 Nicola Petrosillo
Nicola Petrosillo Guido Granata
Guido Granata Maria Adriana Cataldo
Maria Adriana Cataldo
- Clinical and Research Department for Infectious Diseases, Unit Systemic and Immunedepression-Associated Infections, National Institute for Infectious Diseases L. Spallanzani, Rome, Italy
The current picture of Clostridium difficile infection (CDI) is alarming with a mortality rate ranging between 3% and 15% and a CDI recurrence rate ranging from 12% to 40%. Despite the great efforts made over the past 10 years to face the CDI burden, there are still gray areas in our knowledge on CDI management. The traditional anti-CDI antimicrobials are not always adequate in addressing the current needs in CDI management. The aim of our review is to give an update on novel antimicrobials for the treatment of CDI, considering the currently available evidences on their efficacy, safety, molecular mechanism of action, and their probability to be successfully introduced into the clinical practice in the near future. We identified, through a PubMed search, 16 novel antimicrobial molecules under study for CDI treatment: cadazolid, surotomycin, ridinilazole, LFF571, ramoplanin, CRS3123, fusidic acid, nitazoxanide, rifampin, rifaximin, tigecycline, auranofin, NVB302, thuricin CD, lacticin 3147, and acyldepsipeptide antimicrobials. In comparison with the traditional anti-CDI antimicrobial treatment, some of the novel antimicrobials reviewed in this study offer several advantages, i.e., the favorable pharmacokinetic and pharmacodynamic profile, the narrow-spectrum activity against CD that implicates a low impact on the gut microbiota composition, the inhibitory activity on CD sporulation and toxins production. Among these novel antimicrobials, the most active compounds in reducing spore production are cadazolid, ridinilazole, CRS3123, ramoplanin and, potentially, the acyldepsipeptide antimicrobials. These antimicrobials may potentially reduce CD environment spread and persistence, thus reducing CDI healthcare-associated acquisition. However, some of them, i.e., surotomycin, fusidic acid, etc., will not be available due to lack of superiority versus standard of treatment. The most CD narrow-spectrum novel antimicrobials that allow to preserve microbiota integrity are cadazolid, ridinilazole, auranofin, and thuricin CD. In conclusion, the novel antimicrobial molecules under development for CDI have promising key features and advancements in comparison to the traditional anti-CDI antimicrobials. In the near future, some of these new molecules might be effective alternatives to fight CDI.
Introduction
The Gram-positive, anaerobic, spore-forming bacillus Clostridium difficile (CD) represents the most common cause of nosocomial diarrhea worldwide (1–5). According to literature data, a total of 15%–25% of all cases of antibiotic-associated diarrhea result from C. difficile infection (CDI) (1–5).
The current picture is alarming with a CDI mortality rate ranging between 3 and 15% and a CDI recurrence rate ranging from 12 to 40% (6–8). Importantly, after the first recurrence, a risk up to 64% of subsequent recurrences has been reported (6–10).
Despite the great efforts made over the past 10 years to face the CDI burden, there are still gray areas in our knowledge on CDI management. Major issues affecting the management of CDI include the high rate of CDI underdiagnosis and the delay in diagnosing it, the unacceptably high rate of CDI recurrence, and the difficulties faced in reducing the spread of CD among hospitalized patients (11–15).
Recurrences currently represent one of the major challenges in the management of CDI, resulting in higher hospitalization costs and in increased morbidity and mortality rate (16). Of relevance, semi-structured interviews with patients who had experienced CDI showed that this disease affects numerous aspects of patients’ lives and causes patients’ emotional distress (17).
The therapeutical management of CDI is mainly based on discontinuation of unnecessary antibiotics and administration of anti-CD antimicrobials (18).
The currently recommended first-line antimicrobial therapy is represented by oral metronidazole or oral vancomycin for the first episode of mild CDI, and oral vancomycin for severe CDI or subsequent CDI recurrences (13, 18, 19). Oral fidaxomicin is also a treatment of choice for recurrent CDI, especially in those with a high risk of relapse (13, 18, 19).
Metronidazole and vancomycin achieve an end-of-treatment cure rate of approximately 86–95% (19, 20); however, they are not as much effective in assuring sustained and bacteriological cure, defined as the prevention of recurrent CDI and the prevention of CD spread, respectively (20–22).
Recently new innovative approaches, based on non-antimicrobial compounds, i.e., monoclonal anti-toxin antibodies, fecal microbiota transplantation, live bacterial vaccines and CD vaccines, have been developed or are under development. However, further studies are needed to confirm the efficacy and safety of these approaches.
Moreover a number of new agents active against CD have been developed and their use for CDI treatment is under study, and hopefully in the near future, these new antimicrobials will represent effective options to fight CDI.
Finally, old antimicrobial agents approved for the treatment of other infection showed activity against CD, and their utility for CDI is now being studied.
The aim of our review is to give an update on the novel antimicrobials for the treatment of CDI, considering the currently available evidences on their efficacy, safety, molecular mechanism of action, and their probability to be successfully introduced into the clinical practice in the near future.
Materials and Methods
Search Strategy and Selection Criteria
Through a PubMed search with the search terms “novel antimicrobials AND clostridium difficile” and “antimicrobial treatment AND clostridium difficile,” we identified 16 novel antimicrobials for the treatment of CDI: cadazolid, surotomycin, ridinilazole, LFF571, ramoplanin, CRS3123, fusidic acid, nitazoxanide, rifampin, rifaximin, tigecycline, auranofin; NVB302, thuricin CD, lacticin 3147, and acyldepsipeptide antimicrobials.
Published articles from January 2000 to November 2017 reporting the use of these 16 antimicrobials for the treatment of CDI in human patients were identified through computerized literature searches using MEDLINE (National Library of Medicine Bethesda MD) and by reviewing the references of retrieved articles.
Indexed search terms included: “cadazolid AND clostridium difficile” OR “ACT-179811 AND clostridium difficile” OR “Surotomycin AND clostridium difficile” OR “CB-183315 AND clostridium difficile”; “Ridinilazole AND clostridium difficile” OR “SMT19969 AND clostridium difficile” OR “LFF571 AND clostridium difficile” OR “Ramoplanin AND clostridium difficile” OR “CRS3123 AND clostridium difficile” OR “REP3123 AND clostridium difficile” OR “fusidic acid AND clostridium difficile” OR “nitazoxanide AND clostridium difficile” OR “rifampin AND clostridium difficile” OR “NVB302 AND clostridium difficile” OR “thuricin CD AND clostridium difficile” OR “lacticin 3147 AND clostridium difficile” OR “auranofin AND clostridium difficile” OR “acyldepsipeptide AND clostridium difficile” OR “tigecycline AND clostridium difficile” OR “rifaximin AND clostridium difficile”.
No attempt was made to obtain information about unpublished studies. English language restriction was applied.
Results
Our literature search identified 453 studies, an additional 4 articles were identified by reviewing the references of retrieved articles.
Regarding cadazolid, we identified 20 studies, 3 of them were clinical trials: a phase II clinical trial with the main objective to evaluate the susceptibilities of CD isolates to cadazolid and vancomycin (23); a multicenter, randomized, double-blind, phase II trial comparing the clinical cure rate of cadazolid versus vancomycin at 48–24 h after the end of treatment (24); and a single-center, open-label, single oral dose phase I study to investigate systemic cadazolid exposure (25).
Regarding surotomycin, our literature search identified 30 studies, 6 of them were clinical trials: a double-blind, multicentre, phase III trials comparing clinical response, sustained clinical response, and safety of surotomycin and vancomycin (26, 27); a randomized, double-blind, dose-ranging, parallel group, phase II trial to evaluate the effects of surotomycin and vancomycin on CD and microbiota (28); a randomized, double-blind, placebo-controlled, phase I trial to characterize the safety, tolerability, and plasma pharmacokinetic profile of single and multiple ascending oral doses of surotomycin in healthy volunteers (29); a randomized, controlled, double-blind, non-inferiority, multicentre, phase II trial to evaluate the clinical response at end of treatment (30); and a double-blind, randomized, placebo controlled, multiple-dose phase I trial to evaluate the impacts of ascending doses of surotomycin on major organism groups in the gut microbiota of healthy volunteers (31).
Regarding ridinilazole, our literature search identified 10 studies, 2 of them were clinical trials: a randomized, double-blind, active-controlled, non-inferiority, phase II trial comparing the sustained clinical response (defined as clinical cure at the end of treatment and no recurrence within 30 days) of ridinilazole and vancomycin (32) and a double-blind, randomized, placebo-controlled, phase I trial to assess safety and tolerability of single and multiple oral doses of ridinilazole in healthy volunteers (33).
For LFF571, our literature search identified 16 studies, 3 of them were clinical trials: 2 multicenter, randomized, evaluator-blind, active-controlled, parallel-group, phase II trials to compare safety, efficacy and pharmacokinetics of LFF571 to those of vancomycin CDI (34, 35) and a randomized, double-blind, placebo-controlled, single- and multiple-ascending oral dose, phase I trial to determine the safety, tolerability, and pharmacokinetics of LFF571 in healthy subjects (36).
The literature search on CRS3123 and ramoplanin identified 10 and 28 studies, respectively.
For CRS3123, one clinical trial was identified, a double-blind, placebo-controlled, dose escalation, study to evaluate the safety and systemic exposure of CRS3123 after a single oral dose (37).
No clinical trials were identified for ramoplanin.
For fusidic acid, our literature search identified 40 studies, 2 of them were clinical trials: a randomized controlled, double-blind, phase III trial to compare the efficacy of fusidic acid and metronidazole for the treatment of patients with a first CDI episode (38) and a randomized controlled, double-blind, phase III trial to evaluate culture positivity for CD, development of resistance and association with treatment failure or recurrence of CDI after fusidic acid or metronidazole treatment (39).
For nitazoxanide and rifampin, 54 and 44 studies were identified, respectively.
For nitazoxanide, three trials were identified: a double-blind, randomized, controlled phase III trial to compare nitazoxanide with vancomycin for treatment of CDI (40); a preliminary, uncontrolled, open-label phase II study to evaluate response to nitazoxanide treatment in patients with CDI who failed a previous metronidazole treatment (41); and a randomized, double-blind, phase III trial to compare nitazoxanide to metronidazole for the treatment of CDI (42).
For rifampin, a clinical trial was identified; it was a prospective, randomized, single-blinded trial to compare therapy with metronidazole alone versus therapy with metronidazole and rifampin for 10 days to treat primary CDI (43).
The literature search on rifaximin identified 110 studies, 1 of them was a clinical trial. This trial was a randomized, double-blind, placebo-controlled, single-center pilot study to assess rates of recurrent diarrhea in patients with CDI who received rifaximin versus placebo immediately after the standard therapy (44).
The literature search on tigecycline identified 78 studies, none of them was a clinical trial.
The literature search on NVB302, thuricin CD, and lacticin 3147 identified 3, 8, and 3 studies, respectively.
The literature search on auranofin and acyldepsipeptide antimicrobials identified two and one study, respectively. No clinical trials were identified by the literature search on NVB302, thuricin CD, lacticin 3147, auranofin, and acyldepsipeptide antimicrobials.
Table 1 shows the main characteristics of the novel antimicrobials in development for CD, included their inhibition of CD sporulation and toxins production.
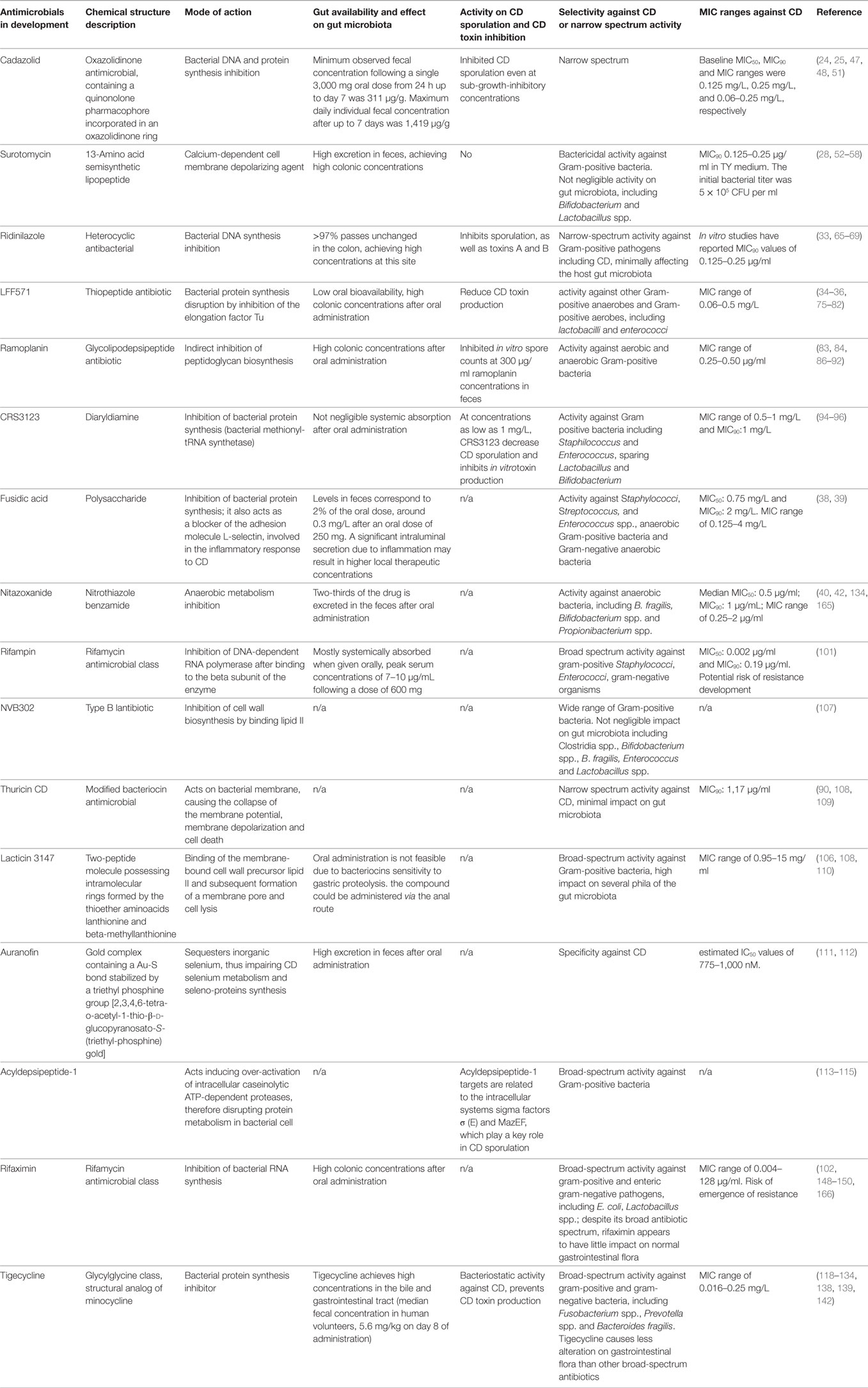
Table 1. Main characteristics and activity on CD spore and toxins production of the novel antimicrobials in development for CD.
Table 2 shows the phase of the latest clinical trials for the novel antimicrobial in development for CD.
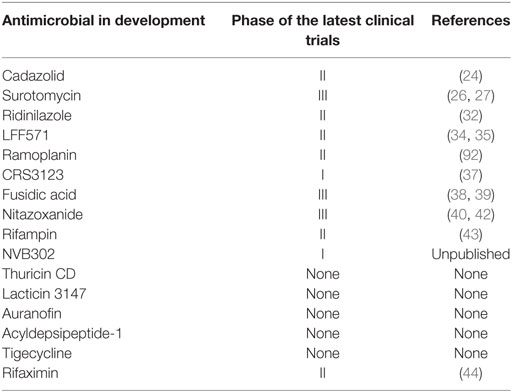
Table 2. Phase of the latest clinical trials for the novel antimicrobial in development for CD.
The main features of the novel anti-CDI molecules are described below.
Cadazolid
Cadazolid, formerly known as ACT-179811, is a novel oxazolidinone antimicrobial characterized by a chemical structure containing a quinonolone pharmacophore incorporated in an oxazolidinone ring (45). The systemic bioavailability of cadazolid is negligible, as well as its absorption from the intestine following oral administration (46). The exact mechanism of action of cadazolid is multifaceted and not still fully elucidated; it has been shown that the quinolone pharmacophore of the cadazolid molecule inhibits both DNA and protein synthesis in the bacterial cell (45, 47). Upon administration, this agent leads to impaired bacterial protein synthesis and consequently to bacterial cell death.
Recently, Locher et al. adopted a macromolecular labeling assay to investigate the site of action of cadazolid in the bacterial cell (48). By monitoring cadazolid incorporation of labeled macromolecules, authors elucidated the cadazolid inhibitory action on the bacterial cell wall synthesis (48). Authors also demonstrated cadazolid influence on bacterial transcription and translation, by means of cell-free coupled transcription/translation assays (48).
Since cadazolid demonstrated to be highly active against CD in vitro as well as in gut and animal models, it has been proposed for the treatment of CDI (24, 49).
Interestingly, in contrast with its wide range of inhibitory effects on bacterial synthesis processes, in a human gut model, cadazolid demonstrated a narrow spectrum antimicrobial activity (50). In this model, cadazolid eliminated CD cells while having a very limited impact on the normal gut microbiota (50).
Cadazolid also showed potent biological effects on CD toxin and spore formation (45, 47, 48).
In an in vitro study comparing vancomycin and cadazolid effect on CD, whereas vancomycin failed to inhibit spore formation, cadazolid markedly inhibited CD sporulation even at sub-growth inhibitory concentrations (25, 49).
The narrow spectrum activity against CD, together with the ability to prevent sporulation, suggests that cadazolid has the potential to reduce CDI recurrence (25).
A single-center, open-label, single oral dose phase I study showed that cadazolid was well tolerated in patients with severe CDI following a single dose of 3,000 mg. Systemic exposure was very low and concentrations in feces were not only very high at peak but remained elevated for several days after single-dose administration (25).
The expectations on this compound have been also supported by a phase II trial reporting lower recurrence rates and higher sustained clinical response rates in patients with CDI treated with cadazolid as compared to those treated with vancomycin (24).
Preclinical and early clinical studies are therefore promising and demonstrate that cadazolid may be an effective option for the treatment of CDI. The results from the ongoing phase III trial will better define the role of cadazolid for the future CDI treatment.
Surotomycin
Surotomycin, previously known as CB-183315, is an orally administered, water-soluble, 13-amino acid semisynthetic lipopeptide (51–56). It was originally obtained from daptomycin after a two-step process of enzymatic cleavage of the decanoyl side chain and its substitution with the (E)-3-(4-pentylphenyl)-but-2-enoyl residue in its molecular structure (56).
Not surprisingly, surotomycin mechanism of action is similar to daptomycin, it acts as a calcium-dependent cell membrane depolarizing agent (31).
Similarly to daptomycin, surotomycin possesses bactericidal activity against Gram-positive bacteria (57). The impact of surotomycin on gut microbiota is not negligible, with bactericidal activity against Gram-positive microbiota components including Bifidobacterium and Lactobacillus spp. but limited effect on Gram-negative species, including Bacteroides (57). An in vitro gut model confirmed limited effects on Bacteroides fragilis after surotomycin administration in comparison with vancomycin (58).
Surotomycin has shown to be effective in CDI animal models (59). A study reported similar survival rate of CD infected hamsters treated with surotomycin or with orally administered vancomycin (59).
Surotomycin proved its efficacy also in human gut models (57, 58, 60, 61).
In summary, evidences from in vitro and animal studies on surotomycin showed a potent in vitro effect on CD and an efficacy to treat CDI similar to that of vancomycin; however, a more sparing effect on gut microbiota has been reported for surotomycin (58, 62).
Preliminary studies on orally administered surotomycinin in humans reported a minimal systemic bioavailability (<1%) and a high excretion in feces, achieving high colonic concentrations (29). Minimal systemic effects have been observed during phase I trials after oral administration (29, 57).
A randomized, double-blind, multicenter phase II trial including 209 CDI patients who received either surotomycin 125 or 250 twice a day for 10 days or vancomycin 125 mg four times daily for 10 days was performed (30). Cure rates at the end of treatment were similar for surotomycin and vancomycin, while recurrence rates and sustained cure rates were higher for surotomycin than for vancomycin (30).
Therefore, two parallel phase III clinical trials were started to demonstrate surotomycin non-inferiority to vancomycin for resolving CDI and surotomycin superiority in preventing recurrence (26, 27).
Unfortunately, the first phase III trial evaluating surotomycin 250 mg twice a day efficacy against CDI in comparison with vancomycin 125 mg four times a day did not meet the study endpoints (27).
Furthermore, a parallel phase III trial was conducted with the primary objective to demonstrate the non-inferiority of surotomycin versus vancomycin in response rates at the end of treatment in adults with CDI. As secondary objectives, this trial aimed to assess clinical response over time and sustained clinical response superiority of surotomycin compared with vancomycin (26). This trial randomized 608 patients to receive twice daily surotomycin 250 mg for 10 days or vancomycin 125 mg four times daily for 10 days (26).
The primary endpoint of the trial was met, and surotomycin demonstrated non-inferiority to vancomycin for the treatment of adults with CDI (clinical response rate at the end of treatment: 83.4% vs. 82.1%) (26). However, surotomycin failed to demonstrate a significant benefit over the existing vancomycin therapy (26).
In conclusion, although surotomycin was generally well tolerated during the conduction of the phase III trials, the published results make doubtful that surotomycin will be introduced for the treatment of CDI (63).
Ridinilazole
Ridinilazole, formerly known as SMT19969, is a new narrow-spectrum synthetic antibiotic (64, 65). The mechanism of action of this novel class of heterocyclic antibacterials has not been fully elucidated, but we know that ridinilazole inhibits DNA bacterial synthesis (64). Moreover, Bassères et al. demonstrated that ridinilazole induces CD cell elongation while inhibiting sporulation, in contrast to other traditional anti-CD antimicrobials (66).
Pharmacokinetics of ridinilazole appears ideal for the treatment of CDI, as it is non-systemically absorbable after oral administration and more than 97% of the antimicrobial passes unchanged in the colon, achieving high concentrations at this site (33).
Ridinilazole showed a narrow-spectrum activity against Gram-positive pathogens, including CD and it minimally affects the host gut microbiota (67–69).
Interestingly, ridinilazole specifically inhibits the growth of CD and has no effect on other Clostridia species; this could have a beneficial effect on the imbalance between CD and Clostridia scindens that has been demonstrated to have an important role in CDI development (67).
Clostridia scindens is one of the few bacterial species able to convert primary bile salts into secondary bile salts in the human gut. A bile acid-dependent, C. scindens-mediated CDI inhibition model has been recently hypothesized (70–72). According to this model, microbiota-mediated modification of bile acids contributes to host resistance to intestinal pathogens such as CD (70–72). In health persons, a microbial network in the gut provides resistance against CDI; exposure to broad-spectrum antibiotics leads to intestinal microflora disruption, including a reduction in C. scindens population. This imbalance between CD and C. scindens gut colonization prevents the metabolism of bile acids increasing the ratio between primary and secondary bile acids, this in turn facilitates CD germination and overgrowth (70–73). Among commensal microbiota components, C. scindens is one of the bacterial species having the ability to convert primary bile salts into secondary bile salts, promoting inhibition of CD vegetative growth. The specific activity of ridinilazole against CD, sparing other clostridia species, may be therefore a promising feature.
In preclinical studies adopting the CD-infected hamster model, ridinilazole has shown to be effective, with an observed survival rate similar to that of vancomycin and fidaxomicin (68, 69).
After the completion of a phase I safety study demonstrating that the compound is well tolerated when assumed orally at a dosage up to 2 g a day over 10 days (33), a phase II double-blind trial (CoDIFy) randomized 100 CDI patients to receive either ridinilazole 200 mg bid or vancomycin 125 mg qid (32).
The trial results demonstrated ridinilazole superiority in achieving response rates at the end of treatment (77.8% and 69.7% for ridinilazole and vancomycin, respectively), in reducing rates of recurrent CDI (14.3% and 34.8% for ridinilazole and vancomycin, respectively) and in obtaining sustained clinical responses (66.7% and 42.4% for ridinilazole and vancomycin, respectively) (32). These results sound promising and support larger phase III clinical trials.
The results from a recently completed randomized trial (NCT02784002) comparing ridinilazole to fidaxomicin for the treatment of CDI have not yet been published (74).
LFF571
LFF571 is a novel semi-synthetic cyclic lipopeptide antibiotic derived from a natural metabolite produced by the actinomycete Planobisporarosea (75, 76). It belongs to a new class of thiopeptide antibiotics and acts disrupting bacterial protein synthesis by inhibiting the elongation factor Tu (EF-Tu), a bacterial factor involved in peptide synthesis (75–78).
LFF571 showed interesting pharmacokinetics features for the treatment of gastrointestinal infections, as it possesses low oral bioavailability and reaches high colonic concentrations (35, 36).
In vitro studies demonstrated LFF571 activity against CD, with a minimal inhibitory concentration (MIC) (79–82). LFF571 also possesses activity against other Gram-positive anaerobes and some Gram-positive aerobes, including lactobacilli and enterococci (79–81).
A study performed in the CDI hamster models showed that LFF571 administration prevented CDI associated mortality (82).
Subsequently, a placebo-controlled phase I trial assessing LFF571 safety and tolerability demonstrated that a single LFF571 dose up to 1,000 mg and repeated doses up to 200 mg four times a day for 10 days were safe and well tolerated in healthy volunteers (36).
Finally, in 2015, a phase II trial has been carried out to compare LFF571 and vancomycin safety and efficacy (34). This evaluator-blind trial randomized 72 patients with moderate severity CDI to receive a 10 days course of either LFF571 200 mg four times daily or vancomycin 125 mg four times daily (34). The trial results showed higher clinical response rates at the end of treatment with LFF571 (90.6% vs. 78.3%), unfortunately also higher recurrence rates were reported with LFF571 (37% vs 31%) (34).
No further phase II or phase III trials for LFF571 have been reported so far.
Ramoplanin
Ramoplanin is a glycolipodepsipeptide antimicrobial that exerts its mechanism of action preventing cell wall peptidoglycan biosynthesis (83). More precisely, ramoplanin indirectly inhibits the transglycosylases responsible for peptidoglycan biosynthesis by sequestering their intermediate substrate Lipid II at the interface between the extracellular environment and the bacterial membrane (83). Binding to the key intermediate moiety lipid II, ramoplanin leads to the disruption of bacterial wall and therefore bacterial death (83, 84).
This compound is non-absorbable orally and achieves high colonic concentrations (85).
Ramoplanin has activity against both aerobic and anaerobic Gram-positive bacteria, including vancomycin-resistant Enterococcus (86, 87).
Regarding its activity against CD, an in vitro model clearly showed that ramoplanin molecule can bind CD spores and also kills vegetative CD cell with high efficacy (87–91).
Ramoplanin activity against CDI has been demonstrated also in animal models (86).
Interestingly, ramoplanin superiority over vancomycin in reducing CD sporulation and spore release has been demonstrated in animal model showing that CD spores were less often recovered from the ramoplanin-treated hamsters as compared to those treated with vancomycin (86).
Subsequently, in a phase II trial, 86 CDI patients were randomized to receive either ramoplanin, 200 mg twice daily or 400 mg twice daily, or vancomycin, 125 mg four times daily for 10 days. The two arms receiving ramoplanin showed similar clinical cure rates at the end of treatment (83% and 85%, respectively, in comparison to 86% of vancomycin), but also higher rate of adverse events, with nausea, vomiting, and diarrhea as the most frequently reported adverse events in the ramoplanin arms (85, 92).
Nonetheless, in the light of the promising features of this drug, a phase III trial on ramoplanin against CDI has been planned and has been recently approved by the FDA (85, 92, 93).
CRS3123
CRS3123, formerly known as REP3123, is a recently developed fully synthetic diaryldiamine antimicrobial (93).
CRS3123 prevents both CD growth and CD spore production by inhibiting bacterial protein synthesis (85, 94, 95). Interestingly, this antimicrobial acts on the bacterial methionyl-tRNA synthetase, has limited effect on the structurally distinct methionyl-tRNA synthetases of Gram-negative bacteria and humans (85, 94, 95), and possesses activity against CD and other Gram positive bacteria including Staphylococcus and Enterococcus. However, CRS3123 is inactive against major intestinal Gram-positive colonizers, including Lactobacillus and Bifidobacterium (95, 96).
Following the evidences of CRS3123 efficacy for CDI treatment obtained from the hamster model (94), the results from a phase I study to assess the safety, tolerability, and systemic exposure of escalating doses of CRS3123 in humans have been recently published (37).
In this single-center, double-blind, placebo-controlled phase I trial, escalating doses of CRS3123 were administered orally to the study participants. The study enrolled 40 participants randomized to receive study product or placebo, with a CRS3123 dose range of 100, 200, 400, 800, and 1,200 mg (37).
Reported adverse events were similar in severity and frequency for participants who received active drug and for those who received placebo, and all the adverse events in the study drug group were mild or moderate (37).
The bioavailability of CRS3123 following oral administration could not be accurately assessed during this trial, because of the absence of standards for the metabolites. Nonetheless, a not negligible fraction of the administered oral dose of CRS3123 was detected systemically (37).
Phase II trials are urgently needed in order to assess CRS3123 efficacy for CDI in humans.
Fusidic Acid
Fusidic acid is a relatively well-known antimicrobial belonging to the class of polysaccharides and originally developed from the fungus Fusidium coccineum (93).
Fusidic acid works by interfering with bacterial protein synthesis, by preventing the translocation of the elongation factor G from the ribosome (93). Of interest for its implication in CDI treatment, fusidic acid also acts as a blocker of L-selectin, an adhesion molecule involved in the inflammatory response to CD (93).
Concerning the economic cost for a course of CDI treatment, fusidic acid may represent one of the cheapest options in US, with a cost similar to metronidazole (38).
So far, a phase III trial by Wullt et al. has been published on fusidic acid for CDI treatment (38, 39). The authors performed a prospective, randomized-controlled, double-blind trial on 131 patients to compare the efficacy of fusidic acid 250 mg orally three times to that of metronidazole 400 mg orally three times daily for 7 days for initial CDI episodes (38). In the fusidic acid group, clinical cure at the end of treatment was achieved in 83% of patients in comparison to 93% in the metronidazole group (P = 0.116) (38). Clinical CDI recurrence was described in 27% and 29% of patients receiving fusidic acid and metronidazole, respectively (39).
Of note, in the group of patients treated with fusidic, the emergence of fusidic acid resistance was reported in the 55% of the CD infecting strain (39). The mechanisms of fusidic acid resistance in CD are still unknown but the emerging fusidic acid resistant CD strains may easily be transmitted between patients, hampering any future wide-spread use of fucidic acid for CDI (39). Nevertheless, fusidic acid monotherapy may still represent a possible future option for selected settings, i.e., units with a high rate of vancomycin-resistant enterococci intestinal colonization or with CDI patients who can not tolerate the standard CDI treatment (39).
At present, there are no further phase III trials on the development of fusidic acid as anti-CDI treatment (93).
Nitazoxanide
Nitazoxanide is a nitrothiazole benzamide that was originally approved as an anti-parasitic drug, but over time, it also showed activity against bacterial enteric pathogens, including CD (97).
After oral administration, nitazoxanide is mostly excreted in feces, and it has been well tolerated in the studies performed in humans, with no reports of serious adverse events, although rare occurrence of elevated creatinine levels and alanine aminotransferase in serum has been reported (98, 99).
So far, clinical trials have compared nitazoxanide use for CDI with both vancomycin and metronidazole.
A preliminary, uncontrolled, open-label phase II study on 35 CDI patients who failed metronidazole treatment for a first CDI episode or who had recurrent CDI assessed a 74% response rates after a 10-day nitazoxanide treatment, but also a 33% recurrence rate (41).
Subsequently, in a phase III, double-blind trial, 110 primary CDI patients were randomized to receive a 10-day treatment course with nitazoxanide or metronidazole, reporting similar clinical response rate at the end of treatment (89.5% and 82.4% for nitazoxanide and metronidazole, respectively) and similar CDI recurrence rates (13.9% and 24% for nitazoxanide and metronidazole, respectively) (42).
Regarding nitazoxanide comparison with vancomycin, a prospective, double-blind randomized trial was conducted in CDI patients (40). The trial compared the efficacy of 10 days of oral nitazoxanide therapy versus 10 days of oral vancomycin in CDI patients (40). Unfortunately, this trial was prematurely stopped for unclear reasons, with a total number of 49 enrolled patients. Even if similar clinical cure rates at the end of treatment (77% for nitazoxanide and 74% for vancomycin) and recurrence rates (5% for nitazoxanide and 7% for vancomycin) were observed, these results did not reach statistical significance (40).
However, nitazoxanide still shows promising features as a future treatment of CDI, and recently its use for severe recurrent cases of CDI has been reported (100).
Rifampin
First synthesized in 1965, rifampin is a well-tolerated antimicrobial compound which exerts its activity by inhibiting bacterial RNA synthesis (101). Although rifampicin cannot be considered a novel antimicrobial, over time its activity against CD leaded to consider its use for CDI treatment (102, 103).
However, in a phase II trial testing rifampin in association with metronidazole, similar initial cure rates were found between rifampin plus metronidazole versus metronidazole alone (63% and 65%, respectively), and the study was ended prematurely because of the non superiority of the combination therapy (43).
A different phase III trial was begun in 2008 and was recently completed (NCT00182429), but no results have been reported so far (93).
Moreover, beside all the uncertainties on its efficacy as a single agent against CDI, the emergence of resistance to rifampin represents a further matter of concern (104, 105).
NVB302
The antimicrobial NVB302 was first isolated from Actinoplanes liguriae (93, 106).
After promising pre-clinical studies in human gut models (107), no results have been published from phase I trials, and no development of phase II or III trials has been reported (106, 107).
Thuricin CD
Thuricin CD is a recently developed, modified bacteriocin antimicrobial, which exhibited excellent narrow spectrum activity against CD (108, 109).
This compound is showing promising features; interestingly, in a human gut research model, it displayed a minimal impact on gut microbiota, sparing Firmicutes, Bacteroides, and Proteobacteria in comparison with traditional anti-CD antibiotics (109).
At present no trials have been conducted on Thuricin CD, and its safety and efficacy for CDI are still to be demonstrated (106).
Lacticin 3147
Lacticin 3147 is a small 2-peptide molecule “lantibiotic” synthesized by Lactococcus lactis (110). Even if it has been demonstrated a high activity of this molecule against CD, its broad-spectrum antimicrobial activity against Gram-positive bacteria and its high impact on several phila of the gut microbiota make unlikely lacticin 3147 future use for CDI treatment (106, 108, 110).
Auranofin
Auranofin is a molecule originally approved for the treatment of rheumatoid arthritis (111). This compound gained attention for a possible treatment in CDI because of its potent in vitro inhibitory activity of CD and its considerable excretion in feces after oral administration (111).
Auranofin acts on CD by sequestering inorganic selenium, thus impairing CD selenium metabolism and seleno-proteins synthesis, essential for the bacterium survival (112).
Interestingly, this mechanism of action should confer to auranofin specificity or quasi-specificity against CD.
At present, despite these promising features, no clinical trials have been published on auranofin use for CDI.
ADEP-1
ADEP-1 (acyldepsipeptide-1) is a novel bactericidal antimicrobial molecule belonging to the acyldepsipeptide antibiotic class (113). These natural antibiotics are originated from the soil bacteria Streptomycetes (113). More precisely, ADEP-1 originates from the bacteria Streptomyces hawaiiensis and showed a potent activity against Gram-positive bacteria (114).
As other acyldepsipeptide antibiotics, ADEP-1 acts disrupting the protein metabolism in bacterial cell, by inducing over-activation of intracellular caseinolytic ATP-dependent proteases (114). Subsequently, the intracellular protein metabolism derangement causes cell division, differentiation, and sporulation impairment, finally leading to bacterial cell death (114, 115).
Interestingly, it has been shown that the CD proteases targeted by acyldepsipeptide antimicrobials are also related to the intracellular systems sigma factors σ (E) and MazEF, which play a key role in CD sporulation (114, 116).
Recently, starting from the consideration of ADEP-1 Gram-positive bactericidal activity and the presence in CD of caseinolytic proteases, potential target of this compound, some authors proposed the use of ADEP-1 and other acyldepsipeptide antimicrobials for the treatment of CDI, alone or in combination with other antimicrobial classes (114).
Even if these authors highlighted the lack of specificity against CD of this compound, in our opinion, the promising feature of ADEP-1, including its bactericidal activity and its potential effect on CD sporulation, urges the design of pre-clinical studies on this molecule.
At present, there are no ongoing clinical trials on acyldepsipeptide antibiotics for CDI treatment.
Tigecycline
Tigecycline is a broad-spectrum antimicrobial of the glycylglycine class (117). It acts as a protein synthesis inhibitor, with activity against Gram-positive, Gram-negative and anaerobe bacteria, including Fusobacterium spp., Prevotella spp., Porphyromonas spp., and Bacteroides fragilis (117, 118).
Tigecycline has been approved for treatment of complicated skin infections and complicated intra-abdominal infections (119).
Tigecycline is not registered for use in CDI; however, it exerts a bacteriostatic activity against CD (119–121).
Importantly, tigecycline achieves high concentrations in the bile and gastrointestinal tract after intravenous administration, with a median fecal concentration in human volunteers of 5.6 mg/kg after 8 days, and has been proposed as an alternative agent for the treatment of CDI in humans (122).
A large pan-European study, conducted between 2011 and 2014 across 22 European countries, reported that tigecycline had in vitro activity against all the CD isolates (2,830) tested for tigecycline susceptibility, with a mean MIC of 0.04 mg/L (123).
Moreover, multi-drug resistant CD strains have been found susceptible to tigecycline, showing a MIC range from 0.016 to 0.25 mg/L (118, 123–135).
Therefore, tigecycline represents a potential antibiotic treatment for CDI (118, 125, 135–137).
Interestingly, the effects of tigecycline on sporulation and toxin production have been evaluated by means of in vitro experiments and animal models, demonstrating that tigecycline is effective in preventing CD overgrowth and CD toxin production (118, 128, 129, 138).
Regarding the impact on gut microbiota, there is evidence that tigecycline causes a significant alteration of the microbiota, including a reduction of Bacteroides spp (118, 132, 139–141). However, Jump et al. demonstrated that, in comparison to other broad-spectrum antibiotics, the use of tigecycline is associated with a lower risk of alteration of colonization resistance to CD (142). In the study, levels of bacterial metabolites in fecal specimens from a mouse model were measured, showing that tigecycline treatment caused a less profound alteration of fecal metabolites in comparison to linezolid, piperacillin/tazobactam, and ceftriaxone (142).
A retrospective cohort study comparing 45 patients receiving tigecycline monotherapy to 45 patients receiving standard treatment for CDI, reported higher clinical cure rates with tigecycline than with vancomycin and metronidazole (143).
However, retrospective studies evaluating the efficacy of tigecycline adjunctive therapy for CDI reported conflicting results (143–147).
Moreover, three retrospective cohort studies failed to demonstrate a beneficial effect of adjunctive tigecycline CDI treatment on patients outcome in comparison to standard treatment alone (145–147).
A phase II trial started in 2011 could have been able to elucidate the role of tigecycline for CDI treatment but unfortunately was discontinued because of a too slow enrollment rate (NCT01401023) (119).
No further phase II or III trials have been started so far (93, 119).
Therefore, randomized controlled trials are needed to fully elucidate tigecycline efficacy and safety for the management of CDI.
A Room for Preventing CDI Recurrence
Rifaximin
Rifaximin is a well-tolerated antimicrobial compound which belongs to the rifamycin class. Similarly to rifampin, rifaximin acts inhibiting bacterial RNA synthesis (117, 118, 148, 149).
Interestingly, rifaximin is minimally absorbed after oral administration, achieving high colonic concentrations (148–150).
Therefore, rifaximin shares with rifampin several interesting features, i.e., a potent activity against CD, but also several drawbacks, including the high risk of emergence of resistance (93, 151).
It was introduced for the treatment of traveler’s diarrhea, rifaximin has also been proposed for CDI treatment (148–150).
Initially, rifaximin has been tested as a “chaser” to augment vancomycin efficacy in the CDI treatment (148–150), however, rifaximin ability to spare enteric microbiota makes it a potentially useful agent for the prevention of CDI recurrence after a first CDI episode.
On this issue, three case series have investigated the use of rifaximin to prevent CDI recurrence so far (152–154).
The first case series reported the administration of a 2-week regimen of rifaximin immediately after the vancomycin standard treatment in eight CDI patients with multiple recurrences (152). During the follow-up after this protocol, only one out of the eight patients experienced CDI (152).
In a report by Garey et al, five patients with multiple CDI recurrences received a rifaximin tapering regimen for 4 weeks (154). None of these subjects experienced CDI recurrence after the rifaximin protocol during a follow-up of at least 54 days (154).
Afterward, a phase II, double-blind, randomized placebo-controlled study was carried out to assess rifaximin efficacy in preventing recurrences in 68 CDI patients (44). Immediately after the standard metronidazole or vancomycin based therapy, the treatment arm received rifaximin at the dose of 1.2 g daily for 20 days (44). The observed rate of recurrence resulted lower in the rifaximin arm in comparison with the placebo (15% vs. 31% of recurrence, respectively), but unfortunately the findings of the study did not reach a significant difference (44).
A new trial to evaluate a rifaximin tapering regimen in a larger cohort of CDI recurrences patients has been recently completed, and results have yet to be published (NCT01670149, available from http://clinicaltrials.gov).
Discussion and Conclusion
In comparison with the traditional anti-CDI antimicrobial treatment, some novel antimicrobials reviewed in this study offer several advantages.
The favorable pharmacokinetic and pharmacodynamic profile, the narrow-spectrum activity and the specificity against CD that implicate a low impact on the gut microbiota composition, and the inhibitory activity on CD sporulation and toxins production are among the most promising features of these compounds.
First, the potential capacity of the new medication to reduce CDI recurrences preserving the human gut microbiota is of major importance.
In fact, beside well-known risk factors for the development of CDI, such as decreased stomach acidity, advanced age, renal disease, and other comorbidities, it has been recently demonstrated that the disruption of the intestinal microbiota is a key factor for CDI development (155–158). Microbiota disruption facilitates CD germination and overgrowth and therefore CDI development (155–158).
Previous studies have demonstrated that the microbiota of CDI patients is characterized by a decrease in species richness, with an association between loss of Bacterioides, Lachnospiraceae, and Ruminococcaceae and the development of CDI (156–158).
A broad spectrum antibiotic treatment given for concomitant infection, usually inactive against CD but very active against bacterial intestinal colonizers, represents one of the main causes of microbiota disruption.
Importantly, also the traditional anti-CD antimicrobial compounds may negatively impact on the indigenous gut flora, paradoxically facilitating the imbalance between “protective” enteric pathogens and CD (159).
In fact, metronidazole possesses bactericidal activity against the protective anaerobic philia of Bifidobacterium and Bacteroides (159); vancomycin has remarkable activity against Enterococcus spp. and Bacteroides (160, 161).
Also fidaxomicin, even if may be considered an advancement in terms of specificity against CD in comparison with metronidazole and vancomycin, has shown relevant activity against Bacillus spp., Enterococcus spp., Lactobacilli, and Bifidobacterium (162, 163).
The imbalance determined in the gut flora mediated by these traditional first-line anti-CD antimicrobials can subsequently facilitate CDI recurrence (159).
Interestingly, it has been observed that the period of major vulnerability for CDI recurrence starts from the end of the traditional anti-CDI treatment, when sub-inhibitory levels of CDI antibiotics are still present in the human gut and further contribute to a misbalance in the gut microflora (157, 163, 164). Therefore, despite the administration of these traditional antimicrobials, CD spores can survive in the gut and be facilitated to germinate by the microbiota disruption, leading to recurrent CDI and trapping the patient in a vicious “recurrent CDI cycle” (157, 163, 164).
In the light of these considerations, the potential narrow-spectrum activity and the specificity against CD showed by some novel antimicrobials in development for CDI appears of crucial importance, allowing to preserve microbiota integrity and thus reducing CDI recurrences.
The novel antimicrobials that better fit with this characteristic are cadazolid, surotomycin, ridinilazole, auranofin, and thuricin CD.
Second, the ecological impact of the new antimicrobials against CD has to be highlighted.
The activity against CD spores and the ability to prevent CD sporulation displayed by several novel antimicrobials suggest that these compounds may not only reduce CDI recurrence rate, but may also potentially reduce CD environment persistence, thus reducing CD spread in the hospital setting and CDI healthcare associated acquisition.
Cadazolid, ridinilazole, CRS3123, ramoplanin, and, potentially, the acyldepsipeptide antimicrobial are, among the antimicrobial reviewed in this paper, the most active compounds in reducing spore production (Table 1).
Along with the potential benefit of new anti-CDI antimicrobials, it is important to emphasize that some new molecules present limitations that could affect the probability of their approval for the CDI treatment.
Main drawbacks include the occurrence of CD resistant strains for rifampicin and fusidic acid; the failure to achieve a significant benefit over the existing traditional antimicrobial CDI treatment for surotomycin; the failure to reduce the CDI recurrence rate at the end of treatment for LFF571, and the relatively high impact on several phila of the gut microbiota for ramoplanin.
In conclusion, the novel antimicrobial molecules under development for CDI present promising key features and advancements in comparison to the traditional anti-CDI antimicrobials. Hopefully, in the near future, these new molecules will be effective alternatives to fight and prevent CDI, a condition which may actually represent a real “spiral of disease.”
Author Contributions
All authors participated in writing the manuscript, revising it, and gave the final approval for publishing the manuscript.
Conflict of Interest Statement
GG and MC declare no conflicts of interest. NP received consulting fees from MSD, Pfizer, Astellas, Zambon, Angelini, Gilead, Achaogen, The Medicine Company, 3 M, Beckton & Dickinson.
Funding
This work was funded by Research of National Center for Diseases Prevention and Control (CCM), Italian Ministry of Health.
References
1. CDC. Annual Report for the Emerging Infections Program for Clostridium difficile Infection. (2013). Available from: https://www.cdc.gov/hai/eip/pdf/2013-Annual-Report-12-8-2015.pdf (Accessed: March 30, 2018).
2. European Centre for Disease Prevention and Control. Point Prevalence Survey of Healthcare-Associated Infections and Antimicrobial Use in European Acute Care Hospitals. Stockholm: ECDC (2013).
3. Bartlett JG, Gerding DN. Clinical recognition and diagnosis of Clostridium difficile infection. Clin Infect Dis (2008) 46:S12–8. doi:10.1086/521863
4. Evans CT, Safdar N. Current trends in the epidemiology and outcomes of Clostridium difficile infection. Clin Infect Dis (2015) 60:S66–71. doi:10.1093/cid/civ140
5. Magill SS, Edwards JR, Bamberg W, Beldavs ZG, Dumyati G, Kainer MA, et al. Multistate point-prevalence survey of health care-associated infections. N Engl J Med (2014) 370(13):1198–208. doi:10.1056/NEJMoa1306801
6. Pepin J, Alary ME, Valiquette L, Raiche E, Ruel J, Fulop K, et al. Increasing risk of relapse after treatment of Clostridium difficile colitis in Quebec, Canada. Clin Infect Dis (2005) 40:1591–7. doi:10.1086/430315
7. Sheitoyan-Pesant C, Abou Chakra CN, Pepin J, Marcil-Heguy A, Nault V, Valiquette L. Clinical and healthcare burden of multiple recurrences of Clostridium difficile infection. Clin Infect Dis (2016) 62:574–80. doi:10.1093/cid/civ958
8. Abou Chakra CN, Pépin J, Sirard S, Valiquette L. Risk factors for recurrence, complications and mortality in Clostridium difficile infection: a systematic review. PLoS One (2014) 9(6):e98400. doi:10.1371/journal.pone.0098400
9. Brandt LJ, Aroniadis OC, Mellow M, Kanatzar A, Kelly C, Park T, et al. Long-term follow-up of colonoscopic fecal microbiota transplant for recurrent Clostridium difficile infection. Am J Gastroenterol (2017) 107(7):1079–87. doi:10.1038/ajg.2012.60
10. Rodriguez C, Taminiau B, Van Broeck J, Delmée M, Daube G. Clostridium difficile infection and intestinal microbiota interactions. Microb Pathog (2015) 89:201–9. doi:10.1016/j.micpath.2015.10.018
11. Davies KA, Ashwin H, Longshaw CM, Burns DA, Davis GL, Wilcox MH, et al. Diversity of Clostridium difficile PCR ribotypes in Europe: results from the European, multicentre, prospective, biannual, point-prevalence study of Clostridium difficile infection in hospitalised patients with diarrhoea (EUCLID), 2012 and 2013. Euro Surveill (2016) 21(29):1–11. doi:10.2807/1560-7917.ES.2016.21.29.30294
12. Bauer MP, Notermans DW, van Benthem BH, Brazier JS, Wilcox MH, Rupnik M, et al. Clostridium difficile infection in Europe: a hospital-based survey. Lancet (2011) 377(9759):63–73. doi:10.1016/S0140-6736(10)61266-4
13. Debast SB, Bauer MP, Kuijper EJ. European society of clinical microbiology and infectious diseases: update of the treatment guidance document for Clostridium difficile infection. Clin Microbiol Infect (2014) 20(Suppl 2):1–26. doi:10.1111/1469-0691.12418
14. Mawer DPC, Eyre DW, Griffiths D, Fawley WN, Martin JSH, Quan TP, et al. Contribution to Clostridium difficile transmission of symptomatic patients with toxigenic strains who are fecal toxin negative. Clin Infect Dis (2017) 64(9):1163–70. doi:10.1093/cid/cix079
15. Hu MY, Katchar K, Kyne L, Maroo S, Tummala S, Dreisbach V, et al. Prospective derivation and validation of a clinical prediction rule for recurrent Clostridium difficile infection. Gastroenterology (2009) 136(4):1206–14. doi:10.1053/j.gastro.2008.12.038
16. Mc Farland LV. Renewed interest in a difficult disease: Clostridium difficile infections—epidemiology and current treatment strategies. Curr Opin Gastroenterol (2009) 25(1):24–35. doi:10.1097/MOG.0b013e32831da7c4
17. Guillemin I, Marrel A, Lambert J, Beriot-Mathiot A, Doucet C, Kazoglou O, et al. Patients’ experience and perception of hospital-treated Clostridium difficile infections: a qualitative study. Patient (2014) 7(1):97–105. doi:10.1007/s40271-013-0043-y
18. Cohen SH, Gerding DN, Johnson S, Kelly CP, Loo VG, McDonald LC, et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol (2010) 31:431–55. doi:10.1086/651706
19. Trubiano JA, Cheng AC, Korman TM, Roder C, Campbell A, May ML, et al. Australasian society of infectious diseases updated guidelines for the management of Clostridium difficile infection in adults and children in Australia and New Zealand. Intern Med J (2016) 46(4):479–93. doi:10.1111/imj.13027
20. Babakhani F, Bouillaut L, Gomez A, Sears P, Nguyen L, Sonenshein AL. Fidaxomicin inhibits spore production in Clostridium difficile. Clin Infect Dis (2012) 55(Suppl 2):S162–9. doi:10.1093/cid/cis453
21. Louie TJ, Emery J, Krulicki W, Byrne B, Mah M. OPT-80 eliminates Clostridium difficile and is sparing of bacteroides species during treatment of C. difficile infection. Antimicrob Agents Chemother (2009) 53(1):261–3. doi:10.1128/AAC.01443-07
22. Biswas JS, Patel A, Otter JA, Wade P, Newsholme W, van Kleef E, et al. Reduction in Clostridium difficile environmental contamination by hospitalized patients treated with fidaxomicin. J Hosp Infect (2015) 90(3):267–70. doi:10.1016/j.jhin.2015.01.015
23. Gerding DN, Hecht DW, Louie T, Nord CE, Talbot GH, Cornely OA, et al. Susceptibility of Clostridium difficile isolates from a phase 2 clinical trial of cadazolid and vancomycin in C. difficile infection. J Antimicrob Chemother (2016) 71(1):213–9. doi:10.1093/jac/dkv300
24. Louie T, Nord CE, Talbot GH, Wilcox M, Gerding DN, Buitrago M, et al. Multicenter, double-blind, randomized, phase 2 study evaluating the novel antibiotic cadazolid in patients with Clostridium difficile infection. Antimicrob Agents Chemother (2015) 59:6266–73. doi:10.1128/AAC.00504-15
25. Gehin M, Desnica B, Dingemanse J. Minimal systemic and high faecal exposure to cadazolid in patients with severe Clostridium difficile infection. Intl J Antimicrob Agents (2015) 46:576–81. doi:10.1016/j.ijantimicag.2015.07.015
26. Daley P, Louie T, Lutz JE, Khanna S, Stoutenburgh U, Jin M, et al. Surotomycin versus vancomycin in adults with Clostridium difficile infection: primary clinical outcomes from the second pivotal, randomized, double-blind, Phase 3 trial. J Antimicrob Chemother (2017) 72(12):3462–70. doi:10.1093/jac/dkx299
27. Boix V, Fedorak RN, Mullane KM, Pesant Y, Stoutenburgh U, Jin M, et al. Primary outcomes from a phase 3, randomized, double-blind, active-controlled trial of surotomycin in subjects with Clostridium difficile infection. Open Forum Infect Dis (2017) 4(1):ofw275. doi:10.1093/ofid/ofw275
28. Cannon K, Byrne B, Happe J, Wu K, Ward L, Chesnel L, et al. Enteric microbiome profiles during a randomized Phase 2 clinical trial of surotomycin versus vancomycin for the treatment of Clostridium difficile infection. J Antimicrob Chemother (2017) 72(12):3453–61. doi:10.1093/jac/dkx318
29. Chandorkar G, Zhan Q, Donovan J, Rege S, Patino H. Pharmacokinetics of surotomycin from phase 1 single and multiple ascending dose studies in healthy volunteers. BMC Pharmacol Toxicol (2017) 18(1):24. doi:10.1186/s40360-017-0123-z
30. Lee CH, Patino H, Stevens C, Rege S, Chesnel L, Louie T, et al. Surotomycin versus vancomycin for Clostridium difficile infection: phase 2, randomized, controlled, double-blind, noninferiority, multicentre trial. J Antimicrob Chemother (2016) 71(10):2964–71. doi:10.1093/jac/dkw246
31. Citron DM, Tyrrell KL, Dale SE, Chesnel L, Goldstein EJ. Impact of surotomycin on the gut microbiota of healthy volunteers in a phase 1 clinical trial. Antimicrob Agents Chemother (2016) 60(4):2069–74. doi:10.1128/AAC.02531-15
32. Vickers RJ, Tillotson GS, Nathan R, Hazan S, Pullman J, Lucasti C, et al. Efficacy and safety of ridinilazole compared with vancomycin for the treatment of Clostridium difficile infection: a phase 2, randomised, double-blind, active-controlled, non-inferiority study. Lancet Infect Dis (2017) 17(7):735–44. doi:10.1016/S1473-3099(17)30235-9
33. Vickers R, Robinson N, Best E, Echols R, Tillotson G, Wilcox M. A randomised phase 1 study to investigate safety, pharmacokinetics and impact on gut microbiota following single and multiple oral doses in healthy male subjects of SMT19969, a novel agent for Clostridium difficile infections. BMC Infect Dis (2015) 15:91. doi:10.1186/s12879-015-0759-5
34. Mullane K, Lee C, Bressler A, Buitrago M, Weiss K, Dabovic K, et al. Multicenter, randomized clinical trial to compare the safety and efficacy of LFF571 and vancomycin for Clostridium difficile infections. Antimicrob Agents Chemother (2015) 59(3):1435–40. doi:10.1128/AAC.04251-14
35. Bhansali SG, Mullane K, Ting LS, Leeds JA, Dabovic K, Praestgaard J, et al. Pharmacokinetics of LFF571 and vancomycin in patients with moderate Clostridium difficile infections. Antimicrob Agents Chemother (2015) 59:1441–5. doi:10.1128/AAC.04252-14
36. Ting LS, Praestgaard J, Grunenberg N, Yang JC, Leeds JA, Pertel P. A first-in-human, randomized, double-blind, placebo-controlled, single- and multiple-ascending oral dose study to assess the safety and tolerability of LFF571 in healthy volunteers. Antimicrob Agents Chemother (2012) 56:5946–51. doi:10.1128/AAC.00867-12
37. Nayak SU, Griffiss JM, Blumer J, O’Riordan MA, Gray W, McKenzie R, et al. Safety, tolerability, systemic exposure, and metabolism of CRS3123, a methionyl-tRNA synthetase inhibitor developed for treatment of Clostridium difficile, in a phase 1 study. Antimicrob Agents Chemother (2017) 61(8):e02760–16. doi:10.1128/AAC.02760-16
38. Wullt M, Odenholt I. A double-blind randomized controlled trial of fusidic acid and metronidazole for treatment of an initial episode of Clostridium difficile-associated dharrhoea. J Antimicrob Chemother (2004) 54:211–6. doi:10.1093/jac/dkh278
39. Norén T, Wullt M, Akerlund T, Bäck E, Odenholt I, Burman LG. Frequent emergence of resistance in Clostridium difficile during treatment of C. difficile-associated diarrhea with fusidic acid. Antimicrob Agents Chemother (2006) 50(9):3028–32. doi:10.1128/AAC.00019-06
40. Musher DM, Logan N, Bressler AM, Johnson DP, Rossignol JF. Nitazoxanide versus vancomycin in Clostridium difficile infection: a randomized, double-blind study. Clin Infect Dis (2009) 48(4):e41–6. doi:10.1086/596552
41. Musher DM, Logan N, Mehendiratta V, Melgarejo NA, Garud S, Hamill RJ. Clostridium difficile colitis that fails conventional metronidazole therapy: response to nitazoxanide. J Antimicrob Chemother (2007) 59:705–10. doi:10.1093/jac/dkl553
42. Musher DM, Logan N, Hamill RJ, Dupont HL, Lentnek A, Gupta A, et al. Nitazoxanide for the treatment of Clostridium difficile colitis. Clin Infect Dis (2006) 43(4):421–7. doi:10.1086/506351
43. Lagrotteria D, Holmes S, Smieja M, Smaill F, Lee C. Prospective, randomized inpatient study of oral metronidazole versus oral metronidazole and rifampin for treatment of primary episode of Clostridium difficile-associated diarrhea. Clin Infect Dis (2006) 43:547–52. doi:10.1086/506354
44. Garey KW, Ghantoji SS, Shah DN, Habib M, Arora V, Jiang ZD, et al. A randomized, double-blind, placebo-controlled pilot study to assess the ability of rifaximin to prevent recurrent diarrhea in patients with Clostridium difficile infection. J Antimicrob Chemother (2011) 66:2850–5. doi:10.1093/jac/dkr377
45. Endres BT, Bassères E, Alam MJ, Garey KW. Cadazolid for the treatment of Clostridium difficile. Expert Opin Investig Drugs (2017) 26(4):509–14. doi:10.1080/13543784.2017.1304538
46. Michalska K, Karpiuk I, Krol M, Tyski S. Recent development of potent analogues of oxazolidinone antibacterial agents. Bioorg Med Chem (2013) 21(3):577–91. doi:10.1016/j.bmc.2012.11.036
47. Locher HH, Caspers P, Bruyère T, Schroeder S, Pfaff P, Knezevic A, et al. Investigations of the mode of action and resistance development of cadazolid, a new antibiotic for treatment of Clostridium difficile infections. Antimicrob Agents Chemother (2014) 58(2):901–8. doi:10.1128/AAC.01831-13
48. Locher HH, Seiler P, Chen X, Schroeder S, Pfaff P, Enderlin M, et al. In vitro and in vivo antibacterial evaluation of cadazolid, a new antibiotic for treatment of Clostridium difficile infections. Antimicrob Agents Chemother (2014) 58(2):892–900. doi:10.1128/AAC.01830-13
49. Chilton CH, Crowther GS, Baines SD, Todhunter SL, Freeman J, Locher HH, et al. In vitro activity of cadazolid against clinically relevant Clostridium difficile isolates and in an in vitro gut model of C. difficile infection. J Antimicrob Chemother (2014) 69(3):697–705. doi:10.1093/jac/dkt411
50. Kali A, Charles MVP, Srirangaraj S. Cadazolid: a new hope in the treatment of Clostridium difficile infection. Australas Med J (2015) 8(8):253–62. doi:10.4066/AMJ.2015.2441
51. Deshpande A, Hurless K, Cadnum JL, Chesnel L, Gao L, Chan L, et al. Effect of surotomycin, a novel cyclic lipopeptide antibiotic, on intestinal colonization with vancomycin-resistant enterococci and Klebsiella pneumoniae in mice. Antimicrob Agents Chemother (2016) 60(6):3333–9. doi:10.1128/AAC.02904-15
52. Adams HM, Li X, Mascio C, Chesnel L, Palmer KL. Mutations associated with reduced surotomycin susceptibility in Clostridium difficile and Enterococcus species. Antimicrob Agents Chemother (2015) 59(7):4139–47. doi:10.1128/AAC.00526-15
53. Bouillaut L, McBride S, Sorg JA, Schmidt DJ, Suarez JM, Tzipori S, et al. Effects of surotomycin on Clostridium difficile viability and toxin production in vitro. Antimicrob Agents Chemother (2015) 59(7):4199–205. doi:10.1128/AAC.00275-15
54. Reigadas E, Alcalá L, Marín M, Pelaéz T, Martin A, Iglesias C, et al. In vitro activity of surotomycin against contemporary clinical isolates of toxigenic Clostridium difficile strains obtained in Spain. J Antimicrob Chemother (2015) 70(8):2311–5. doi:10.1093/jac/dkv092
55. Yin N, Li J, He Y, Herradura P, Pearson A, Mesleh MF, et al. Structure-activity relationship studies of aseries of semisynthetic lipopeptides leading to the discovery of surotomycin, a novel cyclic lipopeptide being developed for thetreatment of Clostridium difficile-associated diarrhea. J Med Chem (2015) 58(12):5137–42. doi:10.1021/acs.jmedchem.5b00366
56. Snydman DR, Jacobus NV, Mcdermott LA. Activity of a novel cycliclipopeptide, CB-183,315, against resistant Clostridium difficile and other gram-positive aerobic and anaerobic intestinal pathogens. Antimicrob Agents Chemother (2012) 56(6):3448–52. doi:10.1128/AAC.06257-11
57. Chilton CH, Crowther GS, Todhunter SL, Nicholson S, Freeman J, Chesnel L, et al. Efficacy of surotomycin in an in vitro gut model of Clostridium difficile infection. J Antimicrob Chemother (2014) 69(9):2426–33. doi:10.1093/jac/dku141
58. Mascio CT, Mortin LI, Howland KT, Van Praagh AD, Zhang S, Arya A, et al. In vitro and in vivo characterization of CB-183,315, a novel lipopeptide antibiotic for treatment of Clostridium difficile. Antimicrob Agents Chemother (2012) 56(10):5023–30. doi:10.1128/AAC.00057-12
59. Knight-Connoni V, Mascio C, Chesnel L, Silverman J. Discovery and development ofsurotomycin for the treatment of Clostridium difficile. J Ind Microbiol Biotechnol (2016) 43(2–3):195–204. doi:10.1007/s10295-015-1714-6
60. Alam MZ, Wu X, Mascio C, Chesnel L, Hurdle JG. Mode of action and bactericidal properties of surotomycin against growing and non growing Clostridium difficile. Antimicrob Agents Chemother (2015) 59(9):5165–70. doi:10.1128/AAC.01087-15
61. Endres BT, Basseres E, Khaleduzzaman M, Alam MJ, Chesnel L, Garey KW. Evaluating the effects of surotomycin treatment on Clostridium difficile toxin A and B production, immune response, and morphological changes. Antimicrob Agents Chemother (2016) 60(6):3519–23. doi:10.1128/AAC.00211-16
62. Slayton ET, Hay AS, Babcock CK, Long TE. New antibiotics in clinical trials for Clostridium difficile. Expert Rev Anti Infect Ther (2016) 25:1–12. doi:10.1080/14787210.2016.1211931
63. Martin J, Wilcox M. New and emerging therapies for Clostridium difficile infection. Curr Opin Infect Dis (2016) 29(6):546–54. doi:10.1097/QCO.0000000000000320
64. Vickers RJ, Tillotson G, Goldstein EJ, Citron DM, Garey KW, Wilcox MH. Ridinilazole: a novel therapy for Clostridium difficile infection. Int J Antimicrob Agents (2015) 48(2):137–43. doi:10.1016/j.ijantimicag.2016.04.026
65. Mann J, Taylor PW, Dorgan CR, Johnson PD, Wilson FX, Vickers R, et al. The discovery of a novel antibiotic for the treatment of Clostridium difficile infections: a story of an effective academic-industrial partnership. Medchemcomm (2015) 6(8):1420–6. doi:10.1039/C5MD00238A
66. Bassères E, Endres BT, Khaleduzzaman M, Miraftabi F, Alam MJ, Vickers RJ, et al. Impact on toxin production and cell morphology in Clostridium difficile by ridinilazole (SMT19969), a novel treatment for C. difficile infection. J Antimicrob Chemother (2016) 71(5):1245–51. doi:10.1093/jac/dkv498
67. Goldstein EJ, Citron DM, Tyrrell KL, Merriam CV. Comparative in vitro activities of SMT19969, a new antimicrobial agent, against Clostridium difficile and 350 gram-positive and gram negative aerobic and anaerobic intestinal flora isolates. Antimicrob Agents Chemother (2013) 57:4872–6. doi:10.1128/AAC.01136-13
68. Sattar A, Thommes P, Payne L, Warn P, Vickers RJ. SMT19969 for Clostridium difficile infection (CDI): in vivo efficacy compared with fidaxomicin and vancomycin in the hamster model of CDI. J Antimicrob Chemother (2015) 70(6):1757–62. doi:10.1093/jac/dkv005
69. Weiss W, Pulse M, Vickers R. In vivo assessment of SMT19969 in a hamster model of Clostridium difficile infection. Antimicrob Agents Chemother (2014) 58:5714–8. doi:10.1128/AAC.02903-14
70. Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature (2015) 517(7533):205–8. doi:10.1038/nature13828
71. Kang DJ, Ridlon JM, Moore DR, Barnes S, Hylemon PB. Clostridium scindens baiCD and baiH genes encode stereo-specific 7α/7β-hydroxy-3-oxo-Δ4-cholenoic acid oxidoreductases. Biochim Biophys Acta (2008) 1781:16–25. doi:10.1016/j.bbalip.2007.10.008
72. Bäumler AJ, Sperandio V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature (2016) 535(7610):85–93. doi:10.1038/nature18849
73. Cataldo MA, Granata G, Petrosillo N. Clostridium difficile infection: new approaches to prevention, non-antimicrobial treatment, and stewardship. Expert Rev Anti Infect Ther (2017) 15(11):1027–40. doi:10.1080/14787210.2017.1387535
74. A Study of Ridinilazole (SMT19969) Compared With Fidaxomicin for the Treatment of Clostridium Difficile Infection (CDI). (2016). Available from: https://clinicaltrials.gov/ct2/show/results/NCT02784002 (Accessed: December 6, 2017).
75. La Marche MJ, Leeds JA, Amaral A, Brewer JT, Bushell SM, Deng G, et al. Discovery of LFF571: an investigational agent for Clostridium difficile infection. J Med Chem (2012) 55(5):2376–87. doi:10.1021/jm201685h
76. Leeds JA. Antibacterials developed to target a single organism: mechanisms and frequencies of reduced susceptibility to the novel anti-Clostridium difficile compounds fidaxomicin and LFF571. Cold Spring Harb Perspect Med (2016) 6:a025445. doi:10.1101/cshperspect.a025445
77. Sachdeva M, Leeds JA. Subinhibitory concentrations of LFF571 reduce toxin production by Clostridium difficile. Antimicrob Agents Chemother (2015) 59(2):1252–7. doi:10.1128/AAC.04436-14
78. Leeds JA, Sachdeva M, Mullin S, Dzink-Fox J, Lamarche MJ. Mechanism of action of and mechanism of reduced susceptibility to the novel anti-Clostridium difficile compound LFF571. Antimicrob Agents Chemother (2012) 56:4463–5. doi:10.1128/AAC.06354-11
79. Citron DM, Tyrrell KL, Merriam CV, Goldstein EJ. Comparative in vitro activities of LFF571 against Clostridium difficile and 630 other intestinal strains of aerobic and anaerobic bacteria. Antimicrob Agents Chemother (2012) 56:2493–503. doi:10.1128/AAC.06305-11
80. Debast SB, Bauer MP, Sanders IM, Wilcox MH, Kuijper EJ, ECDIS Study Group. Antimicrobial activity of LFF571 and three treatment agents against Clostridium difficile isolates collected for a pan-European survey in 2008: clinical and therapeutic implications. J Antimicrob Chemother (2013) 68:1305–11. doi:10.1093/jac/dkt013
81. Beran V, Kuijper EJ, Harmanus C, Sanders IM, van Dorp SM, Knetsch CW, et al. Molecular typing and antimicrobial susceptibility testing to six antimicrobials of Clostridium difficile isolates from three Czech hospitals in Eastern Bohemia in 2011-2012. Folia Microbiol (2017) 62(5):445–51. doi:10.1007/s12223-017-0515-x
82. Trzasko A, Leeds JA, Praestgaard J, Lamarche MJ, McKenney D. Efficacy of LFF571 in a hamster model of Clostridium difficile infection. Antimicrob Agents Chemother (2012) 56:4459–62. doi:10.1128/AAC.06355-11
83. Hamburger JB, Hoertz AJ, Lee A, Senturia RJ, McCafferty DG, Loll PJ. A crystal structure of a dimer of the antibiotic ramoplanin illustrates membrane positioning and a potential lipid II docking interface. Proc Natl Acad Sci U S A (2009) 106(33):13759–64. doi:10.1073/pnas.0904686106
84. Farver DK, Hedge DD, Lee SC. Ramoplanin: a lipoglycodepsipeptide antibiotic. Ann Pharmacother (2005) 39:863–8. doi:10.1345/aph.1E397
85. Bassères E, Endres BT, Dotson KM, Alam MJ, Garey KW. Novel antibiotics in development to treat Clostridium difficile infection. Curr Opin Gastroenterol (2017) 33(1):1–7. doi:10.1097/MOG.0000000000000332
86. Freeman J, Baines SD, Jabes D, Wilcox MH. Comparison of the efficacy of ramoplanin and vancomycin in both in vitro and in vivo models of clindamycin-induced Clostridium difficile infection. J Antimicrob Chemother (2005) 56:717–25. doi:10.1093/jac/dki321
87. Bartoloni A, Colao MG, Orsi A, Dei R, Giganti E, Parenti F. In-vitro activity of vancomycin, teicoplanin, daptomycin, ramoplanin, MDL 62873 and other agents against staphylococci, enterococci and Clostridium difficile. J Antimicrob Chemother (1990) 1990(26):627–33. doi:10.1093/jac/26.5.627
88. Kraus CN, Lyerly MW, Carman RJ. Ambush of Clostridium difficile spores by ramoplanin: activity in an in vitro model. Antimicrob Agents Chemother (2015) 59:2525–30. doi:10.1128/AAC.04853-14
89. Biavasco F, Manso E, Varaldo PE. In vitro activities of ramoplanin and four glycopeptides antibiotics against clinical isolates of Clostridium difficile. Antimicrob Agents Chemother (1991) 35:195–7. doi:10.1128/AAC.35.1.195
90. Mathur H, O’Connor PM, Hill C, Cotter PD, Ross RP. Analysis of anti-Clostridium difficile activity of thuricin CD, vancomycin, metronidazole, ramoplanin, and actagardine, both singly and in paired combinations. Antimicrob Agents Chemother (2013) 57:2882–6. doi:10.1128/AAC.00261-13
91. Peláez T, Alcalá L, Alonso R, Martín-López A, García-Arias V, Marín M, et al. In vitro activity of ramoplanin against Clostridium difficile, including strains with reduced susceptibility to vancomycin or with resistance to metronidazole. Antimicrob Agents Chemother (2005) 49:1157–9. doi:10.1128/AAC.49.3.1157-1159.2005
92. Fulco P, Wenzel RP. Ramoplanin: a topical lipoglycodepsipeptide antibacterial agent. Expert Rev Anti Infect Ther (2006) 4(6):939–45. doi:10.1586/14787210.4.6.939
93. McFarland LV. Therapies on the horizon for Clostridium difficile infections. Expert Opin Investig Drugs (2016) 25(5):541–55. doi:10.1517/13543784.2016.1161025
94. Ochsner UA, Bell SJ, O’Leary AL, Hoang T, Stone KC, Young CL, et al. Inhibitory effect of REP3123 on toxin and spore formation in Clostridium difficile, and in vivo efficacy in a hamster gastrointestinal infection model. J Antimicrob Chemother (2009) 63(5):964–71. doi:10.1093/jac/dkp042
95. Critchley IA, Green LS, Young CL, Bullard JM, Evans RJ, Price M, et al. Spectrum of activity and mode of action of REP3123, a new antibiotic to treat Clostridium difficile infections. J Antimicrob Chemother (2009) 63(5):954–63. doi:10.1093/jac/dkp041
96. Citron DM, Warren YA, Tyrrell KL, Merriam V, Goldstein EJ. Comparative in vitro activity of REP3123 against Clostridium difficile and other anaerobic intestinal bacteria. J Antimicrob Chemother (2009) 63(5):972–6. doi:10.1093/jac/dkp037
97. Anderson VR, Curran MP. Nitazoxanide: a review of its use in the treatment of gastrointestinal infections. Drugs (2007) 67:1947–67. doi:10.2165/00003495-200767130-00015
98. Shah D, Dang MD, Hasbun R, Koo HL, Jiang ZD, DuPont HL, et al. Clostridium difficile infection: update on emerging antibiotic treatment options and antibiotic resistance. Expert Rev Anti Infect Ther (2010) 8(5):555–64. doi:10.1586/eri.10.28
99. Fox LM, Saravolatz LD. Nitazoxanide: a new thiazolide antiparasitic agent. Clin Infect Dis (2005) 40(8):1173–80. doi:10.1086/428839
100. Rafiullah F, Kanwal S, Majeed UM, Korsten MA, Cheema FH, Luthra M, et al. Successful use of nitazoxanide in the treatment of recurrent Clostridium difficile infection. BMJ Case Rep (2011):bcr0420114123. doi:10.1136/bcr.04.2011.4123
101. Chen LF, Kaye D. Current use for old antibacterial agents: polymyxins, rifamycins, and aminoglycosides. Med Clin North Am (2011) 95(4):819–42. doi:10.1016/j.mcna.2011.03.007
102. Garey KW, Salazar M, Shah D, Rodrigue R, DuPont HL. Rifamycin antibiotics for treatment of Clostridium difficile-associated diarrhea. Ann Pharmacother (2008) 42(6):827–35. doi:10.1345/aph.1K675
103. Nomura K, Matsumoto Y, Yoshida N, Taji S, Wakabayashi N, Mitsufuji S, et al. Successful treatment with rifampin for fulminant antibiotics-associated colitis in a patient with non-Hodgkin’s lymphoma. World J GastroenteroI (2004) 10:765–6. doi:10.3748/wjg.v10.i5.765
104. O’Connor JR, Galang MA, Sambol SP, Hecht DW, Vedantam G, Gerding DN, et al. Rifampin and rifaximin resistance in clinical isolates of Clostridium difficile. Antimicrob Agents Chemother (2008) 52:2813–7. doi:10.1128/AAC.00342-08
105. Curry SR, Marsh JW, Shutt KA, Muto CA, O’Leary MM, Saul MI, et al. High frequency of rifampin resistance identified in an epidemic Clostridium difficile clone from a large teaching hospital. Clin Infect Dis (2009) 48:425–9. doi:10.1086/596315
106. Mathur H, Rea MC, Cotter PD, Ross RP, Hill C. The potential for emerging therapeutic options for Clostridium difficile infection. Gut Microbes (2014) 5(6):696–710. doi:10.4161/19490976.2014.983768
107. Crowther GS, Baines SD, Todhunter SL, Freeman J, Chilton CH, Wilcox MH. Evaluation of NVB302 versus vancomycin activity in an in vitro human gut model of Clostridium difficile infection. J Antimicrob Chemother (2013) 68(1):168–76. doi:10.1093/jac/dks359
108. Rea MC, Dobson A, O’Sullivan O, Crispie F, Fouhy F, Cotter PD, et al. Effect of broad- and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc Natl Acad Sci U S A (2011) 108:4639–44. doi:10.1073/pnas.1001224107
109. Rea MC, Sit CS, Clayton E, O’Connor PM, Whittal RM, Zheng J, et al. Thuricin CD, a post translationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc Natl Acad Sci U S A (2010) 107:9352–7. doi:10.1073/pnas.0913554107
110. Rea MC, Clayton E, O’Connor PM, Shanahan F, Kiely B, Ross RP, et al. Antimicrobial activity of lacticin 3147 against clinical Clostridium difficile strains. J Med Microbiol (2007) 56:940–6. doi:10.1099/jmm.0.47085-0
111. Walz DT, Di Martino MJ, Griswold DE, Intoccia AP, Flanagan TL. Biologic actions and pharmacokinetic studies of auranofin. Am J Med (1983) 75:90–108. doi:10.1016/0002-9343(83)90481-3
112. Jackson-Rosario S, Cowart D, Myers A, Tarrien R, Levine RL, Scott RA, et al. Auranofin disrupts selenium metabolism in Clostridium difficile by forming a stable Au-Se adduct. J Biol Inorg Chem (2009) 14:507–19. doi:10.1007/s00775-009-0466-z
113. Clardy J, Fischbach MA, Walsh CT. New antibiotics from bacterial natural products. Nat Biotechnol (2006) 24(12):1541–50. doi:10.1038/nbt1266
114. Gil F, Paredes-Sabja D. Acyldepsipeptide antibiotics as a potential therapeutic agent against Clostridium difficile recurrent infections. Future Microbiol (2016) 11:1179–89. doi:10.2217/fmb-2016-0064
115. Lee BG, Park EY, Lee KE, Jeon H, Sung KH, Paulsen H, et al. Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat Struct Mol Biol (2010) 17(4):471–8. doi:10.1038/nsmb.1787
116. Fimlaid KA, Bond JP, Schutz KC, Putnam EE, Leung JM, Lawley TD, et al. Global analysis of the sporulation pathway of Clostridium difficile. PLoS Genet (2013) 9(8):e1003660. doi:10.1371/journal.pgen.1003660
117. Stein GE, Craig WA. Tigecycline: a critical analysis. Clin Infect Dis (2006) 43:518–24. doi:10.1086/505494
118. Di Bella S, Nisii C, Petrosillo N. Is tigecycline a suitable option for Clostridium difficile infection? Evidence from the literature. Int J Antimicrob Agents (2015) 46(1):8–12. doi:10.1016/j.ijantimicag.2015.03.012
119. Ooijevaar RE, van Beurden YH, Terveer EM, Goorhuis A, Bauer MP, Keller JJ, et al. Update of treatment algorithms for Clostridium difficile infection. Clin Microbiol Infect (2018). doi:10.1016/j.cmi.2017.12.022
120. Olson MW, Ruzin A, Feyfant E, Rush TS III, O’Connell J, Bradford PA. Functional, biophysical, and structural bases for antibacterial activity of tigecycline. Antimicrob Agents Chemother (2006) 50:2156–66. doi:10.1128/AAC.01499-05
121. Hecht DW, Galang MA, Sambol SP, Osmolski JR, Johnson S, Gerding DN. In vitro activities of 15 antimicrobial agents against 110 toxigenic Clostridium difficile clinical isolates collected from 1983 to 2004. Antimicrob Agents Chemother (2007) 51(8):2716–19. doi:10.1128/AAC.01623-06
122. Nord CE, Sillerstrom E, Wahlund E. Effect of tigecycline on normal oropharyngeal and intestinal microflora. Antimicrob Agents Chemother (2006) 50(10):3375–80. doi:10.1128/AAC.00373-06
123. Freeman J, Vernon J, Pilling S, Morris K, Nicholson S, Shearman S, et al. The ClosER study: results from a three-year pan-European longitudinal surveillance of antibiotic resistance among prevalent Clostridium difficile ribotypes, 2011-2014. Clin Microbiol Infect (2017):1–8. doi:10.1016/j.cmi.2017.10.008
124. Fraga EG, Nicodemo AC, Sampaio JL. Antimicrobial susceptibility of Brazilian Clostridium difficile strains determined by agar dilution and disk diffusion. Braz J Infect Dis (2016) 20(5):476–81. doi:10.1016/j.bjid.2016.07.004
125. Nagy E, Dowzicky MJ. In vitro activity of tigecycline and comparators against a European compilation of anaerobes collected as part of the Tigecycline Evaluation and Surveillance Trial (TEST). Scand J Infect Dis (2010) 42:33–8. doi:10.3109/00365540903244543
126. Rashid MU, Lozano HM, Weintraub A, Nord CE. In vitro activity of cadazolid against Clostridium difficile strains isolated from primary and recurrent infections in Stockholm, Sweden. Anaerobe (2013) 20:32–5. doi:10.1016/j.anaerobe.2013.02.003
127. Kundrapu S, Hurless K, Sunkesula VC, Tomas M, Donskey CJ. Tigecycline exhibits inhibitory activity against Clostridium difficile in the intestinal tract of hospitalised patients. Int J Antimicrob Agents (2015) 45:424–6. doi:10.1016/j.ijantimicag.2014.11.016
128. Aldape MJ, Heeney DD, Bryant AE, Stevens DL. Tigecycline suppresses toxin A and B production and sporulation in Clostridium difficile. J Antimicrob Chemother (2015) 70:153–9. doi:10.1093/jac/dku325
129. Garneau JR, Valiquette L, Fortier LC. Prevention of Clostridium difficile spore formation by sub-inhibitory concentrations of tigecycline and piperacillin/tazobactam. BMC Infect Dis (2014) 14:29. doi:10.1186/1471-2334-14-29
130. Lachowicz D, Pituch H, Obuch-Woszczatyński P. Antimicrobial susceptibility patterns of Clostridium difficile strains belonging to different polymerase chain reaction ribotypes isolated in Poland in 2012. Anaerobe (2015) 31:37–41. doi:10.1016/j.anaerobe.2014.09.004
131. Nuding S, Frasch T, Schaller M, Stange EF, Zabel LT. Synergistic effects of antimicrobial peptides and antibiotics against Clostridium difficile. Antimicrob Agents Chemother (2014) 58:5719–25. doi:10.1128/AAC.02542-14
132. Jump RL, Li Y, Pultz MJ, Kypriotakis G, Donskey CJ. Tigecycline exhibits inhibitory activity against Clostridium difficile in the colon of mice and does not promote growth or toxin production. Antimicrob Agents Chemother (2011) 55:546–9. doi:10.1128/AAC.00839-10
133. Lin YC, Huang YT, Tsai PJ, Lee TF, Lee NY, Liao CH, et al. Antimicrobial susceptibilities and molecular epidemiology of clinical isolates of Clostridium difficile in Taiwan. Antimicrob Agents Chemother (2011) 55:1701–5. doi:10.1128/AAC.01440-10
134. Hawser SP. Activity of tigecycline against recent European clinical isolates of Clostridium difficile. Int J Antimicrob Agents (2010) 35:97–8. doi:10.1016/j.ijantimicag.2009.11.006
135. Lu CL, Liu CY, Liao CH, Huang YT, Wang HP, Hsueh PR. Severe and refractory Clostridium difficile infection successfully treated with tigecycline and metronidazole. Int J Antimicrob Agents (2010) 35:311–2. doi:10.1016/j.ijantimicag.2009.11.008
136. Knafl D, Winhofer Y, Lötsch F, Weisshaar S, Steininger C, Burgmann H, et al. Tigecycline as last resort in severe refractory Clostridium difficile infection: a case report. J Hosp Infect (2016) 92(3):296–8. doi:10.1016/j.jhin.2015.11.010
137. Navalkele DB, Lerner AS. Intravenous tigecycline facilitates cure of severe Clostridium difficile infection (CDI) after failure of standard therapy: a case report and literature review of tigecycline use in CDI. Open Forum Infect Dis (2016) 3(2):ofw094. doi:10.1093/ofid/ofw094
138. Baines SD, Saxton K, Freeman J, Wilcox MH. Tigecycline does not induce proliferation or cytotoxin production by epidemic Clostridium difficile strains in a human gut model. J Antimicrob Chemother (2006) 58:1062–5. doi:10.1093/jac/dkl364
139. Bassis CM, Theriot CM, Young VB. Alteration of the murine gastrointestinal microbiota by tigecycline leads to increased susceptibility to Clostridium difficile infection. Antimicrob Agents Chemother (2014) 58(5):2767–74. doi:10.1128/AAC.02262-13
140. Theriot CM, Schumacher CA, Bassis CM, Seekatz AM, Young VB. Effects of tigecycline and vancomycin administration on established Clostridium difficile infection. Antimicrob Agents Chemother (2015) 59(3):1596–604. doi:10.1128/AAC.04296-14
141. Roshan N, Hammer KA, Riley TV. Non-conventional antimicrobial and alternative therapies for the treatment of Clostridium difficile infection. Anaerobe (2018) 49:103–11. doi:10.1016/j.anaerobe.2018.01.003
142. Jump RLP, Kraft D, Hurless K, Polinkovsky A, Donskey CJ. Impact of tigecycline versus other antibiotics on the fecal metabolome and on colonization resistance to Clostridium difficile in mice. Pathog Immun (2017) 2(1):1–20. doi:10.20411/pai.v2i1.159
143. Gergely Szabo B, Kadar B, Szidonia Lenart K, Dezsenyi B, Kunovszki P, Fried K, et al. Use of intravenous tigecycline in patients with severe Clostridium difficile infection: a retrospective observational cohort study. Clin Microbiol Infect (2016) 22:990–5. doi:10.1016/j.cmi.2016.08.017
144. Bishop EJ, Tiruvoipati R, Metcalfe J, Marshall C, Botha J, Kelley PG. The outcome of patients with severe and severe-complicated Clostridium difficile infection treated with tigecycline combination therapy: a retrospective observational study. Intern Med J (2018). doi:10.1111/imj.13742
145. Manea E, Sojo-Dorado J, Jipa RE, Benea SN, Rodriguez-Bano J, Hristea A. The role of tigecycline in the management of Clostridium difficile infection: a retrospective cohort study. Clin Microbiol Infect (2017) 24(2):180–4. doi:10.1016/j.cmi.2017.06.005
146. Thomas A, Khan F, Uddin N, Wallace MR. Tigecycline for severe Clostridium difficile infection. Int J Infect Dis (2014) 26:171–2. doi:10.1016/j.ijid.2014.04.025
147. LaSalvia MT, Branch-Elliman W, Snyder GM, Mahoney MV, Alonso CD, Gold HS, et al. Does adjunctive tigecycline improve outcomes in severe-complicated, nonoperative Clostridium difficile infection? Open Forum Infect Dis (2017) 4(1):ofw264. doi:10.1093/ofid/ofw264
148. Hoover WW, Gerlach EH, Hoban DJ, Eliopoulos GM, Pfaller MA, Jones RN. Antimicrobial activity and spectrum of rifaximin, a new topical rifamycin derivative. Diagn Microbiol Infect Dis (1993) 16:111–8. doi:10.1016/0732-8893(93)90004-Q
149. DuPont HL, Jiang ZD, Okhuysen PC, Ericsson CD, de la Cabada FJ, Ke S, et al. A randomized, double-blind, placebo-controlled trial of rifaximin to prevent travelers’ diarrhea. Ann Intern Med (2006) 142(10):805–12. doi:10.7326/0003-4819-142-10-200505170-00005
150. Mattila E, Arkkila P, Mattila PS, Tarkka E, Tissari P, Anttila VJ. Rifaximin in the treatment of recurrent Clostridium difficile infection. Aliment Pharmacol Ther (2013) 37:122–8. doi:10.1111/apt.12111
151. Huang JS, Jiang ZD, Garey KW, Lasco T, Dupont HL. Use of rifamycin drugs and development of infection by rifamycin-resistant strains of Clostridium difficile. Antimicrob Agents Chemother (2013) 57(6):2690–3. doi:10.1128/AAC.00548-13
152. Johnson S, Schriever C, Galang M, Kelly CP, Gerding DN. Interruption of recurrent Clostridium difficile-associated diarrhea episodes by serial therapy with vancomycin and rifaximin. Clin Infect Dis (2007) 44:846–8. doi:10.1086/511870
153. Johnson S, Schriever C, Patel U, Patel T, Hecht DW, Gerding DN. Rifaximin redux: treatment of recurrent Clostridium difficile infections with rifaximin immediately postvancomycin treatment. Anaerobe (2009) 15(6):290–1. doi:10.1016/j.anaerobe.2009.08.004
154. Garey KW, Jiang ZD, Bellard A, Dupont HL. Rifaximin in treatment of recurrent Clostridium difficile-associated diarrhea: an uncontrolled pilot study. J Clin Gastroenterol (2009) 43:91–3. doi:10.1097/MCG.0b013e31814a4e97
155. Burns DA, Heap JT, Minton NP. Clostridium difficile spore germination: an update. Res Microbiol (2010) 161(9):730–4. doi:10.1016/j.resmic.2010.09.007
156. Bouillaut L, Dubois T, Sonenshein AL, Dupuy B. Integration of metabolism and virulence in Clostridium difficile. Res Microbiol (2015) 166(4):375–83. doi:10.1016/j.resmic.2014.10.002
157. Theriot CM, Koenigsknecht MJ, Carlson PE Jr, Hatton GE, Nelson AM, Li B, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun (2014) 5:3114. doi:10.1038/ncomms4114
158. Schubert AM, Rogers MA, Ring C, Mogle J, Petrosino JP, Young VB, et al. Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile associated diarrhea from healthy controls. MBio (2014) 5(3):e1021–14. doi:10.1128/mBio.01021-14
159. Baines SD, Wilcox MH. Antimicrobial resistance and reduced susceptibility in Clostridium difficile: potential consequences for induction, treatment, and recurrence of C. difficile infection. Antibiotics (Basel) (2015) 4(3):267–98. doi:10.3390/antibiotics4030267
160. Edlund C, Barkholt L, Olsson-Liljequist B, Nord CE. Effect of vancomycin on intestinal flora of patients who previously received antimicrobial therapy. Clin Infect Dis (1997) 25:729–32. doi:10.1086/513755
161. Citron DM, Merriam CV, Tyrrell KL, Warren YA, Fernandez H, Goldstein EJ. In vitro activities of ramoplanin, teicoplanin, vancomycin, linezolid, bacitracin, and four other antimicrobials against intestinal anaerobic bacteria. Antimicrob Agents Chemother (2003) 47:2334–8. doi:10.1128/AAC.47.7.2334-2338.2003
162. Goldstein EJ, Babakhani F, Citron DM. Antimicrobial activities of fidaxomicin. Clin Infect Dis (2012) 55(Suppl 2):S143–8. doi:10.1093/cid/cis339
163. Deshpande A, Pasupuleti V, Thota P, Pant C, Rolston DD, Sferra TJ, et al. Community-associated Clostridium difficile infection and antibiotics: a metaanalysis. J Antimicrob Chemother (2013) 68(9):1951–61. doi:10.1093/jac/dkt129
164. Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog (2012) 8(10):e1002995. doi:10.1371/journal.ppat.1002995
165. Dubreuil L, Houcke I, Mouton Y, Rossignol JF. In vitro evaluation of activities of nitazoxanide and tizoxanide against anaerobes and aerobic organisms. Antimicrob Agents Chemother (1996) 40(10):2266–70.
Keywords: Clostridium difficile infection, novel antimicrobials, Clostridium difficile recurrence, prevention, management
Citation: Petrosillo N, Granata G and Cataldo MA (2018) Novel Antimicrobials for the Treatment of Clostridium difficile Infection. Front. Med. 5:96. doi: 10.3389/fmed.2018.00096
Received: 21 December 2017; Accepted: 26 March 2018;
Published: 16 April 2018
Edited by:
Vitali Sintchenko, University of Sydney, AustraliaReviewed by:
Aleksandra Barac, University of Belgrade, SerbiaAna Afonso, Universidade de São Paulo, Brazil
Copyright: © 2018 Petrosillo, Granata and Cataldo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Petrosillo, bmljb2xhLnBldHJvc2lsbG9AaW5taS5pdA==