 Yangyang Zhang
Yangyang Zhang Dan Zhao
Dan Zhao Lili Jiang
Lili Jiang Xiaoxia Zhao
Xiaoxia Zhao Yujun Du
Yujun Du- Department of Nephrology, The First Hospital of Jilin University, Changchun, China
Renal fibrosis is the common manifestation of the pathogenesis of end-stage renal disease that results from different types of renal insult, and is a hallmark of chronic kidney disease (CKD). The main pathologic characteristics of renal fibrosis are renal interstitial fibroblast hyperplasia and the aberrant and excessive deposition of extracellular matrix, pathologies that lead to the destruction of normal renal tubules and interstitial structures. However, the biological significance of fibrosis during the progression of CKD is not clear, and there are no approved clinical treatments for delaying or reversing renal fibrosis. Studies of the mechanism of renal fibrosis and of potential measures of prevention and treatment have focused on erythropoietin (EPO), a hormone best known as a regulator of red blood cell production. These recent studies have found that EPO may also provide efficient protection against renal fibrosis. Future therapeutic approaches using EPO offer new hope for patients with CKD. The aim of the present review is to briefly discuss the role of EPO in renal fibrosis, to identify its possible mechanisms in preventing renal fibrosis, and to provide novel ideas for the use of EPO in future treatments of renal fibrosis.
Introduction
The prevalence of chronic kidney disease (CKD) has risen significantly during the last several decades, and it is now a major public health issue that poses enormous economic challenges worldwide (1). According to recent epidemiological surveys, more than 500 million people worldwide currently have some stage of CKD (2, 3). The two principal causes of CKD are diabetes and hypertension; several additional conditions often accompany CKD, such as HIV infection, obesity, and primary kidney injury (4). The progression of CKD is accompanied by multiple complications and comorbidities that ultimately lead to renal fibrosis and end-stage renal disease, a condition associated with poor outcome and increased risk of death (5, 6).
Renal fibrosis is a progressive pathophysiological change that is associated with damage and functional loss of the kidney (7). Pathogenesis begins following stimulation by a variety of pathogenic factors, such as trauma, infection, inflammation, blood circulation disorders, and excessive immune responses. These insults damage healthy kidney cells, leading to the deposition and accumulation of large amounts of collagen as disease progresses. During the later stages of pathogenesis, there is a gradual hardening of the renal parenchyma and formation of scar tissue, and then the complete loss of kidney function (8). Renal fibrosis is simply the process of fibrosis and hardening of the formerly healthy cells of the kidney. It is also characterized by the abnormal deposition of extracellular matrix (ECM). Although previous studies have examined the main molecular mediators of renal fibrosis, there are no approved therapies that directly target renal fibrosis (9, 10). Therefore, it is necessary to further study the pathogenesis of renal fibrosis to facilitate the development of new drugs that inhibit renal fibrosis and delay the progression to CKD.
In 1906, Carnot and Deflander first proposed the existence of a hormone primarily produced by the kidneys that regulates red blood cell production. This hormone, subsequently named erythropoietin (EPO), is now known to regulate the formation of red blood cells by stimulating bone marrow, and is widely used for the clinical treatment of anemia caused by various factors (11–13). Recent research in China and elsewhere has examined the non-hematopoietic aspects of EPO (14–16). There is now evidence that EPO is a multifunctional cytokine (17). Thus, in addition to regulating erythropoiesis (which increases tissue oxygen supply) EPO also has anti-apoptosis, anti-inflammatory, antioxidant, angiogenesis, cell proliferation, and anti-tumor effects (18).
The aims of this review were to briefly review the scientific evidence regarding the relationship of the therapeutic use of EPO with renal fibrosis.
EPO and Related Molecules
EPO is an acidic glycoprotein containing sialic acid whose precursor has 193 amino acids. After post-translational processing, the active protein has 165 amino acids and is highly glycosylated (19, 20). The serum EPO level in healthy individuals ranges from 15 to 25 U/L (21), and the oxygenation state of tissues regulates its production. EPO is a glycoprotein hormone secreted by the kidneys whose biological effects occur after it binds to its receptor (EPO-R), which occurs on the plasma membranes of target cells (22). EPO primarily targets hematopoietic cells in the bone marrow, which has high concentrations of EPO, and this is its main site of action (23). The binding of EPO to EPO-R on erythrocytes activates a variety of signaling pathways (24), thus stimulating cell proliferation, differentiation, and maturation (13). EPO can improve anemia by inducing erythropoiesis, thus indirectly ameliorating organ damages. Studies have shown that in predialysis patients with non-severe anemia, the early initiation of erythropoietin significantly slows the progression of kidney disease and delays the initiation of renal replacement therapy (25). Initial studies found EPO-R in erythroid progenitor cells, so researchers considered its function was limited to these cells, and gave little consideration to the role of EPO in the kidney and other tissues (26). More recent studies have shown expression of EPO-R by renal cells (including renal tubular cells, mesangial cells, and collecting duct cells), gastric epithelial cells, and some neurogenic cells (27, 28). Other studies have shown that EPO has cellular protective effects, through its interaction with the EPO-R, in the kidneys, brain, heart, and blood vessels, in that it regulates mitosis, reduces oxidative stress, inhibits apoptosis, promotes vascular repair, and has several other effects (24, 29). The clinical application of erythropoiesis stimulating agents (ESAs) was a milestone in the treatment of renal anemia, because these drugs greatly improve the prognosis of patients with chronic renal failure and provide important renoprotective effects (30–32).
However, due to the poor specificity and sensitivity of existing anti-EPO-R antibodies, there is still controversy about the function of EPO beyond its effect on erythropoiesis. Elliott et al. found that EPO-R protein was below the detection limit in tissues and renal cells and no evidence of EPO-R expression and function in the kidneys (33). A subsequent meta-analysis of clinical trials by Elliott et al. showed that ESAs had no clear renoprotective effect, at least in the included subjects (34). Thus, it is currently controversial whether functional EPO-R occurs on the plasma membranes of renal cells. Moreover, multiple clinical studies showed that ESAs provided no improvement in renal function after transplantation or acute kidney injury (35, 36). However, due to the poor specificity of the EPO-R assay and the discordance of in vitro and in vivo data in animal models, this topic needs further study.
EPO and Renal Fibrosis
Renal fibrosis is a key feature of CKD and is the common pathologic manifestation and pathogenic outcome of end-stage renal disease (37). Renal interstitial fibroblast proliferation and the aberrant and persistent deposition of ECM are the main pathological features of renal fibrosis. This process begins when an inflammatory stimulus accelerates the transformation of epithelial cells and interstitial fibroblasts into myofibroblasts, which together produce excess ECM. Then, because of the decreased activity of matrix proteolytic enzymes and increased activity of protease inhibitors, excessive deposition of ECM occurs, leading to the formation of permanent fibrotic scars, thereby accelerating the progression of tubulointerstitial fibrosis (38, 39).
It is now known that EPO is a major multifunctional glycoprotein hormone that has protective functions in various organs and tissues. For example, an animal study demonstrated that EPO attenuated cardiac dysfunction by inhibiting interstitial fibrosis in diabetic rats (40). Another animal study reported the beneficial effects of EPO in diverse liver injuries, such as carbon tetrachloride-induced hepatic fibrosis (41). EPO also has renoprotective effects (Figure 1) (42, 43), and early treatment of anemia with EPO in CKD patients slows the development in renal dysfunction (44). Extensive research during recent years demonstrated that EPO can improve recovery from acute kidney injury (45). Administration of recombinant human EPO (rhEPO) reduces the production of urinary proteins and biomarkers associated with kidney injury and even with CKD. We therefore believe that EPO may also play a critical role in the development of renal fibrosis, but the mechanism of this effect requires further examination.
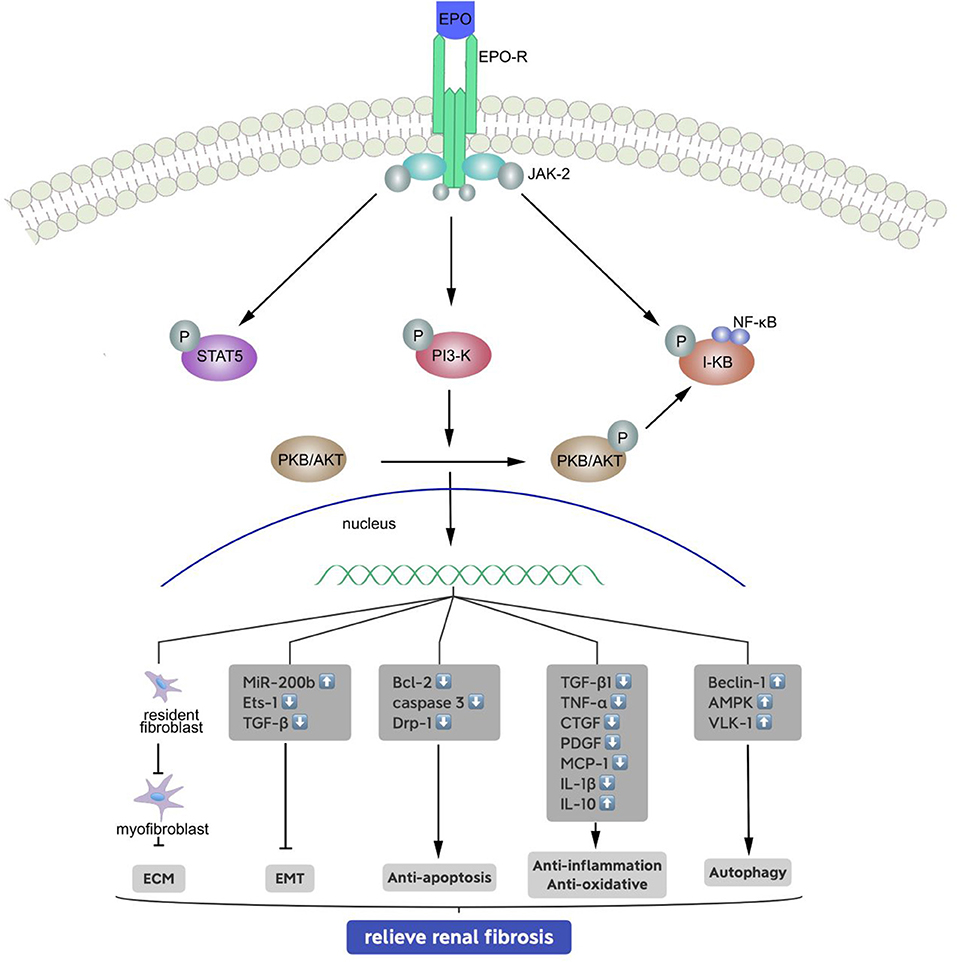
Figure 1. Signal transduction pathways of the erythropoietin receptor and mechanisms of relieving renal fibrosis. Binding of erythropoietin (EPO) causes conformational changes to the EPO receptor, phosphorylation of associated JAK-2, PI-3 kinase and I-kB molecules, and activation of signaling molecules and target genes: (1) inhibits the generation of stromal mesenchymal fibroblasts; (2) inhibits the EMT by upregulating miR-200b, and reducing of Ets-1 and TGF-β; (3) phosphorylates and inactivates proapoptotic molecules; (4) reduces inflammation by inhibiting the release of pro-inflammatory cytokines and anti-oxidative, and (5) enhances autophagy to some extent.
EPO and Myofibroblasts
Myofibroblasts are fibroblasts containing actin, myosin, and other muscle-related proteins that provide these cells with contractile properties. Various stimuli and injuries can induce the activation and proliferation of renal interstitial fibroblasts, leading to the formation of active myofibroblasts. These renal myofibroblasts function as effector cells of the renal interstitial ECM, causing damage to the function of the kidney, and eventually leading to renal failure (46–48). Moreover, the population of novel myofibroblasts present in fibrotic kidneys can derive from renal tubular interstitial resident fibroblasts, bone marrow derived fibrocytes, vascular pericytes, the epithelial-mesenchymal transition (EMT), and the endothelial-mesenchymal transition (EndoMT) (49, 50).
There is an increasing body of evidence suggesting that interstitial myofibroblasts constitutively produce ECM, and that this leads to the development of glomerulosclerosis and tubulointerstitial fibrosis due to activation of TGF-β1 (51). The accumulation of matrix proteins, such as fibronectin and type I and III collagen, is a hallmark of renal fibrosis. Thus, under normal conditions, resident renal fibroblasts produce EPO in response to hypoxic insults to maintain physiological homeostasis. However, under pathologic conditions the resident renal fibroblasts transdifferentiate into myofibroblasts, which promote renal fibrosis by producing large amounts of extracellular matrix proteins rather than EPO (52). A study of mice showed that treatment with rhEPO significantly inhibited the accumulation of fibrocyte by inhibition of α-SMA upregulation, and thereby attenuating renal interstitial fibrosis (53). Another study of transgenic mice found that enhanced signaling mediated by hypoxia-inducible factor (HIF) in myofibroblast-transformed renal EPO-producing cells reactivated the synthesis of EPO, without influencing renal fibrosis or inflammation (54). However, studies of type 2 diabetic mice found that a continuous erythropoietin receptor activator (CERA) enhanced tissue repair by inhibiting the generation of stromal mesenchymal fibroblasts, and thus had a non-hematopoietic and tissue-protective role (55).
EPO and the EMT
During the EMT, epithelial cells undergo a loss of cell-cell adhesion and apical-basal polarity. This is accompanied by increased expression of mesenchymal markers, rearrangement of the cytoskeleton, and increased cell dissociation, all of which contribute to development of the mesenchymal phenotype. The EMT is pathologically reactivated in, and contributes to, the progression of fibrosis (56). Tubulointerstitial fibroblasts, derived from tubular epithelial cells during the EMT, are among the most important effector cells facilitating the progression of renal fibrosis (57, 58). In the past few decades, many studies have examined the role of the EMT during organ fibrosis, wound healing, and cancer metastasis. These studies have demonstrated that TGF-β1 promotes renal fibrosis through the EMT by activation of Smad2/3 (59, 60). In support of this conclusion, animal studies have also shown that the renal EMT contributes to renal fibrosis. For example, Grande et al. concluded that the partial EMT drives renal fibrosis in mice (61). Therefore, it is important to identify interventions or drug therapies that could potentially reverse or inhibit the EMT in the kidney to improve clinical management of these patients.
Previous studies reported that EPO functions as an EMT inhibitor in the kidneys and was effective in ameliorating renal fibrosis (62). Imamura et al. reported that EPO inhibits tubulointerstitial fibrosis in remnant kidney by functioning as an inhibitor of the EMT (63). A recent study using HK-2 cells showed that administration of EPO markedly inhibited hypoxia-induced EMT by upregulating miR-200b expression via the repression of Ets-1. Another study of a unilateral ureteric obstruction (UUO) mouse model reported that rhEPO treatment reduced renal fibrosis by attenuating the EMT (64). Although the role of the EMT in renal fibrosis is not entirely clear, it is nonetheless important to identify therapeutic targets that may allow the prevention or reversal of the EMT and thereby slow the progression of CKD.
EPO and Apoptosis
Apoptosis is the spontaneous and orderly death of cells in multicellular organisms that is regulated by specific genes to maintain the stability of the internal environment. In contrast to the passive process of necrosis, apoptosis is an active process in which there are specific changes in the expression of multiple genes. It is not a phenomenon of self-injury that occurs during pathological conditions, but a process that eliminates cells so that the organism is better adapted to its microenvironment. There are several signal transduction pathways associated with apoptosis; the major pathways are mitochondrial mediated apoptosis, endoplasmic reticulum mediated apoptosis, and death receptor-mediated apoptosis (65).
Renal fibrosis is accompanied by a significant reduction of parenchymal cells, including podocytes, mesangial cells, and tubular epithelial cells, and increased apoptosis also plays an important role. During renal interstitial fibrosis, there is evidence that apoptosis of renal tubular epithelial cells is closely related to renal tubular atrophy, and the increased apoptosis index during this process is an indication of the formation of extracellular matrix (66). Therefore, a targeted inhibition of apoptosis may be an effective strategy to delay or inhibit renal fibrosis.
EPO has many biochemical effects, in addition to its promotion of red blood cell production, and these include anti-inflammatory and anti-apoptotic effects. For example, EPO is the main regulator of erythroid progenitor cell proliferation and differentiation, and these are mediated via its anti-apoptotic effect (67). Many recent animal experiments have shown that EPO may delay the development of CKD and protect the kidneys by reducing the extent of interstitial fibrosis. For example, one study showed that the extent of renal fibrosis correlated positively with the number of apoptotic cells (68). Another study demonstrated that EPO provided a renoprotective and antiapoptotic effects by activation of ERK/p53 signaling (26). A study of renal interstitial fibrosis in a rat model reported that EPO had renoprotective effects due to its due to its down-regulation of dynamin-related protein-1 (Drp-1) (69), a protein with a major role in the mitochondrial mediated apoptosis pathway. A study by Nakazawa et al. showed that EPO inhibited the apoptosis of renal tubular cells, thereby reducing the extent of renal interstitial fibrosis (70). In the context of ischemia-reperfusion (IR) injury, reduced levels of pro-apoptotic genes in the Bcl-2 family can ameliorate the destruction of mitochondrial integrity and tubular cell apoptosis, and consequently inhibit renal injury (71, 72). Sharples et al. studied a rat kidney model of severe IR injury and demonstrated that EPO inhibited the apoptosis of proximal tubular epithelial cells and promoted significant cell proliferation at high doses, despite serum starvation (73).
Anti-inflammatory Effects of EPO
Inflammation is a defensive response of living tissue that involves the vascular system and occurs following injury from factors such as infections, physical, chemical, or antigenic changes, or traumatic damage.
CKD is characterized by persistent renal inflammation that progresses to tubular interstitial fibrosis, renal failure, and end-stage renal disease (74). Elevated levels of inflammatory cytokines are associated with mortality, especially in patients with CKD. Various renal insults can activate tubulointerstitial cells and promote inflammatory cell infiltration, leading to the release of multiple vasoactive molecules and soluble cytokines that promote the progression of fibrosis. These molecules include TGF-β1, TNF-α, connective tissue growth factor, and platelet-derived growth factor. Secretion of these cytokines by tubulointerstitial and inflammatory cells alters the dynamic balance between synthesis and degradation of ECM proteins, eventually leading to the accumulation of ECM components and the development of fibrosis. Renal innate cells (mesangial cells, podocytes, and endothelial cells) also secrete a variety of fibrogenic cytokines such as TGF-β1 and TNF-α.
EPO also has an important non-haematopoietic effects in that it can reduce inflammation caused by injury, toxins, or hypoxia. Mateus et al. reported the anti-inflammatory effect of EPO was due to its inhibition of TNF-α and IL-1β production and stimulation of IL-10 production in an animal model of TNBS-induced colitis (75). Chang et al. found that subcutaneous injection of EPO into rats with UUO significantly reduced the expression of TNF-α and mononuclear cell chemotactic protein-1 (MCP-1), decreased inflammatory cell infiltration, inhibited interstitial fibrosis, and protected kidney function (76). Heme oxygenase-1 (HO-1) is an important antioxidant protein, Katavetin et al. demonstrated that EPO induces expression of HO-1, thereby reducing oxidative stress and delaying CKD progression (77).
EPO and Autophagy
Autophagy is a conserved mechanism of cell self-degradation, in which lysosomes degrade damaged organelles and macromolecules so they can be reused. In general, autophagy is necessary to maintain cell homeostasis, and it has roles in slowing the progress of aging, promoting differentiation and development, increasing immunity and the clearance of microorganisms, and preventing the progression of tumors and other diseases (78, 79). However, excessive autophagy can have pathological effects. For example, Livingston et al., using pharmacological and genetic methods, demonstrated that sustained activation of autophagy in the proximal tubules promoted renal interstitial fibrosis in rats with UUO (80). In agreement, a study of an experimental model of neonatal necrotizing enterocolitis found that EPO reduced excessive autophagy, and contained cell damage (81). EPO also protects against rotenone-induced neurotoxicity in SH-SY5Y cells by enhancing autophagy-related signaling pathways (82). The renoprotective effects of ESAs in animal models may be due to their antiapoptotic effects. A study of kidney IR injury reported that ESAs had renoprotective effects by inducing autophagy (83). The antiapoptotic effects of EPO are dependent on JAK2 signaling and the phosphorylation of Akt by phosphatidylinositol 3-kinase. Increased Akt signaling is associated with suppression of cell apoptosis and promotion of renal fibrosis in IR injury-induced acute kidney injury (84). However, suppression of Akt phosphorylation accelerates tubular repair and inhibits renal fibrosis (85).
Conclusion
The classical role of EPO is the regulation of red blood cell production, but many studies have reported that EPO also has many non-erythroid effects. These findings underline the importance of investigating the global actions of rhEPO and its derivatives in pre-clinical and clinical settings. Thus, EPO may be useful for protection of the kidney because of its effects on multiple pathways, in addition to its effect on red blood cell production. More specifically, EPO can prevent the development of renal failure during the end-stages of various renal diseases, and can actively control the fibrosis of renal interstitial tissue. However, the mechanism of EPO in the prevention and treatment of renal interstitial fibrosis is not fully understood. For this reason, additional studies are required to determine the optimal dose and timing of EPO administration and how to best avoid possible adverse reactions, such as thrombosis. The recent research and development of new types of EPO for treatment of anemia and other conditions may increase the applications of EPO for additional conditions. The results of this new research may also provide new insights into the mechanism by which rhEPO prevents interstitial fibrosis and slows the progression of CKD. Lastly, we believe that the many developments and unremitting efforts from multiple medical disciplines will eventually establish a basis for the use of EPO as a treatment for renal fibrosis and will help guide future research in this area.
Author Contributions
YZ, XZhu, XH, and YD contributed conception and design of the study. XW and DZ organized the database. YZ wrote the first draft of the manuscript. LJ and XZha wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ng JK, Li PK. Chronic kidney disease epidemic: how do we deal with it? Nephrology. (2018) 23:116–20. doi: 10.1111/nep.13464
2. ForniOgna V, Ogna A, Ponte B, Gabutti L, Binet I, Conen D, et al. Prevalence and determinants of chronic kidney disease in the Swiss population. Swiss Med Wkly. (2016) 146:w14313. doi: 10.4414/smw.2016.14313
3. Liyanage T, Ninomiya T, Perkovic V, Woodward M, Stirnadel-Farrant H, Matsushita K, et al. Chronic kidney disease in Asia: protocol for a collaborative overview. Nephrology. (2017) 22:456–62. doi: 10.1111/nep.12821
4. Mehdi U, Toto RD. Anemia, diabetes, and chronic kidney disease. Diabetes Care. (2009) 32:1320–6. doi: 10.2337/dc08-0779
5. De Oliveira Junior WV, Sabino Ade P, Figueiredo RC, Rios DR. Inflammation and poor response to treatment with erythropoietin in chronic kidney disease. J Bras Nefrol. (2015) 37:255–63. doi: 10.5935/0101-2800.20150039
6. Zhang L, Wang F, Wang L, Wang W, Liu B, Liu J, et al. Prevalence of chronic kidney disease in China: a cross-sectional survey. Lancet. (2012) 379:815–22. doi: 10.1016/S0140-6736(12)60033-6
7. Mack M, Yanagita M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. (2015) 87:297–307. doi: 10.1038/ki.2014.287
8. Sun YB, Qu X, Caruana G, Li J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation. (2016) 92:102–7. doi: 10.1016/j.diff.2016.05.008
9. Baues M, Dasgupta A, Ehling J, Prakash J, Boor P, Tacke F. Fibrosis imaging: current concepts and future directions. Adv Drug Deliv Rev. (2017) 121:9–26. doi: 10.1016/j.addr.2017.10.013
10. Chuang PY, Menon MC, He JC. Molecular targets for treatment of kidney fibrosis. J Mol Med. (2013) 91:549–59. doi: 10.1007/s00109-012-0983-z
11. Bai J, Xiao X, Zhang X, Cui H, Hao J, Han J, et al. Erythropoietin inhibits hypoxia-induced epithelial-to-mesenchymal transition via upregulation of miR-200b in HK-2 cells. Cell Physiol Biochem. (2017) 42:269–80. doi: 10.1159/000477327
12. Perreault AA, Venters BJ. Integrative view on how erythropoietin signaling controls transcription patterns in erythroid cells. Curr Opin Hematol. (2018) 25:189–95. doi: 10.1097/MOH.0000000000000415
13. Klingmüller U. The role of tyrosine phosphorylation in proliferation and maturation of erythroid progenitor cells. Eur J Biochem. (1997) 249:637–47. doi: 10.1111/j.1432-1033.1997.t01-1-00637.x
14. Ogunshola OO, Bogdanova AY. Epo and non-hematopoietic cells: what do we know? Methods Mol Biol. (2013) 982:13–41. doi: 10.1007/978-1-62703-308-4_2
15. Kimakova P, Solar P, Solarova Z, Komel R, Debeljak N. Erythropoietin and its angiogenic activity. Int J Mol Sci. (2017) 18:1519. doi: 10.3390/ijms18071519
16. Reinhardt M, Dey S, Noguchi CT, Zhang Y, Krakoff J, Thearle MS. Non-hematopoietic effects of endogenous erythropoietin on lean mass and body weight regulation. Obesity. (2016) 24:1530–36. doi: 10.1002/oby.21537
17. O'Leary OE, Canning P, Reid E, Bertelli PM, McKeown S, Brines M, et al. The vasoreparative potential of endothelial colony-forming cells in the ischemic retina is enhanced by cibinetide, a non-hematopoietic erythropoietin mimetic. Exp Eye Res. (2019) 182:144–55. doi: 10.1016/j.exer.2019.03.001
18. Zheng DH, Han ZQ, Wang XX, Ma D, Zhang J. Erythropoietin attenuates high glucose-induced oxidative stress and inhibition of osteogenic differentiation in periodontal ligament stem cell (PDLSCs). Chem Biol Interact. (2019) 305:40–7. doi: 10.1016/j.cbi.2019.03.007
19. Jelkmann W. Physiology and pharmacology of erythropoietin. Transf Med Hemother. (2013) 40:302–9. doi: 10.1159/000356193
20. Shih HM, Wu CJ, Lin SL. Physiology and pathophysiology of renal erythropoietin-producing cells. J Formos Med Assoc. (2018) 117:955–63. doi: 10.1016/j.jfma.2018.03.017
21. Baker JE. Erythropoietin mimics ischemic preconditioning. Vascul Pharmacol. (2005) 42:233–41. doi: 10.1016/j.vph.2005.02.004
22. Dunlop EA, Percy MJ, Boland MP, Maxwell AP, Lappin TR. Induction of signalling in non-erythroid cells by pharmacological levels of erythropoietin. Neurodegener Dis. (2006) 3:94–100. doi: 10.1159/000092099
23. Cao X, Chen Z, Yu Z, Ge Y, Zeng X. Pharmacokinetics of PEGylated recombinant human erythropoietin in rats. J Anal Methods Chem. (2014) 2014:918686. doi: 10.1155/2014/918686
24. Moore E, Bellomo R. Erythropoietin (EPO) in acute kidney injury. Ann Intensive Care. (2011) 1:3. doi: 10.1186/2110-5820-1-3
25. Gouva C, Nikolopoulos P, Ioannidis JP, Siamopoulos KC. Treating anemia early in renal failure patients slows the decline of renal function: a randomized controlled trial. Kidney Int. (2004) 66:753–60. doi: 10.1111/j.1523-1755.2004.00797.x
26. Zou YR, Zhang J, Wang J, Peng L, Li GS, Wang L. Erythropoietin receptor activation protects the kidney from ischemia/reperfusion-induced apoptosis by activating ERK/p53 signal pathway. Transplant Proc. (2016) 48:217–21. doi: 10.1016/j.transproceed.2016.01.009
27. Westenfelder C, Biddle DL, Baranowski RL. Human, rat, and mouse kidney cells express functional erythropoietin receptors. Kidney Int. (1999) 55:808–20. doi: 10.1046/j.1523-1755.1999.055003808.x
28. Hu MC, Shi M, Cho HJ, Zhang J, Pavlenco A, Liu S, et al. The erythropoietin receptor is a downstream effector of klotho-induced cytoprotection. Kidney Int. (2013) 84:468–81. doi: 10.1038/ki.2013.149
29. Rodrigues CE, Sanches TR, Volpini RA, Shimizu MH, Kuriki PS, Camara NO, et al. Effects of continuous erythropoietin receptor activator in sepsis-induced acute kidney injury and multi-organ dysfunction. PLoS ONE. (2012) 7:e29893. doi: 10.1371/journal.pone.0029893
30. Hung SC, Lin YP, Tarng DC. Erythropoiesis-stimulating agents in chronic kidney disease: what have we learned in 25 years? J Formos Med Assoc. (2014) 113:3–10. doi: 10.1016/j.jfma.2013.09.004
31. Kanbay M, Perazella MA, Kasapoglu B, Koroglu M, Covic A. Erythropoiesis stimulatory agent- resistant anemia in dialysis patients: review of causes and management. Blood Purif. (2010) 29:1–12. doi: 10.1159/000245041
32. Bamgbola O. Resistance to erythropoietin-stimulating agents: etiology, evaluation, and therapeutic considerations. Pediatr Nephrol. (2012) 27:195–205. doi: 10.1007/s00467-011-1839-4
33. Elliott S, Busse L, Swift S, McCaffery I, Rossi J, Kassner P, et al. Lack of expression and function of erythropoietin receptors in the kidney. Nephrol Dial Transplant. (2012) 27:2733–45. doi: 10.1093/ndt/gfr698
34. Elliott S, Tomita D, Endre Z. Erythropoiesis stimulating agents and reno-protection: a meta-analysis. BMC Nephrol. (2017) 18:14. doi: 10.1186/s12882-017-0438-4
35. Fernandez LM, Marcen R, Villafruela J, Teruel JL, Tato A, Rivera M, et al. Effect of rHuEpo therapy in dialysis patients on endogenous erythropoietin synthesis after renal transplantation. Nephron. (1996) 73:54–7. doi: 10.1159/000188998
36. Endre ZH, Walker RJ, Pickering JW, Shaw GM, Frampton CM, Henderson SJ, et al. Early intervention with erythropoietin does not affect the outcome of acute kidney injury (the EARLYARF trial). Kidney Int. (2010) 77:1020–30. doi: 10.1038/ki.2010.25
37. Locatelli F, Del Vecchio L. New strategies for anaemia management in chronic kidney disease. Contrib Nephrol. (2017) 189:184–8. doi: 10.1159/000450779
38. Chen P, Yang Q, Li X, Qin Y. Potential association between elevated serum human epididymis protein 4 and renal fibrosis: a systemic review and meta-analysis. Medicine. (2017) 96:e7824. doi: 10.1097/MD.0000000000007824
39. Park SH, Choi MJ, Song IK, Choi SY, Nam JO, Kim CD, et al. Erythropoietin decreases renal fibrosis in mice with ureteral obstruction: role of inhibiting TGF-β-induced epithelial-to-mesenchymal transition. J Am Soc Nephrol. (2007) 18:1497–507. doi: 10.1681/ASN.2005080866
40. Lu J, Yao YY, Dai QM, Ma GS, Zhang SF, Cao L, et al. Erythropoietin attenuates cardiac dysfunction by increasing myocardial angiogenesis and inhibiting interstitial fibrosis in diabetic rats. Cardiovasc Diabetol. (2012) 11:105. doi: 10.1186/1475-2840-11-105
41. Park SY, Lee JY, Tak WY, Kweon YO, Lee MS. Erythropoietin decreases carbon tetrachloride-induced hepatic fibrosis by inhibiting transforming growth factor-beta. Chin Med J. (2012) 125:3098–103. doi: 10.3760/cma.j.issn.0366-6999.2012.17.024
42. Sharples EJ, Yaqoob MM. Erythropoietin in experimental acute renal failure. Nephron Exp Nephrol. (2006) 104:e83–8. doi: 10.1159/000094546
43. Bagnis C, Beaufils H, Jacquiaud C, Adabra Y, Jouanneau C, Le Nahour G, et al. Erythropoietin enhances recovery after cisplatin-induced acute renal failure in the rat. Nephrol Dial Transplant. (2001) 16:932–8. doi: 10.1093/ndt/16.5.932
44. Togel FE, Ahlstrom JD, Yang Y, Hu Z, Zhang P, Westenfelder C. Carbamylated erythropoietin outperforms erythropoietin in the treatment of AKI-on-CKD and other AKI models. J Am Soc Nephrol. (2016) 27:3394–404. doi: 10.1681/ASN.2015091059
45. Gobe GC, Bennett NC, West M, Colditz P, Brown L, Vesey DA, et al. Increased progression to kidney fibrosis after erythropoietin is used as a treatment for acute kidney injury. Am J Physiol Renal Physiol. (2014) 306:F681–92. doi: 10.1152/ajprenal.00241.2013
46. Yan Y, Ma L, Zhou X, Ponnusamy M, Tang J, Zhuang MA, et al. Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis. Kidney Int. (2016) 89:68–81. doi: 10.1038/ki.2015.293
47. Liang H, Zhang Z, Yan J, Wang Y, Hu Z, Mitch WE, et al. The IL-4 receptor α has a critical role in bone marrow–derived fibroblast activation and renal fibrosis. Kidney Int. (2017) 92:1433–43. doi: 10.1016/j.kint.2017.04.021
48. Saliba Y, Karam R, Smayra V, Aftimos G, Abramowitz J, Birnbaumer L, et al. Evidence of a role for fibroblast transient receptor potential canonical 3 Ca2+ channel in renal fibrosis. J Am Soc Nephrol. (2015) 26:1855–76. doi: 10.1681/ASN.2014010065
49. Pakshir P, Hinz B. The big five in fibrosis: macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. (2018) 68–69:81–93. doi: 10.1016/j.matbio.2018.01.019
50. Diao W, Chen W, Cao W, Yuan H, Ji H, Wang T, et al. Astaxanthin protects against renal fibrosis through inhibiting myofibroblast activation and promoting CD8+ T cell recruitment. Biochim Biophys Acta Gen Subj. (2019) 1863:1360–70. doi: 10.1016/j.bbagen.2019.05.020
51. Nogueira A, Pires MJ, Oliveira PA. Pathophysiological Mechanisms of renal fibrosis: a review of animal models and therapeutic strategies. In Vivo. (2017) 31:1–22. doi: 10.21873/invivo.11019
52. Sato Y, Yanagita M. Resident fibroblasts in the kidney: a major driver of fibrosis and inflammation. Inflam Regen. (2017) 37:17. doi: 10.1186/s41232-017-0048-3
53. Geng XC, Hu ZP, Lian GY. Erythropoietin ameliorates renal interstitial fibrosis via the inhibition of fibrocyte accumulation. Mol Med Rep. (2015) 11:3860–5. doi: 10.3892/mmr.2015.3157
54. Souma T, Nezu M, Nakano D, Yamazaki S, Hirano I, Sekine H, et al. Erythropoietin synthesis in renal myofibroblasts is restored by activation of hypoxia signaling. J Am Soc Nephrol. (2016) 27:428–38. doi: 10.1681/ASN.2014121184
55. Fischer C, Deininger N, Wolf G, Loeffler I. CERA Attenuates kidney fibrogenesis in the db/db mouse by influencing the renal myofibroblast generation. J Clin Med. (2018) 7:E15. doi: 10.3390/jcm7020015
56. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2014) 15:178–96. doi: 10.1038/nrm3758
57. Li J, An C, Kang L, Mitch WE, Wang Y. Recent advances in magnetic resonance imaging assessment of renal fibrosis. Adv Chronic Kidney Dis. (2017) 24:150–53. doi: 10.1053/j.ackd.2017.03.005
58. Marquez-Exposito L, Lavoz C, Rodrigues-Diez RR, Rayego-Mateos S, Orejudo M, Cantero-Navarro E, et al. Gremlin regulates tubular epithelial to mesenchymal transition via VEGFR2: potential role in renal fibrosis. Front Pharmacol. (2018) 9:1195. doi: 10.3389/fphar.2018.01195
59. Ji X, Wang H, Wu Z, Zhong X, Zhu M, Zhang Y, et al. Specific inhibitor of Smad3 (SIS3) attenuates fibrosis, apoptosis, and inflammation in unilateral ureteral obstruction kidneys by inhibition of transforming growth factor beta (TGF-beta)/Smad3 signaling. Med Sci Monit. (2018) 24:1633–41. doi: 10.12659/MSM.909236
60. Zhang D, Sun L, Xian W, Liu F, Ling G, Xiao L, et al. Low-dose paclitaxel ameliorates renal fibrosis in rat UUO model by inhibition of TGFβ/Smad activity. Lab Invest. (2010) 90:436–47. doi: 10.1038/labinvest.2009.149
61. Grande MT, Sanchez-Laorden B, Lopez-Blau C, De Frutos CA, Boutet A, Arevalo M, et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. (2015) 21:989–97. doi: 10.1038/nm.3901
62. Chen CL, Chou KJ, Lee PT, Chen YS, Chang TY, Hsu CY, et al. Erythropoietin suppresses epithelial to mesenchymal transition and intercepts Smad signal transduction through a MEK-dependent mechanism in pig kidney (LLCPK1) cell lines. Exp Cell Res. (2010) 316:1109–18. doi: 10.1016/j.yexcr.2010.02.022
63. Imamura R, Isaka Y, Sandoval RM, Ichimaru N, Abe T, Okumi M, et al. A nonerythropoietic derivative of erythropoietin inhibits tubulointerstitial fibrosis in remnant kidney. Clin Exp Nephrol. (2012) 16:852–62. doi: 10.1007/s10157-012-0647-x
64. Tasanarong A, Kongkham S, Khositseth S. Dual inhibiting senescence and epithelial-to-mesenchymal transition by erythropoietin preserve tubular epithelial cell regeneration and ameliorate renal fibrosis in unilateral ureteral obstruction. Biomed Res Int. (2013) 2013:308130. doi: 10.1155/2013/308130
65. Abate M, Festa A, Falco M, Lombardi A, Luce A, Grimaldi A, et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin Cell Dev Biol. (2019) 98:139–53. doi: 10.1016/j.semcdb.2019.05.022
66. Yang T, Vesey DA, Johnson DW, Wei MQ, Gobe GC. Apoptosis of tubulointerstitial chronic inflammatory cells in progressive renal fibrosis after cancer therapies. Transl Res. (2007) 150:40–50. doi: 10.1016/j.trsl.2007.01.006
68. Izquierdo MC, Sanz AB, Mezzano S, Blanco J, Carrasco S, Sanchez-Nino MD, et al. TWEAK (tumor necrosis factor-like weak inducer of apoptosis) activates CXCL16 expression during renal tubulointerstitial inflammation. Kidney Int. (2012) 81:1098–107. doi: 10.1038/ki.2011.475
69. Zhao XF, Liu YH, Han ZM, Xu YU. Effect of erythropoietin on the expression of dynamin-related protein-1 in rat renal interstitial fibrosis. Exp Ther Med. (2015) 9:2065–71. doi: 10.3892/etm.2015.2419
70. Nakazawa Y, Nishino T, Obata Y, Nakazawa M, Furusu A, Abe K, et al. Recombinant human erythropoietin attenuates renal tubulointerstitial injury in murine adriamycin-induced nephropathy. J Nephrol. (2013) 26:527–33. doi: 10.5301/jn.5000178
71. Sun PP, Yuan F, Xu J, Sai K, Chen J, Guan S. Cryptotanshinone ameliorates hepatic normothermic ischemia and reperfusion injury in rats by anti-mitochondrial apoptosis. Biol Pharm Bull. (2014) 37:1758–65. doi: 10.1248/bpb.b14-00389
72. Xu Y, Li D, Zeng L, Wang C, Zhang L, Wang Y, et al. Proteasome inhibitor lactacystin enhances cisplatin cytotoxicity by increasing endoplasmic reticulum stresseassociated apoptosis in HeLa cells. Mol Med Rep. (2015) 11:189–95. doi: 10.3892/mmr.2014.2683
73. Sharples EJ, Patel N, Brown P, Stewart K, Mota-Philipe H, Sheaff M, et al. Erythropoietin protects the kidney against the injury and dysfunction caused by ischemia-reperfusion. J Am Soc Nephrol. (2004) 15:2115–24. doi: 10.1097/01.ASN.0000135059.67385.5D
74. Morgado-Pascual JL, Marchant V, Rodrigues-Diez R, Dolade N, Suarez-Alvarez B, Kerr B, et al. Epigenetic modification mechanisms involved in inflammation and fibrosis in renal pathology. Mediators Inflamm. (2018) 2018:2931049. doi: 10.1155/2018/2931049
75. Mateus V, Rocha J, Alves P, Mota-Filipe H, Sepodes B, Pinto RMA. Anti-inflammatory effect of erythropoietin in the TNBS-induced colitis. Basic Clin Pharmacol Toxicol. (2017) 120:138–45. doi: 10.1111/bcpt.12663
76. Chang YK, Choi DE, Na KR, et al. Erythropoietin attenuates renal injury in an experimental model of rat unilateral ureteral obstruction via anti-inflammatory and anti-apoptotic effects. J Urol. (2009) 3:1434–43. doi: 10.1016/j.juro.2008.10.105
77. Katavetin P, Inagi R, Miyata T, Shao J, Sassa R, Adler S, et al. Erythropoietin induces heme oxygenase-1 expression and attenuates oxidative stress. Biochem Biophys Res Commun. (2007) 359:928–34. doi: 10.1016/j.bbrc.2007.05.207
78. Ryter SW, Mizumura K, Choi AM. The impact of autophagy on cell death modalities. Int J Cell Biol. (2014) 2014:502676. doi: 10.1155/2014/502676
79. Jing K, Lim K. Why is autophagy important in human diseases? Exp Mol Med. (2012) 44:69–72. doi: 10.3858/emm.2012.44.2.028
80. Livingston MJ, Ding HF, Huang S, Hill JA, Yin XM, Dong Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Basic Res Papers. (2016) 12:976–98. doi: 10.1080/15548627.2016.1166317
81. Yu Y, Shiou SR, Guo Y, Lu L, Westerhoff M, Sun J, Petrof EO, et al. Erythropoietin protects epithelial cells from excessive autophagy and apoptosis in experimental neonatal necrotizing enterocolitis. PLoS ONE. (2013) 8:e69620. doi: 10.1371/journal.pone.0069620
82. Jang W, Kim HJ, Li H, Jo KD, Lee MK, Yang HO. The neuroprotective effect of erythropoietin on rotenone-induced neurotoxicity in SH-SY5Y cells through the induction of autophagy. Mol Neurobiol. (2016) 53:3812–21. doi: 10.1007/s12035-015-9316-x
83. Yang C, Xu Z, Zhao Z, Li L, Zhao T, Peng D, et al. A novel proteolysis-resistant cyclic helix B peptide ameliorates kidney ischemia reperfusion injury. Biochim Biophys Acta. (2014) 1842:2306–17. doi: 10.1016/j.bbadis.2014.09.001
84. Tang J, Liu N, Tolbert E, Ponnusamy M, Ma L, Gong R, et al. Sustained activation of EGFR triggers renal fibrogenesis after acute kidney injury. Am J Pathol. (2013) 183:160–72. doi: 10.1016/j.ajpath.2013.04.005
Keywords: chronic kidney disease, renal fibrosis, erythropoietin, extracellular matrix, myofibroblasts
Citation: Zhang Y, Zhu X, Huang X, Wei X, Zhao D, Jiang L, Zhao X and Du Y (2020) Advances in Understanding the Effects of Erythropoietin on Renal Fibrosis. Front. Med. 7:47. doi: 10.3389/fmed.2020.00047
Received: 25 November 2019; Accepted: 30 January 2020;
Published: 21 February 2020.
Edited by:
Xueying Zhao, Morehouse School of Medicine, United StatesReviewed by:
Norio Suzuki, Tohoku University, JapanNikolina Basic-Jukic, University Hospital Centre Zagreb, Croatia
Copyright © 2020 Zhang, Zhu, Huang, Wei, Zhao, Jiang, Zhao and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yujun Du, a2prZHV5dWp1bkAxMjYuY29t