 Patricia López
Patricia López Javier Rodríguez-Carrio
Javier Rodríguez-Carrio Luis Caminal-Montero2,3
Luis Caminal-Montero2,3- 1Department of Functional Biology, Immunology Area, Faculty of Medicine, University of Oviedo, Oviedo, Spain
- 2Group of Basic and Translational Research in Inflammatory Diseases, Instituto de Investigación Sanitaria del Principado de Asturias (ISPA), Oviedo, Spain
- 3Department of Internal Medicine, Hospital Universitario Central de Asturias, Oviedo, Spain
Objective: To quantify the levels of circulating exosomes derived from T-cells and monocytes and their possible associations with leukocyte subpopulations and cytokine milieu in Systemic Lupus Erythematosus (SLE).
Methods: Total circulating exosomes (CD9+-Ex) and those derived from T-cells (CD3+-Ex) and monocytes (CD14+-Ex) were quantified by flow cytometry in 82 SLE patients and 32 controls. Leukocyte subsets and serum cytokines were analyzed by flow cytometry or by immunoassays. IFN-score was evaluated by real time RT-PCR in whole blood samples from a subgroup of 73 patients and 24 controls.
Results: Activation markers (IFNR1 and BLyS) on monocytes, neutrophils and B-cells correlated inversely with circulating exosomes (CD9+-Ex, CD3+-Ex, and CD14+-Ex) in controls but directly with CD3+-Ex in patients (all p < 0.05). Although CD9+-Ex were increased in SLE, no differences were found in CD3+-Ex, supporting that exosome content accounts for this opposite role. Interestingly, CD4+CD28null cells correlated with CD3+-Ex in patients and controls, and displayed similar associations with leukocyte subsets in both groups. Additionally, CD3+-Ex correlated in patients with the expression of CD25 in CD4+CD28null cells. Furthermore, the activated status of this senescent subset was related to IFNα serum levels in controls and to IFN-score in SLE patients. Finally, patients presenting high IFN-score, in addition to elevated CD25+CD28null cells associated with the activation of myeloid cells, displayed higher levels of inflammatory cytokines and chemokines.
Conclusion: Our results support a relationship between T-cell exosomes and cellular subsets in SLE according to type I IFN-signaling, which could amplify chronic immune activation and excessive cytokine/chemokine response.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with multiple clinical manifestations and numerous cellular and molecular abnormalities in the immune system, including autoantibody production and leukocyte activation (1). Actually, SLE patients display a generalized immune stimulation characterized by the presence of over-activated and senescent lymphocytes, as well as by altered myeloid subpopulations, such as monocytes, dendritic cells (DC), and neutrophils (2, 3). Despite the unknown etiology of SLE, several lines of evidence emphasize the role of the crosstalk among different immune cells as a key mediator of the systemic inflammation and organ damage characteristic of lupus pathogenesis. Altered production of cytokines has largely been considered as the main responsible of cellular communication. However, cytokine blockade in lupus has yielded controversy results, hence suggesting the involvement of other mediators. Therefore, the role of extracellular vesicles has recently emerged as effective vehicles, along with cytokines, of intercellular signaling in physiological and pathological situations (4).
Exosomes are endosomal multivesicular bodies (30–150 nm) formed after fusion with the plasma membrane in many cell types. They can be found in several corporal fluids and carry cell-specific content of bioactive molecules, such as of proteins, lipids, and nucleic acids (5), thus acting as a “fingerprint” of the phenotypic and physiological state of the cell of origin upon exosome shedding. Therefore, cell specific exosomes could serve as a potential signature in pathological conditions. Accordingly, it has been observed that levels of circulating exosomes correlate with disease activity in lupus patients, thus representing potential biomarkers in SLE (6).
Although the biological functions of exosomes are not fully understood, it is known that exosomes can be selectively transferred to target cells to modulate many cellular processes, including immune response. Thus, after release of the exosome cargo in immune cells, both immunostimulatory and regulatory roles have been reported (7, 8). In the context of autoimmunity, and SLE in particular, extracellular vesicles could also represent a source of self-antigens (9) able to participate in the formation of immune-complexes, amplification of autoreactive T-cells activation (10, 11) and secretion of pro-inflammatory cytokines (12).
Among cytokines involved in SLE, type I IFNs plays a central role (13), since IFNα serum levels or expression of IFN-inducible genes are increased in most patients and usually correlated with disease activity and clinical manifestations (14). Binding of IFNα to the type I IFN receptor (IFNAR) on B and T cells, monocytes, macrophages, DCs, and neutrophils stimulates immunoregulatory effects in these cells (15). Thus, IFNα is able to induce the expression of other pathogenic cytokines in SLE, such as BLyS and IL-17, acting in a pathological axis that perpetuates the inflammation and disease progression (2, 16, 17).
In this scenario, the presence of “SLE-specific” exosomes derived from over-activated leukocytes may promote the vicious circle of immune dysregulation. However, the cellular source of exosomes and their immunomodulatory role in SLE are not well-understood. In the current study we quantified the circulating levels of total, T-cell, and monocyte derived exosomes and their possible associations with several leukocyte subsets usually altered in SLE. In addition, the role of IFNα and other relevant cytokines was analyzed.
Patients and Methods
Patients and Controls
Eighty-two SLE patients fulfilling the American College of Rheumatology (ACR) revised criteria for the SLE classification were sequentially recruited from the outpatient clinic of the Autoimmune Disease Unit (Hospital Universitario Central de Asturias, HUCA). Information on clinical manifestations along the disease course was obtained after a retrospective review of their clinical records, whereas parameters of disease activity (anti-dsDNA titer and SLE disease activity index, SLEDAI) and treatments received over the previous 3 months were recorded at the sampling time (Table 1). Thirty-two volunteer without any pathology or treatment were recruited as healthy controls (C). Ethical approval for this study was obtained from the Regional Ethics Committee for Clinical Research (Servicio de Salud del Principado de Asturias), according to the Declaration of Helsinki. Written informed consent was signed from all individuals prior to participation in the study.
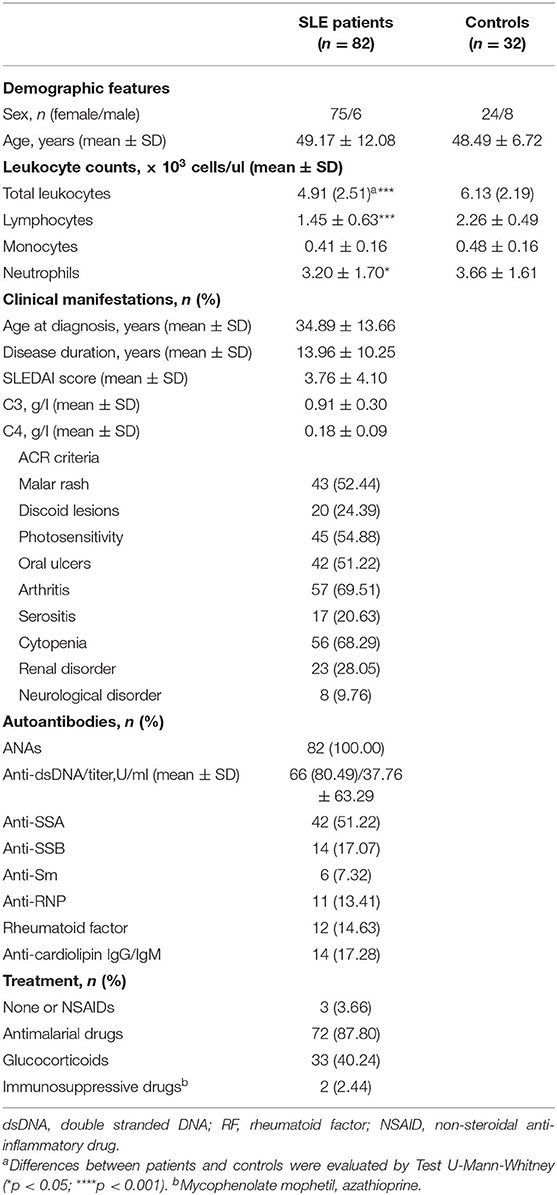
Table 1. Demographic and clinical features of SLE patients.
Exosome Isolation and Quantification by Flow Cytometry
Total and subset-specific circulating exosomes were detected by flow cytometry in serum samples from SLE patients and C by using the ExoStep™ kit (Immunostep) following the manufacturer's recommendations (18). Briefly, serum samples from patients and controls were centrifuged at 3,000 g for 15 min at room temperature (RT). Then, supernatants were incubated overnight at RT with capture beads (polystyrene micro-particles coated with antibodies against CD63 and with discrete fluorescence intensity). Later, bead-bound exosomes were washed with PBS 1% bovine serum albumin (BSA) and collected after centrifugation at 2,500 g-5 min-RT. Bead-bound exosomes were labeled by incubation (60 min, at 4°C in darkness) with primary antibodies against tetraspanins or specific lineage markers conjugated with biotin. Thus, total exosomes were identified by using anti-CD9, exosomes from T-cells by anti-CD3, and exosomes from monocytes by anti-CD14 antibodies (all from Immunostep). Also, additional tubes without exosomes were prepared for background determination. Samples were washed with PBS 1% BSA and centrifuged at 2,500 g-5 min-RT. Finally, exosomes were incubated (30 min, at 4°C in darkness) with streptavidin-PE (Immunostep), washed (2,500 g-5 min-RT) and resuspended in 350 μL of PBS. Then, 2,000 microbeads were immediately acquired using a FACS Canto II flow cytometer (BD). Bead-exosomes population was gated according to their own fluorescence on FL3 vs. FL4 channels and the quantity of exosomes was determined by the MFI (mean fluorescence intensity) in the FL2 channel. Relative Fluorescence Intensity (RFI) was calculated as MFI positive/MFI background.
Flow Cytometry Analysis of Fresh Blood Cells
Peripheral blood samples from SLE patients and controls were collected in ethylene diamine tetra-acetic acid (EDTA)-containing tubes as anticoagulant. BLyS-FITC (eBiosciences) and IFNRA1-PE (R&D Systems) expression was simultaneously quantified on monocytes, B cells, and neutrophils. Monocytes and B cells were identified by expression of CD14-APC-Cy7 (BioLegend), or CD19-CF-Blue (Immunostep), respectively, while neutrophils were identified according to their distinctive forward and side-scatter signals, as previously described (2). To quantify activated CD4+ T-cell subsets (no-Treg CD25+), blood samples were stained with anti-CD4-APC-Cy7, anti-CD25-FITC, and anti-CD127-PE-Cy7 antibodies (all from eBiosciences). Afterward, blood samples were lysed with 2 ml of BD Lysing Solution (BD Biosciences) for 5 min and washed twice with PBS. Then, cells were fixed, permeabilized and intracellularly stained with anti-FOXP3-PE (Foxp3/transcription factor staining buffer set; eBiosciences) to determine the Treg population. The amount of Treg cells was subtracted from the total CD25+CD4+ to obtain activated CD4+ T-lymphocytes. Circulating senescent T-cells (CD4+CD28null) were identified by staining with anti-CD3-PerCP-Cy5.5 (Tonbo Biosciences), anti-CD4-Pacific-Blue (Immunostep), anti-CD25-PE (Immunostep), and anti-CD28-APC-Cy7 (BD Biosciences) antibodies. Isotype fluorochrome-matched control antibodies (eBioscence, USA) were used as negative controls for the cytometry analysis. Cells were stained for 30 min at 4°C with the appropriate monoclonal antibody, washed twice and re-suspended in PBS. Acquisition of 30,000–200,000 events/tube was performed using a FACS Canto II flow cytometer (BD). The analysis was based on cells located the area of dots termed “the living region” which was defined using forward and side scatter. Samples were subsequently analyzed using FlowJo software (Scripps Research Institute). Results were expressed as the percentage of positive cells or MFI.
Cytokine Quantification
Serum samples from patients and controls were maintained at −80°C until cytokine determinations. IFNα, IL-17A, IL-10, IL-6, CCL3 (MIP-1α), and CCL5 (RANTES) concentrations were quantified by Cytometric Bead Arrays Flex Set by using a FACS Canto II flow cytometer (BD) and following the manufacturer's instructions. For IL-10 and IL-6, an Enhanced Sensitivity Flex Set was used. Finally, ELISA kits were used for the quantification of TNFα, CCL2 (MCP-1), CXCL10 (IP-10) (Mini kit, PeproTech), IFNγ (OptEIA kit, BD) and BLyS (Human BAFF Instant ELISA, eBioscience) following the manufacturer's instructions. The lower limits of detection were 1.25 pg/ml for IFNα, 0.30 pg/ml for IL-17A, 0.014 pg/ml for IL-10 and IL-6, 0.20 pg/ml for CCL3, 0.002 pg/ml for CCL5, 3.90 pg/ml for TNFα, 8.00 pg/ml for CCL2, 16.00 pg/ml for CXCL10, 0.58 pg/ml for IFNγ, and 0.13 ng/ml for BLyS.
Gene Expression Assays
Blood samples were immediately mixed with RNA Stabilization Reagent for Blood/Bone Marrow (Roche) and stored at −20°C. Next, samples were thawed at RT and mRNA was isolated using the mRNA Isolation Kit for Blood/Bone Marrow (Roche) and following the manufacturer's instructions. Reverse transcription was performed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression was evaluated in 73 SLE patients and 24 C by Real-Time PCR with pre-designed TaqMan Gene Expression Assays (Applied Biosystems, Germany) for the following interferon regulated genes (IRGs): IFI44 (interferon-induced protein 44, reference Hs00197427_m1), IFI44L (interferon induced protein 44 like, reference Hs00915292_m1), MX1 (MX dynamin like GTPase 1, reference Hs00895608_m1), and IFI6 (interferon-alpha inducible protein 6, reference Hs00242571_m1). Reactions were performed in TaqMan Gene Expression Master Mix (Applied Biosystems). Real-Time quantitative PCR was performed in an ABI Prism HT7900 instrument (Applied Biosystems) and Ct values were analyzed with the software SDS 2.3. All samples were assayed by triplicate and the average was used. Expression level was evaluated by the 2−ΔCt method, using the GAPDH gene expression (glyceraldehyde-3-phosphate dehydrogenase, reference Hs99999905_m1) as housekeeping to normalize Ct values. The expression levels were log-transformed and individual Z-scores were calculated for each gene from the distribution observed in the whole population. All Z-scores were used to determine the composite IFN-score.
Statistical Analysis
Variables were summarized as median (interquartile range, IQR) unless otherwise stated. The Kolmogorov-Smirnov test was used to assess the normal distribution of the data. Differences were assessed by Mann–Whitney U or T-tests as appropriated. Correlations were analyzed by Spearman's rank test and confirmed by multivariate regression analysis. A p < 0.05 was considered statistically significant. Data were analyzed using GraphPad Prism 8 software (GraphPad Software) and SPSS 24 statistical software package (SPSS Inc.).
Results
Exploring the Interactions Between T-Cell and Monocyte-Derived Exosomes and Leukocyte Subsets in SLE
Total exosomes (CD9+-Ex) were elevated in serum from SLE patients compared to controls and a similar trend was observed in monocyte-derived exosomes (CD14+-Ex), whereas no differences were found for those T-cell-derived (CD3+-Ex) (Figure 1A). Exosome levels were not associated with demographic or clinical parameters, and no differences were detected between active (SLEDAI≥7) and non-active patients in any subset (all p > 0.05).
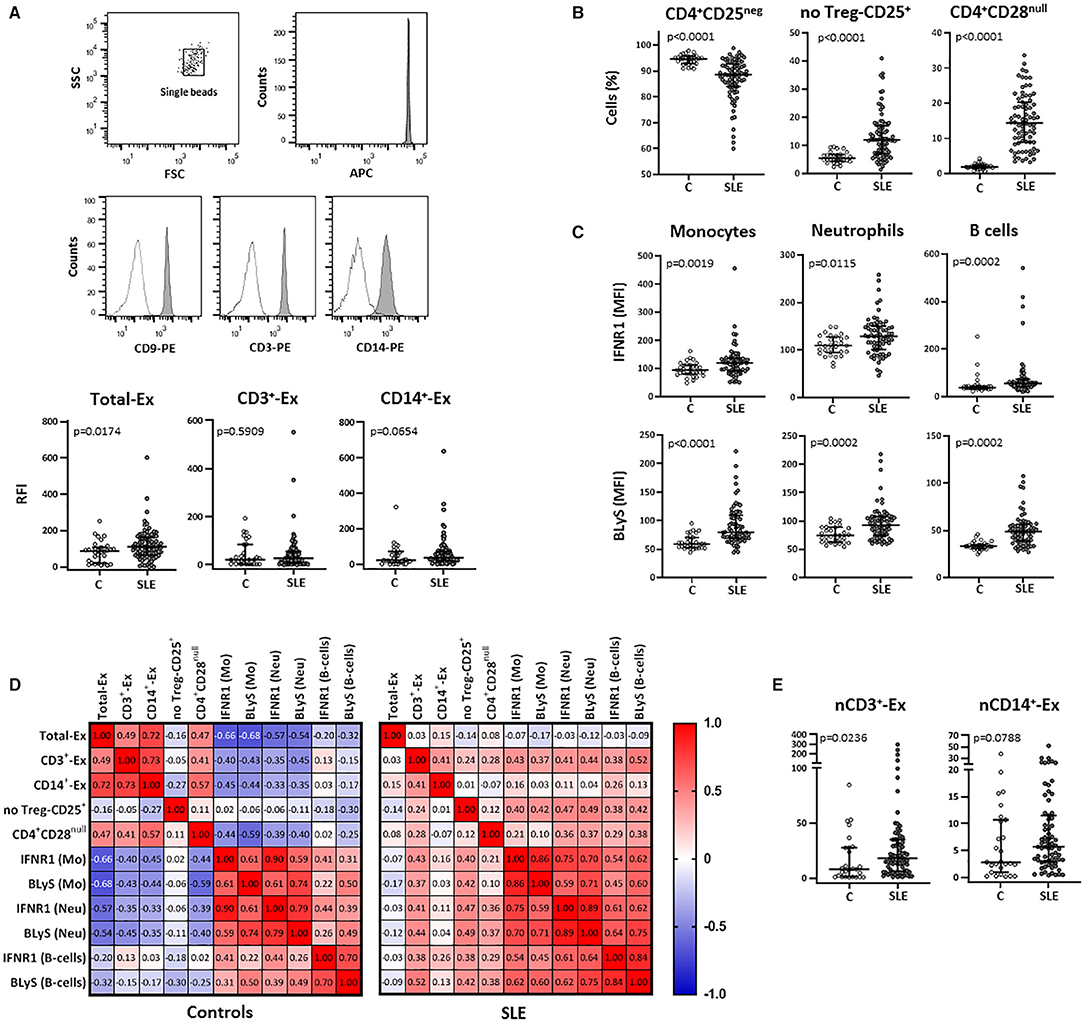
Figure 1. Total and T-cell and monocyte derived circulating exosomes and their relationship with cellular subsets in SLE patients and controls. Total exosomes (CD9+- Ex) and those derived from T-cells (CD3+-Ex) and monocytes (CD14+-Ex), along with several blood cellular subsets, were quantified by flow cytometry in serum samples from SLE patients (SLE) and healthy controls (C). (A) Exosomes immobilized on APC-beads were stained using biotinylated antibody followed by PE-conjugated streptavidin and quantified by flow cytometry. A gate containing single beads was created according to the forward and side scatter (FSC and SSC, respectively) plot, and another gate containing single beads confirmed the APC fluorescence of microbeads (representative dot plots of a SLE patient are shown). Histograms represent CD9–, CD3–, and CD14-PE expression in a SLE patient as an example (shaded) with the respective negative control (dotted line). Scatter plots display RFI values of CD9+-, CD3+-, and CD14+-exosomes from SLE and C. (B) Graphs represent percentage of CD4+CD25neg, no Treg-CD25+ and CD4+CD28null cells in SLE and C. (C) IFNR1 and BLyS levels on surface of monocytes, neutrophils and B-cells from patients and controls. (D) Correlation matrices among different types of circulating exosomes and cellular subsets in SLE and controls, where the color of the tiles is proportional to the strength of the correlation between each pair of variables and the numbers represented in the correlograms are the ρ-coefficients (Spearman tests). (E) Amounts of CD3+- and CD14+-exosomes normalized respect to the absolute T-cell (nCD3+-Ex) and monocyte (nCD14+-Ex) counts in patients and controls. Horizontal lines represent median and interquartile range of RFI values of exosomes in (A), percentage of cells in (B), MFI levels of surface markers in (C), and normalized amounts of exosomes subsets (RFI values of CD3+ or CD14+-exosomes/absolute T-cells or monocytes, respectively) in (E); statistical differences among groups were evaluated by Mann-Whitney U-test.
The analysis of blood cellular subsets revealed higher levels of activated and senescent CD4+ lymphocytes (no-Treg CD25+ and CD28null, respectively) in SLE patients compared to controls, whereas resting T-cells were decreased (Figure 1B) and an increase was observed for FOXP3+ Treg cells [1.34 (0.80) vs. 0.91 (1.27); p = 0.010]. Also, the increased expression of IFNR1 and BLyS observed in monocytes, neutrophils, and B-cells from SLE, supports a higher activation status of these leukocyte subsets (Figure 1C).
Correlation analyses between exosomes and cellular subsets revealed different, and even opposite, findings in controls and patients (Figure 1D). In healthy controls, exosome levels (CD9+-Ex, CD3+-Ex, and CD14+-Ex) were negatively associated with the activation of monocytes and neutrophils, whereas a positive correlation of CD3+-Ex with the activation markers of myeloid cells and lymphocytes (all p < 0.05) was observed in SLE. These associations were maintained after adjusting for sex, age, disease activity, and duration in a linear regression analyses model (Table 2).
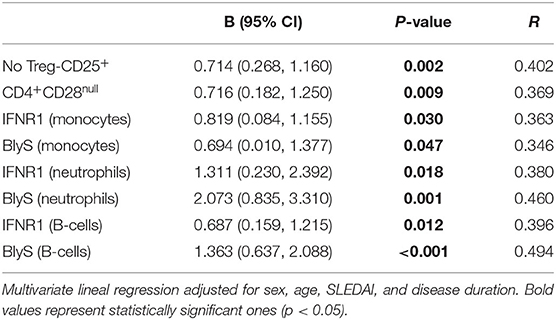
Table 2. Relationship between CD3+-Ex and cellular subsets in SLE patients.
Therefore, the activated and senescent status of T-cells present in SLE patients could generate CD3+-Ex with different effects than those derived from healthy T-cells. However, whereas circulating levels of CD3+-Ex levels were unchanged, T-cell number was significantly reduced in patients [2.22 (0.75) vs. 1.32 (0.77) × 103 cells/μl, p < 0.001]. Actually, after normalizing exosome levels with absolute T-cell and monocyte counts, a significant increase of CD3+-Ex was observed in SLE patients compared with controls (Figure 1E).
Curiously, in spite of the lack of differences between patients and controls, CD3+-Ex levels were directly correlated in both groups with CD4+CD28null cells (p < 0.05), a population strikingly increased in SLE. Moreover, CD4+CD28null cells showed a similar behavior than CD3+-Ex, displaying negative correlations with most activated cell populations in controls, but being positive in SLE patients. Furthermore, CD3+-Ex were correlated in patients with the CD25 expression in CD4+CD28null cells (ρ = 0.261, p = 0.031), independently of disease activity and duration, sex and age [B (95% CI): 1.516 (0.150, 2.882), p = 0.030]. In fact, CD25 expression was significantly increased in CD4+CD28null cells from SLE patients compared to controls (p = 0.008). Accordingly, the SLE group displayed an augmented proportion of CD4+CD28null cells expressing CD25 (CD25+CD28null) compared with controls (9.77 vs. 5.95%, p = 0.002). Interestingly, the percentage of CD25+CD28null cells in controls was related to IFNα serum levels (ρ = 0.369, p = 0.038), thus suggesting that a continuous IFN-signaling could be involved in the activation of senescent-T cells in SLE.
All these results support that qualitative rather than quantitative differences characterize SLE exosomes, especially those derived from T-cells. The activation of senescent CD4+CD28null lymphocytes and the IFNα pathway could play a role in this scenario.
Influence of IFN-Signaling on Exosomes Derived From T Cells
The expression of four IRGs was quantified in patients and controls and the composite IFN-score was calculated (Figure 2A). As expected, the IFN-score was strikingly increased in most of patients, and allowed their classification as IFNneg or IFNpos individuals (P90 in controls). The IFNpos group (72.60% of patients) was more likely to exhibit ENAs (extractable nuclear antigens) positivity, particularly SSA and RNP, lower complement levels and earlier diagnosis, whereas no differences were observed for the rest of clinical features (Table 3). Also, the quantification of circulating cytokines (Table 4) revealed increased levels of IL-6, IFNγ, ICAM-1, CXCL10, CCL2, and CCL3 in IFNpos patients compared to their IFNneg counterparts (Figure 2B), whereas IL-10, BLyS, TNFα, and IFNα amounts were elevated in patients irrespective to IFN status.
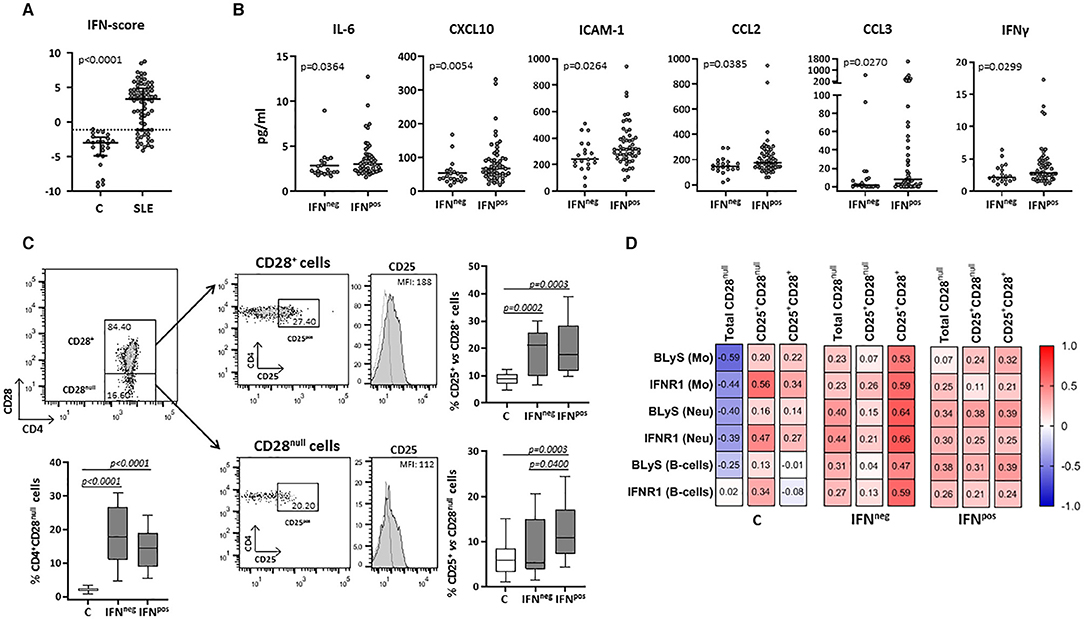
Figure 2. Circulating levels of cytokines and cellular subsets in controls and SLE patients according to their IFN-score. The expression of four IRGs was quantified in blood samples from SLE patients (SLE) and healthy controls (C). (A) Scatter plots display IFN-score in SLE and C. Dotted line represents the P90 of IFN-score in controls, employed to classify SLE patients in IFNneg or IFNpos individuals. (B) Serum levels of IL-6, CXCL10, ICAM-1, CCL2, CCL3, and IFNγ in IFNneg vs. IFNpos patients. (C) Total CD4+CD28null and activated conventional or senescent CD4+ T-cells (CD25+CD28+ and CD25+CD28null, respectively) in IFNneg and IFNpos SLE groups compared with controls. Representative dot plots of CD4 vs. CD28 expression in gated T-cells from a SLE patient are shown. Histograms represent CD25 expression in a SLE patient as an example (shaded) with the respective isotype matched control antibody (dotted line). Numbers in plots indicate the median fluorescence intensity (MFI) with the matched irrelevant control value subtracted (histograms) or percentage of gated cells (dot-plots). Horizontal lines represent median and interquartile range; statistical differences among groups were evaluated by Mann-Whitney U-test. (D) Correlations among activated subsets of CD4+ T-cells and myeloid populations in IFNneg/IFNpos patients and controls, where the color of the tiles is proportional to the strength of the correlation between each pair of variables and the numbers represented in the correlograms are the ρ-coefficients (Spearman tests).
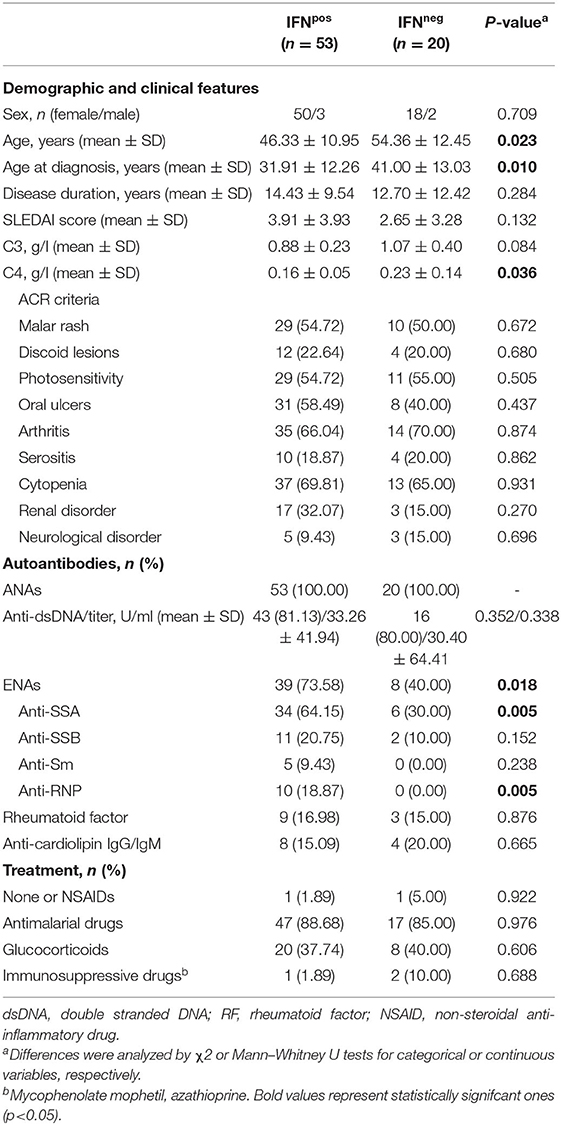
Table 3. Demographic and clinical features of IFNpos vs IFNneg SLE patients.
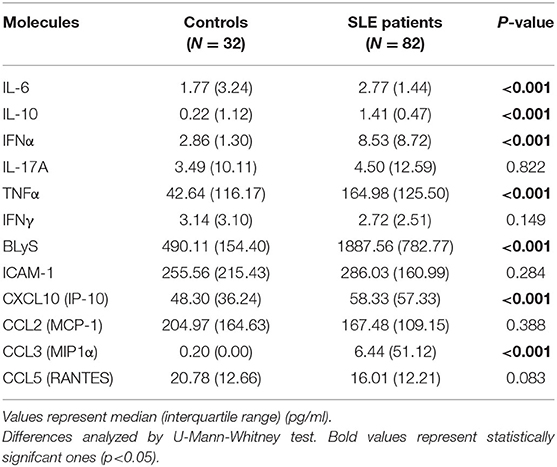
Table 4. Serum levels of inflammatory cytokines on SLE patients and healthy controls.
Total CD4+CD28null and activated conventional CD4+ T-cells (CD25+CD28+) were increased in both IFNneg and IFNpos SLE groups compared with controls (Figure 2C). However, IFNpos patients exhibited an increased proportion of CD25+CD28null cells compared with controls and the IFNneg group, thus supporting a role of IFN-signaling in the senescent-T cells activation.
Interestingly, CD25+CD28null cells were positively associated with the expression of activation markers (especially IFNR1) on monocytes, neutrophils and B-cells from controls, in spite of the opposite effect exerted by the total CD4+CD28null subset (Figure 2D). In SLE, total CD4+CD28null cells were associated with the activation of myeloid cells and lymphocytes in both IFNneg and IFNpos groups, but only in the latter the CD25+CD28null subset accounted for this phenomenon (Figure 2D). Moreover, this activated senescent subset was strongly associated with CCL3 (ρ = 0.522, p < 0.001) and IL-17 (ρ = 0.455, p = 0.001) serum levels in the IFNpos patient group.
On the other hand, in IFNpos patients, but not in those IFNneg, CD3+-Ex were positively correlated with the size of CD4+CD28null and no-Treg CD25+ subsets as well as with the CD25 expression in CD4+CD28null cells (Table 5). However, CD3+-Ex levels showed a no significant reduction in IFNpos compared with IFNneg patients [22.77 (47.03) vs. 41.73 (51.35)]. Actually, multivariate linear regression model including sex, age, disease duration, and activity showed a negative association between IFN-score and CD3+-Ex, disease duration presenting a significant positive effect (Table 6).
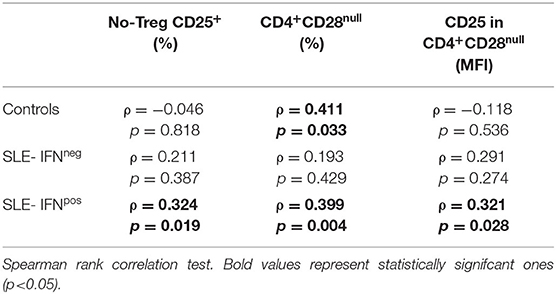
Table 5. Correlation between CD3+-Ex levels and T-cell subsets in healthy controls and SLE patients depending on type I IFN status.
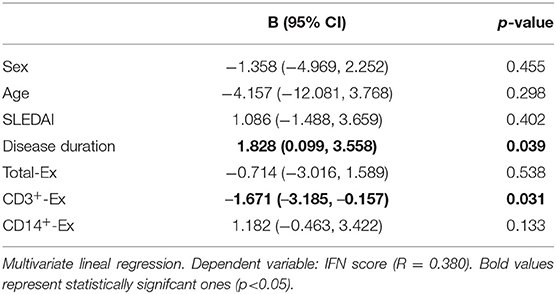
Table 6. IFN score association with CD3+-Ex levels in SLE patients.
Taken together, these results suggest again the existence of qualitative differences among circulating CD3+-Ex depending on the status of parental T-cells, and point out IFN-signaling and activation of senescent CD4+ T-cells as underlying factors of the chronic immune activation and excessive cytokine/chemokine response occurring in SLE, aberrant CD3+-Ex being significant mediators.
Discussion
A compelling body of evidence highlights exosomes as mediators of intercellular communication that could be effectively involved in the establishment, perpetuation and modulation of autoimmune diseases (19). However, almost nothing is known about the presence and possible role of exosomes derived from immune cells in these patients. To the best of our knowledge, the present work quantifies for the first time the circulating exosomes derived from T-cells and monocytes in SLE patients and evaluates their clinical relevance. The results herein reported revealed new findings that suggest the participation of exosomes, especially those derived from T-cells, in the activated status of several blood cells, with a pivotal role of senescent CD4+CD28null lymphocytes. Furthermore, present findings allow us to propose exosomes as relevant players for the effects of type-I IFN pathway in this scenario.
Our results are in line with the idea that circulating exosomes may exert different effects on the immune system depending on physiopathological conditions, ranging from suppressors in tumors to immunostimulators in chronic inflammatory disorders, as a mirror of the altered systemic immune response (6, 20–22). Thus, a relevant breakthrough of this work is the observation of opposed effects on cellular activation accounted for T-cells derived exosomes in controls and SLE patients. In physiological conditions exosomes could prevent the activation of myeloid cells, but this effect seems to be lost in SLE, where T-cells-derived exosomes were associated with the activation status of monocytes, neutrophils, and lymphocytes, markedly increased in these patients (2). An efficient uptake of T-cells exosomes by other immune cells has been reported, mainly monocytes, and DCs, that in turn can modify their normal functions (22, 23). Accordingly, circulating exosomes isolated from SLE patients have been described as promoters of an inflammatory response, being able to trigger the production of pathogenic cytokines by healthy mononuclear cells (6). Conversely, circulating exosomes from healthy controls in the present study were negatively associated or not correlated to the presence of these cellular subsets, supporting their anti-inflammatory or homeostatic profile in normal conditions (20, 21).
Despite of the T-cell activation in SLE, the counts of CD3+-Ex were similar in patients and controls, in contrast to the increased amounts of total exosomes detected in SLE. Nevertheless, this may be due, at least in part, to the lymphopenia present in SLE, since the amount of CD3+-Ex for each individual T-cell was significantly increased in patients. Anyway, given that circulating amounts of CD3+-Ex were similar in patients and controls, we can hypothesize that qualitative differences could be responsible of these opposite associations. As exosome content depends on the status of parental cells (e.g., activation, differentiation, or senescence), these results may be explained by the altered T-cell subsets usually present in lupus. Thus, the striking reduction of resting T-cells displayed by our SLE cohort, especially in lymphopenic conditions, could be responsible of a limited production of homeostatic T-cell derived exosomes, that contrast with the higher amounts of exosomes derived from senescent and activated cells. Unexpectedly, the frequency of circulating CD3+-exosomes in both control and patient groups were associated with the amount of CD4+CD28null cells, a cellular subset associated with immunosencescence. These cells were increased and exert a pathogenic role in SLE and other systemic inflammatory conditions (3, 24). The loss of CD28 expression in CD4+ T cells is a phenotypic change associated to repeated antigenic stimulation that results in drastic alterations in cell activation, proliferation, and survival (25). Therefore, CD4+CD28null cells display potent effector functions such as secretion of inflammatory cytokines, expression of cytotoxic molecules, and resistance to apoptosis and Treg suppression (26–28). But a more surprising result was that CD4+CD28null cells displayed similar opposite associations than CD3-exosomes with the leukocyte activated subsets, i.e., positive in patients and negative in controls. Therefore, we can speculate that exosomes derived from senescent T-cells could amplify their pathogenic role in SLE. These results shed light on the regulatory effects of exosomes in pathogenic conditions and suggest the projection of future in vitro studies oriented to the identification of molecular mediators revealing the presence of altered composition in SLE exosomes and to evaluate the mechanisms underlying their effects in different cellular subsets.
A remarkable finding from our study was the association between CD3-exosomes and the expression of CD25 in CD4+CD28null cells from patients. In physiological conditions, CD4+CD28null cells displayed low or no expression of CD25. It has been proposed that the lack of CD25 expression in CD4+CD28null T-cells contributes to their reduced ability to proliferate and to the prolonged survival and accumulation of CD28null T-cells in the aging immune system (29, 30). However, we not only observed a significant increase of CD28null T-cells in SLE patients, but also a higher CD25 expression in these cells, supporting that senescent lymphocytes could be activated in pathogenic situations. In fact, these cells can be activated in vitro by cytokines or through CD3-stimulation in the absence of costimulatory signals, leading to IL-2 secretion, proliferation, and increased CD25 expression (29, 31). Moreover, the upregulation of pro-inflammatory cytokines and chemokines observed in older compared to younger individuals that includes IL-1, IL-6, TNFα, CCL2, and CXCL10, could be related to the NF-κB transcriptional signature of activated senescent T-cells (32). A novel insight of this work was that CD4+CD28null cells could be also activated in vivo by type I IFNs. In the control group, serum levels of IFNα were associated with the expression of CD25 in CD4+CD28null, whereas lupus patients with high IFN-score (IFNpos) exhibit an increased proportion of activated CD25+CD28null cells, with no differences in the total CD4+CD28null subset compared to the rest of patients. These results are in line with the existing literature, since type I-IFNs are well-known promoters of memory cells and they can also provide survival signals for T cells independently of IL-2 or CD28 engagement (33–35).
All these results allow us to hypothesize that the activated status of senescent T-cells in IFNpos SLE patients may represent a new pathogenic mechanism by which type I IFNs could initiate or perpetuate the inflammatory burden. In this setting, CD3+-derived exosomes can act as enhancers of this response, given their association with the activated CD25+CD28null subset in these patients. Therefore, the inflammatory and immunosenescent milieu found in lupus, hallmarked by sustained type I IFN signaling, may lead to the generation of exosomes with a specific composition and biologic effects which in turn can amplify the abnormal immune response. In line with this, extracellular vesicles from SLE patients exhibit a higher concentration of immunoglobulins, complement components and nucleic acids (like microRNAs) compared to those from healthy controls (36, 37), and these are able to elicit a TLR-dependent inflammatory response (6, 38). The recognition of SLE-exosome components may activate downstream transcription factors, such as NF-κB and IRF3/7, thereby promoting the induction of type I IFNs and other proinflammatory cytokines in various immune cell types (39). According to this, circulating SLE-exosomes have been reported as in vitro inductors of IFNα secretion by pDCs activated through endosomal TLR7/9 (38). Then, the IFN signaling can promote the autocrine activation of DCs and their capacity to stimulate T-cells (40), allowing the subsequent release of (pathogenic) exosomes that could reach and activate immune cells at remote sites via blood circulation, thus boosting a vicious circle of immune stimulation in SLE. Taken together, our findings align with the ability of extracellular vesicles secreted by overstimulated T-cells to activate monocytes and induce inflammatory cytokines (41).
In our SLE cohort, most patients (72.6%) exhibit high IFN-score and were characterized by a poor prognostic, thus confirming previous results in systemic inflammatory conditions displaying elevated type I IFN signature (42). Besides the direct protective effect of type I IFNs against viruses, increased IFN-I signaling for a prolonged period may promote aberrant inflammatory responses during bacterial or viral infections, which can be complicated with (hyper)-cytokinemia or even a cytokine storm in severe forms (43). In the same way, in SLE and other chronic inflammatory diseases, sustained IFN-I signaling can promote aberrant inflammatory responses (42). Accordingly, IFNpos SLE patients, in addition to the activation of senescent T-cells, presented high serum levels of several inflammatory cytokines and chemokines, including IL-6, IFNγ, ICAM-1, CXCL10, CCL2, and CCL3, than their IFNneg counterparts. Moreover, in this patient group, CD25+CD28null cells were correlated with the activated state of myeloid cells and lymphocytes and with serum levels of CCL3 and IL-17.
Therefore, we can speculate that individuals with elevated CD4+CD28null cells and sustained IFN-I signaling may undergo activation of this senescent T-cell subset and secretion of inflammatory mediators, which will be a risk factor for the development of (hyper)-cytokinemia in those with genetics or environmental triggers.
In conclusion, our results draw a picture where CD3+-exosomes and activated CD4+CD28null lymphocytes could mediate a novel pathogenic mechanism by which type I IFN signaling may promote leukocyte over-activation and secretion of inflammatory mediators in SLE. Hence, interventional studies targeting the type-I IFN pathway have shown clinical benefits in immunopathologies associated to IFN-signatures (44). Therefore, we can hypothesize that the specific content of lupus T-cell derived exosomes could be partially responsible of the immunostimulatory effect underlying the leukocyte activation usually present in these patients, thus making them a promising therapeutic tool in SLE.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Regional Ethics Committee for Clinical Research (Servicio de Salud del Principado de Asturias). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
PL participated in the study conduct, cellular and cytokine quantification, data analysis/interpretation, and manuscript preparation. JR-C contributed to experimental procedures. LC-M contributed to sample collection as well as review of demographic, clinical manifestations, disease activity, and therapy of patients. AS contributed to study design/conduct, data interpretation, and manuscript preparation. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the European Union FEDER funds, Fondo de Investigación Sanitaria (FIS PI16/00113) and Plan de Ciencia, Tecnología e Innovación de Asturias (IDI-2018-000152). JR-C was a recipient of a grant from the Sara Borrell program (CD19/00120; from ISCIII).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors acknowledge to SLE patients and Asociación Lúpicos de Asturias for their continuous encouragement.
References
1. Tsokos GC. Systemic lupus erythematosus. N Engl J Med. (2011) 365:2110–21. doi: 10.1056/NEJMra1100359
2. López P, Rodríguez-Carrio J, Caminal-Montero L, Mozo L, Suárez A. A pathogenic IFNα, BLyS and IL-17 axis in Systemic Lupus Erythematosus patients. Sci Rep. (2016) 6:20651. doi: 10.1038/srep20651
3. López P, Rodríguez-Carrio J, Martínez-Zapico A, Caminal-Montero L, Suarez A. Senescent profile of angiogenic T cells from systemic lupus erythematosus patients. J Leukoc Biol. (2016) 99:405–12. doi: 10.1189/jlb.5HI0215-042R
4. Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. (2016) 164:1226–32. doi: 10.1016/j.cell.2016.01.043
5. Simpson RJ, Lim JW, Moritz RL, Mathivanan S. Exosomes: proteomic insights and diagnostic potential. Expert Rev Proteomics. (2009) 6:267–83. doi: 10.1586/epr.09.17
6. Lee JY, Park JK, Lee EY, Lee EB, Song YW. Circulating exosomes from patients with systemic lupus erythematosus induce an proinflammatory immune response. Arthritis Res Ther. (2016) 18:264. doi: 10.1186/s13075-016-1159-y
7. Greening DW, Gopal SK, Xu R, Simpson RJ, Chen W. Exosomes and their roles in immune regulation and cancer. Semin Cell Develop Biol. (2015) 40:72–81. doi: 10.1016/j.semcdb.2015.02.009
8. Lindenbergh MFS, Stoorvogel W. Antigen presentation by extracellular vesicles from professional antigen-presenting cells. Annu Rev Immunol. (2018) 36:435–59. doi: 10.1146/annurev-immunol-041015-055700
9. Buzas EI, György B, Nagy G, Falus A, Gay S. Emerging role of extracellular vesicles in inflammatory diseases. Nat Rev Rheumatol. (2014) 10:356–64. doi: 10.1038/nrrheum.2014.19
10. Sprent J. Direct stimulation of naïve T cells by antigen-presenting cell vesicles. Blood Cells Mol Dis. (2005) 35:17–20. doi: 10.1016/j.bcmd.2005.04.004
11. Obregon C, Rothen-Rutishauser B, Gitahi SK, Gehr P, Nicod LP. Exovesicles from human activated dendritic cells fuse with resting dendritic cells, allowing them to present alloantigens. Am J Pathol. (2006) 169:2127–36. doi: 10.2353/ajpath.2006.060453
12. Berckmans RJ, Nieuwland R, Kraan MC, Schaap MC, Pots D, Smeets TJ, et al. Synovial microparticles from arthritic patients modulate chemokine and cytokine release by synoviocytes. Arthritis Res Therap. (2005) 7:R536. doi: 10.1186/ar1706
13. Pascual V, Farkas L, Banchereau J. Systemic lupus erythematosus: all roads lead to type I interferons. Curr Opin Immunol. (2006) 18:676–82. doi: 10.1016/j.coi.2006.09.014
14. Dall'era MC, Cardarelli PM, Preston BT, Witte A, Davis JC. Type I interferon correlates with serological and clinical manifestations of SLE. Ann Rheum Dis. (2005) 64:1692–7. doi: 10.1136/ard.2004.033753
15. Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Bon AL, Melero I. Direct effects of Type I interferons on cells of the immune system. Clin Cancer Res. (2011) 17:2619–27. doi: 10.1158/1078-0432.CCR-10-1114
16. Axtell RC, Raman C, Steinman L. Type I interferons: beneficial in Th1 and detrimental in Th17 autoimmunity. Clin Rev Allergy Immunol. (2013) 44:114–20. doi: 10.1007/s12016-011-8296-5
17. Mathian A, Weinberg A, Gallegos M, Banchereau J, Koutouzov S. IFN-α induces early lethal lupus in preautoimmune (New Zealand Black × New Zealand White)F1 but not in BALB/c mice. J Immunol. (2005) 174:2499–506. doi: 10.4049/jimmunol.174.5.2499
18. Campos-Silva C, Suárez H, Jara-Acevedo R, Linares-Espinós E, Martinez-Piñeiro L, Yáñez-Mó M, et al. High sensitivity detection of extracellular vesicles immune-captured from urine by conventional flow cytometry. Sci Rep. (2019) 9:2042. doi: 10.1038/s41598-019-38516-8
19. Anel A, Gallego-Lleyda A, de Miguel D, Naval J, Martínez-Lostao L. Role of exosomes in the regulation of T-cell mediated immune responses and in autoimmune disease. Cells. (2019) 8:154. doi: 10.3390/cells8020154
20. Yuana Y, Sturk A, Nieuwland R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. (2013) 27:31–9. doi: 10.1016/j.blre.2012.12.002
21. Beyer C, Pisetsky DS. The role of microparticles in the pathogenesis of rheumatic diseases. Nat Rev Rheumatol. (2010) 6:21–9. doi: 10.1038/nrrheum.2009.229
22. Ventimiglia LN, Alonso MA. Biogenesis and function of T Cell-derived exosomes. Front Cell Dev Biol. (2016) 4:84. doi: 10.3389/fcell.2016.00084
23. Busch A, Quast T, Keller S, Kolanus W, Knolle P, Altevogt P, et al. Transfer of T cell surface molecules to dendritic cells upon CD4+ T cell priming involves two distinct mechanisms. J Immunol. (2008) 181:3965–73. doi: 10.4049/jimmunol.181.6.3965
24. Markovic-Plese S, Cortese I, Wandinger KP, McFarland HF, Martin R. CD4+CD28- costimulation-independent T cells in multiple sclerosis. J Clin Invest. (2001) 108:1185–94. doi: 10.1172/JCI200112516
25. Weng N-P, Akbar AN, Goronzy J. CD28(-) T cells: their role in the age-associated decline of immune function. Trends Immunol. (2009) 30:306–12. doi: 10.1016/j.it.2009.03.013
26. Thewissen M, Somers V, Hellings N, Fraussen J, Damoiseaux J, Stinissen P. CD4+CD28null T cells in autoimmune disease: pathogenic features and decreased susceptibility to immunoregulation. J Immunol. (2007) 179:6514–23. doi: 10.4049/jimmunol.179.10.6514
27. Weyand CM, Brandes JC, Schmidt D, Fulbright JW, Goronzy JJ. Functional properties of CD4+CD28– T cells in the aging immune system. Mech Ageing Dev. (1998) 102:131–47. doi: 10.1016/S0047-6374(97)00161-9
28. Kovalcsik E, Antunes RF, Baruah P, Kaski JC, Dumitriu IE. Proteasome-mediated reduction in proapoptotic molecule Bim renders CD4+CD28null T cells resistant to apoptosis in acute coronary syndrome. Circulation. (2015) 131:709–20. doi: 10.1161/CIRCULATIONAHA.114.013710
29. Park W, Weyand CM, Schmidt D, Goronzy JJ. Co-stimulatory pathways controlling activation and peripheral tolerance of human CD4+CD28- T cells. Eur J Immunol. (1997) 27:1082–90. doi: 10.1002/eji.1830270507
30. Vallejo AN, Schirmer M, Weyand CM, Goronzy JJ. Clonality and longevity of CD4+CD28null T cells are associated with defects in apoptotic pathways. J Immunol. (2000) 165:6301–7. doi: 10.4049/jimmunol.165.11.6301
31. Broux B, Mizee MR, Vanheusden M, Pol S, van der Horssen J, van Wijmeersch BV, et al. IL-15 amplifies the pathogenic properties of CD4+CD28– T cells in multiple sclerosis. J Immunol. (2015) 194:2099–109. doi: 10.4049/jimmunol.1401547
32. Bektas A, Schurman SH, Sen R, Ferrucci L. Human T cell immunosenescence and inflammation in aging. J Leukocyte Biol. (2017) 102:977–88. doi: 10.1189/jlb.3RI0716-335R
33. Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med. (1999) 189:521–30. doi: 10.1084/jem.189.3.521
34. Teijaro JR. Type I interferons in viral control and immune regulation. Curr Opin Virol. (2016) 16:31–40. doi: 10.1016/j.coviro.2016.01.001
35. Davis AM, Ramos HJ, Davis LS, Farrar JD. Cutting edge: a T-bet-independent role for IFN-alpha/beta in regulating IL-2 secretion in human CD4+ central memory T cells. J Immunol. (2008) 181:8204–8. doi: 10.4049/jimmunol.181.12.8204
36. Østergaard O, Nielsen CT, Iversen LV, Tanassi JT, Knudsen S, Jacobsen S, et al. Unique protein signature of circulating microparticles in systemic lupus erythematosus. Arthritis Rheum. (2013) 65:2680–90. doi: 10.1002/art.38065
37. Nielsen CT, Østergaard O, Stener L, Iversen LV, Truedsson L, Gullstrand B, et al. Increased IgG on cell-derived plasma microparticles in systemic lupus erythematosus is associated with autoantibodies and complement activation. Arthritis Rheum. (2012) 64:1227–36. doi: 10.1002/art.34381
38. Salvi V, Gianello V, Busatto S, Bergese P, Andreoli L, D'Oro U, et al. Exosome-delivered microRNAs promote IFN-α secretion by human plasmacytoid DCs via TLR7. JCI Insight. (2018) 3:e98204. doi: 10.1172/jci.insight.98204
39. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
40. Kurche JS, Haluszczak C, McWilliams JA, Sanchez PJ, Kedl RM. Type I IFN-dependent t cell activation is mediated by IFN-dependent dendritic cell OX40 ligand expression and is independent of T cell IFNR Expression. J Immunol. (2012) 188:585–93. doi: 10.4049/jimmunol.1102550
41. Scanu A, Molnarfi N, Brandt KJ, Gruaz L, Dayer J-M, Burger D. Stimulated T cells generate microparticles, which mimic cellular contact activation of human monocytes: differential regulation of pro- and anti-inflammatory cytokine production by high-density lipoproteins. J Leukoc Biol. (2008) 83:921–7. doi: 10.1189/jlb.0807551
42. Jesus AA, de Hou Y, Brooks S, Malle L, Biancotto A, Huang Y, et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J Clin Invest. (2020) 130:1669–82. doi: 10.1172/JCI129301
43. Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, El Jammal T, et al. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun Rev. (2020) 19:102567. doi: 10.1016/j.autrev.2020.102567
Keywords: exosomes, senescent CD4+CD28null T cells, systemic lupus erythematosus, type I interferon (IFN), cytokines
Citation: López P, Rodríguez-Carrio J, Caminal-Montero L and Suárez A (2020) Relationship Between T-Cell Exosomes and Cellular Subsets in SLE According to Type I IFN-Signaling. Front. Med. 7:604098. doi: 10.3389/fmed.2020.604098
Received: 08 September 2020; Accepted: 03 November 2020;
Published: 30 November 2020.
Edited by:
Tadej Avcin, University Medical Centre Ljubljana, SloveniaReviewed by:
Klaus Tenbrock, RWTH Aachen University, GermanyJosé Carlos Crispín, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), Mexico
Copyright © 2020 López, Rodríguez-Carrio, Caminal-Montero and Suárez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patricia López, bG9wZXpwYXRyaWNpYUB1bmlvdmkuZXM=; orcid.org/0000-0002-1843-0653