 Gabriel Lidzbarsky
Gabriel Lidzbarsky Danielle Gutman
Danielle Gutman Huda Adwan Shekhidem
Huda Adwan Shekhidem Lital Sharvit
Lital Sharvit Gil Atzmon
Gil Atzmon- Department of Human Biology, University of Haifa, Haifa, Israel
As average life span and elderly people prevalence in the western world population is gradually increasing, the incidence of age-related diseases such as cancer, heart diseases, diabetes, and dementia is increasing, bearing social and economic consequences worldwide. Understanding the molecular basis of aging-related processes can help extend the organism’s health span, i.e., the life period in which the organism is free of chronic diseases or decrease in basic body functions. During the last few decades, immense progress was made in the understanding of major components of aging and healthy aging biology, including genomic instability, telomere attrition, epigenetic changes, proteostasis, nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and intracellular communications. This progress has been made by three spear-headed strategies: in vitro (cell and tissue culture from various sources), in vivo (includes diverse model and non-model organisms), both can be manipulated and translated to human biology, and the study of aging-like human syndromes and human populations. Herein, we will focus on current repository of genomic “senescence” stage of aging, which includes health decline, structural changes of the genome, faulty DNA damage response and DNA damage, telomere shortening, and epigenetic alterations. Although aging is a complex process, many of the “hallmarks” of aging are directly related to DNA structure and function. This review will illustrate the variety of these studies, done in in vitro, in vivo and human levels, and highlight the unique potential and contribution of each research level and eventually the link between them.
General Introduction
During an organism’s lifetime, cells are constantly exposed to exogenous and endogenous stressful agents. Cells can cope with these stressors by various response mechanisms, or in case of irreversible damage, programmed cell death (apoptosis), or permanent cell-cycle arrest (cellular senescence). Cellular senescence is characterized by a halt in cellular replication, accompanied by a specific molecular phenotype (1–3). This phenotype can be the result of a few factors, such as accumulation of DNA damage, telomere attrition, and various epigenetic alterations (4).
In this review, we will highlight the major efforts to unveil the role of senescence in healthy aging by three main strategies: in vitro, in vivo, and human. Each strategy has advantages and limitations, yet when stratified and combined can elucidate molecular and physiological mechanisms and phenotypes, in general, and in healthy aging in particular.
Cellular Senescence and Physiological Aging
The aging process is a complex trait that combines different biologic levels. Aging at the organism level includes failure to maintain internal environment and regular function, alongside increased susceptibility to diseases. Aging at the tissue level may involve, for example, chronic inflammation, which in turn contributes to cardiovascular and neurodegenerative disorders (5). The mechanisms of aging are affected by cellular and non-cellular pathways. The buildup of chronic stress, for example, is significant for the aging phenotype, but it is an organism-level phenotype (6). Structural deterioration of the body will influence an organism’s ability to forage, resulting in bad nutritional state that in turn will speed the aging process. Cellular senescence is one of the cellular pathways contributing to organismal aging. This process is triggered by several factors such as accumulation of DNA damage, telomere attrition, and various epigenetic alterations and involves the activation of permanent cell-cycle arrest. Yet, unlike quiescence and other kinds of no-proliferation conditions, it is followed by a typical gene expression, metabolic activity, and a senescence-associated secretory phenotype (SASP). Cellular senescence is a multistage path. Once activated, the arrested cells shift from unstable to steady cell-cycle arrest, in a procedure that involves p21, p16Ink4a, and p53 (Figure 1). Next, alterations in chromatin methylation are generated. Senescent cells can accumulate in tissues and organs and can ultimately result in tissue lesions that will cause organ dysfunction (7, 8), and thus the cellular processes can lead to organism-level decay in function and health.
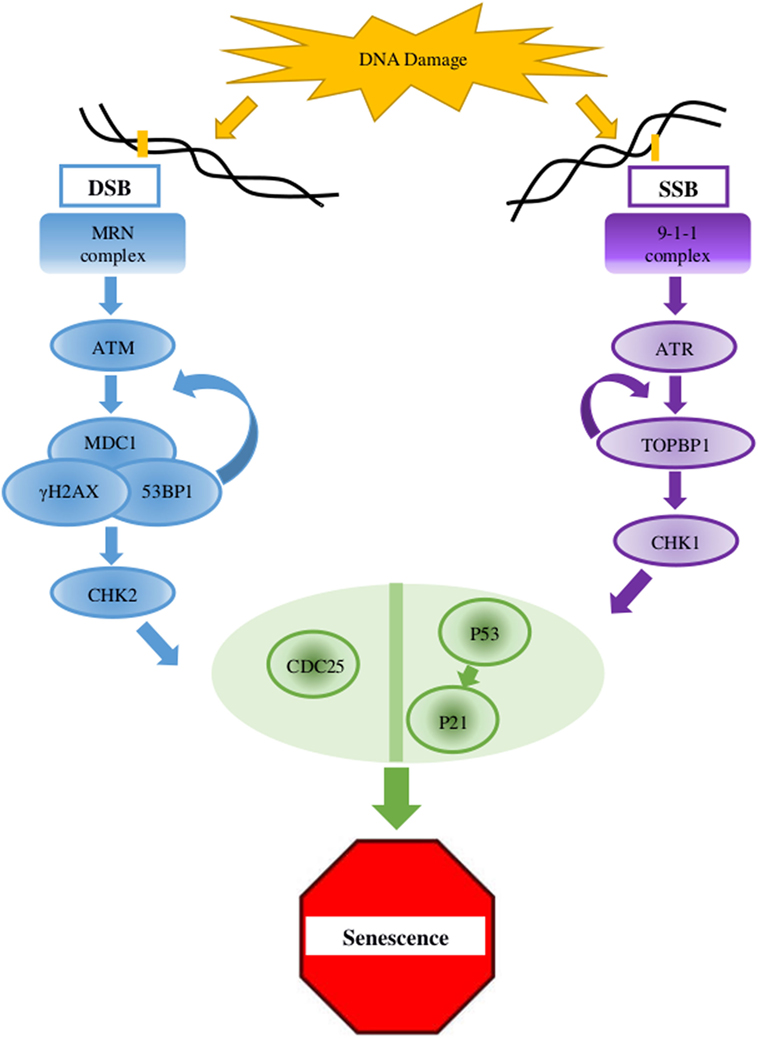
Figure 1. Key elements in the DNA damage response (DDR) pathway. In case of double-strand breaks (DSB), the DNA damage sensor MRN complex recruits the protein kinase ATM which activates γH2AX at the damaged site. γH2AX connects to MDC1, and this complex amplifies the activity of the MRN complex which, in a positive feedback, amplifies the ATM activity and the dispersal of γH2AX along the chromosome. MDC1 and 53BP1 further mediates the activation of CHK2 which carries the signal to distant locations on the genome. For single-strand breaks (SSB), the protein kinase ATR is activated and amplified by the 9-1-1 complex and TOPBP1, which also mediates the activation of CHK1. The signaling pathway cascades toward the key factors p53 and CDC25. When the lesion is repaired, the DDR complexes are dismantled (2, 4, 9).
From Cell Culture to Human Subjects: Strategies in Aging Research
In Vitro
Cell cultures are used in biological research since 1912. Carrel (10) isolated and cultured chicken cells to study aging processes (10). He concluded that the single cell is immortal, and aging and death are multicellular organism-related phenotypes. It was not until 1961 that Hayflick and Moorehead proved that Carrel was wrong and normal cells have limited proliferation capability in culture (10–12), also known as the Hayflick limit. Hayflick and Moorehead also discovered that normal cells looked “old” after they exhausted their replication potential. They speculated that single-celled replicative senescence contributed to the organism’s aging (11), which promoted the use of cell cultures to study aging processes in the full organism (12). Since the study by Hayflick and Moorehead, in vitro studies became the basis for every study in human biology. In vitro studies enable comparisons between many types of cells including mesenchymal stem cells, peripheral blood mononuclear cells, lymphoblast cells, muscle satellite cells (SCs), skin fibroblasts, endothelial cells, and embryonic stem cells, cultures from different organisms and different donor’s ages, enabling use for studying the genetics and biology of aging. Another advantage of in vitro studies is the capability to easily perform manipulations and treatments directly on the cells and to study the responses isolated from the original environment. The biggest limitation of in vitro studies is the translation to a whole organism (13). In culture, cells “behave” differently due to the loss of the cross talk between cells and the extracellular matrix from other regions in the body (such as immune system or hormonal signals). Though it helps with eliminating background pathway signaling noise when investigating certain mechanisms or pathways, it is a setback when trying to translate the effect of a manipulation or treatment to the whole organism. In attempt to compensate for the main in vitro limitation (i.e., translation drawback), researchers turn to in vivo (animal model) studies.
In Vivo
In vivo studies can further test the effect of a manipulation or treatment, either targeted or scattered, on the whole organism. Most of these biological models offer many advantages over humans, for instance, their basic biology and genomes are well documented and are easier to manipulate genetically. Furthermore, they have much shorter life spans than humans, enabling longitudinal studies, while ethical issues, long natural life span, environmental influences, genetic heterogeneity, and various other limiting factors complicate the use of human subjects in aging research. Regardless of the advantages listed earlier and the eminent contribution to our understanding of the aging process, the use of animal models in aging studies has its own limitations. Aging is not a simple process, and there is no genuine agreement about what it is and how to define it (14, 15), despite the agreement on being a multifactorial and complex phenomenon. Additionally, there is conflicting evidence about aging as a process that is similar across all organisms or particular to each species (15, 16). Therefore, it is important to draw attention to the fact that animal models are usually chosen for convenience rather than for specific features applicable to human aging. Hence, choosing the suitable animal model to answer the specific question we aim to understand is of high importance in these types of studies. Among the most prevalent aging model organisms are Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster, and Mus musculus. As a single-celled organism, S. cerevisiae is easily grown, manipulated, and observed; together with a well-characterized genome that bares much resemblance to bigger and more complex organisms, this model organism among others is a convenient platform for the study of the aging phenotype. Another important model system for studying a range of biological processes, including aging, is the nematode C. elegans. C. elegans has a short adult life span of ~2 weeks and a well-documented anatomy which is visible using a microscope. This enables easy observations of aging-related changes in the whole organism, in specific tissues and organs, and even on molecular and cellular levels (17–21). The classic genetic model organism, D. melanogaster, is also used in the study of aging. Studies conducted in these flies have identified single gene mutations that influence their life span. One of the strengths of Drosophila as a model organism is the capability to illustrate how genes that have an established role in regulating organismal life span particularly influence cellular and tissue function, how they work together, and how their tissue-specific functions might be linked (22–25). That said, Drosophila is far from being a good model for human aging as they share only 60% of the human genome. A better similarity is achieved with M. musculus, the mouse. It is the most commonly used model in biological research for various reasons. Mice are small, have a short generation time, and an accelerated life span which means they are not expensive and require only little space and time, compared to larger animal models. Another important reason is the fact that the mouse genome is well documented and can be easily manipulated. In addition, they are biologically similar to humans, exhibiting many of the same diseases and conditions. Nevertheless, mice do not develop several important age-related diseases naturally (e.g., atherosclerosis and diabetes), a fact that limits their potential as an aging model. All the organisms described earlier are short-lived, which is one of their desired traits as model organisms. However, that may not be appropriate for the study of human aging. Thus, in recent years there have been more studies conducted on non-model long-living organisms such as the naked mole rats and bats, which may be more appropriate models in understanding healthy human aging. The naked mole rat (Heterocephalus glaber) is a very important non-model organism in cancer and aging studies. This subterranean, mouse-sized, eusocial rodent is known as the longest-living rodent, living 4–17 years in the wild and with captive individuals demonstrating exceptional longevity that exceeds 30 years (26)—almost an order of magnitude longer than mice. Moreover, until a few years ago no cancer cases were reported in NMRs, and researchers failed to induce tumorigenesis, placing this rodent as a novel model for cancer studies. Bats are the second most speciose mammalian order after rodents. Little brown bats (Myotis) are the smallest bats (3–30 g) with the highest longevity records (Myotis myotis live for 37.1 years and M. brandti live for 41 years). Nevertheless, longevity is generally high in all bat lineages, which makes them an interesting model in biogerontology. One of the most interesting non-model organisms adopted for aging research is the Bowhead whale (Balaena mysticetus), which is estimated to be the longest-living mammal, reaching the age of ~200 years and also one of the biggest species, with length and weight of 20 m and 100 tons (6, 27). Bowhead whales live in arctic environment and are well adapted to these harsh surroundings. They are considered to be resistance to cancer and age-related diseases, and thus, though research is very technically complicated, the study of Bowhead whale in the context of longevity could improve our understanding of molecular mechanisms of healthy aging (27).
Human Aging-Like Syndromes
The limitations of in vitro and in vivo studies, and the great power of inferring from human studies on the human population, lead researchers to focus on aging-like human models. There are obvious moral and ethical limitations when working with human subjects, for this reason, most information on human aging was obtained from various progeroid syndromes, especially Hutchinson–Gilford progeria syndrome (HGPS) and Werner syndrome (28). These genetic conditions offer a glimpse into the molecular and physiological mechanisms of the aging cell and body, yet they do not capture the entire complexity of the aging and senescence phenotypes. Another approach for this purpose is using genome- and epigenome-wide association studies (GWAS and EWAS, respectively), which utilize the great improvement in whole genome sequencing technologies. Such studies have highlighted aging-related genes such as APOE (apolipoprotein E) (29–31) and have alleviated the dependency on in vitro and in vivo models by using direct human samples.
Age-Related DNA Damage and DNA Damage Response (DDR) Activity
Age-related accumulation of DNA damage has been studied thoroughly, showing correlation between age and damage levels or mutation frequency (32, 33). In the presence of DNA lesions or abnormalities, the DDR, a complex multigenic pathway, is activated and can eventually lead to cell cycle arrest (Figure 1) (2, 4, 9). In older organisms, accumulation of DNA damage and loss of regenerative potential consequently increase the number of senescent cells, leading to aging cells, tissues, organs (4), and inevitable death (2, 34, 35). The general term DNA damage encompasses different types of lesions in the DNA, including large chromosomal lesions such as double-strand breaks (DSBs) and small, local lesions such as single-strand breaks (SSBs) and mismatched bases. To prevent the deleterious effect of these lesions, cells have evolved four DNA damage repair mechanisms. For large DSBs, such as the case in DSBs, cells utilize homologous recombination (HR) or non-homologous end joining (NHEJ). SSBs are resolved via the base- or nucleotide-excision repair pathways (BER and NER, respectively) (33, 36), and mismatched bases are corrected by the mismatch repair (MMR) mechanism (37).
BER Reactive Oxygen Species (ROS)-Related DNA Damage Repair Efficiency, In Vitro
Wang et al. (38) tested lens samples isolated from age-related cataract (ARC) patients and age-matched patients with unrelated eye diseases (38). ARC was found to be affected by ROS and oxidative DNA damage, which is repaired by the BER pathway. The study showed that in ARC patients the expression levels of 8-oxoguanine DNA glycosylase (OGG1), a core member of the BER pathway, were significantly low. In addition, hypermethylation was demonstrated in the first exon of OGG1, hinting at the role of faulty DDR in the formation of ARC. Age-related BER activity was also studied with human foreskin fibroblasts derived from 20 to 64-year-old healthy donors, with similar results showing BER efficiency decrease with age. However, among several BER-related factors that were assessed, only Polβ (DNA polymerase beta) and XRCC1 (X-Ray Repair Cross Complementing 1) showed correlation between expression levels and age. In addition, a negative correlation was observed between age and the expression of Sirtuin 6 (SIRT6), which is connected to DNA maintenance and DSB repair (39), demonstrating a correlation between SIRT6 expression levels and BER quality. While overexpression of SIRT6 increased BER activity, SIRT6 knockout decreased BER activity, in the human foreskin fibroblasts (39). Related results were found in young and old rat MSCs. Here, increased cellular ROS production was observed with age. A hinting cause for the increased ROS level was the low superoxide dismutase (SOD) 1 (a central gene in the ROS response pathway) expression suggesting potential DNA damage (40). ROS is a known cause for DNA damage, from single base oxidation to single and DSBs, indicating that high ROS levels have an erroneous effect on genomic integrity (41).
DSB Repair Efficiency, In Vitro and In Vivo
A similar approach was implemented on eyelid fibroblast cells originating from different ages of healthy donors, showing that the efficiency and quality of DNA repair through NHEJ and HR pathways decreased with age (42).
The role of faulty DNA repair machinery in age-related genomic instability was also found in S. cerevisiae and Drosophila. Mutations in the sgs1 and srs2 genes [encoding for RecQ helicase, homologous to the human WRN (43)] shortened S. cerevisiae life span through two distinct pathways: sgs1- and srs2-mutated cells stopped dividing randomly in an age-independent manner that required the RAD9 (cell cycle checkpoint control protein) DNA damage checkpoint, but late-generation sgs1- and srs2-mutated cells exhibited premature aging. The double sgs1/srs2-mutated yeast cells showed a high rate of terminal G2/M arrest. This arrest was suppressed by knockouts of RAD51 (DNA repair protein RAD51 homolog 1), RAD52 (DNA repair protein), and RAD57 (DNA repair protein), hinting for malfunctioning HR. In a similar study, knockout of DNA2, encoding RecQ helicase-like protein, caused premature aging phenotypes including longer cell cycle time, transcriptional silencing, genomic alterations, and eventually shorter life span (44). Shaposhnikov et al. (45) used D. melanogaster to evaluate the effect of overexpression of DNA repair genes in several locations in the body and several time points during the life period on the Drosophila life span. Beneficial effects on life span were observed with overexpression of Hus1 (checkpoint clamp component), mnk (MAPK interacting protein kinases), mei-9 (meiotic 9, D. melanogaster), mus210 (Xeroderma pigmentosum, complementation group C, D. melanogaster), spn-B (spindle B, D. melanogaster), and WRNexo (WRN exonuclease, D. melanogaster), which control the processes of DNA damage recognition and repair (45). Myc, a key regulator protein of cell growth and proliferation, was shown to act as a pro-aging factor, probably by its ability to increase genomic instability. Overexpression of Myc in Drosophila increased the frequency of large genome rearrangements associated with faulty repair of DNA DSBs and decreased adult life span. Myc knockdowns demonstrated reduced mutation rate and extended life span (46). In aged mice, increased levels of DNA breaks or unrepaired DNA damage as illustrated by the formation of γH2AX (phosphorylated variant histone H2A) foci were observed (47–49). A positive effect on longevity was observed with overexpression of the human enzyme hMTH1 (MutT Human Homolog 1), which eliminates oxidized purine18 and deacetylase Sirt6 (50). Overexpression of SIRT6 promotes DSB repair by the activation of PARP1 [Poly (ADP-ribose) polymerase 1] and facilitating the recruitment of Rad51 (51) and NBS1 (Nijmegen Breakage Syndrome 1) (52) to DNA lesions.
Evidence From Omics Experiments, In Vitro
The accumulation of genomic abnormalities is influenced by the quality of the repair pathways, which may also decline with age. Laurie et al. (53) studied age-related DNA damage in peripheral blood cells using single nucleotide polymorphism (SNP) microarray data from over 50,000 individuals. The frequency of detectable genomic abnormalities was low (<0.5%) at birth and rose to 2–3% in 50-year-old donors (53). Peripheral blood cells were also studied using whole-exome sequencing data from DNA of 17,182 individuals lacking hematologic phenotypes. Somatic mutations were rare in young donors (~40 years old) but became more frequent with age. Furthermore, while studying subjects at 70–79 years, compared with 90–108 years, mutation frequency rose from 9.5 to 18.4%, respectively (54). In some cases, the accumulation of damage was noticeable in relatively advanced ages and not as a linear progression. Goronzy et al. (55) found that memory T cells from healthy donors showed steady increase in levels of DNA damage in different ages, up to 65 years (55). All these findings lay the basis for longitudinal in vivo studies in model organisms to decipher the mechanistic view of this phenomenon (i.e., accumulation of DNA damage with age) in a manageable life span.
DNA Repair in Long-Lived Animals
Analysis of two bat genomes showed that DNA repair and DNA damage signaling genes ATMh (human ataxia telangiectasia mutated), TP53 (tumor protein 53), RAD50 (DNA repair protein), and KU70 (XRCC6 protein product) are under selection in bats, suggesting that genome maintenance systems are under selective pressure in longer lived species (56). The study of Bowhead whales in the context of longevity is relatively new, but some insights have already been generated. Keane et al. (27) found duplications in genes linked to DNA damage repair and aging, such as PCNA (proliferating cell nuclear antigen). According to RNA-seq, both the PCNA copies were expressed. Several DNA damage and aging-associated genes, such as ERCC1 and ERCC3 (excision repair cross-complementing rodent repair), had unique mutations (compared to short-living animals) that were found to be under positive selection (27, 57). Mice with deleted ERCC1 suffered from liver dysfunction and died prematurely before weaning, a phenotype that was rescued by overexpression of ERCC1 (58). It is interesting to notice that similar unique mutations in DNA repair genes (including ERCC1 and ERCC3) were also found in naked mole rats and several species of bats (56, 59), hinting again at the role of DDR in longevity.
Comparative Studies of Short-Lived and Long-Lived Animals
Long-lived organisms are suggested to possess more efficient genome maintenance mechanisms than short-lived ones. For instance, in a comparative study conducted on both short- and long-lived wild bats, the MMR system and the levels of DNA damage as well as the antioxidant enzymatic activities were compared (60). By analyzing the DNA MMR proteins MSH2 (DNA MMR protein) and MLH1 (MutL homolog 1) in the liver, lung, and brain of young, adult, and old bats, the study showed that the short-lived bats presented with a decrease in protein levels and an increase in microsatellite instability antioxidant activity with age while the long-lived bats exhibited higher levels of antioxidant enzyme activities. These results suggest that the antioxidant response of those animals is important to attain a long life span. Several genes associated with the repair of DNA damage have been reported as overexpressed in long-lived subterranean rodents than in short-lived surface-dwelling rodents. In addition, when comparing blind mole rats (the genus spalax) to rats, the long-lived spalax showed more transcript abundance in genes that encode for DNA damage repair proteins (61). In another comparative study performed on mice, naked mole rats, and humans, studying the expression levels of DNA repair genes in livers found that humans and naked mole rats exhibit higher levels of expression of DNA repair enzymes that are important for DNA damage sensing and the MMR, NHEJ, and the BER pathways (62). This evidence supports the hypothesis that long-lived organisms have better genome maintenance techniques than short-lived animals.
Antioxidants have been more attentively studied in naked mole rats than in bats. When comparing the activity of antioxidant enzymes such as SODs, catalase, and cGPx (human cellular glutathione peroxidase) in the livers of young, middle-aged, and old naked mole rats with mice, their activity was higher in at least one age class in mole rats (63). More importantly, Csiszar et al. found that relative expression of numerous antioxidant enzymes in naked mole rat blood vessels remained constant with age which may distinguish this species from other short-lived species, such as mice (64). Comparative in vitro studies were performed as well. One recent example of such a comparative study is the study performed by Ma et al. (59) which compared primary skin fibroblasts of 16 different mammalian species and highlighted differences in fibroblast profiles among long- and short-lived species (59). In contrast to these findings, the work of Page et al. (65) did not find correlation between DDR activity and life span. Page and Stuart (65) compared DNA repair rates and life span values by studying BER activity in brain and liver tissues from 15 species including mice, hamster, bat, sheep, dogs, pigs, and two bird species, quail and finch. The BER activity was found to be (negatively) correlated only with body mass (65).
Contradicting Evidence, In Vitro and In Vivo
Despite the body of evidence mentioned here and in other reviews, some studies report contrary results. In a study performed by Schellenberg et al. (66), using long-term cultures of hMSC, Karyotype analyses at early passage and late passage did not reveal age-related chromosomal abnormalities and SNP array analysis did not reveal passage-related changes (66). A similar trend was observed when the efficiency of DNA MMR pathway was studied using CD4+ T cells from 25 to 80-year-old healthy donors. In this study, there was no connection between MMR frequency and donor’s age. Only when mutations were chemically induced, there was a negative correlation between MMR efficiency and age, but only among the younger age groups, 25–40 years old; no such connection was found for the older donors (67). Similar contradictions were also established in in vivo studies. Though there is a documented phenotype of DNA instability in aging yeast cells, it is still under debate whether accumulation of mutations is a cause of aging for yeast. Ijpma and Greider (68) found that chromosome loss was not related to loss of viability (68, 69). Daughter cells produced in early stages of their mother cell life live as long as their progenitors, yet cells produced later had reduced life span. However, the last cell created by a specific mother cell is still capable of bearing offspring. The observed increase in division time, which corresponded with an age-specific decline in reproduction in old mother cells, was only partially passed on to the daughter cells, and they resumed normal division time after a few budding cycles (70, 71). Kaya et al. (72) studied de novo mutations during multiple replications in daughter cells of mother cells at different ages. Mutations were found to increase with age, but their frequency was very low, and no effect on viability was detected (72). All these observations suggest genome integrity conservation through generations and question the role of genomic changes in aging in yeast. A possible explanation for aging-related genomic instability in yeast could be found in extra-chromosomal rDNA circles (ERCs), which were shown to be correlated with premature aging and short life span in yeast. sgs1 mutant accumulated more ERCs than wild-type cells, causing shorter life span (73), while knockouts of FOB1 (DNA replication fork blocking protein) decreased the formation of ERCs and extending life span (74).
Progeroid Diseases as Models for Aging
As mentioned earlier, age-related genomic instabilities in humans are studied through progeroid diseases. The first three genes causally linked to human aging (according to HAGRID) are progeroid phenotype causing genes: LMNA (Lamin A/C), WRN (Werner Syndrome RecQ-Like Helicase), and ERCC8 (DNA excision repair protein) (75). LMNA is a gene coding for a nuclear envelope scaffolding protein, mutations in which lead to genomic instability which in turn cause HGPS. This syndrome serves as a model for human aging since progerin (the mutated LMNA protein) can be found in normally aging cells and is believed to cause cellular toxicity and senescence (76). Mutates WRN (RecQ-like helicase) causes Werner syndrome and is involved in the DNA DSB repair pathway, similar to the S. cerevisiae SGS1 (43, 77). ERCC8, mutated in Cockayne syndrome patients, is a protein involved in the NER pathway, mutations in which cause high sensitivity to UV due to loss of ability to repair UV-induced DNA damage (78). These genes exemplify the effect of the DNA damage repair quality on aging, as brought forth by the previously mentioned in vitro and in vivo studies. Besides these three genes, another, more recently described gene is the SPRTN (SprT-Like N-Terminal Domain) gene whose translated protein product acts in the translation repair pathway, allowing DNA replication despite single nucleotide lesions. Mutations in this genes cause Werner-like progeria, probably due to their disabling effect on this replication pathway (79). Additional support for the importance of genomic integrity in the aging process is 53BP1 (p53 binding protein 1) (76). This protein is crucial for DNA DSB repair mediation and proteins’ recruitment. First described as a p53 binding protein, 53BP1 recognizes DSB histone code and recruits the repair proteins to the site in different mechanisms depending on different stages of the cell cycle (80). The DNA DSB repair is crucial as it is well established that DSBs lead to premature aging and senescence (81, 82).
Telomere Alterations and Cellular Senescence
Besides direct DNA damage, cellular senescence can be induced by diverse mechanisms, the principal among them is telomere attrition. Telomeres are short tandem repeats that serve as “caps” that protect the ends of the chromosomes from being recognized as DSBs and prevent the cascade of DDR in the cell and actively participate in genome maintenance. With every cellular division, the telomeres shorten by several repeats.
Evidence From In Vitro Studies
In most organisms, telomere elongation is controlled by the enzyme telomerase under tight regulation to ensure sufficient number of replications, yet when this number is reached, telomere elongation is seized (2, 83). Once telomeres reach the critical length, the cells undergo senescence and stop proliferating (84). This process is believed to be the trigger for the aging process, according to the telomere theory (11, 85, 86). It is further supported by Bodnar et al. who proved that telomere elongation caused by ectopic expression of telomerase avoids the senescence phenotype (87). His work relied on one of the earliest studies linking telomere shortening to aging which was performed by Harley et al. on human fibroblast cells (88). In their paper, they describe the shortening of telomeres in aging fibroblasts alongside chromosomal abnormalities, specifically the fusion of two chromosomes at the telomeric region and chromosomal rearrangement, while hinting at a biological significance to the shortening process. Since this early study, numerous studies have emerged strengthening this association and aiming to elucidate the exact underlying mechanism of telomere shortening. Murillo-Ortiz et al. (89) studied telomere alterations using T, B, and NK cells from 20 to 25-year-old and 60 to 65-year-old donors. Treatment with concanavalin A (a mitogen of T cells) caused increase in telomere length and number of replications in the samples from the young donors, but did not improve the samples from the older donors, which exhibited loss of telomere parts, decrease in telomere length, and decreased proliferation potential (89). Age-related changes in telomere length were also established in bone marrow hMSC in a long-term in vitro study (90). COMET assay revealed higher levels of damage in cells from older donors (91). Similar results were obtained in the study of CD34− and CD34+ cells isolated from healthy donors of different ages. However, some of the cells exhibited telomere shortening that was not correlated with age. It seems that CD34+ cells from older donor suffer from increased non-telomeric DNA damage, but the variation among the cultures hints for multiple factors contributing to DNA damage (92).
The Question of Telomere-Related Senescence in S. cerevisiae
For S. cerevisiae, various studies were performed on the effect of missing/broken telomere and mutated telomerase on the physiology of the organism. Genetic manipulations of S. cerevisiae cells caused decreased growth, irregular shape, and eventually, cellular senescence (69). Several genes, such as EST1 (telomere elongation protein), EST2 (telomere reverse transcriptase), EST3 (telomere replication protein), TLC1 (template RNA component), RAD9, RAP1 (DNA binding protein), CDC13 (cell division control protein 13), TEL1 (serine/threonine protein kinase), MEC1 (serine/threonine protein kinase), and MRC1 (macrophage mannose receptor 1 precursor) were studied in connection to telomere-related senescence; however, despite the extensive experimental work put into using mutated cells, the role of eroded telomeres in “natural” cellular senescence in yeast remained questionable (93). For example, EST1-4 (ever short telomere) mutants began to lose viability after 60 doublings, but late knockout cultures continued to maintain proliferation potential (94). Cells with mutated telomerase exhibited irregular morphology and short telomeres, but these changes did not cause deadly damage and determinate senescence (95). One hypothesis connects aging to telomere erosion through the transcription of subtelomeric genes. Genes located in subtelomeric regions are affected by transcriptional silencing which was found to change in an age-related manner. Kim et al. (96) found that silencing of genes in subtelomeric regions declined during the cell’s senescence, hinting at a connection between the transcription of subtelomeric regions and cellular senescence in yeast (96). The work of Austriaco and Guarente (97) reinforced this model, as they found that mutated telomerase extended life span (relatively to the wild type), probably by hanging the silencing procedure in the subtelomeric locations (97).
Telomere Alterations in C. elegans
The evidence for the role of telomere attrition in the senescence of C. elegans are contradicting and are influenced by the worm’s unique physiology, as the adult worm go through a short reproductive stage, followed by a “post-mitotic life” with a definite number of steady post-mitotic cells (98, 99). Overexpression of HRP1 (Heterogeneous nuclear Ribonucleo Protein 1) was found to increase telomere length and, subsequently, the life span of transgenic worms. The resulting prolonged life span was reliant on DAF16 (Forkhead box protein O gene, C. elegans) (100), which codes for a FOXO (Forkhead Box protein O) transcription factor and is required also for the effect of the insulin/IGF-1 pathway on life span in C. elegans (98). This connects to the first life span-related gene that was discovered in C. elegans—AGE-1. AGE-1 encodes a phosphatidylinositol-3-kinase that functions in the insulin/IGF-1 signaling pathway. Mutations in this gene cause delay in age-related deterioration of body movement and muscle deterioration a twofold extension of the life span (17, 101, 102). Opposing results were obtained by Raices et al. (103) that found no correlation between telomere length and the life span of daf-2 and daf-16 mutants. Furthermore, a study of different wild-type populations with diverse telomere lengths found again that the length of the telomeres was not correlated with life span (103). Similar phenomena were observed in mutants of TRT-1, a catalytic subunit of telomerase. The mutants reproduced regularly for several generations but eventually became sterile (104). The telomeres shortened by ~125 nucleotides per generation and suffered from sequence abnormalities, but the mutation and other telomere-shortening manipulations did not affect post-mitotic aging (104, 105). Mutations in MRT-2, a gene in the same pathway as TRT-1, caused similar phenotypes including telomere shortening, accumulation of DNA damage, and sterility. Similarly, the mutation had no effect on life span (106).
Relevance of Drosophila and Mice in the Study of Telomere-Related Senescence
While most organisms have a tandem repeat-based telomere and a telomerase for its maintenance, Drosophila telomeres are composed of randomly ordered retrotransposable elements that are maintained by retrotransposition (107–110). Although the length of the drosophila telomere is close to the human telomere (~10–12 kb), its structure is much more complex since each building block contains its own promoter regions, coding sequences, and regulatory elements (110, 111). These might be the reasons why there are no evidence for connection between telomere shortening and aging in Drosophila. Walter et al. (112) found that like C. elegans, the length of the telomeres in Drosophila did not affect life span, but it was correlated with fertility and fecundity (112). Study of age-related transcriptional changes did not find any telomere-related modifications (113). As in C. elegans, the FOXO-mediated insulin/IGF-1 pathway can affect the Drosophila life span (114), but a possible connection to telomere length was not studied.
Similar to Drosophila, the relevance of mice telomeres studies is also debated and unclear. Several studies show that mice with shortened or lengthened telomeres exhibit decreased or increased life span, respectively (115–119). The premature aging of telomerase-deficient mice was reverted when telomerase was genetically reactivated in aged mice (120), and systematic viral transduction of telomerase in adult wild-type mice delayed normal physiological aging (121). Mice with telomerase deficiencies exhibited signs of accelerated aging, but only after several generations and that overexpressing telomerase did not alter aging (122). The delayed phenotype implies that for senescence activation, telomeres need to be shortened extensively, in a manner that might not be realistic during the regular mouse life span. Mice are interesting models for the research of human telomere diseases. Telomerase dysfunction in humans causes a disease called dyskeratosis congenita (DKC), which shares some features with telomerase-deficient mice (123). However, the use of mice as a model for telomere-related human aging and aging-related human diseases is very questionable since the telomeres of most laboratory mice are 5–10 times longer than in humans (~40–50 kb), yet their life span is 30 times shorter (111, 124). Like S. cerevisiae, although genetic manipulations of telomere and telomerase may influence the organism’s life span, this effect might be overlooked while observing naive mice.
Telomere-Related Senescence in Long-Lived Animals
In a study conducted on four wild populations of long-lived bats, telomeres were shown to maintain their length in blood fibroblasts in the M. myotis species, and similar to humans, they also showed no signs of telomerase expression (125). In naked mole rats, genes involved in the function and regulation of telomerase, Tep1 (telomerase-associated protein 1) and Terf1 (telomeric repeat binding factor 1), were found to have undergone positive selection which may contribute to their slow rate of aging, though contradicting results were also published (126). For instance, a different study established that similar to mice (but unlike humans), naked mole rat somatic cells express telomerase, although at lower levels, and are not amenable to telomere-dependent replicative senescence. Gomes et al. (124) studied the telomeres of the bowhead whale lung fibroblast cells and found that the average telomere lengths was ~9 kb, in resemblance to human telomere length (124). The bowhead whale telomerase had repressed activity as well, again, similar to human telomerase (124, 127). Lai et al. (128) tested cultured bowhead whale lung fibroblasts at different population doublings and found age-related telomere shortening (128).
Human Diseases—Telomeropathies
In humans, early telomere attrition or exhaustion leads to telomeropathies (telomere syndromes) and age-related diseases (129). Telomeropathies are divided into two subgroups: primary and secondary telomeropathies. Primary telomeropathies are disorders of impaired telomere maintenance, or in other words, telomere disorders, while secondary telomeropathies are disorders in which the main mutated gene has a role in DNA repair, thus affecting telomere maintenance without actual damage to the telomere maintenance biology (130, 131). As previously mentioned, human genetic diseases are the main mode of “in vivo” research in humans. Almost all secondary telomeropathies, such as Werner syndrome and Hutchinson–Gilford progeria, are associated with premature aging and increased disease risk. Yet, most of the primary telomeropathies, such as the various forms of DKC, do not present with a progeroid phenotype but do have a wide phenotypic range which includes bone marrow failure, hair loss, emphysema, liver cirrhosis, osteoporosis, and pulmonary fibrosis. All these symptoms are also associated with aging, linking once again, the deterioration of bodily functions to shortening telomeres (130). A study conducted on 274 pairs of aged twins concluded that shortened telomeres can forecast death in the elderly (132). There are supporting (133, 134) and contradicting (135–137) evidence for this, yet the authors used intrapair comparisons on same-sex twins in order to eliminate biases of gender, genetic background, and age differences, providing another strong supportive evidence.
Telomere Position Effect—Over Long Distances
An additional effect of telomere shortening is the increase in expression of TPE-OLD (Telomere Position Effect—Over Long Distances) genes. Robin et al. demonstrated, using high-resolution Hi-C (an unbiased 3D chromatin capture technique), that long telomeres form chromatin loops reaching up to 10 Mb away from them. This loop is highly condensed causing epigenetic silencing of the genes in that region (called TPE-OLD genes). When the telomeres shorten, this loop is no longer able to form and in turn, the epigenetic regulation is changed to activation of the TPE-OLD genes. This happens before the telomeres reach the critical length that causes activation of DDR, thus leading to another earlier possible effect of telomere shortening on aging (138, 139). Interestingly, a following study by Kim et al. showed that one of the TPE-OLD sensitive genes is hTERT, the core reverse transcriptase component of telomerase (140). This is also supported by the abovementioned studies of subtelomeric regions performed in yeast.
Senescence-Related Epigenetic Alterations
Epigenetics as a field, and specifically epigenetics of aging, has gained much interest in recent years. According to Pal and Tyler (141), genetics only explain 20–30% of the aging phenomenon and researchers now aim to elucidate the remaining 70–80% mainly through epigenetics. Epigenetics can be broadly defined as changes in gene regulation without changes to the DNA coding sequence. It encompasses a range of possible changes; DNA methylation (142), histone modifications (143), various non-coding RNAs (144), and recently emerging evidence show that change in chromatin structure offers epigenetic regulation as well (145).
DNA Methylation
Age-related epigenetic modifications were shown in long-term cultures of hMSC. DNA methylation profiles of early and later passage were compared and revealed highly consistent senescence-associated (SA) modifications at specific CpG sites (66). Similar results were obtained in a long-term in vitro study of bone marrow hMSC. DNA methylation analysis revealed methylation changes between early and advanced passages. At early passages, 61.6% of all CpG islands were methylated while later, methylation decreased to 44.7% (90). A related phenotype was also observed in skeletal muscle stem cells (SCs) from young and old mice. Epigenetic profiles revealed age-related accumulation of epigenetic changes (145). Additionally, DNA methylation profiles were compared between different passages in order to identify SA changes. 1,702 CpG sites were SA hypermethylated, and 2,116 CpG sites were SA hypomethylated. SA hypermethylation was enriched in inter- and intragenic regions, and in the 3′UTR, while SA hypomethylation was highly enriched in intergenic regions (146).
The gene dDNMT2 (DNA methyltransferase) was found to be necessary for maintenance of the average life span of the flies, as mutants suffered from shorten life span. Overexpression of dDNMT2, however, extended Drosophila life span (147).
DNA methylation is also used as an “aging clock” to predict a person’s age. Horvath has provided a breakthrough “epigenetic clock” in his study from 2013. He used 8,000 samples from 82 publicly available datasets of Illumina DNA methylation arrays, including 51 tissues and cell types. This clock was able to detect the age of the sample using only 353 CpGs (148). This remarkable clock was later further improved, using fresh human blood samples, and now contains just three CpG sites (149).
Age- and Radiation-Related DNA Methylation, In Vitro
Koch et al. (146) studied age-related methylation profile in bone marrow hMSCs under several conditions and after different number of passages. Their results reveal that ionizing radiation (IR), although connected to DNA damage, did not affect age-related methylation profile. Chemical immortalization of the cells increased telomere length, but the cells still exhibited a senescence-related methylation profile. The only treatment that completely inhibited the age-related profile was “reprogramming” the cells back to their pluripotent stage (induced pluripotent stem cells) (146). It seems that although senescence has an epigenetic regulation, IR and immortalization are not connected to this process.
Histone Deacetylation—Sirtuin 2 (SIR2) and RPD3, In Vivo
Epigenetic alterations were also found to play a major role in S. Cerevisiae, C. elegans, and Drosophila life span. The histone deacetylase SIR2 was found to extend yeast life span when overexpressed, as was found in worms and flies (150, 151). A double mutant of the C. elegans SIR2 homolog significantly induced life span, and analysis revealed that the sir-2.1 functions upstream of daf-16 in the insulin-like signaling pathway (152). Also, it was found that during aging, histone H4K16 acetylation increases while H3K56 acetylation decreases (153). This is thought to be a result of the decline of SIR2 that occurs naturally during aging, which leads to H4K16 deacetylation (154). Moreover, all histone protein levels were found to descend with age which has a direct effect on the life span of the cells (155). RPD3, another histone deacetylase targeting H4K16, was also found to affect longevity in several organisms. RPD3 deletion increased S. cerevisiae life span by increasing silencing at three loci, the silent mating type (HMR), subtelomeric, and rDNA loci (96). Similarly, a fractional decrease in the levels of Rpd3 resulted in a 30–50% increase in life span of Drosophila (156, 157). Yet, Drosophila life span was not affected through gene silencing. It seems that in flies, the two deacetylases, SIR2 and RPD3, function opposingly at the euchromatin influencing gene expression and affecting longevity (156).
Age-Related Histone Deacetylation—Sirtuin Family, In Vivo
In mice, numerous sirtuin paralogs were found to improve different characteristics of aging (158, 159). Transgenic overexpression of SIRT1, an ortholog of the histone deacetylase SIR2 in yeast, improved healthy aging but did not increase longevity (160). The mechanisms involved in the beneficial effects of SIRT1 are complex and interconnected, including improved genomic stability (161, 162). Other convincing evidence for the sirtuin role in pro-longevity is the SIRT6 that modulates genomic stability through histone H3K9 deacetylation (163–165). Mutant mice that lack SIRT6 exhibit accelerated aging (166), while overexpression in male transgenic mice leads to longer life span compared to wild-type animals, an effect that is associated with reduced serum IGF-1 (Insulin Growth Factor 1) and other indicators of IGF-1 signaling (50). It has been reported that SIRT3 improves the regenerative ability of aged hematopoietic stem cells (167). Therefore, in mice, SIRT1, SIRT3, and SIRT6 contribute to healthy aging. SIRT6 has been associated with aging and disease protection through repression of aging and cancer-related transcription factors, promotion of chromatin changes essential for DNA repair, maintenance of telomere structure, and thus preventing genomic instability and senescence, in humans as well (168).
Histone Methylation, In Vivo
Greer et al. (169) discovered a crucial role for histone methylation in aging. They examined chromatin in different states and its effect on life span by investigating different enzymatic complexes and performing a targeted RNAi screening in fertile C. elegans. They discovered what is now known as the COMPASS complex, a key regulator of worm life span that acts in germline cells. This complex trimethylates histone H3 at a lysine residue (H3K4me3), and deficiencies in its members including the H3K4 methyltransferase SET2 extend life span (169). On the other hand, loss of function of the H3K4 demethylase RBR2 leads to a decreased life span, which agrees with the key idea that an increase in H3K trimethylation activates chromatin, thus promoting aging. When studying histone marks associated with repressed chromatin, Maures et al. discovered that absence of the demethylase for the repressive H3K27me3 mark—UTX1, increased worm life span separately of the germline. This mark significantly declines with normal aging in soma cell, which means that repressive H3K27me3 levels allow somatic maintenance during aging (170). Related phenotypes for H3K4me3 were also discovered in Drosophila. Overexpression of LID, a RBR2 homolog, extends life span, while its knockdown shortens life span of male flies by 18% (171). Siebold et al. (172) found that heterozygous mutations in two core subunits of PRC2 (Polycomb Repressive Complex 2), the histone H3 lysine 27 (H3K27)-specific methyltransferase E(Z), and the H3 binding protein ESC, enhanced life span and decreased H3K27me3 levels in adults. Mutations in trithorax (trx), an antagonist of Polycomb silencing, reversed the H3K27me3 level of the E(z) mutants and suppressed their enhanced longevity and resistance to oxidative stress and starvation, hinting that the reduced levels of H3K27me3 are connected to longevity and stress resistance in the PRC2 mutants (172). In drosophila, H3K27me3 seems to influence life span in an opposite manner compared to C. elegans. Mutations in H3K27 methyltransferase (PRC2) subunits E(Z) and ESC reduce global levels of H3K27me3 and extend life span of male drosophila by activating target genes Abd-B (abdominal B) and Odc1 (Ornithine Decarboxylase 1) (172).
Large-Scale Chromatin Remodeling, In Vitro
Epigenetic alterations include also genomic organization and large-scale chromatin remodeling which are facilitated by smaller scale epigenetic changes such as DNA methylation and histone post-translational modifications (PTMs). Human MSCs were also used in a recent study performed by Dillinger et al. (173) showing genomic organizational changes associated with senescence. In this study, they show using Hi-C data that there is little change in nucleolus-associated chromosomal domains between proliferating and senescent cells, yet there are large satellite repeat clusters that dissociate from centromeric and pericentromeric regions in the nucleolus during senescence (173). These findings relate back to the established aging-associated genomic instability and chromatin remodeling as discussed earlier.
Chromosomal Rearrangements, In Vivo
An examination of chromatin structure during aging in Drosophila revealed significant age-associated chromosomal rearrangements (174). In young flies, H3K9me3 and HP1 were enriched in the pericentric regions, in chromosome 4, and in heterochromatin islands spread throughout the genome. However, this enrichment decreased in an age-associated manner, equalizing H3K9me3 and HP1 levels in the pericentric regions, chromosome 4, heterochromatin, and euchromatin. Furthermore, single-cell immunohistochemistry showed changes in nuclear distribution of H3K9me3 and HP1 marks with age.
miR’s Activity, In Vivo
miR’s also play a role in aging. Liu et al. (175) showed that miR-34 regulates age-related effects and long-term brain stability in Drosophila. Expression of the drosophila mir-34 exhibits adult-onset, brain-enriched, and age-related phenotypes. While mir-34 loss induced genetic profile of brain aging, late-onset brain degeneration, and a significant decline in life span, mir-34 upregulation extended life span and reduced neurodegeneration evoked by human pathogenic polyglutamine disease protein (175). miRNAs also affect gene expression during the aging process in mice (176) and modulate senescence in human cell lines (177). Studies have found that miRNAs work in groups by modulating gene expression and silencing that can lead to age-dependent disease states or alternatively to longevity (178). Inherited epigenetic effects in miRNA loci cause changes in gene expression that modulate longevity (179), and miRNAs that target the insulin/IGF-1 pathway can foresee up to 47% of life span variations (180). Some loci show positive effects on life span, promoting longevity, while others show the opposite effect, causing a shorter life span (181). Ugalde et al. have reported that alteration in the expression of two miRNAs leads to a progeroid phenotype in a mouse model for a progeria syndrome by effecting key components of the DNA-damage response pathways (182).
Epigenetic Alterations in Long-Lived Animal Model
Only a few studies were conducted on the epigenome of the naked mole rats, especially in the context of aging. Sequencing the naked mole rat genome (183) showed that its genome had relatively low CpG density and higher fraction of CpG dinucleotides within CpG islands compared to the human genome. CpG dinucleotides within CpG islands contribute less to genetic variation because of their lower methylation rate. In a different study of the reprogramming of naked mole rat cells, analyzing the global histone landscape revealed that naked mole rats had higher levels of repressive H3K27 methylation marks and lower levels of activating H3K27 acetylation marks than mice which suggests that naked mole rats display a more stable epigenome that resists de-differentiation contributing to its longevity as well as to its resistant to cancer.
DNA Methylation—Twin Studies
Since the genomic methylation profile of each person is unique, comparative studies are needed. Monozygotic (MZ) twins have identical methylation and epigenetic patterns immediately after birth and in early childhood, making them a perfect platform for the study of methylation and epigenetic changes in general. Such a study performed in 2005 by Fraga et al. has provided many insights on the genomic methylation and gene expression changes in MZ twins of different ages. Fraga et al. were the first to look into epigenetics of MZ twins, and in their paper, they described the changes in methylation with age between the twins as “epigenetic drift.” Epigenetic drift, as they define it, is changes in the methylation profile over time due to accumulating “small defects” in transmitting epigenetic information over successive cell divisions. In other words, changes in the epigenome of an organism over time are due to random changes in methylation (184). The effect of epigenetic drift on the genome can be small or large, depending on where those changes occur. Keeping in mind that hypermethylation of promoter regions is associated with transcriptional repression, epigenetic drift can, and indeed does, cause changes in gene expression. The pattern of elevated methylation with age was also shown for general human populations (not twins) by Horvath (148) and Hannum et al. (185).
Histone PTMs
Human studies of histone PTMs related to aging are emerging and following are a few recent advances. There is accumulating evidence to the role of histones in memory and cognitive functions (186, 187) in the human brain. Hohl et al. showed that the histone methyltransferase SUV39H1 plays a role together with HDAC4 (histone deacetylase 4) in repression of pro-hypertrophic genes in the human heart (188) linking histone PTMs to cardiac stress and aging. Ucar et al. most recently published results indicating association of chromatin condensation with age in 27 histone-related genes. Among those genes were a few coding for histones (HIST1H3D, HIST1H3E, and HIST4H4) and histone modifiers such as EZH1 and SETD7 (189). These results strengthen the previously established patterns of reduction in core histone expression and changes in histone modifications (190).
Conclusion
Healthy aging and cellular senescence are complex processes of great interest to researchers. The multigenic nature of both of them complicates studies and necessitates creative and novel approaches in the path for understanding those phenomena. The three spear-headed strategies implemented for this purpose have brought forth much information and knowledge, yet there is still much to learn in these fields. The doubting and contradicting results in in vivo studies are influenced both by physiological and genetic differences between the model organisms and humans and the differences in the possible research methodologies between in vitro and in vivo studies. In many cases, the age-related phenotypes searched for and studied in vitro are not visible in vivo or not relevant for the model organism (Table 1.).
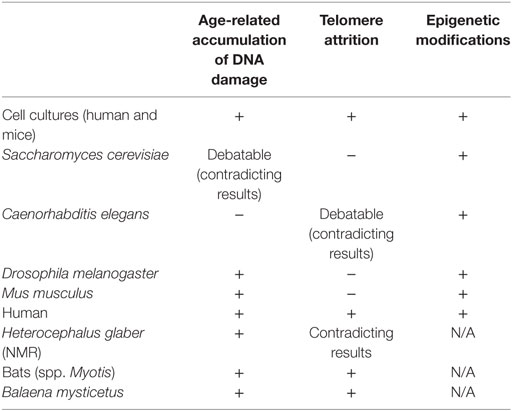
Table 1. Evidence for correlation between DNA damage accumulation, telomeres attrition and epigenetic alterations, and aging in In Vitro, In Vivo, and aging-like human syndromes studies.
Molecular processes such as DNA damage repair, telomere shortening, and epigenetic alterations discussed earlier are the driving forces of the aging process in human, but their significance is varied in other organisms. Many evidence for age-related accumulation of DNA damage were found in in vitro studies, both in human and mice cell cultures. The connection between DNA damage and aging is emphasized by the secretion of senescence-associated proteins during cellular senescence, a phenotype which is activated by DNA damage and is common for both human and mice. Human progeroid diseases also show the connection between early aging and faulty DNA repair. In yeast, flies and mice, however, although some evidence for age-related damage and faulty DNA repair mechanisms were found, contradicting and debating results highlight the complexity of the use of these model organisms in this aging research. The study of telomeres in relation to aging demonstrates the questions derived from both physiological differences between organisms and differences in research approaches. The connection between telomere attrition and aging is very present in human aging (both in in vitro studies and as telomeropathies such as DKC, Werner syndrome, and Hutchinson–Gilford progeria) but not relevant in model organisms. In C. elegans, the evidence are contradicting. In drosophila, maybe because of the unique telomere structure, there are no evidence connecting telomere attrition to aging. In yeast and mice, genetic manipulations enabled the study of telomere-aging relations, but such relations were not seen in wild-type subjects. The study of telomere-related aging in mice especially feature the difficulties of comparing human and model organisms, since the telomeres of most laboratory mice are 5–10 times longer than in humans, but their life span is much shorter.
Interestingly, the only common effector on aging found among cell cultures, different model organisms, and humans is epigenetic modifications. Epigenetic modifications are indeed a part of every genetic response in the cell, but the existence of common age-related modifications and key-players is intriguing. Epigenetic alterations are “core” elements in cellular responses. They play an upstream role to specific cellular processes, and this might be the reason for the relatively joint phenotypes. Furthermore, epigenetic modifications that are related to age-associated chromosomal rearrangements in yeast and flies might be a link to age-related DNA damage, where direct evidence were not found.
Though much progress has been achieved, full understanding of these mechanisms has still a long way to go. New tools such as GWAS and EWAS studies hold the potential to further elucidate the aging phenotype by investigating large datasets obtained from human subjects, but, it is still important and useful to study the above discussed strategies and organisms. However, the selection of those organisms will have to be more conscious and target-based.
Author Contributions
GL, DG, and HS wrote and edited the article; LS and GA edited the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AP and the handling Editor declared their shared affiliation.
References
1. Turinetto V, Vitale E, Giachino C. Senescence in human mesenchymal stem cells: functional changes and implications in stem cell-based therapy. Int J Mol Sci (2016) 17:1164. doi:10.3390/ijms17071164
2. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev (2014) 28:99–114. doi:10.1101/gad.235184.113
3. Vermeij WP, Hoeijmakers JH, Pothof J. Genome integrity in aging: human syndromes, mouse models, and therapeutic options. Annu Rev Pharmacol Toxicol (2016) 56:427–45. doi:10.1146/annurev-pharmtox-010814-124316
4. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153:1194–217. doi:10.1016/j.cell.2013.05.039
5. Moskalev A, Proshkina E, Belyi A, Solovyev I. Genetics of aging and longevity. Russ J Genet Appl Res (2017) 7:369–84. doi:10.1134/S2079059717040074
6. Cohen AA. Aging across the tree of life: the importance of a comparative perspective for the use of animal models in aging. Biochim Biophys Acta (2017). doi:10.1016/j.bbadis.2017.05.028
7. Van Deursen JM. The role of senescent cells in ageing. Nature (2014) 509:439. doi:10.1038/nature13193
8. Bhatia-Dey N, Kanherkar RR, Stair SE, Makarev EO, Csoka AB. Cellular senescence as the causal nexus of aging. Front Genet (2016) 7:13. doi:10.3389/fgene.2016.00013
9. D’adda Di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer (2008) 8:512. doi:10.1038/nrc2440
10. Carrel A. On the permanent life of tissues outside of the organism. J Exp Med (1912) 15:516–28. doi:10.1084/jem.15.5.516
11. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res (1961) 25:585–621. doi:10.1016/0014-4827(61)90192-6
12. Campisi J. From cells to organisms: can we learn about aging from cells in culture? Exp Gerontol (2001) 36:607–18. doi:10.1016/S0531-5565(00)00230-8
13. Hartung T. Look back in anger – what clinical studies tell us about preclinical work. ALTEX (2013) 30:275–91. doi:10.14573/altex.2013.3.275
14. Medvedev ZA. An attempt at a rational classification of theories of ageing. Biol Rev (1990) 65:375–98. doi:10.1111/j.1469-185X.1990.tb01428.x
15. Shefferson RP, Jones OR, Salguero-Gómez R. The Evolution of Senescence in the Tree of Life. Cambridge: Cambridge University Press (2017).
16. Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature (2000) 408:255–62. doi:10.1038/35041700
17. Collins JJ, Huang C, Hughes S, Kornfeld K. The measurement and analysis of age-related changes in Caenorhabditis elegans. WormBook (2007) 1–21. doi:10.1895/wormbook.1.137.1
18. McGee MD, Weber D, Day N, Vitelli C, Crippen D, Herndon LA, et al. Loss of intestinal nuclei and intestinal integrity in aging C. elegans. Aging Cell (2011) 10:699–710. doi:10.1111/j.1474-9726.2011.00713.x
19. Hughes SE, Huang C, Kornfeld K. Identification of mutations that delay somatic or reproductive aging of Caenorhabditis elegans. Genetics (2011) 189:341–56. doi:10.1534/genetics.111.130450
20. Luo S, Kleemann GA, Ashraf JM, Shaw WM, Murphy CT. TGF-β and insulin signaling regulate reproductive aging via oocyte and germline quality maintenance. Cell (2010) 143:299–312. doi:10.1016/j.cell.2010.09.013
21. Golden TR, Hubbard A, Dando C, Herren MA, Melov S. Age-related behaviors have distinct transcriptional profiles in Caenorhabditis elegans. Aging Cell (2008) 7:850–65. doi:10.1111/j.1474-9726.2008.00433.x
22. Biteau B, Karpac J, Hwangbo D, Jasper H. Regulation of Drosophila lifespan by JNK signaling. Exp Gerontol (2011) 46:349–54. doi:10.1016/j.exger.2010.11.003
23. Karpac J, Younger A, Jasper H. Dynamic coordination of innate immune signaling and insulin signaling regulates systemic responses to localized DNA damage. Dev Cell (2011) 20:841–54. doi:10.1016/j.devcel.2011.05.011
24. Karpac J, Jasper H. Insulin and JNK: optimizing metabolic homeostasis and lifespan. Trends Endocrinol Metab (2009) 20:100–6. doi:10.1016/j.tem.2008.11.004
25. Demontis F, Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell (2010) 143:813–25. doi:10.1016/j.cell.2010.10.007
26. Edrey YH, Hanes M, Pinto M, Mele J, Buffenstein R. Successful aging and sustained good health in the naked mole rat: a long-lived mammalian model for biogerontology and biomedical research. ILAR J (2011) 52:41–53. doi:10.1093/ilar.52.1.41
27. Keane M, Semeiks J, Webb AE, Li YI, Quesada V, Craig T, et al. Insights into the evolution of longevity from the bowhead whale genome. Cell Rep (2015) 10:112–22. doi:10.1016/j.celrep.2014.12.008
28. Gordon LB, Rothman FG, López-Otín C, Misteli T. Progeria: a paradigm for translational medicine. Cell (2014) 156:400–7. doi:10.1016/j.cell.2013.12.028
29. Nebel A, Kleindorp R, Caliebe A, Nothnagel M, Blanché H, Junge O, et al. A genome-wide association study confirms APOE as the major gene influencing survival in long-lived individuals. Mech Ageing Dev (2011) 132:324–30. doi:10.1016/j.mad.2011.06.008
30. Broer L, Buchman AS, Deelen J, Evans DS, Faul JD, Lunetta KL, et al. GWAS of longevity in CHARGE consortium confirms APOE and FOXO3 candidacy. J Gerontol A Biomed Sci Med Sci (2014) 70:110–8. doi:10.1093/gerona/glu166
31. Pilling LC, Atkins JL, Bowman K, Jones SE, Tyrrell J, Beaumont RN, et al. Human longevity is influenced by many genetic variants: evidence from 75,000 UK Biobank participants. Aging (Albany NY) (2016) 8:547–60. doi:10.18632/aging.100930
32. Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature (2016) 538:260–4. doi:10.1038/nature19768
33. Pan M, Li K, Lin S, Hung W. Connecting the dots: from DNA damage and repair to aging. Int J Mol Sci (2016) 17:685. doi:10.3390/ijms17050685
34. Fumagalli M, Rossiello F, Mondello C, d’Adda di Fagagna F. Stable cellular senescence is associated with persistent DDR activation. PLoS One (2014) 9:e110969. doi:10.1371/journal.pone.0110969
35. James EL, Lane JA, Michalek RD, Karoly ED, Parkinson EK. Replicatively senescent human fibroblasts reveal a distinct intracellular metabolic profile with alterations in NAD and nicotinamide metabolism. Sci Rep (2016) 6:38489. doi:10.1038/srep38489
36. Lindahl T. New class of enzymes acting on damaged DNA. Nature (1976) 259:64. doi:10.1038/259064a0
37. Lahue RS, Au KG, Modrich P. DNA mismatch correction in a defined system. Science (1989) 245:160–4. doi:10.1126/science.2665076
38. Wang Y, Li F, Zhang G, Kang L, Qin B, Guan H. Altered DNA methylation and expression profiles of 8-oxoguanine DNA glycosylase 1 in lens tissue from age-related cataract patients. Curr Eye Res (2015) 40:815–21. doi:10.3109/02713683.2014.957778
39. Xu Z, Zhang L, Zhang W, Meng D, Zhang H, Jiang Y, et al. SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle (2015) 14:269–76. doi:10.4161/15384101.2014.980641
40. Tan J, Xu X, Tong Z, Lin J, Yu Q, Lin Y, et al. Decreased osteogenesis of adult mesenchymal stem cells by reactive oxygen species under cyclic stretch: a possible mechanism of age related osteoporosis. Bone Res (2015) 3:15003. doi:10.1038/boneres.2015.3
41. Vitale I, Manic G, De Maria R, Kroemer G, Galluzzi L. DNA damage in stem cells. Mol Cell (2017) 66:306–19. doi:10.1016/j.molcel.2017.04.006
42. Li Z, Zhang W, Chen Y, Guo W, Zhang J, Tang H, et al. Impaired DNA double-strand break repair contributes to the age-associated rise of genomic instability in humans. Cell Death Differ (2016) 23:1765–77. doi:10.1038/cdd.2016.65
43. McVey M, Kaeberlein M, Tissenbaum HA, Guarente L. The short life span of Saccharomyces cerevisiae sgs1 and srs2 mutants is a composite of normal aging processes and mitotic arrest due to defective recombination. Genetics (2001) 157:1531–42.
44. Hoopes LL, Budd M, Choe W, Weitao T, Campbell JL. Mutations in DNA replication genes reduce yeast life span. Mol Cell Biol (2002) 22:4136–46. doi:10.1128/MCB.22.12.4136-4146.2002
45. Shaposhnikov M, Proshkina E, Shilova L, Zhavoronkov A, Moskalev A. Lifespan and stress resistance in Drosophila with overexpressed DNA repair genes. Sci Rep (2015) 5:sre15299. doi:10.1038/srep15299
46. Greer C, Lee M, Westerhof M, Milholland B, Spokony R, Vijg J, et al. Myc-dependent genome instability and lifespan in Drosophila. PLoS One (2013) 8:e74641. doi:10.1371/journal.pone.0074641
47. Liu B, Yip RKH, Zhou Z. Chromatin remodeling, DNA damage repair and aging. Curr Genomics (2012) 13:533–47. doi:10.2174/138920212803251373
48. Mah L, El-Osta A, Karagiannis T. γH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia (2010) 24:679–86. doi:10.1038/leu.2010.6
49. Sedelnikova OA, Horikawa I, Redon C, Nakamura A, Zimonjic DB, Popescu NC, et al. Delayed kinetics of DNA double-strand break processing in normal and pathological aging. Aging Cell (2008) 7:89–100. doi:10.1111/j.1474-9726.2007.00354.x
50. Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature (2012) 483:218–21. doi:10.1038/nature10815
51. Mao Z, Tian X, Van Meter M, Ke Z, Gorbunova V, Seluanov A. Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc Natl Acad Sci U S A (2012) 109:11800–5. doi:10.1073/pnas.1200583109
52. Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science (2011) 332:1443–6. doi:10.1126/science.1202723
53. Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet (2012) 44:642–50. doi:10.1038/ng.2271
54. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med (2014) 371:2488–98. doi:10.1056/NEJMoa1408617
55. Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM. Naive T cell maintenance and function in human aging. J Immunol (2015) 194:4073–80. doi:10.4049/jimmunol.1500046
56. Zhang G, Cowled C, Shi Z, Huang Z, Bishop-Lilly KA, Fang X, et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science (2013) 339:456–60. doi:10.1126/science.1230835
57. Tollis M, Schiffman JD, Boddy AM. Evolution of cancer suppression as revealed by mammalian comparative genomics. Curr Opin Genet Dev (2017) 42:40–7. doi:10.1016/j.gde.2016.12.004
58. Selfridge J, Hsia K, Redhead NJ, Melton DW. Correction of liver dysfunction in DNA repair-deficient mice with an ERCC1 transgene. Nucleic Acids Res (2001) 29:4541–50. doi:10.1093/nar/29.22.4541
59. Ma S, Upneja A, Galecki A, Tsai YM, Burant CF, Raskind S, et al. Cell culture-based profiling across mammals reveals DNA repair and metabolism as determinants of species longevity. Elife (2016) 5:e19130. doi:10.7554/eLife.19130
60. Conde-Pérezprina JC, Luna-López A, González-Puertos VY, Zenteno-Savín T, Á León-Galván M, Königsberg M. DNA MMR systems, microsatellite instability and antioxidant activity variations in two species of wild bats: Myotis velifer and Desmodus rotundus, as possible factors associated with longevity. Age (2012) 34:1473–92. doi:10.1007/s11357-012-9399-5
61. Fang X, Seim I, Huang Z, Gerashchenko MV, Xiong Z, Turanov AA, et al. Adaptations to a subterranean environment and longevity revealed by the analysis of mole rat genomes. Cell Rep (2014) 8:1354–64. doi:10.1016/j.celrep.2014.07.030
62. MacRae SL, Croken MM, Calder RB, Aliper A, Milholland B, White RR, et al. DNA repair in species with extreme lifespan differences. Aging (Albany NY) (2015) 7:1171–84. doi:10.18632/aging.100866
63. Andziak B, O’Connor TP, Buffenstein R. Antioxidants do not explain the disparate longevity between mice and the longest-living rodent, the naked mole-rat. Mech Ageing Dev (2005) 126:1206–12. doi:10.1016/j.mad.2005.06.009
64. Csiszar A, Labinskyy N, Orosz Z, Xiangmin Z, Buffenstein R, Ungvari Z. Vascular aging in the longest-living rodent, the naked mole rat. Am J Physiol Heart Circ Physiol (2007) 293:H919–27. doi:10.1152/ajpheart.01287.2006
65. Page MM, Stuart JA. Activities of DNA base excision repair enzymes in liver and brain correlate with body mass, but not lifespan. Age (2012) 34:1195–209. doi:10.1007/s11357-011-9302-9
66. Schellenberg A, Lin Q, Schuler H, Koch CM, Joussen S, Denecke B, et al. Replicative senescence of mesenchymal stem cells causes DNA-methylation changes which correlate with repressive histone marks. Aging (Albany NY) (2011) 3:873–88. doi:10.18632/aging.100391
67. Annett K, Duggan O, Freeburn R, Hyland P, Pawelec G, Barnett Y. An investigation of DNA mismatch repair capacity under normal culture conditions and under conditions of supra-physiological challenge in human CD4 T cell clones from donors of different ages. Exp Gerontol (2005) 40:976–81. doi:10.1016/j.exger.2005.09.001
68. IJpma AS, Greider CW. Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Mol Biol Cell (2003) 14:987–1001. doi:10.1091/mbc.02-04-0057
69. Teixeira MT. Saccharomyces cerevisiae as a model to study replicative senescence triggered by telomere shortening. Front Oncol (2013) 3:101. doi:10.3389/fonc.2013.00101
70. Kennedy BK, Austriaco NR Jr, Guarente L. Daughter cells of Saccharomyces cerevisiae from old mothers display a reduced life span. J Cell Biol (1994) 127:1985–93. doi:10.1083/jcb.127.6.1985
71. Denoth Lippuner A, Julou T, Barral Y. Budding yeast as a model organism to study the effects of age. FEMS Microbiol Rev (2014) 38:300–25. doi:10.1111/1574-6976.12060
72. Kaya A, Lobanov AV, Gladyshev VN. Evidence that mutation accumulation does not cause aging in Saccharomyces cerevisiae. Aging Cell (2015) 14:366–71. doi:10.1111/acel.12290
73. Sinclair DA, Guarente L. Extrachromosomal rDNA circles – a cause of aging in yeast. Cell (1997) 91:1033–42. doi:10.1016/S0092-8674(00)80493-6
74. Defossez P, Prusty R, Kaeberlein M, Lin S, Ferrigno P, Silver PA, et al. Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol Cell (1999) 3:447–55. doi:10.1016/S1097-2765(00)80472-4
75. Tacutu R, Craig T, Budovsky A, Wuttke D, Lehmann G, Taranukha D, et al. Human ageing genomic resources: integrated databases and tools for the biology and genetics of ageing. Nucleic Acids Res (2012) 41:D1027–33. doi:10.1093/nar/gks1155
76. Gonzalo S, Eissenberg JC. Tying up loose ends: telomeres, genomic instability and lamins. Curr Opin Genet Dev (2016) 37:109–18. doi:10.1016/j.gde.2016.03.003
77. Edwards DN, Orren DK, Machwe A. Strand exchange of telomeric DNA catalyzed by the Werner syndrome protein (WRN) is specifically stimulated by TRF2. Nucleic Acids Res (2014) 42:7748–61. doi:10.1093/nar/gku454
78. Aamann MD, Muftuoglu M, Bohr VA, Stevnsner T. Multiple interaction partners for Cockayne syndrome proteins: implications for genome and transcriptome maintenance. Mech Ageing Dev (2013) 134:212–24. doi:10.1016/j.mad.2013.03.009
80. Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol (2014) 15:7–18. doi:10.1038/nrm3719
81. Gorbunova V, Seluanov A. DNA double strand break repair, aging and the chromatin connection. Mutat Res (2016) 788:2–6. doi:10.1016/j.mrfmmm.2016.02.004
82. Ribezzo F, Shiloh Y, Schumacher B. Systemic DNA damage responses in aging and diseases. Semin Cancer Biol (2016) 37:26–35. doi:10.1016/j.semcancer.2015.12.005
83. Deng Z, Glousker G, Molczan A, Fox AJ, Lamm N, Dheekollu J, et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci U S A (2013) 110:E3408–16. doi:10.1073/pnas.1300600110
84. Bernadotte A, Mikhelson VM, Spivak IM. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging (Albany NY) (2016) 8:3–11. doi:10.18632/aging.100871
85. Olovnikov AM. A theory of marginotomy: the incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol (1973) 41:181–90. doi:10.1016/0022-5193(73)90198-7
86. Greider CW, Blackburn EH. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell (1987) 51:887–98. doi:10.1016/0092-8674(87)90576-9
87. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science (1998) 279:349–52. doi:10.1126/science.279.5349.349
88. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature (1990) 345:458–60. doi:10.1038/345458a0
89. Murillo-Ortiz B, Albarrán-Tamayo F, López-Briones S, Martínez-Garza S, Benítez-Bribiesca L, Arenas-Aranda D. Increased telomere length and proliferative potential in peripheral blood mononuclear cells of adults of different ages stimulated with concanavalin A. BMC Geriatr (2013) 13:99. doi:10.1186/1471-2318-13-99
90. Redaelli S, Bentivegna A, Foudah D, Miloso M, Redondo J, Riva G, et al. From cytogenomic to epigenomic profiles: monitoring the biologic behavior of in vitro cultured human bone marrow mesenchymal stem cells. Stem Cell Res Ther (2012) 3:47. doi:10.1186/scrt138
91. Alt EU, Senst C, Murthy SN, Slakey DP, Dupin CL, Chaffin AE, et al. Aging alters tissue resident mesenchymal stem cell properties. Stem Cell Res (2012) 8:215–25. doi:10.1016/j.scr.2011.11.002
92. Rübe CE, Fricke A, Widmann TA, Fürst T, Madry H, Pfreundschuh M, et al. Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS One (2011) 6:e17487. doi:10.1371/journal.pone.0017487
93. Janssens GE, Veenhoff LM. Evidence for the hallmarks of human aging in replicatively aging yeast. Microb Cell (2016) 3:263–74. doi:10.15698/mic2016.07.510
94. Lundblad V, Blackburn EH. An alternative pathway for yeast telomere maintenance rescues est1-senescence. Cell (1993) 73:347–60. doi:10.1016/0092-8674(93)90234-H
95. Becerra SC, Thambugala HT, Erickson AR, Lee CK, Lewis LK. Reversibility of replicative senescence in Saccharomyces cerevisiae: effect of homologous recombination and cell cycle checkpoints. DNA Repair (2012) 11:35–45. doi:10.1016/j.dnarep.2011.10.003
96. Kim S, Villeponteau B, Jazwinski SM. Effect of replicative age on transcriptional silencing near telomeres in Saccharomyces cerevisiae. Biochem Biophys Res Commun (1996) 219:370–6. doi:10.1006/bbrc.1996.0240
97. Austriaco NR Jr, Guarente LP. Changes of telomere length cause reciprocal changes in the lifespan of mother cells in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A (1997) 94:9768–72. doi:10.1073/pnas.94.18.9768
98. Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell (2005) 120:449–60. doi:10.1016/j.cell.2005.02.002
99. Kappei D, Londoño-Vallejo JA. Telomere length inheritance and aging. Mech Ageing Dev (2008) 129:17–26. doi:10.1016/j.mad.2007.10.009
100. Joeng KS, Song EJ, Lee K, Lee J. Long lifespan in worms with long telomeric DNA. Nat Genet (2004) 36:607–11. doi:10.1038/ng1356
101. Klass MR. A method for the isolation of longevity mutants in the nematode Caenorhabditis elegans and initial results. Mech Ageing Dev (1983) 22:279–86. doi:10.1016/0047-6374(83)90082-9
102. Friedman DB, Johnson TE. Three mutants that extend both mean and maximum life span of the nematode, Caenorhabditis elegans, define the age-1 gene. J Gerontol (1988) 43:B102–9. doi:10.1093/geronj/43.4.B102
103. Raices M, Maruyama H, Dillin A, Karlseder J. Uncoupling of longevity and telomere length in C. elegans. PLoS Genet (2005) 1:e30. doi:10.1371/journal.pgen.0010030
104. Meier B, Clejan I, Liu Y, Lowden M, Gartner A, Hodgkin J, et al. trt-1 is the Caenorhabditis elegans catalytic subunit of telomerase. PLoS Genet (2006) 2:e18. doi:10.1371/journal.pgen.0020018
105. Cheung I, Schertzer M, Rose A, Lansdorp PM. High incidence of rapid telomere loss in telomerase-deficient Caenorhabditis elegans. Nucleic Acids Res (2006) 34:96–103. doi:10.1093/nar/gkj417
106. Ahmed S, Hodgkin J. MRT-2 checkpoint protein is required for germline immortality and telomere replication in C. elegans. Nature (2000) 403:159–64. doi:10.1038/35003120
107. Raffa GD, Cenci G, Siriaco G, Goldberg ML, Gatti M. The putative Drosophila transcription factor woc is required to prevent telomeric fusions. Mol Cell (2005) 20:821–31. doi:10.1016/j.molcel.2005.12.003
108. Mason JM, Frydrychova RC, Biessmann H. Drosophila telomeres: an exception providing new insights. Bioessays (2008) 30:25–37. doi:10.1002/bies.20688
109. Jain D, Cooper JP. Telomeric strategies: means to an end. Annu Rev Genet (2010) 44:243–69. doi:10.1146/annurev-genet-102108-134841
110. Casacuberta E. Drosophila: retrotransposons making up telomeres. Viruses (2017) 9:192. doi:10.3390/v9070192
111. Vera E, de Jesus BB, Foronda M, Flores JM, Blasco MA. The rate of increase of short telomeres predicts longevity in mammals. Cell Rep (2012) 2:732–7. doi:10.1016/j.celrep.2012.08.023
112. Walter MF, Biessmann MR, Benitez C, Török T, Mason JM, Biessmann H. Effects of telomere length in Drosophila melanogaster on life span, fecundity, and fertility. Chromosoma (2007) 116:41–51. doi:10.1007/s00412-006-0081-5
113. Pletcher SD, Macdonald SJ, Marguerie R, Certa U, Stearns SC, Goldstein DB, et al. Genome-wide transcript profiles in aging and calorically restricted Drosophila melanogaster. Curr Biol (2002) 12:712–23. doi:10.1016/S0960-9822(02)00808-4
114. Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science (2001) 292:107–10. doi:10.1126/science.1057987
115. Armanios M, Alder JK, Parry EM, Karim B, Strong MA, Greider CW. Short telomeres are sufficient to cause the degenerative defects associated with aging. Am J Hum Genet (2009) 85:823–32. doi:10.1016/j.ajhg.2009.10.028
116. Blasco MA, Lee H, Hande MP, Samper E, Lansdorp PM, DePinho RA, et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell (1997) 91:25–34. doi:10.1016/S0092-8674(01)80006-4
117. Herrera E, Samper E, Martin-Caballero J, Flores JM, Lee HW, Blasco MA. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J (1999) 18:2950–60. doi:10.1093/emboj/18.11.2950
118. Rudolph KL, Chang S, Lee H, Blasco M, Gottlieb GJ, Greider C, et al. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell (1999) 96:701–12. doi:10.1016/S0092-8674(00)80580-2
119. Tomás-Loba A, Flores I, Fernández-Marcos PJ, Cayuela ML, Maraver A, Tejera A, et al. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell (2008) 135:609–22. doi:10.1016/j.cell.2008.09.034
120. Jaskelioff M, Muller FL, Paik J, Thomas E, Jiang S, Adams AC, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature (2011) 469:102–6. doi:10.1038/nature09603
121. Bernardes de Jesus B, Vera E, Schneeberger K, Tejera AM, Ayuso E, Bosch F, et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med (2012) 4:691–704. doi:10.1002/emmm.201200245
122. Espejel S, Klatt P, Menissier-de Murcia J, Martin-Caballero J, Flores JM, Taccioli G, et al. Impact of telomerase ablation on organismal viability, aging, and tumorigenesis in mice lacking the DNA repair proteins PARP-1, Ku86, or DNA-PKcs. J Cell Biol (2004) 167:627–38. doi:10.1083/jcb.200407178
123. Calado RT, Dumitriu B. Telomere dynamics in mice and humans. Semin Hematol (2013) 50:165–74. doi:10.1053/j.seminhematol.2013.03.030
124. Gomes N, Ryder OA, Houck ML, Charter SJ, Walker W, Forsyth NR, et al. Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell (2011) 10:761–8. doi:10.1111/j.1474-9726.2011.00718.x
125. Foley NM, Hughes GM, Huang Z, Clarke M, Jebb D, Whelan CV, et al. Growing old, yet staying young: the role of telomeres in bats’ exceptional longevity. Sci Adv (2018) 4:eaao0926. doi:10.1126/sciadv.aao0926
126. Yu C, Li Y, Holmes A, Szafranski K, Faulkes CG, Coen CW, et al. RNA sequencing reveals differential expression of mitochondrial and oxidation reduction genes in the long-lived naked mole-rat when compared to mice. PLoS One (2011) 6:e26729. doi:10.1371/journal.pone.0026729
127. Aviv A, Shay JW. Reflections on telomere dynamics and ageing-related diseases in humans. Philos Trans R Soc Lond B Biol Sci (2018) 373:20160436. doi:10.1098/rstb.2016.0436
128. Lai T, Zhang N, Noh J, Mender I, Tedone E, Huang E, et al. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nat Commun (2017) 8:1356. doi:10.1038/s41467-017-01291-z
129. Bar C, Blasco MA. Telomeres and telomerase as therapeutic targets to prevent and treat age-related diseases. F1000Res (2016) 5(F1000 Faculty Rev):89. doi:10.12688/f1000research.7020.1
130. Opresko PL, Shay JW. Telomere-associated aging disorders. Ageing Res Rev (2017) 33:52–66. doi:10.1016/j.arr.2016.05.009
131. Holohan B, Wright WE, Shay JW. Cell biology of disease: telomeropathies: an emerging spectrum disorder. J Cell Biol (2014) 205:289–99. doi:10.1083/jcb.201401012
132. Kimura M, Hjelmborg JVB, Gardner JP, Bathum L, Brimacombe M, Lu X, et al. Telomere length and mortality: a study of leukocytes in elderly Danish twins. Am J Epidemiol (2008) 167:799–806. doi:10.1093/aje/kwm380
133. Cawthon RM, Smith KR, O’Brien E, Sivatchenko A, Kerber RA. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet (2003) 361:393–5. doi:10.1016/S0140-6736(03)12384-7
134. Honig LS, Schupf N, Lee JH, Tang MX, Mayeux R. Shorter telomeres are associated with mortality in those with APOE ϵ4 and dementia. Ann Neurol (2006) 60:181–7. doi:10.1002/ana.20894
135. Martin-Ruiz CM, Baird D, Roger L, Boukamp P, Krunic D, Cawthon R, et al. Reproducibility of telomere length assessment: an international collaborative study. Int J Epidemiol (2014) 44:1673–83. doi:10.1093/ije/dyu191
136. Bischoff C, Petersen HC, Graakjaer J, Andersen-Ranberg K, Vaupel JW, Bohr VA, et al. No association between telomere length and survival among the elderly and oldest old. Epidemiology (2006) 17:190–4. doi:10.1097/01.ede.0000199436.55248.10
137. Harris SE, Deary IJ, MacIntyre A, Lamb KJ, Radhakrishnan K, Starr JM, et al. The association between telomere length, physical health, cognitive ageing, and mortality in non-demented older people. Neurosci Lett (2006) 406:260–4. doi:10.1016/j.neulet.2006.07.055
138. Robin JD, Ludlow AT, Batten K, Magdinier F, Stadler G, Wagner KR, et al. Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances. Genes Dev (2014) 28:2464–76. doi:10.1101/gad.251041.114
140. Kim W, Ludlow AT, Min J, Robin JD, Stadler G, Mender I, et al. Regulation of the human telomerase gene TERT by telomere position effect-over long distances (TPE-OLD): implications for aging and cancer. PLoS Biol (2016) 14:e2000016. doi:10.1371/journal.pbio.2000016
142. Zhang Y, Wilson R, Heiss J, Breitling LP, Saum K, Schöttker B, et al. DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat Commun (2017) 8:14617. doi:10.1038/ncomms14617
143. Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci U S A (1964) 51:786–94. doi:10.1073/pnas.51.5.786
144. Coelho-Lima J, Spyridopoulos I. Non-coding RNA regulation of T cell biology: implications for age-associated cardiovascular diseases. Exp Gerontol (2017). doi:10.1016/j.exger.2017.06.014
145. Liu L, Cheung TH, Charville GW, Hurgo BMC, Leavitt T, Shih J, et al. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep (2013) 4:189–204. doi:10.1016/j.celrep.2013.05.043
146. Koch CM, Reck K, Shao K, Lin Q, Joussen S, Ziegler P, et al. Pluripotent stem cells escape from senescence-associated DNA methylation changes. Genome Res (2013) 23:248–59. doi:10.1101/gr.141945.112
147. Lin MJ, Tang LY, Reddy MN, Shen CK. DNA methyltransferase gene dDnmt2 and longevity of Drosophila. J Biol Chem (2005) 280:861–4. doi:10.1074/jbc.C400477200
148. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol (2013) 14:3156. doi:10.1186/gb-2013-14-10-r115
149. Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol (2014) 15:R24. doi:10.1186/gb-2014-15-2-r24
150. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev (1999) 13:2570–80. doi:10.1101/gad.13.19.2570
151. Guarente L. Sirtuins, aging, and metabolism. Cold Spring Harb Symp Quant Biol (2011) 76:81–90. doi:10.1101/sqb.2011.76.010629
152. Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature (2001) 410:227–30. doi:10.1038/35065638
153. Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature (2009) 459:802–7. doi:10.1038/nature08085
154. Suka N, Luo K, Grunstein M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat Genet (2002) 32:378–83. doi:10.1038/ng1017
155. Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, et al. Elevated histone expression promotes life span extension. Mol Cell (2010) 39:724–35. doi:10.1016/j.molcel.2010.08.015
156. Frankel S, Rogina B. Drosophila longevity is not affected by heterochromatin-mediated gene silencing. Aging Cell (2005) 4:53–6. doi:10.1111/j.1474-9726.2005.00143.x
157. Rogina B, Helfand SL, Frankel S. Longevity regulation by Drosophila Rpd3 deacetylase and caloric restriction. Science (2002) 298:1745. doi:10.1126/science.1078986
158. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol (2012) 13:225–38. doi:10.1038/nrm3293
159. Sebastian C, Satterstrom FK, Haigis MC, Mostoslavsky R. From sirtuin biology to human diseases: an update. J Biol Chem (2012) 287:42444–52. doi:10.1074/jbc.R112.402768
160. Herranz D, Muñoz-Martin M, Cañamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, et al. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun (2010) 1:3. doi:10.1038/ncomms1001
161. Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park S, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell (2008) 135:907–18. doi:10.1016/j.cell.2008.10.025
162. Wang R, Sengupta K, Li C, Kim H, Cao L, Xiao C, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell (2008) 14:312–23. doi:10.1016/j.ccr.2008.09.001
163. Kanfi Y, Peshti V, Gil R, Naiman S, Nahum L, Levin E, et al. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell (2010) 9:162–73. doi:10.1111/j.1474-9726.2009.00544.x
164. Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-κB-dependent gene expression and organismal life span. Cell (2009) 136:62–74. doi:10.1016/j.cell.2008.10.052
165. Zhang Z, Lowry SF, Guarente L, Haimovich B. Roles of SIRT1 in the acute and restorative phases following induction of inflammation. J Biol Chem (2010) 285:41391–401. doi:10.1074/jbc.M110.174482
166. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell (2006) 124:315–29. doi:10.1016/j.cell.2005.11.044
167. Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, et al. SIRT3 reverses aging-associated degeneration. Cell rep (2013) 3:319–27. doi:10.1016/j.celrep.2013.01.005
168. Tasselli L, Zheng W, Chua KF. SIRT6: novel mechanisms and links to aging and disease. Trends Endocrinol Metab (2017) 28:168–85. doi:10.1016/j.tem.2016.10.002
169. Greer EL, Maures TJ, Hauswirth AG, Green EM, Leeman DS, Maro GS, et al. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature (2010) 466:383–7. doi:10.1038/nature09195
170. Maures TJ, Greer EL, Hauswirth AG, Brunet A. The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell (2011) 10:980–90. doi:10.1111/j.1474-9726.2011.00738.x
171. Li L, Greer C, Eisenman RN, Secombe J. Essential functions of the histone demethylase lid. PLoS Genet (2010) 6:e1001221. doi:10.1371/journal.pgen.1001221
172. Siebold AP, Banerjee R, Tie F, Kiss DL, Moskowitz J, Harte PJ. Polycomb repressive complex 2 and trithorax modulate Drosophila longevity and stress resistance. Proc Natl Acad Sci U S A (2010) 107:169–74. doi:10.1073/pnas.0907739107
173. Dillinger S, Straub T, Nemeth A. Nucleolus association of chromosomal domains is largely maintained in cellular senescence despite massive nuclear reorganisation. PLoS One (2017) 12:e0178821. doi:10.1371/journal.pone.0178821
174. Wood JG, Hillenmeyer S, Lawrence C, Chang C, Hosier S, Lightfoot W, et al. Chromatin remodeling in the aging genome of Drosophila. Aging Cell (2010) 9:971–8. doi:10.1111/j.1474-9726.2010.00624.x
175. Liu N, Landreh M, Cao K, Abe M, Hendriks G, Kennerdell JR, et al. The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature (2012) 482:519–23. doi:10.1038/nature10810
176. Inukai S, de Lencastre A, Turner M, Slack F. Novel microRNAs differentially expressed during aging in the mouse brain. PLoS One (2012) 7:e40028. doi:10.1371/journal.pone.0040028
177. Smith-Vikos T, Slack FJ. MicroRNAs and their roles in aging. J Cell Sci (2012) 125:7–17. doi:10.1242/jcs.099200
178. Lanceta J, Prough RA, Liang R, Wang E. MicroRNA group disorganization in aging. Exp Gerontol (2010) 45:269–78. doi:10.1016/j.exger.2009.12.009
179. Pang S, Curran SP. Longevity and the long arm of epigenetics: acquired parental marks influence lifespan across several generations. Bioessays (2012) 34:652–4. doi:10.1002/bies.201200046
180. Pincus Z, Smith-Vikos T, Slack FJ. MicroRNA predictors of longevity in Caenorhabditis elegans. PLoS Genet (2011) 7:e1002306. doi:10.1371/journal.pgen.1002306
181. De Lencastre A, Pincus Z, Zhou K, Kato M, Lee SS, Slack FJ. MicroRNAs both promote and antagonize longevity in C. elegans. Curr Biol (2010) 20:2159–68. doi:10.1016/j.cub.2010.11.015
182. Ugalde AP, Español Y, López-Otín C. Micromanaging aging with miRNAs: new messages from the nuclear envelope. Nucleus (2011) 2:549–55. doi:10.4161/nucl.2.6.17986
183. Kim EB, Fang X, Fushan AA, Huang Z, Lobanov AV, Han L, et al. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature (2011) 479:223. doi:10.1038/nature10533
184. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A (2005) 102:10604–9. doi:10.1073/pnas.0500398102
185. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell (2013) 49:359–67. doi:10.1016/j.molcel.2012.10.016
186. Gräff J, Tsai L. Histone acetylation: molecular mnemonics on the chromatin. Nat Rev Neurosci (2013) 14:97–111. doi:10.1038/nrn3427
187. Sanchez-Mut JV, Heyn H, Vidal E, Moran S, Sayols S, Delgado-Morales R, et al. Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl Psychiatry (2016) 6:e718. doi:10.1038/tp.2015.214
188. Hohl M, Wagner M, Reil JC, Muller SA, Tauchnitz M, Zimmer AM, et al. HDAC4 controls histone methylation in response to elevated cardiac load. J Clin Invest (2013) 123:1359–70. doi:10.1172/JCI61084
189. Ucar D, Marquez EJ, Chung CH, Marches R, Rossi RJ, Uyar A, et al. The chromatin accessibility signature of human immune aging stems from CD8+ T cells. J Exp Med (2017) 214:3123–44. doi:10.1084/jem.20170416
Keywords: aging, cellular senescence, DNA damage, telomeres, epigenetics
Citation: Lidzbarsky G, Gutman D, Shekhidem HA, Sharvit L and Atzmon G (2018) Genomic Instabilities, Cellular Senescence, and Aging: In Vitro, In Vivo and Aging-Like Human Syndromes. Front. Med. 5:104. doi: 10.3389/fmed.2018.00104
Received: 07 January 2018; Accepted: 29 March 2018;
Published: 17 April 2018
Edited by:
Emanuele Marzetti, Fondazione Policlinico Universitario A. Gemelli, Università Cattolica del Sacro Cuore, ItalyReviewed by:
Antonella Sgura, Università degli Studi Roma Tre, ItalyAnna Picca, Università Cattolica del Sacro Cuore, Italy
Copyright: © 2018 Lidzbarsky, Gutman, Shekhidem, Sharvit and Atzmon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gil Atzmon, Z2F0em1vbkB1bml2LmhhaWZhLmFjLmls
†These authors have contributed equally to this work.