 Francesc Castro-Giner
Francesc Castro-Giner Manuel C. Scheidmann
Manuel C. Scheidmann Nicola Aceto
Nicola Aceto- 1Department of Biomedicine, Cancer Metastasis Laboratory, University of Basel, University Hospital Basel, Basel, Switzerland
- 2Swiss Institute of Bioinformatics, Lausanne, Switzerland
Circulating tumor cells (CTCs) are defined as those cells that detach from a cancerous lesion and enter the bloodstream. While generally most CTCs are subjected to high shear stress, anoikis signals, and immune attack in the circulatory system, few are able to survive and reach a distant organ in a viable state, possibly leading to metastasis formation. A large number of studies, both prospective and retrospective, have highlighted the association between CTC abundance and bad prognosis in patients with various cancer types. Yet, beyond CTC enumeration, much less is known about the distinction between metastatic and nonmetastatic CTCs, namely those features that enable only some CTCs to survive and seed a cancerous lesion at a distant site. In addition, critical aspects such as CTC heterogeneity, mechanisms that trigger CTC intravasation and extravasation, as well as vulnerabilities of metastatic CTCs subpopulations are poorly understood. In this short review, we highlight recent studies that successfully adopted functional and computational analysis to gain insights into CTC biology. We also discuss approaches to overcome challenges that are associated with CTC isolation, molecular and computational analysis, and speculate regarding few open questions that currently frame the CTC research field.
Introduction
More than 90% of cancer-related deaths are due to metastasis development (1). However, historically, the majority of cancer research has been conducted with a focus on the primary tumor, mostly because of the higher availability of primary tumor specimens compared to biopsies of metastatic lesions, and difficulties to investigate spontaneous metastatic dissemination in vivo (2). As a consequence, our understanding of the vulnerabilities of metastatic cells remains limited, thus hampering the development of effective metastasis-suppressing agents. The metastatic process is commonly thought to be a relatively inefficient, multistep phenomenon that involves the intravasation of cancer cells into the blood circulatory system, their survival in the blood as circulating tumor cells (CTCs), followed by extravasation and seeding of metastatic CTCs into distant sites (3). While it is generally accepted that tumors are able to shed a relatively high number of cancer cells in circulation, it also appears that most CTCs are poised to die in the bloodstream or upon arrival to a distant site, mostly due to high shear forces and anoikis signals in circulation, immune attack or limited capability to adapt to a foreign microenvironment (4). It is therefore critical to dissect features that distinguish those CTCs that are able to survive and initiate metastasis. To this end, liquid biopsy (e.g., blood sampling) represents a minimally invasive yet extraordinary valuable source of CTCs (as well as other tumor-derived material such as circulating tumor DNA, proteins, and exosomes) from virtually all cancer types (5).
CTC Isolation from Blood Samples
In recent years, we have witnessed remarkable improvements in the ability to efficiently isolate CTCs from blood samples. Several CTC isolation technologies are now available and designed to overcome constrains such as CTCs dilution in blood samples and variations in expression levels of cell surface markers that distinguish CTCs from blood cells. While these technologies have been extensively discussed elsewhere (6–10), on the other hand, it is becoming increasingly clear that a consensus is not yet reached within the CTC community regarding which technology should be used as a reference for CTC-related studies. While the CellSearch system has been FDA-cleared for CTC enumeration, a gold-standard technology for live CTC isolation and molecular analysis is not yet defined. Presently, it is often the case that CTC isolation for molecular catheterization is conducted in different laboratories with different technologies, and this may result in biases such as the preferential entrapment of specific CTC subpopulations depending on the technology of choice. Future studies involving large sample numbers will be needed to address specific biases of current individual technologies and indicate a reference tool for CTC isolation to standardize CTC-related studies.
While challenging and still subjected to technology-driven biases, CTC isolation and molecular characterization offers an extraordinary opportunity to investigate the metastatic process in real time and with minimally invasive procedures (i.e., blood sampling). Molecular analysis of CTCs often implies dealing with very limited cell numbers (10) and adapting molecular assays and computational analysis tools toward single-cell resolution (11). In the next paragraphs, we discuss recent studies that adopted molecular and computational analysis of CTCs to dissect features of metastatic cells. We also discuss single-cell analysis-related challenges that are typically encountered in CTC studies.
Molecular Analysis of CTCs
Molecular interrogation of CTCs has been made possible not only by the development of specialized CTC isolation technologies but also with the achievement of single-cell-resolution-sequencing protocols (12) and single-cell-based assays (13, 14). The application of these approaches to CTCs has already generated a number of exciting observations, many of which provide insightful information in regard to features of metastatic precursors, CTC heterogeneity, and patient stratification.
Metastatic Precursors among CTCs
Circulating tumor cells are found in the blood of patients as single cells and as clusters of cells, with the latter being associated to a higher metastatic potential (15–19). Using mouse models with multicolor primary tumors, CTC clusters have been shown to break off from tumors already as clusters and to be composed by an oligoclonal group of cells (18). The oligoclonality of CTC clusters might have relevant implications regarding the fitness, seeding capability, and potential to resist to therapy of cluster-derived metastatic foci; yet, this has to be further investigated. RNA sequencing of CTC clusters from breast cancer patients showed that cells within clusters rely upon the expression of cell–cell junction components such as plakoglobin, and that targeting plakoglobin reduces CTC clustering and metastasis in mouse models (18). Beyond plakoglobin—which is a challenging pharmacological target—it is currently unclear how to prevent or suppress the formation of CTC clusters in vivo, and further studies will be needed to identify cluster-specific vulnerabilities, as well as—more broadly—the vulnerabilities of metastatic precursors.
CTC Heterogeneity
Various studies have tackled the issue of CTC heterogeneity. For example, the phenotypic analysis of CTCs from patients initially diagnosed with ER-positive/HER2-negative breast cancer showed that CTCs are able to acquire HER2 expression in the metastatic setting (20). Using a combination of CTC culturing, single-cell RNA sequencing (scRNA-seq), and a small-scale drug screen combined with mass spectrometry analysis, it was shown that HER2-positive CTCs are more proliferative but not addicted to HER2, while HER2-negative CTCs display the activation of Notch and DNA damage pathways, as well as resistance to cytotoxic therapy (20). Combination treatment with paclitaxel and Notch inhibitors enables the suppression of the tumorigenic potential of both HER2-negative and HER2-positive phenotypes (20). In a separate study, the phenotypic analysis of human breast CTCs using quantitative RNA in situ hybridization (RNA-ISH) revealed a dynamic expression of epithelial versus mesenchymal markers, mostly as a consequence to treatment (21), suggesting that treatment itself may strongly impact the phenotypic heterogeneity of CTCs. In prostate cancer, the RNA expression profile of CTCs revealed a high degree of intra-patient heterogeneity, but even higher diversity across CTCs from different patients (22). For instance, prostate CTCs were shown to contain diverse AR gene mutations and splicing variants, with the activation of Wnt-signaling pathway featuring a subgroup of CTCs belonging to patients who were resistant to anti-androgen therapy (22). Among others, examples of CTC heterogeneity include studies showing Wnt2 expression mediating metastasis-associated survival signals through the TGF-b-activated kinase 1 in a subset of pancreatic CTCs (23), as well as evidence that the expression of specific gene combinations—such as HER2/EGFR/HPSE/Notch1 and EpCAM/CD44/CD47/MET—may confer high metastatic potential to breast CTCs (24, 25).
Patient Stratification through the Analysis of CTCs
Molecular analysis of CTCs has also recently provided very exciting observations regarding patient stratification and the use of CTCs as a surrogate tool to investigate tumor genotypes and response to therapy. In multiple myeloma, when analyzing the mutational landscape of CTCs and matched bone marrow tumor cells, it was found that alterations in oncogenes like KRAS, NRAS, or BRAF were detectable in both specimens, suggesting blood sampling as a reliable and less invasive method for mutational screening of such patients (26). In a similar fashion, comparing whole-exome sequencing data with primary tumor, matched lymph node metastasis and CTCs from a prostate cancer patient revealed that the majority of CTC mutations were present in matched tumor tissue, thus providing a proof-of-concept for the effectiveness of CTC genomics in the clinical setting (27). With regard to patient stratification, CTCs have also proven useful to stratify chemosensitive versus chemorefractory small-cell lung cancer patients (28). Particularly, it was shown how DNA copy number variations (CNVs) can be assessed in small-cell lung CTCs and help predict responsiveness to chemotherapeutic agents, arguing that CTC analysis may be used as a tool to guide treatment decisions in the clinical setting (28). In addition to the analysis of CTCs, interrogation of other blood components including circulating tumor DNA (currently more advanced on the clinical side) allows to detect cancer-associated variants in the blood of patients with good sensitivity and specificity; yet, it may require a more targeted approach (i.e., assessing specific hotspots that are known or expected to carry a mutation) compared to CTC analysis.
Taken together (see also Table 1), these examples of molecular CTC analysis represent a proof of concept of the versatility and potential of CTC-related studies to investigate the metastatic process as well as to influence treatment decisions, complementarily to the analysis of other tumor-derived blood components such as circulating tumor DNA, reviewed elsewhere (5). Hand in hand with the molecular characterization of CTCs, computational analysis represents a major accelerator to take full advantage of CTC-sequencing efforts and to generate actionable hypotheses. Below, we discuss typical challenges of computational CTC data analysis, mostly dealing with single-cell resolution data, and approaches to help overcome current sequencing limitations.

Table 1. Examples of adopted methods to enable a molecular analysis of circulating tumor cells (CTCs).
Computational Analysis of CTC
With a number of recent technological breakthroughs in cell capture and single-cell-sequencing (SCS) protocols, it is now possible to interrogate the genome, transcriptome, and epigenome of CTCs. The application of these technologies is improving our understanding of CTC biology as well as the relationship between CTCs and matched primary or metastatic tumors (12). Yet, despite significant improvements in SCS, the major analytical challenges remain. The main challenge is the interpretation of data in the context of strong stochastic variation and high error rate generated during the amplification of the low amount of starting material derived from a single cell. To address this issue, noise-specific computational methods have been developed. In the following paragraphs, we summarize the current challenges in the analysis of SCS data, with an emphasis on those that are potentially affecting the interpretation of CTC studies (see also Table 2).
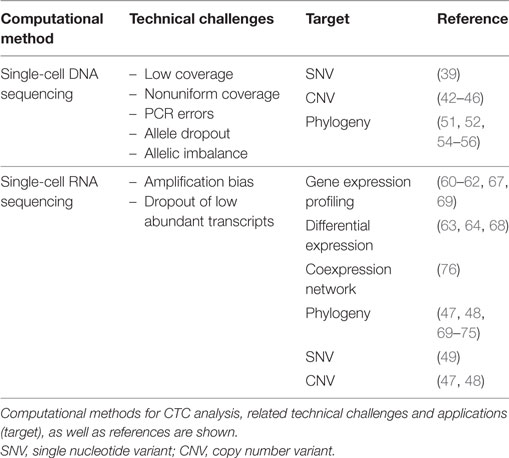
Table 2. Computational methods for circulating tumor cell (CTC) analysis based on single-cell-sequencing approaches.
As a matter of fact, there are specific aspects of CTCs that might contribute to increase the noise of the data. First, CTCs are exposed to different extrinsic stress factors in vivo, such as shear stress, attack by the immune system, and anticancer therapies (10, 29). These phenomena might modify the gene expression profile and induce apoptosis, thus reducing the quality and quantity of the extracted DNA and mRNA. Second, depending on the method used for isolation, CTCs can be admixed with leukocytes, stromal cells, and platelets (30). In RNA-seq data, it is possible to perform a negative selection of samples that show a substantial contamination, for instance, by evaluating the expression of specific leukocyte-associated markers or to digitally remove signatures of genes typically belonging to benign cells (31). In addition, CTC can be found as a single cell or clusters of cells (18). When not carefully controlled for, this introduces variability on the amount of starting material per each sample, possibly leading to systematic biases during amplification steps.
As described below, noise and low coverage on SCS analysis is traditionally compensated by combining data from other cells, usually from dozens to thousands of cells. However, the dilution factor of CTCs in the bloodstream and the limited volume of blood available from cancer patients generally lead to obtain only very few cells per patient (typically between 0 and 10 per tube of blood) (11). This limitation is traditionally addressed by pooling cells from the same individual, but this hampers any conclusions regarding the CTC heterogeneity and the detection of rare CTC subpopulations. Experimental solutions such as CTC-derived explants and the development of cell lines from captured CTC can help to increase the number of cells to investigate (32, 33), at the expense of low efficiency of such processes and possible biases during ex vivo culturing.
Variant Detection
Single-cell DNA sequencing (scDNA-seq) of CTCs followed by mutation calling has enabled to investigate oligoclonality in CTCs and to identify differences compared to primary and metastatic tumors (27, 34–36). Yet, the data obtained after whole-genome amplification (WGA) is generally characterized by low-coverage breath, nonuniform coverage, false-positive (FP) errors introduced by PCR, false-negative (FN) errors due to insufficient coverage, allele dropout (ADO), and allelic imbalance. To date, there are no computational approaches for variant detection that model this noise. As an alternative, most published studies tried to estimate technical variability comparing the variants obtained from single-cell data with bulk sequencing or control samples (27, 37, 38). Currently, only single nucleotide variants (SNVs) and CNVs can be detected accurately from SCS.
Methods developed for SNV detection (39) rely on the mutation frequency across cells to calculate the posterior probability of the variant to be present in at least two cells. This approach reduces the fraction of FP but, as a consequence, mutations observed in only one cell are discarded. More robust methods will be needed to address FN events introduced by ADO, the major contributor to technical errors, affecting 10–50% of mutation sites (40, 41). Current methods for CNV use segmentation algorithm based on GC-normalized coverage (42–46). These methods are heavily affected by the nonuniform coverage obtained after WGA. Although this can be partially improved using amplification protocols that produce more uniform coverage (40), the resolution is limited to megabase-scale CNVs (42, 44). On the other hand, these methods have shown a relatively high specificity in low-coverage data (42, 44, 46). Further, variant detection can also be performed on scRNA-seq data, and a number of studies have attempted to infer copy number profiles by averaging the relative expression levels over large genomic regions (47, 48). RNA-seq can be used for SNV calling, but this approach is limited by RNA-editing events, allele-specific expression, and the small fraction of genes that are expressed at a high level (49).
A specific downstream analysis of variant calling is the reconstruction of single-cell phylogenies. These phylogenies can reconstruct the subclonal compositions of cells, revealing the evolutionary history of the tumor and, in the case of CTC-sequencing studies, specific metastatic-seeding patterns. A variety of approaches have been recently developed that are customized for the characteristics of scDNA-seq data (50–55). Some of these models can accommodate doublet cells into the analysis (51, 56) and are of particular interest for the analysis of CTC clusters. At the moment, current approaches only use SNVs as source information, and additional efforts are required to integrate CNV into the models.
Gene Expression Profiling
Methods of whole transcriptome amplification (WTA) for single cells are well established but also suffer from amplification bias and unwanted variation. To overcome this challenge, some of these protocols integrate unique molecular identifiers that allow to track single molecules through the amplification process, thus reducing amplification bias (57). In addition, spike-ins of known concentration can be used to quantify sensitivity and technical variation (58). However, WTA and subsequent analysis methods struggle with reliable amplification for lowly expressed transcripts (59), leading to dropout events that add uncertainty to downstream analyses. Different methods and workflows have been developed for single-cell quality control that allows to remove batch effects and unwanted biological variability such as cell-cycle variation (60–62). To compare expression across cells, the data need to be normalized to remove cell-specific biases such as read depth and cell capture efficiency. Specific methods are available to normalize scRNA-seq data that account for a high degree of technical noise, low coverage, and a high proportion of dropout events (63–65).
Generally, there are two main downstream analyses of scRNA-seq data: differential expression (DE) and clustering. DE may help to find specific expression signatures associated with metastasis (31, 66) or to study differences between CTC and CTC clusters (18). It is possible to use methods for DE developed for bulk sequencing (67) coupled with the single-cell-specific pre-normalization. However, there are tools specifically developed for scRNA-seq, such as SCDE (64), MAST (63), and BASiC (68), that apply their specific normalization approaches. Second, similar to scDNA-seq phylogenies, unsupervised clustering of cells based on scRNA-seq can help to identify differences across cells and subclonal tumor populations. There is a wide range of methods available for clustering developed for scRNA-seq data, each of them with specific advantages and disadvantages (47, 48, 69–75). Besides DE and clustering, scRNA-seq has been used for network modeling to provide insights about complex transcriptional regulation. The most popular approach that has been already applied in a number of scRNA-seq studies is gene coexpression network analysis (WGCNA) (76). However, network analysis methods require a large sample size, which rarely applies to CTC studies.
Single-Cell Multi-omics
Recent technological breakthroughs now allow simultaneously interrogating RNA and DNA from a single cell, enabling a parallel analysis of gene expression, methylation status, and DNA mutations (77–81). This opens up the possibility to more comprehensively understand cellular processes. For example, it is now feasible to correlate DNA methylation status with gene expression and to directly link genomic variation to transcriptional variability to discover expression quantitative loci (79, 82). To date, there are no computational methods that are tailored to combine multiple molecular layers and at the same time to control for technical variation from single-cell genomics. Computational tools in this field are needed, as future developments in amplification and sequencing techniques will allow to robustly apply single-cell multi-omics to study complex questions in CTC biology.
Concluding Remarks
We have recently witnessed extraordinary advances in understanding CTC biology, including the potential of CTCs to reveal targetable cancer vulnerabilities. For instance, several proof-of-concept studies have begun dissecting the heterogeneity of CTCs and have highlighted characteristics that enable few selected CTCs to seed a metastasis. Other studies have shown a potential for CTCs to drive patient stratification and treatment eligibility choices. However, many questions remain to be answered, several of which relate to the applicability of CTC analysis in the clinical setting (possibly together with the analysis of circulating tumor DNA) and the identification of metastasis-suppressing therapies. To this end, computational analysis plays a pivotal role. Addressing major single-cell-related challenges in computational biology will allow scientists to generate hypotheses and interpret data in a highly reliable manner and to accelerate the discovery process in CTC biology and beyond.
Author Contributions
All authors have contributed to write the manuscript and approve its final version.
Conflict of Interest Statement
NA is listed as an inventor in patent WO2015061091 on “Treating cancer, by measuring level of CTC clusters in sample obtained from patient with breast or epithelial cancer, administering treatment to prevent or reduce metastasis” owned by the Massachusetts General Hospital, Boston, MA, USA, and patent PCT/EP2017/068024 on “Apparatus and methods for disrupting circulating tumor cell clusters” owned by the University of Basel, the University of Linz, and Griesmühle Kleinkraftwerk GmbH. All the other authors declare no potential conflict of interest.
Acknowledgments
We thank all members of the Aceto Lab for feedback and discussions. Research in the Aceto Lab is supported by the European Research Council, the Swiss National Science Foundation, the Swiss Cancer League, the Basel Cancer League, the two Cantons of Basel through the ETH Zürich, and the University of Basel.
References
1. Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res (2010) 70(14):5649–69. doi:10.1158/0008-5472.CAN-10-1040
2. Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncog (2013) 18(1–2):43–73. doi:10.1615/CritRevOncog.v18.i1-2.40
3. Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer (2009) 9(4):274–84. doi:10.1038/nrc2622
4. Pantel K, Speicher MR. The biology of circulating tumor cells. Oncogene (2016) 35(10):1216–24. doi:10.1038/onc.2015.192
5. Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol (2017) 14(9):531–48. doi:10.1038/nrclinonc.2017.14
6. Aceto N, Toner M, Maheswaran S, Haber DA. En route to metastasis: circulating tumor cell clusters and epithelial-to-mesenchymal transition. Trends Cancer (2015) 1(1):44–52. doi:10.1016/j.trecan.2015.07.006
7. Ferreira MM, Ramani VC, Jeffrey SS. Circulating tumor cell technologies. Mol Oncol (2016) 10(3):374–94. doi:10.1016/j.molonc.2016.01.007
8. Gkountela S, Aceto N. Stem-like features of cancer cells on their way to metastasis. Biol Direct (2016) 11:33. doi:10.1186/s13062-016-0135-4
9. Gkountela S, Szczerba B, Donato C, Aceto N. Recent advances in the biology of human circulating tumour cells and metastasis. ESMO Open (2016) 1(4):e000078. doi:10.1136/esmoopen-2016-000078
10. Alix-Panabieres C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer (2014) 14(9):623–31. doi:10.1038/nrc3820
11. Miyamoto DT, Ting DT, Toner M, Maheswaran S, Haber DA. Single-cell analysis of circulating tumor cells as a window into tumor heterogeneity. Cold Spring Harb Symp Quant Biol (2016) 81:269–74. doi:10.1101/sqb.2016.81.031120
12. Navin NE. The first five years of single-cell cancer genomics and beyond. Genome Res (2015) 25(10):1499–507. doi:10.1101/gr.191098.115
13. Ryan D, Ren K, Wu H. Single-cell assays. Biomicrofluidics (2011) 5(2):21501. doi:10.1063/1.3574448
14. Zhang K. Stratifying tissue heterogeneity with scalable single-cell assays. Nat Methods (2017) 14(3):238–9. doi:10.1038/nmeth.4209
15. Fidler IJ. The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. Eur J Cancer (1973) 9(3):223–7. doi:10.1016/S0014-2964(73)80022-2
16. Liotta LA, Saidel MG, Kleinerman J. The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res (1976) 36(3):889–94.
17. Hou JM, Krebs MG, Lancashire L, Sloane R, Backen A, Swain RK, et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J Clin Oncol (2012) 30(5):525–32. doi:10.1200/JCO.2010.33.3716
18. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell (2014) 158(5):1110–22. doi:10.1016/j.cell.2014.07.013
19. Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc Natl Acad Sci U S A (2016) 113(7):E854–63. doi:10.1073/pnas.1508541113
20. Jordan NV, Bardia A, Wittner BS, Benes C, Ligorio M, Zheng Y, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature (2016) 537(7618):102–6. doi:10.1038/nature19328
21. Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science (2013) 339(6119):580–4. doi:10.1126/science.1228522
22. Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, et al. RNA-seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science (2015) 349(6254):1351–6. doi:10.1126/science.aab0917
23. Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature (2012) 487(7408):510–3. doi:10.1038/nature11217
24. Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol (2013) 31(6):539–44. doi:10.1038/nbt.2576
25. Zhang L, Ridgway LD, Wetzel MD, Ngo J, Yin W, Kumar D, et al. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci Transl Med (2013) 5(180):180ra148. doi:10.1126/scitranslmed.3005109
26. Mishima Y, Paiva B, Shi J, Park J, Manier S, Takagi S, et al. The mutational landscape of circulating tumor cells in multiple myeloma. Cell Rep (2017) 19(1):218–24. doi:10.1016/j.celrep.2017.03.025
27. Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, Cruz-Gordillo P, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol (2014) 32(5):479–84. doi:10.1038/nbt.2892
28. Carter L, Rothwell DG, Mesquita B, Smowton C, Leong HS, Fernandez-Gutierrez F, et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med (2017) 23(1):114–9. doi:10.1038/nm.4239
29. Phillips KG, Kuhn P, McCarty OJ. Physical biology in cancer. 2. The physical biology of circulating tumor cells. Am J Physiol Cell Physiol (2014) 306(2):C80–8. doi:10.1152/ajpcell.00294.2013
30. Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol (2011) 192(3):373–82. doi:10.1083/jcb.201010021
31. Ting DT, Wittner BS, Ligorio M, Vincent Jordan N, Shah AM, Miyamoto DT, et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep (2014) 8(6):1905–18. doi:10.1016/j.celrep.2014.08.029
32. Hodgkinson CL, Morrow CJ, Li Y, Metcalf RL, Rothwell DG, Trapani F, et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med (2014) 20(8):897–903. doi:10.1038/nm.3600
33. Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science (2014) 345(6193):216–20. doi:10.1126/science.1253533
34. Dago AE, Stepansky A, Carlsson A, Luttgen M, Kendall J, Baslan T, et al. Rapid phenotypic and genomic change in response to therapeutic pressure in prostate cancer inferred by high content analysis of single circulating tumor cells. PLoS One (2014) 9(8):e101777. doi:10.1371/journal.pone.0101777
35. De Luca F, Rotunno G, Salvianti F, Galardi F, Pestrin M, Gabellini S, et al. Mutational analysis of single circulating tumor cells by next generation sequencing in metastatic breast cancer. Oncotarget (2016) 7(18):26107–19. doi:10.18632/oncotarget.8431
36. Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z, et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc Natl Acad Sci U S A (2013) 110(52):21083–8. doi:10.1073/pnas.1320659110
37. Hou Y, Song L, Zhu P, Zhang B, Tao Y, Xu X, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell (2012) 148(5):873–85. doi:10.1016/j.cell.2012.02.028
38. Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A, et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science (2015) 350(6256):94–8. doi:10.1126/science.aab1785
39. Zafar H, Wang Y, Nakhleh L, Navin N, Chen K. Monovar: single-nucleotide variant detection in single cells. Nat Methods (2016) 13(6):505–7. doi:10.1038/nmeth.3835
40. Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet (2016) 17(3):175–88. doi:10.1038/nrg.2015.16
41. Wang Y, Navin NE. Advances and applications of single-cell sequencing technologies. Mol Cell (2015) 58(4):598–609. doi:10.1016/j.molcel.2015.05.005
42. Garvin T, Aboukhalil R, Kendall J, Baslan T, Atwal GS, Hicks J, et al. Interactive analysis and assessment of single-cell copy-number variations. Nat Methods (2015) 12(11):1058–60. doi:10.1038/nmeth.3578
43. Hou Y, Fan W, Yan L, Li R, Lian Y, Huang J, et al. Genome analyses of single human oocytes. Cell (2013) 155(7):1492–506. doi:10.1016/j.cell.2013.11.040
44. Knouse KA, Wu J, Amon A. Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res (2016) 26(3):376–84. doi:10.1101/gr.198937.115
45. McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, et al. Mosaic copy number variation in human neurons. Science (2013) 342(6158):632–7. doi:10.1126/science.1243472
46. Zhang C, Zhang C, Chen S, Yin X, Pan X, Lin G, et al. A single cell level based method for copy number variation analysis by low coverage massively parallel sequencing. PLoS One (2013) 8(1):e54236. doi:10.1371/journal.pone.0054236
47. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (2014) 344(6190):1396–401. doi:10.1126/science.1254257
48. Tirosh I, Izar B, Prakadan SM, Wadsworth MH II, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science (2016) 352(6282):189–96. doi:10.1126/science.aad0501
49. Piskol R, Ramaswami G, Li JB. Reliable identification of genomic variants from RNA-seq data. Am J Hum Genet (2013) 93(4):641–51. doi:10.1016/j.ajhg.2013.08.008
50. Davis A, Navin NE. Computing tumor trees from single cells. Genome Biol (2016) 17(1):113. doi:10.1186/s13059-016-0987-z
51. Jahn K, Kuipers J, Beerenwinkel N. Tree inference for single-cell data. Genome Biol (2016) 17:86. doi:10.1186/s13059-016-0936-x
52. Kim KI, Simon R. Using single cell sequencing data to model the evolutionary history of a tumor. BMC Bioinformatics (2014) 15:27. doi:10.1186/1471-2105-15-27
53. Kuipers J, Jahn K, Beerenwinkel N. Advances in understanding tumour evolution through single-cell sequencing. Biochim Biophys Acta (2017) 1867(2):127–38. doi:10.1016/j.bbcan.2017.02.001
54. Ross EM, Markowetz F. OncoNEM: inferring tumor evolution from single-cell sequencing data. Genome Biol (2016) 17:69. doi:10.1186/s13059-016-0929-9
55. Yuan K, Sakoparnig T, Markowetz F, Beerenwinkel N. BitPhylogeny: a probabilistic framework for reconstructing intra-tumor phylogenies. Genome Biol (2015) 16:36. doi:10.1186/s13059-015-0592-6
56. Roth A, McPherson A, Laks E, Biele J, Yap D, Wan A, et al. Clonal genotype and population structure inference from single-cell tumor sequencing. Nat Methods (2016) 13(7):573–6. doi:10.1038/nmeth.3867
57. Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol (2016) 34(5):547–55. doi:10.1038/nbt.3520
58. Brennecke P, Anders S, Kim JK, Kolodziejczyk AA, Zhang X, Proserpio V, et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods (2013) 10(11):1093–5. doi:10.1038/nmeth.2645
59. Marinov GK, Williams BA, McCue K, Schroth GP, Gertz J, Myers RM, et al. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res (2014) 24(3):496–510. doi:10.1101/gr.161034.113
60. Lun AT, McCarthy DJ, Marioni JC. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res (2016) 5:2122. doi:10.12688/f1000research.9501.2
61. Scialdone A, Natarajan KN, Saraiva LR, Proserpio V, Teichmann SA, Stegle O, et al. Computational assignment of cell-cycle stage from single-cell transcriptome data. Methods (2015) 85:54–61. doi:10.1016/j.ymeth.2015.06.021
62. Stegle O, Teichmann SA, Marioni JC. Computational and analytical challenges in single-cell transcriptomics. Nat Rev Genet (2015) 16(3):133–45. doi:10.1038/nrg3833
63. Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol (2015) 16:278. doi:10.1186/s13059-015-0844-5
64. Kharchenko PV, Silberstein L, Scadden DT. Bayesian approach to single-cell differential expression analysis. Nat Methods (2014) 11(7):740–2. doi:10.1038/nmeth.2967
65. Lun AT, Bach K, Marioni JC. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol (2016) 17:75. doi:10.1186/s13059-016-0947-7
66. Ramskold D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, et al. Full-length mRNA-seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol (2012) 30(8):777–82. doi:10.1038/nbt.2282
67. Vallejos CA, Risso D, Scialdone A, Dudoit S, Marioni JC. Normalizing single-cell RNA sequencing data: challenges and opportunities. Nat Methods (2017) 14(6):565–71. doi:10.1038/nmeth.4292
68. Vallejos CA, Richardson S, Marioni JC. Beyond comparisons of means: understanding changes in gene expression at the single-cell level. Genome Biol (2016) 17:70. doi:10.1186/s13059-016-0930-3
69. Guo M, Wang H, Potter SS, Whitsett JA, Xu Y. SINCERA: a pipeline for single-cell RNA-seq profiling analysis. PLoS Comput Biol (2015) 11(11):e1004575. doi:10.1371/journal.pcbi.1004575
70. Kiselev VY, Kirschner K, Schaub MT, Andrews T, Yiu A, Chandra T, et al. SC3: consensus clustering of single-cell RNA-seq data. Nat Methods (2017) 14(5):483–6. doi:10.1038/nmeth.4236
71. Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet (2017) 49(5):708–18. doi:10.1038/ng.3818
72. Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature (2016) 539(7628):309–13. doi:10.1038/nature20123
73. Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science (2017) 355(6332). doi:10.1126/science.aai8478
74. Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun (2017) 8:14049. doi:10.1038/ncomms14049
75. Zurauskiene J, Yau C. pcaReduce: hierarchical clustering of single cell transcriptional profiles. BMC Bioinformatics (2016) 17:140. doi:10.1186/s12859-016-0984-y
76. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics (2008) 9:559. doi:10.1186/1471-2105-9-559
77. Dey SS, Kester L, Spanjaard B, Bienko M, van Oudenaarden A. Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol (2015) 33(3):285–9. doi:10.1038/nbt.3129
78. Guo H, Zhu P, Wu X, Li X, Wen L, Tang F. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res (2013) 23(12):2126–35. doi:10.1101/gr.161679.113
79. Macaulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods (2015) 12(6):519–22. doi:10.1038/nmeth.3370
80. Macaulay IC, Ponting CP, Voet T. Single-cell multiomics: multiple measurements from single cells. Trends Genet (2017) 33(2):155–68. doi:10.1016/j.tig.2016.12.003
81. Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods (2014) 11(8):817–20. doi:10.1038/nmeth.3035
Keywords: circulating tumor cells, molecular analysis, computational biology, metastasis, single-cell genomics
Citation: Castro-Giner F, Scheidmann MC and Aceto N (2018) Beyond Enumeration: Functional and Computational Analysis of Circulating Tumor Cells to Investigate Cancer Metastasis. Front. Med. 5:34. doi: 10.3389/fmed.2018.00034
Received: 23 October 2017; Accepted: 30 January 2018;
Published: 19 February 2018
Edited by:
Dolores Di Vizio, Harvard University, United StatesReviewed by:
Salvatore Lorenzo Renne, Istituto Nazionale dei Tumori (IRCCS), ItalyPier Paolo Piccaluga, Università di Bologna, Italy
Copyright: © 2018 Castro-Giner, Scheidmann and Aceto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Aceto, bmljb2xhLmFjZXRvQHVuaWJhcy5jaA==