 Samuel Mon-Wei Yu
Samuel Mon-Wei Yu Pitchaphon Nissaisorakarn1
Pitchaphon Nissaisorakarn1 Irma Husain
Irma Husain Belinda Jim
Belinda Jim- 1Department of Medicine, Jacobi Medical Center, Bronx, NY, United States
- 2Department of Medicine, James J. Peters VA Medical Center, Bronx, NY, United States
- 3Renal Division, Jacobi Medical Center, Bronx, NY, United States
Proteinuric kidney diseases are a group of disorders with diverse pathological mechanisms associated with significant losses of protein in the urine. The glomerular filtration barrier (GFB), comprised of the three important layers, the fenestrated glomerular endothelium, the glomerular basement membrane (GBM), and the podocyte, dictates that disruption of any one of these structures should lead to proteinuric disease. Podocytes, in particular, have long been considered as the final gatekeeper of the GFB. This specialized visceral epithelial cell contains a complex framework of cytoskeletons forming foot processes and mediate important cell signaling to maintain podocyte health. In this review, we will focus on slit diaphragm proteins such as nephrin, podocin, TRPC6/5, as well as cytoskeletal proteins Rho/small GTPases and synaptopodin and their respective roles in participating in the pathogenesis of proteinuric kidney diseases. Furthermore, we will summarize the potential therapeutic options targeting the podocyte to treat this group of kidney diseases.
Introduction
The kidney is dubbed the organ that filters out toxins and reabsorbs essential electrolytes from the blood to maintain homeostasis in our bodies. Podocytes, along with endothelial cells in the capillaries and glomerular basement membrane (GBM), form this filtration apparatus. The podocyte is a highly-differentiated cell that is composed of several structures: a cell body, primary process, secondary, and tertiary foot processes (FP) (1–7). Primary processes and foot processes (FPs) are highly supported by a complex cytoskeletal architecture, which in of itself is intertwined by a network of microtubules, intermediate filaments, and actin (8, 9). This complex network (microtubules, intermediate filaments, and actin) not only accommodate podocytes to the constantly changing environment, they are crucial hubs of signal transduction within podocytes (10). Furthermore, the neighboring FPs form an interdigitating structure lining outside of the capillary walls (11) and are connected by a specialized cell junction called the slit diaphragm (SD) (12–15). The SD and the negatively charged surface on FP restrict protein from filtering through. Either a dysregulated podocyte cytoskeleton or the loss of integrity of the SD can result in podocyte foot process effacement (FPE), the most common pathological finding in podocyte diseases. Proteinuria is, therefore, the pathognomonic phenomenon in podocytopathy (16). Research on podocyte pathophysiology is crucial due to their function and their lack of ability to regenerate, despite more recent data supporting its potential regenerative capacity (17). In this review, we will summarize our current understanding of podocyte signaling pathways via the SD proteins and the actin cytoskeleton and how they cause proteinuric diseases. We will also discuss the latest updates in using the podocyte as a target for drug development.
Slit Diaphragm Proteins and Disease Implications-Nephrin, Podocin, and TRPC
In the past two decades, an increasing number of disease-causing SD proteins has been identified (5). During kidney development, the SD initially incorporates the features of cell-cell junction which eventually matures into a zipper-like structure by molecular cross-linking via proteins such as nephrin, podocin, and Neph1. Other SD-related proteins may either participate in junctional formation (Nck, CD2AP) or not (TRPC). It is now known that the SD not only acts as a filter to prevent proteinuria (18), but serves as a complex signaling hub to divert different chemical and mechanical stimuli to podocytes (19). We will highlight the best-studied proteins on the SD with clinical significance: nephrin, podocin, and transient receptor potential canonical (TRPC) proteins (Figure 1).
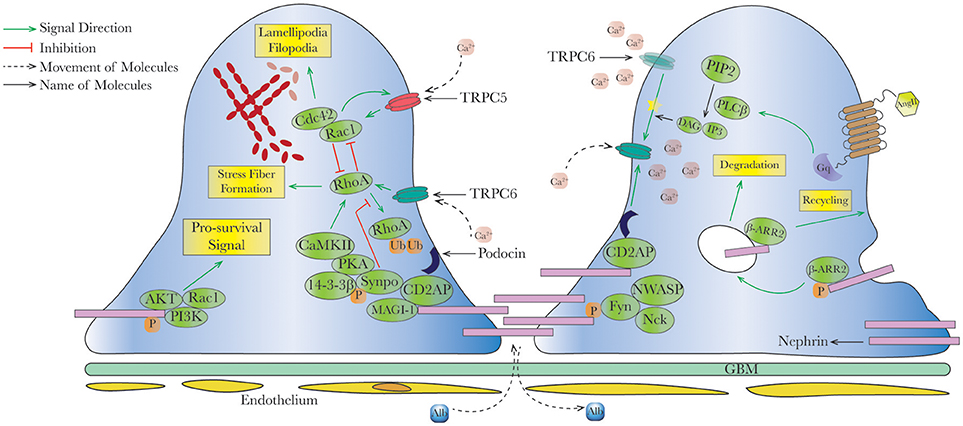
Figure 1. Schematic interaction between slit diaphragm signal cascades and actin dynamics in healthy podocytes. In healthy podocytes, slit diaphragm (SD) is composed by various proteins and acts as a signaling hub, to maintain podocyte survival and regulated actin dynamics. For example, nephrin phosphorylation recruits AKT/PI3K and Nck/NWASP to modulate pro-survival signaling and actin organization, respectively, in concert with other SD proteins such as podocin, CD2AP, and synaptopodin. In physiological status, predominant TRPC6 offers necessary intracellular calcium and activates RhoA, an important small GTPase promoting stress fiber formation. Imbalance between RhoA and Rac1/Cdc42 (activated by TRPC5) leads to dysregulated actin formation. Upon stimulation (for example angiotensin II), G-protein coupled receptors (GPCRs) triggers second messengers' formations, further modulating intracellular calcium concentration via activating TRPC6 (yellow star ). Endocytosis and recycling of proteins on slit diaphragm is controlled by serial phosphorylation and associated proteins such as β-arrestin2. PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; Rac1, ras-related C3 botulinum toxin substrate 1; Cdc42, cell division control protein 42 homolog; RhoA, ras homolog gene family, member A; CaMKII, ca2+/calmodulin-dependent protein kinase II; Synpo, synaptopodin; MAGI-1, membrane-associated guanylate kinase inverted-1; PIP2, phosphatidylinositol bisphosphate; PLCβ, phospholipase Cβ; DAG, diacylglycerol; IP3, inositol 1,4,5-trisphosphate; β-ARR2, β-arrestin 2.
). Endocytosis and recycling of proteins on slit diaphragm is controlled by serial phosphorylation and associated proteins such as β-arrestin2. PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; Rac1, ras-related C3 botulinum toxin substrate 1; Cdc42, cell division control protein 42 homolog; RhoA, ras homolog gene family, member A; CaMKII, ca2+/calmodulin-dependent protein kinase II; Synpo, synaptopodin; MAGI-1, membrane-associated guanylate kinase inverted-1; PIP2, phosphatidylinositol bisphosphate; PLCβ, phospholipase Cβ; DAG, diacylglycerol; IP3, inositol 1,4,5-trisphosphate; β-ARR2, β-arrestin 2.
Nephrin
Most of our understanding of nephrin stems from the successful cloning from patients with Congenital Nephrotic Syndrome of the Finnish type (CNF) (20). Nephrin, a 180-kDa protein encoded by NPHS1, consists of a cytoplasmic C-terminus, a short transmembrane domain, and an extracellular fibronectin type III motif attached with eight distal IgG-like motifs (19, 21). It acts as an intracellular signaling scaffold (22–24) and a podocyte cytoskeleton modulator (25–27). Consistent with the high mortality in children born with CNF, global nephrin-null mice died soon after birth. The technical difficulties of studying its biological function (28, 29) were overcome by the discovery of podocyte-specific promoters (30–33) and the use of short hairpin RNA (34). Of note, although nephrin-null mice are not compatible with life, more than 80–90% successful knockdown of nephrin only resulted in mild proteinuria unless second renal injuries were implemented, suggesting more pivotal role of nephrin in prenatal slit diaphragm development (34). Most of the nephrin signaling occurs via the cytoplasmic tail that is enriched by the highly-conserved tyrosine residues (35) which are phosphorylated by a series of Src-family kinases such as Fyn (23, 26, 36–38). Verma et al. (26) and Jones et al. (27) established the concept of a nephrin-Nck complex as an adaptor after nephrin phosphorylation to regulate actin reorganization/polymerization (39–46), in concert with other signaling pathways such as PI3K/Akt/Rac1 (25, 47, 48).
To date, derangement of nephrin can be largely divided into four mechanisms: (1) decreased phosphorylation, (2) increased endocytosis, (3) loss of function due to genetic mutations, and (4) downregulation at both the mRNA and protein level. This loss of the nephrin-led signaling cascade eventually causes the proteinuric phenotype (27, 49, 50) Nephrin dephosphorylation can be induced by angiotensin II (AngII) which results in the diminished interaction with c-Abl and Akt inactivation, pathways that promote cell survival and growth (51); in addition, it can be caused by the c-maf inducing protein (c-mip) which inhibits interactions between Fyn and the cytoskeletal regulator N-WASP as well as between the adaptor protein Nck and nephrin, resulting in actin cytoskeleton disorganization and foot process effacement (52). Decreased nephrin phosphorylation caused by phosphatases were also observed in various models (53), such as protein tyrosine phosphatase 1B (PTP1B) in rat with puromycin aminonucleoside nephrosis (PAN) (54) and Src homology region 2 domain-containing phosphatase-1 (SHP-1) in human podocytes with hyperglycemia (55). The blockade of phosphatases on nephrin may thus be a therapeutic target in proteinuric diseases and has shown positive results in murine models (lipopolysaccharide[LPS]-induced proteinuria) (56, 57). Nephrin endocytosis through β-arrestin can be stimulated by AngII (58). and hyperglycemia, the latter effect occurs via the interaction of β-arrestin2 with PKC-α which can be abrogated by knocking down PKC-α (59, 60). Interestingly, the loss of phosphorylation of nephrin increases nephrin endocytosis via β-arrestin2 (61), suggesting that nephrin is under a location-specific regulation. Apart from the original mutation described in CNF, there are presently almost 200 disease-associated NPHS1 mutations that have been described (62–66). The phenotypic variability of these mutations has shown us that the nephrotic syndrome manifests not only in the first 90 days of life, but can occur much later on in life as well (67). Furthermore, compound heterozygous or homozygous NPHS1 mutations have also been identified in families with adults with focal segmental glomerulosclerosis (FSGS) (65). In addition to mutations, single nucleotide polymorphisms (SNPs) in NPHS1 and WT-1 have also been identified to confer risk end-stage kidney disease in African Americans (68) and FSGS (69), respectively. Nephrin expression is further regulated by transcription factors such as WT1 (70, 71), NF-κB (72), Sp1 (specificity protein 1) (73), Furthermore, studies have demonstrated that Wilms' tumor 1 (WT1) not only functions as a pivotal gene in kidney development, it also regulates other important SD proteins by binding to the proximal promoters, such as Nphs2, Synaptopodin, and Nck2 (74, 75). Downregulation of nephrin has been observed in many human glomerular diseases by detecting protein and mRNA levels, such as minimal change disease (76), membranous nephropathy, membranoproliferative glomerulonephritis, IgA nephropathy, lupus nephritis (77), diabetic kidney disease (DKD) (78, 79), and preeclampsia (80). Detection of urinary nephrin protein (nephrinuria) or mRNA has already been studied as an early biomarker of disease in DKD, glomerulonephritis, and preeclampsia (81–87) with variable success.
Podocin
After successful cloning of NPHS1, a new mutated gene NPHS2, which encodes for podocin on chromosome 1q25-31, was mapped in patients with autosomal-recessive steroid-resistant nephrotic syndrome (SRNS) (88, 89). Patients' clinical presentations can range from early-onset nephrotic syndrome (typically from 3 months to 6 years of life), FSGS, to end-stage kidney disease (ESKD) by the first decade of life (90, 91). Podocin is a hairpin-like protein including both cytoplasmic N- and C-terminus with a transmembrane portion (92). Almost exclusively expressed on SD of the kidneys (93), podocin interacts with phosho-nephrin and Neph1, another SD protein with a similar structure to nephrin; the podocin-Neph1 interaction is believed to be important for recruitment of nephrin to the lipid raft which augments nephrin signaling (23, 94–99). The prohibin homology (PHB) domain of podocin does not participate in hairpin formation or mediate podocin endocytosis (100), though it may regulate the activity of the TRPC6 channel and act as a mechanic sensor in podocytes (101–103). Among the several types of mutations, nonsense and missense mutations of NPHS2 fail to recruit nephrin into raft domains (94). Other types of nephrotic syndrome (NS), including familial and sporadic steroid resistant nephrotic syndrome (SRNS) (28–40% and 6–19%, respectively) (90, 91, 104–107), adult-onset FSGS (108–110), and non-diabetic ESKD in African Americans (111) have also found to exhibit NPHS2 mutations. The type of mutation, whether heterozygous or homozygous, appears to correlate with a different phenotype. For example, patients with two pathogenic NPHS2 mutations have presented with an early onset of SNRS with a low incidence of post-transplant recurrence, whereas heterozygous NPHS2 variants seem to exhibit a milder, late-onset phenotype with a higher incidence of recurrence (91, 105, 112). Furthermore, mutations of the transcription factors LMX1B and FoxC (113–115) that control podocin expression can also result in disease, such as the nail-patella syndrome or isolated hereditary FSGS (116, 117). Interestingly, the transcription factor WT1 can also regulate LMX1B and FoxC expression, adding an extra layer of modulation in podocin expression (118). Currently, more than 1,000 podocin variants that have been described (https://databases.lovd.nl/shared/genes/NPHS2) (119) and further study will be needed to understand their individual roles, particularly in different genetic backgrounds (120). Lastly, podocin downregulation has also been observed in lupus nephritis (121), pediatric nephrotic syndrome (122), and FSGS (123), but not in preeclampsia (80).
TRPC
Transient receptor potential canonical (TRPC) channels are a group of seven families of non-selective cationic channels (TRPC1 to TRPC7) and largely divided into TRPC3/6/7 and TRPC1/4/5 based on sequence alignment and functionality (124). Among them, TRPC6 and TRPC5 have been identified in podocytes and have been widely researched in the past decade. The impetus for this research started after a gain-of-function mutation was identified in a family of autosomal FSGS (125, 126). Like other TRPCs, TRPC6 can be activated by phospholipase C (PLC)-dependent pathway via the generation of diacylglycerol (DAG) and inositol 1,4,5-phosphate (IP3), resulting in the subsequent Ca2+ influx into podocytes. Winn et al. demonstrated that a gain-of-function mutation in TRPC6 (proline to glutamine substitution at position 112) enhanced calcium signaling via AngII stimulation and cell surface expression of TRPC6 in HEK 293 cells (126), which ultimately causes eventual podocyte apoptosis and detachment (127). Recently, a TRPC6 loss-of-function type of mutation was also found to be associated with human FSGS (128), thus providing evidence that reduced physiological TRPC6-mediated calcium entry may also cause disease. In addition to FSGS, TRPC6 has been reported to affect DKD by increasing TRPC6 expression mediated by AngII as seen in streptozotocin-induced diabetic rats; this suggests a role for TRPC6 in causing podoctyopenia and proteinuria (129, 130). TRPC5 and TRPC6 both affect the actin cytoskeleton differently, i.e., upon activation through AngII, TRPC6 couples with Rho A (see section on Rho/Small GTPases) while TRPC5 couples with Rac1, resulting in stress fiber vs. motile phenotype in the podocyte, respectively. TRPC5-mediated signals have been considered as “disease-type” leading to cytoskeletal collapse (131). The function of TRPC5, however, has recently been put in question. There is evidence that deletion of TRPC5, either genetically or pharmacologically, results in decreased glomerular injury when exposed to LPS or protamine sulfate (PS) (132). Recently, Zhou et al. showed that inhibition of TRPC5 with a small molecule inhibitor (AC1903) prevented podocyte loss in a rat model of AT1R overexpression (model of FSGS) and halted the disease progression in the Dahl salt-sensitive rate model of hypertensive kidney disease (133). These findings, however, were contradicted by another study suggesting that the overexpression TRPC5 in mice does not cause proteinuria nor aggravate LPS-induced albuminuria (134). Nonetheless, these conflicting findings further complicate our current understanding of TRPC channelopathy on podocytes, and in fact, echo some of the contradictory results in GTPases studies in this field (see below).
Podocyte Signaling at Actin Cytoskeletons- Rho/Small GTPases, and Synaptopodin
Derangements in the actin cytoskeleton of podocytes have been widely reported to cause proteinuric kidney diseases. Not only does it maintain the structural integrity of the podocytes, but it also mediates complex signal cascades, endocytic pathways, and interactions outside of podocytes such as the GBM (10). Here we will focus on Rho-family of small GTPases and synaptopodin.
GTPases
The Rho-family of small GTPases such as RhoA, Cdc42, and Rac1 regulate actin cytoskeleton remodeling by acting as molecular switches to coordinate signaling by either promoting cell motility or inhibiting cell migration (135). Each GTPase cycles between an active GTP-bound state and an inactive GDP-bound state. Upon activation, Rho GTPases are able to bind to a range of effector proteins to regulate downstream signaling pathways. Rho-dependent pathways are activated by AngII, platelet-derived growth factor, endothelin-1 (136), and promote contractile phenotype with the formation of stress fibers. On the contrary, Cdc42/Rac1 activation leads to lamellipodia/filopodia formation, an unstable phenotype of podocytes which causes FPE (137). Although initially it seemed that RhoA activation with Cdc42/Rac1 suppression was beneficial to the podocyte, studies in the past decade have shown conflicting results. For example, transgenic mice with constitutive activation of RhoA in podocytes resulted in proteinuria with FPE, podocyte apoptosis, decreasing nephrin expression (138) and histologic features of FSGS (139). Inhibiting RhoA signaling was thus shown to mitigate podocyte injury murine chronic kidney disease model (5/6 nephrectomy) and rats with PAN (140, 141). However, constitutive expression of dominant-negative RhoA in mice also caused proteinuria and loss of stress fiber formation in mouse podocytes (138), while preserving RhoA signaling in mice with LPS-induced proteinuria via synaptopodin (142) showed anti-proteinuric effects (143). Thus, the balance of RhoA signaling is imperative for podocyte, and it is likely to be dependent on the specific disease state. In the Cdc42/Rac1 system, podocyte-specific Rac1 knockout mice were shown to exhibit normal podocyte morphology and protection from PS induced-podocyte injury (144). However, these Rac1 knockout mice were also found to exacerbate the injury from chronic hypertensive glomerulosclerosis. Activation of Rac1 has shown to attenuate podocyte injury and expedite recovery in vitro (145, 146), yet hyperactivation of Rac1 has been linked to diseased podocytes in a dose-dependent manner (147–149). For Cdc42, podocyte-specific deletion in mice led to early-onset severe proteinuria, FPE, glomerulosclerosis (144), and congenital nephropathy (150). Similarly, reduced Cdc42 expression resulted in mouse podocyte apoptosis due to downregulated Yes-Associated Protein (YAP) signaling (151, 152) and promoted filopodia formation. Decreased activity of both Cdc42 and Rac1 via FAT1 mutation has shown to cause human and mouse glomerulotubular nephropathy (153). These results collectively suggest Rac1 signaling might be dispensable for podocyte development as compared with Cdc42 (150), but whether Rac1 deletion is beneficial or detrimental, again, depends on the type of podocyte injury.
Mutations in human genes that interfere with Rho GTPase signaling have been identified in nephrotic syndrome (154), FSGS (155), and DKD (156). For example, mutations in ARHGDIA, which exists in a complex with Rho GTPases, increased levels of Rac1 and Cdc42 (but not RhoA) have been found in individuals with SRNS (Table 1) (154). The human disease mutation in Rac1-GTPase-activating protein (Rac1-GAP) ARHGAP24, which results in elevated Rac1 and CDC42 levels, was also found to be associated with a family with FSGS (155). Hence, we are only beginning to learn more about the significance of these subclasses of the Rho-family of small GTPases as they appear to have divergent roles in specific disease states.
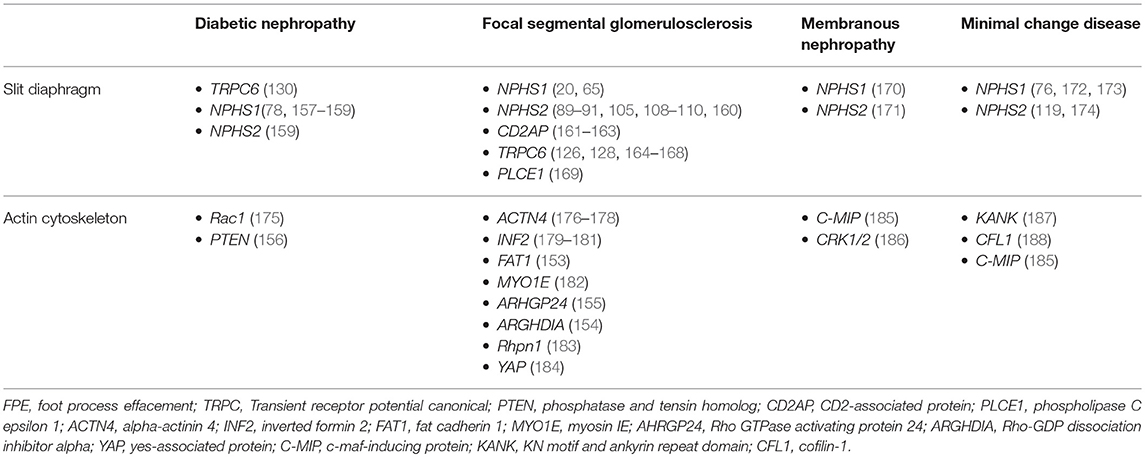
Table 1. Genes of slit diaphragm and actin cytoskeleton that are associated with proteinuric kidney diseases.
Synaptopodin
Synaptopodin is a proline-rich actin-binding protein highly expressed in dynamic cells such as telencephalic dendrites and podocytes. In podocytes, synaptopodin was found to interact with α-actinin-4, an important protein to cross-link between actin filaments (189). Loss of synaptopodin led to delayed stress fiber formation and development of aberrant nonpolarized filopodia. Although the ultrastructure of podocytes in synaptopodin-deficient (synpo−/−) mice appeared to be normal, these mice showed impaired recovery from both PS- and LPS-induced kidney injury (190). Synaptopodin was later revealed to block Smurf1-mediated ubiquitination of RhoA to preserve stress fibers formation by increasing RhoA expression and activation (142). By contrast, synaptopodin inhibits binding of Cdc42 and Mena to IRSp53 and therefore protects aberrant filopodia formation in mouse podocytes (191). Synaptopodin is phosphorylated by PKA/CaMKII and binds 14-3-3ß to promote Rho signaling (143). Under TRPC5-mediated calcium influx, synaptopodin is degraded by calcineurin-mediated phosphorylation and promotes Rac1 signaling and ROS formation (192). Cyclosporine A, a calcineurin inhibitor used in immunosuppression, was found to have an anti-proteinuric effect due to blocking calcineurin-mediated dephosphorylation of synaptopodin (143). Additionally, Yu et al. demonstrated that synaptopodin affects TRPC6 localization and function which suggests that decreased synaptopodin levels under diseased conditions will increase TRPC6-mediated calcium influx and induce podocyte apoptosis (193). Clinically, reduced expression of synaptopodin has been observed in FSGS (176, 194), HIV-associated nephropathy (HIVAN) (194–196), IgA nephropathy (197), and idiopathic nephrotic syndrome of childhood (198). Synaptopodin levels have been shown to correlate with disease severity and response to treatment in human FSGS and minimal change disease (MCD) (199, 200). Urinary synaptopodin protein and mRNA excretion have also been used to assess the decline of renal function and certain glomerular diseases (201, 202).
Clinical Implications
Advances in our understanding of podocyte biology have been a great asset to elucidate underlying pathophysiology in proteinuric kidney diseases. DKD, MCD, FSGS, and membranous nephropathy (MN) were all found to have dysfunction at the SD or actin cytoskeletal system (Figure 2). Although the therapeutic options targeted to podocytes are limited, the concept of restoring SD proteins, stabilizing the cytoskeleton, or stimulating podocyte regeneration is showing promise.
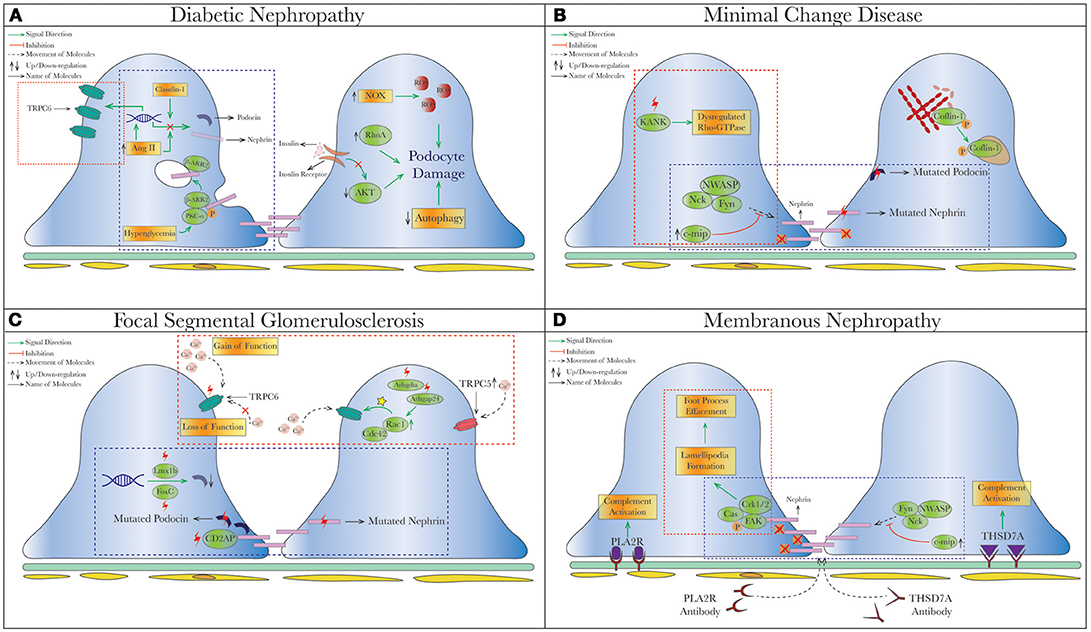
Figure 2. Schematic depiction of pathophysiologic processes in specific proteinuric kidney diseases. Dysregulated slit diaphragm and altered actin dynamics depicted in specific disease states that cause podocyte foot process effacement (FPE) and podocyte loss. The commonalities of pathophysiology in different diseases are grouped into blue  (dysregulated pathways related to SD proteins) or red
(dysregulated pathways related to SD proteins) or red  (cytoskeletal dysregulation via TRPC or Rho-GTPase) dashed square. (A) Diabetic Nephropathy. Under hyperglycemia, nephrin endocytosis can be induced by the interaction of β-arrestin2 with PKC-α (59, 60). Increased AngII (78, 158) and Claudin-1 (159, 203) in diabetes were both shown to reduce expression of nephrin and podocin. On the contrary, increased TRPC6 expression mediated by AngII was seen in streptozotocin-induced diabetic rats causing podocytopenia and proteinuria (129, 130). Hyperglycemia also leads to podocyte apoptosis from production of reactive oxygen species (ROS) through NAPDH oxidase (NOX) (204), so as does insulin resistance due to reduced AKT pro-survival signaling (205). (B) Minimal Change Disease. Mutations of nephrin (NPHS1) and podocin (NPHS2) have been reported in MCD and tended to have higher rates of steroid-resistance (119, 174, 206), as well as recessive mutations of KANK (kidney ankyrin repeating-containing protein) KANK1 and KANK2 identified in a cohort of Arab and European origins (187). Cofilin-1, actin-binding protein necessary for maintaining podocyte architecture (207), is inactivated by phosphorylation seen in human MCD, leading to its redistribution to nucleus in the disease states (188). A novel molecule c-mip noted to be upregulated in human MN/MCD was shown to impair podocyte actin reorganization by inhibiting interaction between Fyn/N-WASP and nephrin/Nck. Podocyte overexpressed with c-mip could result in downregulation of nephrin and synaptopodin (52, 185, 208). (C) Focal Segmental Glomerulosclerosis (FSGS). Multiple gene mutations have been identified in patients with FSGS through genetic studies. Most of them encode various critical podocyte structures or signaling pathways, such as SD complex (NPHS1, NPHS2, CD2AP), SD-related Ca2+ signaling (TRPC6, PLCE1), actin cytoskeleton/endocytosis (ACTN4, INF2, FAT1, MYO1E), and small-GTPases (AHRGP24, ARGHDIA, ARHGEF17) (209–214). A novel mechanism of increased podocin endocytosis via sorting nexin 9 (SNX9), which is seen in human IgA nephropathy, membranous nephropathy and FSGS was also described (171). Blocking the imbalanced TRPC5/6 signaling and increased circulating permeability factors (suPAR/CLC-1) are possible new therapeutic approaches in FSGS. (D) Membranous Nephropathy. Besides the well-known antibody-mediated primary MN (anti-PLA2R/THSd7A), reduced nephrin phosphorylation (170) and increased podocin endocytosis (171) have been described in human MN. Disorganization of actin cytoskeleton from a different cytoskeletal pathway Cas-FAK-Crk1/2 activation (not the typical nephrin-Nck pathway) was also seen in human MCD and MN (186). Mutation (red thunder
(cytoskeletal dysregulation via TRPC or Rho-GTPase) dashed square. (A) Diabetic Nephropathy. Under hyperglycemia, nephrin endocytosis can be induced by the interaction of β-arrestin2 with PKC-α (59, 60). Increased AngII (78, 158) and Claudin-1 (159, 203) in diabetes were both shown to reduce expression of nephrin and podocin. On the contrary, increased TRPC6 expression mediated by AngII was seen in streptozotocin-induced diabetic rats causing podocytopenia and proteinuria (129, 130). Hyperglycemia also leads to podocyte apoptosis from production of reactive oxygen species (ROS) through NAPDH oxidase (NOX) (204), so as does insulin resistance due to reduced AKT pro-survival signaling (205). (B) Minimal Change Disease. Mutations of nephrin (NPHS1) and podocin (NPHS2) have been reported in MCD and tended to have higher rates of steroid-resistance (119, 174, 206), as well as recessive mutations of KANK (kidney ankyrin repeating-containing protein) KANK1 and KANK2 identified in a cohort of Arab and European origins (187). Cofilin-1, actin-binding protein necessary for maintaining podocyte architecture (207), is inactivated by phosphorylation seen in human MCD, leading to its redistribution to nucleus in the disease states (188). A novel molecule c-mip noted to be upregulated in human MN/MCD was shown to impair podocyte actin reorganization by inhibiting interaction between Fyn/N-WASP and nephrin/Nck. Podocyte overexpressed with c-mip could result in downregulation of nephrin and synaptopodin (52, 185, 208). (C) Focal Segmental Glomerulosclerosis (FSGS). Multiple gene mutations have been identified in patients with FSGS through genetic studies. Most of them encode various critical podocyte structures or signaling pathways, such as SD complex (NPHS1, NPHS2, CD2AP), SD-related Ca2+ signaling (TRPC6, PLCE1), actin cytoskeleton/endocytosis (ACTN4, INF2, FAT1, MYO1E), and small-GTPases (AHRGP24, ARGHDIA, ARHGEF17) (209–214). A novel mechanism of increased podocin endocytosis via sorting nexin 9 (SNX9), which is seen in human IgA nephropathy, membranous nephropathy and FSGS was also described (171). Blocking the imbalanced TRPC5/6 signaling and increased circulating permeability factors (suPAR/CLC-1) are possible new therapeutic approaches in FSGS. (D) Membranous Nephropathy. Besides the well-known antibody-mediated primary MN (anti-PLA2R/THSd7A), reduced nephrin phosphorylation (170) and increased podocin endocytosis (171) have been described in human MN. Disorganization of actin cytoskeleton from a different cytoskeletal pathway Cas-FAK-Crk1/2 activation (not the typical nephrin-Nck pathway) was also seen in human MCD and MN (186). Mutation (red thunder ) of proteins, inhibiting (red cross
) of proteins, inhibiting (red cross ) or activating (yellow star) different signal pathways leads to reduced level of phosphorylation, increased intracellular calcium and endocytic process. AngII, Angiotensin II; c-ABL, Abelson murine leukemia viral oncogene homolog 1; RhoA, ras homolog gene family, member A; NOX, NAPDH oxidase; ROS, reactive oxygen species; Cdc42, cell division control protein 42 homolog; Rac1, ras-related C3 botulinum toxin substrate 1; Lmx1b, LIM homeobox transcription factor 1-beta; SNX-9, sorting nexin 9; suPAR, soluble urokinase plasminogen activator receptor; CLC-1, cardiotropin-like cytokine-1; FAK, focal adhesion kinase; PLA2R, phospholipase A2 receptor; THSD7A, thrombospondin Type 1 domain containing 7A; c-mip, c-maf-inducing protein; KANK, kidney ankyrin repeating-containing protein.
) or activating (yellow star) different signal pathways leads to reduced level of phosphorylation, increased intracellular calcium and endocytic process. AngII, Angiotensin II; c-ABL, Abelson murine leukemia viral oncogene homolog 1; RhoA, ras homolog gene family, member A; NOX, NAPDH oxidase; ROS, reactive oxygen species; Cdc42, cell division control protein 42 homolog; Rac1, ras-related C3 botulinum toxin substrate 1; Lmx1b, LIM homeobox transcription factor 1-beta; SNX-9, sorting nexin 9; suPAR, soluble urokinase plasminogen activator receptor; CLC-1, cardiotropin-like cytokine-1; FAK, focal adhesion kinase; PLA2R, phospholipase A2 receptor; THSD7A, thrombospondin Type 1 domain containing 7A; c-mip, c-maf-inducing protein; KANK, kidney ankyrin repeating-containing protein.
Renin-Angiotensin-Aldosterone System Inhibition
Inhibiting AngII has shown to be beneficial in several models of glomerular diseases. In DKD, there is a large body of evidence that shows inhibition of AngII results in the reduction in proteinuria and the slowing of the rate glomerular filtration rate (GFR) decline in humans. In addition to the well-known hemodynamic effects of inhibiting the renin-angiotensin-aldosterone system (RAAS) (215), both angiotensin-converting enzyme inhibitor (ACEI) and AngII type 1 receptor blocker (ARB) have shown to significantly increase nephrin expression in murine diabetic models (157, 216). A similar effect was also observed in rats models by adriamycin-induced nephropathy and PAN (217, 218). Since AngII has a direct TRPC6-mediated calcium signaling (219, 220) which results in ROS expression (221) and proteinuria (222), inhibiting TRPC6 indirectly via AngII blockade is a feasible strategy in proteinuric diseases such as FSGS.
As described above, the concept of podocyte's limited capacity of regeneration has been challenged by novel genetic labeling techniques to trace possible podocyte progenitors. The exact origin of the detected newly-developed podocytes is still under debate; to name a few, glomerular parietal epithelial cells (PEC) and cells of renin lineage (CoRL) have been most investigated to date. Surprisingly, RAAS inhibition has been also shown to increase podocyte numbers either from PEC (223, 224) or CoRL (225, 226), suggesting its unique property and potential therapeutic effect.
Glucocorticoid
Glucocorticoid (GC) is the backbone and initial therapy for several nephrotic syndrome including FSGS (209) and MCD (227). In addition to the effects on immunomodulatory cells, cultured murine podocyte was shown to carry glucocorticoid receptor (GR) complex and respond to GC in a dose- and time-dependent manners at both transcriptional and posttranscriptional levels (228). In an immortalized human podocyte model, the expression of nephrin and other podocyte genes was upregulated after dexamethasone treatment (229, 230). GC was also shown to help to stabilize the actin cytoskeleton system (231), and to increase actin stress fiber formation via overexpression of Krüppel-like factor 15, a downstream target of GC (232). Lastly, in murine model of experimental FSGS (via cytotoxic anti-podocyte antibody), GC might regenerate podocytes from adjacent PECs (233). Therefore, the therapeutic actions of GC in proteinuric diseases may very well be due to its myriad benefits on the podocyte (234).
Inhibition of Rho-Associated Kinases (ROCKs)
With murine diabetic and PAN models, inhibiting downstream effectors Rho-associated kinases (ROCKs) using ROCK inhibitors has shown renoprotective effects (235, 236). Increased Rac1 and mineralocorticoid receptor signaling were also found to be increased in mice lacking Rho GDP-dissociation inhibitor-alpha [Arhgdia(−/−) mice] with heavy albuminuria and podocyte damage. A Rac-specific small-molecule (NSC23766) and eplerenone (mineralocorticoid receptor antagonist) have successfully ameliorated the renal damages seen in Arhgdia(−/−) mice, suggesting the therapeutic effects by suppressing Rac1 or mineralocorticoid receptor signaling (237). Nevertheless, conflicting data in this field demands additional meticulous studies to further illuminate future therapeutic targets.
Vitamin D and Retinoic Acids
Selective vitamin D receptor activation was proven to lower proteinuria in patients with DKD (238); it was later demonstrated that vitamin D3 analogs might ameliorate podocyte injury by reversing the decrease in nephrin expression induced by hyperglycemia (239–241). The combination of vitamin D analog with an ARB has been shown to have synergetic effects on RAAS blockade, thereby boosting its overall therapeutic effect (242, 243). However, as there is currently no recommended clinical dosage of vitamin D analog for use in DKD, long-term and prospective clinical data is required to assess its safety and efficacy (244, 245). Retinoic acids (RAs) are vitamin A derivatives and appear to have renoprotective effects on various murine nephropathy such as HIV-associated nephropathy (HIVAN) (246) and PAN-induced nephrotic rats (247). Recently, Dai et al. reported that RA treatment activated podocyte retinoic acid receptor-α (RAR-α) and increased synaptopodin expression on PEC in wild type mice with nephrotoxic serum-induced glomerulonephritis (GN). The group subsequently identified RAR-α expression in human crescentic GN kidney biopsies, substantiating a possible therapeutic option in treating human crescentic GN (248). RA was also found to induce podocyte markers in PECs in a murine model membranous nephropathy (Heymann nephritis) and FSGS (anti-glomerular antibody model) (249). However, the toxicities of RA have hindered it from wide clinical use and have resulted in difficulties in recruiting patients to a Phase II clinical trial of podocyte diseases (MCD, FSGS, collapsing glomerulopathy) (NCT 00098020) (250). Thus, less toxic alternatives, such as Kruppel-like factor-15 and boronic acid retinoid are being explored (250, 251).
Biological Agents
Emerging evidence also suggests that biological agents might regulate the podocyte SD. Rituximab (RTX), an anti-CD20 antibody which depletes B cells, is widely used in different proteinuric diseases such as FSGS (252–254) and steroid-resistant MCD (255–258). Besides its immunological response, RTX was found to preserve sphingomyelin-phosphodiesterase-acid-like 3b (SMPDL-3b), which in turn regulates acid sphingomyelinase (ASMase) activity in the raft of podocytes (259). SMPDL-3b can partially co-localize with synaptopodin and hence prevent the actin remodeling that occurs after exposure to serum from FSGS patients (259). This concept was supported by a recent clinical observation of the increase in urinary SMPDL-3b in patients at the remission stage after RTX treatment compared with the proteinuric stage (260). Abatacept (CTLA-4-Ig), a costimulatory-inhibitor that targets B7-1 molecule, may be another therapeutic agent to treat nephrotic syndrome via the actin cytoskeleton. Since induction of the T-cell costimulatory molecule B7-1 by LPS has been shown to be associated with nephrotic syndrome and actin reorganization (261), an inhibitor of B7-1 may ameliorate these findings. Though Yu et al. initially reported that treating five FSGS patients with abatacept achieved partial remission (262), these findings have not been successfully replicated (263–266). Presently, there is an ongoing phase II trial of abatacept for patients with FSGS or MCD (NCT02592798).
Conclusion
Our improved understanding of the podocyte in terms of its SDs and cytoskeleton has allowed us to target this cell for new treatments for proteinuric diseases (267, 268). However, there is much more to discover. Novel therapeutic target genes or proteins in podocytes are constantly being discovered as we write this review (269–271). Longstanding concepts, such as the function of the SD and the lack of mitotic ability of the podocyte, are already being challenged (272–274). For instance, the use of glycogen synthase kinases 3 inhibitor has already demonstrated initial promising results of transforming renal progenitor cells into podocytes (275). Growing podocytes from induced pluripotent stem cells on the chip also serves as a great tool to assess new drug development (276). At the same time, newer imaging systems to count podocytes (277) and to study their membrane dynamics have enhanced our ability to study this cell (278). The information that we hope to garner from the field of “-omics” (279, 280) reminds us that data is both tremendously helpful and potentially fraught with error, and hence must be interpreted with great care (281, 282). Thus, persistent endeavors to elucidate complex podocyte biology (283, 284), along with long-term patient follow-up (285), would greatly add to our present understanding of these common yet poorly understood proteinuric diseases.
Author Contributions
SY, PN, IH, and BJ wrote draft of the manuscript. SY contributed to the figures. SY and BJ reviewed and edited the manuscript before submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mundel P, Kriz W. Structure and function of podocytes: an update. Anat Embryol (Berl) (1995) 192:385–97.
2. Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. (2003) 83:253–307. doi: 10.1152/physrev.00020.2002
3. Andrews PM. Investigations of cytoplasmic contractile and cytoskeletal elements in the kidney glomerulus. Kidney Int. (1981) 20:549–62.
4. Ichimura K, Kurihara H, Sakai T. Actin filament organization of foot processes in rat podocytes. J Histochem Cytochem. (2003) 51:1589–600. doi: 10.1177/002215540305101203
6. Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol. (2012) 74:299–323. doi: 10.1146/annurev-physiol-020911-153238
7. Haraldsson B, Nystrom J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. (2008) 88:451–87. doi: 10.1152/physrev.00055.2006
8. Drenckhahn D, Franke RP. Ultrastructural organization of contractile and cytoskeletal proteins in glomerular podocytes of chicken, rat, and man. Lab Invest. (1988) 59:673–82.
9. Andrews PM, Bates SB. Filamentous actin bundles in the kidney. Anat Rec. (1984) 210:1–9. doi: 10.1002/ar.1092100102
10. Perico L, Conti S, Benigni A, Remuzzi G. Podocyte-actin dynamics in health and disease. Nat Rev Nephrol. (2016) 12:692–710. doi: 10.1038/nrneph.2016.127
11. Saleem MA, Zavadil J, Bailly M, McGee K, Witherden IR, Pavenstadt H, et al. The molecular and functional phenotype of glomerular podocytes reveals key features of contractile smooth muscle cells. Am J Physiol Renal Physiol. (2008) 295:F959–70. doi: 10.1152/ajprenal.00559.2007
12. Fukasawa H, Bornheimer S, Kudlicka K, Farquhar MG. Slit diaphragms contain tight junction proteins. J Am Soc Nephrol. (2009) 20:1491–503. doi: 10.1681/ASN.2008101117
13. Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. (2000) 11:1–8.
14. Benzing T. Signaling at the slit diaphragm. J Am Soc Nephrol. (2004) 15:1382–91. doi: 10.1097/01.ASN.0000130167.30769.55
15. Huber TB, Benzing T. The slit diaphragm: a signaling platform to regulate podocyte function. Curr Opin Nephrol Hypertens (2005) 14:211–6.
16. Brinkkoetter PT, Ising C, Benzing T. The role of the podocyte in albumin filtration. Nat Rev Nephrol. (2013) 9:328–36. doi: 10.1038/nrneph.2013.78
17. Anders HJ. Immune system modulation of kidney regeneration–mechanisms and implications. Nat Rev Nephrol. (2014) 10:347–58. doi: 10.1038/nrneph.2014.68
18. Patrakka J, Tryggvason K. New insights into the role of podocytes in proteinuria. Nat Rev Nephrol. (2009) 5:463–8. doi: 10.1038/nrneph.2009.108
19. Grahammer F, Schell C, Huber TB. The podocyte slit diaphragm–from a thin grey line to a complex signalling hub. Nat Rev Nephrol. (2013) 9:587–98. doi: 10.1038/nrneph.2013.169
20. Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell (1998) 1:575–82.
21. Patrakka J, Tryggvason K. Nephrin–a unique structural and signaling protein of the kidney filter. Trends Mol Med. (2007) 13:396–403. doi: 10.1016/j.molmed.2007.06.006
22. Huber TB, Kottgen M, Schilling B, Walz G, Benzing T. Interaction with podocin facilitates nephrin signaling. J Biol Chem. (2001) 276:41543–6. doi: 10.1074/jbc.C100452200
23. Li H, Lemay S, Aoudjit L, Kawachi H, Takano T. SRC-family kinase Fyn phosphorylates the cytoplasmic domain of nephrin and modulates its interaction with podocin. J Am Soc Nephrol. (2004) 15:3006–15. doi: 10.1097/01.ASN.0000146689.88078.80
24. Harita Y, Kurihara H, Kosako H, Tezuka T, Sekine T, Igarashi T, et al. Neph1, a component of the kidney slit diaphragm, is tyrosine-phosphorylated by the Src family tyrosine kinase and modulates intracellular signaling by binding to Grb2. J Biol Chem. (2008) 283:9177–86. doi: 10.1074/jbc.M707247200
25. Zhu J, Sun N, Aoudjit L, Li H, Kawachi H, Lemay S, et al. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int. (2008) 73:556–66. doi: 10.1038/sj.ki.5002691
26. Verma R, Kovari I, Soofi A, Nihalani D, Patrie K, Holzman LB. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest. (2006) 116:1346–59. doi: 10.1172/JCI27414
27. Jones N, Blasutig IM, Eremina V, Ruston JM, Bladt F, Li H, et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature (2006) 440:818–23. doi: 10.1038/nature04662
28. Putaala H, Soininen R, Kilpelainen P, Wartiovaara J, Tryggvason K. The murine nephrin gene is specifically expressed in kidney, brain and pancreas: inactivation of the gene leads to massive proteinuria and neonatal death. Hum Mol Genet. (2001) 10:1–8.
29. Rantanen M, Palmen T, Patari A, Ahola H, Lehtonen S, Astrom E, et al. Nephrin TRAP mice lack slit diaphragms and show fibrotic glomeruli and cystic tubular lesions. J Am Soc Nephrol. (2002) 13:1586–94. doi: 10.1097/01.ASN.0000016142.29721
30. Moeller MJ, Kovari IA, Holzman LB. Evaluation of a new tool for exploring podocyte biology: mouse Nphs1 5′ flanking region drives LacZ expression in podocytes. J Am Soc Nephrol. (2000) 11:2306–14.
31. Moeller MJ, Sanden SK, Soofi A, Wiggins RC, Holzman LB. Two gene fragments that direct podocyte-specific expression in transgenic mice. J Am Soc Nephrol (2002) 13:1561–7. doi: 10.1097/01.ASN.0000015614.68893.0B
32. Juhila J, Lassila M, Roozendaal R, Lehtonen E, Messing M, Langer B, et al. Inducible nephrin transgene expression in podocytes rescues nephrin-deficient mice from perinatal death. Am J Pathol. (2010) 176:51–63. doi: 10.2353/ajpath.2010.080843
33. Juhila J, Roozendaal R, Lassila M, Verbeek SJ, Holthofer H. Podocyte cell-specific expression of doxycycline inducible Cre recombinase in mice. J Am Soc Nephrol. (2006) 17:648–54. doi: 10.1681/ASN.2005050547
34. Li X, Chuang PY, D'Agati VD, Dai Y, Yacoub R, Fu J, et al. Nephrin Preserves Podocyte Viability and Glomerular Structure and Function in Adult Kidneys. J Am Soc Nephrol. (2015) 26:2361–77. doi: 10.1681/ASN.2014040405
35. Putaala H, Sainio K, Sariola H, Tryggvason K. Primary structure of mouse and rat nephrin cDNA and structure and expression of the mouse gene. J Am Soc Nephrol. (2000) 11:991–1001.
36. Lahdenpera J, Kilpelainen P, Liu XL, Pikkarainen T, Reponen P, Ruotsalainen V, et al. Clustering-induced tyrosine phosphorylation of nephrin by Src family kinases. Kidney Int. (2003) 64:404–13. doi: 10.1046/j.1523-1755.2003.00097.x
37. Verma R, Wharram B, Kovari I, Kunkel R, Nihalani D, Wary KK, et al. Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J Biol Chem. (2003) 278:20716–23. doi: 10.1074/jbc.M301689200
38. Harita Y, Kurihara H, Kosako H, Tezuka T, Sekine T, Igarashi T, et al. Phosphorylation of nephrin triggers Ca2+ signaling by recruitment and activation of phospholipase Cγ1. J Biol Chem. (2009) 284:8951–62. doi: 10.1074/jbc.M806851200
39. Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, et al. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. (2005) 168:441–52. doi: 10.1083/jcb.200407076
40. Gruenheid S, DeVinney R, Bladt F, Goosney D, Gelkop S, Gish GD, et al. Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat Cell Biol. (2001) 3:856–9. doi: 10.1038/ncb0901-856
41. Rivera GM, Briceno CA, Takeshima F, Snapper SB, Mayer BJ. Inducible clustering of membrane-targeted SH3 domains of the adaptor protein Nck triggers localized actin polymerization. Curr Biol. (2004) 14:11–22. doi: 10.1016/j.cub.2003.12.033
42. Barda-Saad M, Braiman A, Titerence R, Bunnell SC, Barr VA, Samelson LE. Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat Immunol. (2005) 6:80–9. doi: 10.1038/ni1143
43. Rohatgi R, Nollau P, Ho HY, Kirschner MW, Mayer BJ. Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway. J Biol Chem. (2001) 276:26448–52. doi: 10.1074/jbc.M103856200
44. Blasutig IM, New LA, Thanabalasuriar A, Dayarathna TK, Goudreault M, Quaggin SE, et al. Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol Cell Biol. (2008) 28:2035–46. doi: 10.1128/MCB.01770-07
45. Zhu J, Attias O, Aoudjit L, Jiang R, Kawachi H, Takano T. p21-activated kinases regulate actin remodeling in glomerular podocytes. Am J Physiol Renal Physiol. (2010) 298:F951–61. doi: 10.1152/ajprenal.00536.2009
46. Okrut J, Prakash S, Wu Q, Kelly MJ, Taunton J. Allosteric N-WASP activation by an inter-SH3 domain linker in Nck. Proc Natl Acad Sci USA. (2015) 112:E6436–45. doi: 10.1073/pnas.1510876112
47. Huber TB, Hartleben B, Kim J, Schmidts M, Schermer B, Keil A, et al. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol. (2003) 23:4917–28.
48. Canaud G, Bienaime F, Viau A, Treins C, Baron W, Nguyen C, et al. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med. (2013) 19:1288–96. doi: 10.1038/nm.3313
49. Jones N, New LA, Fortino MA, Eremina V, Ruston J, Blasutig IM, et al. Nck proteins maintain the adult glomerular filtration barrier. J Am Soc Nephrol. (2009) 20:1533–43. doi: 10.1681/ASN.2009010056
50. New LA, Martin CE, Scott RP, Platt MJ, Keyvani Chahi A, Stringer CD, et al. Nephrin tyrosine phosphorylation is required to stabilize and restore podocyte foot process architecture. J Am Soc Nephrol. (2016) 27:2422–35. doi: 10.1681/ASN.2015091048
51. Yang Q, Ma Y, Liu Y, Liang W, Chen X, Ren Z, et al. Angiotensin II down-regulates nephrin-Akt signaling and induces podocyte injury: roleof c-Abl. Mol Biol Cell. (2016) 27:197–208. doi: 10.1091/mbc.E15-04-0223
52. Zhang SY, Kamal M, Dahan K, Pawlak A, Ory V, Desvaux D, et al. c-mip impairs podocyte proximal signaling and induces heavy proteinuria. Sci Signal. (2010) 3:ra39. doi: 10.1126/scisignal.2000678
53. Geraldes P. Protein phosphatases and podocyte function. Curr Opin Nephrol Hypertens (2017). doi: 10.1097/MNH.0000000000000376
54. Aoudjit L, Jiang R, Lee TH, New LA, Jones N, Takano T. Podocyte protein, nephrin, is a substrate of protein tyrosine phosphatase 1B. J Signal Transduct. (2011) 2011:376543. doi: 10.1155/2011/376543
55. Denhez B, Lizotte F, Guimond MO, Jones N, Takano T, Geraldes P. Increased SHP-1 protein expression by high glucose levels reduces nephrin phosphorylation in podocytes. J Biol Chem. (2015) 290:350–8. doi: 10.1074/jbc.M114.612721
56. Hsu MF, Bettaieb A, Ito Y, Graham J, Havel PJ, Haj FG. Protein tyrosine phosphatase Shp2 deficiency in podocytes attenuates lipopolysaccharide-induced proteinuria. Sci Rep. (2017) 7:461. doi: 10.1038/s41598-017-00564-3
57. Kumagai T, Baldwin C, Aoudjit L, Nezvitsky L, Robins R, Jiang R, et al. Protein tyrosine phosphatase 1B inhibition protects against podocyte injury and proteinuria. Am J Pathol. (2014) 184:2211–24. doi: 10.1016/j.ajpath.2014.05.005
58. Konigshausen E, Zierhut UM, Ruetze M, Potthoff SA, Stegbauer J, Woznowski M, et al. Angiotensin II increases glomerular permeability by beta-arrestin mediated nephrin endocytosis. Sci Rep. (2016) 6:39513. doi: 10.1038/srep39513
59. Quack I, Woznowski M, Potthoff SA, Palmer R, Konigshausen E, Sivritas S, et al. PKC alpha mediates beta-arrestin2-dependent nephrin endocytosis in hyperglycemia. J Biol Chem. (2011) 286:12959–70. doi: 10.1074/jbc.M110.204024
60. Tossidou I, Teng B, Menne J, Shushakova N, Park JK, Becker JU, et al. Podocytic PKC-alpha is regulated in murine and human diabetes and mediates nephrin endocytosis. PLoS ONE (2010) 5:e10185. doi: 10.1371/journal.pone.0010185
61. Quack I, Rump LC, Gerke P, Walther I, Vinke T, Vonend O, et al. beta-Arrestin2 mediates nephrin endocytosis and impairs slit diaphragm integrity. Proc Natl Acad Sci USA. (2006) 103:14110–5. doi: 10.1073/pnas.0602587103
62. Sampson MG, Gillies CE, Robertson CC, Crawford B, Vega-Warner V, Otto EA, et al. Using population genetics to interrogate the monogenic nephrotic syndrome diagnosis in a case cohort. J Am Soc Nephrol. (2016) 27:1970–83. doi: 10.1681/ASN.2015050504
63. Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. (2015) 26:1279–89. doi: 10.1681/ASN.2014050489
64. Ovunc B, Ashraf S, Vega-Warner V, Bockenhauer D, Elshakhs NA, Joseph M, et al. Mutation analysis of NPHS1 in a worldwide cohort of congenital nephrotic syndrome patients. Nephron Clin Pract. (2012) 120:c139–46. doi: 10.1159/000337379
65. Santin S, Garcia-Maset R, Ruiz P, Gimenez I, Zamora I, Pena A, et al. Nephrin mutations cause childhood- and adult-onset focal segmental glomerulosclerosis. Kidney Int. (2009) 76:1268–76. doi: 10.1038/ki.2009.381
66. Schoeb DS, Chernin G, Heeringa SF, Matejas V, Held S, Vega-Warner V, et al. Nineteen novel NPHS1 mutations in a worldwide cohort of patients with congenital nephrotic syndrome (CNS). Nephrol Dial Transplant. (2010) 25:2970–6. doi: 10.1093/ndt/gfq088
67. Philippe A, Nevo F, Esquivel EL, Reklaityte D, Gribouval O, Tete MJ, et al. Nephrin mutations can cause childhood-onset steroid-resistant nephrotic syndrome. J Am Soc Nephrol. (2008) 19:1871–8. doi: 10.1681/ASN.2008010059
68. Bonomo JA, Ng MC, Palmer ND, Keaton JM, Larsen CP, Hicks PJ, et al. Coding variants in nephrin. (NPHS1) and susceptibility to nephropathy in African Americans. Clin J Am Soc Nephrol. (2014) 9:1434–40. doi: 10.2215/CJN.00290114
69. Orloff MS, Iyengar SK, Winkler CA, Goddard KA, Dart RA, Ahuja TS, et al. Variants in the Wilms' tumor gene are associated with focal segmental glomerulosclerosis in the African American population. Physiol Genomics. (2005) 21:212–21. doi: 10.1152/physiolgenomics.00201.2004
70. Guo G, Morrison DJ, Licht JD, Quaggin SE. WT1 activates a glomerular-specific enhancer identified from the human nephrin gene. J Am Soc Nephrol. (2004) 15:2851–6. doi: 10.1097/01.ASN.0000143474.91362.C4
71. Wagner N, Wagner KD, Xing Y, Scholz H, Schedl A. The major podocyte protein nephrin is transcriptionally activated by the Wilms' tumor suppressor WT1. J Am Soc Nephrol. (2004) 15:3044–51. doi: 10.1097/01.ASN.0000146687.99058.25
72. Ristola M, Arpiainen S, Saleem MA, Holthofer H, Lehtonen S. Transcription of nephrin-Neph3 gene pair is synergistically activated by WT1 and NF-κB and silenced by DNA methylation. Nephrol Dial Transplant. (2012) 27:1737–45. doi: 10.1093/ndt/gfr576
73. Beltcheva O, Hjorleifsdottir EE, Kontusaari S, Tryggvason K. Sp1 specifically binds to an evolutionarily conserved DNA segment within a region necessary for podocyte-specific expression of nephrin. Nephron Exp Nephrol. (2010) 114:e15–22. doi: 10.1159/000245062
74. Kann M, Ettou S, Jung YL, Lenz MO, Taglienti ME, Park PJ, et al. Genome-wide analysis of Wilms' tumor 1-controlled gene expression in podocytes reveals key regulatory mechanisms. J Am Soc Nephrol. (2015) 26:2097–104. doi: 10.1681/ASN.2014090940
75. Dong L, Pietsch S, Tan Z, Perner B, Sierig R, Kruspe D, et al. Integration of cistromic and transcriptomic analyses identifies Nphs2, Mafb, and Magi2 as Wilms' tumor 1 target genes in podocyte differentiation and maintenance. J Am Soc Nephrol. (2015) 26:2118–28. doi: 10.1681/ASN.2014080819
76. Furness PN, Hall LL, Shaw JA, Pringle JH. Glomerular expression of nephrin is decreased in acquired human nephrotic syndrome. Nephrol Dial Transplant. (1999) 14:1234–7.
77. Huh W, Kim DJ, Kim MK, Kim YG, Oh HY, Ruotsalainen V, et al. Expression of nephrin in acquired human glomerular disease. Nephrol Dial Transplant. (2002) 17:478–84.
78. Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G, et al. Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes. (2003) 52:1023–30. doi: 10.2337/diabetes.52.4.1023
79. Langham RG, Kelly DJ, Cox AJ, Thomson NM, Holthofer H, Zaoui P, et al. Proteinuria and the expression of the podocyte slit diaphragm protein, nephrin, in diabetic nephropathy: effects of angiotensin converting enzyme inhibition. Diabetologia (2002) 45:1572–6. doi: 10.1007/s00125-002-0946-y
80. Garovic VD, Wagner SJ, Petrovic LM, Gray CE, Hall P, Sugimoto H, et al. Glomerular expression of nephrin and synaptopodin, but not podocin, is decreased in kidney sections from women with preeclampsia. Nephrol Dial Transplant. (2007) 22:1136–43. doi: 10.1093/ndt/gfl711
81. Kandasamy Y, Smith R, Lumbers ER, Rudd D. Nephrin-a biomarker of early glomerular injury. Biomark Res. (2014) 2:21. doi: 10.1186/2050-7771-2-21
82. Zhai T, Furuta I, Akaishi R, Kawabata K, Chiba K, Umazume T, et al. Feasibility of nephrinuria as a screening tool for the risk of pre-eclampsia: prospective observational study. BMJ Open (2016) 6:e011229. doi: 10.1136/bmjopen-2016-011229
83. Zhai T, Furuta I, Nakagawa K, Kojima T, Umazume T, Ishikawa S, et al. Second-trimester urine nephrin:creatinine ratio versus soluble fms-like tyrosine kinase-1:placental growth factor ratio for prediction of preeclampsia among asymptomatic women. Sci Rep. (2016) 6:37442. doi: 10.1038/srep37442
84. Wada Y, Abe M, Moritani H, Mitori H, Kondo M, Tanaka-Amino K, et al. Original research: potential of urinary nephrin as a biomarker reflecting podocyte dysfunction in various kidney disease models. Exp Biol Med. (Maywood). (2016) 241:1865–76. doi: 10.1177/1535370216651937
85. Perez-Hernandez J, Olivares MD, Forner MJ, Chaves FJ, Cortes R, Redon J. Urinary dedifferentiated podocytes as a non-invasive biomarker of lupus nephritis. Nephrol Dial Transplant. (2016) 31:780–9. doi: 10.1093/ndt/gfw002
86. Wang Y, Gu Y, Loyd S, Jia X, Groome LJ. Increased urinary levels of podocyte glycoproteins, matrix metallopeptidases, inflammatory cytokines, and kidney injury biomarkers in women with preeclampsia. Am J Physiol Renal Physiol. (2015) 309:F1009–17. doi: 10.1152/ajprenal.00257.2015
87. Jim B, Ghanta M, Qipo A, Fan Y, Chuang PY, Cohen HW, et al. Dysregulated nephrin in diabetic nephropathy of type 2 diabetes: a cross sectional study. PLoS ONE (2012) 7:e36041. doi: 10.1371/journal.pone.0036041
88. Fuchshuber A, Jean G, Gribouval O, Gubler MC, Broyer M, Beckmann JS, et al. Mapping a gene (SRN1) to chromosome 1q25-q31 in idiopathic nephrotic syndrome confirms a distinct entity of autosomal recessive nephrosis. Hum Mol Genet. (1995) 4:2155–8.
89. Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. (2000) 24:349–54. doi: 10.1038/74166
90. Ruf RG, Lichtenberger A, Karle SM, Haas JP, Anacleto FE, Schultheiss M, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol. (2004) 15:722–32. doi: 10.1097/01.ASN.0000113552.59155.72
91. Weber S, Gribouval O, Esquivel EL, Moriniere V, Tete MJ, Legendre C, et al. NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence. Kidney Int. (2004) 66:571–9. doi: 10.1111/j.1523-1755.2004.00776.x
92. Snyers L, Umlauf E, Prohaska R. Oligomeric nature of the integral membrane protein stomatin. J Biol Chem. (1998) 273:17221–6.
93. Roselli S, Gribouval O, Boute N, Sich M, Benessy F, Attie T, et al. Podocin localizes in the kidney to the slit diaphragm area. Am J Pathol. (2002) 160:131–9. doi: 10.1016/S0002-9440(10)64357-X
94. Huber TB, Simons M, Hartleben B, Sernetz L, Schmidts M, Gundlach E, et al. Molecular basis of the functional podocin-nephrin complex: mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum Mol Genet. (2003) 12:3397–405. doi: 10.1093/hmg/ddg360
95. Morrow IC, Parton RG. Flotillins and the PHB domain protein family: rafts, worms and anaesthetics. Traffic. (2005) 6:725–40. doi: 10.1111/j.1600-0854.2005.00318.x
96. Schermer B, Benzing T. Lipid-protein interactions along the slit diaphragm of podocytes. J Am Soc Nephrol. (2009) 20:473–8. doi: 10.1681/ASN.2008070694
97. Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest. (2001) 108:1621–9. doi: 10.1172/JCI12849
98. Saleem MA, Ni L, Witherden I, Tryggvason K, Ruotsalainen V, Mundel P, et al. Co-localization of nephrin, podocin, and the actin cytoskeleton: evidence for a role in podocyte foot process formation. Am J Pathol. (2002) 161:1459–66. doi: 10.1016/S0002-9440(10)64421-5
99. Garg P, Verma R, Nihalani D, Johnstone DB, Holzman LB. Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol Cell Biol. (2007) 27:8698–712. doi: 10.1128/MCB.00948-07
100. Swiatecka-Urban A. Endocytic Trafficking at the Mature Podocyte Slit Diaphragm. Front Pediatr. (2017) 5:32. doi: 10.3389/fped.2017.00032
101. Ising C, Bharill P, Brinkkoetter S, Brahler S, Schroeter C, Koehler S, et al. Prohibitin-2 depletion unravels extra-mitochondrial functions at the kidney filtration barrier. Am J Pathol. (2016) 186:1128–39. doi: 10.1016/j.ajpath.2015.12.018
102. Schurek EM, Volker LA, Tax J, Lamkemeyer T, Rinschen MM, Ungrue D, et al. A disease-causing mutation illuminates the protein membrane topology of the kidney-expressed prohibitin homology (PHB) domain protein podocin. J Biol Chem. (2014) 289:11262–71. doi: 10.1074/jbc.M113.521773
103. Huber TB, Schermer B, Muller RU, Hohne M, Bartram M, Calixto A, et al. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc Natl Acad Sci USA. (2006) 103:17079–86. doi: 10.1073/pnas.0607465103
104. Caridi G, Bertelli R, Di Duca M, Dagnino M, Emma F, Onetti Muda A, et al. Broadening the spectrum of diseases related to podocin mutations. J Am Soc Nephrol. (2003) 14:1278–86. doi: 10.1097/01.ASN.0000060578.79050.E0
105. Hinkes B, Vlangos C, Heeringa S, Mucha B, Gbadegesin R, Liu J, et al. Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J Am Soc Nephrol. (2008) 19:365–71. doi: 10.1681/ASN.2007040452
106. Berdeli A, Mir S, Yavascan O, Serdaroglu E, Bak M, Aksu N, et al. NPHS2 (podicin) mutations in Turkish children with idiopathic nephrotic syndrome. Pediatr Nephrol. (2007) 22:2031–40. doi: 10.1007/s00467-007-0595-y
107. Karle SM, Uetz B, Ronner V, Glaeser L, Hildebrandt F, Fuchshuber A. Novel mutations in NPHS2 detected in both familial and sporadic steroid-resistant nephrotic syndrome. J Am Soc Nephrol. (2002) 13:388–93.
108. Machuca E, Hummel A, Nevo F, Dantal J, Martinez F, Al-Sabban E, et al. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int. (2009) 75:727–35. doi: 10.1038/ki.2008.650
109. Tsukaguchi H, Sudhakar A, Le TC, Nguyen T, Yao J, Schwimmer JA, et al. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J Clin Invest. (2002) 110:1659–66. doi: 10.1172/JCI16242
110. McKenzie LM, Hendrickson SL, Briggs WA, Dart RA, Korbet SM, Mokrzycki MH, et al. NPHS2 variation in sporadic focal segmental glomerulosclerosis. J Am Soc Nephrol. (2007) 18:2987–95. doi: 10.1681/ASN.2007030319
111. Dusel JA, Burdon KP, Hicks PJ, Hawkins GA, Bowden DW, Freedman BI. Identification of podocin (NPHS2) gene mutations in African Americans with nondiabetic end-stage renal disease. Kidney Int. (2005) 68:256–62. doi: 10.1111/j.1523-1755.2005.00400.x
112. Jungraithmayr TC, Hofer K, Cochat P, Chernin G, Cortina G, Fargue S, et al. Screening for NPHS2 mutations may help predict FSGS recurrence after transplantation. J Am Soc Nephrol. (2011) 22:579–85. doi: 10.1681/ASN.2010010029
113. Burghardt T, Kastner J, Suleiman H, Rivera-Milla E, Stepanova N, Lottaz C, et al. LMX1B is essential for the maintenance of differentiated podocytes in adult kidneys. J Am Soc Nephrol. (2013) 24:1830–48. doi: 10.1681/ASN.2012080788
114. He B, Ebarasi L, Zhao Z, Guo J, Ojala JR, Hultenby K, et al. Lmx1b and FoxC combinatorially regulate podocin expression in podocytes. J Am Soc Nephrol. (2014) 25:2764–77. doi: 10.1681/ASN.2012080823
115. Harendza S, Stahl RA, Schneider A. The transcriptional regulation of podocin. (NPHS2) by Lmx1b and a promoter single nucleotide polymorphism. Cell Mol Biol Lett. (2009) 14:679–91. doi: 10.2478/s11658-009-0026-0
116. Boyer O, Woerner S, Yang F, Oakeley EJ, Linghu B, Gribouval O, et al. LMX1B mutations cause hereditary FSGS without extrarenal involvement. J Am Soc Nephrol. (2013) 24:1216–22. doi: 10.1681/ASN.2013020171
117. Andeen NK, Schleit J, Blosser CD, Dorschner MO, Hisama FM, Smith KD. LMX1B-Associated nephropathy with Type III collagen deposition in the glomerular and tubular basement membranes. Am J Kidney Dis. (2017). doi: 10.1053/j.ajkd.2017.09.023
118. Dong L, Pietsch S, Englert C. Towards an understanding of kidney diseases associated with WT1 mutations. Kidney Int. (2015) 88:684–90. doi: 10.1038/ki.2015.198
119. Bouchireb K, Boyer O, Gribouval O, Nevo F, Huynh-Cong E, Moriniere V, et al. NPHS2 mutations in steroid-resistant nephrotic syndrome: a mutation update and the associated phenotypic spectrum. Hum Mutat. (2014) 35:178–86. doi: 10.1002/humu.22485
120. Chernin G, Heeringa SF, Gbadegesin R, Liu J, Hinkes BG, Vlangos CN, et al. Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. (2008) 23:1455–60. doi: 10.1007/s00467-008-0861-7
121. Perysinaki GS, Moysiadis DK, Bertsias G, Giannopoulou I, Kyriacou K, Nakopoulou L, et al. Podocyte main slit diaphragm proteins, nephrin and podocin, are affected at early stages of lupus nephritis and correlate with disease histology. Lupus. (2011) 20:781–91. doi: 10.1177/0961203310397412
122. Guan N, Ding J, Zhang J, Yang J. Expression of nephrin, podocin, alpha-actinin, and WT1 in children with nephrotic syndrome. Pediatr Nephrol. (2003) 18:1122–7. doi: 10.1007/s00467-003-1240-z
123. Koop K, Eikmans M, Baelde HJ, Kawachi H, De Heer E, Paul LC, et al. Expression of podocyte-associated molecules in acquired human kidney diseases. J Am Soc Nephrol. (2003) 14:2063–71. doi: 10.1097/01.ASN.0000078803.53165.C9
124. Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. (2006) 68:619–47. doi: 10.1146/annurev.physiol.68.040204.100431
125. Greka A, Mundel P. Calcium regulates podocyte actin dynamics. Semin Nephrol. (2012) 32:319–26. doi: 10.1016/j.semnephrol.2012.06.003
126. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science (2005) 308:1801–4. doi: 10.1126/science.1106215
127. Ilatovskaya DV, Staruschenko A. TRPC6 channel as an emerging determinant of the podocyte injury susceptibility in kidney diseases. Am J Physiol Renal Physiol. (2015) 309:F393–7. doi: 10.1152/ajprenal.00186.2015
128. Riehle M, Buscher AK, Gohlke BO, Kassmann M, Kolatsi-Joannou M, Brasen JH, et al. TRPC6 G757D Loss-of-Function Mutation Associates with FSGS. J Am Soc Nephrol. (2016) 27:2771–83. doi: 10.1681/ASN.2015030318
129. Sonneveld R, van der Vlag J, Baltissen MP, Verkaart SA, Wetzels JF, Berden JH, et al. Glucose specifically regulates TRPC6 expression in the podocyte in an AngII-dependent manner. Am J Pathol. (2014) 184:1715–26. doi: 10.1016/j.ajpath.2014.02.008
130. Ilatovskaya DV, Levchenko V, Lowing A, Shuyskiy LS, Palygin O, Staruschenko A. Podocyte injury in diabetic nephropathy: implications of angiotensin II-dependent activation of TRPC channels. Sci Rep. (2015) 5:17637. doi: 10.1038/srep17637
131. Wieder N, Greka A. Calcium, TRPC channels, and regulation of the actin cytoskeleton in podocytes: towards a future of targeted therapies. Pediatr Nephrol. (2016) 31:1047–54. doi: 10.1007/s00467-015-3224-1
132. Schaldecker T, Kim S, Tarabanis C, Tian D, Hakroush S, Castonguay P, et al. Inhibition of the TRPC5 ion channel protects the kidney filter. J Clin Invest. (2013) 123:5298–309. doi: 10.1172/JCI71165
133. Zhou Y, Castonguay P, Sidhom EH, Clark AR, Dvela-Levitt M, Kim S, et al. A small-molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science (2017) 358:1332–6. doi: 10.1126/science.aal4178
134. Wang X, Dande RR, Yu H, Samelko B, Miller RE, Altintas MM, et al. TRPC5 does not cause or aggravate Glomerular disease. J Am Soc Nephrol. (2017). doi: 10.1681/ASN.2017060682
135. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature (2002) 420:629–35. doi: 10.1038/nature01148
136. Katz ME, McCormick F. Signal transduction from multiple Ras effectors. Curr Opin Genet Dev. (1997) 7:75–9.
137. Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. (2007) 17:428–37. doi: 10.1016/j.tcb.2007.06.006
138. Wang L, Ellis MJ, Gomez JA, Eisner W, Fennell W, Howell DN, et al. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int. (2012) 81:1075–85. doi: 10.1038/ki.2011.472
139. Zhu L, Jiang R, Aoudjit L, Jones N, Takano T. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J Am Soc Nephrol. (2011) 22:1621–30. doi: 10.1681/ASN.2010111146
140. Babelova A, Jansen F, Sander K, Lohn M, Schafer L, Fork C, et al. Activation of Rac-1 and RhoA contributes to podocyte injury in chronic kidney disease. PLoS ONE (2013) 8:e80328. doi: 10.1371/journal.pone.0080328
141. Shibata S, Nagase M, Fujita T. Fluvastatin ameliorates podocyte injury in proteinuric rats via modulation of excessive Rho signaling. J Am Soc Nephrol. (2006) 17:754–64. doi: 10.1681/ASN.2005050571
142. Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. (2006) 8:485–91. doi: 10.1038/ncb1400
143. Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. (2008) 14:931–8. doi: 10.1038/nm.1857
144. Blattner SM, Hodgin JB, Nishio M, Wylie SA, Saha J, Soofi AA, et al. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int. (2013) 84:920–30. doi: 10.1038/ki.2013.175
145. Attias O, Jiang R, Aoudjit L, Kawachi H, Takano T. Rac1 contributes to actin organization in glomerular podocytes. Nephron Exp Nephrol. (2010) 114:e93-e106. doi: 10.1159/000262317
146. Zhang H, Cybulsky AV, Aoudjit L, Zhu J, Li H, Lamarche-Vane N, et al. Role of Rho-GTPases in complement-mediated glomerular epithelial cell injury. Am J Physiol Renal Physiol. (2007) 293:F148–56. doi: 10.1152/ajprenal.00294.2006
147. Auguste D, Maier M, Baldwin C, Aoudjit L, Robins R, Gupta IR, et al. Disease-causing mutations of RhoGDIalpha induce Rac1 hyperactivation in podocytes. Small GTPases (2016) 7:107–21. doi: 10.1080/21541248.2015.1113353
148. Sun Y, Guo C, Ma P, Lai Y, Yang F, Cai J, et al. Kindlin-2 Association with Rho GDP-dissociation inhibitor alpha suppresses Rac1 activation and podocyte injury. J Am Soc Nephrol. (2017) 28:3545–62. doi: 10.1681/ASN.2016091021
149. Yu H, Suleiman H, Kim AH, Miner JH, Dani A, Shaw AS, et al. Rac1 activation in podocytes induces rapid foot process effacement and proteinuria. Mol Cell Biol. (2013) 33:4755–64. doi: 10.1128/MCB.00730-13
150. Scott RP, Hawley SP, Ruston J, Du J, Brakebusch C, Jones N, et al. Podocyte-specific loss of Cdc42 leads to congenital nephropathy. J Am Soc Nephrol. (2012) 23:1149–54. doi: 10.1681/ASN.2011121206
151. Huang Z, Zhang L, Chen Y, Zhang H, Zhang Q, Li R, et al. Cdc42 deficiency induces podocyte apoptosis by inhibiting the Nwasp/stress fibers/YAP pathway. Cell Death Dis. (2016) 7:e2142. doi: 10.1038/cddis.2016.51
152. Campbell KN, Wong JS, Gupta R, Asanuma K, Sudol M, He JC, et al. Yes-associated protein (YAP) promotes cell survival by inhibiting proapoptotic dendrin signaling. J Biol Chem. (2013) 288:17057–62. doi: 10.1074/jbc.C113.457390
153. Gee HY, Sadowski CE, Aggarwal PK, Porath JD, Yakulov TA, Schueler M, et al. FAT1 mutations cause a glomerulotubular nephropathy. Nat Commun. (2016) 7:10822. doi: 10.1038/ncomms10822
154. Gee HY, Saisawat P, Ashraf S, Hurd TW, Vega-Warner V, Fang H, et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest. (2013) 123:3243–53. doi: 10.1172/JCI69134
155. Akilesh S, Suleiman H, Yu H, Stander MC, Lavin P, Gbadegesin R, et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest. (2011) 121:4127–37. doi: 10.1172/JCI46458
156. Lin J, Shi Y, Peng H, Shen X, Thomas S, Wang Y, et al. Loss of PTEN promotes podocyte cytoskeletal rearrangement, aggravating diabetic nephropathy. J Pathol. (2015) 236:30–40. doi: 10.1002/path.4508
157. Kelly DJ, Aaltonen P, Cox AJ, Rumble JR, Langham R, Panagiotopoulos S, et al. Expression of the slit-diaphragm protein, nephrin, in experimental diabetic nephropathy: differing effects of anti-proteinuric therapies. Nephrol Dial Transplant. (2002) 17:1327–32.
158. Veron D, Bertuccio CA, Marlier A, Reidy K, Garcia AM, Jimenez J, et al. Podocyte vascular endothelial growth factor. (Vegf164) overexpression causes severe nodular glomerulosclerosis in a mouse model of type 1 diabetes. Diabetologia (2011) 54:1227–41. doi: 10.1007/s00125-010-2034-z
159. Gong Y, Sunq A, Roth RA, Hou J. Inducible Expression of Claudin-1 in Glomerular Podocytes Generates Aberrant Tight Junctions and Proteinuria through Slit Diaphragm Destabilization. J Am Soc Nephrol. (2017) 28:106–17. doi: 10.1681/ASN.2015121324
160. Tonna SJ, Needham A, Polu K, Uscinski A, Appel GB, Falk RJ, et al. NPHS2 variation in focal and segmental glomerulosclerosis. BMC Nephrol. (2008) 9:13. doi: 10.1186/1471-2369-9-13
161. Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science (2003) 300:1298–300. doi: 10.1126/science.1081068
162. Lowik MM, Groenen PJ, Pronk I, Lilien MR, Goldschmeding R, Dijkman HB, et al. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int. (2007) 72:1198–203. doi: 10.1038/sj.ki.5002469
163. Gigante M, Pontrelli P, Montemurno E, Roca L, Aucella F, Penza R, et al. CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS). Nephrol Dial Transplant. (2009) 24:1858–64. doi: 10.1093/ndt/gfn712
164. Heeringa SF, Moller CC, Du J, Yue L, Hinkes B, Chernin G, et al. A novel TRPC6 mutation that causes childhood FSGS. PLoS ONE (2009) 4:e7771. doi: 10.1371/journal.pone.0007771
165. Hofstra JM, Lainez S, van Kuijk WH, Schoots J, Baltissen MP, Hoefsloot LH, et al. New TRPC6 gain-of-function mutation in a non-consanguineous Dutch family with late-onset focal segmental glomerulosclerosis. Nephrol Dial Transplant. (2013) 28:1830–8. doi: 10.1093/ndt/gfs572
166. Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. (2005) 37:739–44. doi: 10.1038/ng1592
167. Santin S, Ars E, Rossetti S, Salido E, Silva I, Garcia-Maset R, et al. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol Dial Transplant. (2009) 24:3089–96. doi: 10.1093/ndt/gfp229
168. Schlondorff J, Del Camino D, Carrasquillo R, Lacey V, Pollak MR. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am J Physiol Cell Physiol. (2009) 296:C558–69. doi: 10.1152/ajpcell.00077.2008
169. Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nurnberg G, et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet. (2006) 38:1397–405. doi: 10.1038/ng1918
170. Ohashi T, Uchida K, Asamiya Y, Tsuruta Y, Ohno M, Horita S, et al. Phosphorylation status of nephrin in human membranous nephropathy. Clin Exp Nephrol. (2010) 14:51–5. doi: 10.1007/s10157-009-0241-z
171. Sasaki Y, Hidaka T, Ueno T, Akiba-Takagi M, Oliva Trejo JA, Seki T, et al. Sorting Nexin 9 facilitates podocin endocytosis in the injured podocyte. Sci Rep. (2017) 7:43921. doi: 10.1038/srep43921
172. Lahdenkari AT, Kestila M, Holmberg C, Koskimies O, Jalanko H. Nephrin gene (NPHS1) in patients with minimal change nephrotic syndrome (MCNS). Kidney Int. (2004) 65:1856–63. doi: 10.1111/j.1523-1755.2004.00583.x
173. Uchida K, Suzuki K, Iwamoto M, Kawachi H, Ohno M, Horita S, et al. Decreased tyrosine phosphorylation of nephrin in rat and human nephrosis. Kidney Int. (2008) 73:926–32. doi: 10.1038/ki.2008.19
174. Tory K, Menyhard DK, Woerner S, Nevo F, Gribouval O, Kerti A, et al. Mutation-dependent recessive inheritance of NPHS2-associated steroid-resistant nephrotic syndrome. Nat Genet. (2014) 46:299–304. doi: 10.1038/ng.2898
175. Ishizaka M, Gohda T, Takagi M, Omote K, Sonoda Y, Oliva Trejo JA, et al. Podocyte-specific deletion of Rac1 leads to aggravation of renal injury in STZ-induced diabetic mice. Biochem Biophys Res Commun. (2015) 467:549–55. doi: 10.1016/j.bbrc.2015.09.158
176. Dai S, Wang Z, Pan X, Wang W, Chen X, Ren H, et al. Functional analysis of promoter mutations in the ACTN4 and SYNPO genes in focal segmental glomerulosclerosis. Nephrol Dial Transplant. (2010) 25:824–35. doi: 10.1093/ndt/gfp394
177. Benoit G, Machuca E, Nevo F, Gribouval O, Lepage D, Antignac C. Analysis of recessive CD2AP and ACTN4 mutations in steroid-resistant nephrotic syndrome. Pediatr Nephrol. (2010) 25:445–51. doi: 10.1007/s00467-009-1372-x
178. Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. (2000) 24:251–6. doi: 10.1038/73456
179. Boyer O, Benoit G, Gribouval O, Nevo F, Tete MJ, Dantal J, et al. Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J Am Soc Nephrol. (2011) 22:239–45. doi: 10.1681/ASN.2010050518
180. Gbadegesin RA, Lavin PJ, Hall G, Bartkowiak B, Homstad A, Jiang R, et al. Inverted formin 2 mutations with variable expression in patients with sporadic and hereditary focal and segmental glomerulosclerosis. Kidney Int. (2012) 81:94–9. doi: 10.1038/ki.2011.297
181. Caridi G, Lugani F, Dagnino M, Gigante M, Iolascon A, Falco M, et al. Novel INF2 mutations in an Italian cohort of patients with focal segmental glomerulosclerosis, renal failure and Charcot-Marie-Tooth neuropathy. Nephrol Dial Transplant. (2014) 29(Suppl. 4):iv80–6. doi: 10.1093/ndt/gfu071
182. Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med. (2011) 365:295–306. doi: 10.1056/NEJMoa1101273
183. Lal MA, Andersson AC, Katayama K, Xiao Z, Nukui M, Hultenby K, et al. Rhophilin-1 is a key regulator of the podocyte cytoskeleton and is essential for glomerular filtration. J Am Soc Nephrol. (2015) 26:647–62. doi: 10.1681/ASN.2013111195
184. Schwartzman M, Reginensi A, Wong JS, Basgen JM, Meliambro K, Nicholas SB, et al. Podocyte-Specific Deletion of Yes-Associated Protein Causes FSGS and Progressive Renal Failure. J Am Soc Nephrol. (2016) 27:216–26. doi: 10.1681/ASN.2014090916
185. Sendeyo K, Audard V, Zhang SY, Fan Q, Bouachi K, Ollero M, et al. Upregulation of c-mip is closely related to podocyte dysfunction in membranous nephropathy. Kidney Int. (2013) 83:414–25. doi: 10.1038/ki.2012.426
186. George B, Verma R, Soofi AA, Garg P, Zhang J, Park TJ, et al. Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease. J Clin Invest. (2012) 122:674–92. doi: 10.1172/JCI60070
187. Gee HY, Zhang F, Ashraf S, Kohl S, Sadowski CE, Vega-Warner V, et al. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J Clin Invest. (2015) 125:2375–84. doi: 10.1172/JCI79504
188. Ashworth S, Teng B, Kaufeld J, Miller E, Tossidou I, Englert C, et al. Cofilin-1 inactivation leads to proteinuria–studies in zebrafish, mice and humans. PLoS ONE (2010) 5:e12626. doi: 10.1371/journal.pone.0012626
189. Djinovic-Carugo K, Young P, Gautel M, Saraste M. Structure of the alpha-actinin rod: molecular basis for cross-linking of actin filaments. Cell. (1999) 98:537–46.
190. Asanuma K, Kim K, Oh J, Giardino L, Chabanis S, Faul C, et al. Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J Clin Invest. (2005) 115:1188–98. doi: 10.1172/JCI23371
191. Yanagida-Asanuma E, Asanuma K, Kim K, Donnelly M, Young Choi H, Hyung Chang J, et al. Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am J Pathol. (2007) 171:415–27. doi: 10.2353/ajpath.2007.070075
192. Buvall L, Wallentin H, Sieber J, Andreeva S, Choi HY, Mundel P, et al. Synaptopodin is a coincidence detector of tyrosine versus serine/threonine phosphorylation for the modulation of rho protein crosstalk in podocytes. J Am Soc Nephrol. (2017) 28:837–51. doi: 10.1681/ASN.2016040414
193. Yu H, Kistler A, Faridi MH, Meyer JO, Tryniszewska B, Mehta D, et al. Synaptopodin limits TRPC6 Podocyte surface expression and attenuates proteinuria. J Am Soc Nephrol. (2016) 27:3308–19. doi: 10.1681/ASN.2015080896
194. Barisoni L, Kriz W, Mundel P, D'Agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. (1999) 10:51–61.
195. Lu TC, He JC, Klotman PE. Podocytes in HIV-associated nephropathy. Nephron Clin Pract. (2007) 106:c67–71. doi: 10.1159/000101800
196. Barisoni L, Mokrzycki M, Sablay L, Nagata M, Yamase H, Mundel P. Podocyte cell cycle regulation and proliferation in collapsing glomerulopathies. Kidney Int. (2000) 58:137–43. doi: 10.1046/j.1523-1755.2000.00149.x
197. Hill GS, Karoui KE, Karras A, Mandet C, Duong Van Huyen JP, Nochy D, et al. Focal segmental glomerulosclerosis plays a major role in the progression of IgA nephropathy. I. Immunohistochemical studies. Kidney Int. (2011) 79:635–42. doi: 10.1038/ki.2010.466
198. Srivastava T, Garola RE, Whiting JM, Alon US. Synaptopodin expression in idiopathic nephrotic syndrome of childhood. Kidney Int. (2001) 59:118–25. doi: 10.1046/j.1523-1755.2001.00472.x
199. Hirakawa M, Tsuruya K, Yotsueda H, Tokumoto M, Ikeda H, Katafuchi R, et al. Expression of synaptopodin and GLEPP1 as markers of steroid responsiveness in primary focal segmental glomerulosclerosis. Life Sci. (2006) 79:757–63. doi: 10.1016/j.lfs.2006.02.031
200. Wagrowska-Danilewicz M, Danilewicz M. [Synaptopodin immunoexpression in steroid-responsive and steroid-resistant minimal change disease and focal segmental glomerulosclerosis]. Nefrologia (2007) 27:710–5.
201. Szeto CC, Wang G, Chow KM, Lai FM, Ma TK, Kwan BC, et al. Podocyte mRNA in the urinary sediment of minimal change nephropathy and focal segmental glomerulosclerosis. Clin Nephrol. (2015) 84:198–205. doi: 10.5414/CN108607
202. Kwon SK, Kim SJ, Kim HY. Urine synaptopodin excretion is an important marker of glomerular disease progression. Korean J Intern Med. (2016) 31:938–43. doi: 10.3904/kjim.2015.226
203. Hasegawa K, Wakino S, Simic P, Sakamaki Y, Minakuchi H, Fujimura K, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. (2013) 19:1496–504. doi: 10.1038/nm.3363
204. Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes (2006) 55:225–33. doi: 10.2337/diabetes.55.01.06.db05-0894
205. Tejada T, Catanuto P, Ijaz A, Santos JV, Xia X, Sanchez P, et al. Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death. Kidney Int. (2008) 73:1385–93. doi: 10.1038/ki.2008.109
206. Liapis H. Molecular pathology of nephrotic syndrome in childhood: a contemporary approach to diagnosis. Pediatr Dev Pathol. (2008) 11:154–63. doi: 10.2350/07-11-0375.1
207. Garg P, Verma R, Cook L, Soofi A, Venkatareddy M, George B, et al. Actin-depolymerizing factor cofilin-1 is necessary in maintaining mature podocyte architecture. J Biol Chem. (2010) 285:22676–88. doi: 10.1074/jbc.M110.122929
208. Sahali D, Sendeyo K, Mangier M, Audard V, Zhang SY, Lang P, et al. Immunopathogenesis of idiopathic nephrotic syndrome with relapse. Semin Immunopathol. (2014) 36:421–9. doi: 10.1007/s00281-013-0415-3
209. Rosenberg AZ, Kopp JB. Focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. (2017) 12:502–17. doi: 10.2215/CJN.05960616
210. Chen YM, Liapis H. Focal segmental glomerulosclerosis: molecular genetics and targeted therapies. BMC Nephrol. (2015) 16:101. doi: 10.1186/s12882-015-0090-9
211. D'Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. New Engl J Med. (2011) 365:2398–411. doi: 10.1056/NEJMra1106556
212. Jefferson JA, Shankland SJ. The pathogenesis of focal segmental glomerulosclerosis. Adv Chronic Kidney Dis. (2014) 21:408–16. doi: 10.1053/j.ackd.2014.05.009
213. Rheault MN, Gbadegesin RA. The genetics of nephrotic syndrome. J Pediatr Genet. (2016) 5:15–24. doi: 10.1055/s-0035-1557109
214. Yu H, Artomov M, Brahler S, Stander MC, Shamsan G, Sampson MG, et al. A role for genetic susceptibility in sporadic focal segmental glomerulosclerosis. J Clin Invest. (2016) 126:1067–78. doi: 10.1172/JCI82592
215. Krause MW, Fonseca VA, Shah SV. Combination inhibition of the renin-angiotensin system: is more better? Kidney international. (2011) 80:245–55. doi: 10.1038/ki.2011.142
216. Xu HZ, Wang WN, Zhang YY, Cheng YL, Xu ZG. Effect of angiotensin II type 1 receptor blocker on 12-lipoxygenase activity and slit diaphragm protein expression in type 2 diabetic rat glomeruli. J Nephrol. (2016) 29:775–82. doi: 10.1007/s40620-016-0296-3
217. Wang N, Wei RB, Li P, Li QP, Yang X, Yang Y, et al. Treatment with irbesatan may improve slit diaphragm alterations in rats with adriamycin-induced nephropathy. J Renin Angiotensin Aldosterone Syst. (2016) 17:1470320316646884. doi: 10.1177/1470320316646884
218. Takahashi A, Fukusumi Y, Yamazaki M, Kayaba M, Kitazawa Y, Tomita M, et al. Angiotensin II type 1 receptor blockade ameliorates proteinuria in puromycin aminonucleoside nephropathy by inhibiting the reduction of NEPH1 and nephrin. J Nephrol. (2014) 27:627–34. doi: 10.1007/s40620-014-0147-z
219. Ilatovskaya DV, Palygin O, Chubinskiy-Nadezhdin V, Negulyaev YA, Ma R, Birnbaumer L, et al. Angiotensin II has acute effects on TRPC6 channels in podocytes of freshly isolated glomeruli. Kidney Int. (2014) 86:506–14. doi: 10.1038/ki.2014.71
220. Nijenhuis T, Sloan AJ, Hoenderop JG, Flesche J, van Goor H, Kistler AD, et al. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol. (2011) 179:1719–32. doi: 10.1016/j.ajpath.2011.06.033
221. Roshanravan H, Dryer SE. ATP acting through P2Y receptors causes activation of podocyte TRPC6 channels: role of podocin and reactive oxygen species. Am J Physiol Renal Physiol. (2014) 306:F1088–97. doi: 10.1152/ajprenal.00661.2013
222. Eckel J, Lavin PJ, Finch EA, Mukerji N, Burch J, Gbadegesin R, et al. TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol. (2011) 22:526–35. doi: 10.1681/ASN.2010050522
223. Zhang J, Yanez D, Floege A, Lichtnekert J, Krofft RD, Liu ZH, et al. ACE-inhibition increases podocyte number in experimental glomerular disease independent of proliferation. J Renin Angiotensin Aldosterone Syst. (2015) 16:234–48. doi: 10.1177/1470320314543910
224. Kaverina NV, Eng DG, Schneider RR, Pippin JW, Shankland SJ. Partial podocyte replenishment in experimental FSGS derives from nonpodocyte sources. Am J Physiol Renal Physiol. (2016) 310:F1397–413. doi: 10.1152/ajprenal.00369.2015
225. Lichtnekert J, Kaverina NV, Eng DG, Gross KW, Kutz JN, Pippin JW, et al. Renin-angiotensin-aldosterone system inhibition increases podocyte derivation from cells of renin lineage. J Am Soc Nephrol. (2016) 27:3611–27. doi: 10.1681/ASN.2015080877
226. Eng DG, Kaverina NV, Schneider RRS, Freedman BS, Gross KW, Miner JH, et al. Detection of renin lineage cell transdifferentiation to podocytes in the kidney glomerulus with dual lineage tracing. Kidney Int. (2018) 93:1240–6. doi: 10.1016/j.kint.2018.01.014
227. Vivarelli M, Massella L, Ruggiero B, Emma F. Minimal change disease. Clin J Am Soc Nephrol. (2017) 12:332–45. doi: 10.2215/CJN.05000516
228. Guess A, Agrawal S, Wei CC, Ransom RF, Benndorf R, Smoyer WE. Dose- and time-dependent glucocorticoid receptor signaling in podocytes. Am J Physiol Renal Physiol. (2010) 299:F845–53. doi: 10.1152/ajprenal.00161.2010
229. Xing CY, Saleem MA, Coward RJ, Ni L, Witherden IR, Mathieson PW. Direct effects of dexamethasone on human podocytes. Kidney Int. (2006) 70:1038–45. doi: 10.1038/sj.ki.5001655
230. Jiang L, Hindmarch CC, Rogers M, Campbell C, Waterfall C, Coghill J, et al. RNA sequencing analysis of human podocytes reveals glucocorticoid regulated gene networks targeting non-immune pathways. Sci Rep. (2016) 6:35671. doi: 10.1038/srep35671
231. Liu H, Gao X, Xu H, Feng C, Kuang X, Li Z, et al. alpha-Actinin-4 is involved in the process by which dexamethasone protects actin cytoskeleton stabilization from adriamycin-induced podocyte injury. Nephrology. (Carlton). (2012) 17:669–75. doi: 10.1111/j.1440-1797.2012.01645.x
232. Mallipattu SK, Guo Y, Revelo MP, Roa-Pena L, Miller T, Ling J, et al. Kruppel-like factor 15 mediates glucocorticoid-induced restoration of podocyte differentiation markers. J Am Soc Nephrol. (2017) 28:166–84. doi: 10.1681/ASN.2015060672
233. Zhang J, Pippin JW, Krofft RD, Naito S, Liu ZH, Shankland SJ. Podocyte repopulation by renal progenitor cells following glucocorticoids treatment in experimental FSGS. Am J Physiol Renal Physiol. (2013) 304:F1375–89. doi: 10.1152/ajprenal.00020.2013
234. Ponticelli C, Locatelli F. Glucocorticoids in the Treatment of Glomerular Diseases: Pitfalls and Pearls. Clin J Am Soc Nephrol. (2018) 13:815–22. doi: 10.2215/CJN.12991117
235. Komers R. Rho kinase inhibition in diabetic kidney disease. Br J Clin Pharmacol. (2013) 76:551–9. doi: 10.1111/bcp.12196
236. Wang L, Ellis MJ, Fields TA, Howell DN, Spurney RF. Beneficial effects of the Rho kinase inhibitor Y27632 in murine puromycin aminonucleoside nephrosis. Kidney Blood Press Res. (2008) 31:111–21. doi: 10.1159/000121531
237. Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. (2008) 14:1370–6. doi: 10.1038/nm.1879
238. de Zeeuw D, Agarwal R, Amdahl M, Audhya P, Coyne D, Garimella T, et al. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet (2010) 376:1543–51. doi: 10.1016/S0140-6736(10)61032-X
239. Trohatou O, Tsilibary EF, Charonis A, Iatrou C, Drossopoulou G. Vitamin D3 ameliorates podocyte injury through the nephrin signalling pathway. J Cell Mol Med. (2017) 21:2599–609. doi: 10.1111/jcmm.13180
240. Song Z, Guo Y, Zhou M, Zhang X. The PI3K/p-Akt signaling pathway participates in calcitriol ameliorating podocyte injury in DN rats. Metabolism (2014) 63:1324–33. doi: 10.1016/j.metabol.2014.06.013
241. Deb DK, Wang Y, Zhang Z, Nie H, Huang X, Yuan Z, et al. Molecular mechanism underlying 1,25-dihydroxyvitamin D regulation of nephrin gene expression. J Biol Chem. (2011) 286:32011–7. doi: 10.1074/jbc.M111.269118
242. Deb DK, Sun T, Wong KE, Zhang Z, Ning G, Zhang Y, et al. Combined vitamin D analog and AT1 receptor antagonist synergistically block the development of kidney disease in a model of type 2 diabetes. Kidney Int. (2010) 77:1000–9. doi: 10.1038/ki.2010.22
243. Zhang Z, Zhang Y, Ning G, Deb DK, Kong J, Li YC. Combination therapy with AT1 blocker and vitamin D analog markedly ameliorates diabetic nephropathy: blockade of compensatory renin increase. Proc Natl Acad Sci USA. (2008) 105:15896–901. doi: 10.1073/pnas.0803751105
244. Fernandez-Fernandez B, Ortiz A. Paricalcitol and albuminuria: tread carefully. Lancet Diabetes Endocrinol. (2018) 6:3–5. doi: 10.1016/S2213-8587(17)30361-3
245. Parvanova A, Trillini M, Podesta MA, Iliev IP, Ruggiero B, Abbate M, et al. Moderate salt restriction with or without paricalcitol in type 2 diabetes and losartan-resistant macroalbuminuria. (PROCEED): a randomised, double-blind, placebo-controlled, crossover trial. Lancet Diabetes Endocrinol. (2018) 6:27–40. doi: 10.1016/S2213-8587(17)30359-5
246. He JC, Lu TC, Fleet M, Sunamoto M, Husain M, Fang W, et al. Retinoic acid inhibits HIV-1-induced podocyte proliferation through the cAMP pathway. J Am Soc Nephrol. (2007) 18:93–102. doi: 10.1681/ASN.2006070727
247. Suzuki A, Ito T, Imai E, Yamato M, Iwatani H, Kawachi H, et al. Retinoids regulate the repairing process of the podocytes in puromycin aminonucleoside-induced nephrotic rats. J Am Soc Nephrol. (2003) 14:981–91.
248. Dai Y, Chen A, Liu R, Gu L, Sharma S, Cai W, et al. Retinoic acid improves nephrotoxic serum-induced glomerulonephritis through activation of podocyte retinoic acid receptor alpha. Kidney Int. (2017) 92:1444–57. doi: 10.1016/j.kint.2017.04.026
249. Zhang J, Pippin JW, Vaughan MR, Krofft RD, Taniguchi Y, Romagnani P, et al. Retinoids augment the expression of podocyte proteins by glomerular parietal epithelial cells in experimental glomerular disease. Nephron Exp Nephrol. (2012) 121:e23–37. doi: 10.1159/000342808
250. Mallipattu SK, He JC. The beneficial role of retinoids in glomerular disease. Front Med (Lausanne). (2015) 2:16. doi: 10.3389/fmed.2015.00016
251. Mallipattu SK, Estrada CC, He JC. The critical role of Kruppel-like factors in kidney disease. Am J Physiol Renal Physiol. (2017) 312:F259–65. doi: 10.1152/ajprenal.00550.2016
252. Kronbichler A, Konig P, Busch M, Wolf G, Mayer G, Rudnicki M. Rituximab in adult patients with multi-relapsing/steroid-dependent minimal change disease and focal segmental glomerulosclerosis: a report of 5 cases. Wien Klin Wochenschr. (2013) 125:328–33. doi: 10.1007/s00508-013-0366-7
253. Ochi A, Takei T, Nakayama K, Iwasaki C, Kamei D, Tsuruta Y, et al. Rituximab treatment for adult patients with focal segmental glomerulosclerosis. Intern Med. (2012) 51:759–62. doi: 10.2169/internalmedicine.51.6854
254. Fernandez-Fresnedo G, Segarra A, Gonzalez E, Alexandru S, Delgado R, Ramos N, et al. Rituximab treatment of adult patients with steroid-resistant focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. (2009) 4:1317–23. doi: 10.2215/CJN.00570109
255. Ruggenenti P, Ruggiero B, Cravedi P, Vivarelli M, Massella L, Marasa M, et al. Rituximab in steroid-dependent or frequently relapsing idiopathic nephrotic syndrome. J Am Soc Nephrol. (2014) 25:850–63. doi: 10.1681/asn.2013030251
256. Hogan J, Radhakrishnan J. The treatment of minimal change disease in adults. J Am Soc Nephrol. (2013) 24:702–11. doi: 10.1681/asn.2012070734
257. Ponticelli C, Graziani G. Current and emerging treatments for idiopathic focal and segmental glomerulosclerosis in adults. Expert Rev Clin Immunol. (2013) 9:251–61. doi: 10.1586/eci.12.109
258. Iijima K, Sako M, Nozu K, Mori R, Tuchida N, Kamei K, et al. Rituximab for childhood-onset, complicated, frequently relapsing nephrotic syndrome or steroid-dependent nephrotic syndrome: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet (2014) 384:1273–81. doi: 10.1016/s0140-6736(14)60541-9
259. Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Trans Med. (2011) 3:85ra46. doi: 10.1126/scitranslmed.3002231
260. Watanabe S, Tsugawa K, Tsuruga K, Imaizumi T, Tanaka H. Urinary excretion of sphingomyelinase phosphodiesterase acid-like 3b in children with intractable nephrotic syndrome. Pediatr Int. (2017) 59:1112–5. doi: 10.1111/ped.13355
261. Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardino L, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. (2004) 113:1390–7. doi: 10.1172/jci20402
262. Yu CC, Fornoni A, Weins A, Hakroush S, Maiguel D, Sageshima J, et al. Abatacept in B7-1-positive proteinuric kidney disease. New Engl J Med. (2013) 369:2416–23. doi: 10.1056/NEJMoa1304572
263. Delville M, Baye E, Durrbach A, Audard V, Kofman T, Braun L, et al. B7-1 Blockade does not improve post-transplant nephrotic syndrome caused by recurrent FSGS. J Am Soc Nephrol. (2016) 27:2520–7. doi: 10.1681/asn.2015091002
264. Novelli R, Gagliardini E, Ruggiero B, Benigni A, Remuzzi G. Any value of podocyte B7-1 as a biomarker in human MCD and FSGS? Am J Physiol Renal Physiol. (2016) 310:F335–41. doi: 10.1152/ajprenal.00510.2015
265. Baye E, Gallazzini M, Delville M, Legendre C, Terzi F, Canaud G. The costimulatory receptor B7-1 is not induced in injured podocytes. Kidney Int. (2016) 90:1037–44. doi: 10.1016/j.kint.2016.06.022
266. Kristensen T, Ivarsen P, Povlsen JV. Unsuccessful treatment with abatacept in recurrent focal segmental glomerulosclerosis after kidney transplantation. Case Rep Nephrol Dial. (2017) 7:1–5. doi: 10.1159/000454947
267. Cravedi P, Angeletti A, Remuzzi G. New biologics in the treatment of rare glomerular diseases of childhood. Curr Opin Pharmacol. (2017) 33:27–33. doi: 10.1016/j.coph.2017.03.010
268. Tian X, Ishibe S. Targeting the podocyte cytoskeleton: from pathogenesis to therapy in proteinuric kidney disease. Nephrol Dial Transplant. (2016) 31:1577–83. doi: 10.1093/ndt/gfw021
269. Fan X, Yang H, Kumar S, Tumelty KE, Pisarek-Horowitz A, Rasouly HM, et al. SLIT2/ROBO2 signaling pathway inhibits nonmuscle myosin IIA activity and destabilizes kidney podocyte adhesion. JCI Insight (2016) 1:e86934. doi: 10.1172/jci.insight.86934
270. Long J, Badal SS, Ye Z, Wang Y, Ayanga BA, Galvan DL, et al. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J Clin Invest. (2016) 126:4205–18. doi: 10.1172/JCI87927
271. Yamamoto-Nonaka K, Koike M, Asanuma K, Takagi M, Oliva Trejo JA, Seki T, et al. Cathepsin D in podocytes is important in the pathogenesis of proteinuria and CKD. J Am Soc Nephrol. (2016) 27:2685–700. doi: 10.1681/ASN.2015040366
272. Lawrence MG, Altenburg MK, Sanford R, Willett JD, Bleasdale B, Ballou B, et al. Permeation of macromolecules into the renal glomerular basement membrane and capture by the tubules. Proc Natl Acad Sci USA. (2017) 114:2958–63. doi: 10.1073/pnas.1616457114
273. Schlondorff D, Wyatt CM, Campbell KN. Revisiting the determinants of the glomerular filtration barrier: what goes round must come round. Kidney Int. (2017) 92:533–6. doi: 10.1016/j.kint.2017.06.003
274. Shankland SJ, Freedman BS, Pippin JW. Can podocytes be regenerated in adults? Curr Opin Nephrol Hypertens (2017) 26:154–64. doi: 10.1097/MNH.0000000000000311
275. Lasagni L, Angelotti ML, Ronconi E, Lombardi D, Nardi S, Peired A, et al. podocyte regeneration driven by renal progenitors determines glomerular disease remission and can be pharmacologically enhanced. Stem Cell Rep. (2015) 5:248–63. doi: 10.1016/j.stemcr.2015.07.003
276. Musah S, Mammoto A, Ferrante TC, Jeanty SSF, Hirano-Kobayashi M, Mammoto T, et al. Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary-wall function on a chip. Nat Biomed Eng. (2017) 1. doi: 10.1038/s41551-017-0069
277. Puelles VG, van der Wolde JW, Schulze KE, Short KM, Wong MN, Bensley JG, et al. Validation of a three-dimensional method for counting and sizing podocytes in whole glomeruli. J Am Soc Nephrol. (2016) 27:3093–104. doi: 10.1681/ASN.2015121340
278. Brahler S, Yu H, Suleiman H, Krishnan GM, Saunders BT, Kopp JB, et al. Intravital and kidney slice imaging of podocyte membrane dynamics. J Am Soc Nephrol. (2016) 27:3285–90. doi: 10.1681/ASN.2015121303
279. Rinschen MM, Wu X, Konig T, Pisitkun T, Hagmann H, Pahmeyer C, et al. Phosphoproteomic analysis reveals regulatory mechanisms at the kidney filtration barrier. J Am Soc Nephrol. (2014) 25:1509–22. doi: 10.1681/ASN.2013070760
280. Schena FP, Nistor I, Curci C. Transcriptomics in kidney biopsy is an untapped resource for precision therapy in nephrology: a systematic review. Nephrol Dial Transplant. (2017). doi: 10.1093/ndt/gfx211
281. Kesselheim A, Ashton E, Bockenhauer D. Potential and pitfalls in the genetic diagnosis of kidney diseases. Clin Kidney J. (2017) 10:581–5. doi: 10.1093/ckj/sfx075
282. Weber S, Buscher AK, Hagmann H, Liebau MC, Heberle C, Ludwig M, et al. Dealing with the incidental finding of secondary variants by the example of SRNS patients undergoing targeted next-generation sequencing. Pediatr Nephrol. (2016) 31:73–81. doi: 10.1007/s00467-015-3167-6
283. Schell C, Huber TB. The Evolving Complexity of the Podocyte Cytoskeleton. J Am Soc Nephrol. (2017) 28:3166–74. doi: 10.1681/ASN.2017020143
284. Assady S, Wanner N, Skorecki KL, Huber TB. New Insights into podocyte biology in glomerular health and disease. J Am Soc Nephrol. (2017) 28:1707–15. doi: 10.1681/ASN.2017010027
Keywords: cytoskeleton, nephrin, podocin, podocyte, slit diaphragm, synaptopodin, Rho/small GTPases, TRPC5/6
Citation: Yu SM-W, Nissaisorakarn P, Husain I and Jim B (2018) Proteinuric Kidney Diseases: A Podocyte's Slit Diaphragm and Cytoskeleton Approach. Front. Med. 5:221. doi: 10.3389/fmed.2018.00221
Received: 07 May 2018; Accepted: 18 July 2018;
Published: 11 September 2018.
Edited by:
Robert P. Woroniecki, Stony Brook Children's Hospital, United StatesReviewed by:
Sandeep Mallipattu, Stony Brook University, United StatesDeepak Nihalani, Medical University of South Carolina, United States
Copyright © 2018 Yu, Nissaisorakarn, Husain and Jim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samuel Mon-Wei Yu, c2FtdWVsbXl1QGdtYWlsLmNvbQ==