Abstract
Thyroid hormones (THs) elicit significant effects on numerous physiological processes, such as growth, development, and metabolism. A lack of thyroid hormones is not compatible with normal health. Most THs effects are mediated by two different thyroid hormone receptor (TR) isoforms, namely TRα and TRβ, with the TRβ isoform known to be responsible for the main beneficial effects of TH on liver. In brain, despite the crucial role of TRα isoform in neuronal development, TRβ has been proposed to play a role in the remyelination processes. Consequently, over the past two decades, much effort has been applied in developing thyroid hormone analogs capable of uncoupling beneficial actions on liver (triglyceride and cholesterol lowering) and central nervous system (CNS) (oligodendrocyte proliferation) from deleterious effects on the heart, muscle and bone. Sobetirome (GC-1) and subsequently Eprotirome (KB2115) were the first examples of TRβ selective thyromimetics, with Sobetirome differing from the structure of thyronines because of the absence of halogens, biaryl ether oxygen, and amino-acidic side chain. Even though both thyromimetics showed encouraging actions against hypercholesterolemia, non-alcoholic steatohepatitis (NASH) and in the stimulation of hepatocytes proliferation, they were stopped after Phase 1 and Phase 2–3 clinical trials, respectively. In recent years, advances in molecular and structural biology have facilitated the design of new selective thyroid hormone mimetics that exhibit TR isoform-selective binding, and/or liver- and tissue-selective uptake, with Resmetirom (MGL-3196) and Hep-Direct prodrug VK2809 (MB07811) probably representing two of the most promising lipid lowering agents, currently under phase 2–3 clinical trials. More recently the application of a comprehensive panel of ADME-Toxicity assays enabled the selection of novel thyromimetic IS25 and its prodrug TG68, as very powerful lipid lowering agents both in vitro and in vivo. In addition to dyslipidemia and other liver pathologies, THs analogs could also be of value for the treatment of neurodegenerative diseases, such as multiple sclerosis (MS). Sob-AM2, a CNS- selective prodrug of Sobetirome has been shown to promote significant myelin repair in the brain and spinal cord of mouse demyelinating models and it is rapidly moving into clinical trials in humans. Taken together all these findings support the great potential of selective thyromimetics in targeting a large variety of human pathologies characterized by altered metabolism and/or cellular differentiation.
Introduction
Thyroid hormones (THs), namely 3,3′,5,-triiodo-L-thyronine (T3), 3,3′,5,5′-tetraiodo-L-thyronine (T4) and their metabolites refer to a group of tyrosine-based molecules rich in iodine, which exert a key regulatory role in human metabolism (1, 2). Indeed, THs are involved in fetal tissues differentiation, brain development, cardiovascular homeostasis, skeletal maintenance, and in the control of carbohydrates and lipids metabolism (3–5). Thyroxine (T4) is the prevalent form of TH, produced and released from the thyroid gland in humans. Once released, T4 is converted into T3, the major form of TH, by two enzymes called deiodinases type I (D1) and type II (D2), whereas type III deiodinases (D3) leads to the formation of reverse T3 (rT3), that represents a key step in the process of TH inactivation [(2); Figure 1]. This classical picture in the last decade turned to be simplistic, since different classes of enzymes, namely deiodinases, amine transferases, amine oxidases, decarboxylase, and sulfotransferases, have been demonstrated to act on T4 and T3. As a matter of facts, several derivative metabolites of T4 and T3, active on THs receptors, are produced and identified as novel thyroid hormones. These are 3,5-didiodothyronine (T2); thyronamines, particularly 3-iodothyronamine (T1AM) and non-iodinated thyronamines (T0AM); thyroacetic acids, particularly 3,5,3′,5′-thyroacetic acid (TA4), 3,5,3′-thyroacetic acid (TA3), 3-thyroacetic acid (TA1), which will be not discussed in details in this review (6, 7).
Figure 1
During the past few decades, increasing interest has been directed toward a possible therapeutic use of THs and their analogs in the field of dyslipidemia and liver diseases, with an abundance of data coming from many new promising compounds (8–10). Initially the idea that thyromimetics could be useful in lipid metabolism came from the clinical observation that patients with hyperthyroidism show an important reduction of body weight and serum cholesterol. Unfortunately, they also suffer from many unwanted secondary effects such as cardiovascular impairment, muscle and bone loss and depression (11). Thus, the potential to uncouple the beneficial effects of THs from side effects offers an intriguing challenge.
THs actions (Figure 2) can be mediated by THs receptors (TRs) or not, leading to a wide range of effects, previously described as genomic and non-genomic effects (12, 13). Recently a revision of this over simplified description has been proposed with a novel classification of THs effects according to the involved molecular pathways (14), as reported in the next paragraph. THs receptors (TRs) have been known since 1986 (15, 16). They are part of a larger family of intracellular receptors that includes those for steroid hormones, retinoic acid, vitamin D, and peroxisomal proliferators (17). TRs act as transcriptional factors that regulate a wide variety of genes through the interaction with specific co-activators, co-repressors and DNA sequences (TH response elements—TREs), located in the regulatory regions of target genes [(2, 18, 19); Figure 1].
Figure 2
The two isoforms TRα and TRβ are encoded by two genes, THRA on chromosome 17 and THRB on chromosome 3 in human species (Thra and Thrb in mice), respectively, and various forms of TR proteins are produced, through alternative splicing (20). The distribution of specific isoforms varies between different tissues and species: in human, TRα is proved to be predominant in the heart, brain and bone, whereas TRβ is prevalent in the liver, kidneys, pituitary gland, and brain, and it is responsible for THs effects on metabolism (21–23). Based on this consideration, in the last few decades considerable effort has been devoted to developing compounds endowed of selectivity for the beta isoform of the TRs, that would be effective on the treatment of metabolic and/or on brain disorders, without producing deleterious effects on heart and bone. To date, several molecules have been developed (Figure 3), including Sobetirome (GC-1) and Eprotirome (KB2115), just to mention two compounds that have been extensively investigated for many years. These molecules showed great promise of becoming drugs for the treatment of lipid metabolism disorders, but ultimately failed to reach the therapeutic market because of the onset of unwanted side-effects (23). Noteworthy, in recent years there has been a resurgence of interest in developing TRβ selective thyromimetics, and in particular “tissue-selective prodrugs” able to release the desired active compound at the site of action (24). The aim of the present review is to provide an update regarding the most significant steps toward the obtainment of clinically useful thyromimetics, providing further details on a subject that has already been widely discussed in recent reviews (25, 26).
Figure 3
Effects of THs in the Liver and Central Nervous System
In the liver, THs are actively involved in the regulation of metabolism throughout different pathways (Figure 2). Indeed, THs increase energy expenditure by having a direct effect on ATP consumption in metabolic cycles and membrane permeability as well as by indirect effects on mitochondrial biogenesis and activation (27, 28). In addition, different critical steps in the lipid metabolism are under THs control: cholesterol serum clearance by the low density lipoprotein (LDL) receptor, 3-hydroxy-3-metylgutaryl-coenzyme A reductase, cholesterol biosynthesis and cholesterol 7α-hydroxylase (CYP7A1) (29). Disruption in the normal THs signaling has been also reported to be causative of liver diseases such as non-alcoholic fatty liver disease (NAFLD), a very common disease in Western countries, whereas exogenous T3 administration has been demonstrated to improve the fatty profile in animals with experimental induced NAFLD (30, 31). Finally, neoplastic process in the liver as hepato-carcinoma (HCC) appears to be correlated with THs signaling alterations (32) and some studies demonstrated T3 to be effective in reducing cancer progression and metastasis formation (33). Overall, THs appear significantly beneficial for the liver metabolism. Unfortunately, these positive effects are accompanied by the dangerous effects of thyrotoxicosis, such as tachycardia, muscle wasting, bone impairment, in addition to harmful effects in the brain (3).
Many studies in animal models underlined the importance of TRs for the specific action of THs in the liver [(34); Figure 2]. THs have been demonstrated to regulate 7α-hydroxylase (CYP7A), a crucial liver enzyme in the synthesis of bile acids. TRα1 and TRβ different contribution to this regulation has been studied in TRα1 and TRβ knockout mice treated with 2% dietary cholesterol and T3. The study showed that only TRβ knockout mice were unable to modulate CYP7A1 and that T3 administration was not effective on cholesterol levels in these animals (35), thus suggesting a crucial and specific role of TRβ in liver.
Moreover, this specific role is likely not only dependent on the abundance of TRβ isoform in the liver (which represents the 80% of T3 binding activity in rodent). A distinct zonal expression of the TRs and their respective target genes has been proposed in support of the hepatic target gene specificity by TRs (29). On the other hand, TRα1 is predominant in the myocardium, where it covers 40 −70% of T3 heart-binding capacity, depending on the species (36, 37), and it is responsible for the heart side effects of THs excess.
THs play a very critical role on the central nervous system (CNS) (Figure 2) as it is depicted by the severe effects on neuropsychological, cognitive development, and motor activity of children with congenital hypothyroidism, a condition known as cretinism (38). A perfect synergy between mother's THs actions and normal fetus thyroid development is necessary for neuronal differentiation, migration and proliferation (39). While TRα1 appears to be ubiquitous in CNS regions, TRβ1 is only present in hypothalamus, retina, pituitary gland, and cochlea in later stages of development (40). Indeed, animal studies on TRα1 knockout mice demonstrated delay in the formation of dendrites and axons of GABA-ergic neurons, which are very important for nervous circuits (41). Moreover, THs have been shown to enhance the crucial interaction between Purkinje and granule cells, necessary for the adequate layering of cerebellar cortex: in either TRα1 and TRβ1 knockout mice abnormalities in cerebellum differentiation and function have been described (42). The actions of THs on CNS depend on the availability of the hormones and the controlled transport across blood brain barrier (BBB), which seems to be the preferred route in the adult life. It is not the same in embryo, in which choroid plexus is mainly involved in the transport. Despite the initial belief of a passive diffusion of THs through cell membranes, subsequent data clearly showed that multiple families of THs transporters guarantee a saturable and stereospecific THs uptake (43). The most important carriers in BBB are the monocarboxylate transporter 8 (MCT8) and the Na+ independent organic anion-transporting polypeptide 1C1 (OATP1C1). In mice lacking both MCT8 and OATP1C1, the concentration of THs in brain are significantly affected (44). Transporters of the LAT family LAT1 and LAT2 have been implicated in THs transport across BBB in rodents, whereas LAT1 in human is expressed in brain endothelial cells (45). Once THs have passed BBB, their local availability depends from the activity of the astrocytic type II deiodinase (D2) which converts T4 in T3. T3 is subsequently metabolized into 3,5-diiodothyronine (T2) by type 3 deiodinase (D3) in neurons (39).
It is widely known that THs are required for the proper proceeding of oligodendrocytes differentiation and maturation (46), and a novel role of THs as promoters of remyelination in CNS has been recently suggested, opening a new perspective for the treatment of demyelinating disorders such as multiple sclerosis (MS) (47, 48). In fact, in cells and animal models, THs demonstrated to enhance normal processes of myelination acting on oligodendrocytes development, and a THs deficiency revealed to be associated with a decreasing of the number and function of oligodendrocytes and the expression of myelin basic protein (MBP) in the brain (49). Moreover, THs stimulate the process of remyelination, that occurs after an injury and is triggered by the oligodendrocyte precursor cells (OPCs). These are stem cells that distributed in the CNS and are capable to differentiate in mature myelinating oligodendrocytes (50). This effect is mediated by THs presumably through the induction of neurotrophic expression factors, suppression of apoptosis and OPCs differentiation (51, 52). The full mechanism of THs action on OPCs is not completely understood, but the current hypothesis is that TRβ isoform plays a pivotal role. THs binding to TRβ could act together with RXR activation to promote the expression of a crucial gene for OPCs differentiation, named KLF9 or Kruppel Like Factor 9 (a basic helix loop helix family member) (48). Studies in mice's OPCs showed that the overexpression of TRβ isoform increases the OPCs differentiation, confirming the central role of TRβ (53).
Thyroid Hormone Receptors Actions and Rationale Design of Selective Thyromimetics
In the classification of THs effects proposed by Flamant et al., the “type 1 effects” cover the classical genomic actions of THs, which require TRs and direct interaction with DNA (14) TRs act by binding to their cognate TRE, in the promoters of target genes, thereby stimulating or preventing transcriptional activity (Figure 1). TRs can bind as monomers, homodimers, and heterodimers to TREs (54). TRs have been shown to form heterodimers with several TR auxiliary proteins (TRAPs) that increase TR binding to TREs. Among them, the retinoid X receptors (RXRs) represent the predominant form of TRAPs and thus play a fundamental role in TH-mediated transcription (54). The regulation of transcription by TH involves various regulatory proteins, namely coactivators (i.e., SRC-1, CBP/p300) and corepressors (i.e., SMRT, NCoR), that bind to the receptor–DNA complexes and either promote or repress transcription, respectively (Figure 1). The interaction site of coactivators with nuclear hormone receptors involves a recognition motif characterized by the amino acid sequence (LXXLL) (55). TRs have a well-defined domain organization, common to all nuclear hormone receptors, consisting of an amino-terminal A/B domain, a DNA binding domain (DBD), a hinge region that connects the DBD to the Ligand Binding Domain (LBD), and the C-terminal of the LBD (Figure 4). TRs have a compact LBD, produced by a three-layer antiparallel α-helical sandwich formed by 12 α-helices. The dynamic of the AF-2 region located in helix 12 of the LBD plays a critical role in regulating the interaction of the receptor with coactivators or corepressors following agonist or antagonist binding to the LBD, respectively (56–58). Notably, this compact helical organization generates a “wedge-shaped” structural arrangement into which the hormone binds resulting deeply buried within the receptor (59).
Figure 4
TRs are also involved in “type 2 effects” of THs (14), without direct interaction with DNA. Consistent data are available suggesting that TRs can modulate chromatin by a protein-protein interaction, likely with other transcription factors (60), thus influencing epigenome. TRs have been also demonstrated to be active in the cytoplasm, beyond nuclear action and without interacting with DNA, participating in intracellular signaling cascades (type 3 effects of THs). It is noteworthy that some THs effects exist that seem to be independent from TRs, with different hypothesized mechanisms (use of integrin αVβ3 as membrane receptor, direct allosteric regulation of metabolic enzymes) (14).
TRs are obviously the natural target of any drug with thyromimetic function. The LBD represents the most relevant domain when designing putative thyromimetics (61). The selectivity of the THs effect on metabolism and liver is due to a crucial difference in the crystal structure of the TRα1 and TRβ: one residue in the binding cavity i.e., Asn 331 in TRβ and Ser 277 in TRα1 [(59); Figure 4]. This understanding led to the idea of selective thyromimetics. In 1998 a novel compound named GC-1 (Sobetirome) (Figure 3) was synthesized, by Chiellini et al., which was capable to bind TRβ with the same affinity as T3, but TRα1 with 10-fold lower affinity (62). The structural characteristics responsible for the selectivity of GC-1 are the presence of the oxyacetic acid chain, replacing amino acid side chain, that enhances polar interaction with Arginine residues in the TRβ pocket, and the presence of a diphenylmethane scaffold where the 3,5 di-methyl substitutions allow the ligand to be locked in the active perpendicular conformation with respect to the terminal phenyl ring, while establishing many van der Waals interactions with the lipophilic side chains of numerous amino acid residues present in the ligand binding cavity (LBC). Other profitable lipophilic interactions were also established by the iso-propyl substituent at position 3′ of the outer ring (59). GC-1 demonstrated many beneficial effects on hepatic metabolism, in the absence of heart side effects, and during last years a number of selective thyromimetics have been produced (63, 64), either with a classical thyronine based phenoxyphenyl-structure, including KB141 (10) and KB2115 (65) or with a Sobetirome-like diphenylmethane scaffold, as summarized in the following section (Figure 3).
TRβ -Selective Thyromimetics Showing a Thyronine Based Aryloxyphenyl-Structure
Eprotirome (KB2115)
(3-[[3,5-dibromo-4-[4-hydroxy-3-(1-methylethyl)-phenoxy]-phenyl]-amino]-3-oxopropanoic acid) (Figure 3) is a TH analog that has modestly higher affinity for the TRβ isoform compared with its affinity for the TRα isoform and displays hepatic-selective uptake (65). Data collected from initial short-term studies testing Eprotirome for the treatment of hyperlipidemia in humans appeared very promising. Administration of Eprotirome was found to reduce serum cholesterol (up to 40%) and ApoB without producing undesirable side effects. Notably, no cardiac toxicity was observed [(66); Table 1]. Despite the promising activity on LDL cholesterol, triglyceride, ApoB, and Lp(a) lipoprotein reduction observed in subsequent clinical studies, a Phase 3 trial was terminated because alterations of dog cartilage were observed following chronic treatment (NCT01410383). In addition, reduced T4 levels, as well as liver toxicity, occurred in homozygous patients affected by familiar hypercholesterolemia, after only 6 weeks of treatment with 50 or 100 μg of Eprotirome (69).
Table 1
| Thyromimetic | Structure | Beneficial effects | Side effects/clinical trials |
|---|---|---|---|
| GC-1 (Sobetirome) QuatRx | 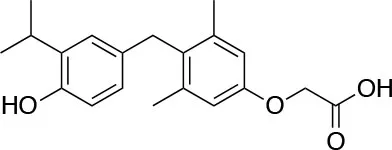 | ↓LDL cholesterol ↓Serum triglycerides ↓Lipoprotein a [Lp(a)] [Scanlan (67); Hartley (68)] | No adverse side effects associated with excess TH Terminated after Phase 1 |
| Eprotirome (KB2115) KaroBio | 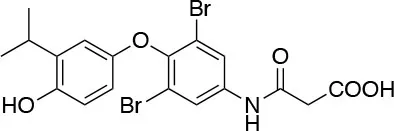 | ↓Total cholesterol ↓LDL cholesterol ↓Lp(a) ↓triglycerides [Berkenstam et al. (66); Sjouke et al. (69)] | Upregulation of LDL receptors and cardio-vascular risks Liver toxicity; increase of AST and ALT levels Cartilage Defect in Dogs Terminated during Phase 3 |
| Resmetirom (MGL-3196) Madrigal Pharmaceutical | 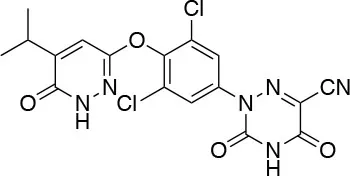 | ↓Hepatic fat in patients with NASH [Kelly et al. (70); Harrison et al. (71)] | No adverse side effects after Phase 2 clinical trails Phase 3 in progress (NCT03900429) |
| VK2809 (MB08711) Viking Therapeutics | 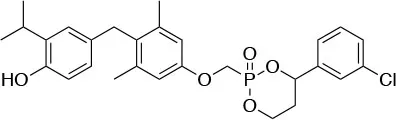 | ↓LDL-C ↓Liver fat content in patient with NAFLD [Erion et al. (72)] | Almost negligible side effects reported Phase 2b in progress (NCT4173065) |
| Sob-AM2 Llama Therapeutics | 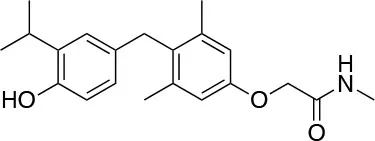 | ↑Myelin repair [Hartley et al. (73)] | Preclinical testing |
| IS25 I.S.D.D. srl | 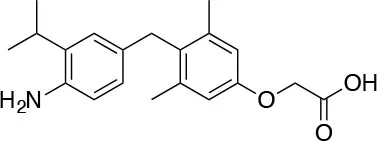 | ↑Lipolysis ↓Triglycerides ↑Hepatocyte proliferation [Runfola et al. (74); Perra et al. (75)] | Preclinical testing |
| TG68 I.S.D.D. srl | 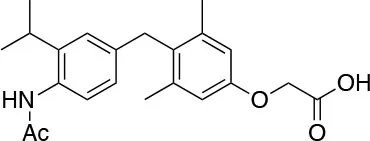 | ↑Lipolysis ↓Triglycerides ↑Hepatocyte proliferation [Runfola et al. (74); Perra et al. (75)] | Preclinical testing |
Effects of TRβ-selective thyromimetics in preclinical studies and human trials.
Resmetirom (MGL-3196)
2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yloxy) phenyl]-3,5-dioxo-2,3,4,5-tetrahydro[1,2,4]triazine-6-carbonitrile (Figure 3) is a recently developed liver-directed and markedly TRβ-selective agonist (EC50s = 0.21 and 3.74 μM for TRβ and TRα, respectively) (70). Preclinical, toxicology, Phase 1, and Phase 2 clinical data suggest Resmetirom has great potential for the treatment of non-alcoholic steatohepatitis (NASH), non-alcoholic fatty liver disease (NAFLD), and associated dyslipidemias (Table 1). Notably, Resmetirom has shown neither suppression of the central thyroid axis nor TRα effects on heart rate or bone, and it reduces elevated liver enzymes in NASH patients. On the basis of positive Phase 2 clinical trial results in patients with NASH (71), which demonstrated significant effects in reduction of hepatic fat and markers of inflammation and fibrosis, as compared to placebo, Madrigal Pharmaceuticals recently started a Phase 3 multinational, double-blind, randomized, placebo-controlled clinical trial of Resmetirom in patients with NASH and liver fibrosis (ClinicalTrials.gov Identifier: NCT03900429).
Sobetirome and Novel Diphenylmethane Structure Based Thyromimetics
Sobetirome (GC-1)
(3,5-Dimethyl-4 (4′-hydroxy-3′-isopropylbenzyl) phenoxy acetic acid) (Figure 3) differs from T3 for the presence of three hydrocarbon residues in place of the iodine atoms, the methylene linkage between the two phenyl rings, and the substitution of the 1-aminopropionic acid with oxyacetic acid. When it was first synthesized and tested, in the late 1990s, the selectivity for TRβ isoform was an unexpected finding (67). At that time, this selectivity was unequivocally proved by running studies in a model of Xenopus laevis tadpole growth. In the frog, metamorphosis occurs as a result of gene expression cascades induced by TH secreted from the developing tadpole's thyroid gland. Xenopus TRα is expressed early in development, long before the larval tadpole have a functional thyroid gland, whereas much lower levels of Xenopus TRβ mRNA are detectable prior to metamorphosis. At metamorphosis, TRβ is strongly induced by TH in larval tissues that will die and resorb such as the tail. In this model, treatment of premetamorphic tadpoles with GC-1 actively stimulated tail and gill resorption, that are a TRβ dependent mechanism, while poorly inducing limb bud development, typically TRα mediated. Notably, the effectiveness of GC-1 in inducing tail resorption and tail gene expression correlates with increasing TRβ levels (76).
GC-1 beneficial effects on lipid profile has been shown firstly in hypothyroid mice and rats with hypercholesterolemia, where it enhanced the lowering of cholesterol and triglycerides, without important side effects in cardiomyocytes and muscle cells (77). GC-1 has direct effects in promoting the so called “reverse cholesterol transport” which describes the clearance of atherogenic cholesterol from extrahepatic tissues to the liver, by increasing the activity of the high density lipoprotein (HDL) and LDL receptor on hepatocytes and by the stimulation of CYP7A1 enzyme (8). GC-1 is also active in preventing atherosclerosis by lowering the levels of cholesteryl esters in the vasculature, as demonstrated in a model of Apolipoprotein E (Apo-E) deficient mice (56, 78), and is effective in stimulating the brown adipose tissue (BAT) thermogenesis, thus reducing fat mass [(79); Table 1].
Although a large body of evidence supports the efficacy of GC-1 as a potent and hepatoselective lipid- lowering agent, in 2008 it was terminated by QuatrX after completed one Phase 1 clinical trial due to lack of funding.
Due to the impact of liver chronic diseases in public health and based on previous findings with T3, GC-1 was also tested in models of NAFLD, HCC, and hepatectomy. In a rodent model of NAFLD, obtained by choline devoid methionine deficient (CMD) diet, GC-1 showed to be effective in preventing or promoting the improvement of the induced hepatic steatosis (80). This and other evidences in the literature confirmed a possible role of GC-1 as antisteatogenic compound, even though some authors advice caution because hyperglycemia and insulin resistance have been observed in some of the experiments (81). An interesting effect of GC-1 has been demonstrated in hepatic neoplastic proliferation: in a rat model of HCC the treatment with GC-1 for 2 weeks caused the regression of pre-neoplastic nodules and the induction of markers of hepatic differentiation (82). The finding that TRβ isoform is downregulated in hepatic neoplasms is another indirect evidence that restoring THs molecular effects, could be of benefit against tumor progression.
Finally, a promising field of investigation for GC-1 is its action as potent inducer of hepatic proliferation: in rodent models of partial hepatectomy, the pre-treatment with GC-1 led to a significant increase in hepatocytes proliferation, without substantial side-effects (83). The mechanism underlying this effect seems to be mediated, at least in part, by the activation of β-catenin pathway, since the potent effect on proliferation is lost in liver specific β-catenin knockout mice (83).
In respect to the action of GC-1 in the CNS, as we already mentioned, GC-1 was tested in an in-vitro system, that allowed to measure quantitatively and qualitatively oligodendrogenesis. In this model, GC-1 was found to promote the differentiation of the OPCs in both in murine and human cells. Moreover, during the oligodendrogenesis promoted by GC-1, TRβ1 was shown to be upregulated. Finally, the same group demonstrated that it was possible to show and monitor the GC-1 induced oligodendrogenesis in the corpus callosum, occipital cortex and optic nerve using an in-vivo transgenic mouse model (73, 84).
VK2809 (MB07811)
Recently, a new strategy to overcome the detrimental effects related to TR activation in extrahepatic tissues has been the development of liver targeting “TR agonist prodrugs.” VK2809 is the most promising of the prodrug candidates and was originally developed by Metabasis Therapeutics, Inc., under the name of MB07811 (2R,4S)-4-(3-chlorophenyl)-2-[(3,5-dimethyl-4-(4′-hydroxy-3′-isopropylbenzyl) phenoxy)methyl]-2-oxido-[1–3]-dioxaphosphonane (24, 72). Notably, from a structural point of view, this new generation of Hep-Direct prodrugs are small aryl-substituted cyclic prodrugs (Figure 3) that are able to generate the active drug after oxidation of the benzylic methine proton produced by the cytochrome P450 (CYP) isoenzyme CYP3A. This leads to the irreversible opening of the ring and the formation of phosphonate, that binds to TRs (72). Indeed, pharmacokinetic studies in rats demonstrated that the prodrug (MB07811) is exposed to first-pass hepatic extraction and after cleavage generates the negatively charged 3,5-dimethyl-4-(4′-hydroxy-3′-isopropylbenzyl)phenoxy)methylphosphonic acid (MB07344), which has a very low tissue distribution and rapidly undergoes to elimination in the bile. The activated form has a more pronounced affinity for TRβ (Ki = 3 nM) than for TRα (Ki = 35 nM), and preclinical studies showed that when administered in hyperlipidemia and normal rodent models, MB07811 was able to produce a significant reduction of total plasma cholesterol and hepatic and plasma triglycerides [(72); Table 1]. Subsequently, MB07811 was used in human trials. Notably, a Phase 1 multiple-ascending dose study in patients with mild hypercholesterolemia, revealed that administration of VK2809 (MB07811) was able to induce a significant reduction in LDL-C, triglycerides and atherogenic proteins. In addition, the results of a Phase 2 trial for the treatment of NAFLD and elevated LDL-C, showed that the administration of VK2809 significantly reduced LDL-C and liver fat content as compared to placebo treated patients. Notably, in both trials VK2809 resulted also to be safe and well-tolerated. At last, few months ago Viking Therapeutics announced the initiation of a Phase 2b clinical trial of VK2809, in patients with biopsy-confirmed NASH (ClinicalTrials.gov identifier: NCT4173065). Given VK2809's promising initial data on both liver fat and plasma lipids, this novel diphenylmethane prodrug may quickly move forward to reach the market as a new drug for the treatment of a large variety of lipid related liver pathologies, including NASH.
Sob-AM2: A CNS-Selective Prodrug of Sobetirome
A novel field of interest is the possible use of selective thyromimetics in demyelinating diseases, such as multiple sclerosis (MS), that have poor prognosis and are in need of new therapeutic approaches. As reported, TRβ isoform seems to play a pivotal role in the remyelination process, that is induced by THs. Consistently, GC-1 (Sobetirome) has been proved to distribute to the CNS in vivo and to induce the differentiation of OPCs in human and rodent in vitro models (84). However, the presence of a carboxylate negatively charged group in the structure makes difficult for GC-1 to cross the blood brain barrier. More recently, with the aim to combine the TRβ selectivity of GC-1 with a better brain tissue selectivity and capacity to cross the blood brain barrier, new prodrugs of GC-1, named Sobetiramides, have been produced by masking the carboxylate group with easily cleavable protecting groups (85).
Sob-AM2
Sob-AM2, the methyl amide derivative of Sobetirome (Figure 3), is an optimized prodrug that significantly increases delivery of Sobetirome to the brain while concomitantly decreasing blood and peripheral organ exposure to the active drug (85). Notably, in mice lacking Mct8 and Dio2, Sob-AM2 treatment has been shown to increase Sobetirome brain content and exert central and peripheral thyromimetic actions by modulating the expression of T3-dependent genes (86). Since a concomitant decrease of circulating T4 and T3 was also observed in the same animal model, the potential for Sob-AM2 to address the cerebral hypothyroidism and the peripheral hyperthyroidism characteristic of MCT8 deficiency has been consistently suggested (86). Using iCKO-Myrf mice, a recently developed genetic mouse model of demyelination and remyelination, where demyelination was induced by tamoxifen treatment of adult Myrffl/fl; Plp1-CreERT mice Hartley et al. (73) and Koenning et al. (87) demonstrated that chronic treatment with Sobetirome (80 μg/kg/d, p.o.) or Sob-AM2 (84 μg/kg/d, p.o.) leads to significant improvement in both clinical signs and remyelination, as assessed by using motor function tests, histology, and MRI techniques (Table 1). Notably, Sob-AM2 is selectively hydrolysed by the fatty acid amide hydrolase (FAAH), a crucial enzyme, highly expressed in the CNS (85, 88, 89), and efficiently converted to Sobetirome. Therefore, when administered systemically, Sob-AM2 leads to increased CNS distribution and decreased peripheral exposure of the active drug Sobetirome (85). However, after oral administration of Sob-AM2 in the iCKO-Myrf model, a significant conversion to Sobetirome was observed to occur in the gastrointestinal tract, conducing to increased Sobetirome and decreased Sob-AM2 concentration in the blood. Furthermore, Sob-AM2 displayed an oral bioavailability ~5-fold lower than that of Sobetirome. As a consequence of these two issues, orally dosed Sob-AM2 resulted in only a modest (~2- to 4-fold) increase of CNS Sobetirome level, as compared with the same oral dose of Sobetirome (85, 90). Consistently, only a moderate improvement in rotarod performance was observed in iCKO-Myrf mice treated with 84 μg/kg/d (p.o.) Sob-AM2 as compared to mice receiving an equivalent dose of Sobetirome. In the same study, it was observed that chronic treatment with TH (hyperthyroidism) inhibited the endogenous myelin repair and aggravated disease. Therefore, these results further underscore the need for selective TH action to promote remyelination in the CNS. To date, Sobetirome, Sob-AM2 (85), and other recently developed Sobetirome derivatives (90–92) are the only thyromimetics reported to distribute to the CNS from systemic administration, and might represent the first class of TH agonists that can be subjected to clinical evaluation in demyelinating diseases such as MS.
Recently, Sobetirome has been proposed for the treatment of X-linked adrenoleukodystrophy (ALD), a rare congenital disease due to mutations in the ALD protein, characterized by adrenal insufficiency and central nervous system (CNS) demyelination. No specific pharmacological therapy is available for ALD. All patients with X-ALD have the biochemical abnormality of elevated blood and tissue levels of very long chain fatty acids (VLCFAs), namely saturated fatty acids with ≥22 carbon atoms. Notably, in a transgenic mouse model of ALD, Sobetirome administration reduced the brain and adrenal content of very-long-chain fatty acids (68). Based on these observations, a clinical trial with sobetirome in X-linked ALD was posted in the NIH database (NCT01787578). Unfortunately, in March 2019 NeuroVia, Inc. withdrew a phase I/II trial prior to enrolment due to no funding in Adrenoleukodystrophy (in children, in adolescents) in Australia, United Kingdom, Chile (PO) (NCT03196765).
Novel 4′-Amino-Benzyl-Phenoxyacetic Acid Thyromimetics: IS25 and TG68
Novel halogen free TRβ selective agonist IS25 and its pro-drug TG68 (Figure 3) have been recently identified (74, 93). Notably, both compounds initially exposed to in vitro analysis of cytotoxicity and ADME-Tox/off-target liability revealed a convincing lack of toxicity (74, 94), supporting their progression in the drug discovery process. In vitro investigation proved that both compounds were able to reduce lipid accumulation in human hepatoma HepG2 cells by promoting lipolysis with a potency comparable to that of T3, convincingly showing that derivative TG68 was capable of efficiently deliver the corresponding parent compound IS25 (Table 1). Moreover, western blot analysis showed that the decreasing of lipid accumulation observed in HepG2 cells after treatment with both compounds was related to the stimulation of AMPK phosphorylation, concomitantly leading to the phosphorylation and consequent inactivation of acetyl coenzyme A carboxylase (ACC), the major regulator of fatty acids synthesis (74). In addition, in vivo studies confirmed the lipolytic action and the apparent lack of toxicity that had already been observed for both compounds with the in vitro experiments. Indeed, reduced triglyceride levels and no liver injury, as assayed by serum levels of ALT, AST, and bilirubin, combined to the absence of cardiac hypertrophy, were observed in F344 rats exposed to a sub-chronic treatment with the two novel TRβ-agonists (74). Nevertheless, additional in vivo studies will be required in order to validate the therapeutic potential of both compounds in fatty liver dysfunctions, including NAFLD and NAFL.
It is widely known that thyroid hormone T3 represents a potent hepatomitogen, and its mitogenic effect seems to be mediated by TRβ (95). Therefore, TRβ-selective thyromimetics devoid of relevant side effects might find a possible use in regenerative medicine (83, 96). Even though still at a preliminary level, ongoing studies have revealed that in F344 rats a sub-chronic treatment with the two novel TRβ-agonists, namely IS25 and TG68, induced hepatocyte proliferation in the absence of any sign of hepatic toxicity and cardiac hypertrophy (75). Notably, hepatocyte proliferation appears to be associated with activation of TR-target genes, such as Dio1 and Spot14, suggesting that the mitogenic effect of these drugs may be due to binding and activation of TRβ. Importantly, the liver proliferative response induced by the two TRβ agonists was not associated with liver damage, as assessed by biochemical determination of serum transaminases and by immunostaining for Caspase-3, but it was the result of a direct effect of these drugs enabling quiescent hepatocytes to re-entry into the cell cycle (75). Hence these newly developed agents may have a significant clinical application for hepatic regenerative therapies or other surgical procedures.
Glucagon/t3 Hybrid: A Promising Alternative to Isoform-Selective Thyromimetics
A new perspective for the therapy of metabolic syndrome has recently emerged from the work of Finan et al. (97). The authors generated a Glucagon/T3 hybrid molecule to release both the beneficial anti-lipid effects of glucagon and the energy expending effects of T3. The most remarkable aspect of this new Glucagon/T3 hybrid is the ability to confer key beneficial metabolic effects of each component, while avoiding the negative side effects observed for each hormone separately. Indeed, the use of Glucagon/T3 in obese mice ameliorates serum dyslipidemia, diminishes adipose mass, reverses NASH, reduces atherosclerotic plaque accumulation, and improves glucose metabolism, while avoiding thyrotoxicosis and the diabetogenic effects of glucagon. Even though additional investigations will be required for the advancement of this compound to clinical trials, the engineered Glucagon/T3 co-agonism holds promise as an alternative to isoform-selective thyromimetics.
Conclusions
TRβ is the predominant isoform of TR in the liver and therefore is primarily responsible for the reduction of cholesterol levels, whereas the adverse effects on heart and bone are mainly connected to TRα. Thus, in the development of clinically useful drug candidates, much effort has been directed toward the design of compounds with isomer-specific activity related to the structure of thyroid hormones, i.e., thyromimetics that are liver and/or TRβ selective. Among the several TRβ-selective T3 analogs generated in the past two decades, Sobetirome (GC-1), Eprotirome (KB2115), Resmetirom (MGL-3196) and the Hep-Direct prodrug VK2809 (MB07811) have demonstrated to mimic most of the beneficial effects of T3, in the absence of adverse side effects. For example, Sobetirome and Eprotirome entered human clinical trials for dyslipidemia, showing promising results in the absence of deleterious effects typically associated with hyperthyroidism [for reviews, see (63, 98)]. Unfortunately, no Phase 2 trials for GC-1 have been performed and a Phase 3 trial with KB2115 was terminated due to harmful effects on cartilage observed in canines. In spite of this disappointing result, Resmetirom (MGL-3196) successfully completed Phase 2 trials showing to potently reduce liver fat content after 12 and 36 weeks of treatment in patients with NASH in the absence of significant side effects, and was recently advanced to Phase 3 trials. Liver and TRβ-selective agonist VK2809 also showed positive results from a Phase 2 trial in patients with hypercholesterolemia and NAFLD, and is currently being evaluated in a Phase 2b clinical trial in patients with biopsy-confirmed NASH. Taken together, all these findings suggest that modern thyromimetics hold promise for dyslipidemia.
Due to their encouraging effects in metabolism control and CNS pathologies, thyromimetics have arisen much interest in the field of medical research; however, the combination of receptor and organ selectivity is now a fundamental requirement for the development of novel compounds.
Sob-AM2, a CNS-selective prodrug of Sobetirome, has been shown to promote brain and spinal cord myelin repair. This effect was associated with a significant improvement in neurological clinical signs that stem from CNS demyelination. Notably, at the present time there is still a lack of approved treatments for MS that stimulate myelin repair, therefore Sob-AM2 holds promise as a effective drug candidate for the treatment of demyelinating disorders, such as MS.
During a drug discovery investigation aimed at identifying novel TRβ-selective agonists, the application of a comprehensive panel of ADME-Toxicity assays enabled the selection of analog IS25 and its prodrug TG68 as the best candidates to progress along the drug discovery pipeline. Their lipid lowering properties have been well-documented by both in vitro and in vivo assays. Importantly, preliminary results from in vivo studies indicate that they are able to potently produce hepatocyte proliferation without causing liver damage or cardiac and renal hypertrophy, classically associated to the treatment with THs. Therefore, these compounds may result clinically useful in the context of regenerative medicine, for the treatment of conditions where a rapid liver cell proliferation is required or when liver regenerative capacity is impaired.
In conclusion, last decade has seen a reawakening of interest in developing TRβ selective TH analogs, and at the present there is renewed hope for novel tissue- and/or isoform-selective thyromimetics to find application in therapy for the treatment of dyslipidemia and liver-related life threatening conditions, as well as for disabling diseases of the CNS, such as MS. Even though still at its infancy as a therapeutic tool, the engineered Glucagon/T3 co-agonism opens a new scenario in the search of a pleiotropic approach to the treatment of diseases that would benefit of T3 tissue and isoform selective actions. It is arguable that in the near future the use of this “combinatorial chemistry” technology will see a further expansion to maximize the benefits and minimize the risks of each component as compared to individual forms of administration.
Statements
Author contributions
GC and SR had the idea of this review, wrote most of the paper, and organized final draft. FS and SS wrote the initial section of the paper and prepared the figures. MR edited and revised the paper. All authors revised the final manuscript and approved the final version.
Funding
This work was supported by University of Pisa grant (PRA 2018-19_83).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
- ACC
acetyl coenzyme A carboxylase
- Apo-E
Apolipoprotein E
- BAT
brown adipose tissue
- CMD
choline devoid methionine deficient
- CNS
central nervous system
- CYP
cytochrome P450
- CYP7A1
7 α-hydroxylase
- D3
type 3 iodothyronine deiodinase
- DBD
DNA binding domain
- FAAH
fatty acid amide hydrolase
- HAT
histone acetyltransferase
- HCC
hepato-carcinoma
- HDAc
Histone Deacetylase
- HDL
high density lipoprotein
- LAT1
L-type amino acid transporter1
- LBC
ligand binding cavity
- LBD
ligand binding domain
- LDL
low density lipoprotein
- KLF9
Kruppel Like Factor 9
- MBP
myelin basic protein
- MS
multiple sclerosis
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- OPCs
oligodendrocyte precursor cells
- RXRs
retinoid X receptors
- T3
3,3′,5-triiodo-L-thyronine
- T4
3,3′,5,5′-tetraiodo-L-thyronine
- THA
thyroid hormone axis
- THs
Thyroid hormones
- TRAPs
TR auxiliary proteins
- TREs
TH response elements
- TRs
THs receptors.
Abbreviations
References
1.
HennemannGDocterRFriesemaECde JongMKrenningEPVisserTJ. Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev. (2001) 22:451–76. 10.1210/edrv.22.4.0435
2.
YenPM. Physiological and molecular basis of thyroid hormone action. Physiol Rev. (2001) 81:1097–142. 10.1152/physrev.2001.81.3.1097
3.
AccorroniASaponaroFZucchiR. Tissue thyroid hormones and thyronamines. Heart Failure Rev. (2016) 21:373–90. 10.1007/s10741-016-9553-8
4.
GreenspanSLGreenspanFS. The effect of thyroid hormone on skeletal integrity. Ann Intern Med. (1999) 130:750–8. 10.7326/0003-4819-130-9-199905040-00016
5.
KöhrleJ. Thyroid hormones and derivatives: endogenous thyroid hormones and their targets. In: Plateroti M, Samarut J, editors. Thyroid Hormone Nuclear Receptor. Methods in Molecular Biology, Vol 1801. New York, NY: Humana Press. (2018). p. 85–104. 10.1007/978-1-4939-7902-8_9
6.
ZucchiRRutiglianoGSaponaroF. Novel thyroid hormones. Endocrine. (2019) 66:95–104. 10.1007/s12020-019-02018-4
7.
LorenziniLLNguyenNMSacripantiGSerniEBorsòMSaponaroFet al. Assay of endogenous 3, 5-diiodo-L-thyronine (3, 5-T2) and 3, 3'-diiodo-L-thyronine (3, 3'-T2) in human serum: a feasibility study. Front Endocrinol. (2019) 10:88. 10.3389/fendo.2019.00088
8.
ColumbanoAChielliniGKowalikMA. GC-1: a thyromimetic with multiple therapeutic applications in liver disease. Gene Exp J Liver Res. (2017) 17:265–75. 10.3727/105221617X14968563796227
9.
CoppolaMCioffiFMorenoMGogliaFSilvestriE. 3, 5-Diiodo-L-thyronine: a possible pharmacological agent?Curr Drug Deliv. (2016) 13:330–8. 10.2174/1567201813666151123124340
10.
GroverGJMellstromKMalmJ. Development of the thyroid hormone receptor β-subtype agonist KB-141: a strategy for body weight reduction and lipid lowering with minimal cardiac side effects. Cardiovasc Drug Rev. (2005) 23:133–48. 10.1111/j.1527-3466.2005.tb00161.x
11.
RossDSBurchHBCooperDSGreenleeMCLaurbergPMaiaALet al. American thyroid association guidelines for diagnosis and management of hyperthyroidism and other causes of thyrotoxicosis. Thyroid. (2016) 26:1343–421. 10.1089/thy.2016.0229
12.
DavisPJLeonardJLDavisFB. Mechanisms of nongenomic actions of thyroid hormone. Front Neuroendocrinol. (2008) 29:211–8. 10.1016/j.yfrne.2007.09.003
13.
DavisPJGogliaFLeonardJL. Nongenomic actions of thyroid hormone. Nat Rev Endocrinol. (2016) 12:111–21. 10.1038/nrendo.2015.205
14.
FlamantFChengSYHollenbergANMoellerLCSamarutJWondisfordFEet al. Thyroid hormone signaling pathways: time for a more precise nomenclature. Endocrinology. (2017) 158:2052–7. 10.1210/en.2017-00250
15.
WeinbergerCThompsonCCOngESLeboRGruolDJEvansRM. The c-erb-A gene encodes a thyroid hormone receptor. Nature. (1986) 324:641–6. 10.1038/324641a0
16.
SapJMuñozADammKGoldbergYGhysdaelJLeutzAet al. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature. (1986) 324:635–40. 10.1038/324635a0
17.
FlamantFBaxterJDForrestDRefetoffSSamuelsHScanlanTSet al. International Union of Pharmacology. LIX. The pharmacology and classification of the nuclear receptor superfamily: thyroid hormone receptors. Pharmacol Rev. (2006) 58:705–11. 10.1124/pr.58.4.3
18.
McKennaNJO'MalleyBW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. (2002) 108:465–74. 10.1016/S0092-8674(02)00641-4
19.
ChengSY. Multiple mechanisms for regulation of the transcriptional activity of thyroid hormone receptors. Rev Endocr Metab Disord. (2000) 1:9–18. 10.1023/A:1010052101214
20.
ChiamoleraMISidhayeARMatsumotoSHeQHashimotoKOrtiga-CarvalhoTMet al. Fundamentally distinct roles of thyroid hormone receptor isoforms in a thyrotroph cell line are due to differential DNA binding. Mol Endocrinol. (2012) 26:926–39. 10.1210/me.2011-1290
21.
PantosCXinarisCMourouzisIPerimenisPPolitiESpanouDet al. Thyroid hormone receptor alpha 1: a switch to cardiac cell “metamorphosis”. J Physiol Pharmacol. (2008) 59:253–69. 10.1016/j.yjmcc.2008.02.076
22.
RibeiroMO. Effects of thyroid hormone analogs on lipid metabolism and thermogenesis. Thyroid. (2008) 18:197–203. 10.1089/thy.2007.0288
23.
JakobssonTVedinLLPariniP. Potential role of thyroid receptor β agonists in the treatment of hyperlipidemia. Drugs. (2017) 77:1613–21. 10.1007/s40265-017-0791-4
24.
ZhouJWaskowiczLRLimALiaoXHLianBMasamuneHet al. A liver-specific thyromimetic, VK2809, decreases hepatosteatosis in glycogen storage disease type Ia. Thyroid. (2019) 29:1158–67. 10.1089/thy.2019.0007
25.
ZucchiR. Thyroid hormone analogues: an update. Thyroid. (2020). 10.1089/thy.2020.0071
26.
SeneseRCioffiFPetitoGGogliaFLanniA. Thyroid hormone metabolites and analogues. Endocrine. (2019) 66:1–10. 10.1007/s12020-019-02025-5
27.
VaitkusJAFarrarJSCeliFS. Thyroid hormone mediated modulation of energy expenditure. Int J Mol Sci. (2015) 16:16158–75. 10.3390/ijms160716158
28.
CioffiFSeneseRLanniAGogliaF. Thyroid hormones and mitochondria: with a brief look at derivatives and analogues. Mol Cell Endocrinol. (2013) 379:51–61. 10.1016/j.mce.2013.06.006
29.
GullbergHRudlingMSaltóCForrestDAngelinBVennströmB. Requirement for thyroid hormone receptor β in T3 regulation of cholesterol metabolism in mice. Mol Endocrinol. (2002) 16:1767–77. 10.1210/me.2002-0009
30.
ChiHCChenCYTsaiMMTsaiCYLinKH. Molecular functions of thyroid hormones and their clinical significance in liver-related diseases. BioMed Res Int. (2013) 2013:601361. 10.1155/2013/601361
31.
FarrellGCLarterCZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. (2006). 43(2 Suppl 1):S99–S112. 10.1002/hep.20973
32.
HassanMMKasebALiDPattYZVautheyJNThomasMBet al. Association between hypothyroidism and hepatocellular carcinoma: a case-control study in the United States. Hepatology. (2009) 49:1563–70. 10.1002/hep.22793
33.
Ledda-ColumbanoGMPerraALoiRShinozukaHColumbanoA. Cell proliferation induced by triiodothyronine in rat liver is associated with nodule regression and reduction of hepatocellular carcinomas. Cancer Res. (2000) 60:603–9.
34.
MariashCN. Thyroid Hormone Actions: To Be ta or Not to Be ta. New Rochelle, NY: Mary Ann Liebert, Inc. (2010).
35.
GullbergHRudlingMForrestDAngelinBVennströmB. Thyroid hormone receptor β-deficient mice show complete loss of the normal cholesterol 7α-hydroxylase (CYP7A) response to thyroid hormone but display enhanced resistance to dietary cholesterol. Mol Endocrinol. (2000) 14:1739–49. 10.1210/mend.14.11.0548
36.
JohanssonCVennströmBThorénP. Evidence that decreased heart rate in thyroid hormone receptor-α1-deficient mice is an intrinsic defect. Am J Physiol. (1998) 275:R640–R6. 10.1152/ajpregu.1998.275.2.R640
37.
GlossBTrostSUBluhmWFSwansonEAClarkRWinkfeinRet al. Cardiac ion channel expression and contractile function in mice with deletion of thyroid hormone receptor α or β. Endocrinology. (2001) 142:544–50. 10.1210/endo.142.2.7935
38.
BiondiBCooperDS. Subclinical hyperthyroidism. New Engl J Med. (2018) 4:680–2. 10.1016/B978-0-12-801238-3.99453-4
39.
PreziosoGGianniniCChiarelliF. Effect of thyroid hormones on neurons and neurodevelopment. Hormone Res Paediatr. (2018) 90:73–81. 10.1159/000492129
40.
BradleyDJTowleHYoungWS. Spatial and temporal expression of alpha-and beta-thyroid hormone receptor mRNAs, including the beta 2-subtype, in the developing mammalian nervous system. J Neurosci. (1992) 12:2288–302. 10.1523/JNEUROSCI.12-06-02288.1992
41.
WallisKSjögrenMVan HogerlindenMSilberbergGFisahnANordströmKet al. Locomotor deficiencies and aberrant development of subtype-specific GABAergic interneurons caused by an unliganded thyroid hormone receptor α1. J Neurosci. (2008) 28:1904–15. 10.1523/JNEUROSCI.5163-07.2008
42.
FlamantFGauthierKRichardS. Genetic investigation of thyroid hormone receptor function in the developing and adult brain. Curr Top Dev Biol. (2017) 125:303–35. 10.1016/bs.ctdb.2017.01.001
43.
LandersKRichardK. Traversing barriers–how thyroid hormones pass placental, blood-brain and blood-cerebrospinal fluid barriers. Mol Cell Endocrinol. (2017) 458:22–8. 10.1016/j.mce.2017.01.041
44.
MayerlSVisserTJDarrasVMHornSHeuerH. Impact of Oatp1c1 deficiency on thyroid hormone metabolism and action in the mouse brain. Endocrinology. (2012) 153:1528–37. 10.1210/en.2011-1633
45.
BraunDKinneABräuerAUSapinRKleinMOKöhrleJet al. Developmental and cell type-specific expression of thyroid hormone transporters in the mouse brain and in primary brain cells. Glia. (2011) 59:463–71. 10.1002/glia.21116
46.
RogisterBBen-HurTDubois-DalcqM. From neural stem cells to myelinating oligodendrocytes. Mol Cell Neurosci. (1999) 14:287–300. 10.1006/mcne.1999.0790
47.
FernandezMGiulianiAPirondiSD'IntinoGGiardinoLAloeLet al. Thyroid hormone administration enhances remyelination in chronic demyelinating inflammatory disease. Proc Natl Acad Sci USA. (2004) 101:16363–8. 10.1073/pnas.0407262101
48.
BaldassarroVAKrezelWFernándezMSchuhbaurBGiardinoLCalzàL. The role of nuclear receptors in the differentiation of oligodendrocyte precursor cells derived from fetal and adult neural stem cells. Stem Cell Res. (2019) 37:101443. 10.1016/j.scr.2019.101443
49.
FernándezMParadisiMIntinoGDel VecchioGSiviliaSGiardinoLet al. A single prenatal exposure to the endocrine disruptor 2,3,7,8-tetrachlorodibenzo-p-dioxin alters developmental myelination and remyelination potential in the rat brain. J Neurochem. (2010) 115:897–909. 10.1111/j.1471-4159.2010.06974.x
50.
LevineJMReynoldsRFawcettJW. The oligodendrocyte precursor cell in health and disease. Trends Neurosci. (2001) 24:39–47. 10.1016/S0166-2236(00)01691-X
51.
PicouFFauquierTChatonnetFFlamantF. A bimodal influence of thyroid hormone on cerebellum oligodendrocyte differentiation. Mol Endocrinol. (2012) 26:608–18. 10.1210/me.2011-1316
52.
MarzialiLNGarciaCIPasquiniJM. Transferrin and thyroid hormone converge in the control of myelinogenesis. Exp Neurol. (2015) 265:129–41. 10.1016/j.expneurol.2014.12.021
53.
BillonNTokumotoYForrestDRaffM. Role of thyroid hormone receptors in timing oligodendrocyte differentiation. Dev Biol. (2001) 235:110–20. 10.1006/dbio.2001.0293
54.
RanjanMWongJShiYB. Transcriptional repression of Xenopus TR beta gene is mediated by a thyroid hormone response element located near the start site. J Biol Chem. (1994) 269:24699–705.
55.
DarimontBDWagnerRLAprilettiJWStallcupMRKushnerPJBaxterJDet al. Structure and specificity of nuclear receptor–coactivator interactions. Genes Dev. (1998) 12:3343–56. 10.1101/gad.12.21.3343
56.
ScanlanTSYoshiharaHNguyenNHChielliniG. Selective thyromimetics: tissue-selective thyroid hormone analogs. Curr Opin Drug Discov Dev. (2001) 4:614–22.
57.
NguyenNHAprilettiJWBaxterJDScanlanTS. Hammett analysis of selective thyroid hormone receptor modulators reveals structural and electronic requirements for hormone antagonists. J Am Chem Soc. (2005) 127:4599–608. 10.1021/ja0440093
58.
SchapiraMRaakaBMDasSFanLTotrovMZhouZet al. Discovery of diverse thyroid hormone receptor antagonists by high-throughput docking. Proc Natl Acad Sci USA. (2003) 100:7354–9. 10.1073/pnas.1131854100
59.
WagnerRLHuberBRShiauAKKellyACunha LimaSTScanlanTSet al. Hormone selectivity in thyroid hormone receptors. Mol Endocrinol. (2001) 15:398–410. 10.1210/mend.15.3.0608
60.
GrøntvedLWaterfallJJKimDWBaekSSungMHZhaoLet al. Transcriptional activation by the thyroid hormone receptor through ligand-dependent receptor recruitment and chromatin remodelling. Nat Commun. (2015) 6:7048. 10.1038/ncomms8048
61.
BleicherLAparicioRNunesFMMartinezLDiasSMGFigueiraACMet al. Structural basis of GC-1 selectivity for thyroid hormone receptor isoforms. BMC Struct Biol. (2008) 8:8. 10.1186/1472-6807-8-8
62.
ChielliniGAprilettiJWAl YoshiharaHBaxterJDRibeiroRCScanlanTS. A high-affinity subtype-selective agonist ligand for the thyroid hormone receptor. Chem Biol. (1998) 5:299–306. 10.1016/S1074-5521(98)90168-5
63.
TancevskiIRudlingMEllerP. Thyromimetics: a journey from bench to bed-side. Pharmacol Ther. (2011) 131:33–9. 10.1016/j.pharmthera.2011.04.003
64.
ElbersLPKasteleinJJSjoukeB. Thyroid hormone mimetics: the past, current status and future challenges. Curr Atherosc Rep. (2016) 18:14. 10.1007/s11883-016-0564-7
65.
BaxterJDWebbP. Thyroid hormone mimetics: potential applications in atherosclerosis, obesity and type 2 diabetes. Nat Rev Drug Discov. (2009) 8:308–20. 10.1038/nrd2830
66.
BerkenstamAKristensenJMellströmKCarlssonBMalmJRehnmarkSet al. The thyroid hormone mimetic compound KB2115 lowers plasma LDL cholesterol and stimulates bile acid synthesis without cardiac effects in humans. Proc Natl Acad Sci USA. (2008) 105:663–7. 10.1073/pnas.0705286104
67.
ScanlanTS. Sobetirome: a case history of bench-to-clinic drug discovery and development. Heart Failure Rev. (2010) 15:177–82. 10.1007/s10741-008-9122-x
68.
HartleyMDKirkemoLLBanerjiTScanlanTS. A thyroid hormone–based strategy for correcting the biochemical abnormality in X-linked adrenoleukodystrophy. Endocrinology. (2017) 158:1328–38. 10.1210/en.2016-1842
69.
SjoukeBLangsletGCeskaRNichollsSJNissenSEÖhlanderMet al. Eprotirome in patients with familial hypercholesterolaemia (the AKKA trial): a randomised, double-blind, placebo-controlled phase 3 study. Lancet Diabetes Endocrinol. (2014) 2:455–63. 10.1016/S2213-8587(14)70006-3
70.
KellyMJPietranico-ColeSLariganJDHaynesN-EReynoldsCHScottNet al. Discovery of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yloxy)phenyl]-3,5-dioxo-2,3,4,5-tetrahydro[1,2,4]triazine-6-carbonitrile (MGL-3196), a highly selective thyroid hormone receptor β agonist in clinical trials for the treatment of dyslipidemia. J Med Chem. (2014) 57:3912–23. 10.1021/jm4019299
71.
HarrisonSABashirMRGuyCDZhouRMoylanCAFriasJPet al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. (2019) 394:2012–24. 10.1016/S0140-6736(19)32517-6
72.
ErionMDCableEEItoBRJiangHFujitakiJMFinnPDet al. Targeting thyroid hormone receptor-β agonists to the liver reduces cholesterol and triglycerides and improves the therapeutic index. Proc Natl Acad Sci USA. (2007) 104:15490–5. 10.1073/pnas.0702759104
73.
HartleyMDBanerjiTTaggeIJKirkemoLLChaudharyPCalkinsEet al. Myelin repair stimulated by CNS-selective thyroid hormone action. JCI Insight. (2019) 4:e126329. 10.1172/jci.insight.126329
74.
RunfolaMSestitoSBellusciLLa PietraVD'AmoreVMKowalikMAet al. Design, synthesis and biological evaluation of novel TRβ selective agonists sustained by ADME-toxicity analysis. Eur J Med Chem. (2020) 188:112006. 10.1016/j.ejmech.2019.112006
75.
PerraAKMCabrasLRunfolaMSestitoSMiglioreCGiordanoSet al. Potential role of two novel agonists of thyroid hormone receptor–β on liver regeneration. Cell Prolif. (2020) 53:e12808. 10.1111/cpr.12808
76.
FurlowJDYangHYHsuMLimWErmioDJChielliniGet al. Induction of larval tissue resorption in Xenopus laevis tadpoles by the thyroid hormone receptor agonist GC-1. J Biol Chem. (2004) 279:26555–62. 10.1074/jbc.M402847200
77.
TrostSUSwansonEGlossBWang-IversonDBZhangHVolodarskyTet al. The thyroid hormone receptor-β-selective agonist GC-1 differentially affects plasma lipids and cardiac activity. Endocrinology. (2000) 141:3057–64. 10.1210/endo.141.9.7681
78.
KannistoKRehnmarkSSlätisKWebbPLarssonLGåfvelsMet al. The thyroid receptor β modulator GC-1 reduces atherosclerosis in ApoE deficient mice. Atherosclerosis. (2014) 237:544–54. 10.1016/j.atherosclerosis.2014.09.035
79.
RibeiroMOCarvalhoSDSchultzJJChielliniGScanlanTSBiancoACet al. Thyroid hormone-sympathetic interaction and adaptive thermogenesis are thyroid hormone receptor isoform-specific. J Clin Invest. (2001) 108:91–105. 10.1172/JCI200112584
80.
PerraASimbulaGSimbulaMPibiriMKowalikMASulasPet al. Thyroid hormone (T3) and TRβ agonist GC-1 inhibit/reverse nonalcoholic fatty liver in rats. FASEB J. (2008) 22:2981–9. 10.1096/fj.08-108464
81.
VatnerDFWeismannDBeddowSAKumashiroNErionDMLiaoXHet al. Thyroid hormone receptor-β agonists prevent hepatic steatosis in fat-fed rats but impair insulin sensitivity via discrete pathways. Am J Physiol Endocrinol Metab. (2013) 305:E89–100. 10.1152/ajpendo.00573.2012
82.
PerraAKowalikMAPibiriMLedda-ColumbanoGMColumbanoA. Thyroid hormone receptor ligands induce regression of rat preneoplastic liver lesions causing their reversion to a differentiated phenotype. Hepatology. (2009) 49:1287–96. 10.1016/S0168-8278(09)60537-7
83.
AlvaradoTFPuligaEPreziosiMPoddarMSinghSColumbanoAet al. Thyroid hormone receptor β agonist induces β-catenin-dependent hepatocyte proliferation in mice: Implications in hepatic regeneration. Gene Exp. (2016) 17:19–34. 10.3727/105221616X691631
84.
BaxiEGSchottJTFairchildANKirbyLAKaraniRUapinyoyingPet al. A selective thyroid hormone β receptor agonist enhances human and rodent oligodendrocyte differentiation. Glia. (2014) 62:1513–29. 10.1002/glia.22697
85.
MeinigJMFerraraSJBanerjiTBanerjiTSanford-CraneHSBourdetteDet al. Targeting fatty-acid amide hydrolase with prodrugs for CNS-selective therapy. ACS Chem Neurosci. (2017) 8:2468–76. 10.1021/acschemneuro.7b00239
86.
Bárez-LópezSHartleyMDGrijota-MartínezCScanlanTSGuadaño-FerrazA. Sobetirome and its amide prodrug Sob-AM2 exert thyromimetic actions in Mct8-deficient brain. Thyroid. (2018) 28:1211–20. 10.1089/thy.2018.0008
87.
KoenningMJacksonSHayCMFauxCKilpatrickTJWillinghamMet al. Myelin gene regulatory factor is required for maintenance of myelin and mature oligodendrocyte identity in the adult CNS. J Neurosci. (2012) 32:12528–42. 10.1523/JNEUROSCI.1069-12.2012
88.
CravattBFGiangDKMayfieldSPBogerDLLernerRAGilulaNB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. (1996) 384:83–7. 10.1038/384083a0
89.
BisognoTPetrocellisLMarzoV. Fatty acid amide hydrolase, an enzyme with many bioactive substrates. Possible therapeutic implications. Curr Pharm Des. (2002) 8:533–47. 10.2174/1381612023395655
90.
FerraraSJMeinigJMPlaczekATBanerjiTMcTiguePHartleyMDet al. Ester-to-amide rearrangement of ethanolamine-derived prodrugs of sobetirome with increased blood-brain barrier penetration. Bioorg Med Chem. (2017) 25:2743–53. 10.1016/j.bmc.2017.03.047
91.
PlaczekATFerraraSJHartleyMDSanford-CraneHSMeinigJMScanlanTS. Sobetirome prodrug esters with enhanced blood–brain barrier permeability. Bioorg Med Chem. (2016) 24:5842–54. 10.1016/j.bmc.2016.09.038
92.
DevereauxJFerraraSJBanerjiTPlaczekATScanlanTS. Increasing thyromimetic potency through halogen substitution. ChemMedChem. (2016) 11:2459–65. 10.1002/cmdc.201600408
93.
ChielliniGRunfolaMBellusciLSestitoSKowalikMPerraAet al. Novel TRβ-selective analog IS25 and its prodrug TG68 demonstrate significant liver regenerative capacity and lipid lowering effects. In: 89th Annual Meeting of the American Thyroid Association 2019 October 30 - November 3. Chicago, IL (2019).
94.
RunfolaMSestitoSGulSChielliniGRapposelliS. Collecting data through high throughput in vitro early toxicity and off-target liability assays to rapidly identify limitations of novel thyromimetics. Data Brief. (2020) 29:105206. 10.1016/j.dib.2020.105206
95.
KowalikMAPerraAPibiriMCoccoMTSamarutJPlaterotiMet al. TRβ is the critical thyroid hormone receptor isoform in T3-induced proliferation of hepatocytes and pancreatic acinar cells. J Hepatol. (2010) 53:686–92. 10.1016/j.jhep.2010.04.028
96.
SzydlowskaMPibiriMPerraAPuligaEMattuSLedda-ColumbanoGMet al. The thyromimetic KB2115 (eprotirome) induces rat hepatocyte proliferation. Gene Exp J Liver Res. (2017) 17:207–18. 10.3727/105221617X695438
97.
FinanBClemmensenCZhuZStemmerKGauthierKMüllerLet al. Chemical hybridization of glucagon and thyroid hormone optimizes therapeutic impact for metabolic disease. Cell. (2016) 167:843–57.e14. 10.1016/j.cell.2016.09.014
98.
MeruvuSAyersSDWinnierGWebbP. Thyroid hormone analogues: where do we stand in 2013?Thyroid. (2013) 23:1333–44. 10.1089/thy.2012.0458
Summary
Keywords
selective thyromimetics, prodrugs, thyroid hormone receptors, dyslipidemia, non-alcoholic steatohepatitis (NASH), non-alcoholic fatty liver disease (NAFLD), myelin, clinical trials
Citation
Saponaro F, Sestito S, Runfola M, Rapposelli S and Chiellini G (2020) Selective Thyroid Hormone Receptor-Beta (TRβ) Agonists: New Perspectives for the Treatment of Metabolic and Neurodegenerative Disorders. Front. Med. 7:331. doi: 10.3389/fmed.2020.00331
Received
27 March 2020
Accepted
04 June 2020
Published
09 July 2020
Volume
7 - 2020
Edited by
Michelina Plateroti, Université Claude Bernard Lyon 1, France
Reviewed by
Carmen Grijota-Martinez, Center for Biomedical Research in Rare Diseases Network (CIBERER), Spain; Frédéric Flamant, Université de Lyon, France; Laurent M. Sachs, Muséum National d'Histoire Naturelle, France
Updates
Copyright
© 2020 Saponaro, Sestito, Runfola, Rapposelli and Chiellini.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Grazia Chiellini grazia.chiellini@unipi.it
This article was submitted to Translational Medicine, a section of the journal Frontiers in Medicine
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.