 Duncan J. Campbell1,2,3*
Duncan J. Campbell1,2,3*- 1Department of Molecular Cardiology, St. Vincent's Institute of Medical Research, Fitzroy, VIC, Australia
- 2Department of Medicine, The University of Melbourne, Parkville, VIC, Australia
- 3St. Vincent's Hospital, Melbourne, VIC, Australia
Bradykinin has important physiological actions related to the regulation of blood vessel tone and renal function, and protection from ischemia reperfusion injury. However, bradykinin also contributes to pathological states such as angioedema and inflammation. Bradykinin is metabolized by many different peptidases that play a major role in the control of bradykinin levels. Peptidase inhibitor therapies such as angiotensin converting enzyme (ACE) and neprilysin inhibitors increase bradykinin levels, and the challenge for such therapies is to achieve the beneficial cardiovascular and renal effects without the adverse consequences such as angioedema that may result from increased bradykinin levels. Neprilysin also metabolizes natriuretic peptides. However, despite the potential therapeutic benefit of increased natriuretic peptide and bradykinin levels, neprilysin inhibitor therapy has only modest efficacy in essential hypertension and heart failure. Initial attempts to combine neprilysin inhibition with inhibition of the renin angiotensin system led to the development of omapatrilat, a drug that combines ACE and neprilysin inhibition. However, omapatrilat produced an unacceptably high incidence of angioedema in patients with hypertension (2.17%) in comparison with the ACE inhibitor enalapril (0.68%), although angioedema incidence was less in patients with heart failure with reduced ejection fraction (HFrEF) treated with omapatrilat (0.8%), and not different from that for enalapril therapy (0.5%). More recently, LCZ696, a drug that combines angiotensin receptor blockade and neprilysin inhibition, was approved for the treatment of HFrEF. The approval of LCZ696 therapy for HFrEF represents the first approval of long-term neprilysin inhibitor administration. While angioedema incidence was acceptably low in HFrEF patients receiving LCZ696 therapy (0.45%), it remains to be seen whether LCZ696 therapy for other conditions such as hypertension is also accompanied by an acceptable incidence of angioedema.
Introduction
Despite decreasing incidence, cardiovascular disease remains a major cause of premature morbidity and mortality (1), and there is a continuing search for new therapies for its prevention and treatment. LCZ696 (Entresto) is the first of a new drug class referred to as ARNI (dual acting angiotensin receptor-neprilysin inhibitor) that contains equimolar amounts of valsartan, a type 1 angiotensin II receptor blocker (ARB) and sacubitril, a prodrug that is hydrolyzed to form LBQ657, a potent inhibitor of neprilysin (Table 1). The approval of LCZ696 as therapy for heart failure with reduced ejection fraction (HFrEF) represents the first approval of long-term neprilysin inhibitor therapy. Neprilysin is a key enzyme in the degradation of natriuretic peptides, and the primary rationale for neprilysin inhibitor therapy in cardiovascular disease was to increase endogenous natriuretic peptide levels, and thereby achieve the vasodilatation and natriuresis these peptides produce. However, neprilysin degrades many other peptides, including bradykinin (17). Bradykinin may contribute not only to the benefits of neprilysin inhibitor therapy but also to the adverse effects of this therapy. Of particular concern for drugs that inhibit bradykinin degradation and thereby increase bradykinin levels is the risk of angioedema, with increased bradykinin levels implicated in both hereditary and drug-induced forms of angioedema (18–21). This review will briefly describe neprilysin, the kallikrein kinin system, and the role of neprilysin in bradykinin metabolism, and then discuss the potential role of kinins in mediating the therapeutic benefits and adverse effects of neprilysin inhibitor therapy.
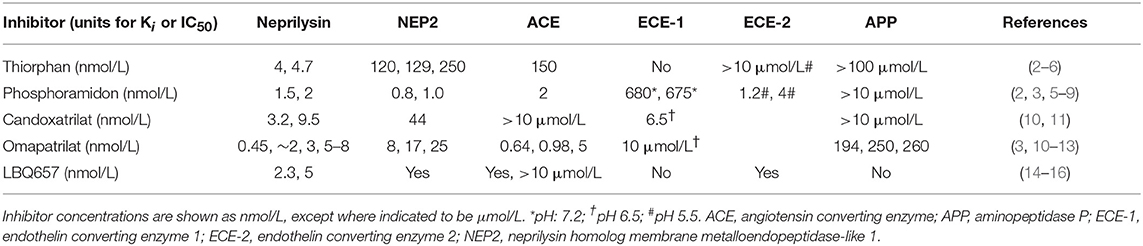
Table 1. Specificity of neprilysin inhibitors (Ki or IC50).
Neprilysin
Neprilysin, also known as neutral endopeptidase 24.11, common acute lymphoblastic leukemia antigen (CALLA), and cluster of differentiation cell surface molecule 10 (CD10), is a member of the neprilysin (M13) family of metallopeptidases. The neprilysin family also includes the neprilysin homolog membrane metalloendopeptidase-like 1 (NEP2) (22), endothelin converting enzymes 1 and 2 (ECE-1 and ECE-2), endothelin converting enzyme-like 1 (ECEL1), phosphate-regulating neutral endopeptidase (PHEX), and the KELL blood group glycoprotein (23, 24). Neprilysin and several other members of the neprilysin family of metallopeptidases degrade bradykinin (Table 2, Figure 1). Neprilysin is a predominantly membrane-bound zinc-dependent metallopeptidase with a broad tissue distribution, including the central nervous system, kidney, and vascular endothelium (39). Neprilysin is expressed at a low level on the membrane of mononuclear cells, and at higher levels by neutrophils, lymphocytes, and lymphoid progenitors (40, 41). A soluble form of neprilysin is found in blood plasma, cerebrospinal fluid, amniotic fluid, and seminal plasma. Neprilysin has a broad substrate selectivity (17), preferentially cleaving peptides on the amino side of the hydrophobic residues phenylalanine, leucine, and methionine (39, 42, 43).
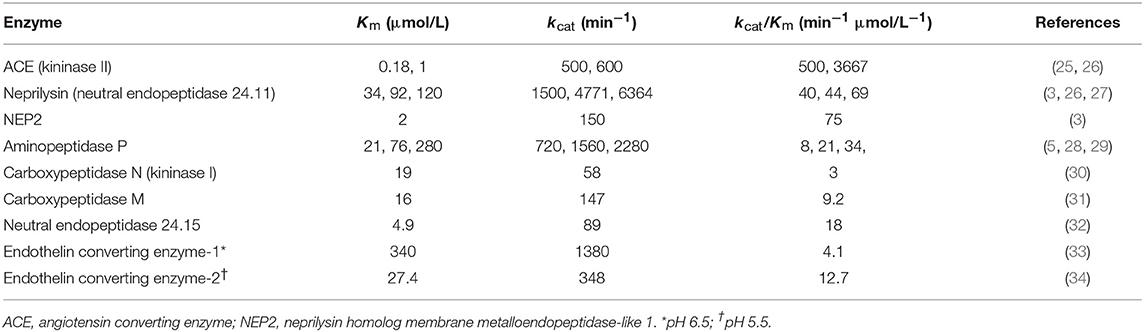
Table 2. Kinetic parameters of bradykinin hydrolysis by different enzymes.
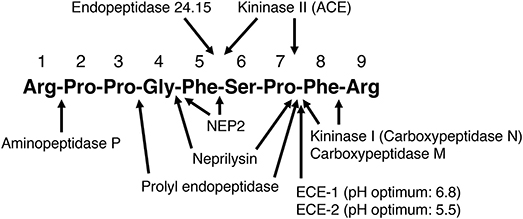
Figure 1. Sites of cleavage of bradykinin by different enzymes. ACE, angiotensin converting enzyme; ECE-1, endothelin converting enzyme-1; ECE-2, endothelin converting enzyme-2; NEP2, neprilysin homolog membrane metalloendopeptidase-like 1. (2–5, 7, 25–28, 30, 32–38).
The Kallikrein Kinin System
The kallikrein kinin system has been reviewed elsewhere (44–46). In humans, plasma kallikrein forms the nonapeptide bradykinin from high molecular weight kininogen, whereas tissue kallikrein forms the decapeptide kallidin (Lys0-bradykinin) from both high and low molecular weight kininogens (Figure 2). Bradykinin is also generated by aminopeptidase-mediated cleavage of kallidin. A proportion of high molecular weight kininogen is hydroxylated on the third proline of the bradykinin sequence, leading to the formation of both hydroxylated and non-hydroxylated bradykinin and kallidin peptides. Hydroxylated and non-hydroxylated kinin peptides are of similar abundance (48–50), and hydroxylated kinins have similar biological activity to non-hydroxylated kinins (46). In the rat, both plasma and tissue kallikrein produce bradykinin, which is not hydroxylated (47).
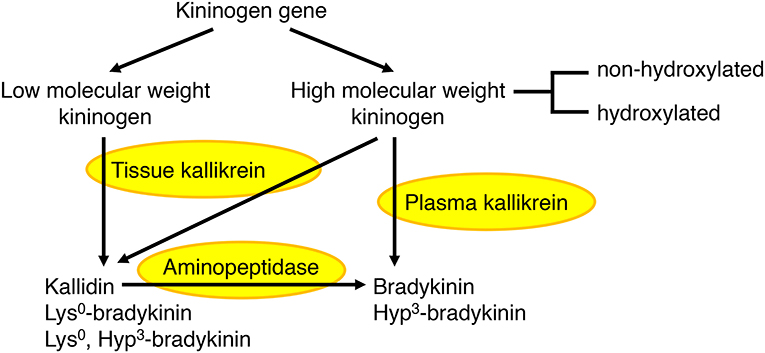
Figure 2. Formation of bradykinin and kallidin peptides. In humans, plasma kallikrein cleaves high molecular weight kininogen to produce bradykinin, whereas tissue kallikrein cleaves both high and low molecular weight kininogens to produce kallidin (Lys0-bradykinin). Bradykinin can also be generated by aminopeptidase-mediated cleavage of kallidin. A proportion of high molecular weight kininogen is hydroxylated on the third proline (Hyp3) of the bradykinin sequence, leading to the formation of both hydroxylated and non-hydroxylated bradykinin and kallidin peptides. In the rat, both plasma and tissue kallikrein produce bradykinin, which is not hydroxylated (44–47).
There are two types of kinin receptor, the type 1 (B1) receptor and the type 2 (B2) receptor. The B2 receptor normally predominates, whereas B1 receptors are induced by tissue injury. Bradykinin and kallidin are more potent on the B2 receptor, whereas the carboxypeptidase N (kininase I) metabolites bradykinin-(1-8) and Lys0-bradykinin-(1-8) are also bioactive and more potent on B1 receptors (46). Kinin peptides have a broad spectrum of activities that include the regulation of blood vessel tone and renal function, and protection from ischemia reperfusion injury (45). However, kinins also participate in inflammation, producing vasodilatation, increased vascular permeability, neutrophil chemotaxis and pain (45).
Tissue Specific Regulation of Kinin Levels
The kallikrein kinin system is primarily a tissue-based system, with tissue kinin levels much higher than blood kinin levels in both humans and in rats (47–49). Evidence for the tissue-specific regulation of the kallikrein kinin system is the marked variation in kinin levels between different tissues of the rat (47). Kinin peptide levels are also higher in atrial tissue than blood of humans (48, 49). Moreover, bradykinin peptide levels are higher than kallidin peptide levels in blood and atrial tissue of humans, whereas kallidin peptide levels are much higher than bradykinin peptide levels in urine (48, 49). Many different enzymes cleave bradykinin and may participate in its metabolism (Figure 1), and peptidase activity plays a major role in the tissue-specific regulation of bradykinin levels (51).
Role of Neprilysin in Bradykinin Metabolism
Several different experimental approaches have been used to study the role of neprilysin in bradykinin metabolism. These include study of the effects of neprilysin gene (MME) deletion and mutation, study of the effects of neprilysin inhibitor administration on physiological bradykinin levels, and study of the metabolism of exogenous (supra-physiological) bradykinin levels and the effect of neprilysin and other peptidase inhibitors on the metabolism of exogenous bradykinin. The effect of inhibition of an enzyme on bradykinin levels depends not only on the specific enzyme's contribution to bradykinin metabolism, relative to other enzymes, but also the baseline degradation rate for bradykinin. This is best illustrated by bradykinin metabolism by the pulmonary circulation, where bradykinin is degraded with approximately 99% efficiency (51). Thus, 1% inhibition of pulmonary inactivation will double the amount of bradykinin surviving pulmonary degradation, and may therefore double the level of bradykinin in arterial blood (Figure 3).
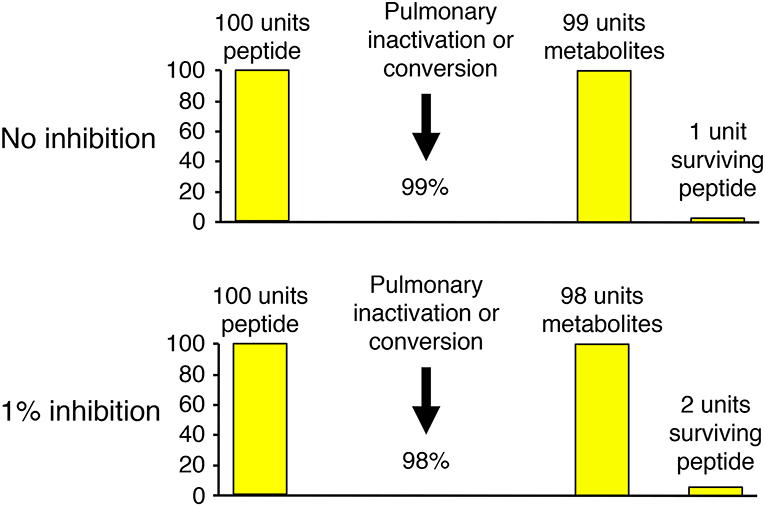
Figure 3. Illustration of how the effect of inhibition of an enzyme on bradykinin levels depends not only on the specific enzyme's contribution to bradykinin metabolism, relative to other enzymes, but also the baseline degradation rate for bradykinin. Pulmonary inactivation of bradykinin is approximately 99% (51). Thus, 1% inhibition of pulmonary inactivation will double the amount of bradykinin surviving pulmonary degradation, and may therefore double the level of bradykinin in arterial blood.
Neprilysin Gene Knockout in Mice
Neprilysin gene knockout in mice causes increased basal vascular permeability, hypotension and reduced heart weight/body weight ratio (52). The reduced heart weight/body weight ratio was attributed to the lower blood pressure. The vascular permeability, but not hypotension, was reversed by administration of recombinant neprilysin, and also by separate administration of SR140333, a substance P (NK1) receptor antagonist, and the bradykinin B2 receptor antagonist icatibant. The increased basal vascular permeability of neprilysin gene knock out was reproduced by administration of the neprilysin inhibitors thiorphan and phosphoramidon (Table 1) to wild-type C57BL/6 mice. These observations indicate an important role for neprilysin in the control of bradykinin- and substance P-mediated regulation of vascular permeability and blood pressure in the mouse. Bradykinin stimulates substance P release from sensory neurons (53), and neprilysin degrades both peptides (54), thereby providing an explanation why neprilysin gene knockout or neprilysin inhibition could increase both bradykinin and substance P levels, and why either a substance P receptor or bradykinin B2 receptor antagonist was able to prevent the increased vascular permeability.
Other consequences of neprilysin gene knockout in the mouse include hyperalgesia and increased susceptibility to inflammation (55, 56), enhanced lethality in response to endotoxin-induced shock (57), shortened ventilatory expiratory time in response to a hypoxic stimulus (58), and improved learning and memory (59). However, apart from hyperalgesia, which was reduced by icatibant (55), the relevance of these consequences of neprilysin gene knockout to bradykinin is unknown.
Neprilysin Gene Deletion and Neprilysin Gene Mutation in Humans
In contrast to the effects of neprilysin gene knockout in the mouse, five women with total neprilysin deficiency due to homozygous truncating mutations of the neprilysin gene had no reported phenotype, although the absence of neprilysin induced an alloimmunization process against neprilysin present in fetal cells, leading to membranous glomerulopathy in their infants (60).
Loss–of-function and missense mutations in the neprilysin gene are associated with polyneuropathy, and also with decreased tissue availability of neprilysin and reduced neprilysin enzymatic activity (61–63), although the relevance of the polyneuropathy to bradykinin is unknown. However, the association of the rs989692 variant of the neprilysin gene with ACE inhibitor-associated angioedema (64) is evidence for a role for neprilysin in the regulation of bradykinin levels in humans.
Effect of Neprilysin Inhibition on Physiological Bradykinin Levels
We examined the effect of the neprilysin inhibitor ecadotril (acetorphan, an orally active prodrug of (S)-thiorphan) on bradykinin levels in Sprague Dawley rats (65). Ecadotril administration produced dose-related occupancy of renal neprilysin, as determined by binding of the neprilysin radioligand 125I-RB104 to kidney sections, and increased total neprilysin levels in plasma, similar to the induction of plasma ACE levels by ACE inhibitor therapy (66, 67). Ecadotril administration produced diuresis, natriuresis and increased urinary excretion of cyclic GMP and bradykinin, indicating a role for neprilysin in bradykinin degradation in renal tubules and/or in urine. However, ecadotril administration did not increase bradykinin levels in blood or renal tissue, although the ACE inhibitor perindopril increased bradykinin levels in both blood and kidney. Ecadotril did, however, increase cardiac bradykinin levels by approximately 2-fold; although the increase in cardiac bradykinin levels did not achieve statistical significance, ecadotril produced a statistically significant reduction in the bradykinin-(1-7)/bradykinin-(1-9) ratio in the heart, consistent with reduced formation of bradykinin-(1-7) by neprilysin-mediated cleavage of bradykinin (Figure 1), and indicating a role for neprilysin in bradykinin metabolism in the heart.
Neprilysin inhibition also increased urinary bradykinin excretion in deoxycorticosterone acetate (DOCA)-salt hypertensive rats, spontaneously hypertensive rats and renovascular hypertensive rats (68). Together, these studies indicate a role for neprilysin in bradykinin metabolism in renal tubules and/or urine, and also in the heart.
Role of Neprilysin in the Metabolism of Supra-Physiological Bradykinin Levels
There is need for caution in the interpretation of studies of the degradation of exogenously administered bradykinin where bradykinin levels may be considerably higher than physiological levels. An enzyme's contribution to bradykinin degradation, and the effect of inhibition of that enzyme on bradykinin degradation, depends not only on the concentration of the enzyme but also on the bradykinin concentration and the Km (Michaelis constant) of the enzyme for bradykinin degradation (Figure 4). Thus, depending on an enzyme's abundance and kcat (turnover number), an enzyme with a low Km, such as angiotensin converting enzyme (ACE) (Table 2), may have a predominant role in bradykinin metabolism when bradykinin levels are low, whereas an enzyme with a higher Km, such as carboxypeptidase M or N, may play a more dominant role when bradykinin levels are high (35).
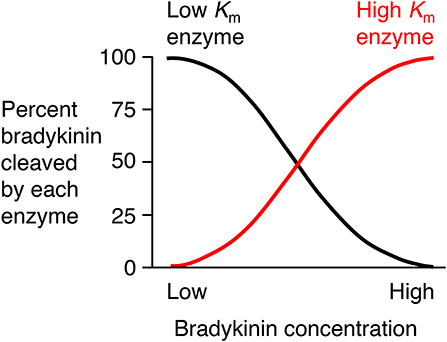
Figure 4. Relative contributions of low Km (Michaelis constant) enzyme and high Km enzyme to bradykinin degradation by a mixture of low and high Km enzymes. An enzyme's contribution to bradykinin degradation, and the effect of inhibition of that enzyme on bradykinin degradation, depends not only on the concentration of the enzyme but also on the bradykinin concentration and the Km of the enzyme for bradykinin degradation. Thus, depending on an enzyme's abundance and kcat (turnover number), an enzyme with a low Km, such as ACE, may have a predominant role in bradykinin metabolism when bradykinin levels are low, whereas an enzyme with a higher Km, such as carboxypeptidase M or N, may play a more dominant role when bradykinin levels are high. Based on data reported by Kuoppala et al. (35).
Two approaches have been used to investigate the metabolism of exogenously administered bradykinin, either examination of the metabolites of bradykinin, or comparison of the effects of different peptidase inhibitors on bradykinin metabolism. Bradykinin-(1-5) was the predominant bradykinin metabolite when bradykinin was infused into human subjects (69), indicative of cleavage by ACE. Moreover, ACE played a predominant role in bradykinin metabolism by human and rat plasma and serum (35, 70–75), with a lesser contribution by carboxypeptidase (kininase I) and aminopeptidase activities. However, carboxypeptidase, a peptidase with higher Km than ACE (Table 2) played a greater role than ACE in bradykinin metabolism when human or rat serum was incubated with ≥μmol/L bradykinin concentrations (76, 77), thereby illustrating how higher bradykinin concentrations can lead to a greater contribution by an enzyme with higher Km to bradykinin metabolism (35).
Another example where a higher concentration of bradykinin led to a greater contribution to bradykinin metabolism by a peptidase with higher Km is the study of bradykinin metabolism by the isolated perfused rat mesenteric arterial bed (78). When bradykinin metabolism was assessed by recovery of bradykinin in the perfusate after injection of ~100 nmol bradykinin, carboxypeptidase inhibition, and to a lesser extent neprilysin inhibition, but not ACE inhibition, reduced bradykinin metabolism (78). These findings were supported by the greater role played by carboxypeptidase B than ACE in the degradation of μmol/L concentrations of bradykinin by mesenteric arterial perfusate (79). However, the opposite result was obtained when bradykinin metabolism was assessed by the vasodilator response of the isolated perfused rat mesenteric arterial bed to ~100 pmol bradykinin, whereby the vasodilator response was potentiated by ACE inhibition, but not by either carboxypeptidase or neprilysin inhibition (78).
ACE played a greater role than neprilysin in bradykinin metabolism by isolated human small resistance vessels (80). Additionally, ACE played a dominant role, with a lesser role for aminopeptidase P, carboxypeptidase, and neprilysin, in bradykinin metabolism by the rat pulmonary vascular bed (10, 51, 77, 81), the isolated perfused rat heart (82–86), and isolated porcine coronary arteries (87), and was the predominant kininase in coronary perfusate, with a lesser role for neprilysin and carboxypeptidase (79). ACE was also the dominant peptidase contributing to bradykinin metabolism by the isolated perfused rat kidney, without evidence for contribution by neprilysin, carboxypeptidase, or aminopeptidase P (88).
A key limitation of the studies of the role of neprilysin in bradykinin metabolism so far described is the failure to address how different peptidases may make different contributions to bradykinin metabolism in different tissue compartments. In support of a tissue compartment-specific role for neprilysin in bradykinin metabolism, studies of lung, cardiac and renal bush border membranes, and urine, demonstrated a contribution by neprilysin that was equal to (89), or greater than (89–91), the contribution of ACE. Further evidence for a tissue compartment-specific role for neprilysin in bradykinin degradation was the metabolism of a bolus of 3H-bradykinin by the isolated perfused rat heart, which showed a delayed release of 3H-bradykinin-(1-7) into the perfusate, consistent with 3H-bradykinin-(1-7) formation in the interstitial compartment of the heart by neprilysin-mediated cleavage of 3H-bradykinin (84).
Role of Kinins in Mediating the Effects of Neprilysin Inhibition
Many studies have used either bradykinin receptor antagonists, anti-bradykinin antibodies, or serine protease (kallikrein) inhibitors to demonstrate a role for bradykinin in mediating the effects of neprilysin inhibitors. Two different mechanisms may account for the potentiation of bradykinin receptor-mediated actions by neprilysin inhibitors (Figure 5). Firstly, neprilysin inhibitors may potentiate bradykinin receptor-mediated actions by inhibiting bradykinin degradation and increasing bradykinin levels in the vicinity of the receptor. Secondly, neprilysin inhibitors may potentiate bradykinin receptor-mediated actions by promoting cross-talk between the neprilysin-inhibitor complex and the bradykinin receptor (92), similar to the cross-talk between the ACE-inhibitor complex and the B2 receptor proposed to mediate ACE inhibitor-induced potentiation of bradykinin receptor-mediated effects (93). Bradykinin receptor antagonists, anti-bradykinin antibodies, and kallikrein inhibitors have different effects on these two mechanisms of neprilysin inhibitor-induced potentiation of bradykinin receptor-mediated actions. A bradykinin receptor antagonist that occupies the bradykinin receptor can prevent both mechanisms of neprilysin inhibitor-induced potentiation of bradykinin receptor-mediated actions. However, bradykinin antibodies that prevent bradykinin binding to its receptor by sequestering bradykinin, and kallikrein inhibitors that prevent bradykinin binding to its receptor by preventing its formation, do not impact on cross-talk between the neprilysin-inhibitor complex and the bradykinin receptor. Therefore, prevention of the effects of neprilysin inhibition by bradykinin antibodies or kallikrein inhibitors indicates that these effects are mediated by increased bradykinin levels consequent to inhibition of neprilysin-mediated bradykinin degradation, and not by cross-talk between the neprilysin-inhibitor complex and the bradykinin receptor.
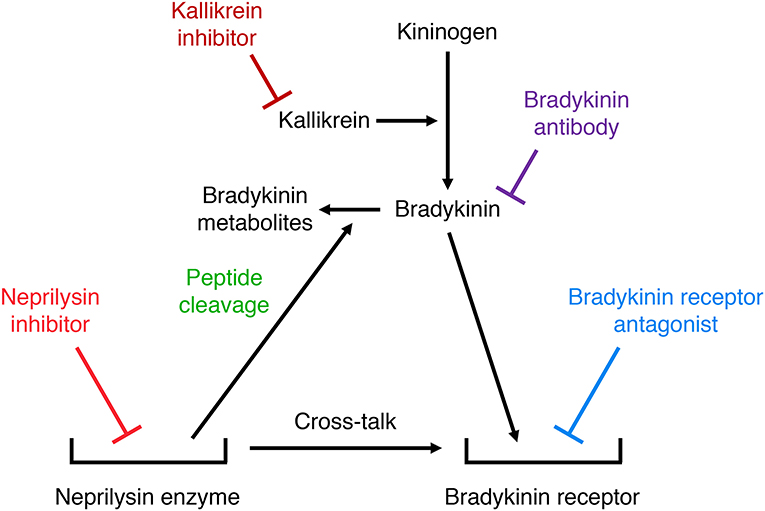
Figure 5. Illustration of two different mechanisms by which neprilysin inhibitors may potentiate bradykinin receptor-mediated actions. Firstly, neprilysin inhibitors may increase bradykinin receptor occupancy by inhibiting bradykinin degradation and increasing bradykinin levels in the vicinity of the receptor. Secondly, neprilysin inhibitors may promote cross-talk between the neprilysin-inhibitor complex and the bradykinin receptor. Bradykinin receptor antagonists, anti-bradykinin antibodies, and kallikrein inhibitors have different effects on these two mechanisms of neprilysin inhibitor-induced potentiation of bradykinin receptor-mediated actions. A bradykinin receptor antagonist that occupies the bradykinin receptor can prevent both mechanisms of neprilysin inhibitor-induced potentiation of bradykinin receptor-mediated actions. However, bradykinin antibodies that prevent bradykinin binding to its receptor by sequestering bradykinin, and kallikrein inhibitors that prevent bradykinin binding to its receptor by preventing its formation, do not impact on cross-talk between the neprilysin-inhibitor complex and the bradykinin receptor.
Role of Kinins in Mediating the Renal Effects of Neprilysin Inhibition
Icatibant prevented the diuretic and natriuretic effects of neprilysin inhibition in normal Sprague Dawley rats (94–96). Bradykinin receptor antagonism also prevented the neprilysin inhibitor-induced potentiation of atrial natriuretic peptide-induced diuresis and natriuresis in rats (97) and in chronic caval dogs (98). Moreover, anti-bradykinin antibodies prevented neprilysin inhibitor-induced potentiation of diuresis, natriuresis and increase in urinary cyclic GMP excretion in volume-expanded rats (99). However, in contrast to studies in normal Sprague Dawley rats, icatibant did not prevent the natriuretic effects of neprilysin inhibition in DOCA-salt hypertensive rats (96, 100), which suggests that the effects of neprilysin inhibition in DOCA-salt hypertensive rats are primarily mediated by increased natriuretic peptide levels consequent to inhibition of natriuretic peptide metabolism.
Role of Kinins in Mediating the Cardiac Effects of Neprilysin Inhibition
Icatibant prevented neprilysin inhibitor-induced reduction in ischemia-reperfusion injury in the rat heart (101), and neprilysin inhibitor-induced potentiation of pre-conditioning-induced reduction in infarct size in the rabbit heart (102). In addition, icatibant prevented neprilysin inhibitor-induced reversal of isoproterenol-induced myocardial hypoperfusion in the rat (103), and neprilysin inhibitor-induced nitric oxide production by isolated canine coronary microvessels (104). Neprilysin inhibitor-induced nitric oxide production by isolated canine coronary microvessels was also prevented by the serine protease (kallikrein) inhibitor dichloroisocoumarin (104).
Omapatrilat and Bradykinin
Despite the potential therapeutic benefits of increased natriuretic peptide and bradykinin levels, neprilysin inhibitor therapy has only modest efficacy in essential hypertension and heart failure, which might be due in part to the inhibition of neprilysin metabolism of the vasoconstrictors angiotensin II and endothelin 1, and the increased plasma angiotensin II, endothelin 1 and noradrenaline levels that accompany neprilysin inhibitor therapy (17). Therefore, to prevent the renin angiotensin system from countering the therapeutic benefits of neprilysin inhibition, neprilysin inhibitor therapy was combined with inhibition of the renin angiotensin system, leading to the development of omapatrilat. Omapatrilat is a single molecule that inhibits both neprilysin and ACE (Table 1). Additionally, omapatrilat inhibits aminopeptidase P, NEP2, and ECE-1 (Table 1). There is currently no information on the effects of omapatrilat on bradykinin levels. However, given that both neprilysin and ACE degrade bradykinin, one would predict higher bradykinin levels with omapatrilat than ACE inhibitor therapy, which no doubt accounts for the higher incidence of angioedema with omapatrilat therapy. The incidence of angioedema was higher for omapatrilat therapy (2.17%) than for enalapril therapy (0.68%) in hypertensive patients (105), and omapatrilat failed to achieve regulatory approval because of the angioedema incidence. However, the incidence of angioedema was lower in patients with HFrEF, without statistically significant difference between omapatrilat therapy (0.8%) and enalapril therapy (0.5%) (106).
The potential consequences of combined neprilysin and ACE inhibition were examined in the rat tracheal plasma extravasation assay (Table 3). Whereas neither ecadotril, sufficient to produce >90% inhibition of renal neprilysin, nor lisinopril, sufficient to produce 83% inhibition of lung ACE, produced plasma extravasation, their combination produced plasma extravasation, suggesting that their combination increased bradykinin (and substance P) levels sufficient to cause extravasation. It is also possible that omapatrilat-induced inhibition of aminopeptidase P, NEP2, and ECE-1 (Table 1) contributed to increased bradykinin (and substance P) levels and the plasma extravasation observed in rats, and angioedema in patients administered this therapy.
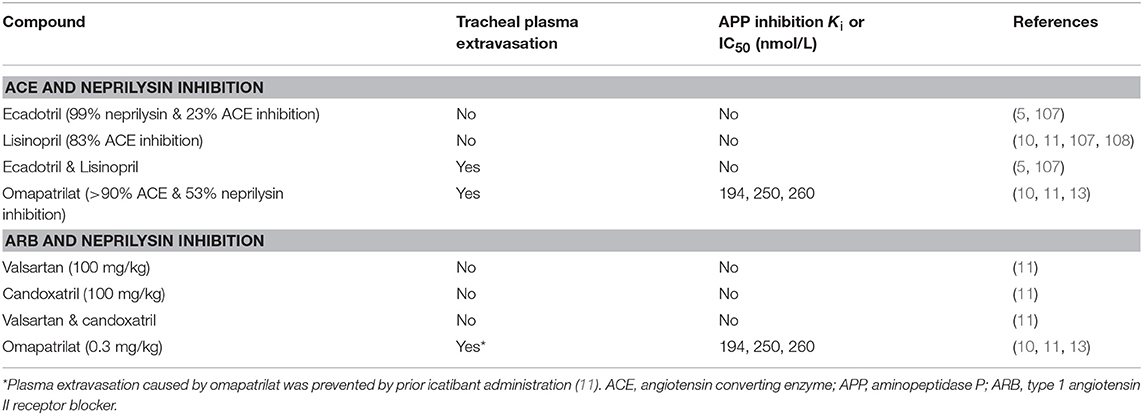
Table 3. Effects of combined renin angiotensin system and neprilysin inhibition on tracheal plasma extravasation in the rat.
LCZ696 and Bradykinin
There is currently no information on the effects of LCZ696, sacubitril or LBQ657 on bradykinin levels. However, several lines of evidence indicate a role for bradykinin in the therapeutic benefits of LCZ696 therapy, and also the angioedema associated with this therapy. Whereas ARBs produce angioedema with an incidence approximately half that of ACE inhibitor therapy in patients without heart failure (109, 110), LCZ696 produces angioedema with an incidence at least equal to that of ACE inhibitor therapy (111). In the Prospective comparison of Angiotensin Receptor-neprilysin inhibitor with Angiotensin converting enzyme inhibitor to Determine Impact on Global Mortality and morbidity in Heart Failure (PARADIGM-HF) study of patients with HFrEF, angioedema was confirmed in 0.45% of patients receiving LCZ696 therapy and 0.24% of patients receiving enalapril therapy (111), a numerical difference that was not statistically significant (P = 0.13). However, the protocol of the PARADIGM–HF study might have resulted in a lower incidence of angioedema in the trial population than might occur in patients naive to LCZ696 therapy. The exclusion criteria for the PARADIGM-HF study included a history of angioedema during treatment with an ACE inhibitor or ARB, and 78 and 22% of participants, respectively, were previously treated with an ACE inhibitor or ARB. Additionally, the study involved a run-in period before randomization during which participants received at least 2 weeks of enalapril therapy, followed by 4–6 weeks of LCZ696 therapy.
ARBs Increase Bradykinin Levels
Losartan increases bradykinin levels approximately 2-fold in arterial blood of patients with hypertension (50), similar to the increase seen with ACE inhibition (112, 113). Eprosartan produced a similar increase in bradykinin levels in the same patients, although the increase did not achieve statistical significance (50). By contrast, neither losartan nor valsartan increased bradykinin levels in rats (114, 115). There are conflicting data on the role of bradykinin in mediating the effects of ARBs. Both animal and human studies implicate kinin peptides and/or the B2 receptor in the actions of ARBs, possibly mediated by AT2 receptor stimulation by the increased angiotensin II levels that accompany ARB therapy (116–124). However, in contrast to the attenuation of the hypotensive effects of ACE inhibition by concomitant icatibant administration (100 μg/kg/h iv for 1 h) in sodium-deplete normotensive and hypertensive subjects (125), and at a higher dose (10 mg infused iv over 15 min) in sodium replete normotensive subjects (126), a lower dose of icatibant (18 μg/kg/h iv for 6 h) did not attenuate the hypotensive effects of either acute or chronic administration of valsartan in sodium-deplete normotensive and hypertensive subjects (127).
LBQ657 Inhibits not Only Neprilysin but Also ACE, NEP2, and ECE-2
In contrast to the plasma transudation seen with combined neprilysin and ACE inhibition in the rat tracheal plasma transudation model (Table 3), no transudation occurred when candoxatril was combined with valsartan (11), suggesting that combined neprilysin inhibitor and ARB therapy may cause less increase in bradykinin levels than combined neprilysin and ACE inhibition. However, LBQ657 may inhibit enzymes other than neprilysin that degrade bradykinin (Table 1). Ksander et al. reported that 10 μmol/L LBQ657 produced <50% inhibition of ACE (14). Moreover, based on information provided by Novartis Europharm Ltd, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency reports that LBQ657 inhibits not only ACE but also NEP2 and ECE-2 (15). It is notable that peak LBQ657 concentrations approximated 37 μmol/L in healthy subjects following 400 mg/day LCZ696, and trough concentrations of LBQ657 (24 h post 400 mg LCZ696) were 4.8 μmol/L. The trough LBQ657 concentration (4.8 μmol/L) is ~2,000 times the Ki of 2.3 nmol/L for neprilysin inhibition by LBQ657 (16), and the peak LBQ657 concentration is correspondingly higher. Thus, recommended doses of LCZ696 (400 mg/day) may produce LBQ657 concentrations sufficient to inhibit ACE and contribute to increased bradykinin levels, given that, as discussed earlier, as little as 1% inhibition of pulmonary inactivation of bradykinin can double bradykinin levels (Figure 3). Furthermore, NEP2 has a much lower Km for bradykinin than NEP (Table 2) and NEP2 inhibition by LBQ657 may also increase bradykinin levels. LBQ657-mediated inhibition of ECE-2 is unlikely to contribute to increased bradykinin levels because ECE-2 is relatively inactive at physiological pH (7, 34).
LCZ696 therapy may therefore potentiate bradykinin-mediated actions by several mechanisms (Figure 6). These include the increase in bradykinin levels with ARB therapy (50), the increase in bradykinin levels consequent to LBQ657-mediated inhibition of neprilysin and possibly ACE and NEP2, and cross-talk between the neprilysin-LBQ657 complex and the bradykinin receptor. Bradykinin-mediated actions will likely contribute to not only the renal and cardioprotective effects but also the angioedema associated with LCZ696 therapy. Given that heart failure is associated with suppression of the kallikrein kinin system (48, 128), and resistance to kinin-mediated cutaneous transudation (129), there is concern that LCZ696 therapy for conditions such as hypertension may be associated with a higher angioedema incidence than observed in patients with HFrEF.
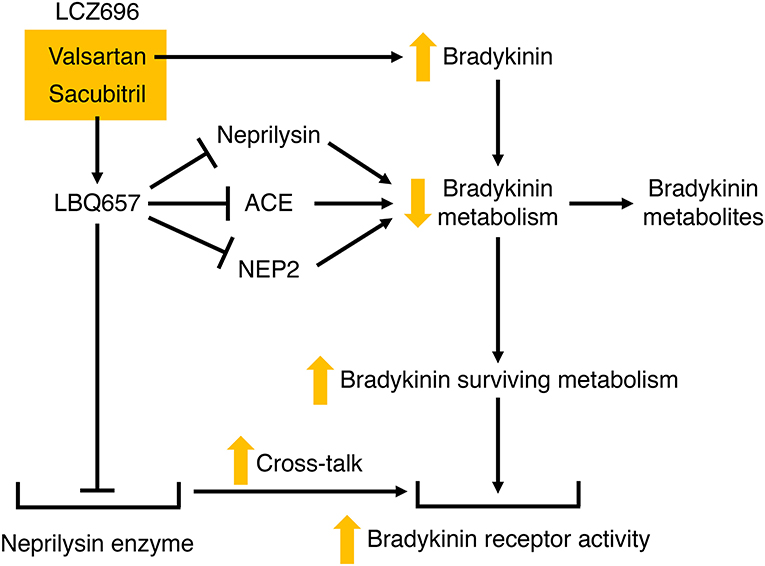
Figure 6. Potential mechanisms by which LCZ696 may potentiate bradykinin receptor-mediated actions. Valsartan may increase bradykinin levels, and LBQ657 may also increase bradykinin levels by inhibiting bradykinin degradation by neprilysin, and possibly angiotensin converting enzyme (ACE) and neprilysin homolog membrane metalloendopeptidase-like 1 (NEP2). In addition, LBQ657 may potentiate bradykinin receptor-mediated actions by cross-talk between the LBQ657-inhibitor complex and the bradykinin receptor.
Summary
Tissue levels of bradykinin are higher than circulating levels and the contribution of neprilysin to bradykinin degradation is specific to the tissue and the tissue compartment. Bradykinin is a likely contributor to the therapeutic benefits of neprilysin inhibitor therapy, particularly the renal and cardioprotective effects. However, bradykinin is also an important contributor to angioedema that may result from peptidase inhibitor therapy, including neprilysin inhibitor therapy, particularly when neprilysin inhibition is combined with ACE inhibitor therapy. LBQ657 inhibits not only neprilysin but also ACE, NEP2, and ECE-2. Although angioedema incidence was acceptable, and similar for LCZ696 and enalapril therapy in HFrEF patients, it remains to be seen whether LCZ696 therapy for other conditions such as hypertension is also accompanied by an acceptable incidence of angioedema.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was funded in part by the National Health and Medical Research Council of Australia (GNT0395508, GNT156723, GNT960229), and the National Heart Foundation of Australia (CR 02M 0829). St Vincent's Institute of Medical Research is supported in part by the Victorian Government's Operational Infrastructure Support Program.
Conflict of Interest Statement
The author declares received research funding from Servier Laboratories, Bayer Pharmaceutical Company, Solvay Pharmaceutical Company and Novartis Institutes for Biomedical Sciences, Inc.
References
1. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation (2018) 137:e67–492. doi: 10.1161/CIR.0000000000000558
2. Voisin S, Rognan D, Gros C, Ouimet T. A three-dimensional model of the neprilysin 2 active site based on the X-ray structure of neprilysin. Identification of residues involved in substrate hydrolysis and inhibitor binding of neprilysin 2. J Biol Chem. (2004) 279:46172–81. doi: 10.1074/jbc.M407333200
3. Rose C, Voisin S, Gros C, Schwartz JC, Ouimet T. Cell-specific activity of neprilysin 2 isoforms and enzymic specificity compared with neprilysin. Biochem J. (2002) 363:697–705. doi: 10.1042/bj3630697
4. Hoang MV, Turner AJ. Novel activity of endothelin-converting enzyme: hydrolysis of bradykinin. Biochem J. (1997) 327:23–6.
5. Simmons WH, Orawski AT. Membrane-bound aminopeptidase P from bovine lung. Its purification, properties, and degradation of bradykinin. J Biol Chem. (1992) 267:4897–903.
6. Ouimet T, Orng SV, Poras H, Gagnidze K, Devi LA, Fournie-Zaluski MC, et al. Identification of an endothelin-converting enzyme-2-specific fluorigenic substrate and development of an in vitro and ex vivo enzymatic assay. J Biol Chem. (2010) 285:34390–400. doi: 10.1074/jbc.M110.120576
7. Emoto N, Yanagisawa M. Endothelin-converting enzyme-2 is a membrane-bound, phosphoramidon-sensitive metalloprotease with acidic pH optimum. J Biol Chem. (1995) 270:15262–8.
8. Fahnoe DC, Knapp J, Johnson GD, Ahn K. Inhibitor potencies and substrate preference for endothelin-converting enzyme-1 are dramatically affected by pH. J Cardiovasc Pharmacol. (2000) 36:S22–5. doi: 10.1097/00005344-200036051-00009
9. Ahn K, Herman SB, Fahnoe DC. Soluble human endothelin-converting enzyme-1: expression, purification, and demonstration of pronounced pH sensitivity. Arch Biochem Biophys. (1998) 359:258–68.
10. Fryer RM, Segreti J, Banfor PN, Widomski DL, Backes BJ, Lin CW, et al. Effect of bradykinin metabolism inhibitors on evoked hypotension in rats: rank efficacy of enzymes associated with bradykinin-mediated angioedema. Br J Pharmacol. (2008) 153:947–55. doi: 10.1038/sj.bjp.0707641
11. Hegde LG, Yu C, Renner T, Thibodeaux H, Armstrong SR, Park T, et al. Concomitant angiotensin AT1 receptor antagonism and neprilysin inhibition produces omapatrilat-like antihypertensive effects without promoting tracheal plasma extravasation in the rat. J Cardiovasc Pharmacol. (2011) 57:495–504. doi: 10.1097/FJC.0b013e318210fc7e
12. Robl JA, Sun CQ, Stevenson J, Ryono DE, Simpkins LM, Cimarusti MP, et al. Dual metalloprotease inhibitors: Mercaptoacetyl-based fused heterocyclic dipeptide mimetics as inhibitors of angiotensin-converting enzyme and neutral endopeptidase. J Med Chem. (1997) 40:1570–7.
13. Sulpizio AC, Pullen MA, Edwards RM, Louttit JB, West R, Brooks DP. Mechanism of vasopeptidase inhibitor-induced plasma extravasation: comparison of omapatrilat and the novel neutral endopeptidase 24.11/angiotensin-converting enzyme inhibitor GW796406. J Pharmacol Exp Ther. (2005) 315:1306–13. doi: 10.1124/jpet.105.084749
14. Ksander GM, Ghai RD, DeJesus R, Diefenbacher CG, Yuan A, Berry C, et al. Dicarboxylic acid dipeptide neutral endopeptidase inhibitors. J Med Chem. (1995) 38:1689–700. doi: 10.1021/jm00010a014
15. Committee for Medicinal Products for Human Use (CHMP). Assessment Report: Entresto. (2015) Available online at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004062/human_med_001929.jsp&mid=WC0b01ac058001d124
16. Brown PC. Center for Drug Evaluation and Research. Application Number 207620Orig1s000. Pharmacology Review (2015). Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/207620Orig1s000TOC.cfm
17. Campbell DJ. Long-term neprilysin inhibition - implications for ARNIs. Nat Rev Cardiol. (2017) 14:171–86. doi: 10.1038/nrcardio.2016.200
18. Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A. Plasma bradykinin in angio-oedema. Lancet (1998) 351:1693–7. doi: 10.1016/S0140-6736(97)09137-X
19. Bas M, Greve J, Stelter K, Havel M, Strassen U, Rotter N, et al. A randomized trial of icatibant in ACE-inhibitor-induced angioedema. N Engl J Med. (2015) 372:418–25. doi: 10.1056/NEJMoa1312524
20. Banerji A, Busse P, Shennak M, Lumry W, Davis-Lorton M, Wedner HJ, et al. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N Engl J Med. (2017) 376:717–28. doi: 10.1056/NEJMoa1605767
21. Schmaier AH. Plasma Prekallikrein: Its Role in Hereditary Angioedema and Health and Disease. Front Med. (2018) 5:3. doi: 10.3389/fmed.2018.00003
22. Whyteside AR, Turner AJ. Human neprilysin-2 (NEP2) and NEP display distinct subcellular localisations and substrate preferences. FEBS Lett. (2008) 582:2382–6. doi: 10.1016/j.febslet.2008.05.046
23. Turner AJ, Brown CD, Carson JA, Barnes K. The neprilysin family in health and disease. Adv Exp Med Biol. (2000) 477:229–40. doi: 10.1007/0-306-46826-3_25
24. Carpentier M, Guillemette C, Bailey JL, Boileau G, Jeannotte L, DesGroseillers L, et al. Reduced fertility in male mice deficient in the zinc metallopeptidase NL1. Mol Cell Biol. (2004) 24:4428–37. doi: 10.1128/MCB.24.10.4428-4437
25. Jaspard E, Wei L, Alhenc-Gelas F. Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J Biol Chem. (1993) 268:9496–503.
26. Gafford JT, Skidgel RA, Erdos EG, Hersh LB. Human kidney “enkephalinase”, a neutral metalloendopeptidase that cleaves active peptides. Biochemistry (1983) 22:3265–71.
27. Matsas R, Kenny AJ, Turner AJ. The metabolism of neuropeptides. The hydrolysis of peptides, including enkephalins, tachykinins and their analogues, by endopeptidase-24.11. Biochem J. (1984) 223:433–40. doi: 10.1042/bj2230433
28. Orawski AT, Simmons WH. Purification and properties of membrane-bound aminopeptidase P from rat lung. Biochemistry (1995) 34:11227–36. doi: 10.1021/bi00035a032
29. Harbeck HT, Mentlein R. Aminopeptidase P from rat brain. Purification and action on bioactive peptides. Eur J Biochem. (1991) 198:451–8.
30. Skidgel RA, Johnson AR, Erdos EG. Hydrolysis of opioid hexapeptides by carboxypeptidase N. Presence of carboxypeptidase in cell membranes. Biochem Pharmacol. (1984) 33:3471–8.
31. Skidgel RA, Davis RM, Tan F. Human carboxypeptidase M. Purification and characterization of a membrane-bound carboxypeptidase that cleaves peptide hormones. J Biol Chem. (1989) 264:2236–41.
32. Lew RA, Hey NJ, Tetaz TJ, Glucksman MJ, Roberts JL, Smith AI. Substrate specificity differences between recombinant rat testes endopeptidase EC 3.4.24.15 and the native brain enzyme. Biochem Biophys Res Commun. (1995) 209:788–95.
33. Johnson GD, Stevenson T, Ahn K. Hydrolysis of peptide hormones by endothelin-converting enzyme-1. A comparison with neprilysin. J Biol Chem. (1999) 274:4053–8.
34. Mzhavia N, Pan H, Che FY, Fricker LD, Devi LA. Characterization of endothelin-converting enzyme-2. Implication for a role in the nonclassical processing of regulatory peptides. J Biol Chem. (2003) 278:14704–11. doi: 10.1074/jbc.M211242200
35. Kuoppala A, Lindstedt KA, Saarinen J, Kovanen PT, Kokkonen JO. Inactivation of bradykinin by angiotensin-converting enzyme and by carboxypeptidase N in human plasma. Am J Physiol Heart Circ Physiol. (2000) 278:H1069–74. doi: 10.1152/ajpheart.2000.278.4.H1069
36. Rosenbaum C, Cardozo C, Lesser M. Degradation of lysylbradykinin by endopeptidase 24.11 and endopeptidase 24.15. Peptides (1995) 16:523–5.
37. Orlowski M, Wilk E, Pearce S, Wilk S. Purification and properties of a prolyl endopeptidase from rabbit brain. J Neurochem. (1979) 33:461–9.
38. Wilk S, Orlowski M. Degradation of bradykinin by isolated neutral endopeptidases of brain and pituitary. Biochem Biophys Res Commun. (1979) 90:1–6.
39. Roques BP, Noble F, Daugé V, Fournié-Zaluski M-C, Beaumont A. Neutral endopeptidase 24.11: Structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev. (1993) 45:87–146.
40. Shipp MA, Vijayaraghavan J, Schmidt EV, Masteller EL, D'Adamio L, Hersh LB, et al. Common acute lymphoblastic leukemia antigen (CALLA) is active neutral endopeptidase 24.11 (“enkephalinase”): direct evidence by cDNA transfection analysis. Proc Natl Acad Sci USA. (1989) 86:297–301. doi: 10.1073/pnas.86.1.297
41. Tran-Paterson R, Boileau G, Giguere V, Letarte M. Comparative levels of CALLA/neutral endopeptidase on normal granulocytes, leukemic cells, and transfected COS-1 cells. Blood (1990) 76:775–82.
42. Kenny AJ. Endopeptidase-24.11: putative substrates and possible roles. Biochem Soc Trans. (1993) 21:663–8. doi: 10.1042/bst0210663
43. Shipp MA, Tarr GE, Chen C-Y, Switzer SN, Hersh LB, Stein H, et al. CD10/neutral endopeptidase 24.11 hydrolyzes bombesin-like peptides and regulates the growth of small cell carcinomas of the lung. Proc Natl Acad Sci USA. (1991) 88:10662–6. doi: 10.1073/pnas.88.23.10662
44. Campbell DJ. Towards understanding the kallikrein-kinin system: insights from measurement of kinin peptides. Braz J Med Biol Res. (2000) 33:665–77. doi: 10.1590/S0100-879X2000000600008
45. Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. (1992) 44:1–80.
46. Regoli D, Rhaleb N-E, Drapeau G, Dion S, Tousignant C, D'Orléans-Juste P, et al. Basic pharmacology of kinins: pharmacologic receptors and other mechanisms. Adv Exp Med Biol. (1989) 247A:399–407. doi: 10.1007/978-1-4615-9543-4_61
47. Campbell DJ, Kladis A, Duncan A-M. Bradykinin peptides in kidney, blood, and other tissues of the rat. Hypertension (1993) 21:155–65. doi: 10.1161/01.HYP.21.2.155
48. Duncan A-M, Kladis A, Jennings GL, Dart AM, Esler M, Campbell DJ. Kinins in humans. Am J Physiol Regul Integr Comp Physiol. (2000) 278:R897–904. doi: 10.1152/ajpregu.2000.278.4.R897
49. Campbell DJ, Duncan A-M, Kladis A. Angiotensin converting enzyme inhibition modifies angiotensin, but not kinin peptide levels in human atrial tissue. Hypertension (1999) 34:171–5. doi: 10.1161/01.HYP.34.2.171
50. Campbell DJ, Krum H, Esler MD. Losartan increases bradykinin levels in hypertensive humans. Circulation (2005) 111:315–20. doi: 10.1161/01.CIR.0000153269.07762.3B
51. Ryan JW, Berryer P, Chung AYK, Sheffy DH. Characterization of rat pulmonary vascular aminopeptidase P in vivo: role in the inactivation of bradykinin. J Pharmacol Exp Ther. (1994) 269:941–7.
52. Lu B, Figini M, Emanueli C, Geppetti P, Grady EF, Gerard NP, et al. The control of microvascular permeability and blood pressure by neutral endopeptidase. Nature Med (1997) 3:904–7.
53. Geppetti P. Sensory neuropeptide release by bradykinin: mechanisms and pathophysiological implications. Regul Pept. (1993) 47:1–23. doi: 10.1016/0167-0115(93)90268-D
54. Skidgel RA, Engelbrecht S, Johnson AR, Erdos EG. Hydrolysis of substance P and neurotensin by converting enzyme and neutral endopeptidase. Peptides (1984) 5:769–76. doi: 10.1016/0196-9781(84)90020-2
55. Fischer HS, Zernig G, Hauser KF, Gerard C, Hersh LB, Saria A. Neutral endopeptidase knockout induces hyperalgesia in a model of visceral pain, an effect related to bradykinin and nitric oxide. J Mol Neurosci. (2002) 18:129–34. doi: 10.1385/JMN:18:1-2:129
56. Kramer HH, He L, Lu B, Birklein F, Sommer C. Increased pain and neurogenic inflammation in mice deficient of neutral endopeptidase. Neurobiol Dis. (2009) 35:177–83. doi: 10.1016/j.nbd.2008.11.002
57. Lu B, Gerard NP, Kolakowski LF Jr, Bozza M, Zurakowski D, Finco O, et al. Neutral endopeptidase modulation of septic shock. J Exp Med. (1995) 181:2271–5. doi: 10.1084/jem.181.6.2271
58. Grasemann H, Lu B, Jiao A, Boudreau J, Gerard NP, De Sanctis GT. Targeted deletion of the neutral endopeptidase gene alters ventilatory responses to acute hypoxia in mice. J Appl Physiol. (1999) 87:1266–71. doi: 10.1152/jappl.1999.87.4.1266
59. Walther T, Albrecht D, Becker M, Schubert M, Kouznetsova E, Wiesner B, et al. Improved learning and memory in aged mice deficient in amyloid ß-degrading neutral endopeptidase. PLoS ONE (2009) 4:e4590. doi: 10.1371/journal.pone.0004590
60. Debiec H, Nauta J, Coulet F, van der Burg M, Guigonis V, Schurmans T, et al. Role of truncating mutations in MME gene in fetomaternal alloimmunisation and antenatal glomerulopathies. Lancet (2004) 364:1252–9. doi: 10.1016/S0140-6736(04)17142-0
61. Higuchi Y, Hashiguchi A, Yuan J, Yoshimura A, Mitsui J, Ishiura H, et al. Mutations in MME cause an autosomal-recessive Charcot-Marie-Tooth disease type 2. Ann Neurol. (2016) 79:659–72. doi: 10.1002/ana.24612
62. Auer-Grumbach M, Toegel S, Schabhuttl M, Weinmann D, Chiari C, Bennett DL, et al. Rare variants in MME, encoding metalloprotease neprilysin, are linked to late-onset autosomal-dominant axonal polyneuropathies. Am J Hum Genet. (2016) 99:607–23. doi: 10.1016/j.ajhg.2016.07.008
63. Depondt C, Donatello S, Rai M, Wang FC, Manto M, Simonis N, et al. MME mutation in dominant spinocerebellar ataxia with neuropathy (SCA43). Neurol Genet. (2016) 2:e94. doi: 10.1212/NXG.0000000000000094
64. Pare G, Kubo M, Byrd JB, McCarty CA, Woodard-Grice A, Teo KK, et al. Genetic variants associated with angiotensin-converting enzyme inhibitor-associated angioedema. Pharmacogenet Genomics (2013) 23:470–8. doi: 10.1097/FPC.0b013e328363c137
65. Campbell DJ, Anastasopoulos F, Duncan AM, James GM, Kladis A, Briscoe TA. Effects of neutral endopeptidase inhibition and combined angiotensin converting enzyme and neutral endopeptidase inhibition on angiotensin and bradykinin peptides in rats. J Pharmacol Exp Ther. (1998) 287:567–77.
66. Boomsma F, De Bruyn JHB, Derkx FHM, Schalekamp MADH. Opposite effects of captopril on angiotensin I-converting enzyme ‘activity’ and ‘concentration’; relation between enzyme inhibition and long-term blood pressure response. Clin Sci (1981) 60:491–8. doi: 10.1042/cs0600491
67. Helin K, Tikkanen I, Hohenthal U, Fyhrquist F. Inhibition of either angiotensin-converting enzyme or neutral endopeptidase induces both enzymes. Eur J Pharmacol. (1994) 264:135–41. doi: 10.1016/0014-2999(94)00450-1
68. Pham I, Gonzalez W, El Amrani A-IK, Fournié-Zaluski M-C, Philippe M, Laboulandine I, et al. Effects of converting enzyme inhibitor and neutral endopeptidase inhibitor on blood pressure and renal function in experimental hypertension. J Pharmacol Exp Ther. (1993) 265:1339–47.
69. Murphey LJ, Hachey DL, Oates JA, Morrow JD, Brown NJ. Metabolism of bradykinin in vivo in humans: identification of BK1-5 as a stable plasma peptide metabolite. J Pharmacol Exp Ther. (2000) 294:263–9.
70. Shima C, Majima M, Katori M. A stable metabolite, Arg-Pro-Pro-Gly-Phe, of bradykinin in the degradation pathway in human plasma. Jpn J Pharmacol. (1992) 60:111–9. doi: 10.1254/jjp.60.111
71. Marshall P, Heudi O, McKeown S, Amour A, Abou-Shakra F. Study of bradykinin metabolism in human and rat plasma by liquid chromatography with inductively coupled plasma mass spectrometry and orthogonal acceleration time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. (2002) 16:220–8. doi: 10.1002/rcm.565
72. Décarie A, Raymond P, Gervais N, Couture R, Adam A. Serum interspecies differences in metabolic pathways of bradykinin and [des-Arg9]BK: Influence of enalaprilat. Am J Physiol Heart Circ Physiol. (1996) 271:H1340–7. doi: 10.1152/ajpheart.1996.271.4.H1340
73. Blais C Jr, Marc-Aurele J, Simmons WH, Loute G, Thibault P, Skidgel RA, et al. Des-Arg9-bradykinin metabolism in patients who presented hypersensitivity reactions during hemodialysis: role of serum ACE and aminopeptidase P. Peptides (1999) 20:421–30. doi: 10.1016/S0196-9781(99)00020-0
74. Cyr M, Lepage Y, Blais C Jr, Gervais N, Cugno M, Rouleau JL, et al. Bradykinin and des-Arg(9)-bradykinin metabolic pathways and kinetics of activation of human plasma. Am J Physiol Heart Circ Physiol. (2001) 281:H275–83. doi: 10.1152/ajpheart.2001.281.1.H275
75. Dendorfer A, Wolfrum S, Wagemann M, Qadri F, Dominiak P. Pathways of bradykinin degradation in blood and plasma of normotensive and hypertensive rats. Am J Physiol Heart Circ Physiol. (2001) 280:H2182–8. doi: 10.1152/ajpheart.2001.280.5.H2182
76. Sheikh IA, Kaplan AP. Mechanism of digestion of bradykinin and lysylbradykinin (kallidin) in human serum. Role of carboxypeptidase, angiotensin-converting enzyme and determination of final degradation products. Biochem Pharmacol. (1989) 38:993–1000. doi: 10.1016/0006-2952(89)90290-6
77. Pesquero JB, Jubilut GN, Lindsey CJ, Paiva ACM. Bradykinin metabolism pathway in the rat pulmonary circulation. J Hypertens. (1992) 10:1471–8.
78. Sivieri DO, Jr, Bispo-da-Silva LB, Oliveira EB, Resende AC, Salgado MC. Potentiation of bradykinin effect by angiotensin-converting enzyme inhibition does not correlate with angiotensin-converting enzyme activity in the rat mesenteric arteries. Hypertension (2007) 50:110–5. doi: 10.1161/HYPERTENSIONAHA.106.085761
79. Oliveira EB, Souza LL, Sivieri DO Jr, Bispo-da-Silva LB, Pereira HJ, Costa-Neto CM, et al. Carboxypeptidase B and other kininases of the rat coronary and mesenteric arterial bed perfusates. Am J Physiol Heart Circ Physiol. (2007) 293:H3550–7. doi: 10.1152/ajpheart.00784.2007
80. Dalzell JR, Seed A, Berry C, Whelan CJ, Petrie MC, Padmanabhan N, et al. Effects of neutral endopeptidase (neprilysin) inhibition on the response to other vasoactive peptides in small human resistance arteries: studies with thiorphan and omapatrilat. Cardiovasc Ther. (2014) 32:13–8. doi: 10.1111/1755-5922.12053
81. Kitamura S, Carbini LA, Simmons WH, Scicli AG. Effects of aminopeptidase P inhibition on kinin-mediated vasodepressor responses. Am J Physiol Heart Circ Physiol. (1999) 276:H1664–71. doi: 10.1152/ajpheart.1999.276.5.H1664
82. Ersahin Ç, Simmons WH. Inhibition of both aminopeptidase P and angiotensin-converting enzyme prevents bradykinin degradation in the rat coronary circulation. J Cardiovasc Pharmacol. (1997) 30:96–101. doi: 10.1097/00005344-199707000-00014
83. Dumoulin MJ, Adam A, Rouleau JL, Lamontagne D. Comparison of a vasopeptidase inhibitor with neutral endopeptidase and angiotensin-converting enzyme inhibitors on bradykinin metabolism in the rat coronary bed. J Cardiovasc Pharmacol. (2001) 37:359–66. doi: 10.1097/00005344-200104000-00002
84. Dendorfer A, Wolfrum S, Wellhöner P, Korsman K, Dominiak P. Intravascular and interstitial degradation of bradykinin in isolated perfused rat heart. Br J Pharmacol. (1997) 122:1179–87. doi: 10.1038/sj.bjp.0701501
85. Ahmad M, Zeitlin IJ, Parratt JR, Pitt AR. Degradation of bradykinin, a cardioprotective substance, during a single passage through isolated rat-heart. Arch Pharm Res. (2006) 29:241–8. doi: 10.1007/BF02969400
86. Koch M, Wendorf M, Dendorfer A, Wolfrum S, Schulze K, Spillmann F, et al. Cardiac kinin level in experimental diabetes mellitus: role of kininases. Am J Physiol Heart Circ Physiol. (2003) 285:H418–23. doi: 10.1152/ajpheart.00677.2002
87. Tom B, Dendorfer A, de Vries R, Saxena PR, Jan Danser AH. Bradykinin potentiation by ACE inhibitors: a matter of metabolism. Br J Pharmacol. (2002) 137:276–84. doi: 10.1038/sj.bjp.0704862
88. Bagate K, Develioglu L, Grima M, De Jong W, Simmons WH, Imbs JL, et al. Vascular catabolism of bradykinin in the isolated perfused rat kidney. Eur J Pharmacol. (2000) 407:317–25. doi: 10.1016/S0014-2999(00)00744-5
89. Ramirez-Molina C, Heudi O, Pullen M, Marshall PS. Study of bradykinin metabolism by rat lung tissue membranes and rat kidney brush border membranes by HPLC with inductively coupled plasma-mass spectrometry and orthogonal acceleration time-of-flight mass spectrometry. J Pept Sci. (2006) 12:220–6. doi: 10.1002/psc.712
90. Kokkonen JO, Kuoppala A, Saarinen J, Lindstedt KA, Kovanen PT. Kallidin- and bradykinin-degrading pathways in human heart: degradation of kallidin by aminopeptidase M-like activity and bradykinin by neutral endopeptidase. Circulation (1999) 99:1984–90. doi: 10.1161/01.CIR.99.15.1984
91. Ura N, Carretero OA, Erdos EG. Role of renal endopeptidase 24.11 in kinin metabolism in vitro and in vivo. Kidney Int. (1987) 32:507–13. doi: 10.1038/ki.1987.239
92. Deddish PA, Marcic BM, Tan F, Jackman HL, Chen Z, Erdos EG. Neprilysin inhibitors potentiate effects of bradykinin on B2 receptor. Hypertension (2002) 39:619–23. doi: 10.1161/hy0202.103298
93. Erdos EG. Kinins, the long march–a personal view. Cardiovasc Res. (2002) 54:485–91. doi: 10.1016/S0008-6363(02)00284-5
94. Ura N, Shimamoto K, Kuroda S, Nomura N, Iwata M, Aoyama T, et al. The role of kinins and atrial natriuretic peptide on the renal effects of neutral endopeptidase inhibitor in rats. Clin Exp Hypertens. (1994) 16:799–808. doi: 10.3109/10641969409078026
95. Ura N, Shimamoto K, Nomura N, Aoyama T, Iwata M, Takagawa Y, et al. The mechanisms of the renal effects of neutral endopeptidase inhibitor in rats. Clin Exp Hypertens. (1995) 17:1183–96. doi: 10.3109/10641969509037403
96. Nomura N, Shimamoto K, Ura N, Iwata M, Aoyama T, Takagawa Y, et al. The role of kinins and atrial natriuretic peptide on the renal effects of neutral endopeptidase inhibitor in normotensive and hypertensive rats. Clin Exp Hypertens. (1995) 17:1219–31. doi: 10.3109/10641969509037405
97. Smits GJ, McGraw DE, Trapani AJ. Interaction of ANP and bradykinin during endopeptidase 24.11 inhibition: renal effects. Am J Physiol Renal Physiol. (1990) 258:F1417–24. doi: 10.1152/ajprenal.1990.258.5.F1417
98. Legault L, Cernacek P, Levy M, Maher E, Farber D. Renal tubular responsiveness to atrial natriuretic peptide in sodium-retaining chronic caval dogs. A possible role for kinins and luminal actions of the peptide. J Clin Invest. (1992) 90:1425–35. doi: 10.1172/JCI116009
99. Bralet J, Mossiat C, Gros C, Schwartz JC. Thiorphan-induced natriuresis in volume-expanded rats: roles of endogenous atrial natriuretic factor and kinins. J Pharmacol Exp Ther. (1991) 258:807–11.
100. Pham I, Gonzalez W, Doucet J, Fournié-Zaluski MC, Roques BP, Michel JB. Effects of angiotensin-converting enzyme and neutral endopeptidase inhibitors: influence of bradykinin. Eur J Pharmacol. (1996) 296:267–76. doi: 10.1016/0014-2999(95)00706-7
101. Yang XP, Liu YH, Peterson E, Carretero OA. Effect of neutral endopeptidase 24.11 inhibition on myocardial ischemia/reperfusion injury: the role of kinins. J Cardiovasc Pharmacol. (1997) 29:250–6.
102. Nakano A, Miura T, Miki T, Nozawa Y, Ichikawa Y, Ura N, et al. Effects of neutral endopeptidase 24.11 inhibition on myocardial infarct size and ischemic preconditioning in rabbits. Naunyn Schmiedebergs Arch Pharmacol. (2002) 366:335–42. doi: 10.1007/s00210-002-0600-8
103. Piedimonte G, Nadel JA, Long CS, Hoffman JIE. Neutral endopeptidase in the heart: neutral endopeptidase inhibition prevents isoproterenol-induced myocardial hypoperfusion in rats by reducing bradykinin degradation. Circ Res. (1994) 75:770–9. doi: 10.1161/01.RES.75.4.770
104. Zhang XP, Nasjletti A, Xu XB, Hintze TH. Neutral endopeptidase and angiotensin-converting enzyme inhibitors increase nitric oxide production in isolated canine coronary microvessels by a kinin-dependent mechanism. J Cardiovasc Pharmacol. (1998) 31:623–9. doi: 10.1097/00005344-199804000-00023
105. Kostis JB, Packer M, Black HR, Schmieder R, Henry D, Levy E. Omapatrilat and enalapril in patients with hypertension: the Omapatrilat Cardiovascular Treatment vs. Enalapril (OCTAVE) trial. Am J Hypertens (2004) 17:103–11. doi: 10.1016/j.amjhyper.2003.09.014
106. Packer M, Califf RM, Konstam MA, Krum H, McMurray JJ, Rouleau JL, et al. Comparison of omapatrilat and enalapril in patients with chronic heart failure: the Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE). Circulation (2002) 106:920–6. doi: 10.1161/01.CIR.0000029801.86489.50
107. Sulpizio AC, Pullen MA, Edwards RM, Brooks DP. The effect of acute angiotensin-converting enzyme and neutral endopeptidase 24.11 inhibition on plasma extravasation in the rat. J Pharmacol Exp Ther. (2004) 309:1141–7. doi: 10.1124/jpet.103.064105
108. Hooper NM, Hryszko J, Oppong SY, Turner AJ. Inhibition by converting enzyme inhibitors of pig kidney aminopeptidase P. Hypertension (1992) 19:281–5.
109. Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, Faire U, et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet (2002) 359:995–1003. doi: 10.1016/S0140-6736(02)08089-3
110. Dickstein K, Kjekshus J. Effects of losartan and captopril on mortality and morbidity in high-risk patients after acute myocardial infarction: the OPTIMAAL randomised trial. optimal trial in myocardial infarction with angiotensin II antagonist losartan. Lancet (2002) 360:752–60. doi: 10.1016/S0140-6736(02)09895-1
111. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. (2014) 371:993–1004. doi: 10.1056/NEJMoa1409077
112. Campbell DJ, Dixon B, Kladis A, Kemme M, Santamaria JD. Activation of the kallikrein-kinin system by cardiopulmonary bypass in humans. Am J Physiol Regul Integr Comp Physiol. (2001) 281:R1059–70. doi: 10.1152/ajpregu.2001.281.4.R1059
113. Zeitz CJ, Campbell DJ, Horowitz JD. Myocardial uptake and biochemical and hemodynamic effects of ACE inhibitors in humans. Hypertension (2003) 41:482–7. doi: 10.1161/01.HYP.0000054976.67487.08
114. Campbell DJ, Kladis A, Valentijn AJ. Effects of losartan on angiotensin and bradykinin peptides, and angiotensin converting enzyme. J Cardiovasc Pharmacol. (1995) 26:233–40. doi: 10.1097/00005344-199508000-00009
115. Koid SS, Ziogas J, Campbell DJ. Aliskiren reduces myocardial ischemia-reperfusion injury by a bradykinin B2 receptor- and angiotensin AT2 receptor-mediated mechanism. Hypertension (2014) 63:768–73. doi: 10.1161/HYPERTENSIONAHA.113.02902
116. Hornig B, Kohler C, Schlink D, Tatge H, Drexler H. AT1-receptor antagonism improves endothelial function in coronary artery disease by a bradykinin/B2-receptor-dependent mechanism. Hypertension (2003) 41:1092–5. doi: 10.1161/01.HYP.0000064942.77814.26
117. Wiemer G, Schölkens BA, Busse R, Wagner A, Heitsch H, Linz W. The functional role of angiotensin II-subtype AT2-receptors in endothelial cells and isolated ischemic rat hearts. Pharm Pharmacol Lett. (1993) 3:24–7.
118. Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E, et al. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure - Role of kinins and angiotensin II type 2 receptors. J Clin Invest. (1997) 99:1926–35. doi: 10.1172/JCI119360
119. Jalowy A, Schulz R, Dorge H, Behrends M, Heusch G. Infarct size reduction by AT1-receptor blockade through a signal cascade of AT2-receptor activation, bradykinin and prostaglandins in pigs. J Am Coll Cardiol. (1998) 32:1787–96. doi: 10.1016/S0735-1097(98)00441-0
120. Zhu P, Zaugg CE, Hornstein PS, Allegrini PR, Buser PT. Bradykinin-dependent cardioprotective effects of losartan against ischemia and reperfusion in rat hearts. J Cardiovasc Pharmacol. (1999) 33:785–90.
121. Sato M, Engelman RM, Otani H, Maulik N, Rousou JA, Flack JE III, et al. Myocardial protection by preconditioning of heart with losartan, an angiotensin II type 1-receptor blocker: implication of bradykinin-dependent and bradykinin-independent mechanisms. Circulation (2000) 102(Suppl. 3):III346-51. doi: 10.1161/01.CIR.102.suppl_3.III-346
122. Carey RM, Wang ZQ, Siragy HM. Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension (2000) 35:155–63. doi: 10.1161/01.HYP.35.1.155
123. Abadir PM, Carey RM, Siragy HM. Angiotensin AT2 receptors directly stimulate renal nitric oxide in bradykinin B2-receptor-null mice. Hypertension (2003) 42:600–4. doi: 10.1161/01.HYP.0000090323.58122.5C
124. Messadi-Laribi E, Griol-Charhbili V, Pizard A, Vincent MP, Heudes D, Meneton P, et al. Tissue kallikrein is involved in the cardioprotective effect of AT1-receptor blockade in acute myocardial ischemia. J Pharmacol Exp Ther. (2007) 323:210–6. doi: 10.1124/jpet.107.124859
125. Gainer JV, Morrow JD, Lovelend A, King DJ, Brown NJ. Effect of bradykinin-receptor blockade on the response to angiotensin-converting-enzyme inhibitor in normotensive and hypertensive subjects. N Engl J Med. (1998) 339:1285–92.
126. Squire IB, O'Kane KP, Anderson N, Reid JL. Bradykinin B(2) receptor antagonism attenuates blood pressure response to acute angiotensin-converting enzyme inhibition in normal men. Hypertension (2000) 36:132–6. doi: 10.1161/hyp.36.1.132
127. LeFebvre J, Shintani A, Gebretsadik T, Petro JR, Murphey LJ, Brown NJ. Bradykinin B(2) receptor does not contribute to blood pressure lowering during AT(1) receptor blockade. J Pharmacol Exp Ther. (2007) 320:1261–7. doi: 10.1124/jpet.106.117259
128. Campbell DJ. The kallikrein-kinin system in humans. Clin Exp Pharmacol Physiol. (2001) 28:1060–5. doi: 10.1046/j.1440-1681.2001.03564.x
Keywords: neprilysin, bradykinin, neprilysin inhibition, angioedema, ARNI
Citation: Campbell DJ (2018) Neprilysin Inhibitors and Bradykinin. Front. Med. 5:257. doi: 10.3389/fmed.2018.00257
Received: 18 July 2018; Accepted: 29 August 2018;
Published: 19 September 2018.
Edited by:
Alvin H. Schmaier, Case Western Reserve University, United StatesReviewed by:
Francois Alhenc-Gelas, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCoen Maas, University Medical Center Utrecht, Netherlands
Nancy J. Brown, Vanderbilt University Medical Center, United States
Copyright © 2018 Campbell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Duncan J. Campbell, ZGNhbXBiZWxsQHN2aS5lZHUuYXU=