 Edward M. Conway
Edward M. Conway- Division of Hematology, Department of Medicine, Faculty of Medicine, Centre for Blood Research, Life Sciences Institute, University of British Columbia, Vancouver, BC, Canada
To sustain life in environments that are fraught with risks of life-threatening injury, organisms have developed innate protective strategies such that the response to wounds is rapid and localized, with the simultaneous recruitment of molecular, biochemical, and cellular pathways that limit bleeding and eliminate pathogens and damaged host cells, while promoting effective healing. These pathways are both coordinated and tightly regulated, as their over- or under-activation may lead to inadequate healing, disease, and/or demise of the host. Recent advances in our understanding of coagulation and complement, a key component of innate immunity, have revealed an intriguing linkage of the two systems. Cell-secreted polyphosphate promotes coagulation, while dampening complement activation, discoveries that are providing insights into disease mechanisms and suggesting novel therapeutic strategies.
Introduction
Two major blood-borne proteolytic cascades, complement and coagulation, are fully integrated to cooperatively fight infections and prevent excessive bleeding from wounds. Interplay between these systems is evolutionarily conserved, as evident in the horseshoe crab, a “living fossil” representing arthropods from 500 million years ago (1–3). The released contents of hemocytes induce clotting and destroy invading pathogens and toxins. Complement and coagulation pathways in mammals now appear more distinct, but interactions are increasingly being recognized. Elucidation of the links is yielding novel treatments, including, eculizumab, an effective anti-complement antibody that prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS) (4, 5). More therapies for other common diseases will undoubtedly enter the clinic in coming years (6).
In this report, I review the complement system, highlighting some key mechanisms by which it is regulated, and how it interfaces with coagulation [Readers are referred to excellent reviews of the coagulation cascade (7–10)]. I then focus on recently uncovered insights into the role of the polyanion polyphosphate (polyP)—known to promote coagulation—in dampening complement activation. This will be followed by a discussion of how such an apparent dichotomy in the function of polyP is physiologically relevant.
Complement Activation
Comprising over 30 soluble and membrane-bound proteins, complement contributes to innate immunity and provides a bridge to adaptive immunity (11–13). Complement activation is triggered by exposure of blood to damage-associated molecular patterns that include, for example, pathogens, host DNA from damaged cells, lipids and oligosaccharides (Figure 1). Three pathways—lectin (LP), classical (CP), and alternative (AP)—converge with generation of C3 convertases that proteolyse C3 into C3b and release the anaphylatoxin C3a. The CP is triggered by C1q recognition of antibodies bound to antigens or microbial surfaces. C1q may also recognize other targets, such as C-reactive protein, apoptotic cells, and microbes. It circulates in complex with zymogens of serine proteases C1r and C1s (C1qr2s2). When C1q binds to its target, C1r autoactivates and activates C1s (15) which in turn cleaves C4, releasing C4a, and C4b, the latter which covalently binds to target surfaces. C2 binds to immobilized C4b and is cleaved by C1s into C2b and C2a, allowing C2a to complex with C4b and form the CP C3 convertase, C4b2a. The LP is similar to the CP (16), but pathogen recognition comprises mannose binding lectin (MBL), ficolins and/or collectin-11. These circulate bound to MBL-associated zymogens of serine proteases MASP1/MASP3 and MASP2 and bind to sugars or N-acetylated groups on micro-organisms. MASP1 autoactivates and cleaves C2 and activates MASP2, while MASP2 cleaves C2 and C4, yielding the C4b2a LP C3 convertase (17).
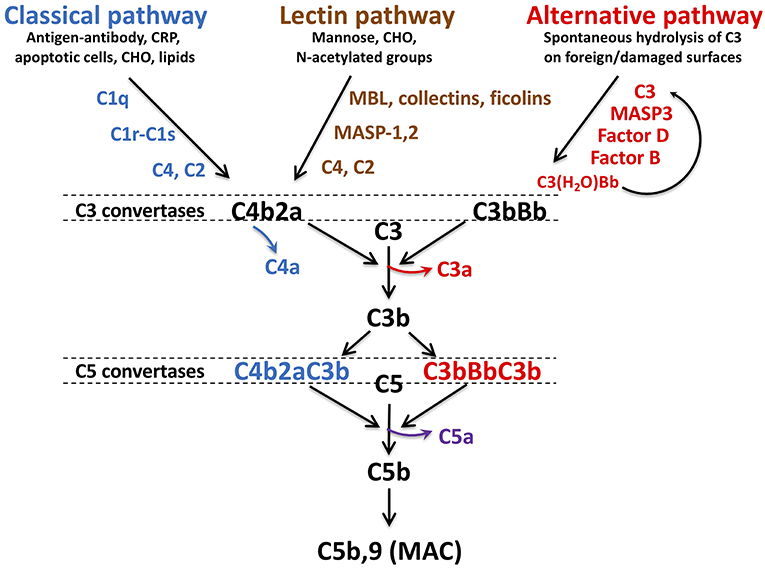
Figure 1. Schematic of complement activation pathways. Complement activation proceeds via the classical, lectin, or alternative pathways, triggered by exposure of surveillance molecules, C1q, MBL, collectins and ficolins, of specific danger signals. The alternative pathway is constitutively “on,” due to spontaneous hydrolysis of C3. The pathways converge to form C3 convertases: C4b2a for the classical and lectin pathways, C3bBb for the alternative pathway. C4a and C3a, are released with cleavage of C4 and C3, respectively. As C3b is further generated, C5 convertases C4bBbC3b and C3bBbC3b are formed, resulting in liberation of the most potent anaphylatoxin C5a, in conjunction with C5b. C5b is the initial component required for spontaneous assembly of the C5b-9 membrane attack complex (MAC) which polymerizes and induces lysis of the cellular target. MBL, mannose binding lectin, CRP, C-reactive protein, CHO, carbohydrate, MASP, MBL associated serine protease. Figure and legend from Conway (14).
The AP is constitutively active, sustained by a “tick-over” mechanism in which small amounts of C3 are hydrolyzed to C3(H2O) (18), exposing a binding site for factor B (FB). Circulating factor D (FD) cleaves FB into Ba and Bb, the latter which binds to C3(H2O) to form a fluid-phase C3 convertase which cleaves C3 to C3b and C3a. With exposure to a pathogen or damaged cell, more C3a and C3b are generated (19), resulting in formation of the AP C3 convertase, C3bBb. Complement is amplified via the AP as the pathways converge and form cell bound C3 convertases. The additional C3b binds to C4b2b and C3bBb, yielding C4bBb(C3b)n and C3bBb(C3b)n, which thus become C5 convertases, cleaving C5 into C5b and C5a. C5b is the trigger for the terminal pathway, which spontaneously proceeds with sequential assembly of C6, C7, C8, and multiple C9 subunits, forming the C5b-9 pore-like, lytic membrane attack complex (MAC) that targets invading pathogens and promotes prothrombinase assembly and tissue factor (TF) activation (20).
C5a is a pleiotropic biologically active peptide, exhibiting potent anaphylatoxin properties. C5a also triggers coagulation and inflammation via TF by endothelial cells and monocytes, release/exposure of VWF and P-selectin by endothelial cells and platelets, secretion of inflammatory cytokines, expression of leukocyte adhesion molecules, and release of platelet granule contents that further promote coagulation and complement activation (21–24).
Regulation of Complement
Complement activation is down-regulated at numerous steps. This ensures a highly localized and temporally appropriate response that spares the host from undesired damage. Acquired or genetic alterations in factors that regulate complement are commonly associated with disease, often featuring varying degrees of vascular-thrombosis. Characterization of these pathways is revealing novel strategies for drug development (6, 25, 26). In the following, I describe a few of the mechanisms by which complement activation is dampened. References for more comprehensive reviews are provided (13, 25, 27–29).
C1-esterase inhibitor (C1-INH) is a serine protease inhibitor that highlights the coordinated regulation of coagulation and complement (10). In coagulation, C1-INH interferes with the proteolytic activities of factor XIa, factor XIIa, and kallikrein, suppressing the contact/intrinsic pathways of coagulation and inflammation. In complement, C1-INH interferes with C1r, C1s, MASP1, and MASP2, preventing formation of the CP/LP convertases. The inhibitory activity of C1-INH is variably potentiated by polyanions, such as heparin (15, 30). Thus, heparin augments C1-INH inhibition of factor XIa and MASP2 (31), but actually dampens C1-INH neutralization of factor XIIa (32), and has almost no effect on C1-INH inhibition of kallikrein, C1r or MASP1 (32, 33). These differential effects of C1-INH that are partly dependent on the cofactor activity of the polyanion, heparin, may help explain why functional deficiencies of C1-INH are not associated clinically with thrombosis.
The major fluid-phase negative regulator of the AP is factor H (FH) (34). Synthesized primarily by the liver, but also by endothelial cells and platelets, FH binds to C3b and glycosaminoglycans of host cells where it suppresses complement by acting as a cofactor for protease factor I (FI) mediated inactivation of C3b to iC3b, accelerating decay of the AP C3 convertase, and competing with FB binding to C3b. Patients with FH mutations are at increased risk of developing aHUS and cardiovascular disease (35–37). FH also binds to VWF in Weibel-Palade bodies and/or facilitates ADAMTS13-mediated proteolysis of ultra large VWF (38–41), providing protection against thrombosis.
The membrane-bound complement receptor (CR)1, glycosylphosphatidylinositol (GPI)-linked CD55, and CD46 are also decay accelerating factors for C3b-containing convertases. Moreover, CR1 and CD46 promote FI-mediated proteolysis of C3b to iC3b (42). Interesting for their distinct clinical presentations, CD46 deficiency is implicated in aHUS (43), while CD55 deficiency is associated with PNH (44).
Assembly and function of the MAC are also regulated to limit host cell damage. The GPI-linked CD59, deficiency of which is also associated with PNH (45), binds to C8 and C9 and prevents C9 polymerization (46). Clusterin binds to C7, C8, and C9, inducing structural changes that reduce integration of C5b-9 into the membrane (47). Vitronectin also prevents C5b-9 membrane binding by promoting formation of a soluble C5b-7 complex (48).
The activities of C3a and C5a are reduced via several coagulation-related enzymes. C5a is proteolysed by plasmin and matrix metalloproteinase 12 (49), while carboxypeptidase B2 (CPB2) [also referred to as activated thrombin-activatable fibrinolysis inhibitor (TAFIa)] (50), reduces the activity of C3a and C5a by cleaving their C-terminal basic amino acids.
PolyP, Coagulation, and Complement
PolyP is a ubiquitously expressed, linear, anionic polymer of monophosphate units, linked by phosphoanhydride bonds (51). Polymer lengths vary from ~25 to 1,000 units in mammalian cells (52), extending to thousands of units in some bacteria (53). It is abundant in the dense granules of platelets (54, 55), released to the cell surface and/or into the circulation upon activation (55, 56), likely in a charge-neutral form, bound to divalent cations (Ca2+, Mg2+) and amines (54). Rather than acting as a calcium ion chelator and anticoagulant, polyP promotes coagulation at multiple steps in the cascade (57–61). Long chain polyP is believed to provide a template for autoactivation of factor XII, thereby also triggering inflammation via factor XIIa-mediated activation of the kallikrein-kinin system (62). Shorter forms of polyP released particularly from platelets, bind directly to thrombin (63), fibrinogen, (64), factors XI and XII, pre/kallikrein, high molecular weight kininogen and VWF (56), amplifying generation of factor XIa and thrombin, enhancing the activation of factor V, inactivating tissue factor pathway inhibitor (TFPI), and integrating into the fibrin clot, rendering it more resistant to fibrinolysis (57). The physiologic relevance of several of these and other polyP-coagulation/fibrinolysis protein interactions remain incompletely understood, but targeting polyP is gaining wide interest as a safe anti-thrombotic (65).
In view of its profound pro-coagulant effects, we predicted that polyP would similarly activate complement. Somewhat surprising, polyP did exactly the opposite (Figure 2). In a C1-INH-dependent manner, polyP dampened C1s-mediated cleavage of C4 and C2 in gel-based assays and cell systems (67). Binding studies revealed that C1-INH directly interacts with the serine protease domain of C1s at a rate that is augmented ~90-fold by the presence of polyP—an effect similar to that seen with heparin. Not formally tested, the data suggested that polyP similarly potentiates C1-INH interactions with MASP2. Interestingly, like heparin, polyP had little potentiating effect on C1r (33). However, these parallels with heparin are limited, since heparin accelerates neutralization of thrombin by antithrombin (AT) >2,000-fold (68), whereas polyP has no effect on the thrombin-AT interaction (57).
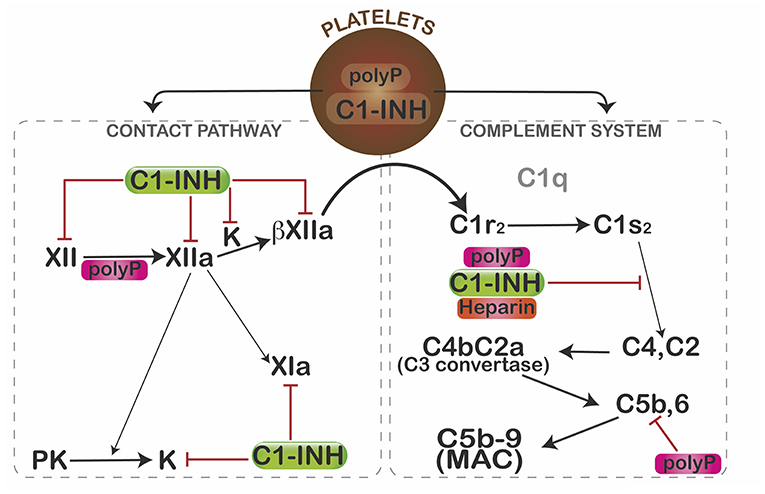
Figure 2. Mechanisms by which polyP regulates complement activation. In resting platelets, polyP and C1-INH are housed in different organelles. After activation, polyP and C1-INH coalesce toward the center of the platelets where they colocalize and are subsequently secreted (66). PolyP triggers a conformational change in factor XII, resulting in generation of XIIa, which can activate prekallikrein (PK) and/or factor XI to XIa. Kallikrein (K) or plasmin (not shown) can further cleave XIIa to generate βXIIa which may activate C1r and thus promote complement activation. C1-INH dampens that pathway by inhibiting factor XII, XIIa, βXIIa, and kallikrein (K). C1s cleaves C4 and C2 to generate the C4b2a C3 convertase, which ultimately leads to formation of the C5b,6 complex, and assembly of the C5b-9 membrane attack complex (MAC). PolyP or heparin potentiate the inhibitory function of C1-INH via direct interactions with C1-INH and the target protease, C1s. PolyP also destabilizes C5b,6, thereby dampening formation of the MAC. Interestingly, in spite of binding to factors XIa and XIIa, reduced levels or function of C1-INH do not cause thrombosis, possibly due in part to differential effects of polyanions (polyP, heparin) on the function of the target enzymes. The over-riding effect of polyP in a serum-based endothelial cell culture system is to suppress complement activation. Figure and legend from Wijeyewickrema et al. (66).
PolyP also significantly interferes with activation of the terminal pathway of complement (69) in a size and concentration-dependent manner. It destabilizes C5b-6, reducing the ability of C5b-7 and C5b-8 to bind to and integrate into the target membrane. Other pathways by which polyP might modulate complement activation have not yet been explored, however, given its highly anionic charge, its stability in a calcium-nanoparticle form (62), and its wide expression profile, it is likely that polyP has multiple effects on this innate immune pathway, analogous to its role in coagulation.
Relevance of polyP in Complement
In the face of its pro-coagulant and pro-inflammatory properties, what might be the physiologic relevance of polyP in complement activation? The complement-dampening effect of polyP does not entirely conflict with the defined role of polyP in other biological systems. PolyP is prominently expressed in several cellular compartments of prokaryotes, where it exhibits pro-survival properties as an energy source, a metal ion chelator, a molecular chaperone, in enhancing pathogenicity (70–72), and in some cases, protecting against complement mediated death (73).
One can speculate on how polyP and C1-INH might co-operate in host protection. C1-INH is synthesized by and found on the surface of endothelial cells (74). Endothelial cells also display abundant glycosaminoglycans on their surface as heparan sulfate. PolyP, released by activated cells and found at low concentrations in the blood of healthy individuals (75, 76) would be available to bind to C1-INH on the endothelium, where it could potentiate the function of C1-INH, allowing the C1-INH:polyanion complex to recruit and neutralize target proteases, such as C1s and/or MASP2. Binding of the polyanion first to C1-INH is required for optimal neutralization of C1s. Such an order of events would best keep complement activation in check. PolyP would also be positioned to dampen generation of MAC on the host cell surface. In such a scenario, the activated endothelial cell would be protected against host-mediated destruction, while retaining its prothrombotic and pro-inflammatory properties.
This model may also apply to other cells. As mentioned, polyP is abundant in platelets and released upon activation (54, 55, 77). C1-INH is also found in platelets, secreted and deposited on the activated platelet membrane (78). Although initially housed in separate granules, platelet activation results in colocalization of polyP and C1-INH in and on the platelet (66). High levels of polyP on activated platelets would therefore readily dampen complement activation by potentiating the inhibitory properties of C1-INH and interfering with the terminal pathway (69), overall protecting the underlying host cells from innate destruction, while allowing the platelets to promote hemostasis-thrombosis and inflammation. Interestingly, polyP also binds to FH (69) and may similar to C1-INH, coat and protect host cells (34) from complement activation, convertase assembly and MAC binding/integration. Disturbances in the release of adequate polyP might therefore be predicted to result in disease. This is in fact evident in patients with dense granule storage pool diseases (79) who have low platelet polyP (80) and exhibit a bleeding diathesis and organ dysfunction secondary to excessive inflammation.
Taking Advantage of the Complement-Dampening Properties of polyP for Therapeutic Purposes
Immediate clinical application of the finding that polyP, a naturally occurring and easily synthesized polyanion that suppresses complement activation, is enticing but not without challenges. Depending on the length, dose and formulation, systemic delivery of polyP may entail risk of thrombosis. However, polyP has been administered safely in vivo, providing protection against endotoxin-induced sepsis in a mouse model (81). If validated, one could envisage using polyP for a wide range of disorders with excess complement activation. Our group has limited in vivo testing of polyP to study its role in protecting against age-related macular degeneration (AMD). AMD is a common cause of blindness where over-activation of complement is a major pathogenic driver (82). In a mouse model of laser-induced AMD, intravitreal administration of polyP dampened the pathologic neovascularization and complement deposition to a similar extent as currently used anti-VEGF targeted therapies (83). No adverse effects of polyP were observed, providing strong rationale for further exploration.
Conclusion
In the last 20–30 years, major inroads have been made in delineating the molecular mechanisms by which complement, coagulation and inflammation intersect. The preceding discussion underlines the unique role that polyP plays in suppressing complement, while promoting coagulation and inflammation. Further understanding of how polyP modulates complement activation through the induction of structural changes in key factors in these different proteolytic cascades, and/or interactions with other proteins and cells, will reveal novel sites for therapeutic intervention for a range of thrombotic and inflammatory disorders.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
EC is supported by grants from the Canadian Institutes of Health Research (CIHR), the Heart and Stroke Foundation of Canada, the Natural Sciences and Engineering Research Council of Canada (NSERC), and the Canada Foundations for Innovation (CFI). He holds a Tier 1 Canada Research Chair in Endothelial Cell Biology and is an Adjunct Scientist with the Canadian Blood Services.
References
1. Krem MM, Di Cera E. Evolution of enzyme cascades from embryonic development to blood coagulation. Trends Biochem Sci. (2002) 27:67–74. doi: 10.1016/S0968-0004(01)02007-2
2. Dodds AW. Which came first, the lectin/classical pathway or the alternative pathway of complement? Immunobiology. (2002) 205:340–54. doi: 10.1078/0171-2985-00137
3. Iwanaga S. The molecular basis of innate immunity in the horseshoe crab. Curr Opin Immunol. (2002) 14:87–95. doi: 10.1016/S0952-7915(01)00302-8
4. Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med. (2009) 360:544–6. doi: 10.1056/NEJMc0809959
5. Hillmen P, Young NS, Schubert J, Brodsky RA, Socie G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. (2006) 355:1233–43. doi: 10.1056/NEJMoa061648
6. Gialeli C, Gungor B, Blom AM. Novel potential inhibitors of complement system and their roles in complement regulation and beyond. Molecul Immunol. (2018) 102:73–83. doi: 10.1016/j.molimm.2018.05.023
7. Swieringa F, Spronk MH H, Heemskerk WMJ. van der Meijden PEJ. Integrating platelet and coagulation activation in fibrin clot formation. Res Pract Thromb Haemost. (2018) 2:450–460. doi: 10.1002/rth2.12107
8. Weidmann H, Heikaus L, Long AT, Naudin C, Schluter H, Renne T. The plasma contact system, a protease cascade at the nexus of inflammation, coagulation and immunity. Biochim Biophys Acta. (2017) 1864:2118–27. doi: 10.1016/j.bbamcr.2017.07.009
9. Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. (2013) 93:327–58. doi: 10.1152/physrev.00016.2011
10. De Maat SZ, Hofman LM, Maas C. Hereditary angioedema: the plasma contact system out of control: reply. J Thromb Haemost. (2018) 16:2349–51. doi: 10.1111/jth.14269
11. Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. (2007) 25:1265–75. doi: 10.1038/nbt1342
12. Mihlan M, Blom AM, Kupreishvili K, Lauer N, Stelzner K, Bergstrom F, et al. Monomeric C-reactive protein modulates classic complement activation on necrotic cells. FASEB J. (2011) 25:4198–210. doi: 10.1096/fj.11-186460
13. Killick J, Morisse G, Sieger D, Astier AL. Complement as a regulator of adaptive immunity. Semin Immunopathol. (2018) 40:37–48. doi: 10.1007/s00281-017-0644-y
14. Conway EM. Complement-coagulation connections. Blood Coagul Fibrinol. (2018) 29:243–51. doi: 10.1097/MBC.0000000000000720
15. Wijeyewickrema LC, Yongqing T, Tran TP, Thompson PE, Viljoen JE, Coetzer TH, et al. Molecular determinants of the substrate specificity of the complement-initiating protease, C1r. J Biol Chem. (2013) 288:15571–80. doi: 10.1074/jbc.M113.451757
16. Degn SE, Jensen L, Hansen AG, Duman D, Tekin M, Jensenius JC, et al. Mannan-binding lectin-associated serine protease (MASP)-1 is crucial for lectin pathway activation in human serum, whereas neither MASP-1 nor MASP-3 is required for alternative pathway function. J Immunol. (2012) 189:3957–69. doi: 10.4049/jimmunol.1201736
17. Dobo J, Schroeder V, Jenny L, Cervenak L, Zavodszky P, Gal P. Multiple roles of complement MASP-1 at the interface of innate immune response and coagulation. Molecul Immunol. (2014) 61:69–78. doi: 10.1016/j.molimm.2014.05.013
18. Lachmann PJ, Hughes-Jones NC. Initiation of complement activation. Springer Semin Immunopathol. (1984) 7:143–62. doi: 10.1007/BF01893018
19. Saggu G, Cortes C, Emch HN, Ramirez G, Worth RG, Ferreira VP. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and C3(H2O). J Immunol. (2013) 190:6457–67. doi: 10.4049/jimmunol.1300610
20. Langer F, Spath B, Fischer C, Stolz M, Ayuk FA, Kroger N, et al. Rapid activation of monocyte tissue factor by antithymocyte globulin is dependent on complement and protein disulfide isomerase. Blood. (2013) 121:2324–35. doi: 10.1182/blood-2012-10-460493
21. Hamad OA, Ekdahl KN, Nilsson PH, Andersson J, Magotti P, Lambris JD, et al. Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J Thromb Haemost. (2008) 6:1413–21. doi: 10.1111/j.1538-7836.2008.03034.x
22. Martel C, Cointe S, Maurice P, Matar S, Ghitescu M, Theroux P, et al. Requirements for membrane attack complex formation and anaphylatoxins binding to collagen-activated platelets. PLoS ONE. (2011) 6:e18812. doi: 10.1371/journal.pone.0018812
23. Oikonomopoulou K, Ricklin D, Ward PA, Lambris JD. Interactions between coagulation and complement–their role in inflammation. Semin Immunopathol. (2012) 34:151–65. doi: 10.1007/s00281-011-0280-x
24. Bosmann M, Grailer JJ, Ruemmler R, Russkamp NF, Zetoune FS, Sarma JV, et al. Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation in acute lung injury. FASEB J. (2013) 27:5010–21. doi: 10.1096/fj.13-236380
25. Harris CL, Pouw RB, Kavanagh D, Sun R, Ricklin D. Developments in anti-complement therapy, from disease to clinical trial. Molecul Immunol. (2018) 102:89–119. doi: 10.1016/j.molimm.2018.06.008
26. Reddy YN, Siedlecki AM, Francis JM. Breaking down the complement system: a review and update on novel therapies. Curr Opin Nephrol Hypertens. (2017) 26:123–8. doi: 10.1097/MNH.0000000000000305
27. Baines AC, Brodsky RA. Complementopathies. Blood Rev. (2017) 31:213–23. doi: 10.1016/j.blre.2017.02.003
28. Liszewski MK, Java A, Schramm EC, Atkinson JP. Complement dysregulation and disease: insights from contemporary genetics. Annual Rev Pathol. (2017) 12:25–52. doi: 10.1146/annurev-pathol-012615-044145
29. Mizuno M, Suzuki Y, Ito Y. Complement regulation and kidney diseases: recent knowledge of the double-edged roles of complement activation in nephrology. Clin Exp Nephrol. (2018) 22:3–14. doi: 10.1007/s10157-017-1405-x
30. Pike RN, Wijeyewickrema LC. The molecular switches controlling the interaction between complement proteases of the classical and lectin pathways and their substrates. Curr Opin Struct Biol. (2013) 23:820–7. doi: 10.1016/j.sbi.2013.07.016
31. Dobo J, Harmat V, Beinrohr L, Sebestyen E, Zavodszky P, Gal P. MASP-1, a promiscuous complement protease: structure of its catalytic region reveals the basis of its broad specificity. J Immunol. (2009) 183:1207–14. doi: 10.4049/jimmunol.0901141
32. Pixley RA, Schmaier A, Colman RW. Effect of negatively charged activating compounds on inactivation of factor XIIa by Cl inhibitor. Arch Biochem Biophys. (1987) 256:490–8. doi: 10.1016/0003-9861(87)90606-0
33. Sim RB, Arlaud GJ, Colomb MG. Kinetics of reaction of human C1-inhibitor with the human complement system proteases C1r and C1s. Biochim Biophys Acta. (1980) 612:433–49. doi: 10.1016/0005-2744(80)90126-6
34. Licht C, Pluthero FG, Li L, Christensen H, Habbig S, Hoppe B, et al. Platelet-associated complement factor H in healthy persons and patients with atypical HUS. Blood. (2009) 114:4538–45. doi: 10.1182/blood-2009-03-205096
35. Ferreira VP, Herbert AP, Cortes C, McKee KA, Blaum BS, Esswein ST, et al. The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J Immunol. (2009) 182:7009–18. doi: 10.4049/jimmunol.0804031
36. Saunders RE, Abarrategui-Garrido C, Fremeaux-Bacchi V, Goicoechea de Jorge E, Goodship TH, Lopez Trascasa M, et al. The interactive Factor H-atypical hemolytic uremic syndrome mutation database and website: update and integration of membrane cofactor protein and Factor I mutations with structural models. Hum Mutat. (2007) 28:222–34. doi: 10.1002/humu.20435
37. Buraczynska M, Ksiazek P, Zukowski P, Benedyk-Lorens E, Orlowska-Kowalik G. Complement factor H gene polymorphism and risk of cardiovascular disease in end-stage renal disease patients. Clin Immunol. (2009) 132:285–90. doi: 10.1016/j.clim.2009.04.005
38. Feng S, Liang X, Cruz MA, Vu H, Zhou Z, Pemmaraju N, et al. The interaction between factor H and Von Willebrand factor. PLoS ONE. (2013) 8:e73715. doi: 10.1371/journal.pone.0073715
39. Rayes J, Roumenina LT, Dimitrov JD, Repesse Y, Ing M, Christophe O, et al. The interaction between factor H and VWF increases factor H cofactor activity and regulates VWF prothrombotic status. Blood. (2014) 123:121–5. doi: 10.1182/blood-2013-04-495853
40. Nolasco L, Nolasco J, Feng S, Afshar-Kharghan V, Moake J. Human complement factor H is a reductase for large soluble von Willebrand factor multimers–brief report. Arterioscler Thromb Vasc Biol. (2013) 33:2524–8. doi: 10.1161/ATVBAHA.113.302280
41. Turner N, Nolasco L, Nolasco J, Sartain S, Moake J. Thrombotic microangiopathies and the linkage between von Willebrand factor and the alternative complement pathway. Semin Thromb Hemost. (2014) 40:544–50. doi: 10.1055/s-0034-1383547
42. Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. (2006) 118:127–36. doi: 10.1016/j.clim.2005.10.014
43. Kavanagh D, Goodship TH, Richards A. Atypical haemolytic uraemic syndrome. Br Med Bull. (2006) 77–78:5–22. doi: 10.1093/bmb/ldl004
44. Hill A, Richards SJ, Hillmen P. Recent developments in the understanding and management of paroxysmal nocturnal haemoglobinuria. Br J Haematol. (2007) 137:181–92. doi: 10.1111/j.1365-2141.2007.06554.x
45. Risitano AM. Paroxysmal nocturnal hemoglobinuria and the complement system: recent insights and novel anticomplement strategies. Adv Exp Med Biol. (2013) 735:155–72. doi: 10.1007/978-1-4614-4118-2_10
46. Ninomiya H, Sims PJ. The human complement regulatory protein CD59 binds to the alpha-chain of C8 and to the “b”domain of C9. J Biol Chem. (1992) 267:13675–80.
47. Falgarone G, Chiocchia G. Chapter 8: clusterin: a multifacet protein at the crossroad of inflammation and autoimmunity. Adv Cancer Res. (2009) 104:139–70. doi: 10.1016/S0065-230X(09)04008-1
48. Podack ER, Kolb WP, Muller-Eberhard HJ. The SC5b-7 complex: formation, isolation, properties, and subunit composition. J Immunol. (1977) 119:2024–9.
49. Bellac CL, Dufour A, Krisinger MJ, Loonchanta A, Starr AE, Auf dem Keller U, et al. Macrophage matrix metalloproteinase-12 dampens inflammation and neutrophil influx in arthritis. Cell Rep. (2014) 9:618–32. doi: 10.1016/j.celrep.2014.09.006
50. Campbell WD, Lazoura E, Okada N, Okada H. Inactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase N. Microbiol Immunol. (2002) 46:131–4. doi: 10.1111/j.1348-0421.2002.tb02669.x
51. Harold FM. Inorganic polyphosphates in biology: structure, metabolism, and function. Bacteriol Rev. (1966) 30:772–94.
52. Kornberg A, Rao NN, Ault-Riche D. Inorganic polyphosphate: a molecule of many functions. Ann Rev Biochem. (1999) 68:89–125. doi: 10.1146/annurev.biochem.68.1.89
53. Brown MR, Kornberg A. The long and short of it - polyphosphate, PPK and bacterial survival. Trends Biochem Sci. (2008) 33:284–90. doi: 10.1016/j.tibs.2008.04.005
54. Ruiz FA, Lea CR, Oldfield E, Docampo R. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J Biol Chem. (2004) 279:44250–7. doi: 10.1074/jbc.M406261200
55. Muller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. (2009) 139:1143–56. doi: 10.1016/j.cell.2009.11.001
56. Montilla M, Hernandez-Ruiz L, Garcia-Cozar FJ, Alvarez-Laderas I, Rodriguez-Martorell J, Ruiz A. Polyphosphate binds to human von Willebrand factor in vivo and modulates its interaction with Glycoprotein Ib. J Thromb Haemost. (2012) 10:2315–23. doi: 10.1111/jth.12004
57. Smith SA, Mutch NJ, Baskar D, Rohloff P, Docampo R, Morrissey JH. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci USA. (2006) 103:903–8. doi: 10.1073/pnas.0507195103
58. Smith SA, Morrissey JH. Polyphosphate enhances fibrin clot structure. Blood. (2008) 112:2810–6. doi: 10.1182/blood-2008-03-145755
59. Smith SA, Choi SH, Davis-Harrison R, Huyck J, Boettcher J, Rienstra CM, et al. Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood. (2010) 116:4353–9. doi: 10.1182/blood-2010-01-266791
60. Smith SA, Morrissey JH. Polyphosphate: a new player in the field of hemostasis. Curr Opin Hematol. (2014) 21:388–94. doi: 10.1097/MOH.0000000000000069
61. Travers RJ, Smith SA, Morrissey JH. Polyphosphate, platelets, and coagulation. Int J Lab Hematol. (2015) 37 (Suppl. 1):31–5. doi: 10.1111/ijlh.12349
62. Maas C, Renne T. Coagulation factor XII in thrombosis and inflammation. Blood. (2018) 131:1903–9. doi: 10.1182/blood-2017-04-569111
63. Mutch NJ, Myles T, Leung LL, Morrissey JH. Polyphosphate binds with high affinity to exosite II of thrombin. J Thromb Haemost. (2010) 8:548–55. doi: 10.1111/j.1538-7836.2009.03723.x
64. Mutch NJ, Engel R, Uitte de Willige S, Philippou H, Ariens RA. Polyphosphate modifies the fibrin network and down-regulates fibrinolysis by attenuating binding of tPA and plasminogen to fibrin. Blood. (2010) 115:3980–8. doi: 10.1182/blood-2009-11-254029
65. Travers RJ, Shenoi RA, Kalathottukaren MT, Kizhakkedathu JN, Morrissey JH. Nontoxic polyphosphate inhibitors reduce thrombosis while sparing hemostasis. Blood. (2014) 124:3183–90. doi: 10.1182/blood-2014-05-577932
66. Wijeyewickrema LC, Lameignere E, Hor L, Duncan RC, Shiba T, Travers RJ, et al. Polyphosphate is a novel cofactor for regulation of complement by a serpin, C1 inhibitor. Blood. (2016) 128:1766–76. doi: 10.1182/blood-2016-02-699561
67. Lameignere E, Wijeyewickrema L, O'Byrne A, Ocariza LM, Shiba T, Smith S, et al. Polyphosphate acts as a cofactor for C1-inhibitor-mediated regulation of the classical pathway of complement. J Thromb Haemost. (2015) (Suppl. 2):616.
68. Olson ST, Richard B, Izaguirre G, Schedin-Weiss S, Gettins PG. Molecular mechanisms of antithrombin-heparin regulation of blood clotting proteinases. A paradigm for understanding proteinase regulation by serpin family protein proteinase inhibitors. Biochimie. (2010) 92:1587–96. doi: 10.1016/j.biochi.2010.05.011
69. Wat J, Foley JH, Krisinger MJ, Ocariza LM, Lei V, Wasney G, et al. Polyphosphate suppresses complement via the terminal pathway. Blood. (2014) 123:768–76. doi: 10.1182/blood-2013-07-515726
70. Castuma CE, Huang R, Kornberg A, Reusch RN. Inorganic polyphosphates in the acquisition of competence in Escherichia coli. J Biol Chem. (1995) 270:12980–3. doi: 10.1074/jbc.270.22.12980
71. Moreno SN, Docampo R. Polyphosphate and its diverse functions in host cells and pathogens. PLoS Pathog. (2013) 9:e1003230. doi: 10.1371/journal.ppat.1003230
72. Gray MJ, Wholey WY, Wagner NO, Cremers CM, Mueller-Schickert A, Hock NT, et al. Polyphosphate is a primordial chaperone. Mol Cell. (2014) 53:689–99. doi: 10.1016/j.molcel.2014.01.012
73. Zhang Q, Li Y, Tang CM. The role of the exopolyphosphatase PPX in avoidance by Neisseria meningitidis of complement-mediated killing. J Biol Chem. (2010) 285:34259–68. doi: 10.1074/jbc.M110.154393
74. Schmaier AH, Murray SC, Heda GD, Farber A, Kuo A, McCrae K, et al. Synthesis and expression of C1 inhibitor by human umbilical vein endothelial cells. J Biol Chem. (1989) 264:18173–9.
75. Schroder HC, Kurz L, Muller WE, Lorenz B. Polyphosphate in bone. Biochemistry. (2000) 65:296–303.
76. Brandt S, Krauel K, Jaax M, Renne T, Helm CA, Hammerschmidt S, et al. Polyphosphates form antigenic complexes with platelet factor 4 (PF4) and enhance PF4-binding to bacteria. Thromb Haemost. (2015) 114:1189–98. doi: 10.1160/TH15-01-0062
77. Labberton L, Long AT, Gendler SJ, Snozek CL, Stavrou EX, Nickel KF, et al. A flow cytometry-based assay for procoagulant platelet polyphosphate. Cytometry. (2018) 94:369–73. doi: 10.1002/cyto.b.21492
78. Schmaier AH, Smith PM, Colman RW. Platelet C1- inhibitor. A secreted alpha-granule protein. J Clin Invest. (1985) 75:242–50. doi: 10.1172/JCI111680
79. Hurford MT, Sebastiano C. Hermansky-pudlak syndrome: report of a case and review of the literature. Int J Clin Exp Pathol. (2008) 1:550–4.
80. Hernandez-Ruiz L, Saez-Benito A, Pujol-Moix N, Rodriguez-Martorell J, Ruiz FA. Platelet inorganic polyphosphate decreases in patients with delta storage pool disease. J Thromb Haemost. (2009) 7:361–3. doi: 10.1111/j.1538-7836.2008.03238.x
81. Terashima-Hasegawa M, Ashino T, Kawazoe Y, Shiba T, Manabe A, Numazawa S. Inorganic polyphosphate protects against lipopolysaccharide-induced lethality and tissue injury through regulation of macrophage recruitment. Biochem Pharmacol. (2018) 159:96–105. doi: 10.1016/j.bcp.2018.11.017
Keywords: innate immunity, thrombosis, inflammation, coagulation, age-related macular degeneration, mouse models, C1-esterase inhibitor, membrane attack complex
Citation: Conway EM (2019) Polyphosphates and Complement Activation. Front. Med. 6:67. doi: 10.3389/fmed.2019.00067
Received: 10 January 2019; Accepted: 18 March 2019;
Published: 04 April 2019.
Edited by:
Alvin H. Schmaier, Case Western Reserve University, United StatesReviewed by:
Wei Li, Marshall University, United StatesGary Eugene Gilbert, Harvard Medical School, United States
Copyright © 2019 Conway. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edward M. Conway, ZWQuY29ud2F5QHViYy5jYQ==