 Yumeng Wen
Yumeng Wen Sapna Shah
Sapna Shah Kirk N. Campbell
Kirk N. Campbell- Division of Nephrology, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, United States
Focal segmental glomerulosclerosis (FSGS) is the most common primary glomerular disease resulting in end-stage renal disease in the USA and is increasing in prevalence worldwide. It is a diverse clinical entity with idiopathic, genetic, metabolic, infectious, and other causes that culminate in a characteristic histologic pattern of injury. Proteinuria is a hallmark of FSGS as well as other primary and secondary glomerular disorders. The magnitude of proteinuria at disease onset and during treatment has prognostic implications for renal survival as well as associated cardiovascular morbidity and mortality. Significant advances over the last two decades have shed light on the molecular architecture of the glomerular filtration barrier. The podocyte is the target cell for injury in FSGS. A growing list of disease-causing gene mutations encoding proteins that regulate podocyte survival and homeostasis has been identified in FSGS patients. Several pathogenic and regulatory pathways have been uncovered that result in proteinuria in rodent models and human FSGS. The recurrence of proteinuria and FSGS after kidney transplantation is supporting evidence for the role of a circulating permeability factor in disease pathogenesis. These advances reviewed herein have significant implications for disease classification and therapeutic drug development for FSGS.
Introduction
The glomerular filtration barrier is composed of fenestrated endothelial cells, the glomerular basement membrane (GBM), and podocytes, terminally differentiated epithelial cells connected by interposed slit diaphragms between actin-rich foot processes (1). Seminal studies by Farquhar and Palade highlight the role of the GBM in excluding molecules of the size and negative charge of albumin from the urine under normal conditions (2). Though proteinuria can result from injury to any of these components, experimental nephrosis was initially described as mainly affecting the visceral epithelium with loss of podocyte foot processes, reduction and modification of the “urinary slits” (interposed slit diaphragm), and intracellular accumulation of vacuoles and protein absorption droplets (3, 4). Positional cloning of the NPHS1 gene encoding nephrin, a vital component of the slit diaphragm, was a landmark event that initiated the era of molecular discovery of the complex molecular architecture of the podocyte (5). Wiggins et al. performed elegant studies showing that in a diphtheria toxin rat model, podocyte loss of more than 20% results in a low-level-sustained proteinuria. Loss greater than 40% directly causes a high-level proteinuria, a decreased renal function, and lesions of focal segmental glomerulosclerosis (FSGS) (6). Podocyte loss triggers a cascade involving tuft adhesions of bare areas of GBM to Bowman’s capsule, a nidus for sclerosis development that can progress to segmental sclerosis (7). Rather than a disease, FSGS should be considered a histologic pattern of glomerular injury resulting from podocyte loss. It describes not only primary podocyte injury but also a lesion that occurs as a secondary process such as hypertensive and diabetic nephropathy (DN). Clinically and genetically heterogeneous, FSGS is characterized by segmental sclerosis of the glomerular capillary tuft, with or without deposition of IgM and complement C3 (8). The pathogenesis of FSGS is multifactorial, thereby complicating classification efforts and therapeutic approaches. The contribution of circulating permeability factors has been suggested for four decades with the recurrence of FSGS after kidney transplantation potentially occurring within hours (9, 10). Separately, many genes identified in patients with familial and sporadic FSGS encode essential podocyte proteins (11). Viral nephritides, HIV in particular, has been associated with collapsing FSGS (12).
This review discusses the knowledge to date of the underlying pathogenesis of podocyte injury leading to proteinuria and FSGS. We focus on the contributions of putative circulating factors, gene mutations, and viral infections.
Circulating Factors
The high recurrence rate of FSGS post transplant (estimated at 30–40%) suggests the presence of a circulating factor in recipients. A pivotal study by Savin et al. showed that plasma from patients with recurrent FSGS could increase the glomerular permeability to albumin in an in vitro assay. Plasmapheresis was associated with a reduced proteinuria as well as a reduced glomerular permeability (13). Gallon presented an interesting case of a kidney-transplant patient with recurrent FSGS (14). The allograft kidney regained function with a reduction in proteinuria after the graft was removed and re-transplanted to another patient whose ESRD was due to diabetes mellitus. In addition, an infant born to a mother with primary FSGS developed transient proteinuria, suggesting a circulating factor crossing the placenta leading to proteinuria (15). These studies well illustrate the existence of a circulating factor, but the nature and source of such a factor remained unclear. Several putative factors have been proposed including soluble urokinase-type plasminogen activator receptor (suPAR), CLC-1, Apolipoprotein A1, active protease, and anti-CD40.
Soluble Urokinase-Type Plasminogen Activator Receptor
Of the putative factors, the role of urokinase-type plasminogen activator receptor (uPAR) and its soluble form (suPAR) have been the most studied (16, 17). uPAR is a glycosylphosphatidylinositol (GPI)-anchored three-domain (DI, DII, and DIII) protein. Apart from being a receptor for urokinase, uPAR also forms signaling complexes with various transmembrane proteins including integrin and is involved in non-proteolytic pathways. The soluble form suPAR can be released by the cleavage of GPI anchor. In Wei’s study, uPAR was upregulated in the glomeruli from both DN and FSGS. Using uPAR knockout (Plaur−/−) mice model, they showed that lipopolysaccharide (LPS)-induced proteinuria was uPAR-mediated, and uPAR upregulation following LPS stimulation led to αvβ3 integrin activation, with subsequent podocyte foot process effacement, increased podocyte motility, and proteinuria (16). The same group subsequently revealed the role of suPAR as a potential pathogenic factor involved in podocyte injury (17). Recombinant suPAR and serum from patients with FSGS recurrence induced αvβ3 integrin activation both in vitro and in vivo. In uPAR-knockout (Plaur−/−) mice, the supraphysiologic level of endogenous suPAR and exogenous suPAR injection induced αvβ3 integrin activation, resulting in podocyte injury and proteinuria. A recent study (18) has shed light on the source of suPAR production. Myeloid progenitor Gr-1lo cell expansion along with suPAR upregulation and proteinuria was observed in different rodent models for proteinuric kidney disease including DN and nephrotoxic serum nephritis. Incubating Gr-1lo − Sca-1 + myeloid progenitor cells with LPS led to uPAR expression and suPAR secretion, and injecting these myeloid progenitor cells could induce proteinuria in NSG mice (deficient in mature lymphocytes and natural killer cells). To translate their findings to human disease, the investigators introduced whole peripheral blood mononuclear cells (human hematopoietic cells), drawn from individuals with recurrent FSGS and injected into NSG mice. This approach led to Gr-1lo cell expansion, suPAR upregulation, and proteinuria. However, CD34+-depleted PBMC from these patients failed to induce any proteinuria. This process was distinct from GVHD pathophysiology since T cell-depleted PBMC yielded similar results. Though human homologs of Gr-1lo − Sca-1 + myeloid progenitor cells have not been identified given the lack of Gr-1 or Sca-1 antigen, these findings implicate a common upstream pathway relevant to general CKD. The same investigators recently linked APOL1-risk alleles to the suPAR–β3 integrin activation pathway (19). In this study, G1/G2-risk alleles were demonstrated to activate β3 integrin synergistically with suPAR, inducing autophagosome formation in podocytes and leading to foot process effacement, podocyte detachment, and proteinuria.
These findings have not been without controversy. Conflicting studies have shown that suPAR does not distinguish FSGS from other causes of nephrotic syndrome and that suPAR expression may be nonspecifically increased in the setting of a low eGFR (20–22). These findings have raised questions about the reliability of current ELISA methodology as a diagnostic biomarker for FSGS (23). It is likely that suPAR’s utility is confined to patients with a preserved eGFR. This is supported by the fact that elevated suPAR levels predicted future CKD development in the 3683 subject Emory Cardiovascular Biobank (24). The fact that suPAR was associated with both proteinuric and non-proteinuric CKD in this cohort also highlights the need for additional mechanistic studies on suPAR targets, production, and mechanism of action.
Other Proposed Circulating Permeability Factors
Cardiotrophin-Like Cytokine Factor 1 (CLCF1)
Proteomic analysis by liquid chromatography tandem mass spectrometry of recurrent FSGS plasma fractions that induce proteinuria in rats and enhance glomerular permeability to albumin led to the identification of CLCF1 (25–27). CLCF1 is a member of the IL-6 cytokine family and is secreted into the circulation as a heterodimer with either cytokine receptor-like factor 1 (CRLF1) or soluble ciliary neurotrophic factor receptor alpha. This heterodimeric coexpression is essential for efficient CLF1 secretion (28, 29). It has been demonstrated that CLCF1 increased glomerular permeability to albumin in a specific manner with anti-CLCF1 monoclonal antibody, blocking this effect and attenuating the effect of FSGS serum (30). CLCF1 appeared to be JAK/STAT dependent with both JAK2 and STAT3 inhibition blocking the ability of CLCF1 to increase glomerular permeability to albumin. Interestingly, heterodimeric CLCF1–CRLF1 also had an inhibitory effect on CLCF1 and FSGS-induced albumin permeability (30). This would suggest that while monomeric CLCF1 is pathogenic, heterodimeric CLCF1 is protective. The study also highlights the need for further exploration of the JAK–STAT pathway in FSGS pathogenesis.
Anti-CD40 Antibody
Recently, Delville et al. (31) screened 9,000 antigens in pretransplant sera from 64 patients with or without recurrent FSGS, compared to 34 non-FSGS CKD patients. A panel of antibodies (CD40, PTPRO, CGB5, FAS, P2RY11, SNRPB2, and APOL2) could predict posttransplant FSGS recurrence with 92% accuracy. Pretransplant elevation anti-CD40 antibody alone had the best accuracy of 78% in predicting FSGS recurrence. Anti-CD40 antibody caused podocyte injury both in cultured human podocytes and in wild-type mice, and the formation of this antibody may be associated with altered immunogenicity of the CD40 protein in serum among patients with recurrent FSGS. Proteinuria was further enhanced by co-injecting full-length suPAR into wild mice, suggesting that uPAR–αvβ3 signaling pathway may also be involved in anti-CD40 antibody-induced podocyte injury (31). This study was limited by a small sample size with validation required in other recurrent FSGS cohorts.
Apolipoprotein A-I
Proteomic analysis of plasma and urine samples performed on patients with recurrent FSGS post transplant identified a high-molecular-weight form of Apolipoprotein A-I, named as ApoA-Ib in 93% of recurrent FSGS urines compared with <5% of those without recurrence, patients with non- FSGS proteinuric diseases, or patients transplanted for familial FSGS. Urinary ApoA-Ib had a sensitivity of 92.8% and a specificity of 98.1% for identifying FSGS relapse (32). Urinary ApoA-Ib warrants additional investigation as a biomarker of FSGS recurrence post transplant. It remains unclear whether it is a cause or a consequence of FSGS recurrence. A potential pathogenic role in causing podocyte injury has not been established.
Active Proteases
There is evidence that plasma proteases may have a pathogenic role in recurrent FSGS. Vasodilator-stimulated phosphoprotein (VASP), a molecule involved in actin cytoskeleton organization is phosphorylated in response to exchange plasma from 10 patients with recurrent FSGS (33). VASP phosphorylation was associated with pathogenic-enhanced podocyte motility. Protease inhibitory drug cocktails and silencing of the protease-activated receptor-1 led to the loss of VASP phosphorylation. The source of increased proteases or whether they are enhanced by protease inhibitor loss remains unclear.
Genetic Etiologies of FSGS
Beginning with nephrin, the positional cloning of patients and relatives with familial FSGS has enabled the identification of numerous disease-causing podocyte genes (Table 1). They encode podocyte proteins localized to the cell membrane (TRPC6), nucleus (WT1), mitochondria (COQ2 and COQ6), lysosomes (LIMP2), and cytosol (PLCE1) (34–43). The most common mutations encode actin cytoskeletal (INF2, ACTN4, MYO1E) and slit diaphragm (NPHS1, NPHS2, CD2AP) (5, 44–49) proteins, thereby highlighting the essential role of these structures in the maintenance of the glomerular filtration barrier. Mutations in the ACTN4 gene are all in the actin-binding domain of the encoded protein and have a much higher affinity for actin filaments than of the wild-type protein (50). This gain of function produces a rigid cytoskeleton more susceptible to stress and actin network breaking. INF2, a member of the formin family, also regulates the actin cytoskeleton but in a different way. INF2 variants occur in the diaphanous inhibitory domain that is essential in the inhibition of Rho activation (51). Unchecked Rho signaling has therefore been suggested as a potential etiology for podocyte injury in INF2 mutation (51). Indeed, the activation of RhoA in podocytes has been shown to lead to albuminuria, podocyte foot process effacement, and histologic lesions of FSGS (52).
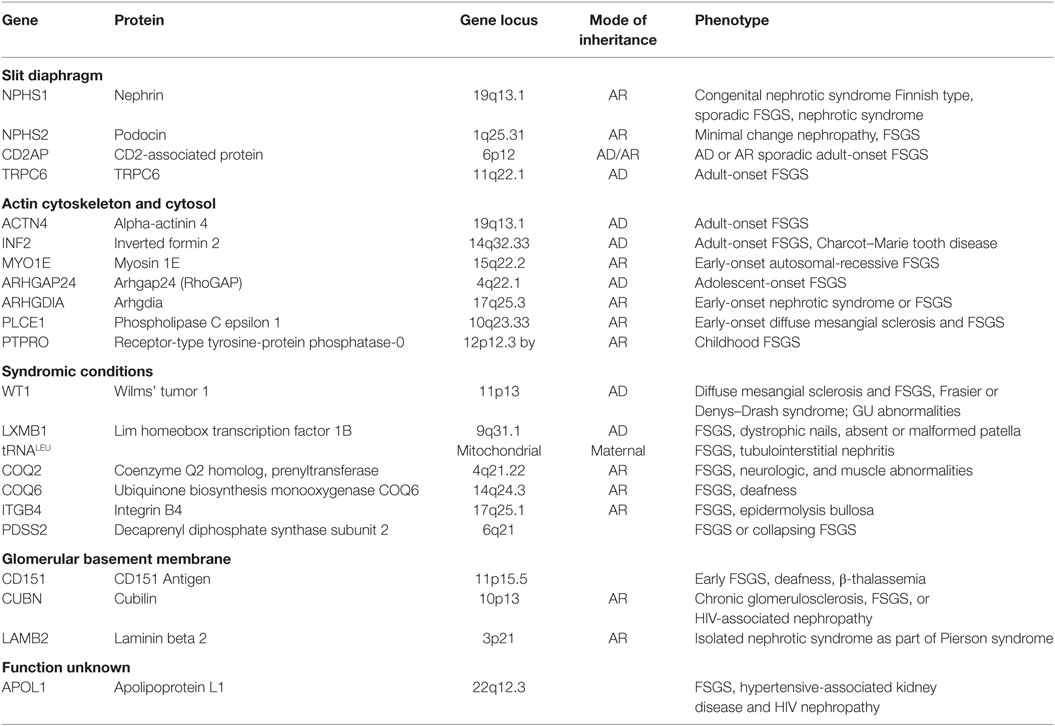
Table 1. Gene mutations linked to focal segmental glomerulosclerosis (FSGS).
The Rho family small GTPases (RhoA, Rac1, and Cdc42) are essential in actin cytoskeletal dynamics, cell morphology, motility, and adhesion. While overactive RhoA can be deleterious, there is an emerging consensus that a relative predominance of RhoA activity relative to Rac1/Cdc42 favors a more stationary podocyte with intact foot processes (53, 54). Conversely, Cdc42/Rac1 activation is associated with a more motile podocyte phenotype and foot process retraction. Loss of function mutations in the Arhgap24 gene associated with familial FSGS cause increased levels of active Rac1 and Cdc42 and increased podocyte motility (55). Likewise, ARHGDIA mutations in patients with steroid-resistant nephrotic syndrome increase active GTP-bound Rac1 and Cdc42 again resulting in increased podocyte motility reversed with Rac1 inhibitors (56). The slit diaphragm is also intricately connected to podocyte actin cytoskeletal dynamics. CD2AP and nephrin are direct binding partners with CD2AP interacting with actin, cortactin, and the alpha-actinin modulating protein synaptopodin (57, 58). Nck adaptor proteins link nephrin to the actin cytoskeleton (59). The cytoplasmic domain of nephrin contains six conserved tyrosine residues that when phosphorylated facilitates binding to the SH2 domains of Nck, leading to actin polymerization (60). Upregulated nephrin tyrosine phosphorylation has been described in glomerular injury (61), but the involved tyrosine kinase remains undefined.
The onset of proteinuria is variable in genetic disease with autosomal-recessive etiologies generally manifesting clinically in childhood and autosomal-dominant traits in adulthood. Commercially available genetic testing is most likely to identify known disease-causing variants in infants and patients with familial and syndromic disease (62). Exomic and genomic sequencing have the potential to uncover rare novel gene variants even in sporadic disease, but confirming causation typically requires experimental modeling with cell- and animal-based models. Identification of disease-causing gene mutations in FSGS has prognostic consequences since these patients are less likely to respond to steroid and calcineurin inhibitor therapy (63). They should perhaps be prioritized for kidney transplantation since disease recurrence is lower (64). Caution is necessary in the clinical application of genetic data, however. It is estimated that only 2.9% of US patients with nephrotic syndrome have a monogenic form of the disease and there is a risk in attributing a pathogenic role to identified variants that could be noncausal (65, 66).
Apolipoprotein L1 (APOL1)
Focal segmental glomerulosclerosis occurs with a higher frequency in African Americans largely due to variations in the Apol1 gene encoding APOL1 (67). APOL1 is a plasma factor that lyses the parasite Trypanosoma b. brucei. Over time, the parasite evolved into a serum resistance-associated protein (SRA) containing T. brucei rhodesiense that cause African sleeping sickness and capable of inactivating and evading Apol1. G1 and G2 Apol1 gene variants evade the SRA and are active against T. brucei rhodesiense. In this sense, this is analogous to malaria where the sickle trait is protective against parasitic infection but causes a hemoglobinopathy. Located on chromosome 22, the G1 and G2 alleles confer an increased risk for developing FSGS, hypertensive-associated kidney disease, and HIV nephropathy (68). In some population-based studies, it has been shown that APOLI1 variants conferred a 17-fold risk of FSGS and a 29-fold risk of HIVAN (69). Interestingly, 9 of 10 (90%) African American patients with collapsing FSGS in one cohort were noted to have at least one APOL1-risk allele (70). Several recently published studies have increased the understanding of how APOL1 G1 and G2 variants induce podocyte injury. Mice with podocyte-specific expression of either allele develop foot process effacement, proteinuria, and azotemia (71). These variants were found to interfere with endosomal trafficking and autophagic flux within podocytes leading to inflammatory-mediated cell death and glomerulosclerosis. Defects in autophagy have been previously shown to induce podocyte injury, proteinuria, and FSGS (72, 73), and manipulation of autophagic pathways could form the basis for therapeutic targeting of Apol1. These findings were distinct from those published by another group in which podocyte-specific APOL1-G2 transgenic mice developed preeclampsia but not kidney disease (74). The difference could be explained by the level-lower APOL1 levels obtained in the latter model. Other potential mechanisms have been implicated in APOL1-induced podocyte injury based on in vitro studies. In a human embryonic kidney cell, Tet-on system expression of G1 and G2 variants enhanced efflux of extracellular potassium and subsequent activation of stress-activated protein kinases (SAPK), p38 MAPK, and JNK. Interestingly, cytotoxicity was abrogated by SAPK inhibition and inhibition of K+ efflux (75). Apol1 has also been postulated to be involved in apoptosis, mitochondrial dysfunction, and energy depletion in other cell-based studies (76–78). Merely carrying high-risk APOL1 variants is not sufficient to cause kidney disease, and significant attention is currently being directed toward identifying second hits in the form of environmental and infectious triggers that promote disease development. The identification of patients lacking APOL1 with a normal phenotype increases the prospect that it can be therapeutically targeted.
Viral-Associated Disease
Podocytes can be injured by viruses either directly or by inflammatory cytokine-mediated targeting of podocyte receptors. In this context, HIV has been best studied. The renal expression of HIV genes has long been recognized as a central role in promoting HIVAN pathogenesis (79). HIV infects podocytes, tubular epithelial cells, infiltrating lymphocytes, as well as macrophages. Early studies of animal models, including the classic transgenic Tg26 mouse lines, studies on reciprocal transplantation between Tg26 and wild-type mice as well as podocyte-specific expression of HIV genes have confirmed the pathogenic role for local HIV gene expression in the kidney (80). Podocyte-predominant infection has been associated with podocyte injury, dedifferentiation, and more rapid loss of kidney function, suggestive of an HIV-driven podocyte injury as a primary pathogenic pathway in HIVAN (81). Moreover, the kidney has been shown to be one of the reservoirs for HIV, allowing active viral replication in this compartment apart from blood (82, 83). In HIVAN and collapsing forms of primary FSGS, injured podocytes were found to revert to a developmental program that includes downregulation of cyclin kinase inhibitors, entry into the cell cycle, upregulation of proliferation maker, and loss of mature phenotypic makers, including CD10/CALLA, C3b receptor, GLEPP-1, podocalyxin, synaptopodin, and importantly, WT1 (84–88). This process, termed podocyte dysregulation or podocyte dedifferentiation, is not observed in other proteinuric kidney disease marked by podocyte injury along with foot process effacement, such as minimal change disease and membranous nephropathy.
Cip/Kip family of cyclin kinase inhibitors acts in both G1 and S phases, while p21 also inhibits G2/M phase complexes. In healthy podocytes, p27 and p57 are expressed but p21 is suppressed (85). However, in patients with HIVAN or idiopathic collapsing FSGS, the expression of p27 and p57 is significantly suppressed, along with an increase in pro-mitotic cyclins (cyclin A), an increase in proliferation marker ki-67, and an expression of p21 (84, 85). Interestingly, the decrease in p27 and p57 was also found in histologically normal glomeruli, suggestive of a decrease of anti-mitotic signals preceding morphological change. Moreover, administering cyclin kinase inhibitors is associated with a restoration of normal podocyte phenotype and attenuated HIVAN presentation without any suppression of HIV gene expression (89, 90). Podocyte dedifferentiation is accompanied by the proliferation of parietal epithelial cells (PECs), which express parietal cell marker such as CK8 and PAX2 and lack podocyte markers. Cell bridges to the CK8-positive parietal lining could be observed without any evidence of coexpression of podocyte markers. These findings suggest that glomerular PECs provide a niche for podocyte progenitor cells (12, 91, 92).
Novel mechanisms of HIV-mediated kidney injury have been recently uncovered. Mammalian target of rapamycin (mTOR) has been found to be critical for p53-induced oxidative cell injury with mTOR inhibition protecting against HIV-induced podocyte apoptosis (93). The protective function of mTOR inhibition in HIVAN could be due to the regulation of microRNAs since rapamycin treatment reverses the downregulation of miR99a, miR-100a, miR-199a, miR-200a, miR-200b, miR-200c, miR-429, and miR-141 caused by HIV infection of human podocytes in culture (94). HIV also compromises the podocyte actin cytoskeleton through downregulation of the vitamin D receptor with associated enhanced deleterious intracellular angiotensin II and cathepsin L expression (95).
Other Viral-Associated FSGS
Collapsing FSGS has long been associated with parvovirus B19 infection (96, 97). The prevalence of parvovirus DNA infection within renal tissue is associated with idiopathic FSGS and collapsing FSGS compared with other diseases (98). Using in situ hybridization techniques, parvovirus B19 DNA was localized at endothelial cells and both visceral and PECs. However, whether this viral infection results in podocyte dysfunction is still a matter of debate (97). CMV has been associated with FSGS (99), but it remains unclear whether this virus directly infects podocytes. The detection of tubuloreticular inclusions in glomerular endothelial cells in CMV infection suggests a pathogenic role for interferon stimulation (100). Susceptible patients may have identifiable genetic risk factors. Indeed, a cohort of patients receiving interferon treatment who developed collapsing FSGS was reported to carry high-risk ApoL1 genotypes. Interferon α, β, and γ treatment of cultured podocytes increased APOL1 expression (101). Upstream of interferons, Toll-like receptor 3 (TLR3) was also shown here to signal through TBK1, NFkB, and JAK kinases in an interferon-independent manner to also increase APOL1 expression where the TLR3 agonist polyl:C promotes the binding of transcription factor IRF1/2 and STAT2 at the APOL1 transcription start site (101). These findings highlight the complex interplay between viruses, antiviral defenses, and the APOL1 genotype in the development of HIV and non-HIV-mediated viral FSGS.
Decreased Nephron Mass
Low birth weight individuals have long been known to be susceptible to a reduced nephron number and resulting abnormal hemodynamic stress on remaining nephrons (102, 103). Low birth weight is more commonly seen in FSGS patients than in the general population (104). Proteinuria has also been recognized in patients with unilateral renal agenesis where the remaining kidney has a reduced nephron number (105). A decreased nephron mass results in glomerular hypertension and hyperfiltration that increase mechanical stretch and injury to podocytes (106, 107). Glomerular hyperfiltration has also been associated with the pathogenesis of obesity-related FSGS (108). Here as with a decreased nephron mass, the activation of the renin–angiotensin–aldosterone system, the upregulation of TGFβ, and glomerulomegaly contribute to the vicious cycle that leads to proteinuria and FSGS (109, 110). Angiotensin blockers are the mainstay of treatment for patients with proteinuria due to hyperfiltration. There is some evidence that bariatric surgery can normalize proteinuria in patients with obesity-related glomerular disease (111).
Targeted Therapy
The identification of molecules essential for maintaining podocyte homeostasis has opened the door to targeted therapeutics for FSGS. Indeed, a number of repurposed drugs currently in clinical use have been shown to act directly on podocytes. Steroids protect podocytes from puromycin aminonucleoside-induced injury (112). Rituximab protects podocytes from injury by preserving sphingomyelin phosphodiesterase acid-like 3b (SMPDL-3b) expression (113). Cyclosporine stabilizes the actin-bundling protein synaptopodin, preventing cathepsin-mediated cleavage (114). Novel target-specific agents being tested at various stages of development include GDC-0879 (115), AC1903 (116), Abatacept (117), Bis-T-23 (118), and cycloRGDfV (17) as summarized in Figure 1.
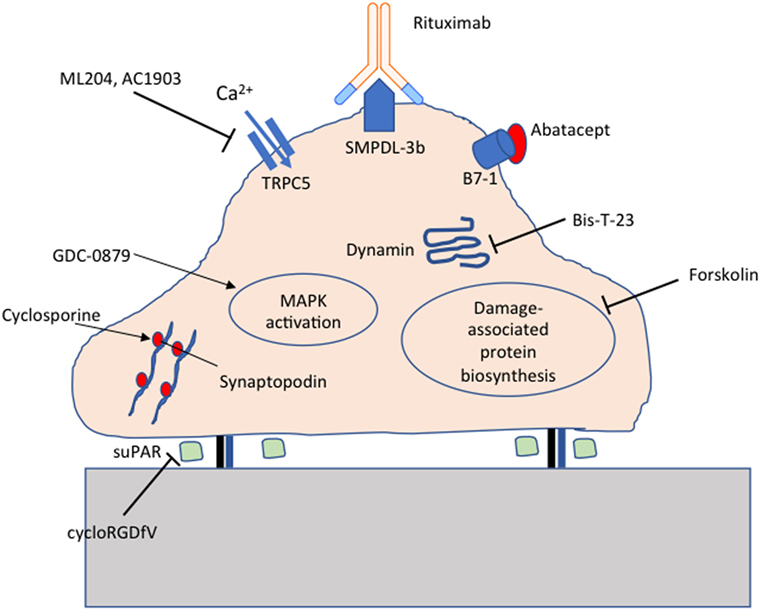
Figure 1. Targeted therapeutics for podocyte protection. Cyclosporine protects synaptopodin from cathepsin-mediated cleavage; cycloRGDfV inhibits soluble urokinase-type plasminogen activator receptor (suPAR) mediated β3 integrin activation; GDC-0879 promotes protective MAPK activation; ML204 and AC1903 inhibit TRPC5 ion channels; Rituximab preserves sphingomyelin phosphodiesterase acid-like 3b (SMPDL-3b) expression; Abatacept inhibits B7-1 mediated podocyte injury; Bis-T-23 promotes actin-dependent dynamin oligomerization; Forskolin inhibits damage-associated protein biosynthesis.
Conclusion
Podocyte injury due to circulating factors, gene mutations, infections, and many diverse etiologies culminate in the clinical manifestation of proteinuria and the histologic finding of FSGS. The evidence for a circulating permeability factor has been evolving in recent years, and various studies have suggested the pathogenic role of suPAR and other factors in the development of FSGS. The composition, origin, identity, and synergistic roles of circulating factor(s) remain unclear, representing an area of high unmet clinical need. Increasingly, podocyte-associated gene mutations are being identified in patients with FSGS. Expanded knowledge of the molecular architecture of the glomerular filtration barrier raises the prospect of a targeted therapeutic drug development.
HIV and other viral infections can induce podocyte injury and FSGS with an increasing interest in the enhanced susceptibility conferred by APOL1-risk alleles. The numerous divergent etiologies and pathogenic mechanisms with resulting variable response rates to therapy and recurrence post transplant speak to the need for a reclassification of the disease into more clinically relevant subtypes. Further exploration of the contributions of JAK/STAT, Rho-GTPase, and ApoL1 to podocyte injury and survival could enhance the quest for novel therapeutic agents.
Author Contributions
YW, SS, and KC conceived the article contents, prepared the manuscript, and endorsed the final draft submitted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by R01 DK103022 to KC.
References
1. Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol (2012) 74:299–323. doi:10.1146/annurev-physiol-020911-153238
2. Farquhar MG, Wissig SL, Palade GE. Glomerular permeability. I. Ferritin transfer across the normal glomerular capillary wall. J Exp Med (1961) 113:47–66. doi:10.1084/jem.113.1.47
3. Farquhar MG. Editorial: the primary glomerular filtration barrier—basement membrane or epithelial slits? Kidney Int (1975) 8:197–211. doi:10.1038/ki.1975.103
4. Farquhar MG, Palade GE. Glomerular permeability. II. Ferritin transfer across the glomerular capillary wall in nephrotic rats. J Exp Med (1961) 114:699–716. doi:10.1084/jem.114.5.699
5. Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell (1998) 1:575–82. doi:10.1016/S1097-2765(00)80057-X
6. Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol (2005) 16:2941–52. doi:10.1681/ASN.2005010055
7. Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int (1998) 54:687–97. doi:10.1046/j.1523-1755.1998.00044.x
8. Woroniecki RP, Kopp JB. Genetics of focal segmental glomerulosclerosis. Pediatr Nephrol (2007) 22:638–44. doi:10.1007/s00467-007-0445-y
9. Raij L, Hoyer JR, Michael AF. Steroid-resistant nephrotic syndrome: recurrence after transplantation. Ann Intern Med (1972) 77:581–6. doi:10.7326/0003-4819-77-4-581
10. Hoyer JR, Vernier RL, Najarian JS, Raij L, Simmons RL, Michael AF. Recurrence of idiopathic nephrotic syndrome after renal transplantation. Lancet (1972) 2:343–8. doi:10.1016/S0140-6736(72)91734-5
11. Pollak MR. The genetic basis of FSGS and steroid-resistant nephrosis. Semin Nephrol (2003) 23:141–6. doi:10.1053/snep.2003.50014
12. Wyatt CM, Klotman PE, D’Agati VD. HIV-associated nephropathy: clinical presentation, pathology, and epidemiology in the era of antiretroviral therapy. Semin Nephrol (2008) 28:513–22. doi:10.1016/j.semnephrol.2008.08.005
13. Savin VJ, Sharma R, Sharma M, McCarthy ET, Swan SK, Ellis E, et al. Circulating factor associated with increased glomerular permeability to albumin in recurrent focal segmental glomerulosclerosis. N Engl J Med (1996) 334:878–83. doi:10.1056/NEJM199604043341402
14. Gallon L, Leventhal J, Skaro A, Kanwar Y, Alvarado A. Resolution of recurrent focal segmental glomerulosclerosis after retransplantation. N Engl J Med (2012) 366:1648–9. doi:10.1056/NEJMc1202500
15. Kemper MJ, Wolf G, Müller-Wiefel DE. Transmission of glomerular permeability factor from a mother to her child. N Engl J Med (2001) 344:386–7. doi:10.1056/NEJM200102013440517
16. Wei C, Moller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med (2008) 14:55–63. doi:10.1038/nm1696
17. Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med (2011) 17:952–60. doi:10.1038/nm.2411
18. Hahm E, Wei C, Fernandez I, Li J, Tardi NJ, Tracy M, et al. Bone marrow-derived immature myeloid cells are a main source of circulating suPAR contributing to proteinuric kidney disease. Nat Med (2017) 23:100–6. doi:10.1038/nm.4242
19. Hayek SS, Koh KH, Grams ME, Wei C, Ko Y-A, Li J, et al. A tripartite complex of suPAR, APOL1 risk variants and [alpha]v[beta]3 integrin on podocytes mediates chronic kidney disease. Nat Med (2017) 23(8):945–53. doi:10.1038/nm.4362
20. Spinale JM, Mariani LH, Kapoor S, Zhang J, Weyant R, Song PX, et al. A reassessment of soluble urokinase-type plasminogen activator receptor in glomerular disease. Kidney Int (2015) 87:564–74. doi:10.1038/ki.2014.346
21. Sinha A, Bajpai J, Saini S, Bhatia D, Gupta A, Puraswani M, et al. Serum-soluble urokinase receptor levels do not distinguish focal segmental glomerulosclerosis from other causes of nephrotic syndrome in children. Kidney Int (2014) 85:649–58. doi:10.1038/ki.2013.546
22. Meijers B, Maas RJ, Sprangers B, Claes K, Poesen R, Bammens B, et al. The soluble urokinase receptor is not a clinical marker for focal segmental glomerulosclerosis. Kidney Int (2014) 85:636–40. doi:10.1038/ki.2013.505
23. Schlondorff D. Are serum suPAR determinations by current ELISA methodology reliable diagnostic biomarkers for FSGS? Kidney Int (2014) 85:499–501. doi:10.1038/ki.2013.549
24. Hayek SS, Sever S, Ko Y-A, Trachtman H, Awad M, Wadhwani S, et al. Soluble urokinase receptor and chronic kidney disease. N Engl J Med (2015) 373:1916–25. doi:10.1056/NEJMoa1506362
25. McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol (2010) 5:2115–21. doi:10.2215/CJN.03800609
26. Sharma M, Sharma R, McCarthy ET, Savin VJ. The focal segmental glomerulosclerosis permeability factor: biochemical characteristics and biological effects. Exp Biol Med (Maywood) (2004) 229:85–98. doi:10.1177/153537020422900111
27. Sharma M, Sharma R, McCarthy ET, Savin VJ. "The FSGS factor:" enrichment and in vivo effect of activity from focal segmental glomerulosclerosis plasma. J Am Soc Nephrol (1999) 10:552–61.
28. Elson GC, Lelievre E, Guillet C, Chevalier S, Plun-Favreau H, Froger J, et al. CLF associates with CLC to form a functional heteromeric ligand for the CNTF receptor complex. Nat Neurosci (2000) 3:867–72. doi:10.1038/78765
29. Plun-Favreau H, Elson G, Chabbert M, Froger J, deLapeyriere O, Lelievre E, et al. The ciliary neurotrophic factor receptor alpha component induces the secretion of and is required for functional responses to cardiotrophin-like cytokine. EMBO J (2001) 20:1692–703. doi:10.1093/emboj/20.7.1692
30. Sharma M, Zhou J, Gauchat JF, Sharma R, McCarthy ET, Srivastava T, et al. Janus kinase 2/signal transducer and activator of transcription 3 inhibitors attenuate the effect of cardiotrophin-like cytokine factor 1 and human focal segmental glomerulosclerosis serum on glomerular filtration barrier. Transl Res (2015) 166:384–98. doi:10.1016/j.trsl.2015.03.002
31. Delville M, Sigdel TK, Wei C, Li J, Hsieh S-C, Fornoni A, et al. A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci Transl Med (2014) 6:256ra136. doi:10.1126/scitranslmed.3008538
32. Lopez-Hellin J, Cantarell C, Jimeno L, Sanchez-Fructuoso A, Puig-Gay N, Guirado L, et al. A form of Apolipoprotein A-I is found specifically in relapses of focal segmental glomerulosclerosis following transplantation. Am J Transplant (2013) 13:493–500. doi:10.1111/j.1600-6143.2012.04338.x
33. Harris JJ, McCarthy HJ, Ni L, Wherlock M, Kang H, Wetzels JF, et al. Active proteases in nephrotic plasma lead to a podocin-dependent phosphorylation of VASP in podocytes via protease activated receptor-1. J Pathol (2013) 229:660–71. doi:10.1002/path.4149
34. Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet (2005) 37:739–44. doi:10.1038/ng1592
35. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science (2005) 308:1801–4. doi:10.1126/science.1106215
36. Li J, Zhao D, Ding J, Xiao H, Guan N, Fan Q, et al. WT1 mutation and podocyte molecular expression in a Chinese Frasier syndrome patient. Pediatr Nephrol (2007) 22:2133–6. doi:10.1007/s00467-007-0579-y
37. Gbadegesin R, Hinkes B, Vlangos C, Mucha B, Liu J, Hopcian J, et al. Mutational analysis of NPHS2 and WT1 in frequently relapsing and steroid-dependent nephrotic syndrome. Pediatr Nephrol (2007) 22:509–13. doi:10.1007/s00467-006-0377-y
38. Tajima T, Sasaki S, Tanaka Y, Kusunoki H, Nagashima T, Nonomura K, et al. 46,XY phenotypic male with focal segmental glomerulosclerosis caused by the WT1 splice site mutation. Horm Res (2003) 60:302–5. doi:10.1159/000074249
39. Denamur E, Bocquet N, Baudouin V, Da Silva F, Veitia R, Peuchmaur M, et al. WT1 splice-site mutations are rarely associated with primary steroid-resistant focal and segmental glomerulosclerosis. Kidney Int (2000) 57:1868–72. doi:10.1046/j.1523-1755.2000.00036.x
40. Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest (2013) 123:5179–89. doi:10.1172/JCI69000
41. Heeringa SF, Chernin G, Chaki M, Zhou W, Sloan AJ, Ji Z, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest (2011) 121:2013–24. doi:10.1172/JCI45693
42. Berkovic SF, Dibbens LM, Oshlack A, Silver JD, Katerelos M, Vears DF, et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet (2008) 82:673–84. doi:10.1016/j.ajhg.2007.12.019
43. Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol (2007) 18:2773–80. doi:10.1681/ASN.2006080833
44. Boyer O, Benoit G, Gribouval O, Nevo F, Tete MJ, Dantal J, et al. Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J Am Soc Nephrol (2011) 22:239–45. doi:10.1681/ASN.2010050518
45. Brown EJ, Schlondorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, et al. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet (2010) 42:72–6. doi:10.1038/ng.505
46. Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet (2000) 24:251–6. doi:10.1038/73456
47. Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med (2011) 365:295–306. doi:10.1056/NEJMoa1101273
48. Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet (2000) 24:349–54. doi:10.1038/74166
49. Gigante M, Pontrelli P, Montemurno E, Roca L, Aucella F, Penza R, et al. CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS). Nephrol Dial Transplant (2009) 24:1858–64. doi:10.1093/ndt/gfn712
50. Weins A, Kenlan P, Herbert S, Le TC, Villegas I, Kaplan BS, et al. Mutational and biological analysis of alpha-actinin-4 in focal segmental glomerulosclerosis. J Am Soc Nephrol (2005) 16:3694–701. doi:10.1681/ASN.2005070706
51. Sun H, Schlondorff JS, Brown EJ, Higgs HN, Pollak MR. Rho activation of mDia formins is modulated by an interaction with inverted formin 2 (INF2). Proc Natl Acad Sci U S A (2011) 108:2933–8. doi:10.1073/pnas.1017010108
52. Zhu L, Jiang R, Aoudjit L, Jones N, Takano T. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J Am Soc Nephrol (2011) 22:1621–30. doi:10.1681/ASN.2010111146
53. Blattner SM, Hodgin JB, Nishio M, Wylie SA, Saha J, Soofi AA, et al. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int (2013) 84(5):920–30. doi:10.1038/ki.2013.175
54. Kistler AD, Altintas MM, Reiser J. Podocyte GTPases regulate kidney filter dynamics. Kidney Int (2012) 81:1053–5. doi:10.1038/ki.2012.12
55. Akilesh S, Suleiman H, Yu H, Stander MC, Lavin P, Gbadegesin R, et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest (2011) 121:4127–37. doi:10.1172/JCI46458
56. Gee HY, Saisawat P, Ashraf S, Hurd TW, Vega-Warner V, Fang H, et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest (2013) 123:3243–53. doi:10.1172/JCI69134
57. Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest (2001) 108:1621–9. doi:10.1172/JCI200112849
58. Lehtonen S, Zhao F, Lehtonen E. CD2-associated protein directly interacts with the actin cytoskeleton. Am J Physiol Renal Physiol (2002) 283:F734–43. doi:10.1152/ajprenal.00312.2001
59. Jones N, Blasutig IM, Eremina V, Ruston JM, Bladt F, Li H, et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature (2006) 440:818–23. doi:10.1038/nature04662
60. Verma R, Kovari I, Soofi A, Nihalani D, Patrie K, Holzman LB. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest (2006) 116:1346–59. doi:10.1172/JCI27414
61. Verma R, Venkatareddy M, Kalinowski A, Patel SR, Salant DJ, Garg P. Shp2 associates with and enhances nephrin tyrosine phosphorylation and is necessary for foot process spreading in mouse models of podocyte injury. Mol Cell Biol (2015) 36:596–614. doi:10.1128/MCB.00956-15
62. Santin S, Tazon-Vega B, Silva I, Cobo MA, Gimenez I, Ruiz P, et al. Clinical value of NPHS2 analysis in early- and adult-onset steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol (2011) 6:344–54. doi:10.2215/CJN.03770410
63. Buscher AK, Kranz B, Buscher R, Hildebrandt F, Dworniczak B, Pennekamp P, et al. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol (2010) 5:2075–84. doi:10.2215/CJN.01190210
64. Jungraithmayr TC, Hofer K, Cochat P, Chernin G, Cortina G, Fargue S, et al. Screening for NPHS2 mutations may help predict FSGS recurrence after transplantation. J Am Soc Nephrol (2011) 22:579–85. doi:10.1681/ASN.2010010029
65. Sampson MG, Gillies CE, Robertson CC, Crawford B, Vega-Warner V, Otto EA, et al. Using population genetics to interrogate the monogenic nephrotic syndrome diagnosis in a case cohort. J Am Soc Nephrol (2016) 27:1970–83. doi:10.1681/ASN.2015050504
66. Crawford BD, Gillies CE, Robertson CC, Kretzler M, Otto E, Vega-Wagner V, et al. Evaluating Mendelian nephrotic syndrome genes for evidence for risk alleles or oligogenicity that explain heritability. Pediatr Nephrol (2017) 32:467–76. doi:10.1007/s00467-016-3513-3
67. Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR. Population-based risk assessment of APOL1 on renal disease. J Am Soc Nephrol (2011) 22:2098–105. doi:10.1681/ASN.2011050519
68. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science (2010) 329:841–5. doi:10.1126/science.1193032
69. Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol (2011) 22:2129–37. doi:10.1681/ASN.2011040388
70. Laurin L-P, Lu M, Mottl AK, Blyth ER, Poulton CJ, Weck KE. Podocyte-associated gene mutation screening in a heterogeneous cohort of patients with sporadic focal segmental glomerulosclerosis. Nephrol Dial Transplant (2014) 29:2062–9. doi:10.1093/ndt/gft532
71. Beckerman P, Bi-Karchin J, Park ASD, Qiu C, Dummer PD, Soomro I, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med (2017) 23:429–38. doi:10.1038/nm.4287
72. Hartleben B, Wanner N, Huber TB. Autophagy in glomerular health and disease. Semin Nephrol (2014) 34:42–52. doi:10.1016/j.semnephrol.2013.11.007
73. Kawakami T, Gomez IG, Ren S, Hudkins K, Roach A, Alpers CE, et al. Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol (2015) 26:1040–52. doi:10.1681/ASN.2013111202
74. Bruggeman LA, Wu Z, Luo L, Madhavan SM, Konieczkowski M, Drawz PE, et al. APOL1-G0 or APOL1-G2 transgenic models develop preeclampsia but not kidney disease. J Am Soc Nephrol (2016) 27:3600–10. doi:10.1681/ASN.2015111220
75. Olabisi OA, Zhang JY, VerPlank L, Zahler N, DiBartolo S III, Heneghan JF, et al. APOL1 kidney disease risk variants cause cytotoxicity by depleting cellular potassium and inducing stress-activated protein kinases. Proc Natl Acad Sci U S A (2016) 113:830–7. doi:10.1073/pnas.1522913113
76. Granado D, Muller D, Krausel V, Kruzel-Davila E, Schuberth C, Eschborn M, et al. Intracellular APOL1 risk variants cause cytotoxicity accompanied by energy depletion. J Am Soc Nephrol (2017) 28:3227–38. doi:10.1681/ASN.2016111220
77. Lan X, Jhaveri A, Cheng K, Wen H, Saleem MA, Mathieson PW, et al. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol (2014) 307:F326–36. doi:10.1152/ajprenal.00647.2013
78. Ma L, Chou JW, Snipes JA, Bharadwaj MS, Craddock AL, Cheng D, et al. APOL1 renal-risk variants induce mitochondrial dysfunction. J Am Soc Nephrol (2017) 28:1093–105. doi:10.1681/ASN.2016050567
79. Kopp JB, Klotman ME, Adler SH, Bruggeman LA, Dickie P, Marinos NJ, et al. Progressive glomerulosclerosis and enhanced renal accumulation of basement membrane components in mice transgenic for human immunodeficiency virus type 1 genes. Proc Natl Acad Sci U S A (1992) 89:1577–81. doi:10.1073/pnas.89.5.1577
80. Rosenstiel P, Gharavi A, D’Agati V, Klotman P. Transgenic and infectious animal models of HIV-associated nephropathy. J Am Soc Nephrol (2009) 20:2296–304. doi:10.1681/ASN.2008121230
81. Canaud G, Dejucq-Rainsford N, Avettand-Fenoël V, Viard J-P, Anglicheau D, Bienaimé F, et al. The kidney as a reservoir for HIV-1 after renal transplantation. J Am Soc Nephrol (2014) 25:407–19. doi:10.1681/ASN.2013050564
82. Winston JA, Bruggeman LA, Ross MD, Jacobson J, Ross L, D’Agati VD, et al. Nephropathy and establishment of a renal reservoir of HIV type 1 during primary infection. N Engl J Med (2001) 344:1979–84. doi:10.1056/NEJM200106283442604
83. Marras D, Bruggeman LA, Gao F, Tanji N, Mansukhani MM, Cara A, et al. Replication and compartmentalization of HIV-1 in kidney epithelium of patients with HIV-associated nephropathy. Nat Med (2002) 8:522–6. doi:10.1038/nm0502-522
84. Barisoni L, Kriz W, Mundel P, D’Agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol (1999) 10:51–61.
85. Shankland SJ, Eitner F, Hudkins KL, Goodpaster T, D’Agati V, Alpers CE. Differential expression of cyclin-dependent kinase inhibitors in human glomerular disease: role in podocyte proliferation and maturation. Kidney Int (2000) 58:674–83. doi:10.1046/j.1523-1755.2000.00213.x
86. Barisoni L, Mokrzycki M, Sablay L, Nagata M, Yamase H, Mundel P. Podocyte cell cycle regulation and proliferation in collapsing glomerulopathies. Kidney Int (2000) 58:137–43. doi:10.1046/j.1523-1755.2000.00149.x
87. Barisoni L, Bruggeman LA, Mundel P, D’Agati VD, Klotman PE. HIV-1 induces renal epithelial dedifferentiation in a transgenic model of HIV-associated nephropathy. Kidney Int (2000) 58:173–81. doi:10.1046/j.1523-1755.2000.00152.x
88. Schwartz EJ, Cara A, Snoeck H, Ross MD, Sunamoto M, Reiser J, et al. Human immunodeficiency virus-1 induces loss of contact inhibition in podocytes. J Am Soc Nephrol (2001) 12:1677–84.
89. Gherardi D, D’Agati V, Chu T-HT, Barnett A, Gianella-Borradori A, Gelman IH, et al. Reversal of collapsing glomerulopathy in mice with the cyclin-dependent kinase inhibitor CYC202. J Am Soc Nephrol (2004) 15:1212–22. doi:10.1097/01.ASN.0000124672.41036.F4
90. Nelson PJ, D’Agati VD, Gries J-M, Suarez J-R, Gelman IH. Amelioration of nephropathy in mice expressing HIV-1 genes by the cyclin-dependent kinase inhibitor flavopiridol. J Antimicrob Chemother (2003) 51:921–9. doi:10.1093/jac/dkg175
91. Dijkman HBPM, Weening JJ, Smeets B, Verrijp KCN, van Kuppevelt TH, Assmann KKJM, et al. Proliferating cells in HIV and pamidronate-associated collapsing focal segmental glomerulosclerosis are parietal epithelial cells. Kidney Int (2006) 70:338–44. doi:10.1038/sj.ki.5001574
92. Bariety J, Mandet C, Hill GS, Bruneval P. Parietal podocytes in normal human glomeruli. J Am Soc Nephrol (2006) 17:2770–80. doi:10.1681/ASN.2006040325
93. Rai P, Plagov A, Lan X, Chandel N, Singh T, Lederman R, et al. mTOR plays a critical role in p53-induced oxidative kidney cell injury in HIVAN. Am J Physiol Renal Physiol (2013) 305:F343–54. doi:10.1152/ajprenal.00135.2013
94. Cheng K, Rai P, Plagov A, Lan X, Mathieson PW, Saleem MA, et al. Rapamycin-induced modulation of miRNA expression is associated with amelioration of HIV-associated nephropathy (HIVAN). Exp Cell Res (2013) 319:2073–80. doi:10.1016/j.yexcr.2013.04.011
95. Chandel N, Sharma B, Husain M, Salhan D, Singh T, Rai P, et al. HIV compromises integrity of the podocyte actin cytoskeleton through downregulation of the vitamin D receptor. Am J Physiol Renal Physiol (2013) 304:F1347–57. doi:10.1152/ajprenal.00717.2012
96. Schwimmer JA, Markowitz GS, Valeri A, Appel GB. Collapsing glomerulopathy. Semin Nephrol (2003) 23:209–18. doi:10.1053/snep.2003.50019
97. Chandra P, Kopp JB. Viruses and collapsing glomerulopathy: a brief critical review. Clin Kidney J (2013) 6:1–5. doi:10.1093/ckj/sft002
98. Tanawattanacharoen S, Falk RJ, Jennette JC, Kopp JB. Parvovirus B19 DNA in kidney tissue of patients with focal segmental glomerulosclerosis. Am J Kidney Dis (2000) 35:1166–74. doi:10.1016/S0272-6386(00)70055-2
99. Dettmar AK, Oh J. Infection-related focal segmental glomerulosclerosis in children. Biomed Res Int (2016) 2016:7351964. doi:10.1155/2016/7351964
100. Bromfield M, McQuillan R, John R, Avila-Casado C. The significance of tubuloreticular inclusions as a marker of systemic stimulation by interferons in a case of focal and segmental glomerulosclerosis associated with cytomegalovirus (CMV) infection. Clin Kidney J (2014) 7:174–8. doi:10.1093/ckj/sft156
101. Nichols B, Jog P, Lee JH, Blackler D, Wilmot M, D’Agati V, et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int (2015) 87:332–42. doi:10.1038/ki.2014.270
102. Hostetter TH, Olson JL, Rennke HG, Venkatachalam MA, Brenner BM. Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. Am J Physiol (1981) 241:F85–93.
103. Luyckx VA, Bertram JF, Brenner BM, Fall C, Hoy WE, Ozanne SE, et al. Effect of fetal and child health on kidney development and long-term risk of hypertension and kidney disease. Lancet (2013) 382:273–83. doi:10.1016/S0140-6736(13)60311-6
104. Ikezumi Y, Suzuki T, Karasawa T, Yamada T, Hasegawa H, Nishimura H, et al. Low birthweight and premature birth are risk factors for podocytopenia and focal segmental glomerulosclerosis. Am J Nephrol (2013) 38:149–57. doi:10.1159/000353898
105. Wikstad I, Celsi G, Larsson L, Herin P, Aperia A. Kidney function in adults born with unilateral renal agenesis or nephrectomized in childhood. Pediatr Nephrol (1988) 2:177–82. doi:10.1007/BF00862585
106. Endlich N, Kress KR, Reiser J, Uttenweiler D, Kriz W, Mundel P, et al. Podocytes respond to mechanical stress in vitro. J Am Soc Nephrol (2001) 12:413–22.
107. Kriz W, Lemley KV. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol (2015) 26:258–69. doi:10.1681/ASN.2014030278
108. Chagnac A, Weinstein T, Korzets A, Ramadan E, Hirsch J, Gafter U. Glomerular hemodynamics in severe obesity. Am J Physiol Renal Physiol (2000) 278:F817–22. doi:10.1152/ajprenal.2000.278.5.F817
109. Ruster C, Wolf G. The role of the renin–angiotensin–aldosterone system in obesity-related renal diseases. Semin Nephrol (2013) 33:44–53. doi:10.1016/j.semnephrol.2012.12.002
110. Riser BL, Ladson-Wofford S, Sharba A, Cortes P, Drake K, Guerin CJ, et al. TGF-beta receptor expression and binding in rat mesangial cells: modulation by glucose and cyclic mechanical strain. Kidney Int (1999) 56:428–39. doi:10.1046/j.1523-1755.1999.00600.x
111. Fowler SM, Kon V, Ma L, Richards WO, Fogo AB, Hunley TE. Obesity-related focal and segmental glomerulosclerosis: normalization of proteinuria in an adolescent after bariatric surgery. Pediatr Nephrol (2009) 24:851–5. doi:10.1007/s00467-008-1024-6
112. Ransom RF, Lam NG, Hallett MA, Atkinson SJ, Smoyer WE. Glucocorticoids protect and enhance recovery of cultured murine podocytes via actin filament stabilization. Kidney Int (2005) 68:2473–83. doi:10.1111/j.1523-1755.2005.00723.x
113. Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med (2011) 3:85ra46. doi:10.1126/scitranslmed.3002231
114. Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med (2008) 14:931–8. doi:10.1038/nm.1857
115. Sieber J, Wieder N, Clark A, Reitberger M, Matan S, Schoenfelder J, et al. GDC-0879, a BRAF(V600E) inhibitor, protects kidney podocytes from death. Cell Chem Biol (2018) 25(175–184):e174. doi:10.1016/j.chembiol.2017.11.006
116. Zhou Y, Castonguay P, Sidhom EH, Clark AR, Dvela-Levitt M, Kim S, et al. A small-molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science (2017) 358:1332–6. doi:10.1126/science.aal4178
117. Yu CC, Fornoni A, Weins A, Hakroush S, Maiguel D, Sageshima J, et al. Abatacept in B7-1-positive proteinuric kidney disease. N Engl J Med (2013) 369(25):2416–23. doi:10.1056/NEJMoa1304572
Keywords: podocyte, focal segmental glomerulosclerosis, HIVAN, soluble urokinase-type plasminogen activator receptor, podocin
Citation: Wen Y, Shah S and Campbell KN (2018) Molecular Mechanisms of Proteinuria in Focal Segmental Glomerulosclerosis. Front. Med. 5:98. doi: 10.3389/fmed.2018.00098
Received: 15 October 2017; Accepted: 26 March 2018;
Published: 16 April 2018
Edited by:
Sandra Merscher, University of Miami, United StatesReviewed by:
Agnieszka Swiatecka-Urban, University of Pittsburgh, United StatesLaura Mariani, University of Michigan, United States
Kimberly Jean Reidy, Montefiore Medical Center, United States
Copyright: © 2018 Wen, Shah and Campbell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kirk N. Campbell, a2lyay5jYW1wYmVsbEBtc3NtLmVkdQ==