamoxifen and trastuzumab, followed by lapatinib in a metastatic ER-positive, HER2-positive BC patient were tracked during 3-years. Stem mutations that were present in all tumor biopsies had highest allelic plasma levels, followed by metastatic-clade and private mutations. Serial changes in ctDNA subclonal private mutations correlated with individual treatment responses of metastatic sites (5). Similarly, primary tumor WGS identified somatic mutations in mBC patients undergoing two phases of chemotherapy (epirubicin and paclitaxel). The dynamics of 10 selected mutations from the primary were followed in serial plasma samples before and after treatment. A sharp decline in AF under therapy and an increase at disease progression were reported (165). Other studies confirmed that ctDNA contains mutations from both primary and metastases in BC (7, 166). The mutation profile of primary tumor, liver metastatic site, and plasma ctDNA of a metastatic ER+/HER2+ BC patient was studied by WES (166): The primary tumor was biopsied 4 months after neoadjuvant chemotherapy. Plasma and liver metastasis were sampled after progression following a two-phase therapy (anastrozole and herceptin). AFs of mutations in cfDNA correlated well with liver metastasis but poorly with the primary tumor. Moreover, the resistance mutation ESR1(p.D538G) in response to estrogen deprivation was detected in cfDNA and metastatic site only, but not in the primary, indicating a subclonal change under selective pressure by the aromatase inhibitor anastrozole. Likewise, the PIK3CA(p.H1047R) mutation was present in the primary only, suggesting that it emerged either after metastatic spread or that it was not present in subpopulation of cells responsible for metastatic seeding (166). Activating mutations in the estrogen receptor 1 (ESR1) gene are acquired under treatment in approximately 20% of patients and drive resistance to antihormonal therapy. Such mutations are predictive of endocrine resistance in mBC. Serial plasma ctDNA samples from 48 ER+ mBC patients receiving antiestrogenic therapy were analyzed by targeted NGS and ddPCR. ESR1 mutations were present in 3/48 patients at baseline with variable AF (p.D538G: 46.3%, p.Y537S: 2.8%, p.E380Q: 24.4%). In four patients, an ESR1 resistance mutation was detected in cfDNA under therapy (76). Comparable data were reported by others (77). However, the abovementioned case studies comprise small patient numbers and the identification of breast cancer driver mutations remains challenging with only a few being identified (HER2, TP53, PIK3CA, and AKT1; amplification of ERBB2 and EGFR). Thus, WGS or WES might aid in identification of unknown oncogenic drivers. To date, such a strategy would lead to a significant cost increase in diagnostics and requires extensive bioinformatics support typically unavailable in routine labs. Despite availability of WGS/WES, discrimination between biologically relevant somatic driver mutations, passengers, and background alterations remains difficult. Large prospective clinical trials are required to evaluate if and to what extent early detection of metastasis by ctDNA monitoring might improve patient outcome. Nevertheless, case study data promote ctDNA testing to facilitate early therapeutic intervention while tumor burden is still low.
Lung Cancer
While surgery offers best curative possibilities in early-stage NSCLC, it is usually not an option in advanced disease. Besides cytostatics, targeted therapies significantly improved clinical outcome in LAC. A number of molecular targets was identified so far in these tumors (EGFR, ALK, HER2, BRAF, MET, ROS1, and RET) but only few in LSCC (FGFR1 and PIK3CA). Targeted therapy choice depends on the molecular profile of the primary. However, despite initial response, almost all tumors become resistant within short time. To date, molecular testing is routinely performed on tumor tissue, typically needle biopsies. 41–62% of LC patients receiving 1st and 2nd generation EGFR TKIs acquire resistance after ~12 months due to the EGFR(p.T790M) mutation (167). Osimertinib is a 3rd generation TKI used to treat metastatic NSCLC patients carrying EGFR(p.T790M). Unfortunately, EGFR-mutated tumors can escape EGFR blockade in several other ways: by amplification of the MET receptor tyrosine kinase or ERBB2, by mutations in PIK3CA and BRAF, as well as by activation of AXL and NFkB (167). KRAS mutations in the tumor are a negative predictor of response to EGFR-TKIs or anti-EGFR antibodies (168). Therefore, continuous monitoring of treatment effects and arising resistance during follow-up is of high clinical relevance. Recently, the first ctDNA screening test (cobas® EGFR Mutation Test v2, Roche, Switzerland) for molecular analysis of ctDNA in metastatic NSCLC patients in whom mutation screening was impossible in tumor tissue was approved by the FDA. This test detects a series of activating EGFR mutations–exon 19 deletions and p.L858R/p.T790M point mutations—in cfDNA, identifying patients who might benefit from TKI therapy in oncological diagnostic routine. cfDNA was analyzed for the presence of p.T790M mutation and patient-specific EGFR activating mutations known from primaries by BEAMing in a cohort of 23 advanced NSCLC cases progressive after EGFR-TKI treatment and 21 advanced NSCLC, EGFR-TKIs-naive patients (99). Most patients had already progressed to stage IV. The p.T790M mutation was detected in 43.5% of EGFR-TKI treated patients at an AF from 0.1 to 1%. Similar findings were reported independently (49, 100). In the phase I AURA study assessing safety, tolerability, and efficacy of osimertinib in EGFR mutant NSCLC progressive under EGFR-TKIs, plasma p.T790M genotyping in 237 patients revealed 70% sensitivity and 69% specificity. ctDNA p.T790M-positive patients had similar outcomes as those with the same mutation detected in tumor tissue. By contrast, p.T790M plasma negativity and tumor tissue positivity correlated with a favorable outcome which, in conclusion, should prompt for a confirmatory tumor biopsy to avoid false-negatives in case p.T790M-negative plasma (101). Correlation between OS and dynamic changes in AFs of EGFR(p.T790M) before and after EGFR-TKI therapy was addressed in a prospective cohort of 103 advanced-stage NSCLC patients (102). Plasma samples before and after EGFR-TKI therapy were analyzed by dPCR array chip (Fluidigm, South San Francisco, CA, USA) and ARMS. Patients with the EGFR(p.T790M) in pre-TKI plasma samples had inferior PFS and OS compared to those without the mutation. Patients with an EGFR-sensitizing mutation and high AF of pre-TKI p.T790M had a shorter PFS (p = 0.001) under EGFR-TKI compared to those with a low AF (102). The resistance mutation EGFR(p.C797S) was identified in cfDNA by NGS of 15 EGFR-TKI ADZ9291-treated patients whose tumors were positive for EGFR(p.T790M) (169). Besides EGFR(p.T790M), a range of driver and resistance mutations/aberrations, including ALK, ROS1, and RET rearrangements, HER2 insertions, and MET amplification have been identified in pretreatment plasma of progressive NSCLC patients by NGS with 100% specificity and 77% sensitivity (170). Similarly, in a study on 102 prospectively enrolled NSCLC patients (81% LAC, 96% stage IV) driver and resistance mutations were identified by ctDNA NGS (92): Concordance between tumor tissue DNA and ctDNA correlates is higher with shorter intervals between tissue and blood sampling (p = 0.038) (92). In sum, plasma ctDNA testing in LC is beneficial for therapy selection and more feasible than serial tissue biopsies.
The aim of the large prospective TRACER× trial (TRAcking non-small lung Cancer Evolution through therapy R[x]) on 842 NSCLC patients (stages I–IIIA) is to monitor clonal evolution from initial diagnosis to death by analysis of multiple genomic regions in tumor tissue, cfDNA, and CTCs (171, 172). Analysis of the first 100 patients detected ctDNA in 48% (46/96) during early stages. Predictors of ctDNA-positivity were non-LAC histology, increased proliferative indices, and lymphovascular invasion (103). Clonal mutations in tumor tissue were identified in 100% and subclonal ones in 68% of ctDNA-positive patients. Tumor volume and clonal variant mean AF correlated directly (103). Hence, prior knowledge of clonal variants for ctDNA screening is more sensitive than tracking subclonal variants. Pre- and postsurgical ctDNA profiling with patient-specific gene panels during follow-up confirmed SNVs in 93% (13/14) of patients with morphological or clinical relapse. The median interval between ctDNA occurrence and later CT-morphological relapse was 70 days (103). High amounts of different mutations in ctDNA of LC patients is associated with poor OS (173).
Colorectal Cancer
Significant proportions of CRCs harbor mutations in KRAS, BRAF, or NRAS that are negative predictors for an EGFR-blockade therapy, making these hotspots an appealing diagnostic cfDNA target (117, 138). Direct correlation between KRAS ctDNA AF and OS was found (4). Metastatic lesion-specific radiographic responses to targeted therapies in CRC can be driven by distinct resistance mechanisms affecting MAPK pathway genes which can arise asynchronously in separate lesions in a single patient (174). Following initial therapy for stage IIIa CRC, a patient experienced relapse and new liver metastases. Upon progression, the patient received an EGFR blockade with cetuximab and panitumumab. While the TP53(p.E171*) mutation was identified in the primary, the resistance mutation MAP2K1(p.K57T) upon EGFR-blockade arose in only one liver metastasis. Response to EGFR-blockade was objectified by a decrease in size of the primary and one liver metastasis. Shortly after, the neighboring liver metastasis increased and ctDNA analysis now identified KRAS(p.Q61H) which was not present in either primary tumor now or in the responding liver metastasis, suggesting that it was present even before EGFR-blockade (174), illustrating that single tumor biopsies may not sufficiently represent tumor heterogeneity. Overall outcome depends on lesion-specific therapy responses. Patients with RAS wild-type CRC primaries and ctDNA positive for KRAS and BRAF mutations were resistant to the EGFR-blockade (4, 121, 122, 163). Such mutations can be detected in the blood of cetuximab or panitumumab treated patients as early as 10 months before clinical disease progression (121, 163). Interestingly, KRAS resistance clones, which emerge in blood during EGFR blockade, can even decline upon withdrawal of anti-EGFR antibodies, allowing for rechallenge that can again lead to response (122).
A multicenter prospective study on 53 metastatic chemotherapy-naive CRC patients analyzed ctDNA level as early therapy response marker. Plasma before, 3 days after surgery, and before the 2nd chemotherapy cycle revealed no significant difference before and 3 days after surgery. Reduction from presurgical to pre-chemotherapy level predicted radiographic response better than absolute levels with no significant correlation between ctDNA fold change and OS (123). ctDNA for MRD detection was prospectively tracked in 18 resected CRC patients. Plasma was serially sampled over 12 weeks postoperatively. Tumor-specific mutations were identified from archival FFPE tissue and individually selected mutations were tracked by BEAMing in plasma. No recurrence was observed if ctDNA was undetectable 2 weeks after surgery. By contrast, ctDNA positivity in postoperative plasma was predictive of relapse. A sharp drop in ctDNA 2 to 10 days after surgery was observed after complete as opposed to incomplete resection (48).
Circulating Tumor Cells
Biological Significance and Origin of CTCs
Circulating tumor cells are cancer cells, detached from tumor tissue, floating in the bloodstream, bearing the potential to seed the disease to other sites, as demonstrated half a century ago (175). Metastatic progression comprises of four steps: local invasion, intravasation, extravasation, and colonization (176, 177). CTC precursors can remodel the surrounding stroma by activation of extracellular proteases allowing them to overcome the basement membrane and extracellular matrix to which they normally adhere (178). A subpopulation of precursor carcinomatous CTCs can undergo partial or complete EMT, e.g., by repressing expression of E-cadherin and cytokeratin as well as inducing vimentin and N-cadherin. Thereby, they detach from epithelial sheets to become invasive and motile. Once they invade blood vessels within the tumor microenvironment they can become CTCs (179). Most CTCs will not survive because of anoikis (apoptosis due to vanished cell–matrix interactions), shearing forces of blood flow, and immune cell attack (180). Once lodged in blood vessels of distant organs, CTCs can extravasate and infiltrate the surroundings (178). During colonization, CTCs resume growth in a distant organ to form a metastasis, undergo cell death or enter dormancy (181). CTC have been isolated from blood as either single cells, or clusters. In breast and prostate cancer, CTC clusters were shown to consist of oligoclonal cells from the primary and are associated with higher metastatic potential than single CTCs (182). CTC clusters contain either just a group of neoplastic cells or are associated with fibroblasts, leukocytes, endothelial cells, and platelets (183). In the next section, we will review the technologies for CTC detection and enrichment, followed by a critical appraisal of the significance of CTC enumeration in breast, lung, and colorectal cancer.
CTC Detection and Enrichment Technologies
The key challenges in CTC isolation are their rarity in blood (1–10 CTCs per 10 ml) and lack of cancer-specific surface markers. Hence, a multitude of enrichment technologies have been developed. CTCs can be positively or negatively enriched based on their biological or physical properties. Methods based on biological properties use antibodies that bind surface markers on CTCs. For detection of epithelial CTCs, antibodies against EpCAM and cytokeratins (CK8, CK18, CK19) are frequently used, while mesenchymal CTCs can be selected by antibodies against N-cadherin and vimentin. Cell Search is so far the only platform approved by the FDA for clinical use. Epithelial CTC enrichment is based on positive selection via EpCAM antibody-coated ferromagnetic beads with subsequent staining with DAPI, anti-CD45, and anti-cytokeratin to identify and enumerate CTCs. CTCs can be negatively enriched by antibodies against CD45 to deplete leukocytes from a blood sample. Alternative enrichment strategies are based on physical properties, namely size, deformability, density, and electric charge. Detailed descriptions of these technologies have already been summarized (184). Epithelial antigen-based positive or negative selection is limited due to their inability to detect carcinoma CTCs after EMT. Hence, these approaches can produce false-negative results as opposed to biophysical methods. However, in the latter, blood cells can have properties similar to CTCs, resulting in high false-positive rates. To date, no single method is able to capture the entire spectrum of CTCs. Due to numerous different isolation approaches and lack of multicenter validation, their robustness, reproducibility, sensitivity, and specificity remain elusive. Currently, the method of choice depends on tumor type and intended downstream analyses: genetic studies require high purity, FISH and immunofluorescence high capture efficiency, and drug testing even viable CTCs. Hence, similar to cfDNA, development of standardized protocols for sample handling, sample storage, enrichment, enumeration, and evaluation are important.
CTC in Cancer Patient Management
The prognostic value of CTC enumeration was demonstrated in BC, LC, CRC, and mCRPC (177). While in patients with BC (78, 185), mCRPC (186), and NSCLC (104) a cut-off value of ≥5 CTC per 7.5 ml of blood indicates worse prognosis, in CRC a cut-off of ≥3 CTC per 7.5 ml of blood is predictive of shorter OS (124). Higher CTC counts in pulmonary vein and mesenteric blood than in peripheral blood of LC and CRC patients, respectively, have been reported (187, 188). In the next section, we will discuss prognostic value of CTC number and use of CTC in monitoring the effect of anticancer therapy.
Breast Cancer
A prospective study on early-stage BC investigated the prognostic value of CTC number for OS in 2,026 patients before and 1,492 after adjuvant chemotherapy using CellSearch. The patients were followed over a median of 35 months. CTCs before chemotherapy were detected in 21.5% and post-therapeutically in 22.5% of patients. Their presence before and after chemotherapy was associated with shorter RFS (p < 0.0001), BC-specific survival (p < 0.008) and OS (p < 0.0002). In metastatic BC, ≥5 CTCs per 7.5 ml of blood before therapy start and at first follow-up was predictive of shorter PFS/OS and correlated with lymph node metastasis (78). The predictive value was independent of time to and site of metastasis, as well as hormone receptor status (79). Other studies confirmed the prognostic value of CTC counts concerning PFS and OS in early-stage and metastatic BC (189, 190). The SWOG S050 randomized clinical trial investigated the value of CTC enumeration in monitoring chemotherapy response in mBC, in particular, if an early switch in first-line regimen would improve OS in those subjects with increasing CTCs under the primary drug (80). Randomization was between continuation of the initial treatment and therapy change. No difference in median OS was observed in the high-risk group despite the change of chemotherapy. Based on this finding, the American Oncology Society clinical practice guidelines for CTCs considered not to use CTC count in mBC management (191). However, median OS for low, moderate, and high-risk groups were 35, 23, and 13 months, respectively. This study confirmed the prognostic but not predictive value of CTC counts in mBC patients receiving first-line chemotherapy (80). The prognostic value of CTCs in 44 HER2-positive mBC patients with cerebral metastases not previously treated with whole brain radiotherapy under HER2-targeted treatment (lapatinib, capecitabine) was investigated as a part of the LANDSCAPE clinical trial. CTC number was analyzed at baseline and at day 21 before the second therapy cycle. Objective CNS response was significantly higher in patients who did not have any CTC detected at day 21 (85). However, others showed the inability of CTCs to predict the risk for cancer dissemination and that the prognostic value of CTC detection depends on the test method (81, 192). Single CTCs and pooled CTCs were analyzed for PIK3CA mutations in 18 mBC patients. Analysis of single CTCs in two patients revealed different PIK3CA mutations in single CTCs (82). If CTCs would reflect biologically relevant clones with regard to treatment, then the heterogeneity between single CTCs in an individual patient limits the usefulness of their genetic analysis in diagnostic settings. CTCs have been frequently detected in HER2+ primary tumors of mBC patients (193). HER2+ CTCs have high metastatic potential (83) and were found in 89% of patients with HER2-negative primaries (84). Residual CTC clusters at days 15 and 29 of therapy (nab-paclitaxel with or without tigatuzumab), but not at baseline, predicted shorter PFS (194).
Lung Cancer
Prognostically relevant cut-off value for CTC counts by CellSearch in NSCLC was defined as ≥5 CTCs/7.5 ml blood, and in SCLC as ≥50 CTCs/7.5 ml (104, 195). In a study on 208 NSCLC patients (stages I–IV), 50% of cases had CTCs detected preoperatively by ISET filtration enrichment. Here, a cut-off of ≥50 CTCs/7.5 ml blood significantly correlated with decreased DFS and OS in early and advanced-stage NSCLC. CTC counts did not correlate with tumor stage, age, gender, tobacco exposure, tumor size, and malignant pleural effusion (105). Serial CTC enumeration showed correlation of a low count with radiographic tumor response, while increased numbers reflected tumor progression. Therefore, CTC number might be used as pharmacodynamic marker where change in CTC count during therapy would inform about response (106).
The prognostic value of CTCs in early NSCLC was assessed in a cohort of 168 chronic obstructive pulmonary disease patients. Five CTC-positive patients (5/168) developed lung nodules 1–4 years after CTC detection, 4/5 were diagnosed with LAC, and 1/5 with LSCC. However, three more formally CTC-positive subjects did not develop any neoplasm during follow-up (107), highlighting a relatively high false-positive rate that limits clinical applicability.
High concordance rates in EGFR mutations in NSCLC patients (84%, 31/37) between primaries and CTCs were observed (108). 92% concordance was found in another study (106). A classifier based on CNVs in single and pooled CTCs from 31 pretreatment SCLCs distinguished chemosensitive from chemorefractory cases (cisplatin and etoposide) with 83.3% accuracy. This study concluded that chemoresistance occurring under therapy differs from de novo resistance since five patients that initially responded and then relapsed had the same CNV profiles before and after relapse (109). CTCs were present in 85% of 97 LSCC patients (range 0–44,896/7.5 ml blood) at baseline before chemotherapy. CTC clusters and CTCs with apoptotic morphology were detected in 32 and 57% of patients, respectively. Baseline CTC count and change in number after one cycle of chemotherapy were independent prognostic factors in SCLC. The numbers of CTCs and CTC clusters correlated with stage, serum LDH, presence of liver metastases, and number of metastatic sites (195).
Colorectal Cancer
A prospective, multicenter study on 430 mCRC patients evaluated the prognostic and predictive value of CTC counts by CellSearch at baseline and after three lines of therapy. ≥ 3 CTCs/7.5 ml blood at baseline or during follow-up were an independent prognostic factor of poor PFS and OS (124). Screening for EMT in CTCs in 1,203 CRC patients by CanPatrol™ enrichment with analysis of epithelial (EpCAM, cytokeratins), and mesenchymal (VIM, TWIST, AKT2, SNAI1) markers revealed three distinct phenotypes: epithelial, mesenchymal, and biphenotypic. CTCs were detectable in 86.9% of patients. Total CTC counts correlated with clinical stage, lymph node, and distant metastases. Biphenotypic and mesenchymal phenotype counts correlated with tumor stage, suggesting that CTCs with EMT have a higher metastatic potential and are more aggressive (125). Preoperative CTC detection (≥1 CTC/7.5ml blood) is an independent prognostic factor for disease progression and OS in patients with non-metastatic CRC (126).
KRAS analysis in primaries is mandatory before starting EGFR-targeted therapy in mCRC. Concordance rate in KRAS state between CTCs and primaries was 77% (128) while only 50% were found in another study (196). This discordance between CTCs and primaries might be due to intratumoral heterogeneity and multiple metastatic clones that disseminate during early disease stages that might remain dormant for years (127).
The usefulness of KRAS testing in CTCs before curative surgery was addressed in 35 mCRC cases at various tumor stages. CTCs were captured based on their size and analyzed for KRAS mutations by ddPCR. 90% of patients harbored at least one CTC/3ml blood; in 7% of cases, CTC clusters were detected. CTC counts correlated with disease stage, but not with serum concentrations of tumor markers, CEA and CA19.9. CTCs of 57% patients were positive for one of the relevant KRAS mutations in codon 12 or 13 (128). Heterogeneity in EGFR expression, mutations in PIK3CA and KRAS between CTCs of the same mCRC patient was observed (127).
Comments and Future Perspectives
cfDNA quantification appears unsuited for cancer-specific questions, as both sensitivity and specificity are low. Numerous studies suggest a clinical usefulness of ctDNA testing as a prognostic, predictive and diagnostic biomarker in various neoplasms. Concordance, though not 100%, in frequencies of mutations found in tumor tissue and cfDNA, with significantly lower allele frequencies in cfDNA, have been described in many studies. Therefore, there is a need for development of highly sensitive analytic methods able to detect genetic alterations at low allelic frequencies in plasma. However, this implies a certain unspecific detection rate, partially impairing diagnostic routine use. As shown in several studies, combination of single or several tumor-associated genetic alterations found in cfDNA with other biomarkers and imaging can increase diagnostic specificity and sensitivity. In clinics, ctDNA analysis will most likely be used complementarily to imaging for monitoring disease burden as it can not deliver precise information about tumor location. Thus, we foresee that a combination of genetic analysis of tumor tissue and ctDNA together with medical imaging may deliver increased accuracy in determination of metastatic growth. Ultimately, tumor surveillance via blood samples, periodically drawn by physicians also in remote locations and sent to the treating center, could be of significant benefit for the patients. Importantly, analytic and clinical validity of ctDNA remains to be demonstrated. The mechanisms of cfDNA release into the bloodstream still remain elusive. Whether all metastatic sites contribute similarly to the ctDNA pool remains unknown. Overall tumor burden appears to be better represented when restricting ctDNA tests to previously known tumor-specific mutant alleles, typically those derived from molecular workup of tumor biopsies with emphasis on driver mutations that are less likely to change during tumor evolution. Qualitative cfDNA analysis may inform of tumor heterogeneity and detect emerging secondary resistance mutations. Detection and quantification of EGFR(p.T790M) mutation in ctDNA might be used as a predictive biomarker under EGFR-TKI treatment of NSCLC patients that could influence treatment changes. However, such clinical evidence has not been demonstrated for other mutations. As with genetic data from tumor tissue, it remains debatable how to deal with aberrations for which no targeted therapies are available. Unfortunately, knowledge about likely progressive disease before deterioration of clinical symptoms may not even be beneficial for quality of life and would not necessarily prolong OS. Most importantly, there is an urgent need to elucidate origin, function, and biological significance of cfDNA before implementing it in routine diagnostics.
The lack of detection at diagnosis or at time of progression in some patients is a key limitation to the use of ctDNA as prognostic marker. Therefore, at this time, we believe that analysis of ctDNA genetic alterations will and should not replace tumor biopsy or radiological evaluation. It can, however, significantly improve clinical follow-up by monitoring treatment efficacy more specifically than imaging. While ctDNA levels might correlate with tumor volume, total cfDNA reflects tumor burden, mirroring overall disease biology. We envision that clinical laboratories will need to provide deep sequencing analysis to define clonal mutation prevalence, focusing on actionable genes and mutations as well as those considered hallmarks of cancer, e.g., KRAS mutations to stratify patients with CRC for anti-EGFR therapy, driver mutations to be tested in plasma for tumor monitoring, and therapy response as well as EGFR mutations to predict benefit from erlotinib, gefitinib, and osimertinib.
Despite technological advance in CTC isolation and analytic methods, the biological fate and significance of these cells remains elusive. It may be expected that they represent a highly heterogeneous population. In fact, antibody-based detection of single tumor cells—not in blood but lymph nodes—has led to substantial effort in routine diagnostics due to the introduction of respective TNM category pN0[i+] (197). However, this category was recently removed (198). Lymph nodes with immunohistochemically detected isolated tumor cells are now only tabulated in the report but do no longer contribute to overall N classification (199). While ctDNA represents a potpourri of many, if not all tumor sites in a patient, CTC-derived nucleic acids are much lower in copies and, hence, may not improve diagnostics over conventional biopsies while ctDNA tests are very likely to proliferate in clinical practice. As for cfDNA, there are several drawbacks in CTC research that limit usefulness of CTC counting in clinics: (i) different methods have been employed for CTC enumeration limiting the comparison of the data across studies. (ii) Reproducibility and sensitivity of these methods has not been thoroughly determined. (iii) All currently available methods are tumor type-specific, i.e., mostly epithelial cancer cells, but not those after EMT are selected while the latter may be of high biological relevance (4). It is still impossible to determine aggressiveness of single CTCs and CTC clusters (5). No universal signatures of CTCs have been identified—should they even exist—that would cover any stage and type of cancer.
In conclusion, we believe that liquid biopsies are likely to become an additional standard for monitoring progressive genomic alterations over tumor evolution during exposure to targeted therapies. They also might prove effective in cases where obtaining tumor tissue would have a high risk of clinical deterioration. For the majority of cases, however, we envision liquid biopsy as second-line diagnostic tool, building on the findings of morphological, genetic, and epigenetic changes derived from classical tissue biopsies, eliminating their shortcoming in holistic, as well as spatio-temporal understanding of each neoplasm in a personalized, patient-oriented manner.
Nomenclature
| AC | adenocarcinoma |
| AF | allelic frequency |
| cfDNA | cell-free DNA |
| ctDNA | circulating tumor DNA |
| CNS | central nervous system |
| CNV | copy number variation |
| CRC | colorectal cancer |
| CTC | circulating tumor cell |
| BAC | bronchoalveolar carcinoma |
| BC | breast cancer |
| BCSS | breast cancer-specific survival |
| DFS | disease-free survival |
| DNA | desoxyribonucleic acid |
| eBC | early-stage breast cancer |
| EMT | epithelial–mesenchymal transition |
| FDA | U.S. Food and Drug Administration |
| dPCR | digital polymerase chain reaction |
| ddPCR | droplet digital PCR |
| iDES | integrated digital error suppression |
| HCC | hepatocellular carcinoma patients |
| HGIN | high-grade intraepithelial lesions |
| LAC | lung adenocarcinoma |
| LC | lung cancer |
| LCC | large cell carcinoma |
| LDH | lactate dehydrogenase |
| LOH | loss of heterozygosity |
| LSCC | lung squamous cell cancer |
| mBC | metastatic breast cancer |
| mCRC | metastatic colorectal cancer |
| mCRPC | metastatic castration-resistant prostate cancer |
| MRD | minimal residual disease |
| NGS | next-generation sequencing, massively parallel sequencing |
| NSCLC | non-small cell lung cancer |
| NSE | neuron-specific enolase |
| OS | overall survival |
| PCR | polymerase chain reaction |
| PFS | progression-free survival |
| PSA | prostate-specific antigen |
| PPV | positive predictive value |
| qPCR | quantitative polymerase chain reaction, also termed real-time PCR |
| RFS | recurrence-free survival |
| SCLC | small cell lung cancer |
| SNV | single-nucleotide variant |
| tDNA | tumor DNA (isolated from tumor tissue directly) |
| TKI | tyrosine kinase inhibitor |
| TNBC | triple-negative (estrogen and progesterone receptor, and Her-2) |
| UID | unique identifiers |
| WES | whole-genome sequencing |
| WGS | whole-exome sequencing |
Author Contributions
IH, JH, and MT have contributed equally to write this manuscript.
Conflict of Interest Statement
The authors declare that the article was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Anker P, Mulcahy H, Chen XQ, Stroun M. Detection of circulating tumour DNA in the blood (plasma/serum) of cancer patients. Cancer Metastasis Rev (1999) 18:65–73. doi:10.1023/A:1006260319913
2. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A (2005) 102:16368–73. doi:10.1073/pnas.0507904102
3. Murtaza M, Dawson S-J, Tsui DWY, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature (2013) 497:108–12. doi:10.1038/nature12065
4. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med (2014) 6:224ra24. doi:10.1126/scitranslmed.3007094
5. Murtaza M, Dawson S-J, Pogrebniak K, Rueda OM, Provenzano E, Grant J, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun (2015) 6:8760. doi:10.1038/ncomms9760
6. Chan KCA, Jiang P, Zheng YWL, Liao GJW, Sun H, Wong J, et al. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem (2013) 59:211–24. doi:10.1373/clinchem.2012.196014
7. De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CKY, Nuciforo P, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol (2014) 25:1729–35. doi:10.1093/annonc/mdu239
8. Sun K, Jiang P, Chan KCA, Wong J, Cheng YKY, Liang RHS, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci U S A (2015) 112:E5503–12. doi:10.1073/pnas.1508736112
9. Stroun M, Lyautey J, Lederrey C, Mulcahy HE, Anker P. Alu repeat sequences are present in increased proportions compared to a unique gene in plasma/serum DNA: evidence for a preferential release from viable cells? Ann N Y Acad Sci (2001) 945:258–64. doi:10.1111/j.1749-6632.2001.tb03894.x
10. Jiang P, Chan CWM, Chan KCA, Cheng SH, Wong J, Wong VW-S, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci U S A (2015) 112:E1317–25. doi:10.1073/pnas.1500076112
11. Mouliere F, El Messaoudi S, Pang D, Dritschilo A, Thierry AR. Multi-marker analysis of circulating cell-free DNA toward personalized medicine for colorectal cancer. Mol Oncol (2014) 8:927–41. doi:10.1016/j.molonc.2014.02.005
12. Jiang P, Lo YMD. The long and short of circulating cell-free DNA and the ins and outs of molecular diagnostics. Trends Genet (2016) 32:360–71. doi:10.1016/j.tig.2016.03.009
13. Mouliere F, Thierry AR. The importance of examining the proportion of circulating DNA originating from tumor, microenvironment and normal cells in colorectal cancer patients. Expert Opin Biol Ther (2012) 12(Suppl 1):S209–15. doi:10.1517/14712598.2012.688023
14. Rykova EY, Morozkin ES, Ponomaryova AA, Loseva EM, Zaporozhchenko IA, Cherdyntseva NV, et al. Cell-free and cell-bound circulating nucleic acid complexes: mechanisms of generation, concentration and content. Expert Opin Biol Ther (2012) 12(Suppl 1):S141–53. doi:10.1517/14712598.2012.673577
15. Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch R-D, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res (2001) 61:1659–65.
16. Heidary M, Auer M, Ulz P, Heitzer E, Petru E, Gasch C, et al. The dynamic range of circulating tumor DNA in metastatic breast cancer. Breast Cancer Res (2014) 16:421. doi:10.1186/s13058-014-0421-y
17. Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell (2016) 164:57–68. doi:10.1016/j.cell.2015.11.050
18. García-Olmo DC, Ruiz-Piqueras R, García-Olmo D. Circulating nucleic acids in plasma and serum (CNAPS) and its relation to stem cells and cancer metastasis: state of the issue. Histol Histopathol (2004) 19:575–83. doi:10.14670/HH-19.575
19. Chen Z, Fadiel A, Naftolin F, Eichenbaum KD, Xia Y. Circulation DNA: biological implications for cancer metastasis and immunology. Med Hypotheses (2005) 65:956–61. doi:10.1016/j.mehy.2005.04.042
20. Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta (2001) 313:139–42. doi:10.1016/S0009-8981(01)00665-9
21. Anker P, Stroun M, Maurice PA. Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res (1975) 35:2375–82.
22. Wang W, Kong P, Ma G, Li L, Zhu J, Xia T, et al. Characterization of the release and biological significance of cell-free DNA from breast cancer cell lines. Oncotarget (2017) 8:43180. doi:10.18632/oncotarget.17858
23. Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res (1977) 37:646–50.
24. van der Vaart M, Pretorius PJ. Is the role of circulating DNA as a biomarker of cancer being prematurely overrated? Clin Biochem (2010) 43:26–36. doi:10.1016/j.clinbiochem.2009.08.027
25. Zhong XY, Ladewig A, Schmid S, Wight E, Hahn S, Holzgreve W. Elevated level of cell-free plasma DNA is associated with breast cancer. Arch Gynecol Obstet (2007) 276:327–31. doi:10.1007/s00404-007-0345-1
26. Frank MO. Circulating cell-free DNA differentiates severity of inflammation. Biol Res Nurs (2016) 18:477–88. doi:10.1177/1099800416642571
27. Tug S, Helmig S, Ricarda Deichmann E, Schmeier-Jürchott A, Wagner E, Zimmermann T, et al. Exercise-induced increases in cell free DNA in human plasma originate predominantly from cells of the haematopoietic lineage. Exerc Immunol Rev (2015) 21:164–73.
28. Thierry AR, Mouliere F, Gongora C, Ollier J, Robert B, Ychou M, et al. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res (2010) 38:6159–75. doi:10.1093/nar/gkq421
29. Yu SCY, Lee SWY, Jiang P, Leung TY, Chan KCA, Chiu RWK, et al. High-resolution profiling of fetal DNA clearance from maternal plasma by massively parallel sequencing. Clin Chem (2013) 59:1228–37. doi:10.1373/clinchem.2013.203679
30. Butler TM, Spellman PT, Gray J. Circulating-tumor DNA as an early detection and diagnostic tool. Curr Opin Genet Dev (2017) 42:14–21. doi:10.1016/j.gde.2016.12.003
31. Jung K, Fleischhacker M, Rabien A. Cell-free DNA in the blood as a solid tumor biomarker – a critical appraisal of the literature. Clin Chim Acta (2010) 411:1611–24. doi:10.1016/j.cca.2010.07.032
32. Swinkels DW, Wiegerinck E, Steegers EA, de Kok JB. Effects of blood-processing protocols on cell-free DNA quantification in plasma. Clin Chem (2003) 49:525–6. doi:10.1373/49.3.525
33. Chan KCA, Yeung S-W, Lui W-B, Rainer TH, Lo YMD. Effects of preanalytical factors on the molecular size of cell-free DNA in blood. Clin Chem (2005) 51:781–4. doi:10.1373/clinchem.2004.046219
34. Parpart-Li S, Bartlett B, Popoli M, Adleff V, Tucker L, Steinberg R, et al. The effect of preservative and temperature on the analysis of circulating tumor DNA. Clin Cancer Res (2017) 23:2471–7. doi:10.1158/1078-0432.CCR-16-1691
35. Malentacchi F, Pizzamiglio S, Verderio P, Pazzagli M, Orlando C, Ciniselli CM, et al. Influence of storage conditions and extraction methods on the quantity and quality of circulating cell-free DNA (ccfDNA): the SPIDIA-DNAplas external quality assessment experience. Clin Chem Lab Med (2015) 53:1935–42. doi:10.1515/cclm-2014-1161
36. Haselmann V, Ahmad-Nejad P, Geilenkeuser WJ, Duda A, Gabor M, Eichner R, et al. Results of the first external quality assessment scheme (EQA) for isolation and analysis of circulating tumour DNA (ctDNA). Clin Chem Lab Med (2017) 56(2):220–8. doi:10.1515/cclm-2017-0283
37. Thijssen MA, Swinkels DW, Ruers TJM, de Kok JB. Difference between free circulating plasma and serum DNA in patients with colorectal liver metastases. Anticancer Res (2002) 22:421–5.
38. Jung M, Klotzek S, Lewandowski M, Fleischhacker M, Jung K. Changes in concentration of DNA in serum and plasma during storage of blood samples. Clin Chem (2003) 49:1028–9. doi:10.1373/49.6.1028
39. Lee TH, Montalvo L, Chrebtow V, Busch MP. Quantitation of genomic DNA in plasma and serum samples: higher concentrations of genomic DNA found in serum than in plasma. Transfusion (2001) 41:276–82. doi:10.1046/j.1537-2995.2001.41020276.x
40. Gautschi O, Bigosch C, Huegli B, Jermann M, Marx A, Chassé E, et al. Circulating deoxyribonucleic acid as prognostic marker in non–small-cell lung cancer patients undergoing chemotherapy. J Clin Oncol (2004) 22:4157–64. doi:10.1200/JCO.2004.11.123
41. Perakis S, Auer M, Belic J, Heitzer E. Advances in circulating tumor DNA analysis. Advances in Clinical Chemistry. Elsevier (2017). p. 73–153. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0065242316301019
42. Thelwell N, Millington S, Solinas A, Booth J, Brown T. Mode of action and application of Scorpion primers to mutation detection. Nucleic Acids Res (2000) 28:3752–61. doi:10.1093/nar/28.19.3752
43. Sorensen BS, Wu L, Wei W, Tsai J, Weber B, Nexo E, et al. Monitoring of epidermal growth factor receptor tyrosine kinase inhibitor-sensitizing and resistance mutations in the plasma DNA of patients with advanced non-small cell lung cancer during treatment with erlotinib: monitoring EGFR Mutations in Plasma DNA. Cancer (2014) 120:3896–901. doi:10.1002/cncr.28964
44. Jenkins S, Yang JC-H, Ramalingam SS, Yu K, Patel S, Weston S, et al. Plasma ctDNA analysis for detection of the EGFR T790M mutation in patients with advanced non-small cell lung cancer. J Thorac Oncol (2017) 12:1061–70. doi:10.1016/j.jtho.2017.04.003
45. Qin L, Zhong W, Zhang L, Li L, Wang M. Comparison of three methods for detecting epidermal growth factor receptor mutations in plasma DNA samples of Chinese patients with advanced non-small cell lung cancer. Chin Med J (Engl) (2011) 124:887–91.
46. Wang W, Song Z, Zhang Y. A Comparison of ddPCR and ARMS for detecting EGFR T790M status in ctDNA from advanced NSCLC patients with acquired EGFR-TKI resistance. Cancer Med (2016) 6(1):154–62. doi:10.1002/cam4.978
47. Newman AM, Bratman SV, To J, Wynne JF, Eclov NCW, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med (2014) 20:548–54. doi:10.1038/nm.3519
48. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med (2008) 14:985–90. doi:10.1038/nm.1789
49. Oxnard GR, Paweletz CP, Kuang Y, Mach SL, O’Connell A, Messineo MM, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res (2014) 20:1698–705. doi:10.1158/1078-0432.CCR-13-2482
50. Beaver JA, Jelovac D, Balukrishna S, Cochran RL, Croessmann S, Zabransky DJ, et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin Cancer Res (2014) 20:2643–50. doi:10.1158/1078-0432.CCR-13-2933
51. Klevebring D, Neiman M, Sundling S, Eriksson L, Darai Ramqvist E, Celebioglu F, et al. Evaluation of exome sequencing to estimate tumor burden in plasma. PLoS One (2014) 9:e104417. doi:10.1371/journal.pone.0104417
52. Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A (2011) 108:9530–5. doi:10.1073/pnas.1105422108
53. Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol (2016) 34:547–55. doi:10.1038/nbt.3520
54. Phallen J, Sausen M, Adleff V, Leal A, Hruban C, White J, et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med (2017) 9:eaan2415. doi:10.1126/scitranslmed.aan2415
55. Leung F, Kulasingam V, Diamandis EP, Hoon DSB, Kinzler K, Pantel K, et al. Circulating tumor DNA as a cancer biomarker: fact or fiction? Clin Chem (2016) 62:1054–60. doi:10.1373/clinchem.2016.260331
56. Xu S, Lou F, Wu Y, Sun D-Q, Zhang J-B, Chen W, et al. Circulating tumor DNA identified by targeted sequencing in advanced-stage non-small cell lung cancer patients. Cancer Lett (2016) 370:324–31. doi:10.1016/j.canlet.2015.11.005
57. Vanni I, Coco S, Truini A, Rusmini M, Dal Bello M, Alama A, et al. Next-generation sequencing workflow for NSCLC critical samples using a targeted sequencing approach by Ion Torrent PGMTM platform. Int J Mol Sci (2015) 16:28765–82. doi:10.3390/ijms161226129
58. Couraud S, Vaca-Paniagua F, Villar S, Oliver J, Schuster T, Blanche H, et al. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never-smokers: a proof-of-concept study from BioCAST/IFCT-1002. Clin Cancer Res (2014) 20:4613–24. doi:10.1158/1078-0432.CCR-13-3063
59. Chen WW, Balaj L, Liau LM, Samuels ML, Kotsopoulos SK, Maguire CA, et al. BEAMing and droplet digital PCR analysis of mutant IDH1 mRNA in glioma patient serum and cerebrospinal fluid extracellular vesicles. Mol Ther Nucleic Acids (2013) 2:e109. doi:10.1038/mtna.2013.28
60. Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med (2012) 4:162ra154. doi:10.1126/scitranslmed.3004742
61. Dietz S, Schirmer U, Mercé C, von Bubnoff N, Dahl E, Meister M, et al. Low input whole-exome sequencing to determine the representation of the tumor exome in circulating DNA of non-small cell lung cancer patients. PLoS One (2016) 11:e0161012. doi:10.1371/journal.pone.0161012
62. Kuderer NM, Burton KA, Blau S, Rose AL, Parker S, Lyman GH, et al. Comparison of 2 commercially available next-generation sequencing platforms in oncology. JAMA Oncol (2017) 3:996–8. doi:10.1001/jamaoncol.2016.4983
63. Gray SW, Hicks-Courant K, Cronin A, Rollins BJ, Weeks JC. Physicians’ attitudes about multiplex tumor genomic testing. J Clin Oncol (2014) 32:1317–23. doi:10.1200/JCO.2013.52.4298
64. Oshiro C, Kagara N, Naoi Y, Shimoda M, Shimomura A, Maruyama N, et al. PIK3CA mutations in serum DNA are predictive of recurrence in primary breast cancer patients. Breast Cancer Res Treat (2015) 150:299–307. doi:10.1007/s10549-015-3322-6
65. Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Inao T, Sueta A, Fujiwara S, et al. Prognostic role of PIK3CA mutations of cell-free DNA in early-stage triple negative breast cancer. Cancer Sci (2015) 106:1582–9. doi:10.1111/cas.12813
66. Chen Y-H, Hancock BA, Solzak JP, Brinza D, Scafe C, Miller KD, et al. Next-generation sequencing of circulating tumor DNA to predict recurrence in triple-negative breast cancer patients with residual disease after neoadjuvant chemotherapy. NPJ Breast Cancer (2017) 3:24. doi:10.1038/s41523-017-0028-4
67. Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med (2015) 7:302ra133. doi:10.1126/scitranslmed.aab0021
68. Shan M, Yin H, Li J, Li X, Wang D, Su Y, et al. Detection of aberrant methylation of a six-gene panel in serum DNA for diagnosis of breast cancer. Oncotarget (2016) 7:18485. doi:10.18632/oncotarget.7608
69. Li Z, Guo X, Tang L, Peng L, Chen M, Luo X, et al. Methylation analysis of plasma cell-free DNA for breast cancer early detection using bisulfite next-generation sequencing. Tumor Biol (2016) 37(10):13111–9. doi:10.1007/s13277-016-5190-z
70. Shaw JA, Page K, Blighe K, Hava N, Guttery D, Ward B, et al. Genomic analysis of circulating cell-free DNA infers breast cancer dormancy. Genome Res (2012) 22:220–31. doi:10.1101/gr.123497.111
71. Higgins MJ, Jelovac D, Barnathan E, Blair B, Slater S, Powers P, et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res (2012) 18:3462–9. doi:10.1158/1078-0432.CCR-11-2696
72. Madic J, Kiialainen A, Bidard F-C, Birzele F, Ramey G, Leroy Q, et al. Circulating tumor DNA and circulating tumor cells in metastatic triple negative breast cancer patients: ctDNA and CTC in metastatic triple negative breast cancer. Int J Cancer (2015) 136:2158–65. doi:10.1002/ijc.29265
73. Liang DH, Ensor JE, Liu Z, Patel A, Patel TA, Chang JC, et al. Cell-free DNA as a molecular tool for monitoring disease progression and response to therapy in breast cancer patients. Breast Cancer Res Treat (2016) 155:139–49. doi:10.1007/s10549-015-3635-5
74. Dawson S-J, Tsui DWY, Murtaza M, Biggs H, Rueda OM, Chin S-F, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med (2013) 368:1199–209. doi:10.1056/NEJMoa1213261
75. Olsson E, Winter C, George A, Chen Y, Howlin J, Tang M-HE, et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol Med (2015) 7:1034–47. doi:10.15252/emmm.201404913
76. Guttery DS, Page K, Hills A, Woodley L, Marchese SD, Rghebi B, et al. Noninvasive detection of activating estrogen receptor 1 (ESR1) mutations in estrogen receptor-positive metastatic breast cancer. Clin Chem (2015) 61:974–82. doi:10.1373/clinchem.2015.238717
77. Chu D, Paoletti C, Gersch C, VanDenBerg DA, Zabransky DJ, Cochran RL, et al. ESR1 mutations in circulating plasma tumor DNA from metastatic breast cancer patients. Clin Cancer Res (2016) 22:993–9. doi:10.1158/1078-0432.CCR-15-0943
78. Rack B, Schindlbeck C, Jückstock J, Andergassen U, Hepp P, Zwingers T, et al. Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J Natl Cancer Inst (2014) 106:dju066. doi:10.1093/jnci/dju066
79. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med (2004) 351:781–91. doi:10.1056/NEJMoa040766
80. Smerage JB, Barlow WE, Hortobagyi GN, Winer EP, Leyland-Jones B, Srkalovic G, et al. Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J Clin Oncol (2014) 32:3483–9. doi:10.1200/JCO.2014.56.2561
81. Müller V, Riethdorf S, Rack B, Janni W, Fasching PA, Solomayer E, et al. Prognostic impact of circulating tumor cells assessed with the CellSearch SystemTM and AdnaTest BreastTM in metastatic breast cancer patients: the DETECT study. Breast Cancer Res (2012) 14:R118. doi:10.1186/bcr3243
82. Pestrin M, Salvianti F, Galardi F, De Luca F, Turner N, Malorni L, et al. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol Oncol (2015) 9:749–57. doi:10.1016/j.molonc.2014.12.001
83. Meng S, Tripathy D, Shete S, Ashfaq R, Haley B, Perkins S, et al. HER-2 gene amplification can be acquired as breast cancer progresses. Proc Natl Acad Sci U S A (2004) 101:9393–8. doi:10.1073/pnas.0402993101
84. Georgoulias V, Bozionelou V, Agelaki S, Perraki M, Apostolaki S, Kallergi G, et al. Trastuzumab decreases the incidence of clinical relapses in patients with early breast cancer presenting chemotherapy-resistant CK-19mRNA-positive circulating tumor cells: results of a randomized phase II study. Ann Oncol (2012) 23:1744–50. doi:10.1093/annonc/mds020
85. Pierga J-Y, Bidard F-C, Cropet C, Tresca P, Dalenc F, Romieu G, et al. Circulating tumor cells and brain metastasis outcome in patients with HER2-positive breast cancer: the LANDSCAPE trial. Ann Oncol (2013) 24:2999–3004. doi:10.1093/annonc/mdt348
86. Guo N, Lou F, Ma Y, Li J, Yang B, Chen W, et al. Circulating tumor DNA detection in lung cancer patients before and after surgery. Sci Rep (2016) 6:33519. doi:10.1038/srep33519
87. Chen K-Z, Lou F, Yang F, Zhang J-B, Ye H, Chen W, et al. Circulating tumor DNA detection in early-stage non-small cell lung cancer patients by targeted sequencing. Sci Rep (2016) 6:31985. doi:10.1038/srep31985
88. Karachaliou N, Mayo-de las Casas C, Queralt C, de Aguirre I, Melloni B, Cardenal F, et al. Association of EGFR L858R mutation in circulating free DNA with survival in the EURTAC trial. JAMA Oncol (2015) 1:149–57. doi:10.1001/jamaoncol.2014.257
89. Zhang Y, Wang R, Song H, Huang G, Yi J, Zheng Y, et al. Methylation of multiple genes as a candidate biomarker in non-small cell lung cancer. Cancer Lett (2011) 303:21–8. doi:10.1016/j.canlet.2010.12.011
90. Sozzi G, Conte D, Mariani L, Lo Vullo S, Roz L, Lombardo C, et al. Analysis of circulating tumor DNA in plasma at diagnosis and during follow-up of lung cancer patients. Cancer Res (2001) 61:4675–8.
91. Tissot C, Toffart A-C, Villar S, Souquet P-J, Merle P, Moro-Sibilot D, et al. Circulating free DNA concentration is an independent prognostic biomarker in lung cancer. Eur Respir J (2015) 46:1773–80. doi:10.1183/13993003.00676-2015
92. Thompson JC, Yee SS, Troxel AB, Savitch SL, Fan R, Balli D, et al. Detection of therapeutically targetable driver and resistance mutations in lung cancer patients by next-generation sequencing of cell-free circulating tumor DNA. Clin Cancer Res (2016) 22:5772–82. doi:10.1158/1078-0432.CCR-16-1231
93. Pécuchet N, Zonta E, Didelot A, Combe P, Thibault C, Gibault L, et al. Base-position error rate analysis of next-generation sequencing applied to circulating tumor DNA in non-small cell lung cancer: a prospective study. PLoS Med (2016) 13:e1002199. doi:10.1371/journal.pmed.1002199
94. Nygaard AD, Holdgaard PC, Spindler K-LG, Pallisgaard N, Jakobsen A. The correlation between cell-free DNA and tumour burden was estimated by PET/CT in patients with advanced NSCLC. Br J Cancer (2014) 110:363–8. doi:10.1038/bjc.2013.705
95. Fournié GJ, Courtin J-P, Laval F, Chalé J-J, Pourrat JP, Pujazon M-C, et al. Plasma DNA as a marker of cancerous cell death. Investigations in patients suffering from lung cancer and in nude mice bearing human tumours. Cancer Lett (1995) 91:221–7. doi:10.1016/0304-3835(95)03742-F
96. Yao Y, Liu J, Li L, Yuan Y, Nan K, Wu X, et al. Detection of circulating tumor DNA in patients with advanced non-small cell lung cancer. Oncotarget (2017) 8:2130–40. doi:10.18632/oncotarget.12883
97. Nygaard AD, Spindler K-LG, Pallisgaard N, Andersen RF, Jakobsen A. The prognostic value of KRAS mutated plasma DNA in advanced non-small cell lung cancer. Lung Cancer (2013) 79:312–7. doi:10.1016/j.lungcan.2012.11.016
98. Camps C, Sirera R, Bremnes R, Blasco A, Sancho E, Bayo P, et al. Is there a prognostic role of K-ras point mutations in the serum of patients with advanced non-small cell lung cancer? Lung Cancer (2005) 50:339–46. doi:10.1016/j.lungcan.2005.06.007
99. Taniguchi K, Uchida J, Nishino K, Kumagai T, Okuyama T, Okami J, et al. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res (2011) 17:7808–15. doi:10.1158/1078-0432.CCR-11-1712
100. Sundaresan TK, Sequist LV, Heymach JV, Riely GJ, Jänne PA, Koch WH, et al. Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood-based analyses. Clin Cancer Res (2016) 22:1103–10. doi:10.1158/1078-0432.CCR-15-1031
101. Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non–small-cell lung cancer. J Clin Oncol (2016) 34:3375–82. doi:10.1200/JCO.2016.66.7162
102. Wang Z, Chen R, Wang S, Zhong J, Wu M, Zhao J, et al. Quantification and dynamic monitoring of EGFR T790M in plasma cell-free DNA by digital PCR for prognosis of EGFR-TKI treatment in advanced NSCLC. PLoS One (2014) 9:e110780. doi:10.1371/journal.pone.0110780
103. Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature (2017) 545:446–51. doi:10.1038/nature22364
104. Krebs MG, Sloane R, Priest L, Lancashire L, Hou J-M, Greystoke A, et al. Evaluation and prognostic significance of circulating tumor cells in patients with non-small-cell lung cancer. J Clin Oncol (2011) 29:1556–63. doi:10.1200/JCO.2010.28.7045
105. Hofman V, Bonnetaud C, Ilie MI, Vielh P, Vignaud JM, Flejou JF, et al. Preoperative circulating tumor cell detection using the isolation by size of epithelial tumor cell method for patients with lung cancer is a new prognostic biomarker. Clin Cancer Res (2011) 17:827–35. doi:10.1158/1078-0432.CCR-10-0445
106. Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med (2008) 359:366–77. doi:10.1056/NEJMoa0800668
107. Ilie M, Hofman V, Long-Mira E, Selva E, Vignaud J-M, Padovani B, et al. “Sentinel” circulating tumor cells allow early diagnosis of lung cancer in patients with chronic obstructive pulmonary disease. PLoS One (2014) 9:e111597. doi:10.1371/journal.pone.0111597
108. Marchetti A, Del Grammastro M, Felicioni L, Malatesta S, Filice G, Centi I, et al. Assessment of EGFR mutations in circulating tumor cell preparations from NSCLC patients by next generation sequencing: toward a real-time liquid biopsy for treatment. PLoS One (2014) 9:e103883. doi:10.1371/journal.pone.0103883
109. Carter L, Rothwell DG, Mesquita B, Smowton C, Leong HS, Fernandez-Gutierrez F, et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med (2016) 23:114–9. doi:10.1038/nm.4239
110. Perrone F, Lampis A, Bertan C, Verderio P, Ciniselli CM, Pizzamiglio S, et al. Circulating free DNA in a screening program for early colorectal cancer detection. Tumori (2014) 100:115–21. doi:10.1700/1491.16389
111. Kopreski MS, Benko FA, Borys DJ, Khan A, McGarrity TJ, Gocke CD. Somatic mutation screening: identification of individuals harboring K-ras mutations with the use of plasma DNA. J Natl Cancer Inst (2000) 92:918–23. doi:10.1093/jnci/92.11.918
112. Frattini M, Gallino G, Signoroni S, Balestra D, Battaglia L, Sozzi G, et al. Quantitative analysis of plasma DNA in colorectal cancer patients: a novel prognostic tool. Ann N Y Acad Sci (2006) 1075:185–90. doi:10.1196/annals.1368.025
113. Frattini M, Gallino G, Signoroni S, Balestra D, Lusa L, Battaglia L, et al. Quantitative and qualitative characterization of plasma DNA identifies primary and recurrent colorectal cancer. Cancer Lett (2008) 263:170–81. doi:10.1016/j.canlet.2008.03.021
114. Reinert T, Schøler LV, Thomsen R, Tobiasen H, Vang S, Nordentoft I, et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut (2016) 65:625–34. doi:10.1136/gutjnl-2014-308859
115. Flamini E, Mercatali L, Nanni O, Calistri D, Nunziatini R, Zoli W, et al. Free DNA and carcinoembryonic antigen serum levels: an important combination for diagnosis of colorectal cancer. Clin Cancer Res (2006) 12:6985–8. doi:10.1158/1078-0432.CCR-06-1931
116. Sato KA, Hachiya T, Iwaya T, Kume K, Matsuo T, Kawasaki K, et al. Individualized mutation detection in circulating tumor DNA for monitoring colorectal tumor burden using a cancer-associated gene sequencing panel. PLoS One (2016) 11:e0146275. doi:10.1371/journal.pone.0146275
117. Spindler K-LG, Pallisgaard N, Vogelius I, Jakobsen A. Quantitative cell-free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin Cancer Res (2012) 18:1177–85. doi:10.1158/1078-0432.CCR-11-0564
118. Tabernero J, Lenz H-J, Siena S, Sobrero A, Falcone A, Ychou M, et al. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: a retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol (2015) 16:937–48. doi:10.1016/S1470-2045(15)00138-2
119. Spindler K-LG, Pallisgaard N, Appelt AL, Andersen RF, Schou JV, Nielsen D, et al. Clinical utility of KRAS status in circulating plasma DNA compared to archival tumour tissue from patients with metastatic colorectal cancer treated with anti-epidermal growth factor receptor therapy. Eur J Cancer (2015) 51:2678–85. doi:10.1016/j.ejca.2015.06.118
120. Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med (2016) 8:346ra92. doi:10.1126/scitranslmed.aaf6219
121. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature (2012) 486(7404):532–6. doi:10.1038/nature11156
122. Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med (2015) 21:795–801. doi:10.1038/nm.3870
123. Tie J, Kinde I, Wang Y, Wong HL, Roebert J, Christie M, et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol (2015) 26:1715–22. doi:10.1093/annonc/mdv177
124. Cohen SJ, Punt CJ, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol (2008) 26:3213–21. doi:10.1200/JCO.2007.15.8923
125. Zhao R, Cai Z, Li S, Cheng Y, Gao H, Liu F, et al. Expression and clinical relevance of epithelial and mesenchymal markers in circulating tumor cells from colorectal cancer. Oncotarget (2017) 8:9293. doi:10.18632/oncotarget.14065
126. Bork U, Rahbari NN, Schölch S, Reissfelder C, Kahlert C, Büchler MW, et al. Circulating tumour cells and outcome in non-metastatic colorectal cancer: a prospective study. Br J Cancer (2015) 112:1306–13. doi:10.1038/bjc.2015.88
127. Gasch C, Bauernhofer T, Pichler M, Langer-Freitag S, Reeh M, Seifert AM, et al. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin Chem (2013) 59:252–60. doi:10.1373/clinchem.2012.188557
128. Denis JA, Patroni A, Guillerm E, Pépin D, Benali-Furet N, Wechsler J, et al. Droplet digital PCR of circulating tumor cells from colorectal cancer patients can predict KRAS mutations before surgery. Mol Oncol (2016) 10:1221–31. doi:10.1016/j.molonc.2016.05.009
129. Gormally E, Vineis P, Matullo G, Veglia F, Caboux E, Le Roux E, et al. TP53 and KRAS2 mutations in plasma DNA of healthy subjects and subsequent cancer occurrence: a prospective study. Cancer Res (2006) 66:6871–6. doi:10.1158/0008-5472.CAN-05-4556
130. Jackson PE, Kuang SY, Wang JB, Strickland PT, Muñoz A, Kensler TW, et al. Prospective detection of codon 249 mutations in plasma of hepatocellular carcinoma patients. Carcinogenesis (2003) 24:1657–63. doi:10.1093/carcin/bgg101
131. Xia L, Li Z, Zhou B, Tian G, Zeng L, Dai H, et al. Statistical analysis of mutant allele frequency level of circulating cell-free DNA and blood cells in healthy individuals. Sci Rep (2017) 7:7526. doi:10.1038/s41598-017-06106-1
132. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med (2014) 20:1472–8. doi:10.1038/nm.3733
133. Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med (2014) 371:2477–87. doi:10.1056/NEJMoa1409405
134. Aravanis AM, Lee M, Klausner RD. Next-generation sequencing of circulating tumor DNA for early cancer detection. Cell (2017) 168:571–4. doi:10.1016/j.cell.2017.01.030
135. Travis WD, Weltgesundheitsorganisation, International Agency for Research on Cancer. editors. WHO Classification of Tumours of Lung, Pleura, Thymus and Heart. 4th ed. Lyon: International Agency for Research on Cancer (2015).
136. Siegel R, DeSantis C, Jemal A. Colorectal cancer statistics, 2014: colorectal cancer statistics, 2014. CA Cancer J Clin (2014) 64:104–17. doi:10.3322/caac.21220
137. Gion M, Mione R, Barioli P, Dittadi R. Dynamic use of tumor markers, rationale-clinical applications and pitfalls. Anticancer Res (1996) 16:2279–84.
138. Spindler K-LG. Methodological, biological and clinical aspects of circulating free DNA in metastatic colorectal cancer. Acta Oncol (2017) 56:7–16. doi:10.1080/0284186X.2016.1253861
139. Gormally E, Hainaut P, Caboux E, Airoldi L, Autrup H, Malaveille C, et al. Amount of DNA in plasma and cancer risk: a prospective study. Int J Cancer (2004) 111:746–9. doi:10.1002/ijc.20327
140. Grossman R, Abel B, Angiuoli S, Barrett J, Bassett D, Bramlett K, et al. Collaborating to compete: blood profiling atlas in cancer (BloodPAC) consortium. Clin Pharmacol Ther (2017) 101:589–92. doi:10.1002/cpt.666
141. Catarino R, Ferreira MM, Rodrigues H, Coelho A, Nogal A, Sousa A, et al. Quantification of free circulating tumor DNA as a diagnostic marker for breast cancer. DNA Cell Biol (2008) 27:415–21. doi:10.1089/dna.2008.0744
142. Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer – a survey. Biochim Biophys Acta (2007) 1775:181–232. doi:10.1016/j.bbcan.2006.10.001
143. Canzoniero JV, Park BH. Use of cell free DNA in breast oncology. Biochim Biophys Acta (2016) 1865:266–74. doi:10.1016/j.bbcan.2016.03.006
144. Zanetti-Dällenbach R, Wight E, Fan AX-C, Lapaire O, Hahn S, Holzgreve W, et al. Positive correlation of cell-free DNA in plasma/serum in patients with malignant and benign breast disease. Anticancer Res (2008) 28:921–5.
145. Huang ZH, Li LH, Hua D. Quantitative analysis of plasma circulating DNA at diagnosis and during follow-up of breast cancer patients. Cancer Lett (2006) 243:64–70. doi:10.1016/j.canlet.2005.11.027
146. Hashad D, Sorour A, Ghazal A, Talaat I. Free circulating tumor DNA as a diagnostic marker for breast cancer. J Clin Lab Anal (2012) 26:467–72. doi:10.1002/jcla.21548
147. Kohler C, Radpour R, Barekati Z, Asadollahi R, Bitzer J, Wight E, et al. Levels of plasma circulating cell free nuclear and mitochondrial DNA as potential biomarkers for breast tumors. Mol Cancer (2009) 8:105. doi:10.1186/1476-4598-8-105
148. Bechmann T, Andersen RF, Pallisgaard N, Madsen JS, Maae E, Jakobsen EH, et al. Plasma HER2 amplification in cell-free DNA during neoadjuvant chemotherapy in breast cancer. J Cancer Res Clin Oncol (2013) 139:995–1003. doi:10.1007/s00432-013-1413-5
149. Umetani N, Giuliano AE, Hiramatsu SH, Amersi F, Nakagawa T, Martino S, et al. Prediction of breast tumor progression by integrity of free circulating DNA in serum. J Clin Oncol (2006) 24:4270–6. doi:10.1200/JCO.2006.05.9493
150. Iqbal S, Vishnubhatla S, Raina V, Sharma S, Gogia A, Deo SSV, et al. Circulating cell-free DNA and its integrity as a prognostic marker for breast cancer. Springerplus (2015) 4:265. doi:10.1186/s40064-015-1071-y
151. Madhavan D, Wallwiener M, Bents K, Zucknick M, Nees J, Schott S, et al. Plasma DNA integrity as a biomarker for primary and metastatic breast cancer and potential marker for early diagnosis. Breast Cancer Res Treat (2014) 146:163–74. doi:10.1007/s10549-014-2946-2
152. Schwarzenbach H. Circulating nucleic acids as biomarkers in breast cancer. Breast Cancer Res (2013) 15:211. doi:10.1186/bcr3446
153. Ai B, Liu H, Huang Y, Peng P. Circulating cell-free DNA as a prognostic and predictive biomarker in non-small cell lung cancer. Oncotarget (2016) 7:44583. doi:10.18632/oncotarget.10069
154. Mao C, Yuan J-Q, Yang Z-Y, Fu X-H, Wu X-Y, Tang J-L. Blood as a substitute for tumor tissue in detecting EGFR mutations for guiding EGFR TKIs treatment of nonsmall cell lung cancer: a systematic review and meta-analysis. Medicine (Baltimore) (2015) 94:e775. doi:10.1097/MD.0000000000000775
155. Shi Y, Au JS-K, Thongprasert S, Srinivasan S, Tsai C-M, Khoa MT, et al. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J Thorac Oncol (2014) 9:154–62. doi:10.1097/JTO.0000000000000033
156. Yung TKF, Chan KCA, Mok TSK, Tong J, To K-F, Lo YM. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin Cancer Res (2009) 15:2076–84. doi:10.1158/1078-0432.CCR-08-2622
157. Dowler Nygaard A, Spindler KL, Pallisgaard N, Andersen RF, Jakobsen A. Levels of cell-free DNA and plasma KRAS during treatment of advanced NSCLC. Oncol Rep (2014) 31(2):969–74. doi:10.3892/or.2013.2906
158. Zhuang R, Li S, Li Q, Guo X, Shen F, Sun H, et al. The prognostic value of KRAS mutation by cell-free DNA in cancer patients: a systematic review and meta-analysis. PLoS One (2017) 12:e0182562. doi:10.1371/journal.pone.0182562
159. Winawer S. Workgroup II: the screening process. UICC international workshop on facilitating screening for colorectal cancer, Oslo, Norway (29 and 30 June 2002). Ann Oncol (2005) 16:31–3. doi:10.1093/annonc/mdi029
160. Spindler K-LG, Boysen AK, Pallisgård N, Johansen JS, Tabernero J, Sørensen MM, et al. Cell-free DNA in metastatic colorectal cancer: a systematic review and meta-analysis. Oncologist (2017) 22(9):1049–55. doi:10.1634/theoncologist.2016-0178
161. Wong A, Lim J, Sinha A, Gopinathan A, Lim R, Tan C-S, et al. Tumour pharmacodynamics and circulating cell free DNA in patients with refractory colorectal carcinoma treated with regorafenib. J Transl Med (2015) 13:57. doi:10.1186/s12967-015-0405-4
162. Basnet S, Zhang Z, Liao W, Li S, Li P, Ge H. The prognostic value of circulating cell-free DNA in colorectal cancer: a meta-analysis. J Cancer (2016) 7:1105–13. doi:10.7150/jca.14801
163. Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature (2012) 486(7404):537–40. doi:10.1038/nature11219
164. Shaw JA, Stebbing J. Circulating free DNA in the management of breast cancer. Ann Transl Med (2014) 2:3. doi:10.3978/j.issn.2305-5839.2013.06.06
165. Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DWY, Kaper F, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med (2012) 4:136ra68. doi:10.1126/scitranslmed.3003726
166. Butler TM, Johnson-Camacho K, Peto M, Wang NJ, Macey TA, Korkola JE, et al. Exome sequencing of cell-free DNA from metastatic cancer patients identifies clinically actionable mutations distinct from primary disease. PLoS One (2015) 10:e0136407. doi:10.1371/journal.pone.0136407
167. Russo A, Franchina T, Ricciardi GRR, Smiroldo V, Picciotto M, Zanghì M, et al. Third generation EGFR TKIs in EGFR-mutated NSCLC: where are we now and where are we going. Crit Rev Oncol Hematol (2017) 117:38–47. doi:10.1016/j.critrevonc.2017.07.003
168. Karapetis CS, Khambata-Ford S, Jonker DJ, O’callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med (2008) 359:1757–65. doi:10.1056/NEJMoa0804385
169. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med (2015) 21:560–2. doi:10.1038/nm.3854
170. Paweletz CP, Sacher A, Raymond CK, Alden RS, O’Connell A, Mach SL, et al. Bias-corrected targeted next-generation sequencing for rapid, multiplexed detection of actionable alterations in cell-free DNA from advanced lung cancer patients. Clin Cancer Res (2015) 22(4):915–22. doi:10.1158/1078-0432.CCR-15-1627-T
171. Jamal-Hanjani M, Hackshaw A, Ngai Y, Shaw J, Dive C, Quezada S, et al. Tracking Genomic Cancer (2014). Available from: http://www.independentcancerpatientsvoice.org.uk/app/download/5804440035/PloS-2014.pdf
172. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the evolution of non–small-cell lung cancer. N Engl J Med (2017) 376:2109–21. doi:10.1056/NEJMoa1616288
173. Yang M, Topaloglu U, Petty WJ, Pagni M, Foley KL, Grant SC, et al. Circulating mutational portrait of cancer: manifestation of aggressive clonal events in both early and late stages. J Hematol Oncol (2017) 10:100. doi:10.1186/s13045-017-0468-1
174. Russo M, Siravegna G, Blaszkowsky LS, Corti G, Crisafulli G, Ahronian LG, et al. Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Cancer Discov (2016) 6:147–53. doi:10.1158/2159-8290.CD-15-1283
175. Watanabe S. The metastasizability of tumor cells. Cancer (1954) 7:215–23. doi:10.1002/1097-0142(195403)7:2<215::AID-CNCR2820070203>3.0.CO;2-6
176. Fidler IJ. The pathogenesis of cancer metastasis: the “seed and soil” hypothesis revisited. Nat Rev Cancer (2003) 3:453–8. doi:10.1038/nrc1098
177. Li Y, Wu S, Bai F. Molecular characterization of circulating tumor cells – from bench to bedside. Semin Cell Dev Biol (2017). doi:10.1016/j.semcdb.2017.09.013
178. Wan L, Pantel K, Kang Y. Tumor metastasis: moving new biological insights into the clinic. Nat Med (2013) 19:1450–64. doi:10.1038/nm.3391
179. Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med (2011) 17:1359–70. doi:10.1038/nm.2537
180. Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature (2004) 430:1034–9. doi:10.1038/nature02765
181. Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer (2004) 4:448–56. doi:10.1038/nrc1370
182. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell (2014) 158:1110–22. doi:10.1016/j.cell.2014.07.013
183. Mohan S, Chemi F, Brady G. Challenges and unanswered questions for the next decade of circulating tumour cell research in lung cancer. Transl Lung Cancer Res (2017) 6:454–72. doi:10.21037/tlcr.2017.06.04
184. Ferreira MM, Ramani VC, Jeffrey SS. Circulating tumor cell technologies. Mol Oncol (2016) 10:374–94. doi:10.1016/j.molonc.2016.01.007
185. Cristofanilli M. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. Semin Oncol (2006) 33:9–14. doi:10.1053/j.seminoncol.2006.03.016
186. de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res (2008) 14:6302–9. doi:10.1158/1078-0432.CCR-08-0872
187. Gallo M, De Luca A, Maiello MR, D’Alessio A, Esposito C, Chicchinelli N, et al. Clinical utility of circulating tumor cells in patients with non-small-cell lung cancer. Transl Lung Cancer Res (2017) 6:486–98. doi:10.21037/tlcr.2017.05.07
188. Deneve E, Riethdorf S, Ramos J, Nocca D, Coffy A, Daures J-P, et al. Capture of viable circulating tumor cells in the liver of colorectal cancer patients. Clin Chem (2013) 59:1384–92. doi:10.1373/clinchem.2013.202846
189. Zhang L, Riethdorf S, Wu G, Wang T, Yang K, Peng G, et al. Meta-analysis of the prognostic value of circulating tumor cells in breast cancer. Clin Cancer Res (2012) 18:5701–10. doi:10.1158/1078-0432.CCR-12-1587
190. Bidard F-C, Peeters DJ, Fehm T, Nolé F, Gisbert-Criado R, Mavroudis D, et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol (2014) 15:406–14. doi:10.1016/S1470-2045(14)70069-5
191. Van Poznak C, Somerfield MR, Bast RC, Cristofanilli M, Goetz MP, Gonzalez-Angulo AM, et al. Use of biomarkers to guide decisions on systemic therapy for women with metastatic breast cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol (2015) 33:2695–704. doi:10.1200/JCO.2015.61.1459
192. Pierga J-Y, Bonneton C, Vincent-Salomon A, de Cremoux P, Nos C, Blin N, et al. Clinical significance of immunocytochemical detection of tumor cells using digital microscopy in peripheral blood and bone marrow of breast cancer patients. Clin Cancer Res (2004) 10:1392–400. doi:10.1158/1078-0432.CCR-0102-03
193. Lv Q, Gong L, Zhang T, Ye J, Chai L, Ni C, et al. Prognostic value of circulating tumor cells in metastatic breast cancer: a systemic review and meta-analysis. Clin Transl Oncol (2016) 18:322–30. doi:10.1007/s12094-015-1372-1
194. Paoletti C, Li Y, Muniz MC, Kidwell KM, Aung K, Thomas DG, et al. Significance of circulating tumor cells in metastatic triple-negative breast cancer patients within a randomized, phase II trial: TBCRC 019. Clin Cancer Res (2015) 21:2771–9. doi:10.1158/1078-0432.CCR-14-2781
195. Hou J-M, Krebs MG, Lancashire L, Sloane R, Backen A, Swain RK, et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J Clin Oncol (2012) 30:525–32. doi:10.1200/JCO.2010.33.3716
196. Fabbri F, Carloni S, Zoli W, Ulivi P, Gallerani G, Fici P, et al. Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs. Cancer Lett (2013) 335:225–31. doi:10.1016/j.canlet.2013.02.015
197. Sobin LH, Gospodarowicz MK, Wittekind C. TNM Classification of Malignant Tumours. 7th ed. New York, NY: John Wiley & Sons (2011).
198. Brierley J, Gospodarowicz MK, Wittekind C, editors. TNM Classification of Malignant Tumours. 8th ed. Chichester, West Sussex, UK; Hoboken, NJ: John Wiley & Sons, Inc (2017).
Keywords: liquid biopsy, cell-free DNA, ctDNA, circulating tumor cell, cancer, screening, blood
Citation: Hench IB, Hench J and Tolnay M (2018) Liquid Biopsy in Clinical Management of Breast, Lung, and Colorectal Cancer. Front. Med. 5:9. doi: 10.3389/fmed.2018.00009
Received: 31 October 2017; Accepted: 15 January 2018;
Published: 30 January 2018
Edited by:
Dolores Di Vizio, Boston Children’s Hospital, Harvard University, United StatesReviewed by:
Dario De Biase, Università di Bologna, ItalyFernando Schmitt, Universidade do Porto, Portugal
Copyright: © 2018 Hench, Hench and Tolnay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivana Bratić Hench, aXZhbmEuYnJhdGljaGVuY2hAdXNiLmNo