 Andrew J. Doyle
Andrew J. Doyle Beverley J. Hunt
Beverley J. Hunt- Thrombosis and Haemophilia Centre, Guy's and St Thomas' NHS Foundation Trust, London, United Kingdom
Extracorporeal membrane oxygenators are used in critical care for the management of severe respiratory and cardiac failure. Activation of the coagulation system is initiated by the exposure of blood to synthetic surfaces and the shear stresses of the circuit, especially from device pumps. Initial fibrinogen deposition and subsequent activation of coagulation factors and complement allow platelets and leucocytes to adhere to oxygenator surfaces and enhance thrombin generation. These changes and others contribute to higher rates of thrombosis seen in these patients. In addition, bleeding rates are also high. Primary haemostasis is impaired by platelet dysfunction and loss of their key adhesive molecules and shear stress causes an acquired von Willebrand defect. In addition, there is also altered fibrinolysis and lastly, administration of systemic anticoagulation is required to maintain circuit patency. Further research is required to fulyl establish the complexities of the haemostatic changes with these devices, and to elucidate the mechanistic changes that are mainly responsible so that plans can be made to reduce their complications and improve management.
Background
Non-biological artificial materials, or biomaterials, are used increasingly to carry blood in medical devices, and much work has gone in to improving morbidity and mortality associated with their use. Extracorporeal circulation is used to manage patients with severe organ dysfunction. Several devices are routinely used with extracorporeal surfaces—extracorporeal membrane oxygenation (ECMO), haemodialysis, left ventricular assist devices (LVAD), and cardiopulmonary bypass (CPB). In all these devices blood comes into constant contact with blood leading to activation of the coagulation system, platelets, leucocytes and complement, which is then returned to the patient. This subsequently may lead to thrombosis, bleeding and device failure, which remain frequent despite increasing knowledge of their pathogenesis. Further understanding of their haemostatic changes will allow the development of improved function and lifespans of the devices and reduce the development of systemic complications for patients.
ECMO is currently used to provide gas exchange in severe respiratory failure with veno-venous (VV) circuits and increasingly with concurrent cardiac failure via a veno-arterial (VA) circuit whilst waiting for organ recovery to occur. The circuit consists of a minimum of two large bore central cannulae which are connected to a membrane oxygenator device, which has a large biomaterial surface area coming into contact with blood, and a pump. The pump can be centrifugal or pulsatile in nature and rates vary due to the type of circuit used causing damage to cells and proteins passing through it. CPB has a similar circuit design to ECMO although with additional reservoirs used for giving therapeutic cardioplegia and return of autologous blood. VAD is a pump with an external extracardiac connection between the ventricle and aorta or pulmonary artery to bypass those with end-stage heart failure. Similar to ECMO and CPB, they have high shear stress but significantly less surface area and have different modes of pulsatility through the pump.
At present, ECMO is associated with significant haemostatic challenges with clinical thrombosis and hemorrhage rates higher than the general critical care population. There is an added complication of circuit loss due to intra-device thrombosis particularly around the oxygenator membrane (Table 1). These problems are superimposed on other haemostatic changes in the patient due to their underlying disease state and treatment which may include the haemostatic effects of renal and hepatic failure, and a procoagulant acute phase response in those with infective and inflammatory illnesses and disseminated intravascular coagulation (DIC). The presence of DIC in patients preceding ECMO has a signficantly increased risk of death (1) (Figure 1). Rates of reported ECMO-associated venous thromboembolism (VTE) range from 18 to 85% in different centers and may be at least partly dependent on anticoagulation regimens (2, 3). Oxygenator thrombosis can occur in around 10–16% of patients depending on the circuit type and age of the patient (4, 5). Severe hemorrhage is reported in nearly 40% and intracranial hemorrhage in 16–21% of patients (6–8). Haemostatic complications are associated with poor patient survival outcomes (4).

Table 1. Clinical manifestations caused by the activation of coagulation and blood components with ECMO and causative changes.
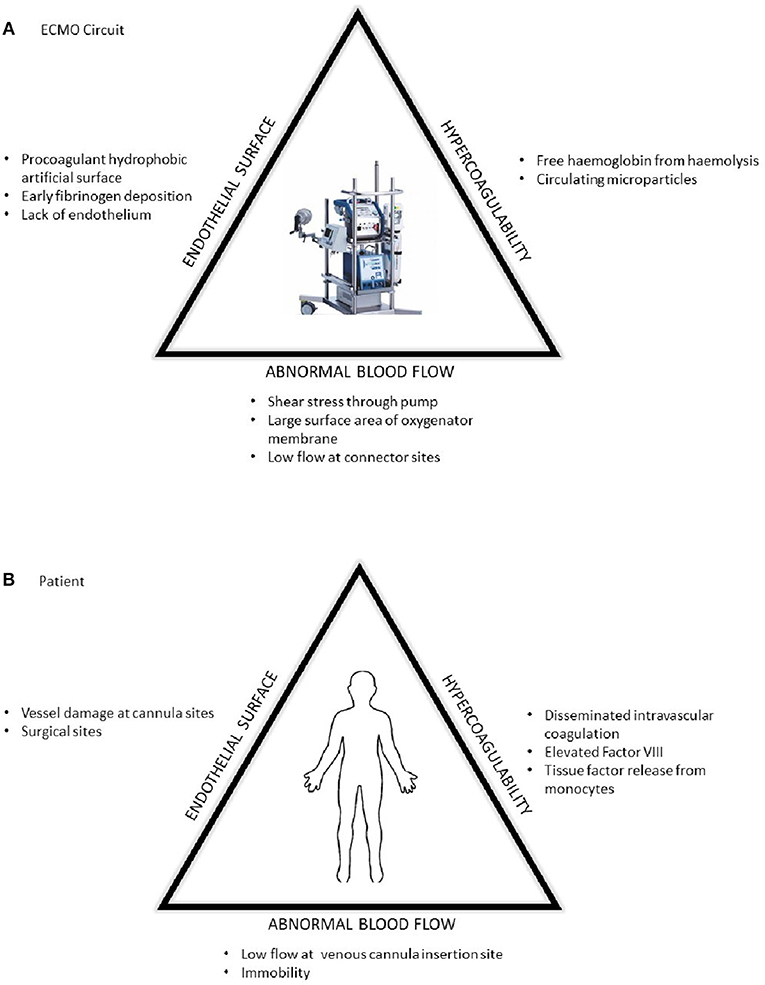
Figure 1. The prothrombotic changes described by Virchow's triad in respect to (A) the ECMO circuit and (B) patient factors.
Extracorporeal surfaces can consist of a variety of manufactured materials including polyethylene, polypropylene and silicone which are designed to withstand the pressures and demands of the associated devices. However, they not haemostatically inert leading to subsequent haemostatic disturbances. There are two approaches to managing this—reducing the surfaces ability to activate haemostasis and using systemic inhibitors of coagulation such as anticoagulation and antiplatelet agents. Today, most ECMO and CPB circuits are heparin-coated which have been shown to reduce platelet, leucocytes and coagulation activation (9).
Modern anticoagulation is principally achieved using an unfractionated heparin infusion for ECMO and CPB although other intravenous agents such as bivalirudin and argatroban are increasingly popular. They must can be considered in those with heparin-induced thrombocytopenia, heparin resistance or allergy but are also used outside of these indications (10, 11). At present, there is wide variation in practice, and no consensus on the administration and monitoring of anticoagulation during ECMO and the management of ECMO-related hemorrhage and VTE (12). The aim of current anticoagulation is to reduce thrombin generation however it also increases the risk of hemorrhage. The ideal therapeutic agent would reduce the thrombotic risk without increasing the rate of bleeding, but this is not possible with current anticoagulants.
ECMO and CPB have similarities in their main function and have a similar exposure to biosurfaces and shear stress enabling extrapolation of data between the two devices. However, there are several differences between them that one must be aware of prior to inferring results. These include the duration of ECMO lasting for day to weeks as opposed to minutes and hours for CPB. Also, the level of anticoagulation is higher during CPB than ECMO with addition of protamine reversal in the former. Furthermore, patients undergoing CPB have therapeutic hypothermia and haemodilution with a large number of surgical sites. The clinical phenotype of the patients undergoing ECMO are those with acute critical illness usually with a generalized inflammatory response and associated coagulopathy whereas most patients have to be fit for anesthesia for CPB but have chronic severe cardiac disease. Patients with VAD have end-stage cardiac failure and are used as a bridge to transplant or to allow for myocardial recovery. They have significantly reduced surface area in comparison to CPB and ECMO for coagulation activation, but the use of a pump still causes shear stress to molecules and cells that pass through them.
We aim to explore the present data suggesting how the haemostatic system becomes activated with ECMO from the current research that is available in comparison to CPB and VAD. The implications that these have upon the risk of thrombosis and hemorrhage in these patients and the impact on their management will be discussed.
Initial Interactions Between Coagulation Factor and Extracorporeal Biosurfaces
The interaction between biomaterial surfaces and blood has been extensively described by Vroman et al. who reviewed the interaction principally between glass surfaces and protein adsorption over 30 years ago (13). The subsequent “Vroman effect” has been described as the sequential adsorption of proteins to a biomaterial surface. Initial immediate adsorption of fibrinogen occurs to the surfaces within minutes of exposure (13). Next, a variety of proteins then bind to the fibrinogen nanosurface (Figure 2). These include the coagulation factors of the contact activation pathway—high molecular weight kininogen (HMWK) and factor XII–as well as high-density lipoprotein (HDL), albumin, immunoglobin G (IgG), and complement component C3 (14). It has been shown by Passmore et al. that fibrinogen levels fall in an ovine lung injury model with the commencement of the ECMO circuit and recover to the baseline level within 48 h in keeping with its adhesion to the biosurface and loss from circulation (15). We have also observed this acquired hypofibrinogenaemia in pediatrics following CPB with increased hemorrhage and reduced clot strength (16).
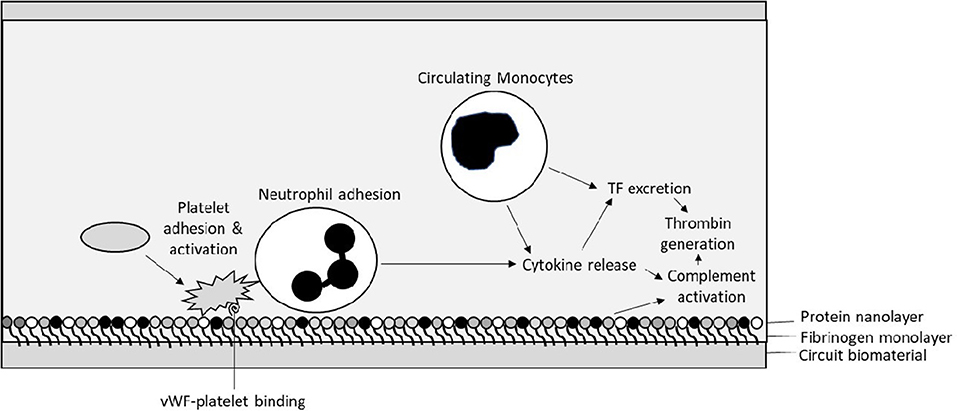
Figure 2. Initial interactions between circuit biosurface, haemostatic factors, and components of blood—Tissue Factor (TF), von Willebrand factor (vWF).
After the formation of a protein-based layer on the surface the ECMO circuit cellular adhesion occurs. Platelets and polymorphonuclear leucocytes (PMN) interact with each other on the protein-covered biomaterial surface. Interaction with the complement system also contributes to their activation. This results in propagating further adhesion and PMN release of cytokines including interleukins-1β, -6, and -8 and tumor necrosis factor-α, potentially contributing further to a pro-inflammatory state (17).
Properties of Biomaterials and Their Interaction With Blood Components
Different components of the ECMO circuit are designed to have varying properties depending on their use such as tubing, oxygenator membranes, and connectors. The oxygenator membranes are of importance as they have a large surface in contact with blood. They are made of materials such as polymethyl pentene and polypropylene, which are hydrophobic. This allows gas exchange to occur but stops blood from passing through the membrane pores. This results in a prothrombotic tendency due to the adsorption of the unwanted proteins, known as protein fouling, which then undergo further conformational changes that also make them more thrombogenic. The nature of protein adsorption depends on the biomaterial surface and alters further protein-cellular interactions and cellular adhesion. For example, HDL adsorption is higher to hydrophobic surfaces with reduced platelet adhesion but surfaces with higher fibrinogen adsorption have more platelet adhesion (14, 18).
To reduce protein fouling and the prothrombotic nature of the ECMO surfaces, small molecules can be incorporated in to the biosurface coating. Initially, molecules such as albumin and silicone have been used to act as a barrier to coagulation factors and complement but still cause a similar degree of thrombocytopenia (19). Further development led to heparin-coating, which is widely used in commercially-available ECMO circuits, providing an additional localized anticoagulant effect. Heparin-coating decreases thrombin generation by binding to circulating antithrombin and demonstrates less platelet and leucocyte activation (20–22). The development of this approach has reduced thrombosis and filter losses although they still occur (23). Currently available ECMO circuits with different coatings do not show any significant difference in haemostatic parameters indicating there is little evidence of heparin leaching off the surfaces (24).
Current developments to further reduce protein fouling include the coating of high density hydrophilic molecules, which bind high amounts of water, and to include polymers with branch chains to stop conformational changes to the adsorbed proteins, known as polymer brushes (25, 26). Obstals et al. have described the development of an polymer brush coated surface for ECMO, which significantly decreased coagulation, platelet, and leucocyte activation ex vivo (27). At present, these are not used clinically due to difficulties in the polymerisation process.
Activation of the Coagulation Cascade in Extracorporeal Circulation
The cell-based model of coagulation suggests that it is initiated by the exposure of subendothelial tissue factor (TF) binding and activating circulating FVII which then through the coagulation cascade causes downstream thrombin generation. In disseminated intravascular coagulation due to sepsis further TF expression on monocytes and macrophages occurs and needs to be considered in patients receiving ECMO as sepsis is a common accompanying problem. The cell-based model does not include the intrinsic “contact activation” pathway coagulation factors (FXI, HMWK, prekalikrein, and FXII) which in vitro react with negatively charged artificial surfaces to cause thrombus formation. There is increasing interest in these coagulation factors and their role in the activation of coagulation by artificial surfaces.
There is a lack of data regarding the extent of TF-FVII as compared to contact activation mediated thrombin generation in ECMO. However, it has been demonstrated in CPB circuits and with synthetic surfaces that there is an increased in monocyte-related TF expression over several hours with an associated increase in procoagulability in vitro (28, 29). This is probably due to yet another mechanism—the adherence and subsequent activation of PMNs of the biomaterial surface with an associated fall in these circulating cells over this period. TF-mediated monocytic procoagulability has additionally been shown to be reduced by the heparin-coating of biomaterials which is now routinely used in most ECMO circuits (30).
Although the contact pathway coagulation factors do not play a principal role in haemostasis as a result of vessel injury, conflicting data indicates that thrombosis with biomaterials is related to Factor XII-mediated activation of coagulation (31, 32). In patients undergoing CPB, there is evidence of reduced circulating FXII, increased FXIIa and prekalikrein levels in association with increasing circulating FXII complexes although this was not replicated by others (33, 34). Plötz et al. showed in 10 neonates receiving ECMO that there was activation of the contact pathway with elevated FXII-C1 esterase inhibitor complexes, decreased kallikrein inhibitory capacity, and increased thrombin-antithrombin complexes (35).
Larsson et al. have described the use of a monoclonal anti-factor XIIa antibody, 3F7, that binds to the active site of FXII reducing its enzymatic activity. 3F7 has been used in an animal model with an ECMO circuit showing a significant reduction in arterial and venous thrombus formation without increasing the rate of bleeding (36). Similarly, May et al. have demonstrated that the use of a FXII inhibitor, rHA-infestin-4, reduced the rate of thrombosis formation similar to that of heparin in animal models using AV-shunts but as expected, it did not impact upon bleeding times that were similar to those without anticoagulation (37). This seems an attractive option for the anticoagulation of ECMO circuits however this has not yet been reproduced in humans in a similar setting (32).
There is minimal data studying other coagulation factors during ECMO. Factor VIII is an acute phase molecule that is significantly elevated in pro-inflammatory states. Passmore et al. showed that FVIII falls in both a lung-injury model and control-animal models on ECMO circuits than those without after 24 h by over 50% (15). This was concurrently noted to be with lower fibrinogen and von Willebrand factor (VWF) levels suggestive of increased consumption of FVIII or decreased VWF-binding ability. This fall of FVIII has not been seen in CPB although decreased level of factor X and prothrombin by 40–50% over 24 h have been shown with a proportional fall in thrombin generation (38). Kreyer et al. showed an association with lower FVII and FX levels in patients receiving ECMO who developed severe bleeding than those who did not (7).
There can be variations in analytical interpretation in the coagulation assays in these groups depending upon the biosurfaces, the anticoagulation regimen used, and the methods used for assessing coagulation. Certainly, increased activated circulating coagulation factors, increased fibrin degradation product, and detectable levels of circulating factor-inhibitor complexes are found but as there is no coherent explanation for the results, more research is needed.
Activation of the Complement System in Extracorporeal Circulation
The complement system is activated by three initiating pathways dependent on the recognition of non-self-antigens: the classical (CP), lectin (LP), and alternative (AP) pathways. The CP and LP are initiated by antibody-antigen complexes and certain carbohydrates, respectively, which can be present in patients receiving ECMO who may have an underlying infectious process. The AP can be activated by the biomaterial surface directly with also a degree of self-activation via the amplification loop. Both immunoglobulin and C3b can bind to the fibrinogen monolayer formed on the surface of the biomaterial leading to activation of the complement activation pathway. These then ultimately trigger the common pathway with the activation of C3 leading to the formation of the membrane attack complex (MAC). As artificial surfaces, unlike the endothelium have no regulatory molecules for supressing the complement system, they lead to the propagation and positive feedback of the complement cascade leading to an excessive inflammatory response and capillary leak syndrome which has been demonstrated as a complication of extracorporeal circuits (17, 39).
Vallhonrat et al. showed a rapid increase in the complement degradation products, C4d, Bb, iC3b and SC5-9b, 1 h after commencing ECMO in two adult patients (40). There was a predominant increase in Bb formation suggestive of complement activation by the AP. These markers subsequently fell within the next few hours and normalized 2 days later in keeping with a significant complement activation from initial exposure to the ECMO circuit. Similar results have been replicated in vivo with neonates and in vitro, although with a prolonged elevated of SC5-9b levels, and in patients on CPB and haemodialysis (41–44). Heparin has an anticomplement effect, so unsurprisingly heparin-bound surfaces in ECMO circuits have shown complement activation is reduced with maintenance of the complement regulatory proteins, Factor H & C1 Inhibitor (39).
The complement degradation products, anaphylatoxins, can either enter the liquid phase causing systemic inflammatory problems for patients or adhere to the ECMO surfaces leading to localized cellular activation principally of the innate immune system. The deposition of anaphylatoxins on the biomaterial surface can then have various implications on haemostasis. iC3b, a degradation product of C3b, can bind to a biosurfaces, and activate a specific granulocyte receptor, CR3, causing adhesion of principally neutrophils and to lesser extent, of monocytes. These then subsequently become activated (45). In keeping with this, it has been demonstrated that there is extensive PMN deposition over the surface of ECMO circuits that had been used in patients with evidence of cellular activation (46).
Complement cleavage products and anaphylatoxins that enter the systemic circulation have multiple interactions with various components of the coagulation system in particular FXI and FXII. They have been shown to activate platelets, increase TF expression, activate of endothelial cells increasing VWF release and enhancie the exposure of P-selectin on both platelets and activated endothelial cells (47, 48). It has also been shown that leucocyte activation occurs within hours of exposure to an ECMO circuit demonstrated by elevation of CD18, L-selectin, and neutrophil-derived elastase which can subsequently result in capillary leakage and pulmonary oedema (29, 49, 50).
Activation of Platelets and Von Willebrand Factor in Extracorporal Circulation
As suggested previously, platelets adhere to the protein-coated monolayer of the biosurface of the ECMO circuit and react with other activated components of the coagulation and complement system systemically. It has been shown that thrombocytopenia is common in patients receiving ECMO although this is not related to its duration (1, 51). Severe thrombocytopenia is associated with multi-organ failure and preceding thrombocytopenia. 22% of patients receiving ECMO can have a severe thrombocytopenia of < 50 × 109/L making adequate anticoagulation difficult and increasing the use of transfusion requirements (51).
Exposure to elevated shear flow from the ECMO circuit has been demonstrated to cause platelet receptor shedding of the key platelet adhesion glycoproteins, GPIbα, and GPVI and an associated loss of high molecular weight VWF multimers (HMWM) (52). The binding of the platelet GP1b-GVI complex and VWF is the main process of platelet adhesion to a fibrin-covered surface. This persists despite platelet transfusions and throughout the period of ECMO use. Subsequently, there are lower levels of platelet aggregation shown by light aggregometry using various agonists including ADP, ristocetin, collagen and epinephrine (52–56). This may contribute to the increased levels of bleeding seen in these patient in addition to the use of anticoagulation and antiplatelet agents. Flow cytometry of platelets in those receiving ECMO showed severely reduced expression of membrane-bound p-selectin and CD63, which modulates platelet spreading (56).
Despite having reduced aggregation and expression of the key platelet adhesion and structural molecules, Cheung et al. showed that there is a time-dependent platelet activation as measured by increased circulating matrix metalloproteinase (MMP)-2 and soluble p-selectin levels. This was not associated with significant activation of the endothelium (53). It suggests that platelet adhesion and aggregation is defective in those receiving ECMO but platelet activation occurs with the release of their granules.
In addition to platelet receptor shedding, the ECMO circuit can potentially induce the formation of thrombi and platelet microparticles. Although this has not been shown in ECMO, Danwanjee et al. have demonstrated in animal models that CPB can contribute to the formation of microthrombi in the liquid phase and deposit in various organ shown at biopsy with indium radio-labeling. No significant macroscopic thrombi nor significant occlusive material within the bypass apparatus were shown (57). These microthrombi are likely to form around the biosurface then subsequently embolise systemically. It has been shown the microthrombi may cause subsequent neurological sequelae and memory impairment in those who have received CPB (58).
Circulating platelet microparticles (PMP) have been demonstrated to be increased in extracorporeal circulation (59). PMP are small cell-derived particles typically 0.1–1 micrometres in size that are produced from activated platelets in situations of shear stress (60). They can act as a prothrombotic surface and due to their size have been shown to be increased systemically at the time of extracorporeal circulation (57). It is not clear whether PMP are important in the pathogenesis of ECMO coagulopathy and at present there is no information if they are correlated to a prothrombotic phenotype.
vWF activity decreases with time on ECMO (61–63). This is due to the loss of high molecular weight vWF multimers (HMWM) due to the effect of shear stresses in the circuit disrupting vWF molecules in the circuit and resolves with cessation. This is also seen with VAD (56, 64). Acquired von Willebrand's syndrome can be demonstrated serologically by a significantly reduced ristocetin cofactor activity and collagen-binding ability as well as a loss of HMWM. This has been correlated to an increased in bleeding typically from the respiratory tract, mucosal surfaces and puncture sites (56). Vincent et al. have shown that devices with a high shear have increased levels of proteolysis without the presence of endothelium in an in vitro circuit. However, in the presence of devices with high pulsality, this stimulates the release of VWF from the recipient's endothelium (65). This is may be a reason why higher bleeding rates with centrifugal pumps are seen in comparison to roller pumps in patients requiring ECMO (26.1 vs. 9.0 events/1000 treatment days, respectively) (66). Kalbhenn et al. have demonstrated after the cessation of ECMO that VWF parameters largely normalize after a few hours and completely within 24 h (55).
Activation of the Fibrinolytic System in Extracorporeal Circulation
Fibrinolysis is essential for haemostatic regulation with the degradation of fibrin complexes once vessel injury has resolved. Hyperfibrinolysis is an overwhelming activation of this system leading to a bleeding phenotype which can be seen in trauma-induced coagulopathy, during liver transplantation and in some cancer patients. The release of tissue plasminogen activator (t-PA) can be stimulated by a number of factors including hypoxia, thrombin, histamine, and vasopressin. t-PA subsequently activates plasminogen to plasmin causing fibrin (and fibrinogen) breakdown.
The role of the fibrinolytic system in ECMO is not well described. McVeen et al. showed that there an initial significant fall in tPA and PAI-1 levels with a subsequent rise in both after several hours of ECMO then normalization over the next few days (67). These elevated fibrinolytic enzymes were also replicated in babies on ECMO >30 days of age with significant bleeding (68). Increases of tPA and D-dimer occur in patients undergoing CPB, with reduced bleeding, and clinical outcomes improved by using antifibrinolytic agents (69, 70).
Effect of Haemolysis on Haemostasis in Extracorporeal Circulation
Red blood cell (RBC) breakdown (haemolysis) is routinely seen with ECMO circuits with 67% of patients showing increased levels of free circulating haemoglobulin (71). This occurs with higher flow rates of typically >147 ml/kg/min. The occurrence of significant haemolysis in ECMO is associated with a higher use of blood products. Free plasma hemoglobin levels of >50 mg/dL after 24 h have been shown to an independent predictor for mortality in ECMO (72).
Haemolysis causes prothrombotic changes as has been shown in other haemolytic conditions including sickle cell disease and autoimmune haemolytic anemia which are also associated with the presence of free hemoglobin and heme. Free hemoglobin is known to bind to nitric oxide further upsetting the haemostatic mileau by removing NO and therefore causing vasoconstriction and increasing platelet reactivity (73). Meyer et al. showed that free hemoglobin increased platelet adhesion to fibrinogen and collagen-coated biosurfaces with vWF. They felt that this was a result of increased binding strength between the vWF A1-domain and the platelet GP1bα receptor (74). Although there is a reduced level of multimeric vWF with ECMO as discussed previously, there are maintained levels of other multimer sizes that may contribute to platelet adhesion to biosurfaces.
In addition, RBC-derived microparticles can be formed as a result of ECMO-mediated haemolysis or from transfused blood products. RBC microparticles can increase thrombin generation as a result of phosphatidylserine exposure (75).
Conclusions
It is apparent that the coagulopathy associated with ECMO is complex and is not fully elucidated. As we have discussed, it is clear that there are multiple interactions with varying defects in different haemostatic mechanisms, which ultimately lead to an increased risk of thrombosis and hemorrhage. Despite developments in the biomaterials with varying molecules embedded into the surface, there is still significant activation of haemostasis and the shear stresses of circuit pump will continue to contribute to the damage of blood-borne cells and large proteins.
Current literature suggests that an acquired von Willebrand syndrome, platelet dysfunction and activation of fibrinolysis are likely to be contributing to the increased hemorrhage rates seen in ECMO in addition to therapeutic anticoagulation. In parallel activation of both the extrinsic and contact-pathway coagulation factors, complement and circulating free hemoglobin promote thrombosis and thus circuit failure and systemic thromboembolism. It is still unclear at what time points in the use of the ECMO circuit that these changes have most clinical effect, and which contribute most significantly to its pathophysiology.
At present, much of what is known about ECMO is extrapolated from other devices in particular CPB but is not fully translatable. Further investigation is need on the haemostatic changes in ECMO to reflect what is known about other extracorporeal circuits. This needs to be applied to patient outcomes to identify patients at risk of thrombosis or bleeding, to guide targeted monitoring and management and develop alternative anticoagulation therapies. With these insights, it will then be possible to improve survival and decrease the morbidity associated with extracorporeal circuits.
Author Contributions
AD performed the relevant literature search and wrote the review article. BH reviewed the article for submission and provided additional points for discussion.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kim HS, Cheon DY, Ha SO, Han SJ, Kim HS, Lee SH, et al. Early changes in coagulation profiles and lactate levels in patients with septic shock undergoing extracorporeal membrane oxygenation. (2018) 10:1418–30. doi: 10.21037/jtd.2018.02.28
2. Cooper E, Burns J, Retter A, Salt G, Camporota L, Meadows CI, et al. Prevalence of venous thrombosis following venovenous extracorporeal membrane oxygenation in patients with severe respiratory failure. Crit Care Med. (2015) 43:e581–4. doi: 10.1097/CCM.0000000000001277
3. Menaker J, Tabatabai A, Rector R, Dolly K, Kufera J, Lee E, et al. Incidence of cannula-associated deep vein thrombosis after veno-venous extracorporeal membrane oxygenation. ASAIO J. (2017) 63:588–91. doi: 10.1097/MAT.0000000000000539
4. Paden ML, Conrad SA, Rycus PT, Thiagarajan RR. Extracorporeal life support organization registry report 2012. ASAIO J. (2013) 59:202–10. doi: 10.1097/MAT.0b013e3182904a52
5. Lubnow M, Philipp A, Foltan M, Bull Enger T, Lunz D, Bein T. No titletechnical complications during venovenous extracorporeal membrane oxygenation and their relevance predicting a system-exchange – retrospective analysis of 265 cases. PLoS ONE (2014) 9:1–22. doi: 10.1371/journal.pone.0112316
6. Fletcher Sandersjöö A, Bartek J, Thelin EP, Eriksson A, Elmi-Terander A, Broman M, et al. Predictors of intracranial hemorrhage in adult patients on extracorporeal membrane oxygenation: an observational cohort study. J Intensive Care (2017) 5:27. doi: 10.1186/s40560-017-0223-2
7. Kreyer S, Muders T, Theuerkauf N, Spitzhüttl J, Schellhaas T, Schewe JC, et al. Hemorrhage under veno-venous extracorporeal membrane oxygenation in acute respiratory distress syndrome patients: a retrospective data analysis. J Thorac Dis. (2017) 9:5017–29. doi: 10.21037/jtd.2017.11.05
8. Lockie CJA, Gillon SA, Barrett NA, Taylor D, Mazumder A, Paramesh K, et al. Severe respiratory failure, extracorporeal membrane oxygenation, and intracranial hemorrhage. Crit Care Med. (2017) 45:1642–9. doi: 10.1097/CCM.0000000000002579
9. Wendel HP, Ziemer G. Coating-techniques to improve the hemocompatibility of artificial devices used for extracorporeal circulation. Eur J Cardio-Thoracic Surg. (2011) 16:342–50. doi: 10.1016/5010-7940(99)00210-9
10. Young G, Yonekawa KE, Nakagawa P, Nugent DJ. Argatroban as an alternative to heparin in extracorporeal membrane oxygenation circuits. Perfusion (2004) 19:283–8. doi: 10.1191/0267659104pf759oa
11. Jyoti A, Maheshwari A, Daniel E, Motihar A, Bhathiwal RS, Sharma D. Bivalirudin in venovenous extracorporeal membrane oxygenation. J Extra Corpor Technol. (2014) 46:94–7.
12. Tay CK, Sung K, Cho YH. ELSO Guidelines for Cardiopulmonary Extracorporeal Life Support. Ann Arbor, MI: Extracorporeal Life Support Organization (2017). p. 15–7.
13. Vroman L, Adams AL, Fischer GC, Munoz PC. Interaction of high molecular weight kininogen, factor XII, and fibrinogen in plasma at interfaces. Blood (1980) 55:156–9.
14. Turbill P, Beugeling T, Poot AA. Proteins involved in the Vroman effect during exposure of human blood plasma to glass and polyethylene. (1996) 17:1279–87. doi: 10.1016/S0142-9612(96)80004-4
15. Passmore MR, Fung YL, Simonova G, Foley SR, Diab SD, Dunster KR, et al. Evidence of altered haemostasis in an ovine model of venovenous extracorporeal membrane oxygenation support. Crit Care (2017) 21:1–9. doi: 10.1186/s13054-017-1788-9
16. Moganasundram S, Hunt BJ, Sykes K, Holton F, Parmar K, Durward A, et al. The relationship among thromboelastography, hemostatic variables, and bleeding after cardiopulmonary bypass surgery in children. Anesth Analg. (2010) 110:995–1002. doi: 10.1213/ANE.0b013e3181cd6d20
17. Millar JE, Fanning JP, McDonald CI, McAuley DF, Fraser JF. The inflammatory response to extracorporeal membrane oxygenation (ECMO): a review of the pathophysiology. Crit Care (2016) 20:1–10. doi: 10.1186/s13054-016-1570-4
18. Poot A, Beugeling T, Cazenave JP, Bantjes A, van Aken WG. Platelet deposition in a capillary perfusion model: quantitative and morphological aspects. Biomaterials (1988) 9:126–32. doi: 10.1016/0142-9612(88)90110-X
19. Watanabe H, Hayashi J, Ohzeki H. Biocompatibility of a silicone-coated polypropylene hollow fiber oxygenator in an in vitro model. Ann Thorac Surg. (1999) 4975:3–7. doi: 10.1016/S0003-4975(99)00213-1
20. Bannan S, Danby A, Cowan D, Ashraf S, Gesinde M, Martin P. Cell activation and thrombin generation in heparin bonded cardiopulmonary bypass circuits using a novel in vitro model. Eur J Cardiothorac Surg. (1997) 12:268–75. doi: 10.1016/S1010-7940(97)01208-6
21. Cohen HC, Joyce EJ, Kao WJ. Biomaterials selectively modulate interactions between human blood-derived polymorphonuclear leukocytes and monocytes. Am J Pathol. (2013) 182:2180–90. doi: 10.1016/j.ajpath.2013.02.022
22. Keuren JF, Wielders SJ, Willems GM, Morra M, Cahalan L, Cahalan P, et al. Thrombogenicity of polysaccharide-coated surfaces. Biomaterials (2003) 24:1917–24. doi: 10.1016/S0142-9612(02)00620-8
23. Wahba A, Philipp A, Behr R, Birnbaum DE. Heparin-coated equipment reduces the risk of oxygenator failure. Ann Thorac Surg. (1998) 65:1310–2. doi: 10.1016/S0003-4975(98)00156-8
24. Malfertheiner MV, Philipp A, Lubnow M, Zeman F, Enger TB, Bein T, et al. Hemostatic changes during extracorporeal membrane oxygenation: a prospective randomized clinical trial comparing three different extracorporeal membrane oxygenation systems. Crit Care Med. (2016) 44:747–54. doi: 10.1097/CCM.0000000000001482
25. Hang K, Lau A, Sileika TS, Park SH, Sousa AML, Burch P, et al. Molecular design of antifouling polymer brushes using sequence-specifi c peptoids. Adv Mater Interface (2014) 2014:1400225. doi: 10.1002/admi.201400225
26. de los Santos Pereira A, Sheikh S, Blaszykowski C, Pop-Georgievski O, Fedorov K, Thompson M, et al. Antifouling polymer brushes displaying antithrombogenic surface properties. Biomacromolecules (2016) 17:1179–185. doi: 10.1021/acs.biomac.6b00019
27. Obstals F, Vorobii M, Riedel T, de los Santos Pereira A, Bruns M, Singh S, et al. Improving hemocompatibility of membranes for extracorporeal membrane oxygenators by grafting nonthrombogenic polymer brushes. Macromol Biosci. (2018) 2018:1700359. doi: 10.1002/mabi.201700359
28. Kappelmayer J, Bernabei A, Edmunds LH, Edgington TS, Colman RW. Tissue factor is expressed on monocytes during simulated extracorporeal circulation. Circ Res. (1993) 72:1075–81. doi: 10.1161/01.RES.72.5.1075
29. Fischer M, Sperling C, Tengvall P, Werner C. The ability of surface characteristics of materials to trigger leukocyte tissue factor expression. Biomaterials (2010) 31:2498–507. doi: 10.1016/j.biomaterials.2009.12.016
30. Barstad RM, Ovrum E, Ringdal MA, Oystese R, Hamers MJ, Veiby OP, et al. Induction of monocyte tissue factor procoagulant activity during coronary artery bypass surgery is reduced with heparin-coated extracorporeal circuit. Br J Haematol. (1996) 94:517–25. doi: 10.1111/j.1365-2141.1996.tb08989.x
31. Yau JW, Stafford AR, Liao P, Fredenburgh JC, Roberts R, Weitz JI. Mechanism of catheter thrombosis: comparison of the antithrombotic activities of fondaparinux, enoxaparin, and heparin in vitro and in vivo. Blood (2011) 118:6667–74. doi: 10.1182/blood-2011-07-364141
32. Tillman B, Gailani D. Inhibition of factors XI and XII for prevention of thrombosis induced by artificial surfaces. Semin Thromb Hemost. (2018) 44:60–9. doi: 10.1055/s-0037-1603937
33. Boisclair MD, Lane DA, Philippou H, Esnouf MP, Sheikh S, Hunt B, et al. Mechanisms of thrombin generation during surgery and cardiopulmonary bypass. Blood (1993) 82:3350–7.
34. Wendel HP, Jones DW, Gallimore MJ. FXII levels, FXIIa-like activities and kallikrein activities in normal subjects and patients undergoing cardiac surgery. Immunopharmacology (1999) 45:141–4.
35. Plötz FB, van Oeveren W, Bartlett RH, Wildevuur CR. Blood activation during neonatal extracorporeal life support. J Thorac Cardiovasc Surg. (1993) 105:823–32.
36. Larsson M, Rayzman V, Nolte MW, Nicke KF, Björkqvist J, Jämsä A, et al. Cardiovascular disease: a factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci Transl Med. (2014) 6: 222ra17. doi: 10.1126/scitranslmed.3006804
37. May F, Krupka J, Fries M, Thielmann I, Pragst I, Weimer T, et al. FXIIa inhibitor rHA-Infestin-4: Safe thromboprotection in experimental venous, arterial and foreign surface-induced thrombosis. Br J Haematol. (2016) 173:769–778. doi: 10.1111/bjh.13990
38. Davidson SJ, Burman JF, Philips SM, Onis SJ, Kelleher AA, De Souza AC, et al. Correlation between thrombin potential and bleeding after cardiac surgery in adults. Blood Coagul Fibrinolysis. (2003) 14:175–9. doi: 10.1097/00001721-200302000-00009
39. Kopp R, Mottaghy K, Kirschfink M. Mechanism of complement activation during extracorporeal blood-biomaterial interaction: Effects of heparin coated and uncoated surfaces. ASAIO J. (2002) 48:598–605. doi: 10.1097/00002480-200211000-00005
40. Vallhonrat H, Swinford RD, Ingelfinger JR, Williams WW, Ryan DP, Tolkoff-Rubin N, et al. Rapid activation of the alternative pathway of complement by ECMO. ASAIO J. (1999) 45:113–4.
41. Graulich J, Sonntag J, Marcinkowski M, Bauer K, Kössel H, Bührer C, et al. Complement activation by in vivo neonatal and in vitro extracorporeal membrane oxygenation. Mediators Inflamm. (2002) 11:69–73. doi: 10.1080/09629350220131908
42. Bruins P, te Velthuis H, Yazdanbakhsh AP, Jansen PG, van Hardevelt FW, de Beaumont EM, et al. Activation of the complement system during and after cardiopulmonary bypass surgery. Circulation (1997) 96:1–14. doi: 10.1161/01.CIR.96.10.3542
43. Ivanovich P, Chenoweth DE, Schmidt R, Klinkmann H, Boxer LA, Jacob HS, et al. Symptoms and activation of granulocytes and complement with two dialysis membranes. Kidney Int. (1983) 24:758–63. doi: 10.1038/ki.1983.224
44. Westfall SH, Stephens C, Kesler K, Connors RH, Tracy TF, Weber TR. Complement activation during prolonged extracorporeal membrane oxygenation. Surgery (1991) 110:887–91.
45. Cheung AK, Hohnholt M, Gilson J. Adherence of neutrophils to hemodialysis membranes: Role of complement receptors. Kidney Int. (1991) 40:1123–33. doi: 10.1038/ki.1991.324
46. Wilm J, Philipp A, Müller T, Bredthauer A, Gleich O, Schmid C, et al. Leukocyte adhesion as an indicator of oxygenator thrombosis during extracorporeal membrane oxygenation therapy? ASAIO J. (2017) 64:24–30. doi: 10.1097/MAT.0000000000000586
47. Keragala CB, Draxler DF, McQuilten ZK, Medcalf RL. Haemostasis and innate immunity – a complementary relationship: a review of the intricate relationship between coagulation and complement pathways. Br J Haematol. (2018) 180:782–98. doi: 10.1111/bjh.15062
48. Delabranche X, Helms J, Meziani F. Immunohaemostasis: a new view on haemostasis during sepsis. Ann Intensive Care (2017) 7:1–14. doi: 10.1186/s13613-017-0339-5
49. Graulich J, Walzog B, Marcinkowski M, Bauer K, Kössel H, Fuhrmann G, et al. Leukocyte and endothelial activation in a laboratory model of extracorporeal membrane oxygenation (ECMO). Pediatr Res. (2000) 48:679–84. doi: 10.1203/00006450-200011000-00021
50. Anderson HL, Coran AG, Drongowski RA, Ha HJ, Bartlett RH. Extracellular fluid and total body water changes in neonates undergoing extracorporal membrane oxygenation. J Pediatr Surg. (1992) 27:1003–8. doi: 10.1016/0022-3468(92)90547-K
51. Abrams D, Baldwin MR, Champion M, Agerstrand C, Eisenberger A, Bacchetta M, et al. Thrombocytopenia and extracorporeal membrane oxygenation in adults with acute respiratory failure: a cohort study. Intensive Care Med. (2016) 42:844–52. doi: 10.1007/s00134-016-4312-9
52. Lukito P, Wong A, Jing J, Arthur JF, Marasco SF, Murphy DA, et al. Mechanical circulatory support is associated with loss of platelet receptors glycoprotein Ibα and glycoprotein VI. J Thromb Haemost. (2016) 14:2253–60. doi: 10.1111/jth.13497
53. Cheung PY, Sawicki G, Salas E, Etches PC, Schulz R, Radomski MW. The mechanisms of platelet dysfunction during extracorporeal membrane oxygenation in critically ill neonates. Crit Care Med. (2000) 28:2584–90. doi: 10.1097/00003246-200007000-00067
54. Hase T, Sirajuddin S, Maluso P, Bangalore R, DePalma L, Sarani B. Platelet dysfunction in critically ill patients. Blood Coagul Fibrinolysis (2017) 28:475–8. doi: 10.1097/MBC.0000000000000625
55. Kalbhenn J, Schlagenhauf A, Rosenfelder S, Schmutz A, Zieger B. Acquired von Willebrand syndrome and impaired platelet function during venovenous extracorporeal membrane oxygenation: Rapid onset and fast recovery. J Hear Lung Transplant. (2018) 37:985–91. doi: 10.1016/j.healun.2018.03.013
56. Kalbhenn J, Schmidt R, Nakamura L, Schelling J, Rosenfelder S, Zieger B. Early Diagnosis of Acquired von Willebrand Syndrome (AVWS) is elementary for clinical practice in patients treated with ECMO therapy. J Atheroscler Thromb. (2015) 22:265–71. doi: 10.5551/jat.27268
57. Dewanjee MK, Wu SM, Kapadvanjwala M, De D, Dewanjee S, Gonzalez L, et al. Emboli from an extraluminal blood flow hollow fiber oxygenator with and without an arterial filter during cardiopulmonary bypass in a pig model. ASAIO J. (1996) 42:1010–8. doi: 10.1097/00002480-199642060-00015
58. Pugsley W, Klinger L, Paschalis C, Aspey B, Newman S, Harrison M, et al. Microemboli and cerebral impairment during cardiac surgery. Vasc Surg. (1990) 24:34–43. doi: 10.1177/153857449002400107
59. Meyer AD, Gelfond JA, Wiles AA, Freishtat RJ, Rais-Bahrami K. Platelet-derived microparticles generated by neonatal extracorporeal membrane oxygenation systems. ASAIO J. (2015) 16:37–42. doi: 10.1097/MAT.0000000000000164
60. Miyazaki Y, Nomura S, Miyake T, Kagawa H, Kitada C, Taniguchi H, et al. High shear stress can initiate both platelet aggregation and shedding of procoagulant containing microparticles. Blood (1996) 88:3456–64.
61. Heilmann C, Geisen U, Beyersdorf F, Nakamura L, Benk C, Trummer G, et al. Acquired von Willebrand syndrome in patients with extracorporeal life support (ECLS). Intensive Care Med. (2012) 38:62–8. doi: 10.1007/s00134-011-2370-6
62. Pasala S, Fiser RT, Stine KC, Swearingen CJ, Prodhan P. Von willebrand factor multimers in pediatric extracorporeal membrane oxygenation support. ASAIO J. (2014) 60:419–23. doi: 10.1097/MAT.0000000000000084
63. Tauber H, Ott H, Streif W, Weigel G, Loacker L, Fritz J, et al. extracorporeal membrane oxygenation induces short-term loss of high-molecular-weight von willebrand factor multimers. Anesth Analg. (2015) 120:730–6. doi: 10.1213/ANE.0000000000000554
64. Adachi I, Kostousov V, Hensch L, Chacon-Portillo MA, Teruya J. Management of hemostasis for pediatric patients on ventricular-assist devices. Semin Thromb Hemost. (2018) 44:30–7. doi: 10.1055/s-0037-1607982
65. Vincent F, Rauch A, Loobuyck V, Robin E, Nix C, Vincentelli A, et al. Arterial pulsatility and circulating von willebrand factor in patients on mechanical circulatory support. J Am Coll Cardiol. (2018) 71:2106–118. doi: 10.1016/j.jacc.2018.02.075
66. Halaweish I, Cole A, Cooley E, Lynch WR, Haft JW. Roller and centrifugal pumps. ASAIO J. (2015) 61:496–501. doi: 10.1097/MAT.0000000000000243
67. McVeen RV, Lorch V, Carroll RC, Goldberg L, Keszler M, Podlasek S, et al. Letter to the editor: Changes in fibrinolytic factors in newborns during extracorporeal membrane oxygenation (ECMO). Am J Hematol. (1991) 38:254–55.
68. Hundalani SG, Nguyen KT, Soundar E, Kostousov V, Bomgaars L, Moise A., et al. Age-based difference in activation markers of coagulation and fibrinolysis in extracorporeal membrane oxygenation. Pediatr Crit Care Med. (2014) 15: e198–205. doi: 10.1097/PCC.0000000000000107
69. Hunt BJ, Parratt RN, Segal HC, Sheikh S, Kallis P, Yacoub M. Activation of coagulation and fibrinolysis during cardiothoracic operations. Ann Thorac Surg. (1998) 65:712–18. doi: 10.1016/S0003-4975(97)01345-3
70. Sigaut S, Tremey B, Outtara A, Couturier R, Tablerlet C, Grassin-Delyle S., et al. Comparison of two doses of tranexamic acid in adults bypass. Anesthesiology. (2018) 2:590–600. doi: 10.1097/ALN.0b013e3182a443e8
71. Lou S, MacLaren G, Best D, Delzoppo C, Butt W. Hemolysis in pediatric patients receiving centrifugal-pump extracorporeal membrane oxygenation: Prevalence, risk factors, and outcomes. Crit Care Med. (2014) 42:1213–20. doi: 10.1097/CCM.0000000000000128
72. Omar HR, Mirsaeidi M, Socias S, Sprenker C, Caldeira C, Camporesi EM, et al. Plasma free hemoglobin is an independent predictor of mortality among patients on extracorporeal membrane oxygenation support. PLOS ONE (2015) 10:e0124034. doi: 10.1371/journal.pone.0124034
73. Marcus, Aaron J, Broekman MJ. Cell-free hemoglobin as an oxygen carrier removes nitric oxide, resulting in defective thromboregulation. Circulation (1996) 92:208–9.
74. Da Q, Teruya M, Guchhait P, Teruya J, Olson JS, Cruz MA. Free hemoglobin increases von Willebrand factor-mediated platelet adhesion in vitro: Implications for circulatory devices. Blood (2015) 126:2338–41. doi: 10.1182/blood-2015-05-648030
Keywords: Haemostasis, ECMO, cardiopulmonary bypass, ventricular assist device, thrombosis, hemorrhage
Citation: Doyle AJ and Hunt BJ (2018) Current Understanding of How Extracorporeal Membrane Oxygenators Activate Haemostasis and Other Blood Components. Front. Med. 5:352. doi: 10.3389/fmed.2018.00352
Received: 17 August 2018; Accepted: 30 November 2018;
Published: 12 December 2018.
Edited by:
Patti Massicotte, University of Alberta, CanadaReviewed by:
Anastasios G. Kriebardis, Technological Educational Institute of Athens, GreeceWei Li, Marshall University, United States
Owen McCarty, Oregon Health & Science University, United States
Copyright © 2018 Doyle and Hunt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beverley J. Hunt, YmV2ZXJsZXkuaHVudEBnc3R0Lm5ocy51aw==