 Ozgur Mete1,2,3*
Ozgur Mete1,2,3* Kai Duan1,2
Kai Duan1,2
- 1Department of Pathology, University Health Network, Toronto, ON, Canada
- 2Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada
- 3Endocrine Oncology Site Group, Princess Margaret Cancer Centre, Toronto, ON, Canada
Adrenal cortical tumors constitute a heterogeneous group of neoplasms with distinct clinical, morphological, and molecular features. Recent discoveries of specific genotype–phenotype correlations in adrenal cortical adenomas have transformed our understanding of their respective endocrine syndromes. Indeed, a proportion of patients with primary aldosteronism are now known to harbor adrenal cortical adenomas with heterogeneous molecular alterations (KCNJ5, ATP1A1, ATP2B3, and CACNA1D) involving the calcium/calmodulin kinase signaling pathway. Several lines of evidence suggest that KCNJ5-mutant aldosterone-producing adenomas have distinct clinicopathological phenotype compared to those harboring ATP1A1, ATP2B3, and CACNA1D mutations. Benign adrenal cortical tumors presenting with Cushing syndrome often have diverse mutations (PRKACA, PRKAR1A, GNAS, PDE11A, and PDE8B) involving the cyclic AMP signaling pathway. In addition to cortisol-producing adenomas, bilateral micronodular adrenocortical disease and primary bilateral macronodular adrenal hyperplasia (PBMAH) have also expanded the spectrum of benign neoplasms causing adrenal Cushing disease. The recent discovery of inactivating ARMC5 germline mutations in PBMAH has challenged the old belief that this disorder is mainly a sporadic disease. Emerging evidence suggests that PBMAH harbors multiple distinct clonal proliferations, reflecting the heterogeneous genomic landscape of this disease. Although most solitary adrenal cortical tumors are sporadic, there is an increasing recognition that inherited susceptibility syndromes may also play a role in their pathogenesis. This review highlights the molecular and morphological heterogeneity of benign adrenal cortical neoplasms, reflected in the diverse presentations of primary aldosteronism and adrenal Cushing syndrome.
Introduction
The past decade has seen tremendous growth in our understanding of the clinical, molecular, and histopathologic characteristics of adrenal cortical neoplasia. Precise clinical and molecular studies have demonstrated that primary adrenal cortical malignancy is not a single disease entity. In fact, it is now well recognized that adrenal cortical carcinomas display a wide spectrum of clinical manifestations with heterogeneous molecular and histopathologic features, as well as distinctive proliferative biology, biomarker expression, and prognostic cluster profiles. While the distinction of carcinomas from adenomas is critical, the recent discovery of specific genotype–phenotype correlations in aldosterone- and glucocorticoid-producing benign adrenal cortical tumors has significant implications for patient management. This review provides an update on the newly described molecular histophenotypic correlates of benign adrenal cortical tumors, resulting in heterogeneous presentations of hyperaldosteronism and hypercortisolism.
Heterogeneity in Aldosterone-Producing Benign Adrenal Cortical Neoplasms
Clinical and Histopathological Heterogeneity
Since the first description by Conn in 1955, much has been learned regarding the clinical, pathological, and molecular properties of primary aldosteronism (1–4). Inappropriate or autonomous aldosterone secretion, often detected through serum aldosterone-to-renin ratio, is attributed to a group of adrenal cortical proliferations that includes adrenal cortical hyperplasia, adenoma, and rare carcinomas (5, 6). Currently, bilateral adrenal cortical hyperplasia is regarded as the most common cause of primary aldosteronism, often diagnosed in older patients with a slight male predilection (1–4, 7). Affected individuals with bilateral disease often present with less severe hypertension and more frequent normokalemia (1–4, 7). In contrast, those with unilateral disease (i.e., aldosterone-producing adenoma) generally have more severe hypertension and more frequent hypokalemia (1–4, 7). Adrenal venous sampling has emerged as an important diagnostic tool to help localize the source of aldosterone excess and to distinguish unilateral from bilateral adrenal disease. Since the latter is treated medically, aldosterone-producing tumors are much more commonly seen in adrenalectomy specimens taken for primary aldosteronism.
Most aldosterone-producing adenomas are composed of tumor cells rich in lipid contents. As a result, adenomas typically exhibit lower attenuation (<10 Hounsfield units) on unenhanced CT imaging studies (8, 9), and their cut surface often appear golden yellow (5, 6). The application of CYP11B2 (aldosterone synthase) immunohistochemistry has enhanced our understanding of CT-detected adrenal nodules, as these may not necessarily represent the source of aldosterone excess, even when ipsilateral lateralization is detected by adrenal vein sampling, likely because additional smaller/microscopic cortical proliferations may be responsible for the hormonal overproduction (10–12). To complicate this conundrum, zona glomerulosa (ZG) hyperplasia or multiple functional micronodules have also been described in patients with primary aldosteronism (11, 13, 14).
Most aldosterone-producing tumors are predominantly composed of clear cells with high lipid content. The neoplastic cells contain mitochondria with lamellar type or plate-like cristae. It should be noted that the cytomorphological details of an individual tumor can be heterogeneous due to variation in four distinct types of tumor cells that have been characterized in aldosterone-producing adenomas: (i) zona fasciculata (ZF)-like clear cells, (ii) ZG-like cells, (iii) zona-reticularis-like compact cells, and (iv) compact cells displaying overlapping cytomorphological features of both ZG- and fasciculata-like cells (5, 6) (Figure 1). Recent evidence suggests that the heterogeneous cytomorphological features of an aldosterone-producing adenoma may be related to its underlying genotype, reflecting different CYP11B2 (aldosterone synthase) and CYP11B1 (11β-hydroxylase that converts 11-deoxycortisol to cortisol) expression profile in the tumor (see below) (14–18). The presence of oncocytic cytomorphology also expands the histopathologic heterogeneity of aldosterone-producing adrenal cortical adenomas, including those with intracytoplasmic globular inclusions due to degenerate mitochondria (19).
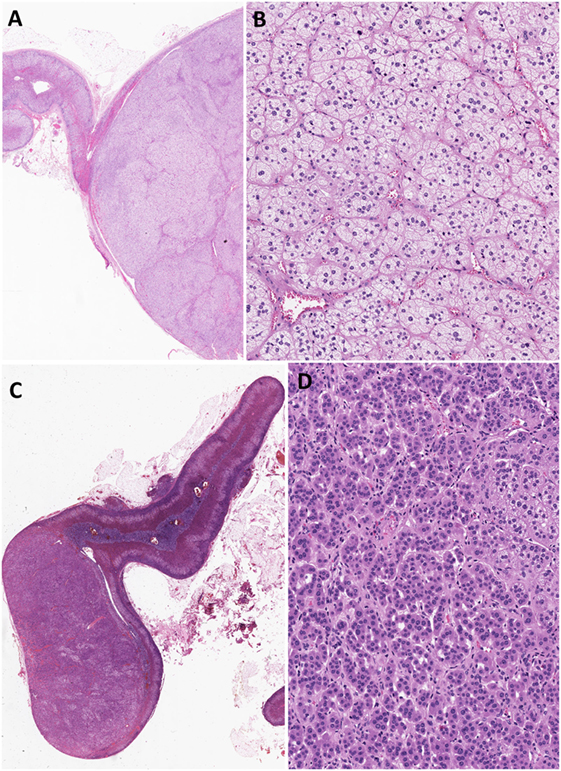
Figure 1. Heterogeneity in aldosterone-producing adrenocortical adenomas. The cytomorphological properties of an individual tumor can be heterogeneous due to variation in the composition of different cell types (A–D). Most aldosterone-producing tumors are predominantly composed of clear cells with high lipid contents (A,B). However, recent evidence suggests that variations in cytomorphological features exist due to the underlying molecular heterogeneity of these neoplasms. For instance, aldosterone-producing adenomas composed mainly of zona fasciculata-like clear cells (A,B) often harbor KCNJ5 mutations. In contrast, tumors that are composed predominantly of compact cells typically show ATP1A1, ATP2B3, and CACNA1D molecular alterations (C,D).
Molecular Characteristics and Pathogenesis
At the molecular level, somatic mutations involving potassium channels (KCNJ5), ATPases (ATP1A1 and ATP2B3), and calcium channels (CACNA1D) account for approximately 60% of sporadic aldosterone-producing adenomas (5, 6) (Figure 2). Among these, activating KCNJ5 mutations implicating G1514 and L168R appear to be the most frequent (5, 20–22). KCNJ5 mutations were identified in approximately 40–50% (range, 30.2–76.8%) of aldosterone-producing adenomas (5, 6, 21). Altered sodium permeability due to KCNJ5 (encoding Kir3.4) mutations results in cellular depolarization and increased intracytoplasmic calcium levels via voltage-gated calcium channels in affected tumor cells. The rates of ATP1A1 (overall rate ~4%, range, 0–25%) (15, 21), ATP2B3 (overall rate ~2%, range, 0–3.1%) (21), and CACNA1D (overall rate ~5%, range, 0–14.3%) (15, 21) mutations were significantly lower in various series. ATP1A1 (Na+/K+ ATPase α-subunit) mutations result in cellular depolarization and subsequent calcium influx, whereas ATP2B3 (Ca2+ATPase) and CACNA1D (Cav1.3) mutations result in impaired intracellular calcium clearance and stimulation of the voltage-gated calcium channels at lower depolarization levels, respectively (5, 6, 20). These four somatic alterations are thought to cause autonomous aldosterone production and cellular proliferation through aberrant activation of the calcium/calmodulin kinase signaling pathway, which is normally implicated in the physiology of aldosterone biosynthesis in ZG cells (5, 6, 20).
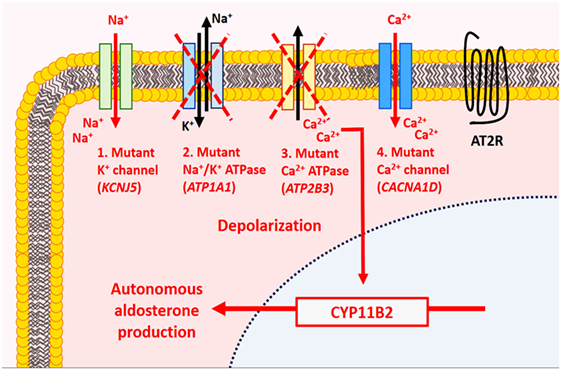
Figure 2. Molecular alterations in aldosterone-producing benign adrenal cortical neoplasms. In normal physiology, aldosterone production by adrenal zona glomerulosa (ZG) cells is mediated by the calcium/calmodulin signaling pathway. In the resting state, ZG cells are hyperpolarized, a process mainly mediated by K+ channels. Activation of the renin–angiotensin system results in the production of angiotensin II, which binds to type 1 angiotensin II receptor (AT2R) causing inhibition of K+ currents and depolarization of cell membrane. The latter activates voltage-gated Ca2+ channel leading to influx of Ca2+ and activation of the calmodulin kinase pathway, which enables aldosterone synthesis. In primary aldosteronism, mutations implicating the K+ channels (KCNJ5), ATPases (ATP1A1, ATP2B3), and Ca2+ channels (CACNA1D) have been identified in ~60% of sporadic aldosterone-producing adenomas. These molecular alterations are thought to result in autonomous aldosterone production and cellular proliferation through aberrant activation of the calcium/calmodulin kinase signaling pathway.
The non-tumorous adrenal cortex often exhibits aldosterone-producing cell clusters (APCCs) and paradoxical ZG layer hyperplasia to justify the unilateral source of primary aldosteronism in the absence of other CYP11B2-expressing adrenal cortical proliferation (11, 18). APCCs typically display a mixture of ZG- and ZF-like cells with both components showing a transcriptome profile similar to that of the ZG layer but with higher CY11B2 expression than that paradoxical ZG layer hyperplasia in a significant proportion of aldosterone-producing adenomas (5, 6, 11, 22) (Figures 1C and 3). The paradoxical ZG layer hyperplasia is typically diagnosed when a continuous ZG layer is identified (5, 6) (Figure 3). In some situations, small microscopic proliferations originating from the ZG layer may extend into the ZF layer, in the setting of a continuous ZG layer (paradoxical hyperplasia with linear growth). There has been recent debate on the biological significance of APCCs, which are generally defined by subcapsular CYP11B2-expressing adrenal cortical cell clusters extending into the ZF (23, 24) (Figure 3). Traditionally, pathologists considered these areas to be part of the spectrum of paradoxical hyperplasia; however, variations in the definition of these microscopic findings persist (25). In some studies, regions of APCCs have been described in the normal adrenals glands of patients without evidence of primary aldosteronism (24). In other studies, APCCs were seen in the normal cortex (24). Using microarray sequencing and NGS approach, the identification of somatic mutations in calcium channels and ATPases in APCCs suggested a potential precursor role in the pathogenesis of primary aldosteronism (24). The lack of somatic KCNJ5 mutations in unilateral hyperplasia and in the paradoxical ZG hyperplasia surrounding KCNJ5-mutant aldosterone-producing adenomas is of significance (22). Further studies are required to establish the precursor role of APCCs and its relationship to ZG paradoxical hyperplasia with linear growth in primary aldosteronism.
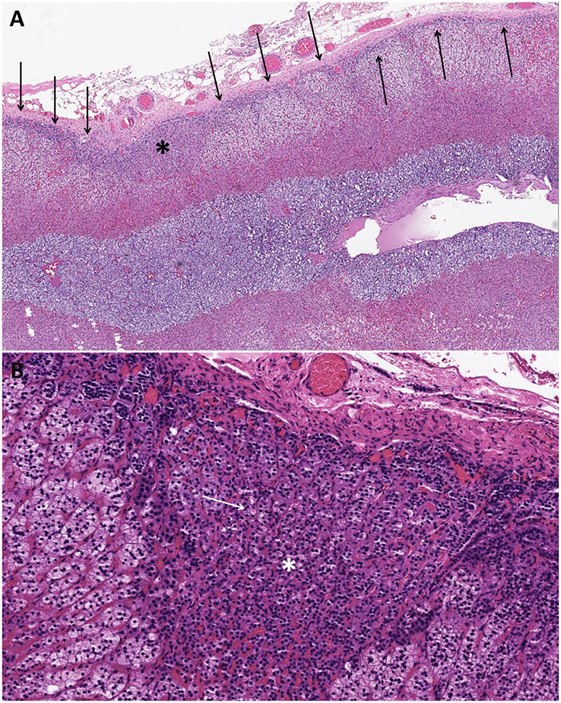
Figure 3. Changes in the non-tumorous adrenal cortex of patients with aldosterone-producing adenomas. The non-tumorous adrenal cortex often exhibits paradoxical zona glomerulosa layer hyperplasia in a significant proportion of aldosterone-producing adenomas. The paradoxical zona glomerulosa layer hyperplasia is typically diagnosed when a continuous zona glomerulosa layer (arrows) is identified (A). In some cases, small microscopic proliferations originating from the zona glomerulosa layer may extend into the zona fasciculata layer [(A); asterisk] in the setting of a continuous zona glomerulosa layer (paradoxical hyperplasia with linear growth). These subcapsular microscopic adrenocortical proliferations extending into the zona fasciculata have been referred to as aldosterone-producing cell clusters [(A,B); asterisk]. Patients treated with spironolactone often display intracytoplasmic eosinophilic inclusions in the aldosterone-producing adenomatous tissue and in the proliferative foci in the adjacent cortex [(B); white arrows indicate one of the spironolactone bodies].
Molecular Heterogeneity and Genotype–Phenotype Correlations
Understanding the functional and cellular correlates of the previously discussed ion channel-related molecular alterations has transformed the field of primary aldosteronism by expanding the role of biomarkers including but not limited to CYP11B2, CD56, and Dab2 (Table 1) (26–28). Several lines of evidence suggest that there is strong genotype (KCNJ5, ATP1A1, ATP2B3, and CACNA1D) and phenotype correlation with respect to patient demographics (16, 22, 29), degree of aldosteronism (29), tumor size (16–18, 22, 29) and focality (11, 22), tumor cytomorphology (11, 14–18, 29), proliferative capacity (15), and expression for CYP11B1, CYP17, and CYP11B2 in aldosterone-producing adenomas (14–18), as well as in APCCs (as discussed above).
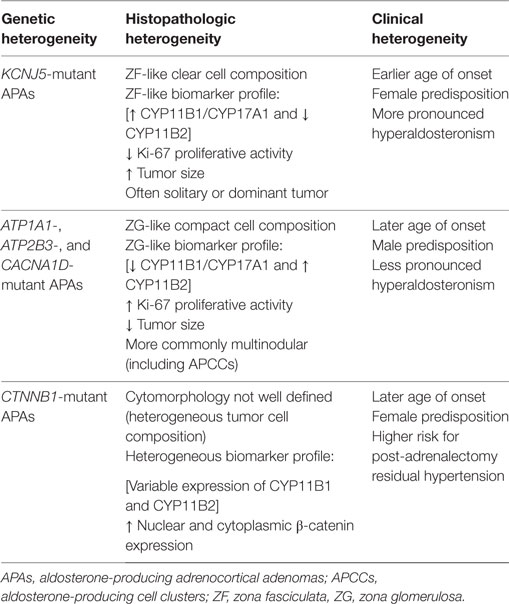
Table 1. Adrenocortical tumor heterogeneity in primary aldosteronism.
Several studies reported that KCNJ5-mutant aldosterone-producing adenomas are associated with younger age, female gender, and larger tumor size than KCNJ5-wild-type aldosterone-producing adenomas (16–18, 22, 29). In contrast, KCNJ5-wild-type adenomas were reported to be more common in older men and smaller in size (16). In keeping with the correlates of KCNJ5-wild-type adenomas, Azizan et al. showed that aldosterone-producing adenomas with CACNA1D or ATP1A1 mutations were generally smaller than 1 cm (17). Furthermore, KCNJ5-mutant aldosterone-producing adenomas tend to be either solitary tumors or dominant tumors (22). Together, these findings may explain why larger solitary nodules are more frequently observed in female patients, whereas multinodular disease appears to be more commonly found in male patients (18). In the study by Dekkers et al., KCNJ5, ATP1A1, and CACNA1D were variably identified in adrenalectomy specimens of patients with solitary or multiple nodules; interestingly, in multinodular glands, only a single nodule was usually found to harbor mutations (11). In that series, no ATP2B3 mutation was noted in adrenals with multiple nodules (11); however, a subsequent series identified a case with ATP2B3 mutation in one nodule and KCNJ5 and ATP2B3 mutation in another distinct nodule within the same adrenal gland (14). A recent meta-analysis on correlates of KCNJ5-mutant tumors also revealed more pronounced hyperaldosteronism in affected patients (29), whereas no significant differences in blood pressure and serum potassium levels were observed (29).
Several lines of evidence suggest that KCNJ5-mutant tumors are indeed different from those harboring mutations in ATPases and CACNA1D. This distinction stems from differences in both cytomorphological features and expression levels for biomarkers of ZG- (CYP11B2) and ZF-(CYP11B1 and CYP17) like phenotypes (11, 14–18, 29). For instance, aldosterone-producing adenomas with KCNJ5 mutations are enriched in tumors that are predominantly composed of ZF-like clear cells (Figures 1A,B) that show higher CYP17A1 and CYP11B1 expression profiles and significantly lower expression profiles for CYP11B2 (11, 15–18). In contrast, tumors that are composed predominantly of ZG-like compact cells (Figures 1C,D) are typically enriched in ATP1A1-, ATP2B3-, and CACNA1D-mutant aldosterone-producing adenomas that show increased and strong CYP11B2 expression and predominantly negative CYP11B1 or CYP17A1 expression profiles (11, 15–18). The series by Chin and colleagues demonstrated that tumors with ZG-like phenotype had an average of 50% compact cells (range, 20–90%) (15). In addition, Monticone et al. demonstrated inverse correlation between tumor size and CYP11B2 expression (18). In fact, this finding is also consistent with the correlates of KCNJ5-mutant tumors. The significant differences between KCNJ5 mutant and wild-type tumors initiated a scientific discussion on the possibility that tumor formation and hyperfunctionality may represent independent processes (14). While these findings question the ZG origin of KCNJ5-mutant adenomas, it also raises the possibility that KCNJ5-mutant tumors may arise from existing adrenal cortical nodules that undergo functional dysregulation, a hypothesis that requires further investigation (27, 28). A recent series reported significantly higher Ki-67 proliferation indices (although all groups had Ki-67 labeling indices <5%) in CACNA1D- and ATP1A1-mutant tumors than in those harboring KCNJ5 mutations (15).
Activation of the Wnt/beta-catenin signaling pathway has also been reported in 60–70% of aldosterone-producing adenomas (30). CTNNB1 mutations were reported in ~5% of these tumors (31, 32). Several studies have confirmed the tumorigenic role of this mutation as it was mutually exclusive to ion channel-related (KCNJ5, ATP1A1, ATP2B3, and CACNA1D) mutations in primary aldosteronism (14, 31, 32). CTNNB1-mutant aldosterone-producing adenomas appear more prevalent in female (31, 32) and older patients with a shorter duration of hypertension (31). Aldosterone-producing adenomas with CTNNB1 mutations have been reported to have higher CYP11B2 mRNA and protein (by immunohistochemistry) expression levels compared to those harboring KCNJ5 mutations (32). These tumors were no different than KCNJ5-mutant adenomas with respect to their tumor size, aldosterone levels, and age at the time of diagnosis (32). In addition, Åkerström et al. did not identify specific cytomorphological correlate of CTNNB1-harboring aldosterone-producing adenomas (32). These adenomas exhibited cytoplasmic and/or nuclear beta-catenin expression (32) as well as a variable expression of CYP11B1 (typically expressed in ZF layer) and CYP11B2 (typically expressed in ZG layer) by immunohistochemistry. In some tumors, diffuse CYP11B2 expression with concomitantly low CYP11B1 expression was seen, whereas in other tumors, heterogeneous expression for both CYP11B1 and CYP11B2 was observed (31, 32). Some tumors also showed CYP11B2-positive and negative regions (14) or diffuse CYP11B1 positivity with low CYP11B2 expression (31, 32). Several researchers have hypothesized that CTNNB1 mutations likely play a role in tumorigenesis rather than in aldosterone production (31); however, others have proposed that CTNNB1 mutations play a role in aldosterone overproduction through aberrant activation of beta-catenin that can result in overexpression of AT1 receptor, as well as certain nuclear receptors (e.g., NURR1 and NURR7) and conversion of progesterone into 11β-deoxycorticosterone (26). The MAPK and PI3K/AKT signaling pathways were also reported to be involved in a proportion of sporadic aldosterone-producing adenomas (27).
Although most aldosterone-producing adrenocortical proliferations are sporadic, three hereditary forms of hyperaldosteronism have been described, accounting for approximately 5% of primary aldosteronism cases (5, 6). Of these, type 3 familial hyperaldosteronism is associated with germline KCNJ5 mutations; therefore, a small proportion of seemingly sporadic aldosterone-producing adenomas with KCNJ5 mutations may in fact be a harbinger of this condition. The other two forms of familial hyperaldosteronism are attributed to gene rearrangements involving CYP11B1/CYP11B2 (type I familial hyperaldosteronism) and potential alterations at 7p22 (type II familial hyperaldosteronism). The identification of a novel germline CACNA1H (M1549V) mutation encoding the low voltage activated T-type calcium channel (Cav3.2) has broadened the genomic landscape of familial hyperaldosteronism (33); the adrenalectomy specimen of one affected individual showed ZG layer hyperplasia (33).
Interestingly, primary aldosteronism was also recently described in African American patients harboring germline mutations in the armadillo repeat containing 5 (ARMC5) (34); however, a more recent study of predominantly Caucasian patients with primary aldosteronism did not identify a pathogenetic ARMC5 mutation (35).
Heterogeneity in Glucocorticoid-Producing Benign Adrenal Cortical Neoplasms
Clinical and Histopathological Heterogeneity
Since the first description in 1932, our understanding of endogenous Cushing syndrome has evolved substantially (36). Similar to primary aldosteronism, the causes of adrenal Cushing syndrome encompass a wide spectrum of adrenal cortical proliferations that exhibit clinical, morphological, and molecular heterogeneity. Adrenal Cushing syndrome can present in patients of all ages, including children (36–41). The clinical presentations range from characteristic biochemical and phenotypical signs and symptoms, as seen in florid Cushing syndrome to much milder or even asymptomatic “subclinical” Cushing syndrome (36–40).
The histopathologic manifestations of adrenal Cushing syndrome include adrenal cortical hyperplasia, adenoma, and carcinoma (36). With the exception of ACTH-dependent bilateral adrenal cortical hyperplasia, which is due to a pituitary corticotroph tumor or ectopic ACTH-secreting neoplasm, the concept of primary adrenal cortical hyperplasia is a misnomer. Indeed, this is further supported by the fact that hyperplasia typically represents a reversible cell proliferation that is driven by a known stimulus. In fact, when the stimulus stops, the hyperplastic cellular proliferation often regresses if no clonal events are superimposed. Unlike hyperplastic processes, neoplastic proliferations are considered to represent cellular proliferations originating from genetically transformed cells. Therefore, in adrenal Cushing syndrome, diagnostic entities falling into the spectrum of primary bilateral nodular (micronodular or macronodular) adrenal cortical hyperplasia do not represent true hyperplastic phenomenon because these nodular proliferations generally include genetically transformed cells (6, 36, 42–45). Consequently, these lesions should be regarded as multiple benign cortical neoplasms. Given the frequent genetic predisposition of micronodular hyperplasia (nodules < 1 cm), the term “primary bilateral micronodular adrenocortical disease” has been used rather than hyperplasia. However, those presenting with bilateral multiple macronodular cortical proliferations (nodules exceeding 1 cm) have been traditionally referred to as “primary bilateral macronodular adrenal hyperplasia” (PBMAH) despite accumulating evidence indicating the diverse clonal nature of the nodules (42, 44).
Primary bilateral micronodular adrenocortical disease frequently presents at a younger age and results in Cushing syndrome. Traditionally, it is divided into two subgroups: (i) a pigmented subtype, which is also referred to as primary pigmented nodular adrenocortical disease (PPNAD), and (ii) a non-pigmented or scarcely pigmented subtype, which is also known as isolated micronodular adrenocortical disease (i-MAD) (36, 45–50). The association between inherited genetic predisposition and the pathogenesis of primary bilateral micronodular adrenocortical disease has been well characterized (see Molecular Characteristics and Pathogenesis) (36, 45). Consequently, PPNAD is further subdivided based on its association with Carney complex: (i) c-PPNAD for those associated with Carney complex, and (ii) i-PPNAD for isolated cases with may be either familial (due to germline mutations) or sporadic (due to somatic alterations) (36, 45, 50). Unlike PBMAH, primary bilateral micronodular adrenocortical disease presents with normal size or slightly enlarged adrenal glands (36, 45–48). Grossly, PPNAD reveals multiple small dark brown nodules typically confined to the adrenal cortex. Histologically, compact cell-rich nodules (with lipofuscin pigment deposition in PPNAD), ranging from 1 to 4 mm, are identified in the deep ZF reticularis (45, 48) (Figure 4). In c-PPNAD, the multiple small pigmented nodules may be associated with variable degree of cortical atrophy and mild intracapsular and epicapsular adrenocortical cell extension (45). A recent retrospective review of patients with Carney complex from the Mayo Clinic also described an attenuated form of PPNAD, which was referred as c-PPNAD variant (45). This c-PPNAD variant was characterized by scattered pigmented micronodules instead of numerous pigmented micronodules, and there was accompanying prominent ZF with intracapsular and epicapsular cortical proliferations (45). These observations highlight the heterogeneity of histophenotypic manifestations in c-PPNAD.
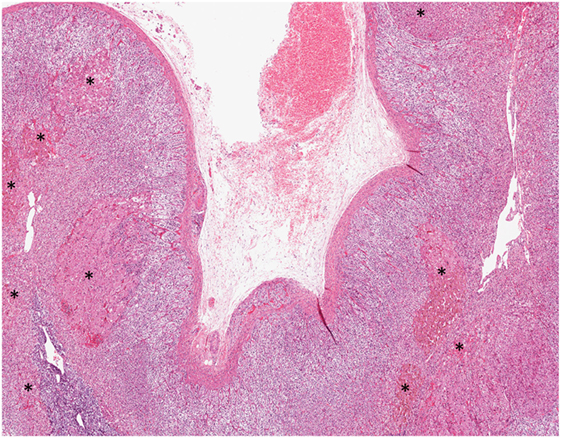
Figure 4. Primary pigmented nodular adrenocortical disease. Multifocal bilateral compact cell-rich pigmented adrenocortical micronodules (asterisk), ranging from 1 to 4 mm, constitute the hallmark of this disorder. The adrenocortical micronodules are often seen in the fasciculata and reticularis layers.
Primary bilateral macronodular adrenal hyperplasia is rare and accounts for less than 1% of cases of endogenous Cushing syndrome (51, 52). In contrast to the micronodular form, most PBMAH cases are diagnosed in adults aged between 40 and 70 years; however, pediatric forms of PBMAH also exist (36, 45, 53, 54). Affected individuals do not always present with overt Cushing syndrome, as milder forms with subclinical Cushing syndrome have also been described (54). For several years, PBMAH was thought to be primarily a sporadic disease. However, recent advances in our understanding of its molecular basis have highlighted hereditary predisposition mechanisms (see Molecular Characteristics and Pathogenesis). The degree of bilateral adrenal enlargement is also heterogeneous in patients with this disease (54). Grossly, the adrenal glands exhibit a lobulated or bosselated appearance due to multiple irregular yellow nodules, each exceeding 1.0 cm in size (6, 36, 48). Histologically, multiple unencapsulated irregular cortical nodular proliferations, composed of lipid-rich clear cells or mixed clear-and-compact cells, are noted (Figure 5). There is significant heterogeneity in both the clinical and histophenotypic manifestations associated with this condition. For instance, patients with McCune-Albright syndrome can also present with PBMAH causing Cushing syndrome in the first year of life. In addition to PBMAH, bilateral primary bimorphic adrenocortical disease and rare examples of adenomas can also occur in the adrenal glands of patients with McCune-Albright syndrome (55).
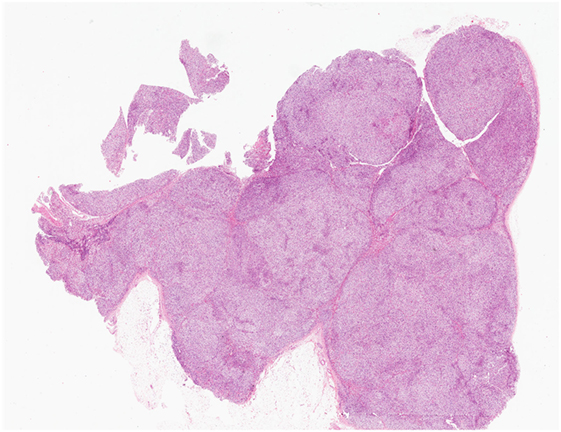
Figure 5. Primary bilateral macronodular adrenocortical hyperplasia. This disease presents with variable degree of bilateral adrenal enlargement. Grossly, the adrenal glands exhibit a lobulated or bosselated appearance due to multiple irregular yellow nodules, each greater than 1.0 cm in size. This photomicrograph illustrates the multiple unencapsulated and irregular cortical nodular proliferations, composed of lipid-rich clear cells.
Glucocorticoid-producing adenomas can occur at all ages, with a slight female predisposition (36, 46, 47). Most of these tumors are well-delineated cortical neoplasms with a yellow appearance, but pigmented variants (termed “black adenomas”) have also been reported (6, 48). The non-tumorous cortex is typically atrophic due to the negative feedback suppression of excess glucocorticoid on the hypothalamic–pituitary–adrenal axis. The tumor cells are composed mainly of clear cells that contain mitochondria with tubulovesicular cristae. Scattered compact cells with variable oncocytic change can occur. While black adenomas are not specific to adrenal Cushing syndrome and can be seen in non-functioning adrenal cortical adenomas, these are composed predominantly of pigmented compact cells (Figure 6). Komiya and colleagues reported that glucocorticoid-producing black adenomas are more frequently associated with lower urinary 17-ketosteroid than glucocorticoid-producing conventional (clear cell rich) adenomas (56). The same study also showed that there are no differences in serum cortisol concentrations and response to dexamethasone suppression between glucocorticoid-producing conventional and black adenomas (56). Interestingly, aldosterone and glucocorticoid co-secreting adenomas have also been described (57, 58).
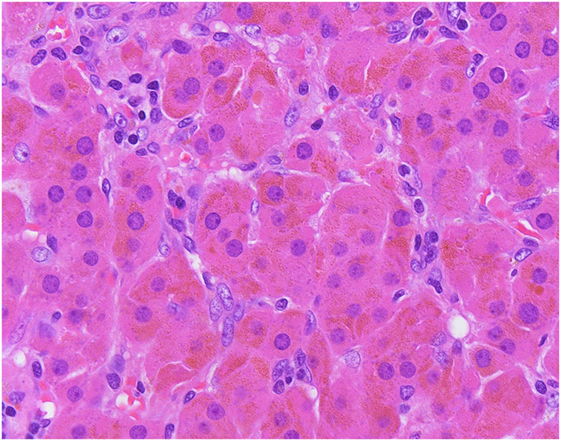
Figure 6. The so-called “black” adenomas expand the morphological spectrum of adrenal cortical neoplasms known to cause adrenal Cushing syndrome. Unlike conventional adrenocortical adenomas, pigmented “black” adenomas are composed mainly of pigmented compact cells.
The radiological findings of patients with ACTH-independent Cushing syndrome vary substantially based on the underlying pathology. For instance, PBMAH typically shows asymmetric, lobulated and bilateral enlargement of adrenal glands due to multiple large nodules ranging from 1 to 5 cm (36, 52). In contrast, those presenting with bilateral multiple micronodular proliferations (e.g., PPNAD) exhibit multiple pigmented hypodense micronodules in a background of slightly enlarged bilateral adrenal glands on CT (59–61). Glucocorticoid-producing conventional adrenal cortical adenomas are often small (<4.0 cm), solitary, and well demarcated with low attenuation values on CT (8, 9, 36). The predominance of compact cells in black adenomas can result in higher density on CT when comparing with conventional lipid-rich adenomas (56). Rare examples of bilateral cortisol-secreting adenomas have also been reported (62).
Molecular Characteristics and Pathogenesis
Alterations in the cyclic adenosine monophosphate (cAMP) or protein kinase A (PKA) signaling pathway have been linked to the pathogenesis of glucocorticoid-producing adrenal cortical neoplasms and bilateral adrenocortical micronodular disease (6, 36, 43, 58, 63) (Figure 7). In normal physiological conditions, cellular proliferation and functional differentiation of glucocorticoid-producing cells require conformational changes of G-protein-coupled receptors due to binding of ACTH to melanocortin-2 receptors (36). G-protein alpha stimulatory subunit is involved in the activation of adenylyl cyclase to generate cAMP from ATP. An increased cytoplasmic level of cAMP activates PKA, by releasing its catalytic subunits (6, 36, 63). These free catalytic subunits phosphorylate downstream elements resulting in gene transcription and enabling cortisol synthesis (63). The PKA has a tetrameric structure consisting of two regulatory and two catalytic subunits (63). Phosphodiesterases (PDEs) regulate this process by hydrolyzing cyclic nucleotides including cAMP. Activating mutations in the stimulatory G-protein alpha subunit (GNAS) and the catalytic subunit of PKA (PRKACA), as well as inactivating mutations in the type 1 alpha regulatory subunit of PKA (PRKAR1A) and cAMP-hydrolyzing PDEs (PDE11A and PDE8B), have been linked with various morphological correlates of ACTH-independent adrenal Cushing syndrome (6, 36, 63, 64). Mutations in CTNNB1 have also been described in glucocorticoid-producing adrenal cortical proliferations including adenomas (58, 65). Aldosterone and glucocorticoid co-secreting adenomas have been reported to harbor KCNJ5 mutations (57, 58). Given the genotype–phenotype correlations in KCNJ5-mutant adenomas (see previous sections), a subset of these tumors may harbor weak glucocorticoid function (21).
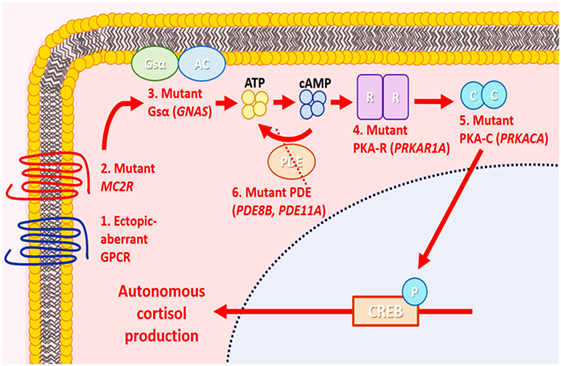
Figure 7. Molecular alterations in cortisol-producing benign adrenal cortical neoplasms. In normal physiology, cortisol production by adrenal zona fasciculata cells is mediated by the cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) signaling pathway. In the resting state, PKA is an inactive tetramer, with two catalytic subunits (PKA-C) bound to two regulatory subunits (PKA-R). Under stress, the pituitary gland produces corticotropin (ACTH), which binds to melanocortin-2 receptor (MC2R), causing activation of adenylyl cyclase (AC) through stimulatory G-protein α stimulatory subunit (Gsα) and generating cAMP from ATP. The cAMP then binds to PKA-R, causing the release of PKA-C and phosphorylating (P) downstream elements, including cAMP response element-binding protein (CREB), enabling cortisol synthesis. Cyclic AMP is hydrolyzed by phosphodiesterase (PDE), and the PKA subunit is reassembled again, returning to its inactive state. In adrenal Cushing syndrome, molecular alterations implicating PKA-C (PRKACA), PKA-R (PRKAR1A), Gsα [G-protein alpha subunit (GNAS)], PDE (PDE8B, PDE11A), MC2R, and aberrant G-protein-coupled receptors (GPCRs) have been identified in cortisol-producing adenomas. These molecular alterations are thought to result in autonomous cortisol production and cellular proliferation through aberrant activation of the cAMP/PKA signaling pathway.
Since the initial description of PBMAH (previously known as ACTH-independent macronodular hyperplasia; AIMAH) by Kirschner et al. in 1964 (66), much has been learned regarding the clinicopathological and molecular heterogeneity of this entity. The aberrant expression of several membrane-bound hormone G-protein-coupled receptors has been implicated in the pathogenesis of PBMAH (67–70). Among these, ACTH-independent steroidogenesis has been explained by the presence of ectopic hormone receptors (e.g., gastric inhibitory polypeptide receptors, beta-adrenergic, 5HT-7 serotonin receptor, V2-V3 vasopressin receptor, and angiotensin receptor “AT1R”) or dysregulation of eutopic receptors (5-HT4 serotonin receptor, V1 vasopressin receptor, luteinizing hormone/human chorionic gonadotropin) (67, 71–73). Subsequent studies from adrenals with PBMAH identified proopiomelanocortin mRNA and ACTH in a subset of adrenocortical cells, suggesting paracrine effect of an intraadrenal ACTH on the regulation of steroidogenesis (74, 75). Bourdeau and Stratakis demonstrated an increased expression of PRKAR2B using real-time PCR and immunohistochemistry in adrenals with PBMAH (76). In addition to postzygotic somatic mosaicism of GNAS in McCune-Albright syndrome, rare examples of hereditary PBMAH have been described in the setting of APC-, MEN1-, FH-, PDE8B-, and PDE11A variant-driven pathogenesis (36, 63, 77–81). Recently, the discovery by Assié et al. that 55% of PBMAH cases harbor inactivating ARMC5 germline mutations has challenged the old belief that PBMAH is mainly a sporadic disease (42). Subsequent studies also supported the frequent association between germline ARMC5-related genetic predisposition and PBMAH (51, 54, 82–85). ARCM5 (16p11.2) appears to function as a tumor suppressor gene (44), and wild-type ARCM5 has been shown to stimulate apoptosis in vitro (42). It has been suggested that PBMAH represents multiple clonal proliferations arising through the propagation of various alterations implicating the cAMP and Wnt signaling pathways (44, 86–88).
Molecular Heterogeneity and Genotype–Phenotype Correlations
Somatic activating mutations of PRKACA are the most common molecular alterations (around 40%; range, 23–57%) in glucocorticoid-producing adrenal cortical adenomas (Table 2) (64, 89–93). PRKACA-altered glucocorticoid-producing adenomas were observed in younger individuals with florid Cushing syndrome (64, 93) and frequently presented with smaller tumor size (93) than that of PRKACA-wild-type glucocorticoid-producing adenomas. The absence of subclinical Cushing syndrome in PRKACA-altered adenomas also supports the role of this mutation on glucocorticoid overproduction (64). In a cohort of PRKACA-wild-type adenomas, glucocorticoid-producing CTNNB1-mutant adrenocortical adenomas were more likely to present with subclinical Cushing syndrome (65). This is also consistent with the finding that larger non-functional adenomas are frequently associated with CTNNB1 mutations (94).
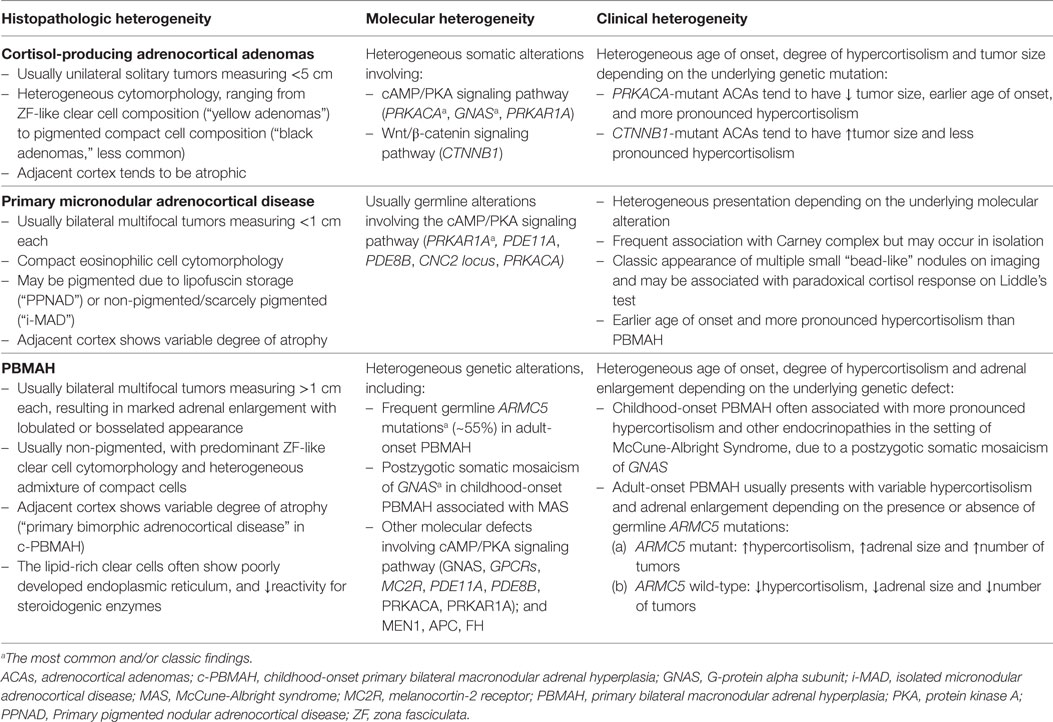
Table 2. Adrenocortical tumor heterogeneity in adrenal Cushing syndrome.
Somatic mosaicism of GNAS has been implicated in the pathogenesis of McCune-Albright syndrome-related adrenal Cushing syndrome manifesting as bilateral macronodular adrenocortical disease (e.g., PBMAH), as well as a bimorphic cortical nodular disease and rare adenomas (55). Somatic activating GNAS mutations and somatic allelic loses of PRKAR1A were seen in up to 17 and 23% of glucocorticoid-producing adenomas, respectively (95, 96). Interestingly, glucocorticoid-producing adenomas with somatic PRKAR1A alterations were more frequently associated with smaller tumor size and paradoxical increase in urinary cortisol levels following dexamethasone suppression (97). The latter is also a pattern shared with Carney complex-associated primary pigmented adrenocortical nodular disease (c-PPNAD) due to enhanced glucocorticoid receptor expression in the lesional cells (97).
Bilateral micronodular adrenocortical disease is also associated with heterogeneous molecular, histophenotypic, and clinical manifestations. Mutations in PRKAR1A, PDE11A, and PDE8B have all been implicated in the pathogenesis of bilateral micronodular adrenocortical disease presenting with Cushing syndrome (36, 43, 63, 98). Germline inactivating mutations involving PRKAR1A accounts for 80% of c-PPNAD (43, 99). A subset of patients with PRKAR1A-wild-type c-PPNAD has been linked to the CNC2 locus at chromosome 2p16; however, little is known with respect to the gene(s) associated with this condition (36, 43). Most of the CNC2 locus-related cases tend to present later in life (43). Germline or sporadic inactivating PRKAR1A, PDE11A, and PDE8B mutations have also been described in i-PPNAD causing adrenal Cushing syndrome (36, 63). PDE11A and PDE8B are also described in the pathogenesis of i-MAD (36, 63).
The role of PDE variants impacting the clinical manifestations of Carney complex patients has also been documented. For instance, a higher frequency of PDE11A variants was noted in male patients with Carney complex presenting with large cell calcifying Sertoli cell tumor (100). Germline PDE11A variant mutation has also been described with adrenal enlargement but without evidence of Cushing syndrome (98). The discovery of PDE11A variants in patients with Carney complex (63) and genomic duplication of the locus of PRKACB (encoding the catalytic C-beta subunit of PKA) in a patient with Carney complex but without evidence of Cushing syndrome support the existence of versatile genomic alterations causing PKA dysregulation (101).
A recent study has broadened our knowledge of pathogenic PRKACA alterations in bilateral micronodular adrenocortical disease presenting with Cushing syndrome (64). In this series, germline duplication of PRKACA was identified in 5 of 35 patients with bilateral nodular cortical disease lacking germline PRKAR1A, PDE11A, PDE8B, and somatic GNAS mutations (31 i-PPNAD, 2 i-MAD, and 2 macronodular type) (64). No significant phenotypic differences were reported between those carrying a germline PRKACA duplication and those lacking this molecular alteration (64). Subsequent review of the adrenocortical morphology of the five patients with germline PRKACA duplication revealed distinct histophenotypes characterized by PPNAD with cortical atrophy in three patients and non-pigmented nodular adrenocortical hyperplasia with extranodular hyperplasia in two patients (102). The variation in phenotypes has been linked to the extent of genomic alteration (103).
In addition to bilateral micronodular adrenocortical disease, PBMAH is also characterized by heterogeneous presentations. Despite significant adrenal gland enlargement, PBMAH is characterized by relatively inefficient and low cortisol overproduction in comparison to its micronodular counterpart, an observation that has attracted interest from the scientific community (36). Poorly developed smooth endoplasmic reticulum in lipid-rich cortical cells and weak reactivity for 3-beta-hydroxysteroid dehydrogenase and other steroidogenic enzymes have been described (36, 48). The low steroidogenic capacity of PBMAH has been well documented on several platforms including gene arrays analyses (76, 87). Significant downregulation of genes encoding enzymes implicated in steroidogenesis (CYP11A, CYP17, and CYP21A2) has also been observed and used to explain this phenomenon in PBMAH (87). Accordingly, the diseased cells favor proliferation over functionality. The heterogeneity of PBMAH is also reflected by the diverse molecular alterations of this disease. Almeida et al. reported that larger nodules often harbored increased expression of bcl-2, E2F-1, c-KIT, MYB, PRKCA, and CTTNB1 when compared with smaller nodules in PBMAH (104). Assié et al. characterized the ARMC5 heterogeneity of individual nodules in PBMAH by demonstrating distinctive secondary somatic mutations arising in the background of germline susceptibility in individual macronodular proliferations (42). This observation combined with the putative tumor suppressor function of ARMC5 may explain the extensive genetic variance of ARCM5-driven PBMAH (44).
The discovery of frequent germline ARCM5 alterations in patients with adrenal Cushing syndrome has advanced our understanding of this disease, especially with regards to its inherent genotype–phenotype correlations. Espiard et al. demonstrated that patients with ARMC5-driven PBMAH presented more frequently with overt Cushing syndrome, higher midnight plasma cortisol, urinary free cortisol, and cortisol after dexamethasone suppression test than those with ARMC5-wild-type disease (54). The same study also demonstrated that ARMC5-wild-type tumors are more frequently associated with subclinical Cushing syndrome and non-functioning status (54). In addition, adrenals with ARMC5-driven PBMAH were significantly bigger and had more nodules than those with ARMC5-wild-type presentations (54, 85).
Conclusion
Aldosterone- and cortisol-producing adrenal cortical tumors encompass a diverse group of neoplasms with distinct genetic and morphological features. The heterogeneous spectrum of molecular alterations seen in these lesions is increasingly reflected in their distinctive demographic, hormonal, cytomorphologic, and immunophenotypic profiles. Although most cases occur sporadically, an increasing number of tumors arise in the setting of familial syndromes with potential implications for gene testing and counseling. As the future of molecular endocrinology becomes more complex, it is anticipated that modern technologies will allow a deeper understanding of the structure, function, and prognosis of these tumors, which will hopefully translate into more precise therapeutic strategies to improve the management of patients with adrenal cortical disease.
Author Contributions
Literature search, writing, and figures: OM and KD. Concept, design, and critical reviews: OM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M, et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab (2008) 93:3266–81. doi:10.1210/jcem.93.9.9998
2. Young WF. Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol (2007) 66:607–18. doi:10.1111/j.1365-2265.2007.02775.x
3. Mattsson C, Young WF. Primary aldosteronism: diagnostic and treatment strategies. Nat Clin Pract Nephrol (2006) 2:198–208. doi:10.1038/ncpneph0151
4. Schirpenbach C, Reincke M. Primary aldosteronism: current knowledge and controversies in Conn’s syndrome. Nat Clin Pract Endocrinol Metab (2007) 3:220–7. doi:10.1038/ncpendmet0430
5. Duan K, Mete O. Clinicopathologic correlates of primary aldosteronism. Arch Pathol Lab Med (2015) 139:948–54. doi:10.5858/arpa.2014-0156-RS
6. Duan K, Giordano TJ, Mete O. Adrenal cortical proliferations. In: Mete O, Asa SL, editors. Endocrine Pathology. UK: Cambridge University Press (2016). p. 602–27.
7. Ganguly A. Primary aldosteronism. N Engl J Med (1998) 339:1828–34. doi:10.1056/NEJM199812173392507
8. Young WF. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med (2007) 356:601–10. doi:10.1056/NEJMcp065470
9. Ilias I, Sahdev A, Reznek RH, Grossman AB, Pacak K. The optimal imaging of adrenal tumours: a comparison of different methods. Endocr Relat Cancer (2007) 14:587–99. doi:10.1677/ERC-07-0045
10. Nanba AT, Nanba K, Byrd JB, Shields JJ, Giordano TJ, Miller BS, et al. Discordance between imaging and immunohistochemistry in unilateral primary aldosteronism. Clin Endocrinol (2017) 87:665–72. doi:10.1111/cen.13442
11. Dekkers T, ter Meer M, Lenders JW, Hermus AR, Schultze Kool L, Langenhuijsen JF, et al. Adrenal nodularity and somatic mutations in primary aldosteronism: one node is the culprit? J Clin Endocrinol Metab (2014) 99:E1341–1135. doi:10.1210/jc.2013-4255
12. Nanba K, Tsuiki M, Sawai K, Mukai K, Nishimoto K, Usui T, et al. Histopathological diagnosis of primary aldosteronism using CYP11B2 immunohistochemistry. J Clin Endocrinol Metab (2013) 98:1567–74. doi:10.1210/jc.2012-3726
13. Yamazaki Y, Nakamura Y, Omata K, Ise K, Tezuka Y, Ono Y, et al. Histopathological classification of cross-sectional image-negative hyperaldosteronism. J Clin Endocrinol Metab (2017) 102:1182–92. doi:10.1210/jc.2016-2986
14. Nanba K, Chen AX, Omata K, Vinco M, Giordano TJ, Else T, et al. Molecular heterogeneity in aldosterone-producing adenomas. J Clin Endocrinol Metab (2016) 101:999–1007. doi:10.1210/jc.2015-3239
15. Tan GC, Negro G, Pinggera A, Tizen Laim NMS, Mohamed Rose I, Ceral J, et al. Aldosterone-producing adenomas: histopathology-genotype correlation and identification of a novel CACNA1D mutation. Hypertension (2017) 70:129–36. doi:10.1161/HYPERTENSIONAHA.117.09057
16. Azizan EA, Lam BY, Newhouse SJ, Zhou J, Kuc RE, Clarke J, et al. Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J Clin Endocrinol Metab (2012) 97:E819–29. doi:10.1210/jc.2011-2965
17. Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet (2013) 45:1055–60. doi:10.1038/ng.2716
18. Monticone S, Castellano I, Versace K, Lucatello B, Veglio F, Gomez-Sanchez CE, et al. Immunohistochemical, genetic and clinical characterization of sporadic aldosterone-producing adenomas. Mol Cell Endocrinol (2015) 411:146–54. doi:10.1016/j.mce.2015.04.022
19. Mete O, Asa SL. Aldosterone-producing adrenal cortical adenoma with oncocytic change and cytoplasmic eosinophilic globular inclusions. Endocr Pathol (2009) 20:182–5. doi:10.1007/s12022-009-9082-2
20. Monticone S, Else T, Mulatero P, Williams TA, Rainey WE. Understanding primary aldosteronism: impact of next generation sequencing and expression profiling. Mol Cell Endocrinol (2015) 399:311–20. doi:10.1016/j.mce.2014.09.015
21. Seidel E, Scholl UI. Intracellular molecular differences in aldosterone compared to cortisol-secreting adrenal cortical adenomas. Front Endocrinol (2016) 7:75. doi:10.3389/fendo.2016.00075
22. Åkerström T, Crona J, Delgado Verdugo A, Starker LF, Cupisti K, Willenberg HS, et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLoS One (2012) 7:e41926. doi:10.1371/journal.pone.0041926
23. Nishimoto K, Nakagawa K, Li D, Kosaka T, Oya M, Mikami S, et al. Adrenocortical zonation in humans under normal and pathological conditions. J Clin Endocrinol Metab (2010) 95:2296–305. doi:10.1210/jc.2009-2010
24. Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, et al. Aldosterone stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci U S A (2015) 112:E4591–9. doi:10.1073/pnas.1505529112
25. Boulkroun S, Samson-Couterie B, Dzib JF, Lefebvre H, Louiset E, Amar L, et al. Adrenal cortex remodeling and functional zona glomerulosa hyperplasia in primary aldosteronism. Hypertension (2010) 56:885–92. doi:10.1161/HYPERTENSIONAHA.110.158543
26. Seccia TM, Caroccia B, Gomez-Sanchez EP, Vanderriele PE, Gomez-Sanchez CE, Rossi GP. Review of markers of zona glomerulosa and aldosterone-producing adenoma cells. Hypertension (2017) 70:867–74. doi:10.1161/HYPERTENSIONAHA.117.09991
27. Zhou J, Lam B, Neogi SG, Yeo GS, Azizan EA, Brown MJ. Transcriptome pathway analysis of pathological and physiological aldosterone-producing human tissues. Hypertension (2016) 68:1424–31. doi:10.1161/HYPERTENSIONAHA.116.08033
28. Maniero C, Garg S, Zhao W, Johnson TI, Zhou J, Gurnell M, et al. NEFM (neurofilament medium) polypeptide, a marker for zona glomerulosa cells in human adrenal, inhibits D1R (dopamine D1 receptor)-mediated secretion of aldosterone. Hypertension (2017) 70:357–64. doi:10.1161/HYPERTENSIONAHA.117.09231
29. Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, Rossi GP. A meta-analysis of somatic KCNJ5 K(+) channel mutations in 1636 patients with an aldosterone-producing adenoma. J Clin Endocrinol Metab (2015) 100:E1089–95. doi:10.1210/jc.2015-2149
30. Berthon A, Drelon C, Ragazzon B, Boulkroun S, Tissier F, Amar L, et al. WNT/β-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum Mol Genet (2014) 23:889–905. doi:10.1093/hmg/ddt484
31. Wu VC, Wang SM, Chueh SJ, Yang SY, Huang KH, Lin YH, et al. The prevalence of CTNNB1 mutations in primary aldosteronism and consequences for clinical outcomes. Sci Rep (2017) 7:39121. doi:10.1038/srep39121
32. Åkerström T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep (2016) 6:19546. doi:10.1038/srep19546
33. Scholl UI, Stölting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife (2015) 4:e06315. doi:10.7554/eLife.06315
34. Zilbermint M, Xekouki P, Faucz FR, Berthon A, Gkourogianni A, Schernthaner-Reiter MH, et al. Primary aldosteronism and ARMC5 variants. J Clin Endocrinol Metab (2015) 100:E900–9. doi:10.1210/jc.2014-4167
35. Mulatero P, Schiavi F, Williams TA, Monticone S, Barbon G, Opocher G, et al. ARMC5 mutation analysis in patients with primary aldosteronism and bilateral adrenal lesions. J Hum Hypertens (2016) 30:374–8. doi:10.1038/jhh.2015.98
36. Duan K, Gomez Hernandez K, Mete O. Clinicopathological correlates of adrenal Cushing’s syndrome. J Clin Pathol (2014) 68:175–86. doi:10.1136/jclinpath-2014-202612
37. Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab (2008) 93:1526–40. doi:10.1210/jc.2008-0125
38. Bertagna X, Guignat L, Groussin L, Bertherat J. Cushing’s disease. Best Pract Res Clin Endocrinol Metab (2009) 23:607–23. doi:10.1016/j.beem.2009.06.001
40. Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet (2006) 367:1605–17. doi:10.1016/S0140-6736(06)68699-6
41. Tabarin A, Perez P. Pros and cons of screening for occult Cushing syndrome. Nat Rev Endocrinol (2011) 7:445–55. doi:10.1038/nrendo.2011.51
42. Assié G, Libé R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Engl J Med (2013) 369:2105–14. doi:10.1056/NEJMoa1304603
43. Correa R, Salpea P, Stratakis CA. Carney complex: an update. Eur J Endocrinol (2015) 173:M85–97. doi:10.1530/EJE-15-0209
44. Correa R, Zilbermint M, Berthon A, Espiard S, Batsis M, Papadakis GZ, et al. The ARMC5 gene shows extensive genetic variance in primary macronodular adrenocortical hyperplasia. Eur J Endocrinol (2015) 173:435–40. doi:10.1530/EJE-15-0205
45. Lowe KM, Young WF Jr, Lyssikatos C, Stratakis CA, Carney JA. Cushing syndrome in Carney complex: clinical, pathologic, and molecular genetic findings in the 17 affected mayo clinic patients. Am J Surg Pathol (2017) 41:171–81. doi:10.1097/PAS.0000000000000748
46. Stratakis CA. Cushing syndrome caused by adrenocortical tumors and hyperplasias (corticotropin-independent Cushing syndrome). Endocr Dev (2008) 13:117–32. doi:10.1159/000134829
47. Stratakis CA, Boikos SA. Genetics of adrenal tumors associated with Cushing’s syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab (2007) 3:748–57. doi:10.1038/ncpendmet0648
48. Lack EE. AFIP Atlas of Tumor Pathology, Fourth Series, Fascicle 8. Tumors of the Adrenal Glands and Extraadrenal Paraganglia. Washington, DC: ARP Press (2007).
49. Mete O, Asa SL. Precursor lesions of endocrine system neoplasms. Pathology (2013) 45:316–30. doi:10.1097/PAT.0b013e32835f45c5
50. Almeida MQ, Stratakis CA. Carney complex and other conditions associated with micronodular adrenal hyperplasias. Best Pract Res Clin Endocrinol Metab (2010) 24:907–14. doi:10.1016/j.beem.2010.10.006
51. Gagliardi L, Schreiber AW, Hahn CN, Feng J, Cranston T, Boon H, et al. ARMC5 mutations are common in familial bilateral macronodular adrenal hyperplasia. J Clin Endocrinol Metab (2014) 99:E1784–92. doi:10.1210/jc.2014-1265
52. Lacroix A. ACTH-independent macronodular adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab (2009) 23:245–59. doi:10.1016/j.beem.2008.10.011
53. Stratakis CA. Cushing syndrome in pediatrics. Endocrinol Metab Clin North Am (2012) 41:793–803. doi:10.1016/j.ecl.2012.08.002
54. Espiard S, Drougat L, Libé R, Assié G, Perlemoine K, Guignat L, et al. ARMC5 mutations in a large cohort of primary macronodular adrenal hyperplasia: clinical and functional consequences. J Clin Endocrinol Metab (2015) 100:E926–35. doi:10.1210/jc.2014-4204
55. Carney JA, Young WF, Stratakis CA. Primary bimorphic adrenocortical disease: cause of hypercortisolism in McCune-Albright syndrome. Am J Surg Pathol (2011) 35:1311–26. doi:10.1097/PAS.0b013e31821ec4ce
56. Komiya I, Takasu N, Aizawa T, Yamada T, Koizumi Y, Hashizume K, et al. Black (or brown) adrenal cortical adenoma: its characteristic features on computed tomography and endocrine data. J Clin Endocrinol Metab (1985) 61:711–7. doi:10.1210/jcem-61-4-711
57. Yamada M, Nakajima Y, Taguchi R, Okamura T, Ishii S, Tomaru T, et al. KCNJ5 mutations in aldosterone- and cortisol-co-secreting adrenal adenomas. Endocr J (2012) 59:735–41. doi:10.1507/endocrj.EJ12-0247
58. Thiel A, Reis AC, Haase M, Goh G, Schott M, Willenberg HS, et al. PRKACA mutations in cortisol-producing adenomas and adrenal hyperplasia: a single-center study of 60 cases. Eur J Endocrinol (2015) 172:677–85. doi:10.1530/EJE-14-1113
59. Rockall AG, Babar SA, Sohaib SA, Isidori AM, Diaz-Cano S, Monson JP, et al. CT and MR imaging of the adrenal glands in ACTH-independent cushing syndrome. Radiographics (2004) 24:435–52. doi:10.1148/rg.242035092
60. Powell AC, Stratakis CA, Patronas NJ, Steinberg SM, Batista D, Alexander HR, et al. Operative management of Cushing syndrome secondary to micronodular adrenal hyperplasia. Surgery (2008) 143:750–8. doi:10.1016/j.surg.2008.03.022
61. Courcoutsakis N, Prassopoulos P, Stratakis CA. CT findings of primary pigmented nodular adrenocortical disease: rare cause of ACTH-independent Cushing syndrome. AJR Am J Roentgenol (2010) 194:W541. doi:10.2214/AJR.09.4056
62. Nomura K, Saito H, Aiba M, Iihara M, Obara T, Takano K. Cushing’s syndrome due to bilateral adrenocortical adenomas with unique histological features. Endocr J (2003) 50:155–62. doi:10.1507/endocrj.50.155
63. Hannah-Shmouni F, Faucz FR, Stratakis CA. Alterations of phosphodiesterases in adrenocortical tumors. Front Endocrinol (2016) 7:111. doi:10.3389/fendo.2016.00111
64. Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A, et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med (2014) 370:1019–28. doi:10.1056/NEJMoa1310359
65. Ronchi CL, Di Dalmazi G, Faillot S, Sbiera S, Assié G, Weigand I, et al. Genetic landscape of sporadic unilateral adrenocortical adenomas without PRKACA p.Leu206Arg mutation. J Clin Endocrinol Metab (2016) 101:3526–38. doi:10.1210/jc.2016-1586
66. Kirschner MA, Powell RD Jr, Lipsett MB. Cushing’s syndrome: nodular cortical hyperplasia of adrenal glands with clinical and pathological features suggesting adrenocortical tumor. J Clin Endocrinol Metabob (1964) 24:947–55. doi:10.1210/jcem-24-10-947
67. Lacroix A, Bourdeau I, Lampron A, Mazzuco TL, Tremblay J, Hamet P. Aberrant G-protein coupled receptor expression in relation to adrenocortical overfunction. Clin Endocrinol (Oxf) (2010) 73:1–15. doi:10.1111/j.1365-2265.2009.03689.x
68. Lacroix A, Ndiaye N, Tremblay J, Hamet P. Ectopic and abnormal hormone receptors in adrenal Cushing’s syndrome. Endocr Rev (2001) 22:75–110. doi:10.1210/edrv.22.1.0420
69. Lacroix A, Baldacchino V, Bourdeau I, Hamet P, Tremblay J. Cushing’s syndrome variants secondary to aberrant hormone receptors. Trends Endocrinol Metab (2004) 8:375–82. doi:10.1016/S1043-2760(04)00188-2
70. Lacroix A, Bourdeau I. Bilateral adrenal Cushing’s syndrome: macronodular adrenal hyperplasia and primary pigmented nodular adrenocortical disease. Endocrinol Metab Clin N Am (2005) 34:441–58. doi:10.1016/j.ecl.2005.01.004
71. Lacroix A, N’Diaye N, Mircescu H, Hamet P, Tremblay J. Abnormal expression and function of hormone receptors in adrenal Cushing’s syndrome. Endocr Res (1998) 24:835–43. doi:10.3109/07435809809032694
72. Messidoro C, Elte JW, Castro Cabezas M, van Agteren M, Lacroix A, de Herder WW. Food-dependent Cushing’s syndrome. Neth J Med (2009) 67:187–90.
73. Mazzuco TL, Thomas M, Martinie M, Cherradi N, Sturm N, Feige JJ, et al. Cellular and molecular abnormalities of a macronodular adrenal hyperplasia causing betablocker-sensitive Cushing’s syndrome. Arq Bras Endocrinol Metab (2007) 51:1452–62. doi:10.1590/S0004-27302007000900007
74. Lefebvre H, Duparc C, Chartrel N, Jegou S, Pellerin A, Laquerriere A, et al. Intraadrenal adrenocorticotropin production in a case of bilateral macronodular adrenal hyperplasia causing Cushing’s syndrome. J Clin Endocrinol Metab (2003) 88:3035–42. doi:10.1210/jc.2002-030014
75. Louiset E, Duparc C, Young J, Renouf S, Tetsi Nomigni M, Boutelet I, et al. Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med (2013) 369:2115–25. doi:10.1056/NEJMoa1215245
76. Bourdeau I, Stratakis CA. Cyclic AMP-dependent signaling aberrations in macronodular adrenal disease. Ann N Y Acad Sci (2002) 968:240–55. doi:10.1111/j.1749-6632.2002.tb04339.x
77. Libé R, Fratticci A, Coste J, Tissier F, Horvath A, Ragazzon B, et al. Phosphodiesterase 11A (PDE11A) and genetic predisposition to adrenocortical tumors. Clin Cancer Res (2008) 14:4016–24. doi:10.1158/1078-0432.CCR-08-0106
78. Matyakhina L, Freedman RJ, Bourdeau I, Wei MH, Stergiopoulos SG, Chidakel A, et al. Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical Cushing syndrome: a clinical and molecular genetic investigation. J Clin Endocrinol Metab (2005) 90:3773–9. doi:10.1210/jc.2004-2377
79. Hsiao HP, Kirschner LS, Bourdeau I, Keil MF, Boikos SA, Verma S, et al. Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab (2009) 94:2930–7. doi:10.1210/jc.2009-0516
80. Gaujoux S, Pinson S, Gimenez-Roqueplo AP, Amar L, Ragazzon B, Launay P, et al. Inactivation of the APC gene is constant in adrenocortical tumors from patients with familial adenomatous polyposis but not frequent in sporadic adrenocortical cancers. Clin Cancer Res (2010) 16:5133–41. doi:10.1158/1078-0432.CCR-10-1497
81. Yoshida M, Hiroi M, Imai T, Kikumori T, Himeno T, Nakamura Y, et al. A case of ACTH-independent macronodular adrenal hyperplasia associated with multiple endocrine neoplasia type 1. Endocr J (2011) 58:269–77. doi:10.1507/endocrj.K10E-218
82. Faucz FR, Zilbermint M, Lodish MB, Szarek E, Trivellin G, Sinaii N, et al. Macronodular adrenal hyperplasia due to mutations in an armadillo repeat containing 5 (ARMC5) gene: a clinical and genetic investigation. J Clin Endocrinol Metab (2014) 99:E1113–9. doi:10.1210/jc.2013-4280
83. Elbelt U, Trovato A, Kloth M, Gentz E, Finke R, Spranger J, et al. Molecular and clinical evidence for an ARMC5 tumor syndrome: concurrent inactivating germline and somatic mutations are associated with both primary macronodular adrenal hyperplasia and meningioma. J Clin Endocrinol Metab (2015) 100:E119–28. doi:10.1210/jc.2014-2648
84. Alencar GA, Lerario AM, Nishi MY, Mariani BM, Almeida MQ, Tremblay J, et al. ARMC5 mutations are a frequent cause of primary macronodular adrenal Hyperplasia. J Clin Endocrinol Metab (2014) 99:E1501–9. doi:10.1210/jc.2013-4237
85. Albiger NM, Regazzo D, Rubin B, Ferrara AM, Rizzati S, Taschin E, et al. A multicenter experience on the prevalence of ARMC5 mutations in patients with primary bilateral macronodular adrenal hyperplasia: from genetic characterization to clinical phenotype. Endocrine (2017) 55:959–68. doi:10.1007/s12020-016-0956-z
86. Bourdeau I, Matyakhina L, Stergiopoulos SG, Sandrini F, Boikos S, Stratakis CA. 17q22-24 chromosomal losses and alterations of protein kinase a subunit expression and activity in adrenocorticotropin-independent macronodular adrenal hyperplasia. J Clin Endocrinol Metab (2006) 91:3626–32. doi:10.1210/jc.2005-2608
87. Bourdeau I, Antonini SR, Lacroix A, Kirschner LS, Matyakhina L, Lorang D, et al. Gene array analysis of macronodular adrenal hyperplasia confirms clinical heterogeneity and identifies several candidate genes as molecular mediators. Oncogene (2004) 23:1575–85. doi:10.1038/sj.onc.1207277
88. Bimpaki EI, Iliopoulos D, Moraitis A, Stratakis CA. MicroRNA signature in massive macronodular adrenocortical disease and implications for adrenocortical tumourigenesis. Clin Endocrinol (2010) 72:744–51. doi:10.1111/j.1365-2265.2009.03725.x
89. Berthon AS, Szarek E, Stratakis CA. PRKACA: the catalytic subunit of protein kinase A and adrenocortical tumors. Front Cell Dev Biol (2015) 3:26. doi:10.3389/fcell.2015.00026
90. Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet (2014) 46:613–7. doi:10.1038/ng.2956
91. Di Dalmazi G, Kisker C, Calebiro D, Mannelli M, Canu L, Arnaldi G, et al. Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. J Clin Endocrinol Metab (2014) 99:E2093–100. doi:10.1210/jc.2014-2152
92. Nakajima Y, Okamura T, Gohko T, Satoh T, Hashimoto K, Shibusawa N, et al. Somatic mutations of the catalytic subunit of cyclic AMP-dependent protein kinase (PRKACA) gene in Japanese patients with several adrenal adenomas secreting cortisol [Rapid Communication]. Endocr J (2014) 61:825–32. doi:10.1507/endocrj.EJ14-0282
93. Sato Y, Maekawa S, Ishii R, Sanada M, Morikawa T, Shiraishi Y, et al. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science (2014) 344:917–20. doi:10.1126/science.1252328
94. Bonnet S, Gaujoux S, Launay P, Baudry C, Chokri I, Ragazzon B, et al. Wnt/β-catenin pathway activation in adrenocortical adenomas is frequently due to somatic CTNNB1-activating mutations, which are associated with larger and nonsecreting tumors: a study in cortisol-secreting and -nonsecreting tumors. J Clin Endocrinol Metab (2011) 96:E419–26. doi:10.1210/jc.2010-1885
95. Fragoso MC, Domenice S, Latronico AC, Martin RM, Pereira MAA, Zerbini MCN, et al. Cushing’s syndrome secondary to adrenocorticotropin-indepen-dent macronodular adrenocortical hyperplasia due to activating mutations of GNAS1 gene. J Clin Endocrinol Metab (2003) 88:2147–51. doi:10.1210/jc.2002-021362
96. Bertherat J, Groussin L, Sandrini F, Matyakhina L, Bei T, Stergiopoulos S, et al. Molecular and functional analysis of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res (2003) 63:5308–19.
97. Louiset E, Stratakis CA, Perraudin V, Griffin KJ, Libé R, Cabrol S, et al. The paradoxical increase in cortisol secretion induced by dexamethasone in primary pigmented nodular adrenocortical disease involves a glucocorticoid receptor-mediated effect of dexamethasone on protein kinase A catalytic subunits. J Clin Endocrinol Metab (2009) 94:2406–13. doi:10.1210/jc.2009-0031
98. Carney JA, Gaillard RC, Bertherat J, Stratakis CA. Familial micronodular adrenocortical disease, Cushing syndrome, and mutations of the gene encoding phosphodiesterase 11A4 (PDE11A). Am J Surg Pathol (2010) 34:547–55. doi:10.1097/PAS.0b013e3181d31f49
99. Groussin L, Cazabat L, René-Corail F, Jullian E, Bertherat J. Adrenal pathophysiology: lessons from the Carney complex. Horm Res (2005) 64:132–9. doi:10.1159/000088586
100. Libé R, Horvath A, Vezzosi D, Fratticci A, Coste J, Perlemoine K, et al. Frequent phosphodiesterase 11A gene (PDE11A) defects in patients with Carney complex (CNC) caused by PRKAR1A mutations: PDE11A may contribute to adrenal and testicular tumors in CNC as a modifier of the phenotype. J Clin Endocrinol Metab (2011) 96:E208–2014. doi:10.1210/jc.2010-1704
101. Forlino A, Vetro A, Garavelli L, Ciccone R, London E, Stratakis CA, et al. PRKACB and Carney complex. N Engl J Med (2014) 370:1065–7. doi:10.1056/NEJMc1309730
102. Carney JA, Lyssikatos C, Lodish MB, Stratakis CA. Germline PRKACA amplification leads to Cushing syndrome caused by 3 adrenocortical pathologic phenotypes. Hum Pathol (2015) 46:40–9. doi:10.1016/j.humpath.2014.09.005
103. Lodish MB, Yuan B, Levy I, Braunstein GD, Lyssikatos C, Salpea P, et al. Germline PRKACA amplification causes variable phenotypes that may depend on the extent of the genomic defect: molecular mechanisms and clinical presentations. Eur J Endocrinol (2015) 172:803–11. doi:10.1530/EJE-14-1154
104. Almeida MQ, Harran M, Bimpaki EI, Hsiao HP, Horvath A, Cheadle C, et al. Integrated genomic analysis of nodular tissue in macronodular adrenocortical hyperplasia: progression of tumorigenesis in a disorder associated with multiple benign lesions. J Clin Endocrinol Metab (2011) 96:E728–38. doi:10.1210/jc.2010-2420
Keywords: adrenocortical adenoma, primary pigmented nodular adrenocortical disease, tumor heterogeneity, endocrinology, primary aldosteronism, Cushing syndrome
Citation: Mete O and Duan K (2018) The Many Faces of Primary Aldosteronism and Cushing Syndrome: A Reflection of Adrenocortical Tumor Heterogeneity. Front. Med. 5:54. doi: 10.3389/fmed.2018.00054
Received: 21 December 2017; Accepted: 15 February 2018;
Published: 12 March 2018
Edited by:
Stefano La Rosa, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandReviewed by:
Silvia Uccella, University of Insubria, ItalyPierlorenzo Pallante, Consiglio Nazionale Delle Ricerche (CNR), Italy
Copyright: © 2018 Mete and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ozgur Mete, b3pndXIubWV0ZTJAdWhuLmNh