 Lisa M. Hillen1*
Lisa M. Hillen1* Joost Van den Oord2Milan S. Geybels3Jürgen C. Becker4
Joost Van den Oord2Milan S. Geybels3Jürgen C. Becker4 Axel zur Hausen1
Axel zur Hausen1 Véronique Winnepenninckx1*
Véronique Winnepenninckx1*- 1Department of Pathology, GROW School for Oncology and Developmental Biology, Maastricht University Medical Center, Maastricht, Netherlands
- 2Laboratory for Translational Cell and Tissue Research, Department of Pathology, KU Leuven, Leuven, Belgium
- 3Department of Epidemiology, GROW School for Oncology and Developmental Biology, Maastricht University, Maastricht, Netherlands
- 4Institute for Translational Skin Cancer Research, German Cancer Consortium (DKTK), Partner Site Essen, University Hospital Essen, Essen, Germany
Spitzoid neoplasms are a distinct group of melanocytic proliferations characterized by epithelioid and/ or spindle shaped melanocytes. Intermediate forms that share features of both benign Spitz nevi (SN) and Spitz melanoma, i.e., malignant Spitz tumor (MST) represent a diagnostically and clinically challenging group of melanocytic lesions. A multitude of descriptive diagnostic terms exist for these ambiguous lesions with atypical Spitz tumor (AST) or Spitz tumor of uncertain malignant potential (STUMP) just naming two of them. This diagnostic gray zone creates confusion and high insecurity in clinicians and in patients. Biological behavior and clinical course of this intermediate group still remains largely unknown, often leading to difficulties with uncertainties in clinical management and prognosis. Consequently, a better stratification of Spitzoid neoplasms in benign and malignant forms is required thereby keeping the diagnostic group of AST/STUMP as small as possible. Ancillary diagnostic techniques such as immunohistochemistry, comparative genomic hybridization, fluorescence in situ hybridization, next generation sequencing, micro RNA and mRNA analysis as well as mass spectrometry imaging offer new opportunities for the distinct diagnosis, thereby allowing the best clinical management of Spitzoid neoplasms. This review gives an overview on these additional diagnostic techniques and the recent developments in the field of molecular genetic alterations in Spitzoid neoplasms. We also discuss how the recent findings might facilitate the diagnosis and stratification of atypical Spitzoid neoplasms and how these findings will impact the diagnostic work up as well as patient management. We suggest a stepwise implementation of ancillary diagnostic techniques thereby integrating immunohistochemistry and molecular pathology findings in the diagnosis of challenging ambiguous Spitzoid neoplasms. Finally, we will give an outlook on pending future research objectives in the field of Spitzoid melanocytic lesions.
Introduction and Background
Melanocytic neoplasms comprise a heterogeneous group of tumors that are characterized by their distinct histopathological appearance, their clinical features and their genetic aberrations. Roughly, melanocytic neoplasms can be distinguished into three major groups of (i) conventional melanocytic neoplasms including benign acquired and congenital nevocellular nevi and cutaneous malignant melanoma, (ii) dendritic, i.e., blue melanocytic neoplasms and (iii) Spitzoid melanocytic neoplasms. Each group is characterized by a distinct histomorphological appearance what is also reflected by the spectrum of underlying genetic aberrations (Figure 1). In the group of conventional melanocytic neoplasms, which by far comprises the largest group, a clear-cut distinction between a benign nevocellular nevus and a malignant melanoma is usually possible on histological grounds.
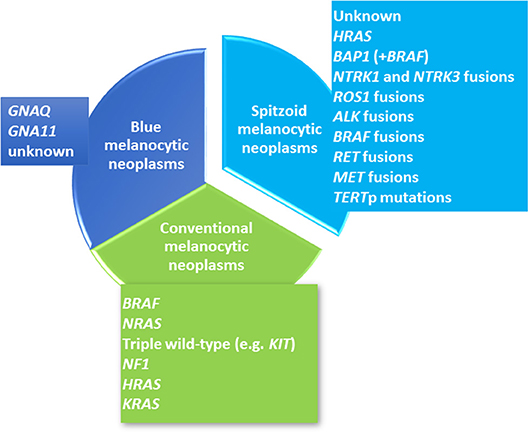
Figure 1. Overview of the Molecular Landscape of Melanocytic Neoplasms. Cutaneous melanocytic neoplasms can be clustered into three major lines including conventional melanocytic neoplasms (colored in green), Spitzoid melanocytic neoplasms (colored in turquoise) and blue melanocytic neoplasms (colored in blue). Each group shows distinct molecular genetic alterations. Conventional melanocytic neoplasms (green color) include common melanocytic nevocellular nevi, congenital nevocellular nevi and cutaneous malignant melanoma. Common nevocellular nevi harbor activating hotspot BRAF mutations at codon 600 [~80%, (1)]. The majority of congenital nevocellular nevi harbor activating NRAS hotspot mutations [~75%, (2)]. Cutaneous malignant melanoma has been clustered into four molecular subtypes (3) with the largest subgroup harboring a BRAFV600E mutation (~50%) and the second largest group with an activating NRAS mutation (~25%) as well as HRAS and KRAS mutations (~1%). The third subgroup shows NF1 inactivation (~10%). The last molecular subgroup lacks BRAF, N/H/KRAS or NF1 mutations and represents a heterogeneous so called “triple wild-type subtype”. The second major line of cutaneous melanocytic neoplasms comprises blue melanocytic neoplasms which are also called dermal dendritic melanocytic neoplasms (blue color) and have been shown to harbor activating GNAQ and GNA11 mutations. GNAQ dominates in cutaneous proliferations while GNAQ and GNA11 is found in almost the half of uveal melanomas (4). The third group refers to Spitzoid melanocytic neoplasms (turquoise color) and shows quite different molecular genetic alterations which are rarely observed in the two other groups. Receptor tyrosine kinase translocations involving ALK, ROS1, NTRK1, NTRK3, BRAF, RET, MET (5–7) as well as HRAS (5, 8, 9), BAP1 (10), and TERTp mutations (11–13) have been described in Spitzoid neoplasms.
According to the Cancer Genome Atlas (TCGA) cutaneous malignant melanoma comprises four molecular subtypes, i.e., melanomas with BRAF V600E mutation, melanomas with NRAS activating mutation, those with NF1 mutation and the “triple wild-type” lacking mutations in the afore mentioned genes [Figure 1, (3)]. The second melanocytic group comprises blue melanocytic neoplasms which are characterized by a proliferation of dendritic, spindled and/ or ovoid dermal melanocytes and therefore are also named dermal dendritic melanocytic neoplasms (14). They show mutations in GNAQ and GNA11 [Figure 1, (4)]. The third group of melanocytic neoplasms encompasses Spitzoid lesions revealing distinct histological features which set them apart from other melanocytic neoplasms. Of interest, this is also reflected in their underlying diverging molecular genetic profile [Figure 1 (15)].
Spitzoid neoplasms are composed of large epithelioid and/ or spindle-shaped melanocytes with large nuclei that contain vesicular chromatin and often prominent nucleoli and are usually arranged in a distinctive architectural configuration (Figure 2). Spitzoid neoplasms represent uncommon melanocytic lesions and account for only about 1% of resected melanocytic neoplasms (16–18). The American pathologist Sophie Spitz was the first, who described them in 1948 as “juvenile melanomas” or as “melanomas of childhood” because they often appear in children or adolescents and show lymphotropic behavior with spread to locoregional lymph nodes (19). However, these lesions can also occur later in life and therefore were renamed as Spitz nevi (SN) to indicate their benign nature (20, 21). The term Spitz melanoma or malignant Spitz tumor (MST) was reserved to Spitzoid neoplasms with marked atypia and an aggressive clinical course eventually leading to metastasis and death. In daily surgical pathology practice Spitzoid melanocytic neoplasms often lead to diagnostic difficulties because they can reveal conflicting histomorphology intermediate of SN and MST. In these ambiguous cases morphology is often incapable to predict biological potential and thereby clinical outcome [(22), see case 12]. The diagnostic difficulties might stem from the idea that the morphological criteria which are applied for conventional melanocytic neoplasms also apply for Spitzoid melanocytic neoplasms. However, it should be considered that the group of blue melanocytic neoplasms, i.e., dermal dendritic melanocytic neoplasms is well recognized to have their own morphological criteria (14). Thus, it is quite likely that Spitzoid melanocytic neoplasms also have peculiar morphological criteria with their exact definition and nomenclature remaining an ongoing matter of debate and a pending area of future research (14, 22). The apparent diagnostic dilemma leads to high insecurity in daily practice with either under-diagnosis as benign SN or over-diagnosis as MST. Consequently, the diagnostic gray zone of ambiguous Spitzoid neoplasms has led to the introduction of a multitude of descriptive diagnostic terms such as atypical Spitz tumor (AST), Spitzoid tumor of unknown malignant potential (STUMP), melanocytic lesion of uncertain malignant potential (MELTUMP), superficial atypical melanocytic proliferation of uncertain significance (SAMPUS), dysplastic Spitzoid neoplasm, or borderline Spitzoid neoplasm. The multitude of these, often only vaguely defined descriptive histological terms hampers the ease of diagnostic and clinical work up, complicates interdisciplinary communication and increases uncertainty in treating dermatologists, surgical pathologists and last but not least in patients. Of note, the descriptive terms MELTUMP and SAMPUS are also used in the context of ambiguous non-Spitzoid melanocytic neoplasms that are characterized by conflicting criteria (14). In this review the term atypical Spitz tumor (AST) will be used for lesions exhibiting a Spitzoid morphology but also harboring features associated with malignancy according to the recently launched WHO 2018 classification of skin tumors (14). In this review we follow a three-tiered classification comprising the benign SN-, the intermediate AST- and the malignant MST category (14).
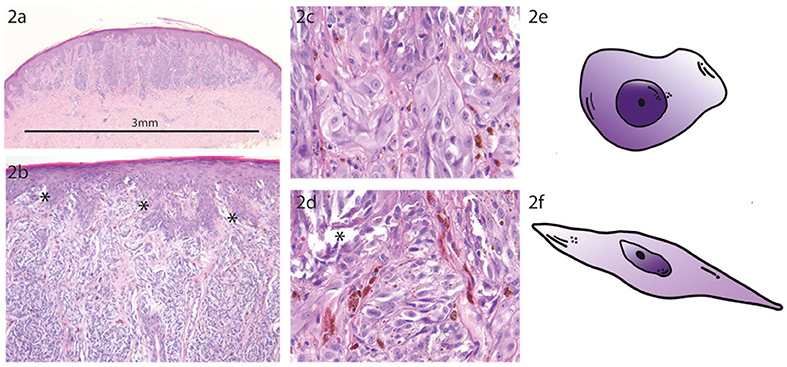
Figure 2. Prototypic Spitz Nevus. (a) The compound Spitz nevus (SN) has a small size of 3 mm with a symmetric dome shaped morphology and shows sharp lateral demarcation (H.E. staining, magnification factor 2.5x). (b) Higher magnification from A shows regular epidermal hyperplasia with vertically orientated fascicles of spindled and epithelioid melanocytes. Note the numerous epidermal junctional clefts (*) which are formed due to discohesion of the melanocytes (H.E. staining, magnification factor 10x). (c) Higher magnification from B with uniform characteristic epithelioid melanocytes at the epidermal junction (H.E. staining, magnification factor 40x). (d) Higher magnification from B showing regular spindle shaped melanocytes at the epidermal junction with formation of junctional clefts (*) due to discohesion of the Spitzoid melanocytes (H.E. staining, magnification factor 40x). (e) Schematic sketch of a prototypic epithelioid shaped melanocyte which is characteristic for a SN. (f) Schematic sketch of a prototypic spindle shaped melanocyte which is characteristic for a SN.
The unclear etiology and pathogenesis of Spitzoid neoplasms as well as the diagnostic difficulties has led to numerous additional studies aiming to better understand pathogenesis and to identify biomarkers for use in daily diagnostics.
In this review, we will briefly describe the histopathological features of Spitzoid neoplasms and give a detailed overview of the current knowledge of the molecular genetic landscape of Spitzoid neoplasms. We also discuss the usefulness and the limitations of ancillary methods in ambiguous Spitzoid neoplasms to achieve a more accurate and distinct diagnosis. A reasonable integration of histopathology and stepwise performance of ancillary immunohistochemical and/ or moleculargenetic techniques should aim to minimize the gray zone in between SN and MST thereby improving diagnostic accuracy and patient management. Finally, we will give an outlook on potential future research questions and unresolved research objectives in the field of Spitzoid melanocytic lesions.
Histopathological Features of Spitzoid Neoplasms
The diagnosis of a Spitzoid neoplasm is primarily based on histopathological grounds. However, the diagnosis should always be made in the context of the clinical features, especially including the patient's age with lesions in adults harboring a higher suspicion for malignancy [Table 1, first row, (24)]. Spitzoid neoplasms may be junctional, compound or intradermal. Intradermal localization is mainly seen in adults (18, 25, 26). The most distinct feature of a Spitzoid neoplasm is the presence of enlarged epithelioid and/ or spindle-shaped melanocytes [Figures 2C–F, (16, 27)]. In addition to cell type other histopathological features are required to render the diagnosis of a Spitzoid neoplasm which are briefly summarized in Table 1.
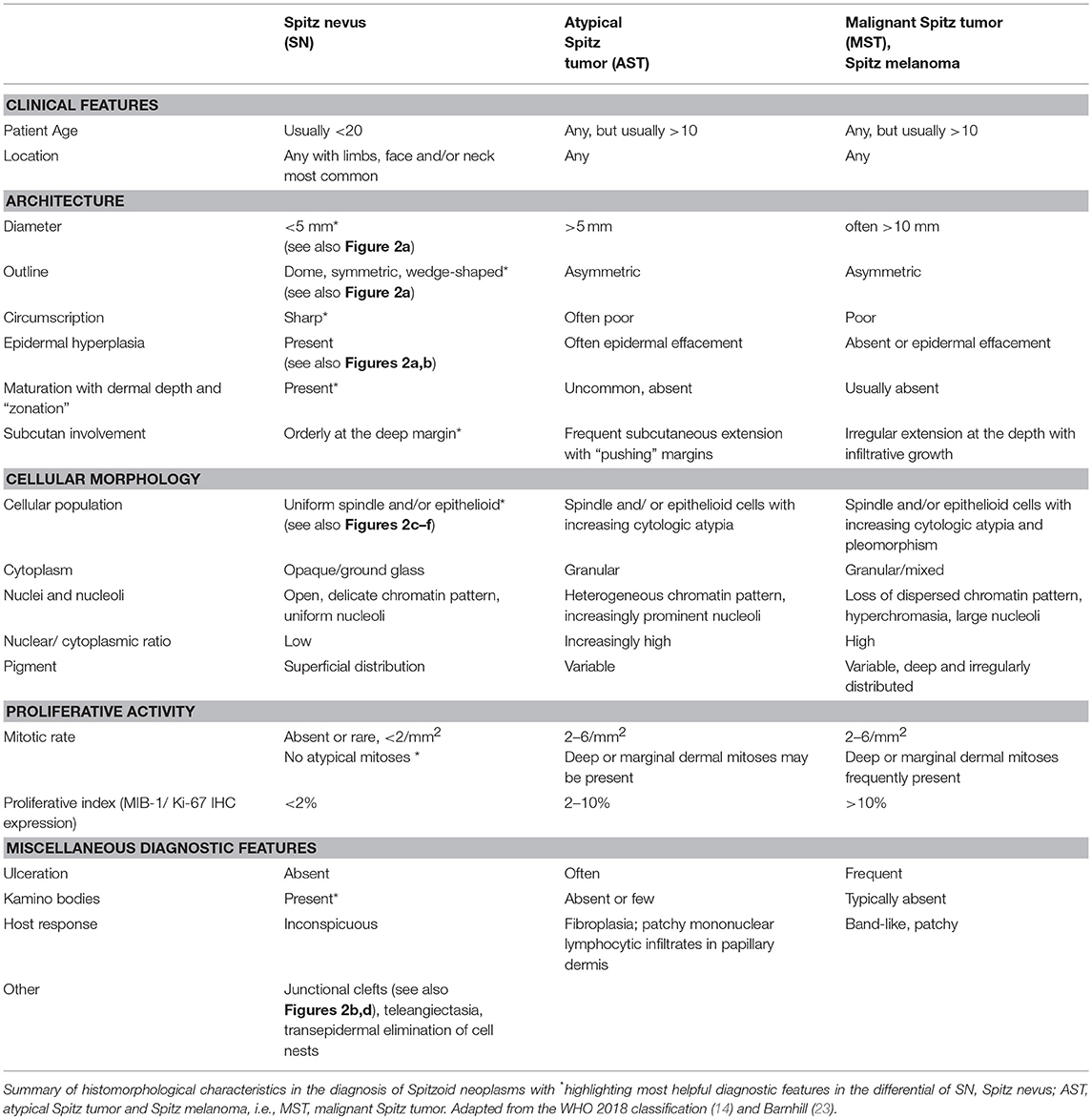
Table 1. Clinical and histopathological features of spitzoid neoplasms.
A SN is usually >5 mm in diameter, shows a dome or wedge shaped symmetrical outline with sharp lateral circumscription, maturation, and zonation to the depth of the lesion and epidermal hyperplasia [Figure 2A, (18, 26, 28)]. There should be no ulceration, absence or only random cytonuclear atypia, no involvement of subcutaneous tissue and no or only few mitoses (Table 1). Zonation of cells with large cells in the superficial layers and maturation of melanocytes from the surface to the depth with decrease in amount of cell nests, cell size, nuclei, and nucleoli is seen in benign SN in contrast to their atypical or malignant counterparts (16, 27). However, the frequency of maturation in Spitzoid neoplasms varies considerably among published studies (16, 18, 21). The distribution of pigment, if present, may be zonal as well and localized subjacent to the epidermis. Kamino bodies constitute an important diagnostic feature of Spitzoid neoplasms and favor a benign nature of the lesion (17, 29, 30). Ultra-structurally Kamino bodies are composed of amorphous filaments and they stain for basement membrane components, including collagen types IV, VII, and laminin on immunohistochemistry [IHC, (15, 31, 32)]. They do not show evidence of apoptosis (33) and should not be confused with Civatte or colloid bodies. Single-cell upward spread of melanocytes is less common in Spitzoid lesions compared to conventional melanocytic malignant neoplasms (18). If pagetoid spread of Spitzoid cells is present, it most often occurs in bundles or nests of cells. At the dermoepidermal junction, the fascicles of Spitzoid cells often show vertical orientation with loss of cohesion and form retraction spaces from the adjacent epidermis somehow resembling “bundles of bananas” or a “raining down” pattern; a feature often found in Spitzoid lesions and less common in melanoma (27). SN often show adnexotropism with involvement of hair follicles and eccrine ducts what should not raise undue concern about melanoma (34). Additionally the distribution of the inflammatory infiltrate in SN can be a helpful diagnostic feature, which is quite often perivascular and diffusely in between the collagenous material (27). In SMM the inflammatory infiltrate is usually found at the base of the lesion.
AST present with intermediate histological features of SN and MST. AST show increase in at least one worrisome histological feature i.e., ulceration, size >5 mm, infiltrative growth into subcutaneous tissue with “pushing” margins, increased cytonuclear atypia, increase in cell density with confluent growth, >2 dermal mitoses, absence of junctional clefts, few or no Kamino bodies and more extensive pagetoid extension (Table 1, second column). However, these histological features to distinguish AST from SN or SMM have often shown to be ineffective due to poor reproducibility (22). Therefore, assessment of an ambiguous Spitzoid neoplasm requires ancillary diagnostic techniques such as IHC as well as molecular pathology analysis, which will be outlined and discussed in the following.
Finally, MST is diagnosed on the basis of the following histological criteria: ulceration, asymmetrical architecture, infiltrative growth, severe and/ or confluent cytonuclear atypia, dermal mitoses, pushing borders, epidermal effacement and pagetoid extension (Table 1, last column).
Underlying Molecular Alterations and Ancillary Studies in Spitzoid Neoplasms
The aim of many additional studies has been to identify criteria that enable assignment of Spitzoid lesions to a correct and distinct diagnosis of SN or MST and thus to keep the ambiguous diagnostic category of AST as small as possible. The major interest of these studies was to identify parameters that predict malignant behavior in Spitzoid neoplasms with increased risk of recurrence, risk for metastasis, and risk for death. In the following, the findings from IHC studies in Spitzoid neoplasms including cell cycle and apoptosis regulator related markers as well as application of melanocytic markers will be outlined. Recent findings from next generation sequencing (NGS) which have identified telomerase reverse transcriptase promotor (TERTp) mutations, BRCA1 associated protein 1 (BAP1) mutations, HRAS mutations and receptor tyrosine kinase alterations (ALK, ROS, NTRK1, NTRK3, RET, MET) will be discussed. Furthermore, results from molecular pathology analysis using comparative genomic hybridization (CGH) and fluorescence in situ hybridization (FISH) will be reviewed and their diagnostic applicability will be discussed. We also discuss recent findings from mRNA and micro RNA (miRNA) expression studies. Finally, we will briefly focus on recent results from mass spectrometry imaging (MSI), which potentially represents a promising adjunct technique. The additional diagnostic techniques and molecular genetic targets which have been reported in the literature for Spitzoid neoplasms are summarized in Table 2. Their usefulness for the differential diagnosis of AST vs. MST or AST vs. SN is commented in the second last column of Table 2.
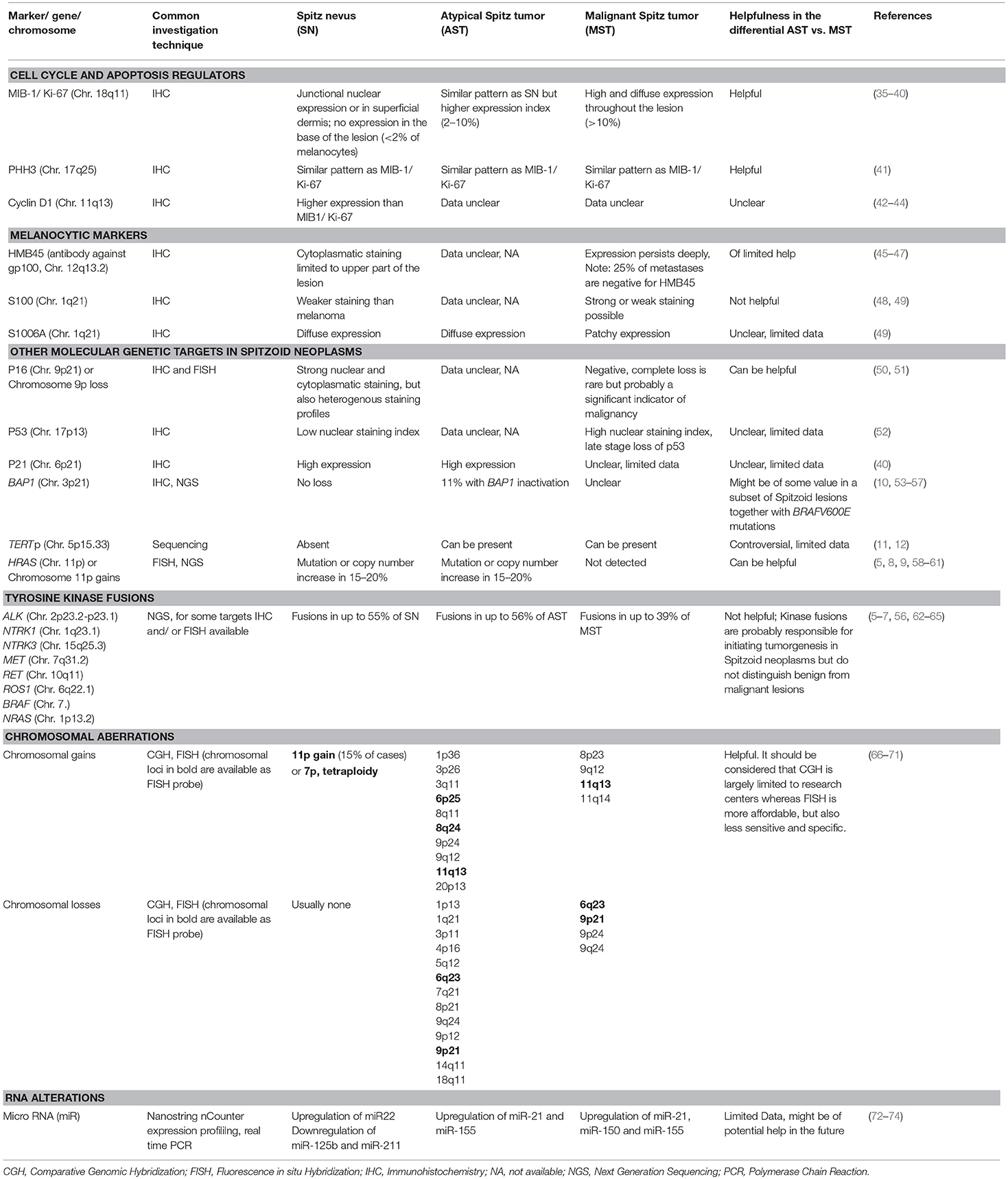
Table 2. Synopsis of ancillary investigations and molecular genetic targets reported in the literature in SN, AST and MST.
Cell Cycle and Apoptosis Regulators
Assessment of IHC expression of the nuclear protein MIB-1/ Ki-67, which is upregulated by cells in the active phase of the cell cycle represents a widely used ancillary technique in the diagnosis of Spitzoid neoplasms (35–40, 42, 75). In SN IHC expression for MIB-1/Ki-67 is largely restricted to the upper parts of the lesion. These findings contrast with MST, in which MIB-1/Ki-67 expression is generally higher and also present in the depth of the lesion (Tables 1, 2). Phosphohistone-H3 (PHH3) IHC reflects mitotic activity and has also been studied in melanocytic lesions (41). The expression of PHH3 is more sparse compared to MIB-1/ Ki-67 because PHH3 IHC expression is restricted to mitotic figures from early prophase through metaphase, anaphase, and telophase whereas MIB-1/Ki-67 stains the whole replicative cycle including G1- and G2 phase. There are only few data for Cyclin-D1 IHC in Spitzoid lesions (42–44). Unlike in conventional malignant melanoma there seems to be a dissociation between Cyclin D1 overexpression and cell proliferation in Spitzoid neoplasms (43). Therefore, Cyclin D1 IHC is not recommended as adjunctive test in the diagnostic work up of Spitzoid neoplasms.
Mutations in p16 (syn. cyclin dependent kinase inhibitor 2A, CDKN2A) which is localized on Chr. 9p21 have been associated with an aggressive clinical behavior in AST (76). FISH analysis for CDKN2A represents an adjunctive tool (77) and loss of 9p21 is associated with malignant behavior in Spitzoid neoplasms. Although p16 IHC is of added value in the differential diagnosis of ambiguous Spitzoid neoplasms, its expression alone is not sufficient to discriminate between SN, AST, and MST (50, 51, 77). The use of p16 IHC to approach p16 mutational status in Spitzoid lesions remains to be interpreted with caution, because p16 is often heterogeneously expressed in these lesions and the customized antibodies for p16 IHC are not mutation specific.
Melanocytic Markers
Spitzoid melanocytic neoplasms have been evaluated with a variety of melanocytic markers including HMB45, S100, S100A6, MITF, Mart-1, Melan A, and SOX10. They are widely used to establish melanocytic differentiation and are helpful to better assess the growth pattern in cases where melanocytes are poorly identifiable on H.E. histology. HMB45 (antibody against Gp100) is a melanogenesis related protein and useful in the assessment of a lesions maturation toward the depth. HMB45 IHC shows cytoplasmatic staining and diminishes in the depth of a SN, whereas in MST positivity has been described to be trans-lesional and more uniform without limitation to the upper parts of the lesion (45, 47). For AST the expression pattern is not very well described in the literature and seems to be of limited additional value in the differential diagnosis in between AST and MST. S100 IHC shows diffuse expression in Spitzoid melanocytic neoplasms independent from benign or malignant behavior and thus has not been proven as helpful in the differential diagnosis of ambiguous Spitzoid lesions (48, 49). The S100 subtype S100A6 has been proposed as potential IHC marker in the differential diagnosis of SN from MST with strong expression in SN and diminished expression in malignant lesions (49), but conformational studies are pending to validate its usefulness in the routine examination of Spitzoid neoplasms.
Intracellular Signal Receptor Molecules and Receptor Tyrosine Kinase Fusions
The HRAS gene is located on the short arm (p) of chromosome 11 and copy number gains of 11p as well as activating HRAS mutations are found in approximately 20% of Spitzoid neoplasms (5, 8, 58, 78). This is in contrast to conventional melanoma where HRAS mutations are found in >1% of cases (3). Mutations in HRAS usually occur in exon 3, causing replacement of the glutamine at amino acid position 61 with a lysine (HRASQ61K) or an arginine (HRASQ61R). Mutations in exon 2 are infrequent. The amino acid exchange leads to constitutive activation of the MAP/ERK and the PI3K/AKT/mTOR pathway. It has been described that tumors with 11p gains often show an intradermal localization, a desmoplastic morphology with stromal sclerosis and infiltrative margins; proliferative activity is low and most often the prognosis is favorable (5, 59). Currently, it is not known why HRAS mutations occur almost exclusively in Spitz tumors. Wiesner et al. suggested that HRAS mutations lead to stronger activation of the PI3K/AKT/mTOR pathway than BRAF mutations do in common nevi, a feature that might explain why the melanocytes have a larger cell size in Spitz tumors with HRAS mutations (56). Although the current data are derived from a limited number of cases with only limited long term follow up the results suggest that HRAS mutations in Spitzoid lesions can be attributed to a benign biological behavior.
BRAF and NRAS mutations are commonly found in conventional melanoma [~70%, (79)] but are rare in Spitzoid lesions. Initial studies did not report BRAF and/ or NRAS mutations in SN (5, 80–82), while later studies detected BRAF mutations in up to 20% and NRAS mutations in up to 5% of Spitzoid neoplasms (5, 6, 60, 62, 83–85).
Fusions such as translocations of the receptor tyrosine kinases including ALK, ROS1, NTRK1, NTRK3, RET, and MET are found in up to 50% of Spitz tumors and are infrequent in conventional melanocytic lesions with BRAF mutations (6, 56, 64, 65).
ALK gene fusions are found in 10% of Spitzoid neoplasms (6, 64). The most prominent fusion partners are TPM3 and DCTN1 and a distinct morphology has been attributed to them with the majority being amelanotic, dome-shaped and with presence of plexiform intersecting melanocytic fascicles (63, 86). Spitzoid neoplasms with ALK alterations are often localized on the extremities (64). Recently, Farah and colleagues reported a case of a plexiform MST with ALK copy number gain instead of ALK rearrangement (87). In summary, ALK mutations appear to be associated with a characteristic plexiform growth pattern, but seem to be present in the whole spectrum of Spitzoid lesions. Therefore, alterations in ALK appear to be of limited help in distinguishing benign from malignant Spitzoid lesions.
ROS1 gene fusions are observed in 17% of Spitzoid neoplasms, and often also occur on the extremities (6). Wiesner et al. found Spitzoid neoplasms with ROS1 fusions to show a similar morphology as those with ALK fusions (63). As according to ALK gene fusions, ROS gene fusions are also found in benign and malignant Spitzoid neoplasms. Therefore, they seem to be of limited value to discriminate benign and malignant lesions.
Fusions of the neurotrophic receptor tyrosine kinase (NTRK) 1 gene have been described in up to 16% of Spitzoid neoplasms spanning a spectrum from benign to malignant (6). No distinct morphology has been linked to NTRK1 rearranged Spitzoid lesions. These fusions have also been confirmed by IHC analysis (6). Another group found NTRK3 fusions in Spitzoid neoplasms with a higher incidence in AST (6/8 cases) than in SN [2/8 cases, (7)].
Wiesner et al. described RET gene fusions in >5% of Spitz tumors. In analogy to other receptor tyrosine kinase fusions the MAPK/ERK and PI3/AKT/mTOR pathways are activated (6). Studies in mice have shown that RET overexpression results in melanocytic proliferation, nevus formation and progression to malignant melanoma (88). Since RET-positive Spitzoid neoplasms are rare, only sporadic data is available on the predictive value with regard to biological and clinical behavior.
MET gene fusions activate the MAPK/ERK and PI3K/AKT/MTOR pathways and have been described in six Spitzoid neoplasms in a study from (65).
In summary, the above described kinase gene fusions (including ALK, ROS, NTRK1, NTRK3, RET, and MET) occur in a mutually exclusive pattern and are not found in BAP1 (see below) or HRAS mutated Spitzoid lesions (56). Furthermore, if present they are detected across the entire spectrum of Spitzoid lesions [i.e., in 55% of SN, 56% of AST and in 39% of MST, (6)], thereby suggesting that the fusions occur early in the pathogenesis of the tumors. Hence, diagnostic applicability in the differential diagnosis in between a benign or a malignant Spitzoid neoplasms seems to be of limited value (56, 57, 62, 84).
BRCA1 Associated Protein 1 (BAP1)
BAP1 is a deubiquitinating enzyme and its inactivation is found in approximately 5% of Spitzoid neoplasms (10). The BAP-1 mutated lesions have been called “Wiesner tumor” or “BAPoma” and often also show BRAFV600E mutations, which is not typical in Spitzoid lesions (10, 53, 54). IHC showing loss of nuclear BAP1 expression seems to be a good surrogate marker for BAP1 inactivation (10).
BAP1 mutations can be associated with a hereditary tumor predisposition syndrome (OMIM #614327) and lesions often show a histologically distinct epithelioid phenotype (6, 10, 55). It has been described that tumors with sporadic BAP1 loss have the same histological appearance as those seen in patients with germline BAP1 mutations. Although infrequent, inactivation of BAP1 together with BRAFV600E mutations might predict a benign clinical course in Spitzoid lesions (6, 53, 57, 64). However, it remains to be considered that the to date available data are based on studies with a limited number of cases with lack of long term follow up data and require conformational studies.
Telomerase Reverse Transcriptase Promotor (TERTp)
Upregulation of TERTp in order to overcome shortening of telomers and consequently senesce has been reported in Spitzoid lesions (11, 12). Mutations in TERTp have been found in subgroups of AST and MST and were associated with an aggressive clinical behavior and distant metastasis (11). However, data are conflicting with other groups that report no association with poor prognosis in Spitzoid lesions (12). More studies are needed to better evaluate the usefulness of TERTp mutations in the differential diagnosis of AST from MST.
Chromosomal Investigation Techniques
CGH is a molecular technique that analyses entire genome copy number alterations by comparing tumor DNA to normal reference DNA. SN usually lack copy number alterations except for gains in 11p (attributed to HRAS activation) in desmoplastic SN (9, 78, 89) and very rarely isolated loss of chromosome 3 in epithelioid melanocytic tumors with BAP1 loss (10). Mutational activation in HRAS does not occur in other nevi and is extremely rare in conventional melanomas (58, 90). As mentioned above HRAS activation in Spitzoid neoplasms might point to a benign behavior of the lesions (58).
AST reveal more genomic aberrations than SN, but fewer than MST. The identification of copy number alterations with CGH has evolved into FISH assays, which are less labor intensive and require only basic laboratory equipment. The most extensively studied FISH assay comprises 4 loci including chromosome 6 with the RREB1 region (6p26), chromosome 6 with the MYB region (6q23), centromere 6 (Cep6) and/ or chromosome 11 with the CCND1 region (11q13), and/or chromosome 9 with the CDKN2A region [9p21; (66, 91, 92)]. The so called “melanoma FISH” which targets 6p26, 6q23, Cep6, and 11q13 shows high sensitivity and specificity in unequivocal conventional melanocytic neoplasms (66, 93), but was less reliable in cases of Spitzoid melanocytic neoplasms (67). Adaptation of chromosomal targets with probes against 9p21, 11q13, and 8q24 (targeting the c-MYC region) has led to improvement in the diagnosis of equivocal Spitzoid neoplasms (66, 68, 70). Table 2 summarizes the currently identified chromosomal targets in Spitzoid neoplasms. It has been shown that homozygous deletions of 9p21 and/ or gains in 6p26 and or 11q13 are associated with a worse clinical behavior in Spitzoid neoplasms whereas deletions in 6q23 favor a benign behavior (67, 70).
Although at first sight FISH analysis seems to be more cost saving than CGH it is important to note that FISH analysis of melanocytic lesions has a comparatively high false negative rate. With FISH analysis only 4–6 genomic loci are investigated, and mutations, small insertions or deletions as well as genomic rearrangements cannot be detected. Additionally, FISH can also show false positive results due to tetraploidy in Spitz nevi occuring in 5–10% of SN (94). Tetraploidy is seen in melanocytes when they are stimulated to divide and replicate their DNA but subsequently do not divide as a result of a halting of the process by regulators of the cell cycle. Hence, the cells are trapped and typically undergo senescence. In summary, tetraploidy can be seen in conventional melanocytic neoplasms and/ or Spitzoid neoplasms and is therefore non-diagnostic. Thus, the diagnostic utility of FISH analysis in the evaluation of equivocal Spitzoid neoplasms remains a subject of intense discussion (69, 95).
miRNA Expression Profiling
Until now only few studies have investigated the miRNA profile of Spitzoid melanocytic tumors (72–74). MiRNA represent 19–22 nucleotide, non-coding RNA molecules that epigenetically regulate gene expression at the post-transcriptional level by either degradation or translational blockade of target mRNA (96). Deregulation of miRNA with oncogenic and tumor-suppressive functions has attracted much attention and it has been shown that the expression profile of miRNA varies across neoplastic lesions at different stages of malignancy (97, 98).
Grignol et al. investigated the expression of miR-21, miR-155, and miR-211 in conventional malignant melanoma and borderline melanocytic lesions (including 22 AST samples) with real-time PCR and compared them to benign common nevi (72). The conventional malignant melanoma group and the borderline melanocytic lesion group (including the AST group) showed upregulation of miR-21 and miR-155 when compared to benign nevi (72). SN or MST samples were not investigated in this study. Latchana et al. have identified distinct miRNA expression profiles in the whole spectrum of Spitzoid melanocytic neoplasms including SN, AST, and MST with the NanoString nCounter Gene Expression platform (73) and investigated a selection of 12 reversely transcribed miRNAs by real time PCR (73). In their study benign SN showed decreased expression of miR-125b and miR-211, and upregulation of miR-22, compared to benign nevi (74). MST showed overexpression of miR-21, miR-150, and miR-155 when compared to benign nevi and also when compared to the AST group. Summarizing, these studies indicate that upregulation of miR-21 and miR155 might be attributed to a malignant behavior in Spitzoid melanocytic neoplasms (72, 74). Summarizing, the analysis of miRNA expression represents a promising additional approach to the categorization of indeterminate Spitzoid melanocytic lesions and may be used as a diagnostic adjunct or eventually therapeutic target in the future.
mRNA Expression Profiling
Characterization of the transcriptome of Spitzoid melanocytic neoplasms by mRNA sequencing has gained increasing attention in the last years (13, 99, 100). Wu et al. analyzed the landscape of fusion transcripts in metastatic MST and biologically indeterminate AST with positive locoregional lymph nodes by RNA sequencing (13). They successfully investigated six out of seven samples and identified novel fusion transcripts involving TPM3-NTRK1 (2 samples), rearrangements in TPM3, ALK and IL6R (1 sample), BAIAP2L1-BRAF (1 sample), EML4-BRAF (1 sample), and two samples containing a second fusion gene, ARID1B-SNX9 or PTPRZ1-NFAM1. Two tumor samples with metastatic disease harbored a TERTp mutation which was also confirmed with mRNA in situ hybridization (ISH).
Jansen et al. investigated RNA expression differences in long intergeneic non-coding RNA 518 (LINC) and preferentially expressed antigen in melanoma (PRAME) from SN and compared them to conventional malignant melanoma (100). From the 23 investigated samples nine samples comprised pediatric SN. While PRAME showed significantly higher expression in malignant melanoma cases in relation to SN cases, LINC expression was not different in both groups. AST or MST were not investigated in this study.
Our group recently investigated the mRNA expression profile of SN in comparison to common nevocellular nevi (99). A significant difference in the mRNA signature in between these two groups was shown even when SN and nevocellular nevi samples were obtained from the same individuals. We could identify a molecular genetic signature of 15 top ranked genes with upregulation of CAPN2, CDC25B, ITGA3, LIG4, HSPA6, DUSP10, and JAK3 and downregulation of IGFR1, IDH1, HES1, BAIAP3, SUV39H2, SHC2, FGF2, and MSH6 transcripts in SN when compared to NCN. We are currently expanding the mRNA expression analysis to AST and MST samples to investigate whether there is a differential mRNA expression profile in the whole spectrum of Spitzoid melanocytic lesions (SN vs. AST vs. MST). We are also evaluating whether the identified transcripts can be identified on translational level with immunohistochemistry.
Although no definitive conclusions can be drawn from the to date performed mRNA expression studies because of the limited number of studies and the relatively small numbers of investigated samples, the results from mRNA analysis are likely to reflect the actual functional state of the neoplastic cells and future studies should expand upon transcriptome sequencing in Spitzoid melanocytic neoplasms.
Mass Spectrometry Imaging (MSI)
MSI is a powerful new methodology to analyze proteomic signatures in fresh frozen and also in formalin-fixed paraffin embedded (FFPE) tissue samples with spatial fidelity. Few studies have implemented MSI in order to assess its value in the differential diagnosis of melanocytic lesions. Hardesty and colleagues found peptides that discriminate prognostic subgroups in melanoma patients (101). Lazova and colleagues could discriminate SN with 97% sensitivity and 90% specificity from MST based on a mass spectral proteomic signature of five heterogeneously expressed peptides. The identified signature consisted of five distinct MSI peaks with the specific mass-to-charge (m/z) values 976.49, 1060.18, 1336.72, 1410.74, and 1428.77. For two of the five distinct values the corresponding peptides could successfully be identified, which are Actin (m/z = 976.49) and Vimentin (m/z = 1428.61). In a subsequent study the identified proteomic MSI signature could correctly classify 102 AST samples with an adverse clinical outcome to the MST signature and those with benign outcome to the SN signature (102). Interestingly, in a following study there was no detectable differential expression of Vimentin or Actin with IHC or immunofluorescence analysis (103) in between SN and MST samples. Although conformational studies are pending, these findings strongly suggest that there may be some characteristics on molecular level that are detected by MSI to better distinguish the gray zone AST category beyond the information provided by histopathology or immunohistochemistry.
Diagnosis, Classification and Management of Spitzoid Neoplasms
After all, the histopathological evaluation remains the initial step and gold standard in diagnosing a Spitzoid neoplasm (Figure 3, first column). Clinical features such as patient's age should always be considered with adults harboring higher suspicion for malignancy (24).
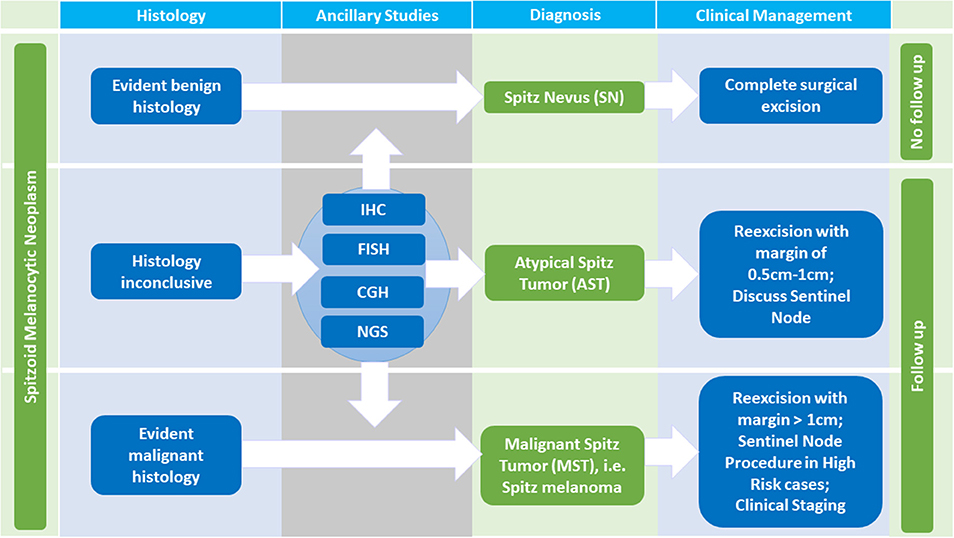
Figure 3. Diagnosis and Management of Spitzoid Melanocytic Neoplasms. Histopathologic evaluation is the initial step in diagnosing a Spitzoid neoplasm. In case of evident benign histologic characteristics, the diagnosis of a Spitz nevus (SN) can be made. If the lesion has been completely excised a clinical follow up is not necessary. In cases of evident malignant histopathology, the diagnosis of Spitz melanoma, i.e., malignant Spitz tumor (MST) is made and clinical management according to conventional malignant melanoma protocols should be followed. The diagnosis of MST necessitates a re-excision with a margin of at least 1 cm, in high risk cases a sentinel node procedure, clinical staging and follow up. If the histopathology is ambiguous stepwise ancillary studies such as immunohistochemistry (IHC), comparative genomic hybridization (CGH), fluorescence in situ hybridization (FISH) and next generation sequencing (NGS) should be performed. If histopathology as well as ancillary studies still remain inconclusive the diagnosis per exclusionem of atypical Spitz tumor (AST) is made. A reexcision with a margin of 0.5–1 cm should follow. The value of a sentinel node procedure, the follow up time, and the extent of clinical staging in this setting remains a topic of ongoing debate.
In cases of evident benign histomorphological characteristics with absence of any suspicious criteria (i.e., diameter <5 mm, symmetrical architecture, sharp circumscription, maturation toward the dermal depth, no subcutan involvement, no striking cytonuclear atypia, no increase in proliferative activity with <2 mitosis/mm2 and absence of ulceration, Table 1, first column) the diagnosis of a SN should be rendered. In patients diagnosed with SN a clinical follow up is not required if the lesion has been completely excised [Figure 3, first line, (104)]. If the excision shows positive margins on histopathological examination, we advise a re-excision or at least clinical follow up to monitor potential locoregional recurrence.
In cases of evident malignant histopathology, the diagnosis of MST is made and clinical management according to conventional malignant melanoma protocols should be followed with a re-excision comprising a margin of at least 1 cm, in high risk cases a sentinel node procedure, clinical staging, and follow up (Figure 3, third line).
If the histopathology is ambiguous one should not hesitate to ask for a second internal and/ or external consultation. In addition, ambiguous cases justify stepwise ancillary studies such as IHC, CGH, FISH, and NGS which are illustrated in Figure 4, of which IHC stands out as first step due to its technical ease and availability in pathology laboratories. An IHC panel including MIB-1/ Ki67, HMB45, and p16 can be useful [(14, 105, 106), Figure 4, see also Table 2 for details]. In special cases with a plexiform morphology ALK IHC as surrogate marker to address ALK gene rearrangement can be of help as well (86) and in cases with a distinct epitheloid phenotype BAP1 IHC as surrogate for BAP1 gene inactivation can be applied (10). A MIB-1 IHC index <2% with diminished expression in the deep dermal component as well as diminished HMB45 IHC expression in the deep dermal component and patchy to strong p16 IHC expression throughout the lesion favors the diagnosis of SN. Cases with MIB-1 IHC expression >10%, persisting HMB45 IHC expression in the deep dermal component as well as loss of p16 IHC expression argue for the diagnosis of MST although exceptions of this staining pattern are possible.
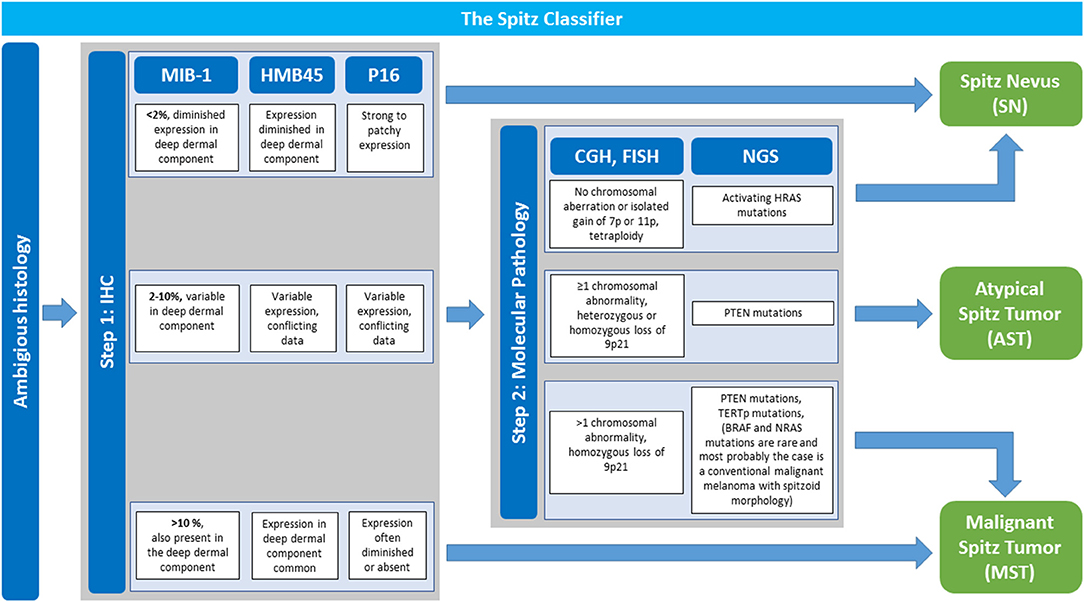
Figure 4. The Spitz classifier: Stepwise Implementation of Immunohistochemistry and Molecular Pathology in Ambiguous Spitzoid Neoplasms. In cases with ambiguous histopathological features that give suspicion for malignancy stepwise ancillary studies are performed to render the diagnosis of a Spitz nevus (SN), atypical Spitz tumor (AST) or malignant Spitz tumor (MST) with more security. Step 1 is immunohistochemistry (IHC) with a panel of MIB-1-, HMB45-, and p16 antibody. In dependence of the obtained result a diagnosis of SN or MST can be made. A MIB-1 IHC index <2% with diminished expression in the deep dermal component as well as diminished HMB45 IHC expression in the deep dermal component and patchy to strong p16 IHC expression throughout the lesion favors the diagnosis of SN. Cases with MIB-1 IHC expression >10%, persisting HMB45 IHC expression in the deep dermal component as well as loss of p16 IHC expression favor the diagnosis of MST although exceptions of this rule are possible. If results of IHC investigation are intermediate with a MIB-1 IHC index of 2–10%, and/or variable HMB45- and p16 IHC expression one should proceed to step 2 with implementation of molecular pathology. Comparative genomic hybridization (CGH) analysis or if not available fluorescence in situ hybridization (FISH) are performed. Next generation sequencing (NGS) is of help as well. No detectable chromosomal aberration or isolated gain of Chr. 7p or 11p as well as activating HRAS mutation favor the diagnosis of a SN. The presence of at least one chromosomal abnormality (excluding Chr. 7p or 11p gain), especially homozygous loss of Chr. 9p11 as well as PTEN and TERTp mutations argue for MST. If the afore mentioned findings are only partly present with ≥1 chromosomal abnormality (including heterozygous and very rarely homozygous loss of Chr. 9p21), or PTEN mutations AST as a diagnosis per exclusionem is to be considered. Adapted from WHO 2018 classification (14).
If results from step one with IHC investigation are intermediate showing a MIB-1 IHC index of 2–10%, and/ or variable HMB45- and p16 IHC expression step two should be followed with implementation of molecular pathology (Figure 4). Since molecular pathology techniques such as CGH, FISH, and NGS are not always available in all pathology laboratories it is recommendable to refer the patients including histopathology tissue to a reference center in ambiguous cases. No detectable chromosomal aberration or isolated gain of Chr. 7p or 11p as well as activating HRAS mutation favor the diagnosis of a SN. The presence of at least one chromosomal abnormality (excluding 7p or 11p gain), especially homozygous loss of Chr. 9p11, loss of Chr. 6q23 as well as PTEN and TERTp mutations argue for MST.
It remains to be noted that interpretation of NGS results is an issue of current intense investigation and there exists no standardized NGS panel recommendation for Spitzoid neoplasms yet.
Investigation of the BRAF gen, either with the mutation specific BRAFV600E antibody or with sequencing analysis are of help in the differential diagnosis to which group the melanocytic lesion should be ascribed. Presence of a BRAFV600E mutation most likely rules out a Spitzoid melanocytic neoplasms and assigns the atypical lesion to the group of conventional melanocytic neoplasms. Thus, most probably the diagnosis would be a “conventional” melanoma harboring a Spitzoid epithelioid and- or spindle shaped morphology.
If the afore mentioned findings from molecular pathology investigation are only partly present with ≥1 chromosomal abnormality (including heterozygous and very rarely homozygous loss of 9p21), or PTEN mutations are present, AST as a diagnosis per exclusionem is to be considered. A patient with the diagnosis of the vaguely defined category AST should be managed with a re-excision with a margin of 0.5–1 cm and follow up is necessary (Figure 3, second line). The value of a sentinel node procedure, the extend of clinical staging and the exact follow up time in this setting remain unclear and a topic of ongoing debate (104, 107, 108). Unlike conventional melanoma, dissemination to regional lymph nodes in AST, in general, seems not to be predictive for distant metastasis or patient survival (104, 107–109).
Over time there have been numerous approaches to classify Spitzoid neoplasms which are summarized in Figure 5. The initial description “juvenile melanoma” by Sophie Spitz (19) was revised by Ackermann and colleagues who proposed a two-tiered classification system (20, 21, 106). This classification system differentiated between benign SN and MST as the malignant counterpart (Figure 5, third line). The ambiguous intermediate category of AST was not considered. From on the 1990s the two-tiered classification system was opposed by a three-tiered categorization including SN, AST and MST [Figure 5, fourth line, (22, 23)]. The international Melanoma Pathology Study group even suggested a four-tiered classification system which subclassifies AST in low grade and high grade lesions [Figure 5, fifth line, (84, 110)]. The proposed three- and four- tiered classification systems best reflect the existence of an ambiguous group of Spitzoid neoplasms with intermediate features in between complete benign SN and clear-cut malignant MST. Recently Tetzlaff and colleagues have emphasized the need toward a molecular based classification system for the morphologically ambiguous and biologically unpredictable group of AST [Figure 5, sixth line, (57)]. The application of ancillary diagnostic techniques and the implementation of the recent molecular genetic findings in the categorization of Spitzoid lesions is also suggested by the recently published WHO 2018 Classification of Skin Tumors (14) and might provide a framework to keep the uncertain diagnostic and therapeutic category of AST at a minimum thereby benefiting patient management.
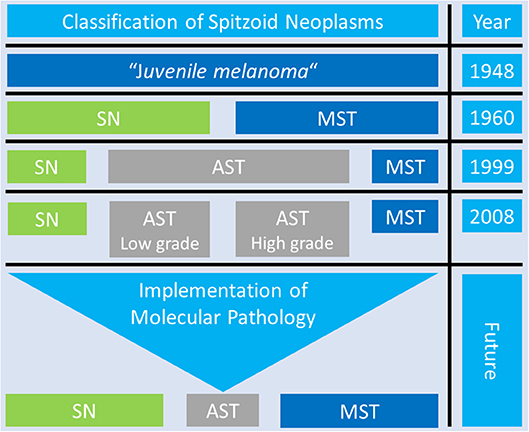
Figure 5. Overview of the Classification Systems of Spitzoid Neoplasms. Over time there have been many different diagnostic approaches to classify Spitzoid neoplasms. The proposed three- and four- tiered classification system best reflect the existence of the group op ambiguous Spitzoid neoplasms with atypical Spitz tumor (AST, gray color) with intermediate features in between complete benign Spitz nevi (SN, green color) and clear cut malignant Spitz melanoma, i.e., malignant Spitz tumor (MST, blue color). The application of ancillary diagnostic techniques and implementation of findings from molecular pathology might help to reduce the uncertain diagnostic category of AST to a minimum and improve and clarify patient management in the future.
Future Research Objectives
The findings from the to date identified molecular alterations have not led to emergence of one stand-alone or a clearly defined distinct set of biomarkers that can reliably distinguish whether an ambiguous Spitzoid lesion will behave benign or malignant in the future. The urgent need remains to extend research projects and further immerge into the search of predictive biomarkers.
First, the analysis of the transcriptome and further proteomic studies, which are likely to represent the actual functional state of a cell, represent a promising strategy for identification of potential robust biomarkers. In first pilot studies the miRNA profile (72–74) and the mRNA profile (13, 99, 100) of Spitzoid melanocytic lesions have been investigated. The results are promising and might motivate surgical pathologists, clinicians and basic scientists to extend their investigations to this research field with larger study numbers and correlation of results with available long term follow up data.
Secondly, Spitzoid melanocytic neoplasms are well-known to harbor prominent collagenous or fibrotic stroma with fine vascularization and presence of infiltrating lymphocytes (16, 18, 111–113). Despite these obvious histological characteristics only little is known about the underlying immunological cellular subtypes, especially the nature of the infiltrating T-lymphocytes and the dermal stromal microenvironment in Spitzoid melanocytic lesions (18, 114). Even less is known about the microenvironment of the locoregional lymph nodes, which often harbor metastatic Spitzoid cells, but have been shown to be not predictive for aggressive biological behavior such as in conventional melanoma (104). We recently identified upregulated gene pathways in SN with increased expression of mRNA transcripts that were related to immunomodulatory, inflammatory, and extracellular matrix interactions as well as angiogenesis associated processes (99). Others have identified a proteomic discriminatory MSI signature, which is based on the stromal environment around SN and MST (115, 116). One may speculate that the immunological and stromal microenvironment might be a potential discriminator in the differential diagnosis of AST vs. MST. We hope that these observations and the preliminary findings will stimulate future research efforts to better characterize the dermal and lymphonodular microenvironment of Spitzoid melanocytic neoplasms.
Finally, the lack of objective and reproducible histologic criteria to determine the malignant potential of Spitzoid tumors remains a major diagnostic challenge. Very often the morphology does not reliably reflect the biological potential of a Spitzoid lesion. Apparently, the established histopathologic criteria which are used to differentiate nevocellular nevi from conventional malignant melanoma do not account for the group of Spitzoid melanocytic neoplasms (22, 117, 118). Thus, a critical revision of the to date applied histopathological criteria and the current classification of Spitzoid melanocytic neoplasms with uniformization of terminology is required. The recently launched WHO 2018 classification gives a first outline for a molecular based classification of Spitzoid melanocytic neoplasms (14) upon which diagnostic algorithmic approaches should be validated in the future.
Conclusion
In summary, the diagnosis of a Spitzoid lesion requires accurate correlation of the clinical, histological and molecular features. Yet, there are no isolated clinical, histological or molecular criteria nor a clearly defined diagnostic pattern of criteria that can distinguish AST from SN or MST with adequate certainty (56, 57, 119–121). A proper differentiation between AST, SN, and MST remains one of the most challenging issues in the field of dermatopathology with each diagnosis having quite different consequences for patient treatment, clinical management and follow up. Rigorous characterization of sufficient numbers of Spitzoid neoplasms and long-term follow up is therefore necessary for the formulation of optimal guidelines for the care of patients with these lesions.
In conclusion, the results from the current ancillary techniques and molecular genetic findings should be interpreted together with the clinical and histopathological data to formulate a firm integrative diagnosis and to keep the uncertain diagnostic category of AST as small as possible.
Author Contributions
VW, JV, and LH designed and developed the outline and concept of the manuscript. VW, JV, LH, and AzH supervised the writing of the manuscript and edited the figures and tables. LH, JV, MG, JB, AzH, and VW wrote, discussed and commented the manuscript. All authors approved the final manuscript.
Funding
This work was supported financially by the academic incentive fund from the Maastricht University Hospital Center, MUMC+, the Netherlands.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We want to thank the reviewers for their expert input with very useful, constructive and critical comments, which substantially helped us to revise the manuscript.
References
1. Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, et al. High frequency of BRAF mutations in nevi. Nat Genet. (2003) 33:19–20. doi: 10.1038/ng1054
2. Bauer J, Curtin JA, Pinkel D, Bastian BC. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J Invest Dermatol. (2007) 127:179–82. doi: 10.1038/sj.jid.5700490
3. Cancer Genome Atlas N. Genomic classification of cutaneous melanoma. Cell (2015) 161:1681–96. doi: 10.1016/j.cell.2015.05.044
4. Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O'Brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature (2009) 457:599–602. doi: 10.1038/nature07586
5. van Dijk MC, Bernsen MR, Ruiter DJ. Analysis of mutations in B-RAF, N-RAS, and H-RAS genes in the differential diagnosis of Spitz nevus and spitzoid melanoma. Am J Surg Pathol. (2005) 29:1145–51. doi: 10.1097/01.pas.0000157749.18591.9e
6. Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. (2014) 5:3116. doi: 10.1038/ncomms4116
7. Yeh I, Tee MK, Botton T, Shain AH, Sparatta AJ, Gagnon A, et al. NTRK3 kinase fusions in Spitz tumours. J Pathol. (2016) 240:282–90. doi: 10.1002/path.4775
8. Ali L, Helm T, Cheney R, Conroy J, Sait S, Guitart J, et al. Correlating array comparative genomic hybridization findings with histology and outcome in spitzoid melanocytic neoplasms. Int J Clin Exp Pathol. (2010) 3:593–9.
9. Bastian BC, LeBoit PE, Pinkel D. Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am J Pathol. (2000) 157:967–72. doi: 10.1016/S0002-9440(10)64609-3
10. Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. (2011) 43:1018–21. doi: 10.1038/ng.910
11. Lee S, Barnhill RL, Dummer R, Dalton J, Wu J, Pappo A, et al. TERT Promoter Mutations Are Predictive of Aggressive Clinical Behavior in Patients with Spitzoid Melanocytic Neoplasms. Sci Rep. (2015) 5:11200. doi: 10.1038/srep11200
12. Requena C, Heidenreich B, Kumar R, Nagore E. TERT promoter mutations are not always associated with poor prognosis in atypical spitzoid tumors. Pigment Cell Melanoma Res. (2017) 30:265–8. doi: 10.1111/pcmr.12565
13. Wu G, Barnhill RL, Lee S, Li Y, Shao Y, Easton J, et al. The landscape of fusion transcripts in spitzoid melanoma and biologically indeterminate spitzoid tumors by RNA sequencing. Mod Pathol. (2016) 29:359–69. doi: 10.1038/modpathol.2016.37
15. Skelton HG, Miller ML, Lupton GP, Smith KJ. Eosinophilic globules in spindle cell and epithelioid cell nevi: composition and possible origin. Am J Dermatopathol. (1998) 20:547–50. doi: 10.1097/00000372-199812000-00002
16. Weedon D, Little JH. Spindle and epithelioid cell nevi in children and adults. A review of 211 cases of the Spitz nevus. Cancer (1977) 40:217–25. doi: 10.1002/1097-0142(197707)40:1<217::AID-CNCR2820400134>3.0.CO;2-2
17. Arbuckle S, Weedon D. Eosinophilic globules in the Spitz nevus. J Am Acad Dermatol. (1982) 7:324–7. doi: 10.1016/S0190-9622(82)80313-7
18. Requena C, Requena L, Kutzner H, Sanchez Yus E. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. (2009) 31:107–16. doi: 10.1097/DAD.0b013e3181934218
20. Sulzberger MB, Kopf AW, Witten VH. Pigmented nevi, benign juvenile melanoma and circumscribed precancerous melanosis. Postgrad Med. (1959) 26:617–31. doi: 10.1080/00325481.1959.11712677
21. Kernen JA, Ackerman LV. Spindle cell nevi and epithelioid cell nevi (so-called juvenile melanomas) in children and adults: a clinicopathological study of 27 cases. Cancer (1960) 13:612–25. doi: 10.1002/1097-0142(196005/06)13:3<612::AID-CNCR2820130324>3.0.CO;2-G
22. Barnhill RL, Argenyi ZB, From L, Glass LF, Maize JC, Mihm MCJr, et al. Atypical Spitz nevi/tumors: lack of consensus for diagnosis, discrimination from melanoma, and prediction of outcome. Hum Pathol. (1999) 30:513–20. doi: 10.1016/S0046-8177(99)90193-4
23. Barnhill RL. The Spitzoid lesion: rethinking Spitz tumors, atypical variants, ‘Spitzoid melanoma' and risk assessment. Mod Pathol. (2006) 19(Suppl. 2):S21–33. doi: 10.1038/modpathol.3800519
24. Spatz A, Calonje E, Handfield-Jones S, Barnhill RL. Spitz tumors in children: a grading system for risk stratification. Arch Dermatol. (1999) 135:282–5. doi: 10.1001/archderm.135.3.282
25. Merot Y, Frenk E. Spitz nevus (large spindle cell and/or epithelioid cell nevus). Age-related involvement of the suprabasal epidermis Virchows Arch A Pathol Anat Histopathol. (1989) 415:97–101. doi: 10.1007/BF00784346
26. Binder SW, Asnong C, Paul E, Cochran AJ. The histology and differential diagnosis of Spitz nevus. Semin Diagn Pathol. (1993) 10:36–46.
27. Weedon D, Little JH. The Spitz naevus. Aust N Z J Surg. (1978) 48:21–2. doi: 10.1111/j.1445-2197.1978.tb05798.x
28. Scott G, Chen KT, Rosai J. Pseudoepitheliomatous hyperplasia in Spitz nevi. A possible source of confusion with squamous cell carcinoma. Arch Pathol Lab Med. (1989) 113:61–3.
29. Kamino H, Flotte TJ, Misheloff E, Greco MA, Ackerman AB. Eosinophilic globules in Spitz's nevi. New findings and a diagnostic sign. Am J Dermatopathol. (1979) 1:319–24. doi: 10.1097/00000372-197900140-00005
30. Ritter A, Tronnier M, Vaske B, Mitteldorf C. Reevaluation of established and new criteria in differential diagnosis of Spitz nevus and melanoma. Arch Dermatol Res. (2018) 310:329–42. doi: 10.1007/s00403-018-1818-8
31. Havenith MG, van Zandvoort EH, Cleutjens JP, Bosman FT. Basement membrane deposition in benign and malignant naevo-melanocytic lesions: an immunohistochemical study with antibodies to type IV collagen and laminin. Histopathology (1989) 15:137–46. doi: 10.1111/j.1365-2559.1989.tb03061.x
32. Wistuba I, Gonzalez S. Eosinophilic globules in pigmented spindle cell nevus. Am J Dermatopathol. (1990) 12:268–71. doi: 10.1097/00000372-199006000-00007
33. Wesselmann U, Becker LR, Brocker EB, LeBoit PE, Bastian BC. Eosinophilic globules in spitz nevi: no evidence for apoptosis. Am J Dermatopathol. (1998) 20:551–4. doi: 10.1097/00000372-199812000-00003
34. Barnhill RL. Pathology of melanocytic nevi and melanoma, 3rd edn. Heidelberg: Springer-Verlag (2016).
35. Kanter-Lewensohn L, Hedblad MA, Wejde J, Larsson O. Immunohistochemical markers for distinguishing Spitz nevi from malignant melanomas. Mod Pathol. (1997) 10:917–20.
36. Li LX, Crotty KA, McCarthy SW, Palmer AA, Kril JJ. A zonal comparison of MIB1-Ki67 immunoreactivity in benign and malignant melanocytic lesions. Am J Dermatopathol. (2000) 22:489–95. doi: 10.1097/00000372-200012000-00002
37. Bergman R, Malkin L, Sabo E, Kerner H. MIB-1 monoclonal antibody to determine proliferative activity of Ki-67 antigen as an adjunct to the histopathologic differential diagnosis of Spitz nevi. J Am Acad Dermatol. (2001) 44:500–4. doi: 10.1067/mjd.2001.111635
38. Chorny JA, Barr RJ, Kyshtoobayeva A, Jakowatz J, Reed RJ. Ki-67 and p53 expression in minimal deviation melanomas as compared with other nevomelanocytic lesions. Mod Pathol. (2003) 16:525–9. doi: 10.1097/01.MP.0000072747.08404.38
39. Vollmer RT. Use of Bayes rule and MIB-1 proliferation index to discriminate Spitz nevus from malignant melanoma. Am J Clin Pathol. (2004) 122:499–505. doi: 10.1309/MFFF06D5CYXR2F8T
40. Kapur P, Selim MA, Roy LC, Yegappan M, Weinberg AG, Hoang MP. Spitz nevi and atypical Spitz nevi/tumors: a histologic and immunohistochemical analysis. Mod Pathol. (2005) 18:197–204. doi: 10.1038/modpathol.3800281
41. Tapia C, Kutzner H, Mentzel T, Savic S, Baumhoer D, Glatz K. Two mitosis-specific antibodies, MPM-2 and phospho-histone H3 (Ser28), allow rapid and precise determination of mitotic activity. Am J Surg Pathol. (2006) 30:83–9. doi: 10.1097/01.pas.0000183572.94140.43
42. Ewanowich C, Brynes RK, Medeiros L, McCourty A, Lai R. Cyclin D1 expression in dysplastic nevi: an immunohistochemical study. Arch Pathol Lab Med. (2001) 125:208–10. doi: 10.1043/0003-9985(2001)125<0208:CDEIDN>2.0.CO;2
43. Nagasaka T, Lai R, Medeiros LJ, Brynes RK, McCourty A, Harada T, et al. Cyclin D1 overexpression in Spitz nevi: an immunohistochemical study. Am J Dermatopathol. (1999) 21:115–20. doi: 10.1097/00000372-199904000-00001
44. Stefanaki C, Stefanaki K, Antoniou C, Argyrakos T, Patereli A, Stratigos A, et al. Cell cycle and apoptosis regulators in Spitz nevi: comparison with melanomas and common nevi. J Am Acad Dermatol. (2007) 56:815–24. doi: 10.1016/j.jaad.2006.09.015
45. Tom WL, Hsu JW, Eichenfield LF, Friedlander SF. Pediatric STUMP lesions: evaluation and management of difficult atypical Spitzoid lesions in children. J Am Acad Dermatol. (2011) 64:559–72. doi: 10.1016/j.jaad.2009.12.063
46. Palazzo J, Duray PH. Typical, dysplastic, congenital, and Spitz nevi: a comparative immunohistochemical study. Hum Pathol. (1989) 20:341–6. doi: 10.1016/0046-8177(89)90043-9
47. Bergman R, Dromi R, Trau H, Cohen I, Lichtig C. The pattern of HMB-45 antibody staining in compound Spitz nevi. Am J Dermatopathol. (1995) 17:542–6. doi: 10.1097/00000372-199512000-00002
48. Rode J, Williams RA, Jarvis LR, Dhillon AP, Jamal O. S100 protein, neurone specific enolase, and nuclear DNA content in Spitz naevus. J Pathol. (1990) 161:41–5. doi: 10.1002/path.1711610108
49. Ribe A, McNutt NS. S100A6 protein expression is different in Spitz nevi and melanomas. Mod Pathol. (2003) 16:505–11. doi: 10.1097/01.MP.0000071128.67149.FD
50. Hilliard NJ, Krahl D, Sellheyer K. p16 expression differentiates between desmoplastic Spitz nevus and desmoplastic melanoma. J Cutan Pathol. (2009) 36:753–9. doi: 10.1111/j.1600-0560.2008.01154.x
51. Al Dhaybi R, Agoumi M, Gagne I, McCuaig C, Powell J, Kokta V. p16 expression: a marker of differentiation between childhood malignant melanomas and Spitz nevi. J Am Acad Dermatol. (2011) 65:357–63. doi: 10.1016/j.jaad.2010.07.031
52. Bergman R, Shemer A, Levy R, Friedman-Birnbaum R, Trau H, Lichtig C. Immunohistochemical study of p53 protein expression in Spitz nevus as compared with other melanocytic lesions. Am J Dermatopathol. (1995) 17:547–50. doi: 10.1097/00000372-199512000-00003
53. Wiesner T, Murali R, Fried I, Cerroni L, Busam K, Kutzner H, et al. A distinct subset of atypical Spitz tumors is characterized by BRAF mutation and loss of BAP1 expression. Am J Surg Pathol. (2012) 36:818–30. doi: 10.1097/PAS.0b013e3182498be5
54. Busam KJ, Sung J, Wiesner T, von Deimling A, Jungbluth A. Combined BRAF(V600E)-positive melanocytic lesions with large epithelioid cells lacking BAP1 expression and conventional nevomelanocytes. Am J Surg Pathol. (2013) 37:193–9. doi: 10.1097/PAS.0b013e318263648c
55. Busam KJ, Wanna M, Wiesner T. Multiple epithelioid Spitz nevi or tumors with loss of BAP1 expression: a clue to a hereditary tumor syndrome. JAMA Dermatol. (2013) 149:335–9. doi: 10.1001/jamadermatol.2013.1529
56. Wiesner T, Kutzner H, Cerroni L, Mihm MCJr, Busam KJ, Murali R. Genomic aberrations in spitzoid melanocytic tumours and their implications for diagnosis, prognosis and therapy. Pathology (2016) 48:113–31. doi: 10.1016/j.pathol.2015.12.007
57. Tetzlaff MT, Reuben A, Billings SD, Prieto VG, Curry JL. Toward a Molecular-Genetic Classification of Spitzoid Neoplasms. Clin Lab Med. (2017) 37:431–48. doi: 10.1016/j.cll.2017.05.003
58. Bastian BC, Wesselmann U, Pinkel D, Leboit PE. Molecular cytogenetic analysis of Spitz nevi shows clear differences to melanoma. J Invest Dermatol. (1999) 113:1065–9. doi: 10.1046/j.1523-1747.1999.00787.x
59. van Engen-van Grunsven AC, van Dijk MC, Ruiter DJ, Klaasen A, Mooi WJ, Blokx WA. HRAS-mutated Spitz tumors: a subtype of Spitz tumors with distinct features. Am J Surg Pathol. (2010) 34:1436–41. doi: 10.1097/PAS.0b013e3181f0a749
60. Massi D, Cesinaro AM, Tomasini C, Paglierani M, Bettelli S, Dal Maso L, et al. Atypical Spitzoid melanocytic tumors: a morphological, mutational, and FISH analysis. J Am Acad Dermatol. (2011) 64:919–35. doi: 10.1016/j.jaad.2010.05.043
61. Harvell JD, Bastian BC, LeBoit PE. Persistent (recurrent) Spitz nevi: a histopathologic, immunohistochemical, and molecular pathologic study of 22 cases. Am J Surg Pathol. (2002) 26:654–61. doi: 10.1097/00000478-200205000-00012
62. Fullen DR, Poynter JN, Lowe L, Su LD, Elder JT, Nair RP, et al. BRAF and NRAS mutations in spitzoid melanocytic lesions. Mod Pathol. (2006) 19:1324–32. doi: 10.1038/modpathol.3800653
63. Busam KJ, Kutzner H, Cerroni L, Wiesner T. Clinical and pathologic findings of Spitz nevi and atypical Spitz tumors with ALK fusions. Am J Surg Pathol. (2014) 38:925–33. doi: 10.1097/PAS.0000000000000187
64. Yeh IA, de la Fouchardiere P D., Mully TW, Garrido MC, Vemula SS, Busam KJ, et al. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am J Surg Pathol. (2015) 39:581–91. doi: 10.1097/PAS.0000000000000387
65. Yeh I, Botton T, Talevich E, Shain AH, Sparatta AJA, de la Fouchardiere MTW, et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun. (2015) 6:7174. doi: 10.1038/ncomms8174
66. Shahbain H, Cooper C, Gerami P. Molecular diagnostics for ambiguous melanocytic tumors. Semin Cutan Med Surg. (2012) 31:274–8. doi: 10.1016/j.sder.2012.09.001
67. Gammon B, Beilfuss B, Guitart J, Gerami P. Enhanced detection of spitzoid melanomas using fluorescence in situ hybridization with 9p21 as an adjunctive probe. Am J Surg Pathol. (2012) 36:81–8. doi: 10.1097/PAS.0b013e31822d5ff8
68. Gerami P, Guitart J, Martini M, Wayne JD, Kuzel T. Cyclin D1 homogeneous staining regions by fluorescent in situ hybridization: a possible indicator of aggressive behavior in melanomas. Arch Dermatol. (2008) 144:1235–6. doi: 10.1001/archderm.144.9.1235-b
69. Ferrara G, De Vanna AC. Fluorescence in situ hybridization for melanoma diagnosis: a review and a reappraisal. Am J Dermatopathol. (2016) 38:253–69. doi: 10.1097/DAD.0000000000000380
70. Gerami P, Scolyer RA, Xu X, Elder DE, Abraham RM, Fullen D, et al. Risk assessment for atypical spitzoid melanocytic neoplasms using FISH to identify chromosomal copy number aberrations. Am J Surg Pathol. (2013) 37:676–84. doi: 10.1097/PAS.0b013e3182753de6
71. Gerami P, Cooper C, Bajaj S, Wagner A, Fullen D, Busam K, et al. Outcomes of atypical spitz tumors with chromosomal copy number aberrations and conventional melanomas in children. Am J Surg Pathol. (2013) 37:1387–94. doi: 10.1097/PAS.0b013e31828fc283
72. Grignol V, Fairchild ET, Zimmerer JM, Lesinski GB, Walker MJ, Magro CM, et al. miR-21 and miR-155 are associated with mitotic activity and lesion depth of borderline melanocytic lesions. Br J Cancer (2011) 105:1023–9. doi: 10.1038/bjc.2011.288
73. Latchana N, Regan K, Howard JH, Aldrink JH, Ranalli MA, Peters SB, et al. Global microRNA profiling for diagnostic appraisal of melanocytic Spitz tumors. J Surg Res. (2016) 205:350–8. doi: 10.1016/j.jss.2016.06.085
74. Latchana N, Del Campo SE, Grignol VP, Clark JR, Albert SP, Zhang J, et al. Classification of Indeterminate Melanocytic Lesions by MicroRNA Profiling. Ann Surg Oncol. (2017) 24:347–54. doi: 10.1245/s10434-016-5476-9
75. Garrido-Ruiz MC, Requena L, Ortiz P, Perez-Gomez B, Alonso SR, Peralto JL. The immunohistochemical profile of Spitz nevi and conventional (non-Spitzoid) melanomas: a baseline study. Mod Pathol. (2010) 23:1215–24. doi: 10.1038/modpathol.2010.102
76. Mason A, Wititsuwannakul J, Klump VR, Lott J, Lazova R. Expression of p16 alone does not differentiate between Spitz nevi and Spitzoid melanoma. J Cutan Pathol. (2012) 39:1062–74. doi: 10.1111/cup.12014
77. Harms PW, Hocker TL, Zhao L, Chan MP, Andea AA, Wang M, et al. Loss of p16 expression and copy number changes of CDKN2A in a spectrum of spitzoid melanocytic lesions. Hum Pathol. (2016) 58:152–60. doi: 10.1016/j.humpath.2016.07.029
78. Bastian BC, LeBoit PE, Hamm H, Brocker EB, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. (1998) 58:2170–5.
79. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature (2002) 417:949–54. doi: 10.1038/nature00766
80. Mihic-Probst D, Perren A, Schmid S, Saremaslani P, Komminoth P, Heitz PU. Absence of BRAF gene mutations differentiates spitz nevi from malignant melanoma. Anticancer Res. (2004) 24:2415–8.
81. Palmedo G, Hantschke M, Rutten A, Mentzel T, Hugel H, Flaig MJ, et al. The T1796A mutation of the BRAF gene is absent in Spitz nevi. J Cutan Pathol. (2004) 31:266–70. doi: 10.1111/j.0303-6987.2003.00179.x
82. Saldanha G, Purnell D, Fletcher A, Potter L, Gillies A, Pringle JH. High BRAF mutation frequency does not characterize all melanocytic tumor types. Int J Cancer (2004) 111:705–10. doi: 10.1002/ijc.20325
83. Indsto JO, Kumar S, Wang L, Crotty KA, Arbuckle SM, Mann GJ. Low prevalence of RAS-RAF-activating mutations in Spitz melanocytic nevi compared with other melanocytic lesions. J Cutan Pathol. (2007) 34:448–55. doi: 10.1111/j.1600-0560.2006.00646.x
84. Da Forno PD, Pringle JH, Fletcher A, Bamford M, Su L, Potter L, et al. BRAF, NRAS and HRAS mutations in spitzoid tumours and their possible pathogenetic significance. Br J Dermatol. (2009) 161:364–72. doi: 10.1111/j.1365-2133.2009.09181.x
85. Emley A, Yang S, Wajapeyee N, Green MR, Mahalingam M. Oncogenic BRAF and the tumor suppressor IGFBP7 in the genesis of atypical spitzoid nevomelanocytic proliferations. J Cutan Pathol. (2010) 37:344–9. doi: 10.1111/j.1600-0560.2009.01433.x
86. Kiuru M, Jungbluth A, Kutzner H, Wiesner T, Busam KJ. Spitz tumors: comparison of histological features in relationship to immunohistochemical staining for ALK and NTRK1. Int J Surg Pathol. (2016) 24:200–6. doi: 10.1177/1066896916630375
87. Farah M, Nagarajan P, Curry JL, Tang Z, Kim TB, Aung PP, et al. Spitzoid melanoma with histopathologic features of ALK gene rearrangement exhibiting ALK copy number gain: a novel mechanism of ALK activation in spitzoid neoplasia. Br J Dermatol. (2018). doi: 10.1111/bjd.16881. [Epub ahead of print].
88. Kato M, Takahashi M, Akhand AA, Liu W, Dai Y, Shimizu S, et al. Transgenic mouse model for skin malignant melanoma. Oncogene (1998) 17:1885–8. doi: 10.1038/sj.onc.1202077
89. Bastian BC, Olshen AB, LeBoit PE, Pinkel D. Classifying melanocytic tumors based on DNA copy number changes. Am J Pathol. (2003) 163:1765–70. doi: 10.1016/S0002-9440(10)63536-5
90. Jiveskog S, Ragnarsson-Olding B, Platz A, Ringborg U. N-ras mutations are common in melanomas from sun-exposed skin of humans but rare in mucosal membranes or unexposed skin. J Invest Dermatol. (1998) 111:757–61. doi: 10.1046/j.1523-1747.1998.00376.x
91. Morey AL, Murali R, McCarthy SW, Mann GJ, Scolyer RA. Diagnosis of cutaneous melanocytic tumours by four-colour fluorescence in situ hybridisation. Pathology (2009) 41:383–7. doi: 10.1080/00313020902915875
92. Gerami P, Zembowicz A. Update on fluorescence in situ hybridization in melanoma: state of the art. Arch Pathol Lab Med. (2011) 135:830–7. doi: 10.1043/2011-0048-RAIR.1
93. Gerami P, Jewell SS, Morrison LE, Blondin B, Schulz J, Ruffalo TP, et al. Fluorescence in situ hybridization (FISH) as an ancillary diagnostic tool in the diagnosis of melanoma. Am J Surg Pathol. (2009) 33:1146–56. doi: 10.1097/PAS.0b013e3181a1ef36
94. Isaac AK, Lertsburapa T, Pathria Mundi J, Martini M, Guitart J, Gerami P. Polyploidy in spitz nevi: a not uncommon karyotypic abnormality identifiable by fluorescence in situ hybridization. Am J Dermatopathol. (2010) 32:144–8. doi: 10.1097/DAD.0b013e3181b72d6f
95. McCalmont TH. Fillet of fish. J Cutan Pathol. (2011) 38:327–8. doi: 10.1111/j.1600-0560.2011.01675.x
96. Finnegan EF, Pasquinelli AE. MicroRNA biogenesis: regulating the regulators. Crit Rev Biochem Mol Biol. (2013) 48:51–68. doi: 10.3109/10409238.2012.738643
97. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer (2006) 6:857–66. doi: 10.1038/nrc1997
98. Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer (2006) 6:259–69. doi: 10.1038/nrc1840
99. Hillen LM, Geybels MS, Rennspiess D, Spassova I, Ritter C, Becker JC, et al. Molecular profiling of Spitz nevi identified by digital RNA counting. Melanoma Res. (2018) 28:510–20. doi: 10.1097/CMR.0000000000000495
100. Jansen B, Hansen D, Moy R, Hanhan M, Yao Z. Gene expression analysis differentiates melanomas from Spitz Nevi. J Drugs Dermatol. (2018) 17:574–6. doi: 10.1016/j.jaad.2017.04.559
101. Hardesty WM, Kelley MC, Mi D, Low RL, Caprioli RM. Protein signatures for survival and recurrence in metastatic melanoma. J Proteomics (2011) 74:1002–14. doi: 10.1016/j.jprot.2011.04.013
102. Lazova R, Seeley EH, Kutzner H, Scolyer RA, Scott G, Cerroni L, et al. Imaging mass spectrometry assists in the classification of diagnostically challenging atypical Spitzoid neoplasms. J Am Acad Dermatol. (2016) 75:1176–86.e1174. doi: 10.1016/j.jaad.2016.07.007
103. Alomari AK, Klump V, Neumeister V, Ariyan S, Narayan D, Lazova R. Comparison of the expression of vimentin and actin in Spitz nevi and Spitzoid malignant melanomas. Am J Dermatopathol. (2015) 37:46–51. doi: 10.1097/DAD.0000000000000147
104. Lallas A, Kyrgidis A, Ferrara G, Kittler H, Apalla Z, Castagnetti F, et al. Atypical Spitz tumours and sentinel lymph node biopsy: a systematic review. Lancet Oncol. (2014) 15:e178–183. doi: 10.1016/S1470-2045(13)70608-9
105. Uguen A, Talagas M, Costa S, Duigou S, Bouvier S, De Braekeleer M, et al. A p16-Ki-67-HMB45 immunohistochemistry scoring system as an ancillary diagnostic tool in the diagnosis of melanoma. Diagn Pathol. (2015) 10:195. doi: 10.1186/s13000-015-0431-9
106. Mones JM, Ackerman AB. Atypical Spitz's nevus, malignant Spitz's nevus, and metastasizing Spitz's nevus: a critique in historical perspective of three concepts flawed fatally. Am J Dermatopathol. (2004) 26:310–33. doi: 10.1097/00000372-200408000-00008
107. Ludgate MW, Fullen DR, Lee J, Lowe L, Bradford C, Geiger J, et al. The atypical Spitz tumor of uncertain biologic potential: a series of 67 patients from a single institution. Cancer (2009) 115:631–41. doi: 10.1002/cncr.24047
108. Ghazi B, Carlson GW, Murray DR, Gow KW, Page A, Durham M, et al. Utility of lymph node assessment for atypical spitzoid melanocytic neoplasms. Ann Surg Oncol. (2010) 17:2471–5. doi: 10.1245/s10434-010-1022-3
109. Hung T, Piris A, Lobo A, Mihm MCJr, Sober AJ, Tsao H, et al. Sentinel lymph node metastasis is not predictive of poor outcome in patients with problematic spitzoid melanocytic tumors. Hum Pathol. (2013) 44:87–94. doi: 10.1016/j.humpath.2012.04.019
110. Zhao G, Lee KC, Peacock S, Reisch LM, Knezevich SR, Elder DE, et al. The utilization of spitz-related nomenclature in the histological interpretation of cutaneous melanocytic lesions by practicing pathologists: results from the M-Path study. J Cutan Pathol. (2017) 44:5–14. doi: 10.1111/cup.12826
111. Paniago-Pereira C, Maize JC, Ackerman AB. Nevus of large spindle and/or epithelioid cells (Spitz's nevus). Arch Dermatol. (1978) 114:1811–23. doi: 10.1001/archderm.1978.01640240053018
112. Cesinaro AM, Foroni M, Sighinolfi P, Migaldi M, Trentini GP. Spitz nevus is relatively frequent in adults: a clinico-pathologic study of 247 cases related to patient's age. Am J Dermatopathol. (2005) 27:469–75. doi: 10.1097/01.dad.0000185249.21805.d3
113. Verardino GC, Rochael MC. Spitz nevi in the classic histopathological pattern–lamb in wolf‘s clothing. An Bras Dermatol. (2015) 90:91–5. doi: 10.1590/abd1806-4841.20153310
114. Mourmouras V, Fimiani M, Rubegni P, Epistolato MC, Malagnino V, Cardone C, et al. Evaluation of tumour-infiltrating CD4+CD25+FOXP3+ regulatory T cells in human cutaneous benign and atypical naevi, melanomas and melanoma metastases. Br J Dermatol. (2007) 157:531–9. doi: 10.1111/j.1365-2133.2007.08057.x
115. Lazova R, Seeley EH, Keenan M, Gueorguieva R, Caprioli RM. Imaging mass spectrometry–a new and promising method to differentiate Spitz nevi from Spitzoid malignant melanomas. Am J Dermatopathol. (2012) 34:82–90. doi: 10.1097/DAD.0b013e31823df1e2
116. Lazova R, Yang Z, El Habr C, Lim Y, Choate KA, Seeley EH, et al. Mass Spectrometry Imaging Can Distinguish on a Proteomic Level Between Proliferative Nodules Within a Benign Congenital Nevus and Malignant Melanoma. Am J Dermatopathol. (2017) 39:689–95. doi: 10.1097/DAD.0000000000000849
117. Cerroni L, Barnhill R, Elder D, Gottlieb G, Heenan P, Kutzner H, et al. Melanocytic tumors of uncertain malignant potential: results of a tutorial held at the XXIX Symposium of the International Society of Dermatopathology in Graz, October 2008. Am J Surg Pathol. (2010) 34:314–26. doi: 10.1097/PAS.0b013e3181cf7fa0
118. Gerami P, Busam K, Cochran A, Cook MG, Duncan LM, Elder DE, et al. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol. (2014) 38:934–40. doi: 10.1097/PAS.0000000000000198
119. Cho-Vega JH. A diagnostic algorithm for atypical spitzoid tumors: guidelines for immunohistochemical and molecular assessment. Mod Pathol. (2016) 29:656–70. doi: 10.1038/modpathol.2016.70
120. McCormack CJ, Conyers RK, Scolyer RA, Kirkwood J, Speakman D, Wong N, et al. Atypical Spitzoid neoplasms: a review of potential markers of biological behavior including sentinel node biopsy. Melanoma Res. (2014) 24:437–47. doi: 10.1097/CMR.0000000000000084
Keywords: spitzoid neoplasms, spitz nevi, malignant spitz tumor, atypical spitz tumor, spitz melanoma, molecular, genetic, patient management
Citation: Hillen LM, Van den Oord J, Geybels MS, Becker JC, zur Hausen A and Winnepenninckx V (2018) Genomic Landscape of Spitzoid Neoplasms Impacting Patient Management. Front. Med. 5:344. doi: 10.3389/fmed.2018.00344
Received: 18 September 2018; Accepted: 23 November 2018;
Published: 13 December 2018.
Edited by:
H. Peter Soyer, The University of Queensland, AustraliaReviewed by:
Gerardo Ferrara, Ospedale Generale Provinciale Macerata, ItalyMitchell Stark, The University of Queensland, Australia
Copyright © 2018 Hillen, Van den Oord, Geybels, Becker, zur Hausen and Winnepenninckx. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa M. Hillen, bGlzYS5oaWxsZW5AbXVtYy5ubA==
Véronique Winnepenninckx, di53aW5uZXBlbm5pbmNreEBtdW1jLm5s