 Vik Meadows
Vik Meadows Lindsey Kennedy
Lindsey Kennedy Debjyoti Kundu
Debjyoti Kundu Gianfranco Alpini
Gianfranco Alpini Heather Francis
Heather Francis- 1Richard L. Roudebush VA Medical Center, Indiana University School of Medicine, Indianapolis, IN, United States
- 2Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, IN, United States
- 3Division of Gastroenterology and Hepatology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, United States
In the past ten years, our understanding of the importance of bile acids has expanded from fat absorption and glucose/lipid/energy homeostasis into potential therapeutic targets for amelioration of chronic cholestatic liver diseases. The discovery of important bile acid signaling mechanisms, as well as their role in metabolism, has increased the interest in bile acid/bile acid receptor research development. Bile acid levels and speciation are dysregulated during liver injury/damage resulting in cytotoxicity, inflammation, and fibrosis. An increasing focus to target bile acid receptors, responsible for bile acid synthesis and circulation, such as Farnesoid X receptor and apical sodium-dependent bile acid transporter to reduce bile acid synthesis have resulted in clinical trials for treatment of previously untreatable chronic liver diseases such as non-alcoholic steatohepatitis and primary sclerosing cholangitis. This review focuses on current bile acid receptor mediators and their effects on parenchymal and non-parenchymal cells. Attention will also be brought to the gut/liver axis during chronic liver damage and its treatment with bile acid receptor modulators. Overall, these studies lend evidence to the importance of bile acids and their receptors on liver disease establishment and progression.
Introduction
Focal studies of hepatic secretion led to the critical analysis and understanding of bile and its circulation connecting the liver and intestine (1). Bile acids (BAs) are heterogenous compounds whose chemical and amphipathic properties result from the enzymatic breakdown of insoluble cholesterol (2). Since their isolation from bile, the field of BA chemistry has provided intensive study of their complex chemical nature and the physiological effects of their dynamic composition in circulating bile (2–5).
Hepatic BA build up leads to inflammation, necrosis, and apoptosis of various liver cells which then affects BA synthesis and transport perpetuating BA-induced damage (2, 3). Due to the extensive knowledge and research concerning hepatic BA formation and secretion, it is natural to assume BAs contribute to chronic liver damage. The increased synthesis of BAs in combination with interrupted BA signaling can lead to adverse effects in patients of chronic liver diseases. It is estimated that 1.5 billion people worldwide suffer from chronic liver diseases whose complications, cirrhosis and liver cancer, result in 2 million deaths globally (6). Chronic liver diseases have an increased burden in the health care system due to the frequent hospital readmissions, accompanying hepatic decompensation, and risk for infection as liver damage progresses (6). Many chronic liver diseases, such as primary sclerosing cholangitis (PSC) and non-alcoholic fatty liver disease (NAFLD), have minimal treatment options requiring liver transplantation as the only permanent remedy (6, 7).
Various therapeutics have been FDA-approved for clinical trials aiming to improve liver function and relieve adverse effects of disrupted BA signaling. This article serves as a brief review of BA signaling and function in various chronic liver diseases and their regulation of the gut microbiome.
Circulation, Modification, and Function of Bile Acids
Bile Acid Synthesis and Transport
The catabolism of cholesterol into BAs in the parenchyma is tightly regulated by over 17 enzymes, preferentially expressed in the liver, that are involved in the synthesis and alteration of BAs into bile salts (8, 9). Impairment of these mechanisms can result in cholestasis, liver damage, impaired lipid metabolism, and other maladies (3, 5, 8). Modifications and conjugations of BAs affect their solubility, hydrophobicity, and receptor binding affinity (8, 10). BA synthesis and conjugations are summarized in Figure 1 (reprinted with permission from Molinaro et al. Trends Endocrinol Metab).
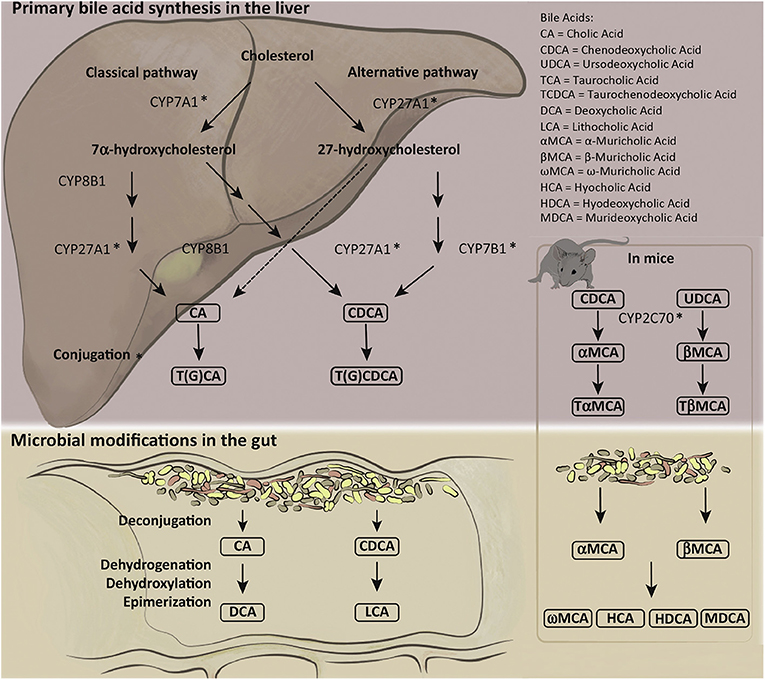
Figure 1. Human and mouse bile acid synthesis and conjugation. Cholesterol catabolism results in the creation of primary bile acids (BAs) through either the classical pathway, accomplished by Cytochrome P450 7A1 (CYP7A1), or the alternative/acidic pathway, conducted by Cytochrome P450 27A1 (CYP27A1). Alteration by various cytochrome P450 family of enzymes allows for the creation of cholic acid (CA) and chenodeoxycholic acid (CDCA). Primary BAs can become conjugated to glycine or taurine prior to secretion to the biliary ductules. Deconjugation and reconjugation occurs in the distal ileum through bacterial intervention creating secondary BAs: lithocholic acid (LCA) or deoxycholic acid (DCA). Mice have additional primary BAs: Ursodeoxycholic acid (UDCA) and α- and β-muricholic acid. The murine specific primary BAs created by cytochrome P450 2C70 (CYP2C70) can also be conjugated to glycine or taurine prior to secretion into bile duct and alteration by gut microbiota into secondary BAs. Figure reprinted with permission from Molinaro et al. Trends Endocrinol Metab.
Bile acids are secreted into the bile canaliculi by hepatocytes, draining to the bile ducts located in the portal triad. Cholangiocytes are the epithelial cells lining the bile ducts and assist in BA modification and circulation by cholehepatic shunting, the process in which BAs are reabsorbed from the bile and returned to hepatocytes (11–14). The intrahepatic bile duct system allows BAs to flow into the intestinal lumen through the common hepatic duct in response to food ingestion to assist in emulsification, metabolism and absorption of dietary lipids and fat-soluble vitamins (A, D, E, and K). Alternatively, BAs are deposited in the gallbladder for storage and prevention of cholesterol-crystallization and gallstone formation (10, 15). Approximately 95% of BAs are reabsorbed in the distal ileum by the apical sodium-dependent bile acid transporter (ASBT; alternatively known as ileal bile acid transporter, IBAT) and delivered to the liver through the portal system via enterohepatic or portal circulation (16–18). The gut microbiome is capable of deconjugating primary bile acids and converting them to secondary bile acids prior to absorption or fecal excretion which affects gut microbiome community, BA pool, and liver health (8, 19, 20).
Aside from their important roles in digestion, BAs can behave as signaling molecules in carbohydrate and lipid metabolism, energy expenditure, and hepatic disease (20–22). BAs and activation of their downstream targets including G-protein-coupled bile acid receptor (TGR5), transforming growth factor-α (TGF-α) and sphingosine-1-phosphate receptor-2 (S1PR2) stimulate cholangiocyte proliferation and contribute to the progression of cholangiocarcinoma [CCA, in vivo and in vitro (21–25)]. Alternatively, Farnesoid X Receptor (FXR) is down regulated in hepatocellular carcinoma (HCC) (26). It has been shown that FXR via increased CYP450 epoxygenase activity suppress NF-κB signaling thereby reducing hepatic inflammation (27, 28). Further exploration into the anti-inflammatory role of FXR and assessment of BA direct or indirect targets may provide understanding of chronic cholestatic disease establishment and progression.
Intrahepatic and Extrahepatic Bile Acid Modification
The catabolism of cholesterol results in the formation of the primary BAs, cholic acid (CA) or chenodeoxycholic acid (CDCA), through the major (classical) pathway or the minor (alternative/acidic) pathway, respectively (29). Cholehepatic shunting alters the BA pool via biliary ASBT transport, multidrug resistance cassette 3 (MDR3, human; multidrug resistance cassette 2, mice), and organic solute transporter α-β (OSTα-β) BA secretion into the peribiliary plexus prior to reaching the hepatic sinusoids (30). Ileal bile acid binding protein (IBABP) is expressed in large cholangiocytes to sequester BAs preventing biliary cytotoxicity (30, 31). CA and CDCA/Ursodeoxycholic acid (UDCA) are converted to deoxycholic acid (DCA) and lithocholic acid (LCA), respectively, via 7α/β-dihydroxylation by various species of the commensal gut microbiota in the gastrointestinal tract (32). Human secondary BAs (DCA and LCA) are capable of being circulated back to the liver via enterohepatic circulation leading to an increased hepatic levels of damaging hydrophobic BAs (32).
Dysregulation of Bile Acids in Chronic Liver Diseases
PSC and PBC
PSC and Primary Biliary Cholangitis (PBC) are rare cholestatic liver diseases that affect the biliary system. PSC is an idiopathic disease with cholestasis, inflammation and eventual fibrosis resulting from strictures of intra- and extrahepatic bile ducts (33). PSC is one of the most common causes for liver transplantation (LT) (33). Due to its heterogenous and spontaneous progression, effective medical therapies have not yet been developed (33). Fat-soluble vitamin deficiency can occur in PSC patients as a result of decreased bile flow and secretion. It has also been shown that PSC has a positive correlation with ulcerative colitis (UC), a form of inflammatory bowel disease (IBD). PSC/IBD patients display altered BA fecal excretion and decreased gut microbiome diversity compared to healthy or IBD patient controls (34). Patients with PSC have decreased expression of hepatic FXR, TGR5, and S1PR2 (35). The multidrug resistance cassette 2 knock-out mouse (MDR2−/−) is a mouse model utilized to mimic the PSC phenotype including increased cholestasis, intrahepatic bile duct mass and hepatic inflammation due to hepatic BA build up (36, 37). This murine model has been useful for identifying effects of potential therapeutics, such as UDCA. Meng et al. reported that UDCA treatment in Mdr2−/− mice reduced serum TBA, elevated hepatic expression of BA transporters, and reduced hepatic inflammation and collagen deposition (36).
PBC is a chronic auto-immune disease, predominantly affecting middle-aged women, that results in biliary ductopenia and cholestasis. Li et al. reported elevated serum levels of total BAs (TBA) and FGF19 in cirrhotic PBC patients compared to healthy controls and non-cirrhotic PBC patients (38). Similarly, Trottier et al. demonstrated elevated BAs in serum samples from both PBC and PSC compared to healthy controls (39). Ursodeoxycholic acid (UDCA), an epimer of CDCA, was the first FDA-approved treatment for PBC. Despite increased bile flow, lower liver enzyme levels, and decreased serum BA levels, one in three PBC patients will have a limited or no response to treatment, strengthening the need for effective therapeutic intervention of PBC progression (33, 40–45).
NAFLD
NAFL and non-alcoholic steatohepatitis (NASH) are two of the most common hepatic diseases worldwide due to an increase of sedentary lifestyle and consumption of a high-fat/high-cholesterol diet (46, 47). Its prevalence has demonstrated positive correlation with an increasing number of obese and type II diabetic patients (46). Currently, there are no approved therapies for the treatment of NAFL and NASH aside from a change of diet and exercise for gradual weight loss.
BA signaling is disrupted in NAFL and NASH patients yielding great interest in the search for exogenous methods of BA regulation (48, 49). Mouzaki et al. uncovered greater fecal BA secretion and increased primary to secondary BA ratio in NASH patients compared to healthy controls (49). Ferslew et al. found elevated serum BAs in NASH patients, compared to healthy controls, with an increase of taurine- and glycine-conjugated BAs (48). Benedict and Zhang proposed that FXR suppression of hepatic inflammation may ameliorate NAFLD progression (50).
HCC and CCA
HCC is currently the third leading cause of cancer deaths worldwide. HCC affects parenchymal cells in the liver, which make up to 70% of the liver tissue. HCC develops in chronic liver disease or cirrhotic liver patients and is usually detected through various imaging methods prior to diagnosis. Patients with HCC can be asymptomatic or present with a range of symptoms including cirrhosis-related pain. The underlying chronic liver injury, and difficulty in diagnosis, both contribute to HCCs high mortality. CCA is a rare but devastating cancer with poor prognosis. Patients present with jaundice, pruritus (intense itch) and acholic (pale) stool due to reduced bile and bilirubin excretion. Due to the intimate relationship between hepatocytes, cholangiocytes and BAs it is important to investigate BA signaling in the establishment and progression of HCC and CCA.
Demonstrating a shift in the BA pool during cancer development, Changbumrung et al. reported elevated ratios for trihydroxy to dihydroxy BAs and for glycine-conjugated to taurine-conjugated BAs in patients with HCC and CCA compared to healthy patients (51). Luo et al. demonstrated a similar trend with elevated glycine-conjugated BAs in hepatic injury patients, ranging from hepatitis B viral infection to cirrhosis, compared to healthy controls suggesting taurine-conjugated BAs as a potential sensitive biomarker for liver injury (52). This increase in BA pool size is due to reduced inhibition of BA synthesis. FXR activation has been indicated to have anti-cancer properties, with decreased expression in progressing human HCC lesions, since its downstream effects include inhibition of BA synthesis and cell proliferation (26, 53). Guo et al. reported decreased FXR expression in HCC tumor lesions, indicating a hindering role in HCC development and progression (26). Wolfe et al. found a decrease of FXR expression in HCC tumor lesions, compared to normal liver tissue, with increasing tumor development stage (53). Similarly, Liu et al. reported decreased small heterodimer partner (SHP) and FXR expression in human HCC compared to paired healthy control (54). Erice et al. demonstrated FXR expression in CCA samples negatively correlates with advanced cancer progression and lymph node invasion. In contrast to FXR, TGR5 expression is elevated in CCA tumors compared to controls (25). This discovery is likely due to cholangiocyte requirement of TGR5 for BA-induced proliferation and anti-apoptosis signaling (55, 56). Additional BA receptors, such as S1PR2, have been found to have elevated expression in human CCA tumors, as shown by Liu et al. (57). Taken together these studies indicate that altered BAs may serve to enhance HCC and CCA progression during disease development.
Current Bile Acid-Related Therapies of Chronic Liver Diseases
Bile Acid Therapies
The use of synthetic or naturally occurring BAs to reduce gallstone formation, aid in lipid absorption following gastric surgeries, and assist in reduction of cholestasis has increased in recent years. UDCA is a hydrophilic BA conjugated to undergo enterohepatic circulation or deconjugated and converted into LCA by the gut microbiome and excreted into feces. UDCA was the first FDA-approved therapy for PBC patients exhibiting altered serum liver enzyme levels (41). UDCA has been a staple therapy due to its ability to prevent gallstone formation, increasing bicarbonate secretion to prevent acidification of bile, and positive side effect profile as compared to treatment with CDCA (18, 41, 58). In many PBC patients, long-term UDCA treatment when administered at early stage of disease increases survival rate, lowers liver enzyme serum levels, and improves liver histology (41, 43, 59, 60). Regardless of its beneficial effects, 30–40% of PBC patients do not respond to UDCA treatment.
UDCA treatment can improve liver histology and serum ALT/ASP levels in PSC patients, however, data supporting any long-term efficacy or long-term survival are lacking (61). Moreover, UDCA prescribed at high doses increased medical complications and mortality in PSC patients (61–64). PSC patients treated with norUDCA, a side chain shortened homolog of UDCA, exhibited reduced ALP serum levels compared to placebo in a dose-dependent manner, however norUDCA's effects on PSC progression, long-term survival, and mortality have not been investigated (65).
Bile Acid Receptor/Transporter Agonists and Antagonists
Obeticholic acid (OCA, Ocaliva) is an FDA-approved, synthetic derivative of CDCA and is a high affinity ligand for the nuclear bile acid receptor, FXR (40). FXR is a nuclear BA receptor that when expressed is capable of reducing BA synthesis, increasing expression of BA transporters and modulating lipoprotein metabolism (66). OCA has been studied as a potential therapeutic drug in the treatment of various chronic liver diseases (40, 44). Following Phase II and Phase III clinical trials, it has been suggested that OCA treatment can be delivered in conjunction with UDCA in PBC, or as a monotherapy in patients who do not tolerate UDCA, respectively (67, 68). Adverse events including pruritus occurred in nearly all patients and was observed to occur in a dose dependent manner (67, 68). The beneficial effects of OCA as a monotherapy are currently being investigated in various clinical trials (7, 67, 69). The use of OCA has promising effects on improving liver enzyme levels, lowering plasma bilirubin and IgM levels, and has recently been shown to improve or stabilize liver fibrosis and biliary injury in PBC patients with cirrhosis (68, 69). Despite promising outcomes in PBC patients, long-term effects of OCA on disease progression and patient survival are still being investigated.
In the Farnesoid X Receptor Ligand Obeticholic Acid in NASH Treatment (FLINT) clinical trial, NASH patients treated with OCA presented with improved fibrosis scores, elevated low-density lipoprotein (LDL) and increased average weight loss compared to placebo control (46, 70). NASH patients treated with OCA also had increased ALP levels compared to placebo control (46). The effects of OCA on insulin resistance and the observed improvement of lipid absorption were not sustained following termination of OCA treatment (46). Further investigation into the short-term and long-term effects of OCA on liver function and injury are warranted and are actively being explored in the Randomized Global Phase III Study to Evaluate the Impact on NASH with Fibrosis of Obeticholic Acid Treatment (REGENERATE) clinical trial assessing FXR activation in the treatment of NASH (71). Translational Research and Evolving Alcoholic hepatitis Treatment (TREAT) consortium conducted phase II clinical trial utilizing OCA on patients with moderately severe alcoholic hepatitis (AH) that was completed in 2018, however currently no data is available on this study (72). The Phase 3 Study of Obeticholic Acid in Patients with Primary Biliary Cirrhosis (POISE) double-blind, placebo controlled clinical trial has been one of the few to publish results demonstrating OCA's long-term safety and effectiveness (69). POISE study data showed that three-year OCA treatment reduced or stabilized hepatic collagen deposition and ductular injury in PBC patients with cirrhosis (69). The Combination OCA and Statins for Monitoring of Lipids (CONTROL) clinical trial study found increased LDL in NASH patients treated with OCA (5, 10, or 25 mg) indicating altered lipid metabolism (73). The observed increase in LDL cholesterol was reduced in NASH patients concurrently treated with OCA and atorvastatin, a statin utilized to reduce endogenous cholesterol production (73). The authors noted that these two drugs were well-tolerated when utilized together, addressing the observed increase in LDL cholesterol following OCA treatment in NASH patients, but remained inconclusive with respect to the combinatorial effect on hepatic injury (70, 73). Eaton et al. observed that decompensated patients with cirrhotic PSC and PBC, OCA treatment led to the development of jaundice and elevated liver enzyme levels (74). While the long-term benefits of OCA are still being evaluated in various liver diseases, it is still being explored as a potential.
During normal enterohepatic circulation, IBAT/ASBT is responsible for the reabsorption of BAs in the intestinal tract prior to secretion into the portal blood system (75). Additionally, ASBT is responsible for cholehepatic shunting of BAs between cholangiocytes and hepatocytes, which ultimately increases hepatic BA pool. A common symptom of chronic cholestatic liver disease is pruritus, intense itching of the dermis due to increased BA deposition (75). IBAT/ASBT inhibitors are potential therapeutic candidates for this detrimental symptom (76). Pilot studies conducted with various synthetic IBAT/ASBT inhibitors have demonstrated variable results. The Al-Dury et al. study reported patients who continued through A4250 (synthetic IBAT/ASBT inhibitor) received relief from itching, but pruritus returned during wash-out and BA sequestrant treatment. Despite improvement of pruritus and lowered serum BAs, many patients in the A4250 study dropped out early due to abdominal pain and diarrhea (76). Similar benefits were identified in the pilot study of GSK2330672 (synthetic IBAT/ASBT inhibitor), PBC patients resulted with lowered serum BAs and increased serum FGF19. The most common adverse events in this treatment cohort were headaches and diarrhea, which provides reason to limit the treatment length in patients with pruritus (77).
BA Receptor Agonist Effects on the Gut/Liver Axis During Liver Disease
BA species affect and regulate gut microbial species composition (32). BA species and concentrations in different portions of the gastrointestinal tract can result in increased side effects including increased intestinal permeability and BA-induced diarrhea (78). Elevated hydrophobic BAs in the colon are capable of inducing inflammation which is reduced following CDCA treatment alleviating the increased toxicity from insoluble BA concentrations and mast cell secretory factors (i.e., histamine or nerve growth factor NGF) (78, 79).
A small cohort of PBC patients exhibited altered gut microbiome composition, compared to healthy controls, which was partially reversed following 6-month UDCA treatment (80). This study found eight-PBC associated genera and a reduction in normal gut microbiome associated microbial members including Faecalibacterium, Bacteroides, Sutterella, and Oscillospira spp. A study from Selmi et al. implicated increased N. aromaticivorans population is responsible for the induction of PBC (81). Tang et al. reported increased epithelial infiltration of Enterobacteriaceae in PBC patients indicating increased permeability and decreased gastrointestinal immune response in diseased patients (80).
The gastrointestinal epithelial barrier plays an important role in maintaining BA enterohepatic circulation homeostasis and reduced inflammation from lipopolysaccharide (LPS) leakage. BA circulation is necessary to avoid damaging increases in colonic BAs that result in inflammation and intestinal damage (78). Song et al. found that CDCA was capable of reducing paracellular permeability and increased cell-to-cell tight junctions via FXR activation in mice (82). Removal of commensal gut microbiota in Mdr2−/− mice resulted in increased serum liver enzyme levels, cholangiocyte senescence, and circulating primary BAs (37). Alternatively, Li et al. discovered a depletion of BA induced damage to the colon and reduced mast cell activation and degranulation in FXR−/− mice or Z-guggulsterone treated mast cells (79).
The role of the gut microbiome in BA homeostasis is still being investigated. The disruption of the gut-liver axis can allow infiltration of microbes, or their metabolic products, into the enterohepatic circulation to prolong disease (83). Chronic liver disease patients all maintain their own gut microbiome signature that inherently play a role in disease development. Future studies of BA-associated therapeutics should consider effects on the gut microbiome and their metabolites in relation to changing BA pool/circulation and chronic liver disease.
Conclusion
BA signaling, and therapeutics that alter them, have effects on the gut microbiome and organs outside of the liver. The use of BA receptor agonists and antagonists and their indirect effects (BA pool, synthesis, circulation, and the gut microbiome composition) is summarized in Figure 2. The human and mouse gut microbiome fluctuates with the increase or decrease of BAs, which can lead to increased inflammation and intestinal malabsorption (19, 37, 80, 84–86). In depth exploration of BA signaling and circulation during chronic liver diseases may provide insight to disease amelioration or treatment strategies. Understanding the role of the gut microbiome on BA modification and circulation may allow for innovative concurrent treatment for the reductions of inopportune side effects of the currently investigated treatments, OCA and UDCA.
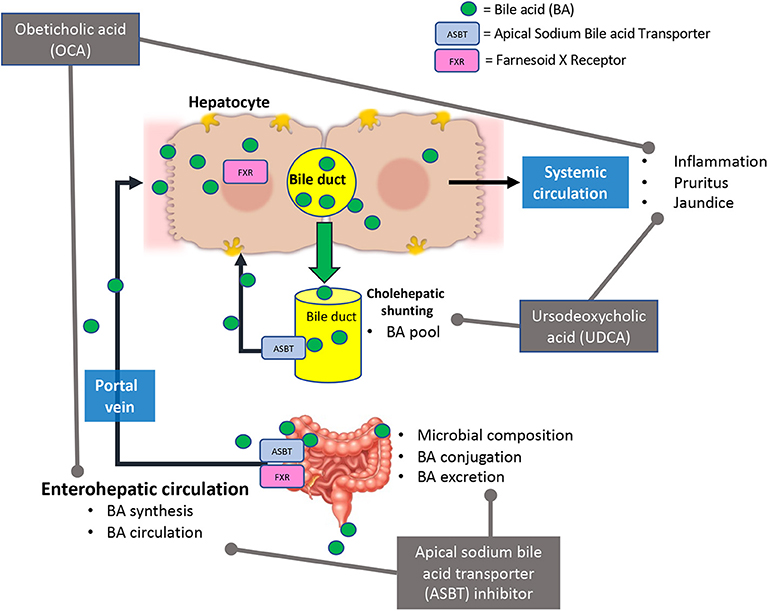
Figure 2. Current bile acid receptor therapeutics and their effects in bile acid signaling. Briefly, Obeticholic acid (OCA) reduces bile acid (BA) synthesis, circulating BA levels, and hepatic inflammation. This treatment may cause adverse effects such as pruritus and jaundice (in decompensated PBC patients). Ursodeoxycholic acid (UDCA) alters BA pool and reduced hepatic inflammation and damage. In some cases, UDCA treatment has led to development of pruritus. Apical sodium bile acid transporter (ASBT) inhibitors affect bile acid synthesis, circulation, secretion, and microbial composition in the intestine.
Author Contributions
VM and HF drafted manuscript. VM, LK, DK, GA, and HF edited and approved final version of manuscript. VM and HF arranged figures.
Funding
The studies were supported by a SRCS Award to GA, VA Merit Awards (1I01BX003031, HF; 4I01BX000574, GA) from the United States Department of Veteran's Affairs, Biomedical Laboratory Research and Development Service and NIH NIDDK grants (DK108959 and DK119421, HF) along with funds from Indiana University School of Medicine (GA and HF), Indianapolis, Indiana.
Disclaimer
The content is the responsibility of the authors alone and does not necessarily reflect the views or policies of the Department of Veterans Affairs or the United States Government.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ALKP/ALP, Alkaline phosphatase; ASBT, apical sodium bile acid transporter; AST, aspartate aminotransferase; Bas, bile acids; FXR, Farnesoid X receptor; CA, cholic acid; CCA, cholangiocarcinoma; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; HCC, hepatocellular carcinoma; IBABP, ileal bile acid binding protein; IBD, irritable bowel disease; IBAT, ileal bile acid transporter; LCA, lithocholic acid; LDL, low density lipoprotein; MDR2, multidrug resistance cassette 2; MDR3, multidrug resistance cassette 3; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; OCA, Obeticholic acid; OSTα-β, organic solute transporter α-β; PBC, primary biliary cholangitis; PSC, primary sclerosing cholangitis; TBA, total bile acid; TGR5, Takeda G protein coupled receptor 5; UDCA, Ursodeoxycholic acid.
References
1. Reuben A. The biliary cycle of Moritz Schiff. Hepatology. (2005) 42:500–5. doi: 10.1002/hep.20823
2. Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. (2008) 65:2461–83. doi: 10.1007/s00018-008-7568-6
3. Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. (2009) 50:1955–66. doi: 10.1194/jlr.R900010-JLR200
4. Chiang JY. Bile acid metabolism and signaling. Compr Physiol. (2013) 3:1191–212. doi: 10.1002/cphy.c120023
5. Hofmann AF, Hagey LR. Key discoveries in bile acid chemistry and biology and their clinical applications: history of the last eight decades. J Lipid Res. (2014) 55:1553–95. doi: 10.1194/jlr.R049437
6. Moon AM, Singal AG, Tapper EB. Contemporary epidemiology of chronic liver disease and cirrhosis. Clin Gastroenterol Hepatol. (2019). doi: 10.1016/j.cgh.2019.07.060. [Epub ahead of print].
7. Jhaveri MA, Kowdley KV. New developments in the treatment of primary biliary cholangitis - role of obeticholic acid. Ther Clin Risk Manag. (2017) 13:1053–60. doi: 10.2147/TCRM.S113052
8. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. (2003) 72:137–74. doi: 10.1146/annurev.biochem.72.121801.161712
9. Liu Y, Rong Z, Xiang D, Zhang C, Liu D. Detection technologies and metabolic profiling of bile acids: a comprehensive review. Lipids Health Dis. (2018) 17:121. doi: 10.1186/s12944-018-0774-9
10. de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab. (2013) 17:657–69. doi: 10.1016/j.cmet.2013.03.013
11. Hofmann AF. Current concepts of biliary secretion. Dig Dis Sci. (1989) 34: 16S−20. doi: 10.1007/BF01536657
12. Glaser SS, Alpini G. Activation of the cholehepatic shunt as a potential therapy for primary sclerosing cholangitis. Hepatology. (2009) 49:1795–7. doi: 10.1002/hep.22969
13. Hofmann AF, Zakko SF, Lira M, Clerici C, Hagey LR, Lambert KK, et al. Novel biotransformation and physiological properties of norursodeoxycholic acid in humans. Hepatology. (2005) 42:1391–8. doi: 10.1002/hep.20943
14. Alpini G, Glaser S, Baiocchi L, Francis H, Xia X, Lesage G. Secretin activation of the apical Na+-dependent bile acid transporter is associated with cholehepatic shunting in rats. Hepatology. (2005) 41:1037–45. doi: 10.1002/hep.20653
15. Cai JS, Chen JH. The mechanism of enterohepatic circulation in the formation of gallstone disease. J Membr Biol. (2014) 247:1067–82. doi: 10.1007/s00232-014-9715-3
16. Chiang JY. Recent advances in understanding bile acid homeostasis. F1000Res. (2017) 6:2029. doi: 10.12688/f1000research.12449.1
17. Chiang JY. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. J Hepatol. (2004) 40:539–51. doi: 10.1016/j.jhep.2003.11.006
18. Tonin F, Arends I. Latest development in the synthesis of ursodeoxycholic acid (UDCA): a critical review. Beilstein J Org Chem. (2018) 14:470–83. doi: 10.3762/bjoc.14.33
19. Kang JD, Myers CJ, Harris SC, Kakiyama G, Lee IK, Yun BS, et al. Bile acid 7α-dehydroxylating gut bacteria secrete antibiotics that inhibit clostridium difficile: role of secondary bile acids. Cell Chem Biol. (2019) 26:27–34 e4. doi: 10.1016/j.chembiol.2018.10.003
20. Jia ET, Liu ZY, Pan M, Lu JF, Ge QY. Regulation of bile acid metabolism-related signaling pathways by gut microbiota in diseases. J Zhejiang Univ Sci B. (2019) 20:781–92. doi: 10.1631/jzus.B1900073
21. Zhou H, Hylemon PB. Bile acids are nutrient signaling hormones. Steroids. (2014) 86:62–8. doi: 10.1016/j.steroids.2014.04.016
22. Dawson PA. Roles of Ileal ASBT and OSTalpha-OSTbeta in regulating bile acid signaling. Dig Dis. (2017) 35:261–6. doi: 10.1159/000450987
23. Werneburg NW, Yoon JH, Higuchi H, Gores GJ. Bile acids activate EGF receptor via a TGF-alpha-dependent mechanism in human cholangiocyte cell lines. Am J Physiol Gastrointest Liver Physiol. (2003) 285:G31–6. doi: 10.1152/ajpgi.00536.2002
24. Deutschmann K, Reich M, Klindt C, Droge C, Spomer L, Haussinger D, et al. Bile acid receptors in the biliary tree: TGR5 in physiology and disease. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1319–25. doi: 10.1016/j.bbadis.2017.08.021
25. Erice O, Labiano I, Arbelaiz A, Santos-Laso A, Munoz-Garrido P, Jimenez-Aguero R, et al. Differential effects of FXR or TGR5 activation in cholangiocarcinoma progression. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1335–44. doi: 10.1016/j.bbadis.2017.08.016
26. Guo F, Xu Z, Zhang Y, Jiang P, Huang G, Chen S, et al. FXR induces SOCS3 and suppresses hepatocellular carcinoma. Oncotarget. (2015) 6:34606–16. doi: 10.18632/oncotarget.5314
27. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. (2008) 48:1632–43. doi: 10.1002/hep.22519
28. Gai Z, Visentin M, Gui T, Zhao L, Thasler WE, Hausler S, et al. Effects of farnesoid X receptor activation on arachidonic acid metabolism, NF-kB signaling, and hepatic inflammation. Mol Pharmacol. (2018) 94:802–11. doi: 10.1124/mol.117.111047
29. Molinaro A, Wahlstrom A, Marschall HU. Role of bile acids in metabolic control. Trends Endocrinol Metab. (2018) 29:31–41. doi: 10.1016/j.tem.2017.11.002
30. Xia X, Francis H, Glaser S, Alpini G, LeSage G. Bile acid interactions with cholangiocytes. World J Gastroenterol. (2006) 12:3553–63. doi: 10.3748/wjg.v12.i22.3553
31. Luo ZL, Cheng L, Wang T, Tang LJ, Tian FZ, Xiang K, et al. Bile acid transporters are expressed and heterogeneously distributed in rat bile ducts. Gut Liver. (2019) 13:569–75. doi: 10.5009/gnl18265
32. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. (2014) 30:332–8. doi: 10.1097/MOG.0000000000000057
33. Tabibian JH, Bowlus CL. Primary sclerosing cholangitis: a review and update. Liver Res. (2017) 1:221–30. doi: 10.1016/j.livres.2017.12.002
34. Vaughn BP, Kaiser T, Staley C, Hamilton MJ, Reich J, Graiziger C, et al. A pilot study of fecal bile acid and microbiota profiles in inflammatory bowel disease and primary sclerosing cholangitis. Clin Exp Gastroenterol. (2019) 12:9–19. doi: 10.2147/CEG.S186097
35. Song WS, Park HM, Ha JM, Shin SG, Park HG, Kim J, et al. Discovery of glycocholic acid and taurochenodeoxycholic acid as phenotypic biomarkers in cholangiocarcinoma. Sci Rep. (2018) 8:11088. doi: 10.1038/s41598–018-29445-z
36. Meng F, Kennedy L, Hargrove L, Demieville J, Jones H, Madeka T, et al. Ursodeoxycholate inhibits mast cell activation and reverses biliary injury and fibrosis in Mdr2(-/-) mice and human primary sclerosing cholangitis. Lab Invest. (2018) 98:1465–77. doi: 10.1038/s41374-018-0101-0
37. Tabibian JH, O'Hara SP, Trussoni CE, Tietz PS, Splinter PL, Mounajjed T, et al. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology. (2016) 63:185–96. doi: 10.1002/hep.27927
38. Li Z, Lin B, Lin G, Wu Y, Jie Y, Li X, et al. Circulating FGF19 closely correlates with bile acid synthesis and cholestasis in patients with primary biliary cirrhosis. PLoS ONE. (2017) 12:e0178580. doi: 10.1371/journal.pone.0178580
39. Trottier J, Bialek A, Caron P, Straka RJ, Heathcote J, Milkiewicz P, et al. Metabolomic profiling of 17 bile acids in serum from patients with primary biliary cirrhosis and primary sclerosing cholangitis: a pilot study. Dig Liver Dis. (2012) 44:303–10. doi: 10.1016/j.dld.2011.10.025
40. Bowlus CL. Obeticholic acid for the treatment of primary biliary cholangitis in adult patients: clinical utility and patient selection. Hepat Med. (2016) 8:89–95. doi: 10.2147/HMER.S91709
41. Floreani A, Mangini C. Primary biliary cholangitis: old and novel therapy. Eur J Intern Med. (2018) 47:1–5. doi: 10.1016/j.ejim.2017.06.020
42. P.Jansen LM. New therapies target the toxic consequences of cholestatic liver disease. Expert Rev Gastroenterol Hepatol. (2018) 12:277–85. doi: 10.1080/17474124.2018.1424538
43. ter Borg PC, Schalm SW, Hansen BE, van Buuren HR, Dutch PBC Study Group. Prognosis of ursodeoxycholic acid-treated patients with primary biliary cirrhosis. results of a 10-yr cohort study involving 297 patients. Am J Gastroenterol. (2006) 101:2044–50. doi: 10.1111/j.1572-0241.2006.00699.x
44. Trivedi PJ, Hirschfield GM, Gershwin ME. Obeticholic acid for the treatment of primary biliary cirrhosis. Expert Rev Clin Pharmacol. (2016) 9:13–26. doi: 10.1586/17512433.2015.1092381
45. Zhang Y, Jiang R, Zheng X, Lei S, Huang F, Xie G, et al. Ursodeoxycholic acid accelerates bile acid enterohepatic circulation. Br J Pharmacol. (2019) 176:2848–63. doi: 10.1111/bph.14705
46. Makri E, Cholongitas E, Tziomalos K. Emerging role of obeticholic acid in the management of nonalcoholic fatty liver disease. World J Gastroenterol. (2016) 22:9039–43. doi: 10.3748/wjg.v22.i41.9039
47. Benedict M, Zhang X. Non-alcoholic fatty liver disease: an expanded review. World J Hepatol. (2017) 9:715–32. doi: 10.4254/wjh.v9.i16.715
48. Ferslew BC, Xie G, Johnston CK, Su M, Stewart PW, Jia W, et al. Altered bile acid metabolome in patients with nonalcoholic steatohepatitis. Dig Dis Sci. (2015) 60:3318–28. doi: 10.1007/s10620-015-3776-8
49. Mouzaki M, Wang AY, Bandsma R, Comelli EM, Arendt BM, Zhang L, et al. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLoS ONE. (2016) 11:e0151829. doi: 10.1371/journal.pone.0151829
50. Armstrong LE, Guo GL. Role of FXR in liver inflammation during nonalcoholic steatohepatitis. Curr Pharmacol Rep. (2017) 3:92–100. doi: 10.1007/s40495-017-0085-2
51. Changbumrung S, Tungtrongchitr R, Migasena P, Chamroenngan S. Serum unconjugated primary and secondary bile acids in patients with cholangiocarcinoma and hepatocellular carcinoma. J Med Assoc Thai. (1990) 73:81–90.
52. Luo L, Aubrecht J, Li D, Warner RL, Johnson KJ, Kenny J, et al. Assessment of serum bile acid profiles as biomarkers of liver injury and liver disease in humans. PLoS ONE. (2018) 13:e0193824. doi: 10.1371/journal.pone.0193824
53. Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL, Apte U. Increased activation of the Wnt/β-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J Pharmacol Exp Ther. (2011) 338:12–21. doi: 10.1124/jpet.111.179390
54. Liu N, Meng Z, Lou G, Zhou W, Wang X, Zhang Y, et al. Hepatocarcinogenesis in FXR-/- mice mimics human HCC progression that operates through HNF1α regulation of FXR expression. Mol Endocrinol. (2012) 26:775–85. doi: 10.1210/me.2011–1383
55. Reich M, Deutschmann K, Sommerfeld A, Klindt C, Kluge S, Kubitz R, et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut. (2016) 65:487–501. doi: 10.1136/gutjnl-2015-309458
56. Masyuk AI, Huang BQ, Radtke BN, Gajdos GB, Splinter PL, Masyuk TV, et al. Ciliary subcellular localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. Am J Physiol Gastrointest Liver Physiol. (2013) 304:G1013–24. doi: 10.1152/ajpgi.00383.2012
57. Liu R, Zhao R, Zhou X, Liang X, Campbell DJ, Zhang X, et al. Conjugated bile acids promote cholangiocarcinoma cell invasive growth through activation of sphingosine 1-phosphate receptor 2. Hepatology. (2014) 60:908–18. doi: 10.1002/hep.27085
58. Magouliotis DE, Tasiopoulou VS, Svokos AA, Svokos KA, Chatedaki C, Sioka E, et al. Ursodeoxycholic acid in the prevention of gallstone formation after bariatric surgery: an updated systematic review and meta-analysis. Obes Surg. (2017) 27:3021–30. doi: 10.1007/s11695–017-2924-y
59. Pares A, Caballeria L, Rodes J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology. (2006) 130:715–20. doi: 10.1053/j.gastro.2005.12.029
60. Burman BE, Jhaveri MA, Kowdley KV. An update on the treatment and follow-up of patients with primary biliary cholangitis. Clin Liver Dis. (2017) 21:709–23. doi: 10.1016/j.cld.2017.06.005
61. Stiehl A. Ursodeoxycholic acid therapy in treatment of primary sclerosing cholangitis. Scand J Gastroenterol Suppl. (1994) 204:59–61. doi: 10.3109/00365529409103626
62. Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology. (2009) 50:808–14. doi: 10.1002/hep.23082
63. Eaton JE, Silveira MG, Pardi DS, Sinakos E, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Am J Gastroenterol. (2011) 106:1638–45. doi: 10.1038/ajg.2011.156
64. Rudolph G, Gotthardt DN, Kloeters-Plachky P, Kulaksiz H, Schirmacher P, Stiehl A. In PSC with colitis treated with UDCA, most colonic carcinomas develop in the first years after the start of treatment. Dig Dis Sci. (2011) 56:3624–30. doi: 10.1007/s10620–011-1763-2
65. Fickert P, Hirschfield GM, Denk G, Marschall HU, Altorjay I, Farkkila M, et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol. (2017) 67:549–58. doi: 10.1016/j.jhep.2017.05.009
66. Xu Y, Li F, Zalzala M, Xu J, Gonzalez FJ, Adorini L, et al. Farnesoid X receptor activation increases reverse cholesterol transport by modulating bile acid composition and cholesterol absorption in mice. Hepatology. (2016) 64:1072–85. doi: 10.1002/hep.28712
67. Clinical Review Report. Obeticholic Acid (Ocaliva): (Intercept Pharmaceuticals Canada, Inc.): Indication: for the Treatment of Primary Biliary Cholangitis (Pbc) in Combination with Ursodeoxycholic Acid (UDCA) in Adults with an Inadequate Response to UDCA or as Monotherapy in Adults Unable to Tolerate UDCA, Ottawa, ON (2017).
68. Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon SC, Mayo M, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. (2015) 148:751–61.e8. doi: 10.1053/j.gastro.2014.12.005
69. Bowlus CL, Pockros PJ, Kremer AE, Pares A, Forman LM, Drenth JPH, et al. Long-term obeticholic acid therapy improves histological endpoints in patients with primary biliary cholangitis. Clin Gastroenterol Hepatol. (2019) doi: 10.1016/j.cgh.2019.09.050. [Epub ahead of print].
70. Hameed B, Terrault NA, Gill RM, Loomba R, Chalasani N, Hoofnagle JH, et al. Clinical and metabolic effects associated with weight changes and obeticholic acid in non-alcoholic steatohepatitis. Aliment Pharmacol Ther. (2018) 47:645–56. doi: 10.1111/apt.14492
71. Ratziu V, Sanyal AJ, Loomba R, Rinella M, Harrison S, Anstee QM, et al. REGENERATE: design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemp Clin Trials. (2019) 84:105803. doi: 10.1016/j.cct.2019.06.017
72. Comerford M, Lourens S, Liangpunsakul S, Chalasani NP, Sanyal AJ, Shah VH, et al. Challenges in patient enrollment and retention in clinical studies for alcoholic hepatitis: experience of the TREAT consortium. Alcohol Clin Exp Res. (2017) 41:2000–6. doi: 10.1111/acer.13515
73. Pockros PJ, Fuchs M, Freilich B, Schiff E, Kohli A, Lawitz EJ, et al. CONTROL: a randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. (2019) 39:2082–93. doi: 10.1111/liv.14209
74. Eaton JE, Vuppalanchi R, Reddy R, Sathapathy S, Ali B, Kamath PS. Liver injury in patients with cholestatic liver disease treated with obeticholic acid. Hepatology. (2019) doi: 10.1002/hep.31017. [Epub ahead of print].
75. Al-Dury S, Marschall HU. Ileal bile acid transporter inhibition for the treatment of chronic constipation, cholestatic pruritus, and NASH. Front Pharmacol. (2018) 9:931. doi: 10.3389/fphar.2018.00931
76. Al-Dury S, Wahlstrom A, Wahlin S, Langedijk J, Elferink RO, Stahlman M, et al. Pilot study with IBAT inhibitor A4250 for the treatment of cholestatic pruritus in primary biliary cholangitis. Sci Rep. (2018) 8:6658. doi: 10.1038/s41598–018-25214-0
77. Hegade VS, Kendrick SF, Dobbins RL, Miller SR, Thompson D, Richards D, et al. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: a double-blind, randomised, placebo-controlled, crossover, phase 2a study. Lancet. (2017) 389:1114–23. doi: 10.1016/S0140-6736(17)30319-7
78. Gelbmann CM, Schteingart CD, Thompson SM, Hofmann AF, Barrett KE. Mast cells and histamine contribute to bile acid-stimulated secretion in the mouse colon. J Clin Invest. (1995) 95: 2831–9. doi: 10.1172/JCI117988
79. Li WT, Luo QQ, Wang B, Chen X, Yan XJ, Qiu HY, et al. Bile acids induce visceral hypersensitivity via mucosal mast cell-to-nociceptor signaling that involves the farnesoid X receptor/nerve growth factor/transient receptor potential vanilloid 1 axis. FASEB J. (2019) 33:2435–50. doi: 10.1096/fj.201800935RR
80. Tang R, Wei Y, Li Y, Chen W, Chen H, Wang Q, et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut. (2018) 67:534–41. doi: 10.1136/gutjnl-2016-313332
81. Selmi C, Balkwill DL, Invernizzi P, Ansari AA, Coppel RL, Podda M, et al. Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic-metabolizing bacterium. Hepatology. (2003) 38:1250–7. doi: 10.1053/jhep.2003.50446
82. Song M, Ye J, Zhang F, Su H, Yang X, He H, et al. Chenodeoxycholic acid (CDCA) protects against the lipopolysaccharide-induced impairment of the intestinal epithelial barrier function via the FXR-MLCK pathway. J Agric Food Chem. (2019) 67:8868–74. doi: 10.1021/acs.jafc.9b03173
83. Tabibian JH, Talwalkar JA, Lindor KD. Role of the microbiota and antibiotics in primary sclerosing cholangitis. Biomed Res Int. (2013) 2013:389537. doi: 10.1155/2013/389537
84. Carino A, Biagioli M, Marchiano S, Fiorucci C, Zampella A, Monti MC, et al. Ursodeoxycholic acid is a GPBAR1 agonist and resets liver/intestinal FXR signaling in a model of diet-induced dysbiosis and NASH. Biochim Biophys Acta Mol Cell Biol Lipids. (2019) 1864:1422–37. doi: 10.1016/j.bbalip.2019.07.006
85. Friedman ES, Li Y, Shen TD, Jiang J, Chau L, Adorini L, et a. FXR-dependent modulation of the human small intestinal microbiome by the bile acid derivative obeticholic acid. Gastroenterology. (2018) 155:1741–52 e5. doi: 10.1053/j.gastro.2018.08.022
Keywords: bile acid, chronic liver disease, obeticholic acid, ursodeoxycholic acid, bile acid receptor
Citation: Meadows V, Kennedy L, Kundu D, Alpini G and Francis H (2020) Bile Acid Receptor Therapeutics Effects on Chronic Liver Diseases. Front. Med. 7:15. doi: 10.3389/fmed.2020.00015
Received: 30 October 2019; Accepted: 13 January 2020;
Published: 29 January 2020.
Edited by:
Pedro M. Baptista, Aragon Health Research Institute, University of Zaragoza, SpainReviewed by:
Paul Dawson, Emory University, United StatesLuciano Adorini, Intercept Pharmaceuticals, United States
Copyright © 2020 Meadows, Kennedy, Kundu, Alpini and Francis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heather Francis, aGVhZnJhbmNAaXUuZWR1