 Kei Nagai
Kei Nagai- Department of Nephrology, Faculty of Medicine, University of Tsukuba, Tsukuba, Japan
Autoimmune glomerulonephritis occurs as a consequence of autoantibodies and T-cell effector functions that target autoantigens. Co-signaling through cell surface receptors profoundly influences the optimal activation of T cells. The scope of this review is signaling mechanisms and the functional roles of representative T-cell co-inhibitory receptors in the regulation of autoimmune glomerulonephritis, along with current therapeutic challenges mainly on preclinical trials. Co-inhibitory receptors utilize both shared and unique signaling pathway, suggesting specialized functions that provide the rationale behind therapies for autoimmune glomerulonephritis by targeting these inhibitory receptors. These receptors largely suppress Th1 immunity, modify Th17 and Th2 immune response, and enhance Treg function. Anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA4) immunoglobulin (Ig), which is able to block both activating CD28 and inhibitory CTLA4 signaling, has been shown in preclinical and clinical investigations to have effects on glomerular disease. Other inhibitory receptors for treating glomerulonephritis have not been clinically tested, and efficacy of manipulating these pathways requires further preclinical investigation. While immune checkpoint inhibition using anti-CTLA4 antibodies and anti-programmed cell death 1 (PD-1)/PD-L1 antibodies has been approved for the treatment of several cancers, blockade of CTLA4 and PD-1/PD-L1 is associated with adverse effects that resemble autoimmune disorders, including systemic vasculitis. A renal autoimmune vasculitis model features an initial Th17 dominancy followed later by a Th1-dominant outcome and Treg cells that attenuate autoreactive T-cell function. Toward the development of effective therapies for T-cell-mediated autoimmune glomerulonephritis, it would be preferable to pay attention to the impact of the inhibitory pathways in immunological renal disease settings.
Introduction
T cells are key effectors of the adaptive immune response, playing important roles in the elimination of pathogens and in the development of autoimmune disease. Autoimmune glomerulonephritis occurs as a consequence of autoantibodies and T-cell effector functions that target either antigens intrinsic to the glomeruli [for example, as occurs in anti-glomerular basement membrane (GBM) nephropathy] or non-specific antibodies that become trapped and accumulate in the glomeruli [for example, as occurs in immunoglobulin (Ig) A nephropathy and anti-neutrophil cytoplasmic autoantibody (ANCA)-associated glomerulonephritis] (1, 2). In this regard, peripheral regulation of T-cell responses is crucial to preventing inappropriate responses to self-antigens leading to autoimmune glomerulonephritis (1).
The optimal activation of T cells is profoundly influenced by co-signaling through cell surface receptors (3). The common feature that identifies receptors as members of the inhibitory class is their ability to attenuate activation signals initiated by other receptors that are often members of the immunoreceptor tyrosine-based activation motif (ITAM) class (4). Loss of inhibitory signaling is often associated with autoreactivity and unchecked inflammatory responses, illustrating the essential role that this system plays in immune regulation (4, 5). Though the human genome is estimated to encode over 300 immunoreceptor tyrosine-based inhibitory motif (ITIM)-containing molecules, of which only a minority has been characterized (6), most reviews discussing the co-receptor signaling pathway as a potential target in autoimmunity have focused on blockade of co-stimulatory receptor signaling (7, 8). The scope of this review is signaling mechanisms and the functional roles of some representative T-cell co-inhibitory receptors in the regulation of autoimmune glomerulonephritis, along with current therapeutic challenges.
Classical and Non-classical Inhibitory Signaling in T-cell Receptor Pathway
TCR Signaling Pathway and Co-signaling
The primary signal for conventional T cells is mediated through T-cell receptor (TCR) engagement. TCRs recognize small antigenic peptides presented in the groove of the self-major histocompatibility complex (MHC) (9). As a result of this recognition, TCR complexes aggregate on T-cell surfaces to form stable contacts, resulting in the formation of immunological synapses on antigen-presenting cells (APCs) (9). This aggregation evokes intracellular signaling that involves the activation of Src protein tyrosine kinase, leading to the phosphorylation of CD3- and ζ chain-localized ITAM. Subsequently, the ζ-associated protein of 70 kD (ZAP-70) is recruited, resulting in a series of downstream phosphorylation events (10) (Figure 1). Another kinase pathway in T cells involves the activation of phosphatidylinositol-3 kinase (PI3K), which phosphorylates a specific membrane-associated inositol lipid. This enzyme is recruited to the TCR complex and generates phosphatidylinositol triphosphate (PIP3) and diacylglycerol (DAG) from membrane phosphatidylinositol biphosphate (PIP2). PIP3 activates signaling enzymes such as PLCγ (phospholipase Cγ) and PKCθ (protein kinase Cθ). However, the primary signal itself does not decide the fate of the immune response (11). Instead, co-stimulatory and co-inhibitory receptors on T cells direct the function and fate. These co-signaling receptors often co-localize with TCR molecules, such that the co-signaling receptors synergize with TCR signaling to promote or inhibit T-cell activation and function (11, 12).
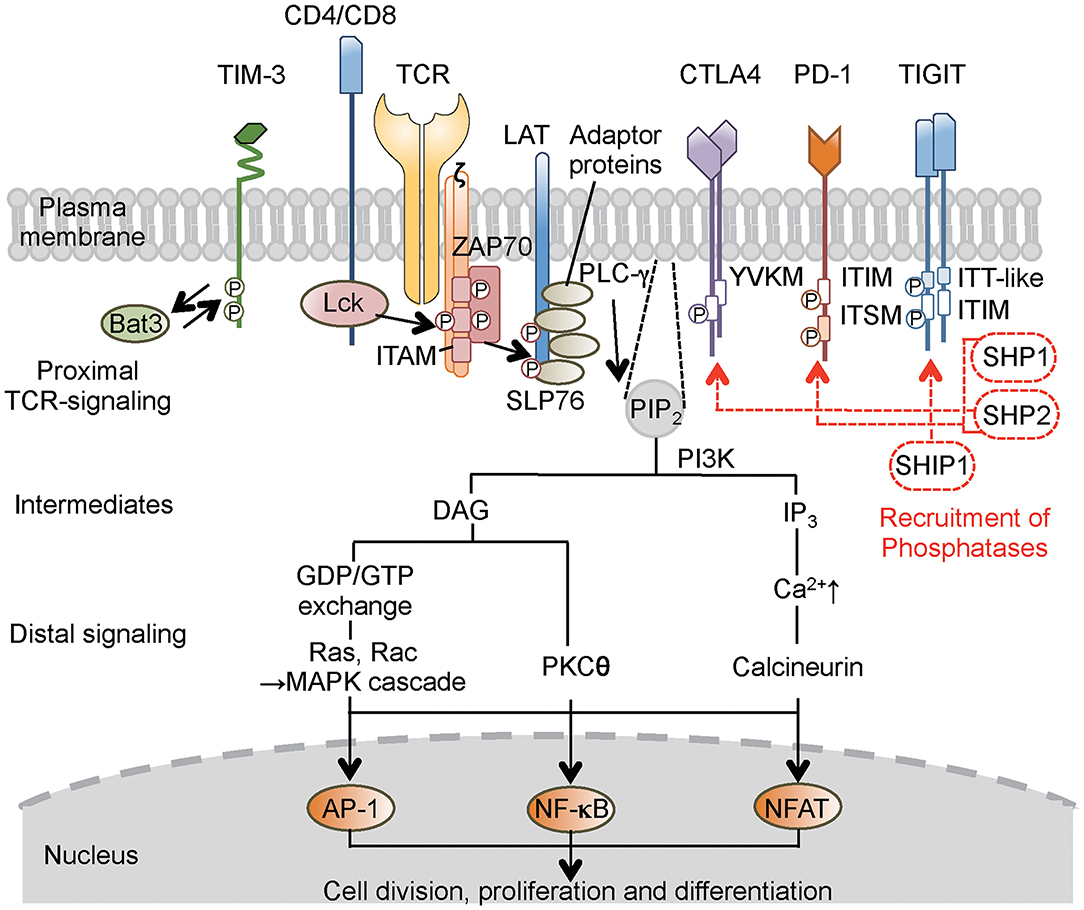
Figure 1. Concise outline of the intracellular signaling events during T-cell activation and roles of co-inhibitory receptors. Binding of the TCR and co-receptors to peptide–MHC (major histocompatibility complex) complexes on the antigen-presenting cells (APCs) initiates proximal signaling events, which result in phosphorylation of the ζ chain, binding and activation of ZAP-70, phosphorylation of LAT and adaptor proteins, production of biochemical intermediates, and activation of distal signaling cascades. MAPK, PKC, and calcineurin are enzymes that activate transcription factors, thereby stimulating the expression of various genes involved in T-cell response. Most inhibitory receptors have an inhibitory motif, represented by ITIM, in their cytoplasmic tails. Ligand binding to these receptors results in the recruitment of phosphatases (SHP-1, SHP-2, or SHIP1), which alter proximal and distal TCR signals. These consequently transmit activating signal (black solid line arrow) and inhibitory signal (red broken line and arrow) in T cells. In the case of TIM-3, which does not have a classical signaling motif, Bat-3 (HLA-B associated transcript 3) binds to the TIM-3 tail and blocks binding of another adaptor molecule under steady state. Ligand binding triggers the dissociation of Bat-3 from the cytoplasmic tail of TIM-3, thus allowing another adaptor molecule to bind and promote the inhibitory function of TIM-3. CTLA4, cytotoxic T-lymphocyte-associated protein 4; PD-1, programmed cell death 1; TIM-3, T-cell immunoglobulin 3; TIGIT, T-cell immunoglobulin and ITIM domain; LAT, linker for activation of T cells; TCR, T-cell receptor; ZAP-70, ζ-associated protein of 70 kD; SLP-76, SH2-binding leukocyte phosphoprotein of 76 kD; ITIM, immunoreceptor tyrosine-based inhibitory motif; ITSM, immunoreceptor tyrosine-based switch motif; ITT, immunoglobulin tail tyrosine; SHP-1,-2, Src-homology-2 domain-containing protein tyrosine phosphatase 1, 2; SHIP1, SH2 domain-containing inositol-5-phosphatase 1; PIP2, phosphatidylinositol biphosphate; PI3K, phosphatidylinositol-3 kinase; DAG, diacylglycerol; IP3, phosphatidylinositol triphosphate; GDP/GTP, guanosine diphosphate/triphosphate; MAPK, mitogen-activated protein kinase; PLC, phospholipase C; PKC, protein kinase C; AP-1, activation protein 1; NF-κB, nuclear factor-κB; NFAT, nuclear factor of activated T cells.
ITIM, ITSM, ITT, and Other Mechanisms
Intracellular protein–protein interaction during cell signaling and activities of cellular enzymes are often regulated by phosphorylation of tyrosine residues. For countering action of phosphorylation by tyrosine kinase, protein tyrosine phosphatases are enzymes that remove phosphate moieties from tyrosine residues to limit and terminate cellular responses that are no longer required (4, 13). One family of immune inhibitory receptors is defined by the presence of a consensus amino acid sequence, the ITIM motif, in the cytoplasmic domain of the proteins (13). The six-amino acid ITIM motif consists of the sequence (Ile/Val/Leu/Ser)-X-Tyr-X-X-(Leu/Val), where X denotes any amino acid (4). Ligand binding induces clustering of the inhibitory receptors and results in tyrosine phosphorylation that provides a docking site for the recruitment of cytoplasmic phosphatases that have Src-homology-2 (SH2) domains, including SHP-1 (SH2 domain-containing protein tyrosine phosphatase 1) and SHP-2. These phosphatases remove phosphate from tyrosine residues in the activated receptor and adaptors, such as SH2-binding leukocyte phosphoprotein of 76 kD (SLP-76), linker for activation of T cells (LAT), and CD3ζ (14) (Figure 1).
CTLA4 carries an ITIM-like YVKM motif, which associates with SHP-2 and reduces proximal TCR signaling through dephosphorylation of targets such as the TCR–CD3ζ complex, LAT, and ZAP-70 (15, 16), thereby inhibiting cell cycle progression and cytokine production. PD-1 has an ITIM motif as well as an immunoreceptor tyrosine-based switch motif (ITSM) (17). Both motifs appear to be phosphorylated following interaction with ligands, resulting in the recruitment of SHP-2 and possibly SHP-1; the co-localization of PD-1 with TCR microclusters induces dephosphorylation of CD3ζ, ZAP70, and PKC (17, 18). The TIGIT (T-cell immunoglobulin and ITIM domain) protein contains an ITIM motif and an immunoglobulin tail tyrosine (ITT)-like motif; phosphorylation of the tyrosine residue in either of these motifs is sufficient for signal transduction and inhibitory activity (19, 20). T-cell immunoglobulin-3 (TIM-3) does not have a classical signaling motif in its cytoplasmic tail (21). To simplify the descriptions in the present review, I am avoiding mention of several other remarkable signaling pathways that employ other phosphatases and intracellular motifs of the CTLA4, PD-1, TIM-3, and TIGIT co-inhibitory receptors; those topics have been covered in a number of excellent detailed reviews (3, 4, 11, 22).
Collectively, co-inhibitory receptors regulate T-cell immunity by using both shared and unique signaling pathways (3), suggesting the specialized functions and providing the rationale for therapies that treat autoimmune glomerulonephritis by targeting these inhibitory receptors. The following subsection addresses the expression on T cells, function in vivo, and potential role for regulating autoimmune glomerular diseases of CTLA4, PD-1, TIM-3, and TIGIT, summarized in Figure 2 and Table 1.
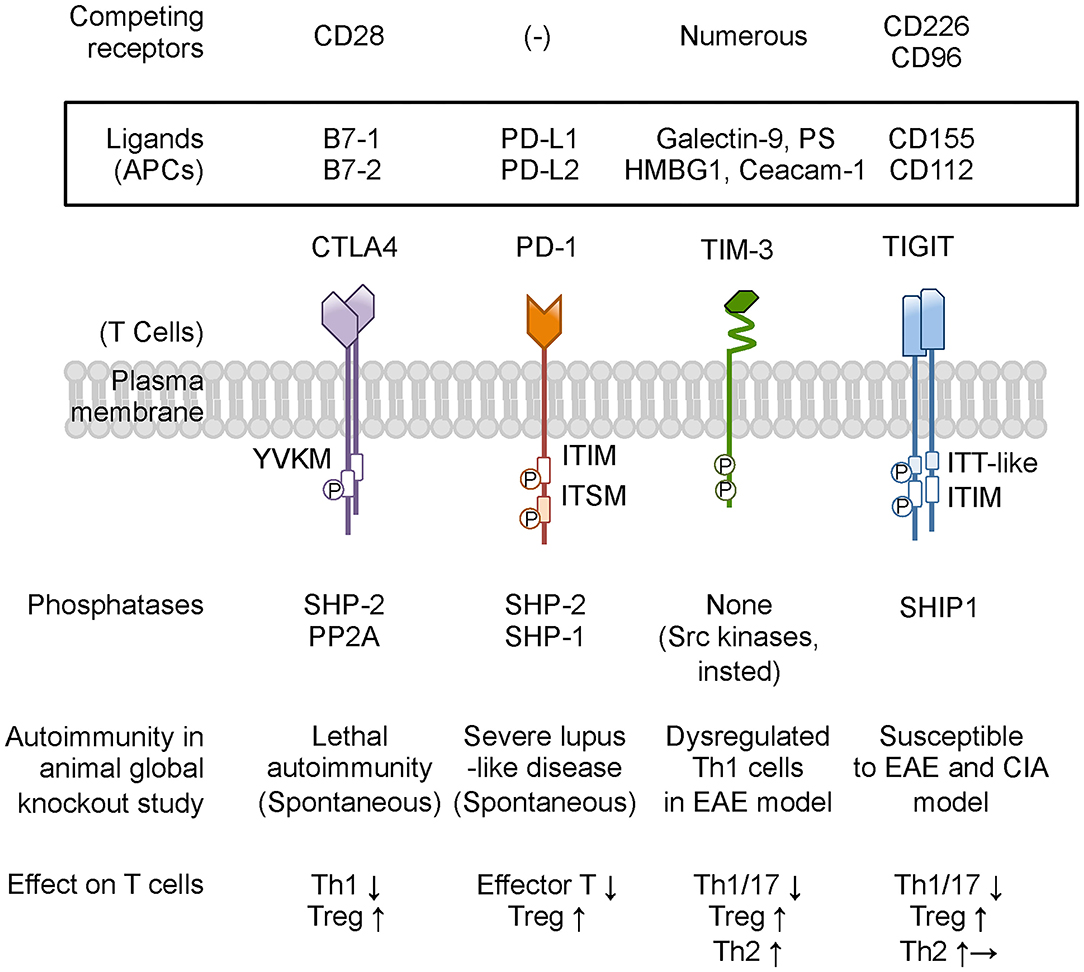
Figure 2. Co-inhibitory receptors on T cells and ligands, phosphatase, and modulation of T-cell function. Co-inhibitory receptors have a variety of physiological and pathological ligands, and the binding is occasionally competed by other immune receptors on T cells. TIM-3 does not have a classical signaling motif in its cytoplasmic tail. However, the cytoplasmic domain of TIM-3 contains tyrosine residues to be targets for phosphorylation and promoting TIM-3-mediated T-cell inhibition by allowing binding of SH2 domain-containing Src kinases. Based on animal studies, CTLA4 and PD-1 are predicted to be associated with more susceptibility to autoimmune disease, while neither TIM-3-deficient nor TIGIT-deficient mice present any spontaneous autoimmune disease phenotype. Co-inhibitory receptors on T cells commonly suppress effector T-cell functions by down-modulating Th1 cells and Th17 cells and by enhancing Treg cells. In addition, TIM-3 and TIGIT shift the cytokine balance to Th2 immunity. PS, phosphatidyl serine; HMGB1, high-mobility group protein B1; Ceacam-1, carcinoembryonic antigen-related cell adhesion molecule 1; PP2A, protein phosphatase 2A; EAE, experimental autoimmune encephalomyelitis; CIA, collagen-induced arthritis. Other abbreviations are shown in Figure 1 caption.
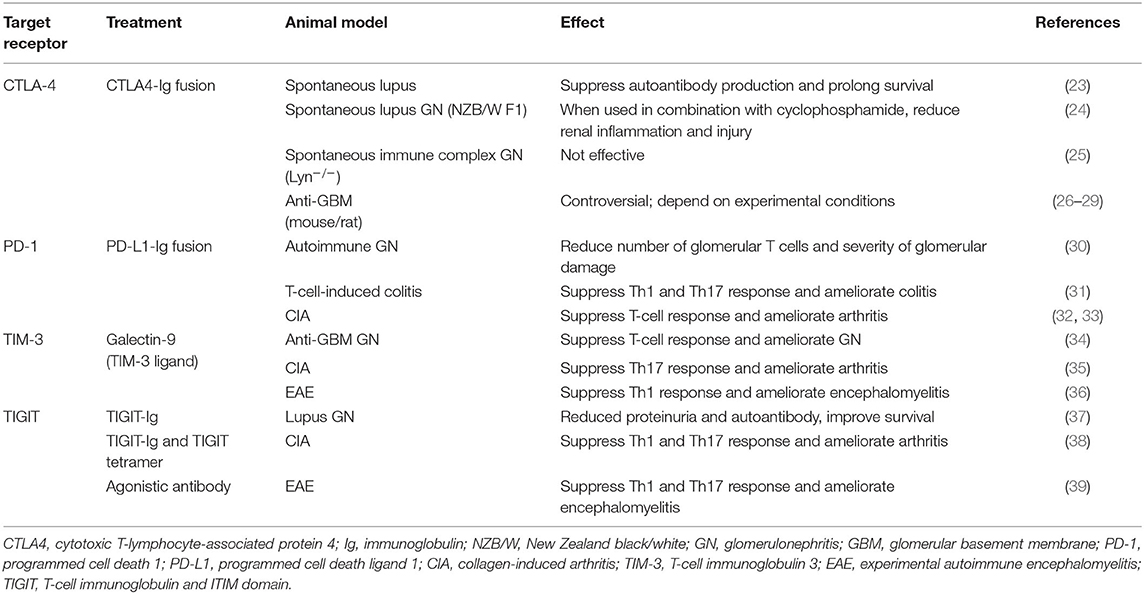
Table 1. Experimental treatment models targeting T-cell co-inhibitory signaling in autoimmune diseases.
CTLA-4
Expression, Ligands, and General Function
CTLA4 is a potent negative regulator of T-cell response and was identified as a member of the CD28 family (40). Although CD28 is constitutively expressed on all naive CD4+ and CD8+ T cells and Treg cells, CTLA4 is transiently expressed on the surface of activated T cells (41, 42). The B7 family of proteins, B7-1 and B7-2, provides the major co-stimulatory signal for augmenting and sustaining T-cell responses through interaction with CD28 (42). B7-1 and B7-2 are shared ligands of CTLA4; the interaction of CTLA4 with these ligands leads to co-inhibitory signaling (43). In other words, the inhibitory mechanisms of CTLA4 include CD28 out-competition and blockade of intracellular signaling pathways, as CTLA4 has a 10-fold higher affinity than CD28 for B7-1 binding (43).
CTLA4-Ig (“abatacept”) is a biological that binds to B7-1 and B7-2, blocking both activating CD28-mediated signaling and inhibitory CTLA4-mediated signaling (44, 45) and effectively inhibiting naive antigen-specific CD4+ T-cell responses (46, 47). While the total CD4+ memory T-cell response was effectively attenuated by administration of CTLA4-Ig (48, 49), examinations of subsets of CD4+ helper T cells revealed that interleukin (IL)-17-secreting CCR6+ memory Th17 cells were resistant to CD28 and CTLA4 blockade (50).
Involvement in Autoimmunity and Glomerular Diseases and Therapeutic Model
CTLA4-deficient mice exhibit severe lymphoproliferative disease, with infiltration of activated T cells into various organs and death within a few weeks of birth (51–53). Given the promising results of CD28 and CTLA4 blockade in small animal models (23), strategies to target this pathway were developed in several clinical trials for the treatment of autoimmunity. CTLA4-Ig has been used clinically for the effective treatment of rheumatoid arthritis (RA) and juvenile idiopathic arthritis, as reviewed in (54), and has been tested against allergen-induced airway inflammation (55), ulcerative colitis (56), systemic lupus erythematosus (SLE) (57, 58), and other autoimmune diseases, as reviewed in (59). The results of those trials indicated that abatacept did not alter the inflammatory response to allergen challenge or show any efficacy in ameliorating colitis symptoms (59). Collectively, abatacept might be efficacious in the treatment of Th1-mediated autoimmune disease, such as RA, but remain less effective in the treatment of Th2- or Th17-mediated autoimmune disease such as asthma and inflammatory bowel disease (7). However, several studies have indicated that CTLA4-Ig protein attenuates glomerular injury in experimental models of crescentic glomerulonephritis (26–29). As a result, abatacept has been tested clinically in patients with granulomatosis with polyangiitis; the drug was well-tolerated in this population, providing a high frequency of disease remission and steroid discontinuation (60). A further clinical trial of abatacept in ANCA-associated vasculitis (AAV) has been progressed (https://clinicaltrials.gov/).
PD-1
Expression, Ligands, and General Function
PD-1 originally was identified as an inducible surface receptor during programed cell death (61) and was shown to be expressed on stimulated T, B, and myeloid cells (62). PD-L1 and PD-L2 are two independent ligands for PD-1. PD-L2 expression is largely confined to dendritic cells (DCs) and monocytes/macrophages, but PD-L1 is more widely distributed on leukocytes and non-hematopoietic cells (63, 64). Expression of PD-L in peripheral tissue may regulate the behavior of infiltrating leukocytes (65). The PD-1/PD-L1 pathway exerts important inhibitory function in primary T-cell proliferation, cytokine production, cytotoxic activity, and cell survival (66, 67). This pathway also promotes development and function of Treg cells (68) and negatively regulates effector T-cell reactivation and function (69).
Involvement in Autoimmunity and Glomerular Diseases and Therapeutic Model
PD-1-deficient mice develop autoantibody-induced disease in a strain-dependent fashion; this autoimmune disease includes lupus-like glomerulonephritis leading to late death (70–72), although the phenotype in these mice is much milder than that of CTLA4-deficient mice. Consistent with human evidence of polymorphisms associated with SLE, T1D (Type 1 diabetes mellitus), and RA, experimental animal models of T1D-prone mice (73) and collagen-induced arthritis (CIA) (32) indicate that PD-1 activation attenuates autoimmune disease.
In the kidney, renal DCs have been shown to express PD-L1 and can be involved in suppressing CD4+ T-cell proliferation (74). Studies in experimental autoimmune glomerulonephritis have shown that blockade of PD-1/PD-1L interaction aggravates glomerular injury and cellular infiltration (64) and that activation of PD-1 using a PD-L1 fusion protein leads to a reduction in disease severity (30). Moreover, recent work with PD-L1−/− mice showed that dosing with PD-L1 provided protection in a crescentic glomerulonephritis model via Treg-mediated suppression of the Th1 immune response (75). However, studies on immune-complex-mediated glomerulonephritis (induced by immunization with a foreign antigen) showed that blockade of the PD-1/PD-L axis (by antibody administration) did not reveal any significant pathological changes (76). This result suggests the need for careful interpretation of the roles of PD-1/PD-L in experimental autoimmune glomerulonephritis. Indeed, clinical trials testing the treatment of glomerulonephritis with PD-1 have not been reported. Thus, the clinical efficacy of modifying this pathway still requires further preclinical investigation.
TIM-3
Expression, Ligands, and General Function
TIM-3 was identified as molecule expressed specifically in interferon (IFN)-γ-producing Th1 and CD8+ cytotoxic T cells, but not in naive T cells (77). Galectin-9 is a soluble S-type lectin that is widely expressed on immune and non-immune cells and has been shown to bind to the IgV domain of TIM-3, resulting in negative regulation of Th1 immunity (36). In addition to galectin-9, phosphatidyl serine, high-mobility group protein B1 (HMGB1), and carcinoembryonic antigen-related cell adhesion molecule 1 (Ceacam-1) have been identified as TIM-3 ligands (78–80). It remains to be determined whether the triggering of TIM-3 by individual ligands or by combinations thereof has distinct impacts on TIM-3 function.
Involvement in Autoimmunity and Glomerular Diseases and Therapeutic Model
TIM-3 can be protective in autoimmunity but often is sparsely expressed; in contrast, the protein is highly expressed in cancer and chronic viral infection, resulting in the dampening of protective immunity (22). Even so, multiple reports have shown that TIM-3 blockade results in abrogation of peripheral tolerance of Th1-cell-mediated responses. Anti-TIM-3 blocking murine model develops hyper-acute experimental autoimmune encephalomyelitis (EAE) (77); treatment with soluble TIM-3-Ig results in T-cell hyper-activation and IFN-γ production (81). In addition to its role in regulating effector T-cell responses, TIM-3 also may have a role in regulating the function of Foxp3+ Treg cells (82). Several studies have shown that TIM-3+ Treg cells have superior suppressive function when compared to TIM-3− Treg cells (82, 83).
Galectin-9 is a rare example of agonistic treatment based on a natural ligand. Administration of galectin-9 as a soluble protein in mouse ameliorates EAE and CIA (35, 36). The indiscriminate nature of galectin avidity, such that the molecule binds to sugars on multiple different glycoproteins, makes it difficult to definitively attribute these effects to TIM-3 signaling rather than to the manipulation of another galectin-9 binding partner (84). Nevertheless, administration of galectin-9 ameliorates experimental anti-GBM glomerulonephritis; this protective role is associated with inhibition of Th1 and Th17-cell-mediated immune responses and enhanced Th2 immunity in the kidney (34).
Human studies suggest that renal TIM-3 and galectin-9 expression levels are higher in immune-complex-mediated glomerulonephritis, such as IgA nephropathy (85) and lupus nephritis (86) compared to the control group. Some investigations have examined the expression of TIM-3 on peripheral blood cells and in the serum of patients with glomerular diseases (87, 88), but there is little evidence of a role for TIM-3 in other types of autoimmune glomerulonephritis. Clinical trials evaluating the treatment of autoimmune glomerulonephritis using an agent targeting TIM-3 have not been reported. Although the targeting of TIM-3 signaling holds potential for the treatment of T-cell-mediated glomerulonephritis, further preclinical investigation will be required to elucidate effects both on different immune cells and on ligand binding partners other than galectin-9.
TIGIT
Expression, Ligands, and General Function
TIGIT was discovered as a novel member of the CD28 protein family (89, 90). TIGIT is expressed on activated T cells, memory T cells, a subset of Treg cells, and follicular helper T (Tfh) cells, and binds to two ligands, CD155 and CD112, that are expressed on APCs (19, 89, 90). CD226 and CD96 bind to the same ligands, and the CD226–CD155 interaction mediates a co-stimulatory response in cytotoxic T cells (89). TIGIT competes with CD226 by binding with greater affinity to CD155–CD112 to disrupt that co-stimulatory effect, thereby resulting in a dominant inhibitory effect (22). In this regard, the pathway formed by CD226, TIGIT, and their ligands resembles the B7-CD28/CTLA4 pathway: in both cases, a pair of receptors—one positive, one negative–share ligands expressed on APCs (22).
Involvement in Autoimmunity and Glomerular Diseases and Therapeutic Model
In human, genomic analyses showed that a polymorphism in CD226 (Gly307Ser) is linked to multiple autoimmune diseases, including T1D, multiple sclerosis, and RA (91, 92). Although TIGIT-deficient mice do not develop spontaneous autoimmunity, these animals display augmented T-cell responses upon immunization (93). The function of TIGIT was examined in EAE and CIA models, with results suggesting that TIGIT is protective for the pro-inflammatory Th1 and Th17 cellular response and contributes to peripheral tolerance (38, 93). In addition to its direct inhibitory role in effector T cells, TIGIT also inhibits immune responses by promoting Treg function and IL-10 production (22). Curiously, several lines of evidence indicates that TIGIT signaling can shift the cytokine balance away from a Th1- and Th17-cell-dominated response and toward a Th2-cell-like response, concurrently with TIGIT-binding-mediated CD155 signaling on APCs (89, 94).
Given the similarity between the B7-CD28/CTLA4 and CD155/CD112-TIGIT/CD226 signaling pathways with regard to their co-signaling frameworks, TIGIT-Ig theoretically should block both activating CD226 signaling and inhibitory TIGIT signaling in a manner similar to that of CTLA-4-Ig. In addition, TIGIT-Ig induces CD155 signaling in cultured DCs in vitro and decreases IL-10 production by Th1 cells in vivo (89). The specific difference between these two pathways is that B7 is expressed primarily in professional APCs, while CD155 is expressed by a variety of non-professional APCs such as the vascular endothelium, fibroblasts, and tumor cells (95). When autoimmune disease occurs, the tissue that is infiltrated by T cells contains mainly non-professional APCs, and the CD155/CD112-TIGIT/CD226 pathway might be involved in tissue damage. Still, in both human and animal models, few studies have examined the role of TIGIT signaling in renal-specific disease. Although the treatment of a murine lupus model (NZB/NZW F1 mice) using TIGIT-Ig significantly improved survival, inflammatory responses, and glomerular damage (37), preclinical studies on other glomerular diseases will be needed to permit clinical use of TIGIT-Ig.
The Development of Autoimmune Glomerulonephritis Caused by Immune Checkpoint Inhibitors
In the past decade, cancer therapy has been revolutionized by the development of drugs that promote immune-mediated tumor destruction (96). CTLA-4 and PD-1/PD-L1 are the two best-studied co-inhibitory pathways (97); the use of antibodies as immune checkpoint inhibitors, anti-CTLA4 antibodies, and anti-PD-1/PD-L1 antibodies has been approved for the treatment of several cancers (98–100). While these immunotherapies have shown striking success, blockade of CTLA-4 and PD-1/PD-L1 are associated with adverse effects that resemble autoimmune disorders, including SLE, RA, thyroiditis, and T1D (59, 101). Additionally, renal vasculitis, immune-complex-mediated glomerulonephritis, and pauci-immune glomerulonephritis recently have been reported (102–108). Most systemic vasculitis cases resolved with either holding the immune checkpoint inhibitors and/or administering glucocorticoids (109). These evidences imply relationship between interventional blocking co-inhibitory receptor signaling and development of renal vasculitis, suggesting that this pathway may be a therapeutic target.
Rationale for Targeting Th1/17 Effector and Regulatory T Cells in Autoimmune Vasculitis
As mentioned before, blockade of inhibitory receptors occasionally has resulted in renal vasculitis as well as lupus-like autoimmunity. While autoantibodies play a role in a number of forms of glomerulonephritis, renal vasculitis in humans features the infiltration of T cells and macrophages (110, 111), suggesting a delayed hypersensitivity reaction in kidney. Given that autoreactive CD4+ and CD8+ cells are present in vasculitis patients (112–115), experimental passive transfer studies have defined a role for CD4+ and CD8+ cells in AAV (116, 117). CD4+ effector T cells, particularly upon differentiation to Th17 cells, mediate production of neutrophil chemoattractants by tissue cells via release of IL-17A and renal injury (118, 119). Studies using mice deficient in Th1- and Th17-defining cytokines have shown an initial Th17-dominant lesion followed later by a Th1-dominant outcome (120). Moreover, as human studies implicate abnormal CD4+ Foxp3+ Treg number and function in AAV patients (121–124), depletion of Treg cells led to more anti-neutrophil cytoplasmic protein-specific T cells and more severe glomerulonephritis (125). Approaches for targeting inhibitory receptors might (in theory) include inhibitory receptor-Ig fusion proteins, ligand-Ig fusion proteins, artificial ligands, and agonistic antibodies, as well as the use of bi-specific antibodies to co-ligate inhibitory and activating receptors (59). Among these approaches, as shown in Table 1, TIGIT-Ig protein, agonistic anti-TIGIT antibodies, and TIM-3 ligands (e.g., galectin-9), along with PD-L1-Ig and CTLA4-Ig proteins, should be considered candidates for development as bench-to-bedside therapeutics for treatment of T-cell-mediated autoimmune glomerulonephritis through regulation of the function of Th1/Th17 and Treg cells.
Conclusion
The studies in knockout mice and clinical experiences of vasculitis caused by immune checkpoint inhibitors treatment give numerous indications that the loss of a functional co-inhibitory receptor leads to sensitivity for autoimmune disease. Clinical utilization of co-inhibitory axes has not progressed in autoimmune disease as it has in cancer. Except for CTLA4-Ig, no clinical trial on co-inhibitory targeted therapy for autoimmune vasculitis and lupus nephritis has been progressed until the middle of 2020. Nevertheless, some recent studies have shown preclinical evidence for the utility of targeting co-inhibitory receptors in lupus and glomerulonephritis. As effector T cells and Treg function have a pivotal role in the development of autoimmune vasculitis, currently available CTLA4-Ig has been tested to evaluate the efficacy of achieving glucocorticoid-free remission in patients with relapsing vasculitis. Clinical efficacy of therapy targeting at PD-1, TIM-3, and TIGIT is expected to be not always consistent because their ligands are different from each other and each co-inhibitory receptor utilizes unique cell signaling as well as shared pathway. Therefore, clinical trials on modulating co-inhibitory signaling by Ig-fusion protein, agonistic antibody, and natural ligands should be carefully designed after sufficient preclinical investigations with clinically relevant animal models show that effector T cells are precisely involved in antigen-specific disease development. Toward the development of effective therapies for T-cell-mediated autoimmune glomerulonephritis, it would be preferable to pay attention to the impact and features of these inhibitory pathways in immunological renal disease settings.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This article was supported in part by JSPS Grant Numbers 19K17729 and 18KK0431.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Holdsworth SR, Gan PY, Kitching AR. Biologics for the treatment of autoimmune renal diseases. Nat Rev Nephrol. (2016) 12:217–31. doi: 10.1038/nrneph.2016.18
2. Suarez-Fueyo A, Bradley SJ, Klatzmann D, Tsokos GC. T cells and autoimmune kidney disease. Nat Rev Nephrol. (2017) 13:329–43. doi: 10.1038/nrneph.2017.34
3. Zhu Y, Yao S, Chen L. Cell surface signaling molecules in the control of immune responses: a tide model. Immunity. (2011) 34:466–78. doi: 10.1016/j.immuni.2011.04.008
4. Lanier LL. Face off–the interplay between activating and inhibitory immune receptors. Curr Opin Immunol. (2001) 13:326–31. doi: 10.1016/S0952-7915(00)00222-3
5. Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. (2000) 13:277–85. doi: 10.1016/S1074-7613(00)00027-3
6. Daeron M, Jaeger S, Du Pasquier L, Vivier E. Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol Rev. (2008) 224:11–43. doi: 10.1111/j.1600-065X.2008.00666.x
7. Ford ML, Adams AB, Pearson TC. Targeting co-stimulatory pathways: transplantation and autoimmunity. Nat Rev Nephrol. (2014) 10:14–24. doi: 10.1038/nrneph.2013.183
8. Zhang Q, Vignali DA. Co-stimulatory and co-inhibitory pathways in autoimmunity. Immunity. (2016) 44:1034–51. doi: 10.1016/j.immuni.2016.04.017
9. Huppa JB, Davis MM. T-cell-antigen recognition and the immunological synapse. Nat Rev Immunol. (2003) 3:973–83. doi: 10.1038/nri1245
10. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. (2009) 27:591–619. doi: 10.1146/annurev.immunol.021908.132706
11. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
12. Saito T, Yokosuka T, Hashimoto-Tane A. Dynamic regulation of T cell activation and co-stimulation through TCR-microclusters. FEBS Lett. (2010) 584:4865–71. doi: 10.1016/j.febslet.2010.11.036
13. Bolland S, Ravetch JV. Inhibitory pathways triggered by ITIM-containing receptors. Adv Immunol. (1999) 72:149–77. doi: 10.1016/S0065-2776(08)60019-X
14. Ravetch JV, Lanier LL. Immune inhibitory receptors. Science. (2000) 290:84–9. doi: 10.1126/science.290.5489.84
15. Schneider H, Smith X, Liu H, Bismuth G, Rudd CE. CTLA-4 disrupts ZAP70 microcluster formation with reduced T cell/APC dwell times and calcium mobilization. Eur J Immunol. (2008) 38:40–7. doi: 10.1002/eji.200737423
16. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. (2009) 229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x
17. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741
18. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. (2004) 574:37–41. doi: 10.1016/j.febslet.2004.07.083
19. Stanietsky N, Rovis TL, Glasner A, Seidel E, Tsukerman P, Yamin R, et al. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur J Immunol. (2013) 43:2138–50. doi: 10.1002/eji.201243072
20. Liu S, Zhang H, Li M, Hu D, Li C, Ge B, et al. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. (2013) 20:456–64. doi: 10.1038/cdd.2012.141
21. Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol. (2011) 31:3963–74. doi: 10.1128/MCB.05297-11
22. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
23. Finck BK, Linsley PS, Wofsy D. Treatment of murine lupus with CTLA4Ig. Science. (1994) 265:1225–7. doi: 10.1126/science.7520604
24. Cunnane G, Chan OT, Cassafer G, Brindis S, Kaufman E, Yen TS, et al. Prevention of renal damage in murine lupus nephritis by CTLA-4Ig and cyclophosphamide. Arthritis Rheum. (2004) 50:1539–48. doi: 10.1002/art.20147
25. Oracki SA, Tsantikos E, Quilici C, Light A, Schmidt T, Lew AM, et al. CTLA4Ig alters the course of autoimmune disease development in Lyn-/- mice. J Immunol. (2010) 184:757–63. doi: 10.4049/jimmunol.0804349
26. Okano K, Nitta K, Ogawa S, Horita S, Habiro K, Nihei H, et al. Effects of double blockade of CD28 and inducible-costimulator signaling on anti-glomerular basement membrane glomerulonephritis. J Lab Clin Med. (2004) 144:183–92. doi: 10.1016/j.lab.2004.06.004
27. Li S, Holdsworth SR, Tipping PG. B7.1 and B7.2 co-stimulatory molecules regulate crescentic glomerulonephritis. Eur J Immunol. (2000) 30:1394–401. doi: 10.1002/(SICI)1521-4141(200005)30:5<1394::AID-IMMU1394>3.0.CO;2-O
28. Kitching AR, Huang XR, Ruth AJ, Tipping PG, Holdsworth SR. Effects of CTLA4-Fc on glomerular injury in humorally-mediated glomerulonephritis in BALB/c mice. Clin Exp Immunol. (2002) 128:429–35. doi: 10.1046/j.1365-2249.2002.01859.x
29. Reynolds J, Tam FW, Chandraker A, Smith J, Karkar AM, Cross J, et al. CD28-B7 blockade prevents the development of experimental autoimmune glomerulonephritis. J Clin Invest. (2000) 105:643–51. doi: 10.1172/JCI6710
30. Reynolds J, Sando GS, Marsh OB, Salama AD, Evans DJ, Cook HT, et al. Stimulation of the PD-1/PDL-1 T-cell co-inhibitory pathway is effective in treatment of experimental autoimmune glomerulonephritis. Nephrol Dial Transplant. (2012) 27:1343–50. doi: 10.1093/ndt/gfr529
31. Song MY, Hong CP, Park SJ, Kim JH, Yang BG, Park Y, et al. Protective effects of Fc-fused PD-L1 on two different animal models of colitis. Gut. (2015) 64:260–71. doi: 10.1136/gutjnl-2014-307311
32. Raptopoulou AP, Bertsias G, Makrygiannakis D, Verginis P, Kritikos I, Tzardi M, et al. The programmed death 1/programmed death ligand 1 inhibitory pathway is up-regulated in rheumatoid synovium and regulates peripheral T cell responses in human and murine arthritis. Arthritis Rheum. (2010) 62:1870–80. doi: 10.1002/art.27500
33. Wang G, Hu P, Yang J, Shen G, Wu X. The effects of PDL-Ig on collagen-induced arthritis. Rheumatol Int. (2011) 31:513–9. doi: 10.1007/s00296-009-1249-0
34. Zhang Q, Luan H, Wang L, He F, Zhou H, Xu X, et al. Galectin-9 ameliorates anti-GBM glomerulonephritis by inhibiting Th1 and Th17 immune responses in mice. Am J Physiol Renal Physiol. (2014) 306:F822–32. doi: 10.1152/ajprenal.00294.2013
35. Seki M, Oomizu S, Sakata KM, Sakata A, Arikawa T, Watanabe K, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol. (2008) 127:78–88. doi: 10.1016/j.clim.2008.01.006
36. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. (2005) 6:1245–52. doi: 10.1038/ni1271
37. Liu S, Sun L, Wang C, Cui Y, Ling Y, Li T, et al. Treatment of murine lupus with TIGIT-Ig. Clin Immunol. (2019) 203:72–80. doi: 10.1016/j.clim.2019.04.007
38. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol. (2011) 41:902–15. doi: 10.1002/eji.201041136
39. Dixon KO, Schorer M, Nevin J, Etminan Y, Amoozgar Z, Kondo T, et al. Functional anti-TIGIT antibodies regulate development of autoimmunity and antitumor immunity. J Immunol. (2018) 200:3000–7. doi: 10.4049/jimmunol.1700407
40. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. (1991) 174:561–9. doi: 10.1084/jem.174.3.561
41. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. (1994) 1:405–13. doi: 10.1016/1074-7613(94)90071-X
42. Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. (2001) 19:565–94. doi: 10.1146/annurev.immunol.19.1.565
43. Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, et al. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature. (2001) 410:608–11. doi: 10.1038/35069118
44. Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. (2001) 19:225–52. doi: 10.1146/annurev.immunol.19.1.225
45. Linsley PS, Wallace PM, Johnson J, Gibson MG, Greene JL, Ledbetter JA, et al. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science. (1992) 257:792–5. doi: 10.1126/science.1496399
46. Trambley J, Bingaman AW, Lin A, Elwood ET, Waitze SY, Ha J, et al. Asialo GM1(+) CD8(+) T cells play a critical role in costimulation blockade-resistant allograft rejection. J Clin Invest. (1999) 104:1715–22. doi: 10.1172/JCI8082
47. Ford ML, Wagener ME, Hanna SS, Pearson TC, Kirk AD, Larsen CP. A critical precursor frequency of donor-reactive CD4+ T cell help is required for CD8+ T cell-mediated CD28/CD154-independent rejection. J Immunol. (2008) 180:7203–11. doi: 10.4049/jimmunol.180.11.7203
48. Adams AB, Williams MA, Jones TR, Shirasugi N, Durham MM, Kaech SM, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. (2003) 111:1887–95. doi: 10.1172/JCI200317477
49. Ndejembi MP, Teijaro JR, Patke DS, Bingaman AW, Chandok MR, Azimzadeh A, et al. Control of memory CD4 T cell recall by the CD28/B7 costimulatory pathway. J Immunol. (2006) 177:7698–706. doi: 10.4049/jimmunol.177.11.7698
50. Yuan X, Paez-Cortez J, Schmitt-Knosalla I, D'Addio F, Mfarrej B, Donnarumma M, et al. A novel role of CD4 Th17 cells in mediating cardiac allograft rejection and vasculopathy. J Exp Med. (2008) 205:3133–44. doi: 10.1084/jem.20081937
51. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
52. Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity. (1997) 7:885–95. doi: 10.1016/S1074-7613(00)80406-9
53. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. (1995) 270:985–8. doi: 10.1126/science.270.5238.985
54. Ceeraz S, Nowak EC, Burns CM, Noelle RJ. Immune checkpoint receptors in regulating immune reactivity in rheumatic disease. Arthritis Res Ther. (2014) 16:469. doi: 10.1186/s13075-014-0469-1
55. Parulekar AD, Boomer JS, Patterson BM, Yin-Declue H, Deppong CM, Wilson BS, et al. A randomized controlled trial to evaluate inhibition of T-cell costimulation in allergen-induced airway inflammation. Am J Respir Crit Care Med. (2013) 187:494–501. doi: 10.1164/rccm.201207-1205OC
56. Sandborn WJ, Colombel JF, Sands BE, Rutgeerts P, Targan SR, Panaccione R, et al. Abatacept for Crohn's disease and ulcerative colitis. Gastroenterology. (2012) 143:62–9.e4. doi: 10.1053/j.gastro.2012.04.010
57. Merrill JT, Burgos-Vargas R, Westhovens R, Chalmers A, D'Cruz D, Wallace DJ, et al. The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. (2010) 62:3077–87. doi: 10.1002/art.27601
58. Furie R, Nicholls K, Cheng TT, Houssiau F, Burgos-Vargas R, Chen SL, et al. Efficacy and safety of abatacept in lupus nephritis: a twelve-month, randomized, double-blind study. Arthritis Rheumatol. (2014) 66:379–89. doi: 10.1002/art.38260
59. van der Vlist M, Kuball J, Radstake TR, Meyaard L. Immune checkpoints and rheumatic diseases: what can cancer immunotherapy teach us? Nat Rev Rheumatol. (2016) 12:593–604. doi: 10.1038/nrrheum.2016.131
60. Langford CA, Monach PA, Specks U, Seo P, Cuthbertson D, McAlear CA, et al. An open-label trial of abatacept (CTLA4-IG) in non-severe relapsing granulomatosis with polyangiitis (Wegener's). Ann Rheum Dis. (2014) 73:1376–9. doi: 10.1136/annrheumdis-2013-204164
61. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. (1992) 11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
62. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. (1996) 8:765–72. doi: 10.1093/intimm/8.5.765
63. Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. (2001) 193:839–46. doi: 10.1084/jem.193.7.839
64. Menke J, Lucas JA, Zeller GC, Keir ME, Huang XR, Tsuboi N, et al. Programmed death 1 ligand (PD-L) 1 and PD-L2 limit autoimmune kidney disease: distinct roles. J Immunol. (2007) 179:7466–77. doi: 10.4049/jimmunol.179.11.7466
65. Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. (2003) 33:2706–16. doi: 10.1002/eji.200324228
66. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
67. Riley JL. PD-1 signaling in primary T cells. Immunol Rev. (2009) 229:114–25. doi: 10.1111/j.1600-065X.2009.00767.x
68. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
69. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. (2006) 203:883–95. doi: 10.1084/jem.20051776
70. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. (2001) 291:319–22. doi: 10.1126/science.291.5502.319
71. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
72. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. (1999) 11:141–51. doi: 10.1016/S1074-7613(00)80089-8
73. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci USA. (2005) 102:11823–8. doi: 10.1073/pnas.0505497102
74. Edgtton KL, Kausman JY, Li M, O'Sullivan K, Lo C, Hutchinson P, et al. Intrarenal antigens activate CD4+ cells via co-stimulatory signals from dendritic cells. J Am Soc Nephrol. (2008) 19:515–26. doi: 10.1681/ASN.2007030386
75. Neumann K, Ostmann A, Breda PC, Ochel A, Tacke F, Paust HJ, et al. The co-inhibitory molecule PD-L1 contributes to regulatory T cell-mediated protection in murine crescentic glomerulonephritis. Sci Rep. (2019) 9:2038. 9:2038. doi: 10.1038/s41598-018-38432-3
76. Ooi JD, Li M, Kourkoutzelos K, Yagita H, Azuma M, Holdsworth SR, et al. Programmed death 1 and its ligands do not limit experimental foreign antigen-induced immune complex glomerulonephritis. Nephrology. (2015) 20:892–8. doi: 10.1111/nep.12532
77. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. (2002) 415:536–41. doi: 10.1038/415536a
78. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. (2015) 517:386–90. doi: 10.1038/nature13848
79. Cao E, Zang X, Ramagopal UA, Mukhopadhaya A, Fedorov A, Fedorov E, et al. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity. (2007) 26:311–21. doi: 10.1016/j.immuni.2007.01.016
80. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. (2012) 13:832–42. doi: 10.1038/ni.2376
81. Sabatos CA, Chakravarti S, Cha E, Schubart A, Sanchez-Fueyo A, Zheng XX, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. (2003) 4:1102–10. doi: 10.1038/ni988
82. Gupta S, Thornley TB, Gao W, Larocca R, Turka LA, Kuchroo VK, et al. Allograft rejection is restrained by short-lived TIM-3+PD-1+Foxp3+ Tregs. J Clin Invest. (2012) 122:2395–404. doi: 10.1172/JCI45138
83. Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced suppressor function of TIM-3+ FoxP3+ regulatory T cells. Eur J Immunol. (2014) 44:2703–11. doi: 10.1002/eji.201344392
84. Cooper DN, Barondes SH. God must love galectins; he made so many of them. Glycobiology. (1999) 9:979–84. doi: 10.1093/glycob/9.10.979
85. Yang X, Hu Z, Xia X, Zhen J, Zhuang X, Peng T. Expression of human T cell immunoglobulin domain and mucin-3 on kidney tissue from immunoglobulin A nephropathy patients. Immunol Res. (2014) 60:85–90. doi: 10.1007/s12026-014-8550-z
86. Guo L, Yang X, Xia Q, Zhen J, Zhuang X, Peng T. Expression of human T cell immunoglobulin domain and mucin-3 (TIM-3) on kidney tissue from systemic lupus erythematosus (SLE) patients. Clin Exp Med. (2014) 14:383–8. doi: 10.1007/s10238-013-0264-3
87. Pan HF, Zhang N, Li WX, Tao JH, Ye DQ. TIM-3 as a new therapeutic target in systemic lupus erythematosus. Mol Biol Rep. (2010) 37:395–8. doi: 10.1007/s11033-009-9833-7
88. Cai XZ, Liu N, Qiao Y, Du SY, Chen Y, Chen D, et al. Decreased TIM-3 mRNA expression in peripheral blood mononuclear cells from nephropathy patients. Genet Mol Res. (2015) 14:6543–8. doi: 10.4238/2015.June.12.7
89. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. (2009) 10:48–57. doi: 10.1038/ni.1674
90. Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG, et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol. (2009) 39:695–703. doi: 10.1002/eji.200839116
91. Hafler JP, Maier LM, Cooper JD, Plagnol V, Hinks A, Simmonds MJ, et al. CD226 Gly307Ser association with multiple autoimmune diseases. Genes Immun. (2009) 10:5–10. doi: 10.1038/gene.2008.82
92. Maiti AK, Kim-Howard X, Viswanathan P, Guillen L, Qian X, Rojas-Villarraga A, et al. Non-synonymous variant (Gly307Ser) in CD226 is associated with susceptibility to multiple autoimmune diseases. Rheumatology. (2010) 49:1239–44. doi: 10.1093/rheumatology/kep470
93. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol. (2011) 186:1338–42. doi: 10.4049/jimmunol.1003081
94. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
95. Sakisaka T, Takai Y. Biology and pathology of nectins and nectin-like molecules. Curr Opin Cell Biol. (2004) 16:513–21. doi: 10.1016/j.ceb.2004.07.007
96. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
97. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
98. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
99. Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. (2015) 16:908–18. doi: 10.1016/S1470-2045(15)00083-2
100. Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. (2015) 16:375–84. doi: 10.1016/S1470-2045(15)70076-8
101. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. (2018) 378:158–68. doi: 10.1056/NEJMra1703481
102. Gallan AJ, Alexander E, Reid P, Kutuby F, Chang A, Henriksen KJ. Renal vasculitis and pauci-immune glomerulonephritis associated with immune checkpoint inhibitors. Am J Kidney Dis. (2019) 74:853–6. doi: 10.1053/j.ajkd.2019.04.016
103. Ashour T, Nakhoul G, Patil P, Funchain P, Herlitz L. Immune check point inhibitor-associated glomerulonephritis. Kidney Int Rep. (2019) 4:355–9. doi: 10.1016/j.ekir.2018.10.017
104. Kishi S, Minato M, Saijo A, Murakami N, Tamaki M, Matsuura M, et al. IgA nephropathy after nivolumab therapy for postoperative recurrence of lung squamous cell carcinoma. Intern Med. (2018) 57:1259–63. doi: 10.2169/internalmedicine.9814-17
105. Jung K, Zeng X, Bilusic M. Nivolumab-associated acute glomerulonephritis: a case report and literature review. BMC Nephrol. (2016) 17:188. doi: 10.1186/s12882-016-0408-2
106. Fadel F, El Karoui K, Knebelmann B. Anti-CTLA4 antibody-induced lupus nephritis. N Engl J Med. (2009) 361:211–2. doi: 10.1056/NEJMc0904283
107. van den Brom RR, Abdulahad WH, Rutgers A, Kroesen BJ, Roozendaal C, de Groot DJ, et al. Rapid granulomatosis with polyangiitis induced by immune checkpoint inhibition. Rheumatology. (2016) 55:1143–5. doi: 10.1093/rheumatology/kew063
108. Heo MH, Kim HK, Lee H, Ahn MJ. Antineutrophil cytoplasmic antibody-associated rapid progressive glomerulonephritis after pembrolizumab treatment in thymic epithelial tumor: a case report. J Thorac Oncol. (2017) 12:e103–5. doi: 10.1016/j.jtho.2017.03.011
109. Daxini A, Cronin K, Sreih AG. Vasculitis associated with immune checkpoint inhibitors -a systematic review. Clin Rheumatol. (2018) 37:2579–84. doi: 10.1007/s10067-018-4177-0
110. Neale TJ, Tipping PG, Carson SD, Holdsworth SR. Participation of cell-mediated immunity in deposition of fibrin in glomerulonephritis. Lancet. (1988) 2:421–4. doi: 10.1016/S0140-6736(88)90413-8
111. Cunningham MA, Huang XR, Dowling JP, Tipping PG, Holdsworth SR. Prominence of cell-mediated immunity effectors in “pauci-immune” glomerulonephritis. J Am Soc Nephrol. (1999) 10:499–506.
112. Seta N, Tajima M, Kobayashi S, Kawakami Y, Hashimoto H, Kuwana M. Autoreactive T-cell responses to myeloperoxidase in patients with antineutrophil cytoplasmic antibody-associated vasculitis and in healthy individuals. Mod Rheumatol. (2008) 18:593–600. doi: 10.3109/s10165-008-0109-1
113. Griffith ME, Coulthart A, Pusey CD. T cell responses to myeloperoxidase (MPO) and proteinase 3 (PR3) in patients with systemic vasculitis. Clin Exp Immunol. (1996) 103:253–8. doi: 10.1046/j.1365-2249.1996.d01-629.x
114. Nogueira E, Hamour S, Sawant D, Henderson S, Mansfield N, Chavele KM, et al. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol Dial Transplant. (2010) 25:2209–17. doi: 10.1093/ndt/gfp783
115. Brouwer E, Stegeman CA, Huitema MG, Limburg PC, Kallenberg CG. T cell reactivity to proteinase 3 and myeloperoxidase in patients with Wegener's granulomatosis (WG). Clin Exp Immunol. (1994) 98:448–53. doi: 10.1111/j.1365-2249.1994.tb05511.x
116. Ooi JD, Chang J, Hickey MJ, Borza DB, Fugger L, Holdsworth SR, et al. The immunodominant myeloperoxidase T-cell epitope induces local cell-mediated injury in antimyeloperoxidase glomerulonephritis. Proc Natl Acad Sci USA. (2012) 109:E2615–24. doi: 10.1073/pnas.1210147109
117. Chang J, Eggenhuizen P, O'Sullivan KM, Alikhan MA, Holdsworth SR, Ooi JD, et al. CD8+ T cells effect glomerular injury in experimental anti-myeloperoxidase GN. J Am Soc Nephrol. (2017) 28:47–55. doi: 10.1681/ASN.2015121356
118. Gan PY, Steinmetz OM, Tan DS, O'Sullivan KM, Ooi JD, Iwakura Y, et al. Th17 cells promote autoimmune anti-myeloperoxidase glomerulonephritis. J Am Soc Nephrol. (2010) 21:925–31. doi: 10.1681/ASN.2009070763
119. Summers SA, Steinmetz OM, Li M, Kausman JY, Semple T, Edgtton KL, et al. Th1 and Th17 cells induce proliferative glomerulonephritis. J Am Soc Nephrol. (2009) 20:2518–24. doi: 10.1681/ASN.2009030337
120. Gan PY, Chan A, Ooi JD, Dick J, Nagai K, O'Sullivan KM, et al. Biologicals targeting T helper cell subset differentiating cytokines are effective in the treatment of murine anti-myeloperoxidase glomerulonephritis. Kidney Int. (2019) 96:1121–33. doi: 10.1016/j.kint.2019.05.012
121. Morgan MD, Day CJ, Piper KP, Khan N, Harper L, Moss PA, et al. Patients with Wegener's granulomatosis demonstrate a relative deficiency and functional impairment of T-regulatory cells. Immunology. (2010) 130:64–73. doi: 10.1111/j.1365-2567.2009.03213.x
122. Abdulahad WH, Stegeman CA, van der Geld YM, Doornbos-van der Meer B, Limburg PC, Kallenberg CG. Functional defect of circulating regulatory CD4+ T cells in patients with Wegener's granulomatosis in remission. Arthritis Rheum. (2007) 56:2080–91. doi: 10.1002/art.22692
123. Rimbert M, Hamidou M, Braudeau C, Puechal X, Teixeira L, Caillon H, et al. Decreased numbers of blood dendritic cells and defective function of regulatory T cells in antineutrophil cytoplasmic antibody-associated vasculitis. PLoS ONE. (2011) 6:e18734. doi: 10.1371/journal.pone.0018734
124. Free ME, Bunch DO, McGregor JA, Jones BE, Berg EA, Hogan SL, et al. Patients with antineutrophil cytoplasmic antibody-associated vasculitis have defective Treg cell function exacerbated by the presence of a suppression-resistant effector cell population. Arthritis Rheum. (2013) 65:1922–33. doi: 10.1002/art.37959
Keywords: glomerulonephritis, therapeutics, ITIM, PD-1, CTLA4, renal vasculitis
Citation: Nagai K (2020) Co-inhibitory Receptor Signaling in T-Cell-Mediated Autoimmune Glomerulonephritis. Front. Med. 7:584382. doi: 10.3389/fmed.2020.584382
Received: 17 July 2020; Accepted: 22 September 2020;
Published: 04 November 2020.
Edited by:
Yuji Nozaki, Kindai University Hospital, JapanReviewed by:
Amod Ashok Sarnaik, Moffitt Cancer Center, United StatesFlorian Grahammer, University Medical Center Hamburg-Eppendorf, Germany
Copyright © 2020 Nagai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kei Nagai, a25hZ2FpQG1kLnRzdWt1YmEuYWMuanA=