 Cécile Lambert
Cécile Lambert Jérémie Zappia
Jérémie Zappia Christelle Sanchez
Christelle Sanchez Antoine Florin
Antoine Florin Jean-Emile Dubuc
Jean-Emile Dubuc Yves Henrotin1,3
Yves Henrotin1,3- 1MusculoSKeletal Innovative Research Lab, University of Liège, Institute of Pathology, CHU Sart-Tilman, Liège, Belgium
- 2Orthopaedic Department, University Clinics St. Luc, Brussels, Belgium
- 3Physical Therapy and Rehabilitation Department, Princess Paola Hospital, Vivalia, Marche-en-Famenne, Belgium
During the osteoarthritis (OA) process, activation of immune systems, whether innate or adaptive, is strongly associated with low-grade systemic inflammation. This process is initiated and driven in the synovial membrane, especially by synovium cells, themselves previously activated by damage-associated molecular patterns (DAMPs) released during cartilage degradation. These fragments exert their biological activities through pattern recognition receptors (PRRs) that, as a consequence, induce the activation of signaling pathways and beyond the release of inflammatory mediators, the latter contributing to the vicious cycle between cartilage and synovial membrane. The primary endpoint of this review is to provide the reader with an overview of these many molecules categorized as DAMPs and the contribution of the latter to the pathophysiology of OA. We will also discuss the different strategies to control their effects. We are convinced that a better understanding of DAMPs, their receptors, and associated pathological mechanisms represents a decisive issue for degenerative joint diseases such as OA.
Introduction
Osteoarthritis (OA) is the most common joint disease affecting more than 70 million people across the United States (CDC: Arthritis: At a Glance) and Europe (1). As underlined by many authors, it has long been considered as “a wear and tear disease” of cartilage associated with age, it is in reality a complex disorder affecting the “whole joint” (2) and the pro-inflammatory pathways of immunity that can culminate in illness (3–5).
During the osteoarthritis (OA) process, activation of immune systems, whether innate or adaptive, is strongly associated with low-grade systemic inflammation (4, 6–10) (Figure 1). This process was initiated and driven in the synovial membrane, especially by damage-associated molecular patterns (DAMPs) released from the extracellular matrix (ECM) to the joint cavity during cartilage degradation (4, 11–13). Briefly, these fragments released into the synovial cavity stimulate the production and release of inflammatory mediators (cytokines, chemokines, lipid mediators, and DAMPs themselves) by the synovial cells (macrophages and fibroblasts) into the synovial fluid. These mediators, in turn, activate chondrocytes that produce metalloproteinase, resulting in a vicious cycle between cartilage and synovial membrane (12).
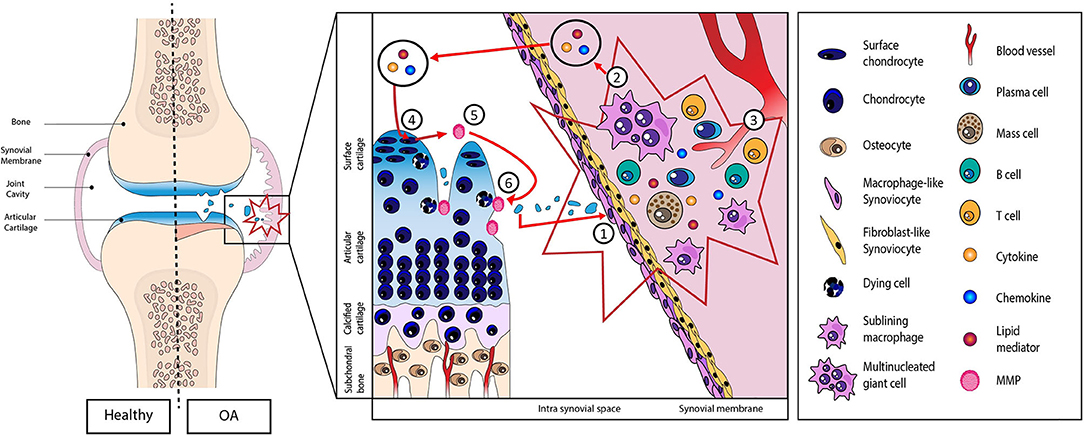
Figure 1. Schematic representation of the role of damage-associated molecular patterns (DAMPs) in the initiation and perpetuation of the low-grade systemic inflammation. (1) DAMPs released from extracellular matrix to the joint cavity during cartilage degradation. (2) Proliferation and hyperplasia of the lining cells along with inflammatory cell infiltration and (3) neoangiogenesis. (4) Production of inflammatory mediators (cytokines, chemokines, lipid mediators, and DAMPs themselves) into the synovial fluid. (5) These mediators then activate chondrocytes that in turn produce metalloproteinase resulting in a vicious cycle (6) between cartilage and synovial membrane.
DAMPs are defined as endogenous stimuli that are released either from ECM or from dying cells (14). “Intracellular” DAMPs consist of a set of immunogenic molecules released from the breakdown of necrotic and apoptotic cells such as calcium-binding protein S-100, high-mobility group box protein 1 (HMGB1), or uric acid, while “extracellular” DAMPs correspond to the ECM components (glycoproteins, proteoglycans, or glycosaminoglycans). The biological activity of these DAMPs goes through pattern recognitions receptors (PRRs) including Toll-like receptors (TLRs), NOD-like receptors (NLRs), and Receptor for Advanced Glycosylation End products (RAGEs) (15). These PRRs have been identified, notably, on the surface of immune cells, chondrocytes, osteoblasts, and synoviocytes. The binding of DAMPs to these receptors initiates downstream signaling cascades leading to the activation of several transcription factors, such as notably, the nuclear factor-κB (NF-κB), an inflammatory response key regulator (16). This activation leads to the release of various factors like catabolic factors [matrix metalloproteinase (MMP)-1,−3,−9, and−13], cytokines [tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6], chemokines [C-C motif chemokine ligand (CCL)-2,−5,−7,−8], cathepsins (B, K, and L), and complement cascade (17), factors described as essential in OA pathogenesis.
The aim of this review is to focus on the roles of DAMPs in the pathogenesis of OA. We have also researched the ways to block DAMP activity and summarized the current therapeutic approaches targeting DAMPs activity.
In this context, the literature search was performed using the Pubmed/Medline database between January 2010 and April 2020. All original papers, systematic and narrative reviews, were included. Searches were performed using the search terms “osteoarthritis,” “cartilage,” “synovium,” “DAMP,” and “immunity.” Papers published in English and reporting on the search criteria were included in this manuscript, while duplicates were removed from the selection. As a consequence, 98 articles were analyzed, and their relevant data were included in this narrative review.
Extracellular Damage-Associated Molecular Patterns From Cartilage Extracellular Matrix
Current evidences indicate that endogenous molecular products derived from ECM disruption can function as DAMPs to activate PRRs (14, 18). MMPs and/or aggrecanases [a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-4 and−5] are able to cleave a large number of ECM molecules (Table 1), leading to the exposure of cryptic epitopes and recognition with ligand receptors (18). Inflammatory mediators produced may in turn stimulate the production of cartilage-degrading enzymes and recruitment of inflammatory cells, thus establishing a vicious cycle between cartilage and synovial membrane that contributes to OA progression.
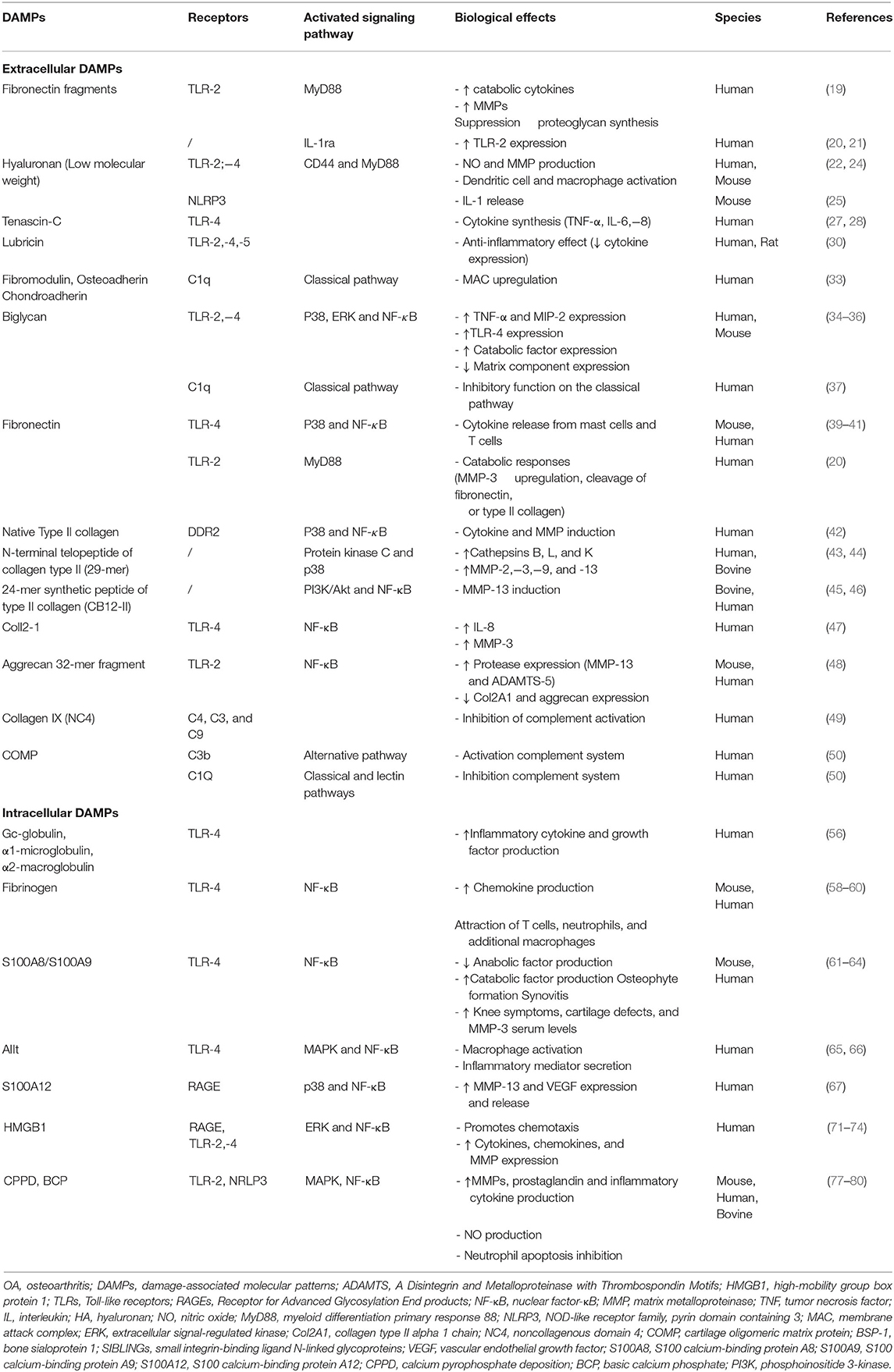
Table 1. Overview of DAMPs and their implications in the OA pathogenesis.
Homandberg and Hui (19) suggested that ECM breakdown fragments may promote inflammation and cartilage loss. So, during cartilage degradation, proteolytic cleavage of fibronectin (Fn) generates fibronectin fragments with cartilage chondrolytic activities. These are exercised through the increase of MMP expression, the suppression of proteoglycan synthesis, or the increase of cytokines. They highlighted that an amino-terminal 29-KDa fibronectin fragment (Fn-f) was able to induce, in human articular cartilage explant cultures, the production of not only pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-1α, but also MMPs, MMP-1 and−3. In human chondrocytes, Hwang et al. (20) also demonstrated that Fn-f was able to regulate cartilage catabolism through TLR-2. Furthermore, Fn-f is also able to upregulate TLR-2 expression through IL-1ra, suggesting an autocrine/paracrine regulation of IL-1 activity (21).
Hyaluronan (HA) can be described as a non-sulfated component of the ECM, commonly and abundantly found in the synovial fluid. Exogenous HA is injected in knee joints with the aim to treat joint inflammation through a mechanical effect leading to the inhibition of inflammatory pathways, stimulation of cartilage anabolism, and reduction of free radical production (22). However, HA action seems related to its molecular mass, HA of high molecular weight being anti-inflammatory and inversely for low-molecular-weight HA (23). In this context, low-molecular-weight HA, resulting from the HA degradation at sites of inflammation and tissue injury, induced nitric oxide (NO) and MMP production by mechanisms dependent on CD44 and myeloid differentiation factor 88 (MyD88) through TLR-2,−4 (22). The fragmentation products of hyaluronic acid containing sugar units of 4–16 oligosaccharide size have also been demonstrated to act as potent activators of dendritic cells and macrophages via TLR-4 (24). Yamasaki et al. (25) also demonstrated that small HA oligosaccharides activate inflammasome through NOD-like receptor family, pyrin domain containing 3 (NLRP3) and release of IL-1β.
Tenascin-C (TN-C) belongs to the ECM glycoprotein family. It is involved in tissue injury and repair. In OA, its expression is upregulated in cartilage and synovium. TN-C is also elevated in OA synovial fluid when compared to healthy one. Sofat et al. (26) demonstrated that TN-C fragments [the epidermal growth factor-like (EGF-L) and Fn type III domains 3–8 of TN-C] contributes to cartilage matrix degradation by inducing aggrecanase activity. Recently, Midwood et al. (27) also highlighted that TN-C induces cytokine production (TNF-α, IL-6, and IL-8) through the activation of TLR-4 in human macrophages and synovial fibroblasts. Zuliani-Alvarez et al. (28) have identified three distinct sites within the C-terminal fibrinogen-like globe (FBG) domain of TN-C contributing to TLR-4 activation.
Lubricin/proteoglycan 4 (PRG4) is a mucin-like glycoprotein. It is present at the surface of articular cartilage and contributes to the maintenance and integrity of the joint. Decreased expression of PRG4 is associated with OA progression (29). However, recently, Iqbal et al. (30) demonstrated in synovial cells that the full-length recombinant human PRG4 can regulate the immune response via TLRs (TLR-2,−4, and−5) and, therefore, modifies cytokine and chemokine secretion. Thus, the PRG4/TLR binding activating the NF-κB pathway is involved in maintaining the homeostatic state of the cell. However, when TLR-2,−4, or−5 is bound to another agonist, in turn, PRG4 activates inflammatory responses via an alternative pathway that does not appear to be nuclear factor NF-κB dependent (30).
Decorin, biglycan, fibromodulin, lumican, PRELP (proline-arginine-rich-end-leucine-rich repeat protein), chondroadherin, and osteoadherin are members of the small leucine-rich repeat protein (SLRP) family, as reviewed by Zappia et al. (31). Fibromodulin is a keratan sulfate proteoglycan found in cartilage and tendon. Sjöberg et al. (32) showed that fibromodulin triggers complement activation. Sjöberg et al. (33) revealed that fibromodulin upregulated the membrane attack complex (MAC) from human OA sera. In addition, these authors also demonstrated that osteoadherin and chondroadherin, like fibromodulin binds C1q and activates classical pathway (33). In macrophages, biglycan, a small leucine-rich proteoglycan, has been demonstrated to act as an endogenous ligand of TLR-4 and TLR-2. This binding results in a rapid activation of p38, extracellular signal-regulated kinase (ERK), and NF-κB and, subsequently, the stimulation of TNF-α and macrophage inflammatory protein-2 (MIP-2) expression (34). Barreto et al. (35) also demonstrated that soluble biglycan is commonly detected in knee synovial fluid of patients with advanced knee OA or rheumatoid arthritis (RA). Soluble biglycan upregulates TLR-4 expression in human OA chondrocytes, increases both expression and concentrations of catabolic factors (ADAMTS-4, ADAMTS-5, MMPs, NO, cathepsin K, IL-6, and IL-8), and decreases the expression of matrix components (collagen type II, aggrecan), globally resulting in net loss of cartilage (35). Recently, Avenoso et al. (36) also reported that human chondrocytes treated with biglycan produces several inflammatory mediators (IL-1β, IL-6, MMP-13, and IL-17) and activates NF-κB and TLR-4 (36). Conversely, biglycan and decorin can also bind to C1q and then inhibit the classical pathway (37).
Fn, whose fragments were found increased in OA cartilage and synovial fluid, was also identified as an activator of TLR (38). Two Fn domains have been identified as TLR activators: the extra Type III domain and FnEDA. These domains stimulate TLR-4-dependent cytokine release from mast cells and T cells (39, 40). Kelsh et al. (41) also identified NF-κB and p38 signaling pathways as transducers of Fn-f/TLR signals. Hwang et al. (20) demonstrated in human chondrocytes the probable involvement of MyD88-dependent TLR-2 signaling pathway in Fn fragment release and mediated cartilage catabolic responses.
Type II collagen-derived peptides also seem to act as potent activators of innate immunity. In human chondrocytes, Klatt et al. (42) have observed the collagen II-dependent induction of both cytokines (IL-1β,−6, and−8) and MMPs (MMP-1,−3,−13, and−14) involved in p38 and NF-κB signaling. In human articular chondrocytes, an N-terminal fragment of type II collagen (29-mer fragment) stimulated the production of cathepsins B, L, and K through the activation of protein kinase C and p38 mitogen-activated protein kinase (MAPK) (43). Fichter et al. (44) demonstrated that mRNA and protein levels of MMP-2,−3,−9, and−13 were also upregulated by this 29-mer peptide. In a cartilage explant culture model, Tchetina et al. (45) reported that a 24-mer synthetic peptide of type II collagen (named CB12-II) was able to stimulate type II collagen cleavage through MMP-13 induction. Subsequently, in a study conducted in human OA chondrocytes, Yasuda (46) demonstrated that CB12-II stimulated phosphoinositide 3-kinase (PI3K)/Akt, leading to NF-κB activation. Recently, our team demonstrated that Coll2-1, a synthetic peptide located in the triple helical part of the type II collagen molecule and currently used as a biomarker of cartilage degradation, activates synoviocytes to produce IL-8 and chondrocytes to produce MMP-3. We also demonstrated that these Coll2-1 effects were mediated through TLR-4 and NF-κB signaling pathway activation (47).
Lees et al. (48) also examined the bio-activity of an aggrecan 32-mer fragment. They reported that it increased MMP-13 and ADAMTS-5 mRNA expression and decreased Col2A1 and aggrecan mRNA through TLR-2- and NF-κB-dependent signaling.
Type IX Collagen is located at the surface of fibrils formed by collagen II, playing roles in tissue stability and integrity. Collagen IX cleavage and loss of the N-terminal non-collagenous domain 4 (NC4) precede major damage of collagen II fibrils and can therefore be considered as key early steps in cartilage degradation. Kalchishkova et al. (49) showed that NC4 is able to bind C4, C3, and C9 and to directly inhibit C9 polymerization and MAC formation and can therefore be considered as a complement system inhibitor. NC4 interactions with fibromodulin and osteoadherin also inhibited complement activation by these proteins (49).
The cartilage oligomeric matrix protein (COMP), detected with abnormally high levels in OA synovial fluid, can also fix the complement system via C3b and C9 through an alternative complement pathway. COMP is also able to inhibit classical and lectin pathways through its interaction with C1q and mannose-binding lectin (50). The same observation is reported with cartilage fragments decorin and biglycan (51).
The bone sialoprotein I (BSP-1) is described as a non-collagenous ECM protein, member of the small integrin-binding ligand N-linked glycoproteins (SIBLINGs) family, expressed by many cell types among which are osteoblasts, osteoclasts, chondrocytes, synoviocytes, macrophages, and activated T cells (52). BSP-1 levels are increased in OA joint (synovial fluid and articular cartilage) compared to healthy controls, and these levels are correlated with the severity of joint lesion and the inflammatory status of patients (53). Furthermore, elevated levels of BSP-1 activate both an increase of MMP-13 expression and NF-κB activation and, consequently, the increased production of cytokines and chemokines, leading to NO, prostaglandin E2 (PGE2), IL-6, and IL-8 production and imbalance the cartilage homeostasis (54). Moreover, BSP regulates T cell development, increases Th1 differentiation, suppresses Th2, and supports Th17 differentiation. Tardelli et al. (55) also demonstrated that BSP-1 has a key role not only in monocyte chemotaxis and macrophage differentiation but also in 4 macrophage proliferation.
Intracellular Damage-Associated Molecular Patterns
Plasma Proteins
Sohn et al. (56) have recently identified by mass spectrometry in synovial fluid three plasma proteins of interest: Gc-globulin (vitamin D-binding protein), α1-microglobulin, and α2-macroglobulin. They showed that these plasma proteins induced TLR-4-dependent production of a large number of inflammatory cytokines and growth factors like IL-1β, IL-6, TNF-α, and vascular endothelial growth factor (VEGF). Fibrinogen, also found with increased levels in OA synovial fluid (57) and whose amount of fibrin deposited in the synovial membrane positively correlates with the severity of OA, is able to stimulate the production of chemokines [IL-8, monocyte chemoattractant protein (MCP)-1, …] by macrophages in a TLR-4-dependent manner, promoting attraction of T cells, neutrophils, and additional macrophages (58–60).
Alarmins
Large amounts of S100A8 and its binding partner S100A9 are released by neutrophils, monocytes, and activated macrophages. This heterodimer is highly expressed by synovial tissue in experimental OA models and involved in synovitis and cartilage destruction. Furthermore, high levels may predict joint destruction in humans (61). Recently, in human OA tissue, Schelbergen et al. (62) also demonstrated that S100A8/S100A9 levels were closely associated with cartilage loss and that they stimulate chondrocytes to produce more MMPs and cytokines (catabolic factors) but less type II collagen and aggrecan (anabolic factors). This effect was triggered by TLR-4. These authors also highlighted the role of S100A8/S100A9 in osteophyte formation and synovial activation in collagenase-induced OA and destabilized medial meniscus OA (62). In a study conducted in patients with knee OA, Ruan et al. (63) also demonstrated the association between serum levels S100A8/S100A9 and increased knee symptoms, cartilage defects, and MMP-3 serum levels. Finally, the canonical Wnt signaling pathway plays a key role in S100A8/S100A9 complex activity (64).
S100A10 forms with annexin II, a heterotetrameric complex called AIIt. This last activates human macrophages, which in turn secretes a number of inflammatory mediators including TNF via TLR-4 (65). Moreover, Song et al. (66) also demonstrated that the production of cytokines (TNF, IL-1β, and IL-10) in human chondrocytes was dependent on S100A10 through MAPK and NF-κB pathways.
Recently, S100A12 expression was found to be increased in OA cartilage and to contribute to the development of OA through an increase of MMP-13 and VEGF expression resulting from p38 MAPK and NF-κB pathway activation (67). Wang et al. (68) has also demonstrated that S100A12 levels in synovial fluid may correlate to clinical severity of patients with primary knee OA. In OA synovial fluid, S100A12 is significantly overexpressed, and Meijer et al. (69) highlighted this role in the innate and acquired inflammatory responses. This role in this innate immunity would be linked to RAGE receptors (70).
HMGB1 is released by necrotic cells or secreted by macrophages and other myeloid cells in response to inflammatory cytokines (IL-1β and TNF). Magna et al. (71) highlighted its role as alarmin binding to a lot of receptors, cytokines, and chemokines to stimulate the innate immune system. Since then, through cytokine production via TLR-4, HMGB1 promotes chemotaxis. HMGB1 was found overexpressed in the synovial fluid and cartilage of OA patients (72, 73). Thus, several authors reported that HMGB1 and RAGE are expressed in OA cartilage, and the activation of OA chondrocytes triggers ERK and NF-κB phosphorylation as well as MMP expression. García-Arnandis et al. (74) also reported that in OA synoviocytes, HMGB1 cooperates with IL-1β to amplify the inflammatory response resulting in the production of cytokines, chemokines, and MMPs. It can also trigger and prolong inflammatory responses via TLR-2,−4 but also RAGE.
Crystals
Microcrystals associated with joint diseases trigger inflammation and beyond innate immunity responses through both inflammasome-dependent and inflammasome-independent pathways (75, 76). Rosenthal (77) highlighted that calcium-containing crystals [calcium pyrophosphate dehydrate (CPPD) and basic calcium phosphate (BCP)] contribute to OA pathogenesis. Thus, these crystals exert direct effects both on synoviocytes and chondrocytes through the production of MMPs, prostaglandins, and inflammatory cytokines and this, via NF-κB, MAPK signals, and NO-dependent pathways. Furthermore, these crystals, combined with uric acid presence, are also able to interact with NLRP3 (78, 79) and subsequent IL-1β and IL-18 activation. Liu-Bryan et al. (80) showed also that CPPD crystals induced NO production in a TLR-2-dependent manner. Rosenthal (77) also report that these calcium-containing crystals directly affect inflammatory cells. For example, CPPD crystals can inhibit neutrophil apoptosis and extend the inflammatory response.
Cellular Receptors Involved in Damage-Associated Molecular Patterns Activity
DAMPs exert their biological activities through receptors TLR, NLR, and RAGE. Actually, 10 functional TLRs were identified in humans numbered TLR1–10. TLR-1,−2,−4,−5,−6, and−10 are located at the cell surface, while TLR-3,−7,−8, and−9 are present at the endolysosomal membrane (81). The signaling pathways activated by TLR involve the recruitment of adapter proteins such as MyD88, TIR domain-containing adaptor-inducing interferon (TRIF), TRIF-related adaptor molecule (TRAM), MyD88-adaptor like (Mal), and the activation of nuclear factors among which NF-κB. TLR also initiates distinct parallel signaling pathways leading to MAPK and PI3K activation (82). These latter regulate the transcription, mRNA stability, and translation of pro-inflammatory cytokine genes (TNF-α, IL-1β, or IL-6) and cell membrane-bound co-stimulatory molecules [intercellular adhesion molecule (ICAM)-1]. TLR-2 and−4 play a key role in OA pathogenesis since their expressions were demonstrated to be increased particularly at sites of cartilage lesions and inflammatory synovial membranes (83, 84). TLR-4 is expressed by numerous cell types in the joint including immune cells, chondrocytes, osteoblasts, and synoviocytes (83). Activation of TLR-4 leads to upregulation of IL-1β, MMP expression, NO release, and PGE2 synthesis, as well as downregulation of aggrecan core protein and type II collagen synthsesis (84, 85). Recently, comparing human cartilage from carpometacarpal (CMC)-I and knee joints, Barreto et al. (86) have observed that TLRs, and specially TLR-4, are differentially expressed depending on cartilage origin. Soluble forms of TLR-2 and−4 were also detected in the OA synovial fluid with sTLR-4 being elevated in OA knee comparing to healthy knee. Studies also highlighted that TLR1–7 and−9 expression was upregulated in the synovium of OA patients. Increased concentrations of several DAMPs (Fn, HA, Tn-C, PRG4, biglycan, or S100 family) are found in the OA synovial joint fluids and tissues and are able to activate TLRs; among them, Fn, HA, Tn-C, PRG4, biglycan, or S100 family.
NLRs are intracellular sensors of pathogen-associated or endogenous danger-associated molecular patterns (87). NLR system counts 22 cytoplasmic proteins including the nucleotide-binding oligomerization domains (NOD) and Nacht domain-containing, leucine-rich repeat-containing and pyrin domain-containing protein (NALP) subfamilies. The best characterized NLR is NLRP3, highly expressed in macrophages, chondrocytes, synoviocytes, and osteoblasts (76). Once activated, NLRP3 forms an oligomer able to interact with adapter proteins, C-terminal caspase recruitment domain (ASC), and Cardinal, creating a complex able to recruit procaspase-1. In turn, it is activated and the result is a multimeric structure named “the inflammasome,” which is capable of inducing maturation and secretion of pro-inflammatory cytokines (such as IL-1β, IL-1α, IL-18) (88, 89). In OA, NLRP3 has been associated with crystal-induced inflammation triggered by uric acid, calcium pyrophosphate, and hydroxyapatite crystals (76). These microcrystals are interpreted as DAMPs by the innate immune system and cause inflammation (75).
RAGE, a transmembrane receptor, which belongs to the immunoglobulin gene superfamily (90), is also bound by DAMPs. RAGE is composed of three distinct regions including an extracellular region responsible for ligand interaction through its V domain, a transmembrane domain, and a cytoplasmic domain responsible for downstream signaling. Activation of RAGE leads to the activation of NF-κB and MAPK pathways, which themselves induce the expression of pro-inflammatory and catabolic genes. Initially identified as a receptor for advanced glycation end-products (AGEs), it can also be bound by several DAMPs including HMGB1, S100 proteins, or amyloid-β protein (90, 91).
Damage-Associated Molecular Patterns, Perspectives, and Target Therapeutics
Several strategies have been suggested especially to control TLR-4 signaling. TLR-4 signaling activities may be downregulated by agonist blockers, activators of antagonist pathways, or new molecules. Among the agonist blockers, high-molecular-weight hyaluronic acid acts as a dressing blocking TLR access to short HA oligosaccharides (HA 4-mers) (92). Another agonist is the blocking peptide, Pep-1. The latter, a 12-mer peptide, inhibits low-molecular-weight HA binding to TLR-4. In a mouse chondrocyte model, Campo et al. (93) hypothesize that hydrophobic and/or polar residues of Pep-1 function as primary binding sites to HA, therefore reducing its binding to TLR-4 and subsequently the pro-inflammatory responses associated with TLR-4 activation.
Another strategy is the activation of antagonist pathways. Among these, peroxisome proliferator-activated receptor γ (PPARγ), PGD2, vasoactive intestinal peptide (VIP), adenosine 2A receptor (A2AR), and bone morphogenic protein 7 (BMP-7) are reported to be the most promising targets. PPARγ has been well characterized as intracellular receptor and transcription factor with anti-inflammatory functions in cartilage. In this context, molecules such as rosiglitazone and pioglitazone, defined as PPARγ agonists, have been proposed to block TLR-4 signaling pathway. Thus, the stimulation of human chondrocytes and synovial fibroblasts by rosiglitazone inhibits TLR4 activation, leading to inhibition of TLR-4-induced catabolism and inflammation mediated by serum amyloid A. Serum amyloid proteins are major acute-phase proteins, detected in OA serum and able to trigger via TLR-2 and−4 stimulating cytokines (IL-6, IL-8, CXCL-1) and metalloproteinase expression (94). Pioglitazone inhibits TLR-4-mediated effects of AGEs including the induction of cyclooxygenase (Cox-2), HMGB1, IL-6, and MMP-13 (95). Besides PPARγ, PGD2 is another candidate pathway and innate immune inhibitor. It inhibits PGE2-dependent induction of TLR-4 and, subsequently, the IL-6 synthesis by chondrocytes (96). Finally, VIP, a neuropeptide produced by immune cells, is also able to inhibit in OA synoviocytes TLR-4-mediated effects including pro-inflammatory responses and TLR-4 expression (97).
Among the new compounds developed to target TLR-4 in joint tissues, we can cite the promising 6-Shogoal that was demonstrated to reduce both TLR-4-mediated innate immune responses and the catabolic TLR-4 signaling pathway in mouse and human chondrocytes (98).
Among the other receptors implicated in innate immunity, the TLR-2 is another potential therapeutic target. In the collagen-induced arthritis model in mice, TLR-2 monoclonal antibody (mAb) reduced the pro-inflammatory cytokine production (IL-12 and TNF-α) as well as the development of clinical parameters (99). Alquraini et al. (100) also evaluated the binding of PRG4 with TLR-2 and−4. It appears that PRG4 binds to these two receptors, highlighting an anti-inflammatory role for PRG4 in OA synovial fluid. With promising in vivo effects, we can also cite RAGE and its soluble receptor, sRAGE. This last acts as a competitive inhibitor of RAGE, inhibiting downstream signaling and integrin binding (101).
Complement system can also be a therapeutic target. So, eculizumab, a humanized monoclonal antibody, is an inhibitor of terminal complement pathway (102). It binds specifically to the complement C5 protein, inhibiting the terminal complex, MAC. The effects of methylprednisolone on complement activation in patients undergoing total knee arthroplasty are currently clinically evaluated (ClinicalTrials.Gov Identifier: NCT02332616).
Another approach is to block the biological activity of DAMPs using a specific ligand. Promising examples are found in the literature. In mouse models, blockage of the pro-inflammatory effects of S100A8/A9 using an anti-carboxylate glycan antibody has also been concluding (12). Neutralizing HMGB1 antibodies or truncated HMGB1-derived A-box proteins are currently evaluated in collagen-induced arthritis rodent models (103). Targeting NLRP3 also looks promising (76). MCC950, a small-molecule chemical inhibitor, selectively inhibits activation of NLRP3 and IL-1β production by preventing NLRP3-induced ASC oligomerization (104). Finally, within our research unit, we demonstrate that Coll2-1, a synthetic peptide, is an actor of synovitis (47). Neutralized Coll2-1 with a humanized mAb may also represent an original approach in the control of OA progression.
In addition to the therapeutic aspect, the question arises as to the clinical utility of DAMPs. A lot of authors suggest the possibility that these DAMPs could be used as diagnostic and prognostic biomarkers of OA. Thus, soluble biglycan in inflammatory renal diseases, HMGB1 in systemic lupus erythematosus, or S100 proteins in several inflammatory conditions are some examples (105, 106).
Conclusion
Numerous pieces of evidence highlight the close link between immune response and the inflammation in OA process. The DAMPs are key actors. The list of these is constantly growing and represents interesting targets for future immunotherapy by blocking DAMP activities or their receptors. A better of understanding of DAMPs, their receptors, and associated pathological mechanisms represents an issue for degenerative joint diseases such as OA.
Author Contributions
CL, JZ, and YH contributed to drafting the manuscript. CL, CS, AF, and YH contributed to revising the manuscript content. CL, JZ, CS, AF, J-ED, and YH contributed to approving the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
OA, osteoarthritis; DAMPs, damage-associated molecular patterns; ECM, extracellular matrix; HMGB1, high-mobility group box protein 1; PRRs, pattern recognition receptors; TLRs, Toll-like receptors; NLRs, NOD-like receptors; RAGEs, Receptor for Advanced Glycosylation End products; NF-κB, nuclear factor-κB; MMP, matrix metalloproteinase; TNF, tumor necrosis factor; CCL, C-C motif chemokine ligand; ADAMTS, A Disintegrin And Metalloproteinase with Thrombospondin Motifs; Fn, fibronectin; IL, interleukin; HA, hyaluronan; NO, nitric oxide; MyD88, myeloid differentiation primary response 88; NLRP3, NOD-like receptor family, pyrin domain containing 3; TN-C, tenascin-C; EGF-L, epidermal growth factor-like; FBG, fibrinogen-like globe; PRG4, lubricin/proteoglycan 4; PRELP, proline-arginine-rich-end-leucine-rich repeat protein; SLRP, small leucine-rich repeat protein; MAC, membrane attack complex; ERK, extracellular signal-regulated kinase; RA, rheumatoid arthritis; FnEDA, fibronectin extra domain A isoform; Col2A1, collagen type II alpha 1 chain; NC4, non-collagenous domain 4; COMP, cartilage oligomeric matrix protein; BSP-1, bone sialoprotein 1; SIBLINGs, small integrin-binding ligand N-linked glycoproteins; PGE2, prostaglandin E2; VEGF, vascular endothelial growth factor; MCP-1, monocyte chemoattractant protein-1; S100A8, S100 calcium-binding protein A8; S100A9, S100 calcium-binding protein A9; S100A12, S100 calcium-binding protein A12; CPPD, calcium pyrophosphate deposition; BCP, basic calcium phosphate; TRIF, TIR domain-containing adaptor-inducing interferon; TRAM, TRIF-related adaptor molecule; Mal, MyD88-adaptor like; PI3K, phosphoinositide 3-kinase; ICAM-1, intercellular adhesion molecule 1; CMC-I, carpometacarpal-I; NODs, nucleotide-binding oligomerization domains; NALP, Nacht domain-containing, leucine-rich repeat-containing and pyrin domain-containing protein; ASC, C-terminal caspase recruitment domain; AGEs, advanced glycation end-products; PPARγ, peroxisome proliferator-activated receptor γ; PGD2, prostaglandin D2; CXCL-1, chemokine (C-X-C motif) ligand 1; Cox-2, cyclooxygenase 2; mAb, monoclonal antibody; sRAGE, soluble RAGE.
References
1. Kingsbury SR, Gross HJ, Isherwood G, Conaghan PG. Osteoarthritis in Europe: impact on health status, work productivity and use of pharmacotherapies in five European countries. Rheumatol. (2014) 53:937–47. doi: 10.1093/rheumatology/ket463
2. Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. (2012) 64:1697–707. doi: 10.1002/art.34453
3. Orlowsky EW, Kraus VB. The role of innate immunity in osteoarthritis: when our first line of defense goes on the offensive. J Rheumatol. (2015) 42:363–71. doi: 10.3899/jrheum.140382
4. Lopes EBP, Filiberti A, Husain SA, Humphrey MB. Immune contributions to osteoarthritis. Curr Osteoporos Rep. (2017) 15:593–600. doi: 10.1007/s11914-017-0411-y
5. Kraus VB, Blanco FJ, Englund M, Karsdal MA, Lohmander LS. Call for standardized definitions of osteoarthritis and risk stratification for clinical trials and clinical use. Osteoarthr Cartil. (2015) 23:1233–41. doi: 10.1016/j.joca.2015.03.036
6. de Lange-Brokaar BJ, Ioan-Facsinay A, van Osch GJ, Zuurmond AM, Schoones J, Toes RE, Huizinga TW, et al. Synovial inflammation, immune cells and their cytokines in osteoarthritis: a review. Osteoarthr Cartil. (2012) 20:1484–99. doi: 10.1016/j.joca.2012.08.027
7. Liu-Bryan R. Synovium and the innate inflammatory network in osteoarthritis progression. Curr Rheumatol Rep. (2013) 15:323. doi: 10.1007/s11926-013-0323-5
8. Scanzello CR, Goldring SR. The role of synovitis in osteoarthritis pathogenesis. Bone. (2012) 51:249–57. doi: 10.1016/j.bone.2012.02.012
9. Wang H, Wang Q, Yang M, Yang L, Wang W, Ding H, et al. Histomorphology and innate immunity during the progression of osteoarthritis: does synovitis affect cartilage degradation? J Cell Physiol. (2018) 233:1342–58. doi: 10.1002/jcp.26011
10. Robinson WH, Lepus CM, Wang Q, Raghu H, Mao R, Lindstrom TM, et al. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat Rev Rheumatol. (2016) 12:580–92. doi: 10.1038/nrrheum.2016.136
11. Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. (2007) 213:626–34. doi: 10.1002/jcp.21258
12. Nefla M, Holzinger D, Berenbaum F, Jacques C. The danger from within: alarmins in arthritis. Nat Rev Rheumatol. (2016) 12:669–83. doi: 10.1038/nrrheum.2016.162
13. Millerand M, Berenbaum F, Jacques C. Danger signals and inflammaging in osteoarthritis. Clin Exp Rheumatol. (2019) 37:48–56.
14. Frevert CW, Felgenhauer J, Wygrecka M, Nastase M V, Schaefer L. Danger-associated molecular patterns derived from the extracellular matrix provide temporal control of innate immunity. J Histochem Cytochem. (2017) 66:213–27. doi: 10.1369/0022155417740880
15. Chung HY, Kim DH, Lee EK, Chung KW, Chung S, Lee B, et al. Redefining chronic inflammation in aging and age-related diseases: proposal of the senoinflammation concept. Aging Dis. (2019) 10:367–82. doi: 10.14336/AD.2018.0324
16. O'Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. (2013) 13:453–60. doi: 10.1038/nri3446
17. Rahmati M, Mobasheri A, Mozafari M. Inflammatory mediators in osteoarthritis: a critical review of the state-of-the-art, current prospects, and future challenges. Bone. (2016) 85:81–90. doi: 10.1016/j.bone.2016.01.019
18. Sofat N. Analysing the role of endogenous matrix molecules in the development of osteoarthritis. Int J Exp Pathol. (2009) 90:463–79. doi: 10.1111/j.1365-2613.2009.00676.x
19. Homandberg GA, Hui F. Association of proteoglycan degradation with catabolic cytokine and stromelysin release from cartilage cultured with fibronectin fragments. Arch Biochem Biophys. (1996) 334:325–31. doi: 10.1006/abbi.1996.0461
20. Hwang HS, Park SJ, Cheon EJ, Lee MH, Kim HA. Fibronectin fragment-induced expression of matrix metalloproteinases is mediated by MyD88-dependent TLR-2 signaling pathway in human chondrocytes. Arthritis Res Ther. (2015) 17:320. doi: 10.1186/s13075-015-0833-9
21. Su SL, Tsai CD, Lee CH, Salter DM, Lee HS. Expression and regulation of Toll-like receptor 2 by IL-1beta and fibronectin fragments in human articular chondrocytes. Osteoarthr Cartil. (2005) 13:879–86. doi: 10.1016/j.joca.2005.04.017
22. Avenoso A, D'Ascola A, Scuruchi M, Mandraffino G, Calatroni A, Saitta A, et al. Hyaluronan in experimental injured/inflamed cartilage: in vivo studies. Life Sci. (2018) 193:132–40. doi: 10.1016/j.lfs.2017.11.006
23. Marcellin E, Steen JA, Nielsen LK. Insight into hyaluronic acid molecular weight control. Appl Microbiol Biotechnol. (2014) 98:6947–56. doi: 10.1007/s00253-014-5853-x
24. Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. (2002) 195:99–111. doi: 10.1084/jem.20001858
25. Yamasaki K, Muto J, Taylor KR, Cogen AL, Audish D, Bertin J, et al. NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J Biol Chem. (2009) 284:12762–71. doi: 10.1074/jbc.M806084200
26. Sofat N, Robertson SD, Hermansson M, Jones J, Mitchell P, Wait R. Tenascin-C fragments are endogenous inducers of cartilage matrix degradation. Rheumatol Int. (2012) 32:2809–17. doi: 10.1007/s00296-011-2067-8
27. Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. (2009) 15:774–80. doi: 10.1038/nm.1987
28. Zuliani-Alvarez L, Marzeda AM, Deligne C, Schwenzer A, McCann FE, Marsden BD, et al. Mapping tenascin-C interaction with toll-like receptor 4 reveals a new subset of endogenous inflammatory triggers. Nat Commun. (2017) 8:1595. doi: 10.1038/s41467-017-01718-7
29. Chavez RD, Sohn P, Serra R. Prg4 prevents osteoarthritis induced by dominant-negative interference of TGF-ß signaling in mice. PLoS ONE. (2019) 14:587 doi: 10.1371/journal.pone.0210601
30. Iqbal SM, Leonard C, S CR, De Rantere D, Tailor P, Ren G, et al. Lubricin/Proteoglycan 4 binds to and regulates the activity of toll-like receptors in vitro. Sci Rep. (2016) 6:18910. doi: 10.1038/srep18910
31. Zappia J, Joiret M, Sanchez C, Lambert C, Geris L, Muller M, et al. From translation to protein degradation as mechanisms for regulating biological functions: a review on the SLRP family in skeletal tissues. Biomolecules. (2020) 10:80. doi: 10.3390/biom10010080
32. Sjoberg A, Onnerfjord P, Morgelin M, Heinegard D, Blom AM. The extracellular matrix and inflammation: fibromodulin activates the classical pathway of complement by directly binding C1q. J Biol Chem. (2005) 280:32301–8. doi: 10.1074/jbc.M504828200
33. Sjoberg AP, Manderson GA, Morgelin M, Day AJ, Heinegard D, Blom AM. Short leucine-rich glycoproteins of the extracellular matrix display diverse patterns of complement interaction and activation. Mol Immunol. (2009) 46:830–9. doi: 10.1016/j.molimm.2008.09.018
34. Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. (2005) 115:2223–33. doi: 10.1172/JCI23755
35. Barreto G, Soininen A, Ylinen P, Sandelin J, Konttinen YT, Nordstrom DC, et al. Soluble biglycan: a potential mediator of cartilage degradation in osteoarthritis. Arthritis Res Ther. (2015) 17:379. doi: 10.1186/s13075-015-0902-0
36. Avenoso A, D'Ascola A, Scuruchi M, Mandraffino G, Calatroni A, Saitta A, et al. The proteoglycan biglycan mediates inflammatory response by activating TLR-4 in human chondrocytes: inhibition by specific siRNA and high polymerized Hyaluronan. Arch Biochem Biophys. (2018) 640:75–82. doi: 10.1016/j.abb.2018.01.007
37. Silawal S, Triebel J, Bertsch T, Schulze-Tanzil G. Osteoarthritis and the complement cascade. Clin Med Insights Arthritis Musculoskelet Disord. (2018) 11:1179544117751430. doi: 10.1177/1179544117751430
38. Blasioli DJ, Kaplan DL. The roles of catabolic factors in the development of osteoarthritis. Tissue Eng Part B Rev. (2014) 20:355–s63. doi: 10.1089/ten.teb.2013.0377
39. Gondokaryono SP, Ushio H, Niyonsaba F, Hara M, Takenaka H, Jayawardana ST, et al. The extra domain A of fibronectin stimulates murine mast cells via toll-like receptor 4. J Leukoc Biol. (2007) 82:657–65. doi: 10.1189/jlb.1206730
40. McFadden JP, Basketter DA, Dearman RJ, Kimber IR. Extra domain A-positive fibronectin-positive feedback loops and their association with cutaneous inflammatory disease. Clin Dermatol. (2011) 29:257–65. doi: 10.1016/j.clindermatol.2010.11.003
41. Kelsh R, You R, Horzempa C, Zheng M, McKeown-Longo PJ. Regulation of the innate immune response by fibronectin: synergism between the III-1 and EDA domains. PLoS ONE. (2014) 9:e102974. doi: 10.1371/journal.pone.0102974
42. Klatt AR, Paul-Klausch B, Klinger G, Kuhn G, Renno JH, Banerjee M, et al. A critical role for collagen II in cartilage matrix degradation: collagen II induces pro-inflammatory cytokines and MMPs in primary human chondrocytes. J Orthop Res. (2009) 27:65–70. doi: 10.1002/jor.20716
43. Ruettger A, Schueler S, Mollenhauer JA, Wiederanders B. Cathepsins B, K, and L are regulated by a defined collagen type II peptide via activation of classical protein kinase C and p38 MAP kinase in articular chondrocytes. J Biol Chem. (2008) 283:1043–51. doi: 10.1074/jbc.M704915200
44. Fichter M, Korner U, Schomburg J, Jennings L, Cole AA, Mollenhauer J. Collagen degradation products modulate matrix metalloproteinase expression in cultured articular chondrocytes. J Orthop Res. (2006) 24:63–70. doi: 10.1002/jor.20001
45. Tchetina EV, Kobayashi M, Yasuda T, Meijers T, Pidoux I, Poole AR. Chondrocyte hypertrophy can be induced by a cryptic sequence of type II collagen and is accompanied by the induction of MMP-13 and collagenase activity: implications for development and arthritis. Matrix Biol. (2007) 26:247–58. doi: 10.1016/j.matbio.2007.01.006
46. Yasuda T. Type II collagen peptide stimulates Akt leading to nuclear factor-kappaB activation: its inhibition by hyaluronan. Biomed Res. (2014) 35:193–9. doi: 10.2220/biomedres.35.193
47. Lambert C, Borderie D, Dubuc JE, Rannou F, Henrotin Y. Type II collagen peptide Coll2-1 is an actor of synovitis. Osteoarthr Cartil. (2019) 27:1680–91. doi: 10.1016/j.joca.2019.07.009
48. Lees S, Golub SB, Last K, Zeng W, Jackson DC, Sutton P, et al. Bioactivity in an Aggrecan 32-mer fragment is mediated via toll-like receptor 2. Arthritis Rheumatol. (2015) 67:1240–9. doi: 10.1002/art.39063
49. Kalchishkova N, Furst CM, Heinegard D, Blom AM. NC4 Domain of cartilage-specific collagen IX inhibits complement directly due to attenuation of membrane attack formation and indirectly through binding and enhancing activity of complement inhibitors C4B-binding protein and factor H. J Biol Chem. (2011) 286:27915–26. doi: 10.1074/jbc.M111.242834
50. Happonen KE, Saxne T, Aspberg A, Morgelin M, Heinegard D, Blom AM. Regulation of complement by cartilage oligomeric matrix protein allows for a novel molecular diagnostic principle in rheumatoid arthritis. Arthritis Rheum. (2010) 62:3574–83. doi: 10.1002/art.27720
51. Groeneveld TWL, Oroszlán M, Owens RT, Faber-Krol MC, Bakker AC, Arlaud GJ, et al. Interactions of the extracellular matrix proteoglycans decorin and biglycan with C1q and collectins. J Immunol. (2005) 175:4715–23. doi: 10.4049/jimmunol.175.7.4715
52. Bellahcene A, Castronovo V, Ogbureke KU, Fisher LW, Fedarko NS. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nat Rev Cancer. (2008) 8:212–6. doi: 10.1038/nrc2345
53. Gao SG, Li KH, Zeng KB, Tu M, Xu M, Lei GH. Elevated osteopontin level of synovial fluid and articular cartilage is associated with disease severity in knee osteoarthritis patients. Osteoarthr Cartil. (2010) 18:82–7. doi: 10.1016/j.joca.2009.07.009
54. Ding F, Wang J, Zhu G, Zhao H, Wu G, Chen L. Osteopontin stimulates matrix metalloproteinase expression through the nuclear factor-kappaB signaling pathway in rat temporomandibular joint and condylar chondrocytes. Am J Transl Res. (2017) 9:316–29.
55. Tardelli M, Zeyda K, Moreno-Viedma V, Wanko B, Grun NG, Staffler G, et al. Osteopontin is a key player for local adipose tissue macrophage proliferation in obesity. Mol Metab. (2016) 5:1131–7. doi: 10.1016/j.molmet.2016.09.003
56. Sohn DH, Sokolove J, Sharpe O, Erhart JC, Chandra PE, Lahey LJ, et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4. Arthritis Res Ther. (2012) 14:R7. doi: 10.1186/ar3555
57. Gobezie R, Kho A, Krastins B, Sarracino DA, Thornhill TS, Chase M, et al. High abundance synovial fluid proteome: distinct profiles in health and osteoarthritis. Arthritis Res Ther. (2007) 9:R36. doi: 10.1186/ar2172
58. Loeuille D, Chary-Valckenaere I, Champigneulle J, Rat AC, Toussaint F, Pinzano-Watrin A, et al. Macroscopic and microscopic features of synovial membrane inflammation in the osteoarthritic knee: correlating magnetic resonance imaging findings with disease severity. Arthritis Rheum. (2005) 52:3492–501. doi: 10.1002/art.21373
59. Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcgamma receptor. Arthritis Rheum. (2011) 63:53–62. doi: 10.1002/art.30081
60. Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. (2001) 167:2887–94. doi: 10.4049/jimmunol.167.5.2887
61. van Lent PL, Blom AB, Schelbergen RF, Sloetjes A, Lafeber FP, Lems WF, et al. Active involvement of alarmins S100A8 and S100A9 in the regulation of synovial activation and joint destruction during mouse and human osteoarthritis. Arthritis Rheum. (2012) 64:1466–76. doi: 10.1002/art.34315
62. Schelbergen RF, Blom AB, van den Bosch MH, Sloetjes A, Abdollahi-Roodsaz S, Schreurs BW, Mort JS, et al. Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on Toll-like receptor 4. Arthritis Rheum. (2012) 64:1477–87. doi: 10.1002/art.33495
63. Ruan G, Xu J, Wang K, Zheng S, Wu J, Bian F, et al. Associations between serum IL-8 and knee symptoms, joint structures, and cartilage or bone biomarkers in patients with knee osteoarthritis. Clin Rheumatol. (2019) 38:3609–17. doi: 10.1007/s10067-019-04718-8
64. van den Bosch MH, Blom AB, Schelbergen RF, Vogl T, Roth JP, Sloetjes AW, et al. Induction of canonical Wnt signaling by the alarmins S100A8/A9 in murine knee joints: implications for osteoarthritis. Arthritis Rheumatol. (2016) 68:152–63. doi: 10.1002/art.39420
65. Swisher JF, Burton N, Bacot SM, Vogel SN, Feldman GM. Annexin A2 tetramer activates human and murine macrophages through TLR4. Blood. (2010) 115:549–58. doi: 10.1182/blood-2009-06-226944
66. Song C, Zhou X, Dong Q, Fan R, Wu G, Ji B, et al. Regulation of inflammatory response in human chondrocytes by lentiviral mediated RNA interference against S100A10. Inflamm Res. (2012) 61:1219–27. doi: 10.1007/s00011-012-0519-6
67. Nakashima M, Sakai T, Hiraiwa H, Hamada T, Omachi T, Ono Y, et al. Role of S100A12 in the pathogenesis of osteoarthritis. Biochem Biophys Res Commun. (2012) 422:508–14. doi: 10.1016/j.bbrc.2012.05.036
68. Wang LC, Zhang HY, Shao L, Chen L, Liu ZH, He X, et al. S100A12 levels in synovial fluid may reflect clinical severity in patients with primary knee osteoarthritis. Biomarkers. (2013) 18:216–20. doi: 10.3109/1354750X.2013.766262
69. Meijer B, Gearry RB, Day AS. The role of S100A12 as a systemic marker of inflammation. Int J Inflam. (2012) 2012:907078. doi: 10.1155/2012/907078
70. Baillet A, Trocme C, Berthier S, Arlotto M, Grange L, Chenau J, et al. Synovial fluid proteomic fingerprint: S100A8, S100A9 and S100A12 proteins discriminate rheumatoid arthritis from other inflammatory joint diseases. Rheumatology. (2010) 49:671–82. doi: 10.1093/rheumatology/kep452
71. Magna M, Pisetsky DS. The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol Med. (2014) 20:138–46. doi: 10.2119/molmed.2013.00164
72. Ke X, Jin G, Yang Y, Cao X, Fang R, Feng X, et al. Synovial fluid HMGB-1 levels are associated with osteoarthritis severity. Clin Lab. (2015) 61:809–18. doi: 10.7754/Clin.Lab.2015.141205
73. Terada C, Yoshida A, Nasu Y, Mori S, Tomono Y, Tanaka M, et al. Gene expression and localization of high-mobility group box chromosomal protein-1 (HMGB-1)in human osteoarthritic cartilage. Acta Med Okayama. (2011) 65:369–77. doi: 10.18926/AMO/47262
74. Garcia-Arnandis I, Guillen MI, Gomar F, Pelletier JP, Martel-Pelletier J, Alcaraz MJ. High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1beta in osteoarthritic synoviocytes. Arthritis Res Ther. (2010) 12:R165. doi: 10.1186/ar3124
75. Busso N, So A. Microcrystals as DAMPs and their role in joint inflammation. Rheumatology. (2012). 51:1154–60. doi: 10.1093/rheumatology/ker524
76. McAllister MJ, Chemaly M, Eakin AJ, Gibson DS, McGilligan VE. NLRP3 as a potentially novel biomarker for the management of osteoarthritis. Osteoarthr Cartil 756. (2018) 26:612–19. doi: 10.1016/j.joca.2018.02.901
77. Rosenthal AK. Crystals, inflammation, and osteoarthritis. Curr Opin Rheumatol. (2011) 23:170–3. doi: 10.1097/BOR.0b013e3283432d1f
78. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. (2006) 440:237–41. doi: 10.1038/nature04516
79. Denoble AE, Huffman KM, Stabler T V, Kelly SJ, Hershfield MS, McDaniel GE, et al. Uric acid is a danger signal of increasing risk for osteoarthritis through inflammasome activation. Proc Natl Acad Sci USA. (2011) 108:2088–93. doi: 10.1073/pnas.1012743108
80. Liu-Bryan R, Pritzker K, Firestein GS, Terkeltaub R. TLR2 signaling in chondrocytes drives calcium pyrophosphate dihydrate and monosodium urate crystal-induced nitric oxide generation. J Immunol. (2005) 174:5016–23. doi: 10.4049/jimmunol.174.8.5016
81. Barreto G, Manninen M, Eklund KK. Osteoarthritis and toll-like receptors: when innate immunity meets chondrocyte apoptosis. Biology. (2020) 9:772 doi: 10.3390/biology9040065
82. Parker LC, Prince LR, Sabroe I. Translational mini-review series on toll-like receptors: networks regulated by Toll-like receptors mediate innate and adaptive immunity. Clin Exp Immunol. (2007) 147:199–207. doi: 10.1111/j.1365-2249.2006.03203.x
83. Gomez R, Villalvilla A, Largo R, Gualillo O, Herrero-Beaumont G. TLR4 signalling in osteoarthritis–finding targets for candidate DMOADs. Nat Rev Rheumatol. (2015) 11:159–70. doi: 10.1038/nrrheum.2014.209
84. Kim HA, Cho M-L, Choi HY, Yoon CS, Jhun JY, Oh HJ, et al. The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum. (2006) 54:2152–63. doi: 10.1002/art.21951
85. Rosenberg JH, Rai V, Dilisio MF, Agrawal DK. Damage-associated molecular patterns in the pathogenesis of osteoarthritis: potentially novel therapeutic targets. Mol Cell Biochem. (2017) 434:171–9. doi: 10.1007/s11010-017-3047-4
86. Barreto G, Sandelin J, Salem A, Nordstrom DC, Waris E. Toll-like receptors and their soluble forms differ in the knee and thumb basal osteoarthritic joints. Acta Orthop. (2017) 88:326–33. doi: 10.1080/17453674.2017.1281058
87. McCormack WJ, Parker AE, O'Neill LA. Toll-like receptors and NOD-like receptors in rheumatic diseases. Arthritis Res Ther. (2009) 11:243. doi: 10.1186/ar2729
88. Mullen LM, Chamberlain G, Sacre S. Pattern recognition receptors as potential therapeutic targets in inflammatory rheumatic disease. Arthritis Res Ther. (2015) 17:122. doi: 10.1186/s13075-015-0645-y
89. Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, et al. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. (2012) 36:388–400. doi: 10.1016/j.immuni.2012.01.018
90. Xie J, Mendez JD, Mendez-Valenzuela V, Aguilar-Hernandez MM. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal. (2013) 25:2185–97. doi: 10.1016/j.cellsig.2013.06.013
91. Sun XH, Liu Y, Han Y, Wang J. Expression and significance of high-mobility group protein B1 (HMGB1) and the Receptor for Advanced Glycation End-Product (RAGE) in knee osteoarthritis. Med Sci Monit. (2016) 22:2105–12. doi: 10.12659/MSM.895689
92. Campo GM, Avenoso A, D'Ascola A, Prestipino V, Scuruchi M, Nastasi G, et al. Hyaluronan differently modulates TLR-4 and the inflammatory response in mouse chondrocytes. BioFactors. (2012) 38:69–76. doi: 10.1002/biof.202
93. Campo GM, Avenoso A, D'Ascola A, Scuruchi M, Prestipino V, Nastasi G, et al. Adenosine A2A receptor activation and hyaluronan fragment inhibition reduce inflammation in mouse articular chondrocytes stimulated with interleukin-1beta. FEBS J. (2012) 279:2120–33. doi: 10.1111/j.1742-4658.2012.08598.x
94. de Seny D, Cobraiville G, Charlier E, Neuville S, Esser N, Malaise D, et al. Acute-phase serum amyloid A in osteoarthritis: regulatory mechanism and proinflammatory properties. PLoS ONE. (2013) 8:815 doi: 10.1371/journal.pone.0066769
95. Roman-Blas JA, Bizzi E, Largo R, Migliore A, Herrero-Beaumont G. An update on the up and coming therapies to treat osteoarthritis, a multifaceted disease. Expert Opin Pharmacother. (2016) 17:1745–56. doi: 10.1080/14656566.2016.1201070
96. Wang P, Zhu F, Konstantopoulos K. Interleukin-6 synthesis in human chondrocytes is regulated via the antagonistic actions of prostaglandin (PG)E2 and 15-deoxy-delta(12,14)-PGJ2. PLoS ONE. (2011) 6:e27630. doi: 10.1371/journal.pone.0027630
97. Juarranz Y, Gutierrez-Canas I, Arranz A, Martinez C, Abad C, Leceta J, et al. VIP decreases TLR4 expression induced by LPS and TNF-alpha treatment in human synovial fibroblasts. Ann N Y Acad Sci. (2006) 1070:359–64. doi: 10.1196/annals.1317.045
98. Villalvilla A, da Silva JA, Largo R, Gualillo O, Vieira PC, Herrero-Beaumont G, et al. 6-Shogaol inhibits chondrocytes' innate immune responses and cathepsin-K activity. Mol Nutr Food Res. (2014) 58:256–66. doi: 10.1002/mnfr.201200833
99. Komai-Koma M, Li D, Wang E, Vaughan D, Xu D. Anti-Toll-like receptor 2 and 4 antibodies suppress inflammatory response in mice. Immunology. (2014) 143:354–62. doi: 10.1111/imm.12312
100. Alquraini A, Garguilo S, D'Souza G, Zhang LX, Schmidt TA, Jay GD, et al. The interaction of lubricin/proteoglycan 4 (PRG4) with toll-like receptors 2 and 4: an anti-inflammatory role of PRG4 in synovial fluid. Arthritis Res Ther. (2015) 17:353. doi: 10.1186/s13075-015-0877-x
101. Peng Y, Park H-S, Tang LA, Horwitz N, Lin L. Generation of sRAGEhigh transgenic mice to study inflammaging. Front Biosci. (2019) 24:555–63. doi: 10.2741/4735
102. Patriquin CJ, Kuo KHM. Eculizumab and beyond: the past, present, and future of complement therapeutics. Transfus Med Rev. (2019) 33:256–65. doi: 10.1016/j.tmrv.2019.09.004
103. Schierbeck H, Lundbäck P, Palmblad K, Klevenvall L, Erlandsson-Harris H, Andersson U, Ottosson L. Monoclonal anti-HMGB1 (high mobility group box chromosomal protein 1) antibody protection in two experimental arthritis models. Mol Med. (2011) 17:1039–44. doi: 10.2119/molmed.2010.00264
104. Tapia-Abellán A, Angosto-Bazarra D, Martínez-Banaclocha H, de Torre-Minguela C, Cerón-Carrasco JP, Pérez-Sánchez H, Arostegui JI, et al. MCCcloses the active conformation of NLRP3 to an inactive state. Nat Chem Biol. (2019) 15:560–4. doi: 10.1038/s41589-019-0278-6
105. Hsieh LT, Nastase M V, Zeng-Brouwers J, Iozzo R V, Schaefer L. Soluble biglycan as a biomarker of inflammatory renal diseases. Int J Biochem Cell Biol. (2014) 54:223–35. doi: 10.1016/j.biocel.2014.07.020
Keywords: osteoarthritis, cartilage, immunity, inflammation, synovitis
Citation: Lambert C, Zappia J, Sanchez C, Florin A, Dubuc J-E and Henrotin Y (2021) The Damage-Associated Molecular Patterns (DAMPs) as Potential Targets to Treat Osteoarthritis: Perspectives From a Review of the Literature. Front. Med. 7:607186. doi: 10.3389/fmed.2020.607186
Received: 16 September 2020; Accepted: 11 November 2020;
Published: 18 January 2021.
Edited by:
George Bertsias, University of Crete, GreeceReviewed by:
Mohit Kapoor, University of Toronto, CanadaChanghai Ding, Southern Medical University, China
Copyright © 2021 Lambert, Zappia, Sanchez, Florin, Dubuc and Henrotin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cécile Lambert, Y2VjaWxlLmxhbWJlcnRAdWxpZWdlLmJl