 Peter Stanko1†
Peter Stanko1† Tomas Baka1†
Tomas Baka1† Kristina Repova1
Kristina Repova1 Silvia Aziriova1Kristina Krajcirovicova1Andrej Barta2Pavol Janega2,3
Silvia Aziriova1Kristina Krajcirovicova1Andrej Barta2Pavol Janega2,3 Michaela Adamcova4
Michaela Adamcova4 Ludovit Paulis1,2
Ludovit Paulis1,2 Fedor Simko1,5,6*
Fedor Simko1,5,6*- 1Institute of Pathophysiology, Faculty of Medicine, Comenius University, Bratislava, Slovakia
- 2Institute of Normal and Pathological Physiology, Centre of Experimental Medicine, Slovak Academy of Sciences, Bratislava, Slovakia
- 3Institute of Pathological Anatomy, Faculty of Medicine, Comenius University, Bratislava, Slovakia
- 4Department of Physiology, School of Medicine, Charles University, Prague, Czechia
- 53rd Department of Internal Medicine, Faculty of Medicine, Comenius University, Bratislava, Slovakia
- 6Institute of Experimental Endocrinology, Biomedical Research Center, Slovak Academy of Sciences, Bratislava, Slovakia
Hypertension-induced renal injury is characterized by structural kidney alterations and function deterioration. Therapeutics for kidney protection are limited, thus novel renoprotectives in hypertension are being continuously sought out. Ivabradine, an inhibitor of the If current in the sinoatrial node reducing heart rate (HR), was shown to be of benefit in various cardiovascular pathologies. Yet, data regarding potential renoprotection by ivabradine in hypertension are sparse. Thirty-six adult male Wistar rats were divided into non-diseased controls and rats with NG-nitro-L-arginine methyl ester (L-NAME)-induced hypertension to assess ivabradine's site-specific effect on kidney fibrosis. After 4 weeks of treatment, L-NAME increased the average systolic blood pressure (SBP) (by 27%), decreased glomerular density (by 28%) and increased glomerular tuft area (by 44%). Moreover, L-NAME induced glomerular, tubulointerstitial, and vascular/perivascular fibrosis by enhancing type I collagen volume (16-, 19- and 25-fold, respectively). L-NAME also increased the glomerular type IV collagen volume and the tubular injury score (3- and 8-fold, respectively). Ivabradine decreased average SBP and HR (by 8 and 12%, respectively), increased glomerular density (by 57%) and reduced glomerular tuft area (by 30%). Importantly, ivabradine decreased type I collagen volume at all three of the investigated sites (by 33, 38, and 72%, respectively) and enhanced vascular/perivascular type III collagen volume (by 67%). Furthermore, ivabradine decreased the glomerular type IV collagen volume and the tubular injury score (by 63 and 34%, respectively). We conclude that ivabradine attenuated the alterations of glomerular density and tuft area and modified renal fibrosis in a site-specific manner in L-NAME-hypertension. It is suggested that ivabradine may be renoprotective in hypertensive kidney disease.
Introduction
Chronic kidney disease (CKD), determined by a decline of total glomerular filtration rate and albuminuria persisting >3 months, is a severe health and social problem. CKD afflicts almost 15% of the global population and significantly worsens life expectancy (1). Although a considerable number of etiologic factors may come into force, diabetes mellitus, and arterial hypertension clearly dominate (2). CKD in hypertension is characterized by a persistent systemic blood pressure overload gradually exceeding auto-regulatory mechanisms for maintaining adequate glomerular filtration pressure. Glomerular hypertension and hyperfiltration are associated with compensatory glomerular hypertrophy, albuminuria, and persistent inflammation (3). Progressive necrotic or apoptotic cell death is followed by glomerulosclerosis, tubular atrophy, and tubulointerstitial fibrosis representing the principle pathologic components and therapeutic targets of CKD (4).
Ivabradine, the heart rate (HR)-reducing selective inhibitor of the sinoatrial If current, was proved to attenuate morbidity in heart failure (5). The HR-reducing effect of ivabradine was declared the principle mechanism of its therapeutic benefit. However, several of ivabradine's pleiotropic effects have recently emerged, which suggests the possibility of using ivabradine in yet off-label indications such as endothelial dysfunction (6), hypertensive heart disease (7), and hypertension with elevated or non-dipping HR (8, 9).
This study aimed to show whether ivabradine is able to modify kidney alterations in NG-nitro-L-arginine methyl ester (L-NAME)-induced hypertension. We investigated ivabradine's potential antifibrotic effect in a site-specific manner as glomerulosclerosis (glomerular fibrosis), tubulointerstitial fibrosis, and arteriosclerosis (vascular fibrosis) with perivascular fibrosis.
Materials and Methods
Thirty-six 3-month-old male Wistar rats (Department of Toxicology and Laboratory Animals Breeding, Slovak Academy of Sciences, Dobra Voda, Slovakia) were divided into 4 groups (n = 9 animals per group) and treated for 4 weeks as follows: control (C; untreated), ivabradine (Iva; 10 mg/kg/day; Servier, Suresnes, France), L-NAME (LN; 40 mg/kg/day; Sigma-Aldrich Chemie, Munich, Germany) and L-NAME plus ivabradine in corresponding doses (LN+Iva). Both L-NAME and ivabradine were dissolved in drinking water and their concentrations were adjusted to daily water consumption to ensure the correct dosage. The rats were individually housed and maintained under standard laboratory conditions (12:12-h light-dark cycle at 22 ± 2°C temperature and 55 ± 10% humidity) with free access to food and water. The study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The protocol was approved by the ethical committee of the Institute of Pathophysiology, Faculty of Medicine, Comenius University, Bratislava, Slovakia (approval number: 1306/14-221).
Systolic blood pressure (SBP) and heart rate (HR) were measured once a week in each animal by non-invasive tail-cuff plethysmography (Hugo-Sachs Elektronic, Freiburg, Germany). After 4 weeks of treatment, the rats were euthanized by isoflurane inhalation and left kidneys were used for subsequent histopathological analysis. The kidney samples were fixed in 4% formaldehyde for 24 h, embedded in paraffin and cut in 5 μm sections. Three sets of deparaffinized and rehydrated sections were stained with: (i) hematoxylin-eosin (H-E) for glomerular morphometry and tubular injury scoring; (ii) picrosirius red (PSR; 0.1% sirius red F3BA in a saturated water solution of picric acid for 90 min and washed in 0.01 N HCl for 2 min) for a quantitative analysis of kidney fibrosis; and (iii) type IV collagen immunostaining (anti-collagen IV antibody; ab6586; Abcam, Cambridge, UK was used for immunostaining conforming the manufacturer's protocol: a heat-mediated antigen retrieval was followed by overnight incubation with primary anti-collagen IV antibody at 4°C; a horseradish peroxidase-conjugated secondary anti-rabbit IgG antibody with a 3,3′-diaminobenzidine chromogen and hematoxylin counterstain was used for visualization; ab205718; Abcam, Cambridge, UK) to determine type IV collagen volume in glomeruli. Histopathological observations were performed using transmitted or polarized light microscopy on a NIKON Eclipse Ti C2+ microscope (NIKON, Tokyo, Japan). The rendered images were analyzed by NIKON NIS-Elements Analysis software (NIKON, Tokyo, Japan) and ImageJ version 1.52p for Windows (National Institutes of Health, Bethesda, MD, USA). All histopathological analyses were performed by an experienced examiner blinded to the group identity.
For glomerular morphometry, H-E-stained sections were analyzed at 10x magnification using transmitted light microscopy and NIKON NIS-Elements Analysis software as follows: (i) to assess glomerular numerical density per area, well-preserved glomeruli were counted in a digital frame of 1 mm2 placed over the kidney cortex in 10 microscopic fields per animal (i.e., 90 per group; n = 9 animals per group); (ii) to assess glomerular tuft area, perpendicular maximum and minimum diameters (dmax and dmin, respectively) of 10 random glomerular tufts per animal (i.e., 90 per group; n = 9 animals per group) were measured to subsequently calculate tuft ellipse areas by using the formula: glomerular tuft area = π(dmax/2)(dmin/2) (10, 11).
In order to obtain a quantitative analysis of kidney fibrosis, PSR-stained sections were analyzed at 100x magnification using polarized light microscopy and ImageJ software as follows: PSR increases birefringence of collagen fibers type-dependently, thus visualizing thick type I collagen (Col-I, 1.6-2.4 μm in diameter) in red-orange shades and thin type III collagen (Col-III, <0.8 μm in diameter) in green-yellow shades; by setting the appropriate “hue” thresholds of the color spectrum, the red-orange and green-yellow shaded areas were expressed as the percentage of the total area of interest (AOI) by ImageJ processing. To particularly detail kidney fibrosis, three AOIs were determined by employing a method described previously (12): (i) to assess glomerular fibrosis, 50 AOIs per animal (i.e., 450 per group; n = 9 animals per group) of 50 × 50 μm each, placed at intraglomerular space were examined (Figure 2A1); (ii) to assess tubulointerstitial fibrosis, 50 AOIs per animal (i.e., 450 per group; n = 9 animals per group) of 72 × 192 μm each, placed at interstitial cortex space including no glomeruli or vessels were examined (Figure 2B1); (iii) to assess vascular/perivascular fibrosis, 5 AOIs per animal (i.e., 45 per group; n = 9 animals per group) were selected by the tight-cropping of an artery between 50 and 100 μm in diameter (corresponding to interlobar, arcuate, and interlobular arteries) in each; only cross-sectionally captured arteries were considered (Figure 2C1).
In order to determine type IV collagen (Col-IV) volume in glomeruli, anti-Col-IV-immunostained sections were analyzed at 200x magnification using transmitted light microscopy and ImageJ software as follows (13): the anti-Col-IV-immunostain visualizes Col-IV in brown shades (Figure 3A); by setting the appropriate “hue” threshold of the color spectrum, the brown shaded area was expressed as the percentage of the total glomerular AOI by ImageJ processing. Ten glomerular AOIs per animal (i.e., 90 per group; n = 9 animals per group) of 50 × 50 μm each, placed at intraglomerular space were examined.
Tubular injury was determined as tubular injury score by employing a method described previously (14). Briefly, 20 cortical fields per animal (i.e., 180 per group; n = 9 animals per group) in H-E-stained sections were analyzed at 100x magnification using transmitted light microscopy. Tubular injury was defined as tubular dilatation, atrophy, cast formation, sloughing of tubular epithelial cells, or thickening of the tubular basement membrane (Figure 4A). The tubular injury was semi-quantitatively scored using the following scoring system: Score 0, no tubular injury; Score 1, <10% of tubules injured; Score 2, 10–25% of tubules injured; Score 3, 26–50% of tubules injured; Score 4, 51–75% of tubules injured; Score 5, >75% of tubules injured.
The results are presented as the mean ± SEM. A Shapiro-Wilk normality test was used to determine data distribution. The one-way two-tailed analysis of variance (ANOVA) followed by a Holm-Sidak post-hoc test was used for statistical analysis. A Spearman correlation was used to analyze the relationship between glomerular tuft area and glomerular Col-IV volume, and tubular injury score and tubulointerstitial fibrosis. Statistical significance was defined as P < 0.05. The statistical analysis was conducted using GraphPad Prism 8 for Windows (GraphPad Software, La Jolla, CA, USA).
Results
The SBP and HR averaged over the 4-week-course of treatment were 122.6 ± 1.05 mmHg and 356.0 ± 2.75 bpm in controls; L-NAME increased (P < 0.01) average SBP by 27% and decreased (P < 0.01) average HR by 8%. In the L-NAME group, ivabradine decreased both average SBP and HR by 8% (P < 0.05) and 12% (P < 0.01), respectively; in controls, ivabradine decreased (P < 0.01) average HR by 16% and had no effect on average SBP (Figures 1A,B).
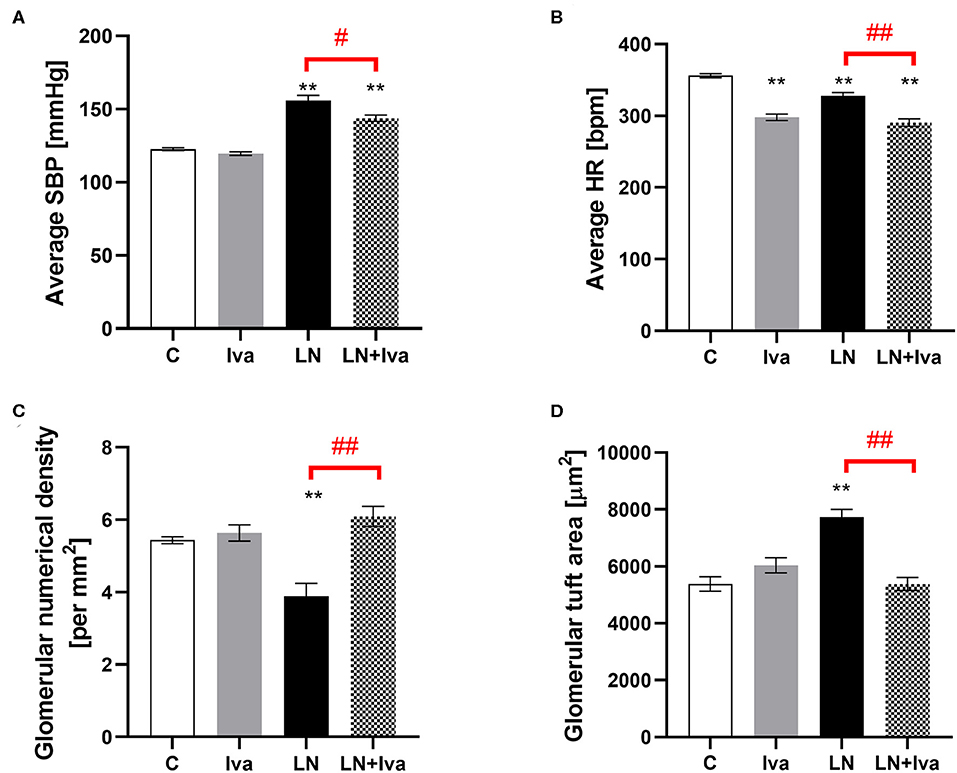
Figure 1. Effect of ivabradine on average systolic blood pressure (SBP) (A), average heart rate (HR) (B), glomerular numerical density per area (C), and glomerular tuft area (D) in L-NAME-induced hypertension after 4 weeks of treatment. C, controls; Iva, ivabradine; LN, L-NAME; n = 9 animals per group. One-way two-tailed ANOVA followed by Holm–Sidak post-hoc test; **P < 0.01 vs. C; #P < 0.05 vs. LN; ##P < 0.01 vs. LN.
The glomerular numerical density per area was 5.43 ± 0.1 per mm2 in controls and L-NAME decreased (P < 0.01) it by 28%. In the L-NAME group, ivabradine increased (P < 0.01) the glomerular numerical density per area by 57% (Figure 1C). The glomerular tuft area was 5,383 ± 256 μm2 in controls and L-NAME increased (P < 0.01) it by 44%. In the L-NAME group, ivabradine decreased (P < 0.01) the glomerular tuft area by 30% (Figure 1D).
Quantitative analysis of glomerular fibrosis: in controls, the volume of Col-I and Col-III in intraglomerular AOI were 0.68 ± 0.11 and 2.71 ± 0.38%, respectively; L-NAME increased (P < 0.0001) the proportion of Col-I by 1,584%. In the L-NAME group, ivabradine decreased (P < 0.05) the proportion of Col-I by 33%. The Col-I:Col-III ratio was 0.29 ± 0.06 in controls and L-NAME increased (P < 0.05) it by 3,880%; ivabradine had no effect on the ratio (Figure 2A).
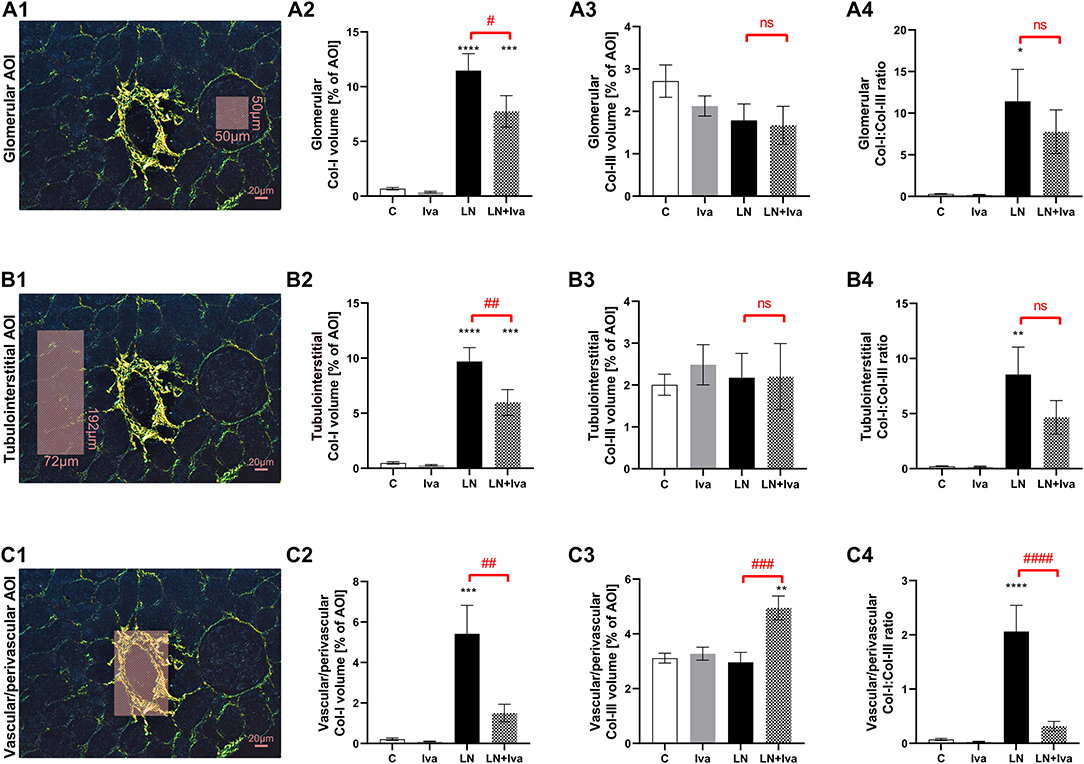
Figure 2. Effect of ivabradine on kidney fibrosis detailed as glomerular (A), tubulointerstitial (B), and vascular/perivascular fibrosis (C) in L-NAME-induced hypertension after 4 weeks of treatment. PSR-stained sections at 100x magnification using polarized light microscopy. AOI, area of interest depicted as shaded rectangle; C, controls; Col-I, type I collagen; Col-III, type III collagen; Iva, ivabradine; LN, L-NAME; n = 9 animals per group. One-way two-tailed ANOVA followed by Holm–Sidak post-hoc test; *P < 0.05 vs. C; **P < 0.01 vs. C; ***P < 0.001 vs. C; ****P < 0.0001 vs. C; #P < 0.05 vs. LN; ##P < 0.01 vs. LN; ###P < 0.001 vs. LN; ####P < 0.0001 vs. LN; ns, non-significant.
Quantitative analysis of tubulointerstitial fibrosis: in controls, the volume of Col-I and Col-III in tubulointerstitial AOI were 0.49 ± 0.11 and 2.01 ± 0.25%, respectively; L-NAME increased (P < 0.0001) the proportion of Col-I by 1,894%. In the L-NAME group, ivabradine decreased (P < 0.01) the proportion of Col-I by 38%. The Col-I:Col-III ratio was 0.22 ± 0.04 in controls and L-NAME increased (P < 0.01) it by 3,734%; ivabradine had no effect on the ratio (Figure 2B).
Quantitative analysis of vascular/perivascular fibrosis: in controls, the volume of Col-I and Col-III in vascular/perivascular AOI were 0.21 ± 0.06 and 3.11 ± 0.18%, respectively; L-NAME increased (P < 0.001) the proportion of Col-I by 2,487%. In the L-NAME group, ivabradine decreased (P < 0.01) the proportion of Col-I by 72% and increased (P < 0.001) the proportion of Col-III by 67%. The Col-I:Col-III ratio was 0.07 ± 0.02 in controls and L-NAME increased (P < 0.0001) it by 2,796%; ivabradine decreased (P < 0.0001) the ratio by 85% (Figure 2C).
The volume of Col-IV in glomerular AOI was 2.80 ± 0.90% in controls and L-NAME increased (P < 0.01) it by 245%. In the L-NAME group, ivabradine decreased (P < 0.01) glomerular Col-IV volume by 63% (Figures 3A,B). The glomerular Col-IV volume significantly (P < 0.001) correlated with glomerular tuft area (Spearman r = 0.51) (Figure 3C).
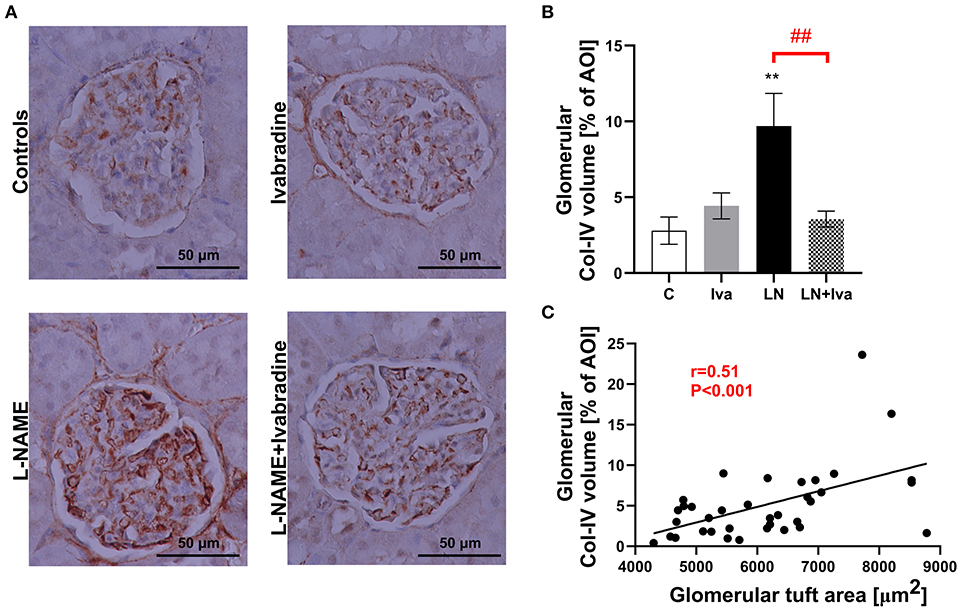
Figure 3. Effect of ivabradine on type IV collagen volume in glomeruli (A,B) and the relationship between glomerular tuft area and glomerular type IV collagen volume (C) in L-NAME-induced hypertension after 4 weeks of treatment. For (A): anti-collagen IV-immunostained sections at 200x magnification using transmitted light microscopy. For (B): C, controls; Iva, ivabradine; LN, L-NAME; n = 9 animals per group. One-way two-tailed ANOVA followed by Holm–Sidak post-hoc test; **P < 0.01 vs. C; ##P < 0.01 vs. LN. For (C): AOI, area of interest; Col-IV, type IV collagen. Spearman correlation; n = 9 animals per group.
The tubular injury score was 0.45 ± 0.02 in controls and L-NAME increased (P < 0.001) it by 754%. In the L-NAME group, ivabradine decreased (P < 0.01) the tubular injury score by 34% (Figures 4A,B). The tubular injury score significantly (P < 0.001) correlated with the sum of Col-I and Col-III volume in tubulointerstitial AOI (Spearman r = 0.81) (Figure 4C).
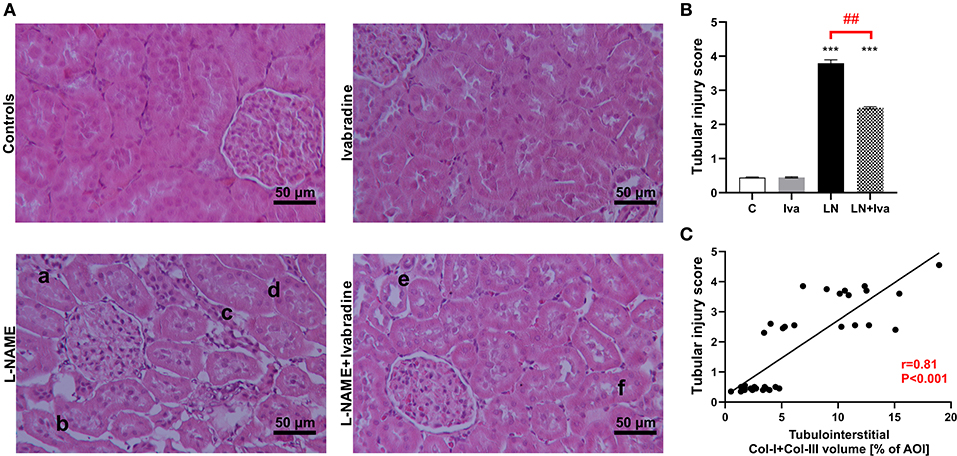
Figure 4. Effect of ivabradine on tubular injury (A,B) and the relationship between tubular injury score and tubulointerstitial fibrosis (C) in L-NAME-induced hypertension after 4 weeks of treatment. For (A): H-E-stained sections at 100x magnification using transmitted light microscopy; a, tubular cast; b, tubular atrophy and interstitial thickening; c, interstitial cellular infiltration; d, tubular basal membrane thickening; e, tubular dilatation; f, tubular cell sloughing. For (B): C, controls; Iva, ivabradine; LN, L-NAME; n = 9 animals per group. One-way two-tailed ANOVA followed by Holm–Sidak post-hoc test; ***P < 0.001 vs. C; ##P < 0.01 vs. LN. For (C): AOI, area of interest; Col-I, type I collagen; Col-III, type III collagen. Spearman correlation; n = 9 animals per group.
Discussion
The increased SBP, induced by a 4-week L-NAME administration, was associated with decreased glomerular density, increased glomerular tuft area, tubular injury, and profound renal fibrosis. Besides reducing the average HR and SBP, ivabradine increased glomerular density and reduced glomerular tuft area. Furthermore, ivabradine ameliorated the L-NAME-induced kidney fibrosis site-dependently: it decreased Col-I volume in glomerular, interstitial, and vascular/perivascular fibrosis and increased Col-III volume in vascular/perivascular fibrosis. Ivabradine also mitigated the increase of both the glomerular Col-IV volume and the tubular injury score in L-NAME-hypertension. To the best of our knowledge, this is the first study analysing L-NAME-induced kidney fibrosis and renoprotection by ivabradine in a site-specific manner.
L-NAME inhibits nitric oxide (NO) synthase activity and decreases cyclic guanosine monophosphate (cGMP) concentration shown in various tissues including heart, aorta, brain, and kidney, thus resulting in NO-deficient hypertension (15–19). NO's waning vasodilative and antiproliferative effects associated with concomitant neurohumoral activation (20) were shown to result in target organ damage (7, 18, 19). In kidneys, L-NAME-hypertension induces glomerulosclerosis, tubulointerstitial fibrosis, and tubular atrophy associated with a deteriorated glomerular filtration rate and increased urinary protein excretion (21–23); these alterations are administered by a cluster of pro-inflammatory and pro-proliferative hormones, cytokines and growth factors (24).
The renal antifibrotic effect of ivabradine observed in this study was similar to previous findings within the heart: ivabradine reduced cardiac collagen and improved left ventricular function in post-myocardial infarction (MI) rats (25) and cholesterol-fed rabbits (26). Antiremodeling by ivabradine may be associated with some of its pleiotropic effects. Indeed, ivabradine improved endothelial function in ApoE knockout mice by reducing vascular oxidative stress and preventing endothelial NO synthase uncoupling (27, 28) and in patients with coronary artery disease after complete revascularisation (6). Increased NO-availability by various interventions was previously shown to reduce the L-NAME's proliferative effect not only in the heart and aorta, but also in the brain and kidneys (17, 18, 28). Besides improving NO-availability, ivabradine was shown to reduce serum angiotensin II in both post-MI rats (25) and hypercholesterolemic rabbits (26), and to decrease serum aldosterone in L-NAME-induced hypertension (7). The potential inhibition of the renin-angiotensin-aldosterone system might contribute to the antiremodeling effect of ivabradine. Indeed, previous data in L-NAME-hypertension indicated an angiotensin converting enzyme inhibitor to mitigate remodeling of the heart, aorta (19, 20, 29–31) and kidneys (21). Furthermore, one of the principle factors of CKD management is the reduction of hemodynamic overload. The recommended target SBP values in hypertensive patients with CKD are below 130 vs. 140 mmHg in hypertensive patients without kidney disease (32). Presumably, HR-reduction by ivabradine may be renoprotective via diminishing the hemodynamic burden by both the rate-pressure working product decline and vascular shear stress modulation (33). Here, in line with our previous experiments (7, 8, 34), ivabradine reduced both the average SBP and HR, which indeed might have contributed to the renoprotection. The HR reduction by L-NAME found in this study is consistent with previous results in L-NAME-hypertension by our laboratory (7, 8, 15) and others (35, 36). Several plausible mechanisms of heart rate reduction in L-NAME-hypertension were suggested, including the baroreceptor-mediated modulation of the autonomic nervous system (37, 38) and the direct effect of NO-deficiency on cardiac function (39, 40). Yet, ivabradine is an open-channel If-blocker, i.e., the ivabradine molecule is able to access its binding site in the f-channel only when the channel is open. This can underlie ivabradine's use-dependence, i.e., a blocking action that is more pronounced the more frequently the f-channel is open, implying that the higher the HR the larger the HR-reducing effect of ivabradine (41, 42). This might explain our finding that ivabradine reduced HR in controls by 16%, but only 12% in L-NAME-hypertension, since the HR in L-NAME-hypertension was already decreased by L-NAME below the values seen in controls.
Col-I and Col-III are the most abundant collagen types in the extracellular matrix (43, 44). In kidneys, they were found co-expressed at all three of the investigated sites, i.e., glomeruli, tubulointerstitium and vasculature (45), and gradually deposited from the early stages of kidney fibrosis (45, 46). Col-I and Col-III co-expression is considered to provide a tissue with high tensile strength, but also contribute to its extensile properties (44, 46). Indeed, Col-I exerts high tensile strength and its expression is associated with tissue stiffness, whilst Col-III is more distensible and its expression refers to tissue elasticity, distensibility, and softness (43, 44). Thus, a high Col-I:Col-III ratio was found in tissues with high mechanical stiffness and low elasticity such as bones, and low Col-I:Col-III ratio was found in tissues with high elasticity, distensibility, or softness such as lung, bladder, and blood vessels (44). In cardiovascular remodeling, the Col-I:Col-III ratio is considered a marker of tissue stiffness determining mechanical properties and was shown to be associated with adverse outcomes (47). Indeed, an elevated Col-I:Col-III ratio was associated with increased myocardial stiffness and electrical instability of the myocardium (48), and increased stiffness of vessels including the aorta and arteries (49, 50). Increased stiffness of the remodeled vasculature was shown to be prognostically unfavorable in hypertension (51). We previously found an up-ward shift of the Col-I:Col-III ratio in a remodeled heart (52) and aorta (49) in a model of continuous light-induced hypertension. Reducing the Col-I:Col-III ratio in vessels was associated with improved hemodynamics in continuous light-induced hypertension (49) and pulmonary arterial hypertension (53). In this study, we dosed L-NAME for only 4 weeks (reaching a moderate increase in 4-week average systolic blood pressure) to assess early hypertensive kidney damage and its potential reversibility with ivabradine. Although Col-I and Col-III are deposited from early stages of kidney fibrosis (45, 46), NO deficiency in L-NAME-hypertension was found to specifically up-regulate collagen I expression in kidneys at an early stage even preceding the increase in blood pressure (54). This might explain the increased Col-I expression (early activation) and unchanged Col-III expression (activation lagging) observed in early hypertensive kidney damage in this study. Furthermore, in L-NAME-treated rats, ivabradine increased Col-III volume solely in the vascular/perivascular fibrosis while a profound drop in Col-I volume prevailed at all three of the investigated sites. Previously, ivabradine was shown to increase aortic compliance in apolipoprotein E-deficient mice (55), improve carotid pulsatile arterial hemodynamics in spontaneously hypertensive rats (56), restore acetylcholine-induced maximal dilatation of renal and cerebral arteries in dyslipidaemic mice (57), and most importantly, improve myocardial perfusion in post-MI rats by ameliorating perivascular fibrosis in small resistant coronary arteries (25). Therefore, by virtue of Col-III's elastic properties, the vascular/perivascular Col-III enhancement associated with the reduction of the Col-I:Col-III ratio by ivabradine observed in our study implies improved arterial compliance and pulsatile hemodynamics (49).
Col-IV, a main component of the glomerular basement membrane, is considered to play a critical role in glomerular pathology (58). Indeed, capillary expansion and mesangial cell stretching by increased intraglomerular pressure, often seen in hypertension, were found to provoke increased mesangial extracellular matrix (including Col-IV) production and deposition (59). Therefore, increased Col-IV protein expression was found in kidneys in various animal models of hypertension such as spontaneously hypertensive rats (60), angiotensin II-induced hypertension (61) or 2 kidneys, 1 clip model of hypertension (62), and also in patients with preeclampsia or other hypertensive syndromes in pregnancy (63). In L-NAME-hypertension, in particular, an exaggerated Col-IV gene and protein expression within the renal vasculature associated with glomerulosclerosis was found (64). Nonetheless, to the best of our knowledge, this is the first study determining Col-IV volume specifically in glomeruli in L-NAME-hypertension, where ivabradine mitigated the L-NAME-induced increase of glomerular Col-IV volume, thus supporting ivabradine's beneficial effect on glomerulosclerosis in L-NAME-hypertension.
Furthermore, in this study, L-NAME-hypertension induced tubular injury that correlated with tubulointerstitial fibrosis. This is in line with findings from other animal models of hypertension such as spontaneously hypertensive rats (65), angiotensin II-induced hypertension (66), and the Dahl salt-sensitive rat model of hypertension (67). Yet, mechanisms underlying tubular injury in hypertension are puzzling. Indeed, there were suggested (i) hemodynamics-dependent mechanisms, including tubular atrophy following glomerulotubular disconnection associated with glomerular injury (68), and (ii) hemodynamics-independent mechanisms, including renal oxidative stress and inflammation (69). In this study, ivabradine mitigated tubular injury and decreased tubulointerstitial fibrosis in L-NAME-hypertension, which were presumably associated with ivabradine's effects on both hemodynamics-dependent and independent mechanisms of tubular injury.
Recently, the plasma and urinary markers of Col-III (70), Col-IV and Col-VI (43) turnover have been shown to be a proxy for kidney fibrosis correlating with kidney function deterioration, and severity and the prognosis of CKD, thus holding promise as a novel, non-invasive diagnostic and prognostic tool to monitor kidney fibrosis in CKD (70, 71). This histopathological study was designed to directly and site-specifically determine collagen volumes in L-NAME-induced kidney fibrosis. It may be of interest to correlate histopathology and plasma or urinary markers of kidney fibrosis and function. Yet, this was beyond the scope and possibilities of the present histopathological study.
We conclude that ivabradine mitigated alterations to glomerular density and tuft area and site-specifically modified renal fibrosis in L-NAME-hypertension. These results suggest that ivabradine may be renoprotective in hypertensive kidney disease.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by The Ethical Committee of the Institute of Pathophysiology, Faculty of Medicine, Comenius University, Bratislava, Slovakia.
Author Contributions
FS and TB conceived and designed the study and drafted the manuscript. PS, TB, KR, SA, and KK carried out animal experiments. PS, AB, PJ, MA, and LP conducted histopathological analysis. All authors participated in data analysis and interpretation, manuscript revision, and approved the submitted version.
Funding
This experiment was supported by research Grants VEGA 1/0035/19, VEGA 2/0112/19, UK/45/2020, and Program PROGRES Q40/5.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mills KT, Xu Y, Zhang W, Bundy JD, Chen CS, Kelly TN, et al. A systematic analysis of worldwide population-based data on the global burden of chronic kidney disease in 2010. Kidney Int. (2015) 88:950–7. doi: 10.1038/ki.2015.230
2. Hrenak J, Paulis L, Repova K, Aziriova S, Nagtegaal EJ, Reiter RJ, et al. Melatonin and renal protection: novel perspectives from animal experiments and human studies (review). Curr Pharm Des. (2015) 21:936–49. doi: 10.2174/1381612820666140929092929
3. Schlondorff DO. Overview of factors contributing to the pathophysiology of progressive renal disease. Kidney Int. (2008) 74:860–6. doi: 10.1038/ki.2008.351
4. Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: implications for therapy. Hypertension. (2004) 44:595–601. doi: 10.1161/01.HYP.0000145180.38707.84
5. Swedberg K, Komajda M, Böhm M, Borer JS, Ford I, Dubost-Brama A, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. (2010) 376:875–85. doi: 10.1016/S0140-6736(10)61198-1
6. Mangiacapra F, Colaiori I, Ricottini E, Balducci F, Creta A, Demartini C, et al. Heart rate reduction by IVabradine for improvement of ENDothELial function in patients with coronary artery disease: the RIVENDEL study. Clin Res Cardiol. (2017) 106:69–75. doi: 10.1007/s00392-016-1024-7
7. Simko F, Baka T, Poglitsch M, Repova K, Aziriova S, Krajcirovicova K, et al. Effect of ivabradine on a hypertensive heart and the renin-angiotensin-aldosterone system in L-NAME-induced hypertension. Int J Mol Sci. (2018) 19:E3017. doi: 10.3390/ijms19103017
8. Baka T, Simko F. Ivabradine reversed nondipping heart rate in rats with l-NAME-induced hypertension. Clin Exp Pharmacol Physiol. (2019) 46:607–10. doi: 10.1111/1440-1681.13075
9. Simko F, Baka T. Chronotherapy as a potential approach to hypertensive patients with elevated heart rate? Br J Clin Pharmacol. (2019) 85:1861–2. doi: 10.1111/bcp.14020
10. Hrenak J, Arendasova K, Rajkovicova R, Aziriova S, Repova K, Krajcirovicova K, et al. Protective effect of captopril, olmesartan, melatonin and compound 21 on doxorubicin-induced nephrotoxicity in rats. Physiol Res. (2013) 62 (Suppl. 1):S181–9.
11. Pechanova O, Matuskova J, Capikova D, Jendekova L, Paulis L, Simko F. Effect of spironolactone and captopril on nitric oxide and S-nitrosothiol formation in kidney of L-NAME-treated rats. Kidney Int. (2006) 70:170–6. doi: 10.1038/sj.ki.5001513
12. Seccia TM, Maniero C, Belloni AS, Guidolin D, Pothen P, Pessina AC, et al. Role of angiotensin II, endothelin-1 and L-type calcium channel in the development of glomerular, tubulointerstitial and perivascular fibrosis. J Hypertens. (2008) 26:2022–9. doi: 10.1097/HJH.0b013e328309f00a
13. Hartig SM. Basic image analysis and manipulation in ImageJ. Curr Protoc Mol Biol. (2013) Chapter 14:Unit14.15. doi: 10.1002/0471142727.mb1415s102
14. Pichler RH, Franceschini N, Young BA, Hugo C, Andoh TF, Burdmann EA, et al. Pathogenesis of cyclosporine nephropathy: roles of angiotensin II and osteopontin. J Am Soc Nephrol. (1995) 6:1186–96.
15. Simko F, Baka T, Krajcirovicova K, Repova K, Aziriova S, Zorad S, et al. Effect of melatonin on the renin-angiotensin-aldosterone system in l-NAME-induced hypertension. Molecules. (2018) 23:E265. doi: 10.3390/molecules23020265
16. Bernatova I, Pechanova O, Simko F. Captopril prevents NO-deficient hypertension and left ventricular hypertrophy without affecting nitric oxide synthase activity in rats. Physiol Res. (1996) 45:311–6.
17. Pechanova O, Bernatova I, Pelouch V, Simko F. Protein remodelling of the heart in NO-deficient hypertension: the effect of captopril. J Mol Cell Cardiol. (1997) 29:3365–74. doi: 10.1006/jmcc.1997.0566
18. Bernatova I, Pechanova O, Simko F. Effect of captopril in L-NAME-induced hypertension on the rat myocardium, aorta, brain and kidney. Exp Physiol. (1999) 84:1095–105. doi: 10.1111/j.1469-445X.1999.01890.x
19. Simko F, Matuskova J, Luptak I, Krajcirovicova K, Kucharska J, Gvozdjakova A, et al. Effect of simvastatin on remodeling of the left ventricle and aorta in L-NAME-induced hypertension. Life Sci. (2004) 74:1211–24. doi: 10.1016/j.lfs.2003.07.032
20. Simko F, Simko J. The potential role of nitric oxide in the hypertrophic growth of the left ventricle. Physiol Res. (2000) 49:37–46.
21. Hropot M, Grötsch H, Klaus E, Langer KH, Linz W, Wiemer G, et al. Ramipril prevents the detrimental sequels of chronic NO synthase inhibition in rats: hypertension, cardiac hypertrophy and renal insufficiency. Naunyn Schmiedebergs Arch Pharmacol. (1994) 350:646–52. doi: 10.1007/BF00169370
22. Ndisang JF, Chibbar R. Heme oxygenase improves renal function by potentiating podocyte-associated proteins in Nω-nitro-l-arginine-methyl ester (l-NAME)-induced hypertension. Am J Hypertens. (2015) 28:930–42. doi: 10.1093/ajh/hpu240
23. Ikeda H, Tsuruya K, Toyonaga J, Masutani K, Hayashida H, Hirakata H, et al. Spironolactone suppresses inflammation and prevents L-NAME-induced renal injury in rats. Kidney Int. (2009) 75:147–55. doi: 10.1038/ki.2008.507
24. Suehiro T, Tsuruya K, Ikeda H, Toyonaga J, Yamada S, Noguchi H, et al. Systemic aldosterone, but not angiotensin II, plays a pivotal role in the pathogenesis of renal injury in chronic nitric oxide-deficient male rats. Endocrinology. (2015) 156:2657–66. doi: 10.1210/en.2014-1369
25. Dedkov EI, Zheng W, Christensen LP, Weiss RM, Mahlberg-Gaudin F, Tomanek RJ. Preservation of coronary reserve by ivabradine-induced reduction in heart rate in infarcted rats is associated with decrease in perivascular collagen. Am J Physiol Heart Circ Physiol. (2007) 293:H590–8. doi: 10.1152/ajpheart.00047.2007
26. Busseuil D, Shi Y, Mecteau M, Brand G, Gillis MA, Thorin E, et al. Heart rate reduction by ivabradine reduces diastolic dysfunction and cardiac fibrosis. Cardiology. (2010) 117:234–42. doi: 10.1159/000322905
27. Custodis F, Baumhäkel M, Schlimmer N, List F, Gensch C, Böhm M, et al. Heart rate reduction by ivabradine reduces oxidative stress, improves endothelial function, and prevents atherosclerosis in apolipoprotein E-deficient mice. Circulation. (2008) 117:2377–87. doi: 10.1161/CIRCULATIONAHA.107.746537
28. Kröller-Schön S, Schulz E, Wenzel P, Kleschyov AL, Hortmann M, Torzewski M, et al. Differential effects of heart rate reduction with ivabradine in two models of endothelial dysfunction and oxidative stress. Basic Res Cardiol. (2011) 106:1147–58. doi: 10.1007/s00395-011-0227-3
29. Simko F, Pechanova O, Pelouch V, Krajcirovicova K, Mullerova M, Bednarova K, et al. Effect of melatonin, captopril, spironolactone and simvastatin on blood pressure and left ventricular remodelling in spontaneously hypertensive rats. J Hypertens Suppl. (2009) 27:S5–10. doi: 10.1097/01.hjh.0000358830.95439.e8
30. Simko F, Pechanova O, Pelouch V, Krajcirovicova K, Celec P, Palffy R, et al. Continuous light and L-NAME-induced left ventricular remodelling: different protection with melatonin and captopril. J Hypertens. (2010) 28 (Suppl. 1):S13–8. doi: 10.1097/01.hjh.0000388489.28213.08
31. Simko F, Pechanova O, Repova K, Aziriova S, Krajcirovicova K, Celec P, et al. Lactacystin-induced model of hypertension in rats: effects of melatonin and captopril. Int J Mol Sci. (2017) 18:E1612. doi: 10.3390/ijms18081612
32. Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, et al. 2018 ESC/ESH guidelines for the management of arterial hypertension. Eur Heart J. (2018) 39:3021–104. doi: 10.1097/HJH.0000000000001940
33. Luong L, Duckles H, Schenkel T, Mahmoud M, Tremoleda JL, Wylezinska-Arridge M, et al. Heart rate reduction with ivabradine promotes shear stress-dependent anti-inflammatory mechanisms in arteries. Thromb Haemost. (2016) 116:181–90. doi: 10.1160/TH16-03-0214
34. Aziriova S, Repova K, Krajcirovicova K, Baka T, Zorad S, Mojto V, et al. Effect of ivabradine, captopril and melatonin on the behaviour of rats in L-nitro-arginine methyl ester-induced hypertension. J Physiol Pharmacol. (2016) 67:895–902.
35. Küng CF, Moreau P, Takase H, Lüscher TF. L-NAME hypertension alters endothelial and smooth muscle function in rat aorta. Prevention by trandolapril and verapamil. Hypertension. (1995) 26:744–51. doi: 10.1161/01.HYP.26.5.744
36. Jones AM, Wilkerson DP, Campbell IT. Nitric oxide synthase inhibition with L-NAME reduces maximal oxygen uptake but not gas exchange threshold during incremental cycle exercise in man. J Physiol. (2004) 560:329–38. doi: 10.1113/jphysiol.2004.065664
37. Scrogin KE, Hatton DC, Chi Y, Luft FC. Chronic nitric oxide inhibition with L-NAME: effects on autonomic control of the cardiovascular system. Am J Physiol. (1998) 274:R367–74. doi: 10.1152/ajpregu.1998.274.2.R367
38. Vasquez EC, Cunha RS, Cabral AM. Baroreceptor reflex function in rats submitted to chronic inhibition of nitric oxide synthesis. Braz J Med Biol Res. (1994) 27:767–74.
39. Sherman AJ, Davis CA III, Klocke FJ, Harris KR, Srinivasan G, Yaacoub AS, et al. Blockade of nitric oxide synthesis reduces myocardial oxygen consumption in vivo. Circulation. (1997) 95:1328–34. doi: 10.1161/01.CIR.95.5.1328
40. Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, et al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA. (1996) 93:13176–81. doi: 10.1073/pnas.93.23.13176
41. DiFrancesco D. Cardiac pacemaker I(f) current and its inhibition by heart rate-reducing agents. Curr Med Res Opin. (2005) 21:1115–22. doi: 10.1185/030079905X50543
42. Bucchi A, Baruscotti M, DiFrancesco D. Current-dependent block of rabbit sino-atrial node I(f) channels by ivabradine. J Gen Physiol. (2002) 120:1–13. doi: 10.1085/jgp.20028593
43. Rasmussen DGK, Boesby L, Nielsen SH, Tepel M, Birot S, Karsdal MA, et al. Collagen turnover profiles in chronic kidney disease. Sci Rep. (2019) 9:16062. doi: 10.1038/s41598-019-51905-3
44. Asgari M, Latifi N, Heris HK, Vali H, Mongeau L. In vitro fibrillogenesis of tropocollagen type III in collagen type I affects its relative fibrillar topology and mechanics. Sci Rep. (2017) 7:1392. doi: 10.1038/s41598-017-01476-y
45. Bülow RD, Boor P. Extracellular matrix in kidney fibrosis: more than just a scaffold. J Histochem Cytochem. (2019) 67:643–61. doi: 10.1369/0022155419849388
46. Genovese F, Manresa AA, Leeming DJ, Karsdal MA, Boor P. The extracellular matrix in the kidney: a source of novel non-invasive biomarkers of kidney fibrosis? Fibrogenesis Tissue Repair. (2014) 7:4. doi: 10.1186/1755-1536-7-4
47. Weber KT. Targeting pathological remodeling: concepts of cardioprotection and reparation. Circulation. (2000) 102:1342–5. doi: 10.1161/01.CIR.102.12.1342
48. Xu J, Cui G, Esmailian F, Plunkett M, Marelli D, Ardehali A, et al. Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation. Circulation. (2004) 109:363–8. doi: 10.1161/01.CIR.0000109495.02213.52
49. Repova-Bednarova K, Aziriova S, Hrenak J, Krajcirovicova K, Adamcova M, Paulis L, et al. Effect of captopril and melatonin on fibrotic rebuilding of the aorta in 24 hour light-induced hypertension. Physiol Res. (2013) 62 (Suppl. 1):S135–41.
50. Dobrin PB, Mrkvicka R. Failure of elastin or collagen as possible critical connective tissue alterations underlying aneurysmal dilatation. Cardiovasc Surg. (1994) 2:484–8.
51. Schiffrin EL. Vascular remodeling in hypertension: mechanisms and treatment. Hypertension. (2012) 59:367–74. doi: 10.1161/HYPERTENSIONAHA.111.187021
52. Paulis L, Vazan R, Simko F, Pechanova O, Styk J, Babal P, et al. Morphological alterations and NO-synthase expression in the heart after continuous light exposure of rats. Physiol Res. (2007) 56(Suppl. 2):S71–6.
53. Liu P, Yan S, Chen M, Chen A, Yao D, Xu X, et al. Effects of baicalin on collagen I and collagen III expression in pulmonary arteries of rats with hypoxic pulmonary hypertension. Int J Mol Med. (2015) 35:901–8. doi: 10.3892/ijmm.2015.2110
54. Chatziantoniou C, Boffa JJ, Ardaillou R, Dussaule JC. Nitric oxide inhibition induces early activation of type I collagen gene in renal resistance vessels and glomeruli in transgenic mice. Role of endothelin. J Clin Invest. (1998) 101:2780–9. doi: 10.1172/JCI2132
55. Custodis F, Fries P, Müller A, Stamm C, Grube M, Kroemer HK, et al. Heart rate reduction by ivabradine improves aortic compliance in apolipoprotein E-deficient mice. J Vasc Res. (2012) 49:432–40. doi: 10.1159/000339547
56. Albaladejo P, Carusi A, Apartian A, Lacolley P, Safar ME, Bénétos A. Effect of chronic heart rate reduction with ivabradine on carotid and aortic structure and function in normotensive and hypertensive rats. J Vasc Res. (2003) 40:320–8. doi: 10.1159/000072696
57. Drouin A, Gendron ME, Thorin E, Gillis MA, Mahlberg-Gaudin F, Tardif JC. Chronic heart rate reduction by ivabradine prevents endothelial dysfunction in dyslipidaemic mice. Br J Pharmacol. (2008) 154:749–57. doi: 10.1038/bjp.2008.116
58. Miner JH. The glomerular basement membrane. Exp Cell Res. (2012) 318:973–8. doi: 10.1016/j.yexcr.2012.02.031
59. Riser BL, Cortes P, Zhao X, Bernstein J, Dumler F, Narins RG. Intraglomerular pressure and mesangial stretching stimulate extracellular matrix formation in the rat. J Clin Invest. (1992) 90:1932–43. doi: 10.1172/JCI116071
60. Guo Z, Sun H, Zhang H, Zhang Y. Anti-hypertensive and renoprotective effects of berberine in spontaneously hypertensive rats. Clin Exp Hypertens. (2015) 37:332–9. doi: 10.3109/10641963.2014.972560
61. Castoldi G, Carletti R, Ippolito S, Colzani M, Barzaghi F, Stella A, et al. Renal anti-fibrotic effect of sodium glucose cotransporter 2 inhibition in angiotensin II-dependent hypertension. Am J Nephrol. (2020) 51:119–29. doi: 10.1159/000505144
62. Li L, Wang C, Gu Y. Collagen IV, a promising serum biomarker for evaluating the prognosis of revascularization in a 2-kidney, 1-clip hypertensive rat model. Interact Cardiovasc Thorac Surg. (2020) 30:483–90. doi: 10.1093/icvts/ivz275
63. Foidart JM, Nochy D, Nusgens B, Foidart JB, Mahieu PR, Lapiere CM, et al. Accumulation of several basement membrane proteins in glomeruli of patients with preeclampsia and other hypertensive syndromes of pregnancy. possible role of renal prostaglandins and fibronectin. Lab Invest. (1983) 49:250–9.
64. Boffa JJ, Lu Y, Placier S, Stefanski A, Dussaule JC, Chatziantoniou C. Regression of renal vascular and glomerular fibrosis: role of angiotensin II receptor antagonism and matrix metalloproteinases. J Am Soc Nephrol. (2003) 14:1132–44. doi: 10.1097/01.ASN.0000060574.38107.3B
65. Ofstad J, Iversen BM. Glomerular and tubular damage in normotensive and hypertensive rats. Am J Physiol Renal Physiol. (2005) 288:F665–72. doi: 10.1152/ajprenal.00226.2004
66. Polichnowski AJ, Cowley AW Jr. Pressure-induced renal injury in angiotensin II versus norepinephrine-induced hypertensive rats. Hypertension. (2009) 54:1269–77. doi: 10.1161/HYPERTENSIONAHA.109.139287
67. Wei SY, Wang YX, Zhang QF, Zhao SL, Diao TT, Li JS, et al. Multiple mechanisms are involved in salt-sensitive hypertension-induced renal injury and interstitial fibrosis. Sci Rep. (2017) 7:45952. doi: 10.1038/srep45952
68. Chevalier RL, Forbes MS. Generation and evolution of atubular glomeruli in the progression of renal disorders. J Am Soc Nephrol. (2008) 19:197–206. doi: 10.1681/ASN.2007080862
69. Polichnowski AJ, Lu L, Cowley AW Jr. Renal injury in angiotensin II+L-NAME-induced hypertensive rats is independent of elevated blood pressure. Am J Physiol Renal Physiol. (2011) 300:F1008–16. doi: 10.1152/ajprenal.00354.2010
70. Genovese F, Boor P, Papasotiriou M, Leeming DJ, Karsdal MA, Floege J. Turnover of type III collagen reflects disease severity and is associated with progression and microinflammation in patients with IgA nephropathy. Nephrol Dial Transplant. (2016) 31:472–9. doi: 10.1093/ndt/gfv301
Keywords: ivabradine, L-NAME, hypertension, fibrosis, nephroprotection
Citation: Stanko P, Baka T, Repova K, Aziriova S, Krajcirovicova K, Barta A, Janega P, Adamcova M, Paulis L and Simko F (2020) Ivabradine Ameliorates Kidney Fibrosis in L-NAME-Induced Hypertension. Front. Med. 7:325. doi: 10.3389/fmed.2020.00325
Received: 06 March 2020; Accepted: 03 June 2020;
Published: 10 July 2020.
Edited by:
Jennifer Sullivan, Augusta University, United StatesReviewed by:
Hee-Seong Jang, University of Nebraska Medical Center, United StatesSwastika Sur, University of California San Francisco, United States
Copyright © 2020 Stanko, Baka, Repova, Aziriova, Krajcirovicova, Barta, Janega, Adamcova, Paulis and Simko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fedor Simko, ZmVkb3Iuc2lta29AZm1lZC51bmliYS5zaw==
†These authors have contributed equally to this work