 Ha-Yeun Chung
Ha-Yeun Chung Ralf A. Claus
Ralf A. Claus- 1Section Translational Neuroimmunology, Department of Neurology, Jena University Hospital, Jena, Germany
- 2Center for Sepsis Control and Care, Jena University Hospital, Jena, Germany
- 3Department for Anaesthesiology and Intensive Care, Jena University Hospital, Jena, Germany
Breakdown of the inert and constitutive membrane building block sphingomyelin to the highly active lipid mediator ceramide by extracellularly active acid sphingomyelinase is tightly regulated during stress response and opens the gate for invading pathogens, triggering the immune response, development of remote organ failure, and tissue repair following severe infection. How do one enzyme and one mediator manage all of these affairs? Under physiological conditions, the enzyme is located in the lysosomes and takes part in the noiseless metabolism of sphingolipids, but following stress the protein is secreted into circulation. When secreted, acid sphingomyelinase (ASM) is able to hydrolyze sphingomyelin present at the outer leaflet of membranes to ceramide. Its generation troubles the biophysical context of cellular membranes resulting in functional assembly and reorganization of proteins and receptors, also embedded in highly conserved response mechanisms. As a consequence of cellular signaling, not only induction of cell death but also proliferation, differentiation, and fibrogenesis are affected. Here, we discuss the current state of the art on both the impact and function of the enzyme during host response and damage control. Also, the potential role of lysosomotropic agents as functional inhibitors of this upstream alarming cascade is highlighted.
Introduction—Why Is Consideration of ASM Important In The Context of Infection and Host Response?
During severe infection and sepsis, a stress-responsive enzyme becomes present in circulation, which is known to be essentially involved in membrane repair, internalization of pathogens, maturation of phagolysosomes, and mitochondrial dysfunction. The function of the circulating enzyme is held responsible for rapid and transient formation of the highly bioactive lipid mediator ceramide from the inert membrane constituent sphingomyelin (SM), which is localized at the outer leaflet of cellular membranes. Under physiological conditions, there is a very consequent segregation of the enzyme from its substrate. But why it is important for a better understanding of the pathophysiology of organ dysfunction development, that there is a resolution of the spatial separation? Is there an action of the enzyme for a remote signaling to regulate tissue damage apart from the infectious focus? In this review, we summarize the functions of acid sphingomyelinase in order to contribute to the question whether acid sphingomyelinase (ASM) is our friend or foe in the course of sepsis and severe infection, and further discuss the significance of both, an intended and an unattended inhibition for interpretation of results from preclinical and clinical studies with septic patients.
The first clear description of an enzymatic activity cleaving SM into ceramide with an acidic pH optimum was given by Gatt (1). Twenty-five years later, the protein was isolated and purified from human urine and biochemically characterized (2). Surprisingly, the urine obtained from patients with peritonitis was identified as a rich source for purifying human acid sphingomyelinase (3), which was at that time used as a reagent for in vitro experimentation of cellular membranes. Tissue, i.e., brain tissue, was also identified as an appropriate starting material useful for purification procedures (4).
ASM is Our Buddy
ASM Profile
The plasma membrane with an asymmetric distribution of phospho- and sphingolipids as well as lateral segregation of SM and cholesterol has—beyond separating cellular compartments—an important function with respect to signal transduction and a plethora of other essential cellular processes (5, 6). The outer leaflet of plasma membranes is enriched in SM, phosphatidylcholines, and glycosphingolipids, whereas in the inner leaflet phosphatidylinositoles, phosphatidylserine, phosphatidylethanolamines, and phosphatidic acid are abundant (5, 7). Metabolism of SM is the entry point in a unique and highly interconnected universe of sphingolipids with a plethora of compounds differing in physicochemical properties and diverse functions regarding cellular signaling and membrane organization (8). The first step—hydrolysis of SM to ceramides and phosphatidylcholine—is catalyzed by the pacemaking enzyme sphingomyelinase, of which so far five different isoforms are known (8, 9). These sphingomyelinases can be distinguished according to primary structure, triggering of activity, cation dependence, and subcellular localization (10, 11). In this review, we focus on acid sphingomyelinase, which is primarily localized in lysosomes (12). Other sources of ceramide formation (de novo synthesis, synthesis from sphingosine and fatty acid, as well as hydrolysis of glucosylated or phosphorylated specimen) are playing a minor role during stress response and severe infection (13). A short overview on in vitro determination of ASM activity is given in Box 1.
Box 1. Determination of ASM activity.
For activity determination, there is a broad range of assays, using either naturally occurring or labeled substrates for in vitro measurements up to quenched SM probes: highest structural similarity is given by radiolabelled sphingomyelin (either in the backbone or in acylated fatty acid), when the decrease of SM of formation of ceramide is determined in vivo or in vitro. This procedure is also suitable for in-situ assays without any need for detergent or any other artificial condition [5]. Next, substrates with fluorescently labeled fatty acids in SM were hydrolyzed by ASM, the generated corresponding ceramide derivative is separated using thin-layer chromatography TLC and determined using a CCD camera in a high-throughput format [6]. In addition, SM substrates with radioactively labeled phosphocholine are used following extraction of the water-soluble reaction product [7]. An artificial SM substrate with short chain fatty acids are used with highly specific mass spectrometric analysis of corresponding ceramides resulting in improved sensitivity [8]. From naturally occurring sphingomyelin, another opportunity for determination of the second reaction product (phosphocholine) is oxidation of a pro-fluorescence dye following hydrolysis and oxidation to betaine, where formation of a resorufin analog by released hydrogen peroxide is used as a reporter system. The strength of this assay format is the transfer to a multi-well-format without need of any separation step [9]. A most recent review on strengths and pitfalls of ASM assays is given by Nikolova-Karakashian [10].
Quenched fluorescent SM probes based on FAM/BODIPY dyes are allowing real-time analysis to monitor relative sphingomyelinase activities and ceramide formation of intact, living cells by techniques of flow cytometry. At the end, these probes are acting as a biosensor in a non-invasive manner and in native cellular environment with high spatial and temporal resolution [11, 12]. The specific profile of generated ceramides in affected cells is quantified by methods of mass spectrometry following lipid extraction and chromatographical separation [13].
Almost 30 years ago (1991), the full sequence of ASM was firstly described (14, 15). The genetic locus was identified on the short arm of chromosome 11 (15), interestingly in close proximity to the locus of other lysosomal proteins such as cathepsin D and acid phosphatase (13). The sequence of ASM is highly conserved among mammals (16) and the ASM locus [systematically sphingomyelin phosphodiesterase 1 (SMPD1)] is undergoing epigenetic regulation by paternal imprinting (17). During protein synthesis, ASM undergoes extensive posttranslational modification. In this process, glycosylation at all of the six potential sites is of major importance to ensure correct folding, sorting, and/or proper stability within the proteolytic milieu of the lysosome (18, 19). By mannose-6-phosphate-receptor shuttling, the protein is transported via the endosomal system to its final destination, the lysosome (20).
Along the primary subcellular localization, the purified protein favors an optimum pH around 5.0, but sphingolytic activity is retained also at neutral pH (21): regarding parameter of enzymatic activity, despite the pH shift up to 7.45, the maximum turnover velocity (vmax) remains constant (22, 23). The decrease in affinity to the substrate (Km) is of no relevance due to the enormous excess of SM at the outer leaflet of cell membranes (24, 25). The persistence of enzymatic activity at neutral pH values is of great importance as discussed later.
In vitro experiments showed us that enzyme activity is also dependent on lipid environment (26) and addition of a detergent in order to overcome dependence from activation by specific proteins (26). Sensitivity to reducing agents such as dithiothreitol is underlining the relevance of disulfide bridges within the mature protein structure for hydrolyzing activity (27, 28).
Resolving the crystal structure of mammalian ASM (with 88% identity to the human protein) confirmed that ASM-mediated hydrolysis of SM is functioning in a canonical mechanism, where phosphoesterases are utilizing a nucleophilic attack of a zinc (Zn++)-activated water molecule and protonation of the leaving group for release of phosphocholine and ceramide (29). Two saposin domains are relevant for determination and stabilization of either a closed or open conformation of the enzyme. In the latter one, the enzyme is able to bind and dock to membranes, and extract SM therefrom for subsequent hydrolysis (29). Direct inhibitors of the enzyme (i.e., bisphosphonic acid derivatives) are competing with Zn++ binding within the active center (29). A short overview on milestones in translational research regarding ASM activity is given in Table 1.

Table 1. Milestones for ASM in translational sepsis research.
Under physiological conditions, ASM is fulfilling essential house-keeping functions in the lysosomes, whereas genetic deficiency leads to extensive accumulation and deposition of SM resulting in organ abnormalities as described in Niemann-Pick disease (NBD), types A and B (30). The moment of glory has come to ASM as soon as response to external or internal signals of stress is much needed. Since ceramide generation is the common final pathway of most stressors, the panel of ASM stimulators in a large variety of cell types goes far beyond those previously described for any other molecular switch of the SM pathway and are summarized in Table 2. Activation of the ASM-sensitive pathway leads to a rapid and transient translocation of the enzyme toward the cell membrane ranging from seconds to hours (60, 61).
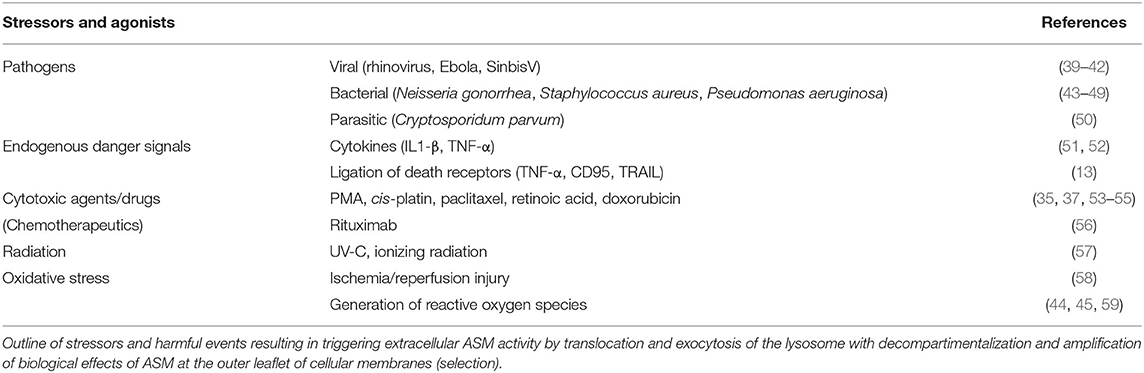
Table 2. Stressors triggering ASM activity and translocation (selection).
In order to highlight the importance of ceramide generation in the course of a severe systemic disease, it is noteworthy that ASM-deficient fibroblasts or mice are resistant to radiation-induced cell death (57). In addition, specific ceramide-binding antibodies rescued mice from lethal radiation gastrointestinal syndrome by preventing signaling platform formation (see below) and inhibition of endothelial cell death (62).
Maturation of Phagolysosome, Translocation, and Role in Raft Modeling
ASM is essential for proper fusion of late phagosomes with lysosomes, which is crucial for efficient transfer of lysosomal antibacterial hydrolases into phagosomes (63, 64). A significant role of ASM in the phagolysosomal compartment for the defense against infection with intracellular pathogens was shown by a dramatically increased susceptibility to Listeria monocytogenes in ASM-deficient mice (ASMKO). Although ASM-deficient immune cells showed intact production of reactive nitrogen intermediates and oxidative burst, they are completely incapable of restricting and controlling the intracellular growth of L. monocytogenes in vitro (65). Similar findings were obtained from mouse peritoneal macrophages infected with the obligate intracellular protozoan, Leishmania donovani. Increase of intracellular ceramide was not only a consequence of ASM-triggered activity but also from de novo synthesis, which resulted in upregulation of Ca++-independent atypical protein kinase C (PKC)-ζ. Surprisingly, suppression of formation of reactive nitrogen species (i.e., nitric oxide) facilitated the survival of leishmanial parasites in the intramacrophageal milieu (66).
Accumulation of reactive oxygen species (ROS), as a consequence of altered redox status followed by ASM activation, ceramide generation, and subsequent clustering of CD95 in ceramide-enriched lipid rafts is a common and early event in neutrophil apoptosis, which are abundant, and short-lived leukocytes. Their death by apoptosis is central to hemostasis and the resolution of inflammation (67).
Extralysosomal Activity—Circulation in Plasma?
In 1996, Tabas et al. described an additional, plasma-secreted, circulating product of the sphingomyelin phosphodiesterase 1 (SMPD1) gene with a similar glycosylation pattern, but increased dependency and susceptibility to Zn++ ions (31, 68). Differential protein trafficking was held responsible for the regularly observed activity increase in men, mice, and cell culture experiments upon stimulation, but the underlying mechanisms remain unknown. There is an ongoing debate on the origin of the ASM isoform circulating in plasma, since the mechanisms for a differentiated intracellular trafficking upon stimulation are hard to explain. Phosphorylation of serine residue 508 (S508) by PKC-δ upon stimulation with phorbol ester or UV light was considered to be essential for activation and translocation (35). But to the best of our knowledge, there is no evidence of phosphorylated ASM in disease models or clinical samples.
However, recent studies with the protozoan parasite Trypanosoma cruzi revealed that conventional lysosomes are regularly fusing with the plasma membrane in response to increased intracellular Ca++ concentration with subsequent triggering of exocytosis (69) and release of the intralysosomal content to the extracellular space, eventually also into circulation. As a result, exocytosed ASM is capable to act at the outer leaflet of the membrane. This mechanism is thought to define the role of extracellular ASM as well as to represent the major source of extracellular form of the protein (38). The findings, that a plethora of harmful, stress/injury-triggering events induced lysosomal exocytosis including interaction with pathogens (59, 70, 71) offers a plausible perspective of the possible origin of extracellular and circulating ASM activity without any need for postulating differential trafficking (38). An overview on ASM release as a feature of stress response is given in Figure 1.
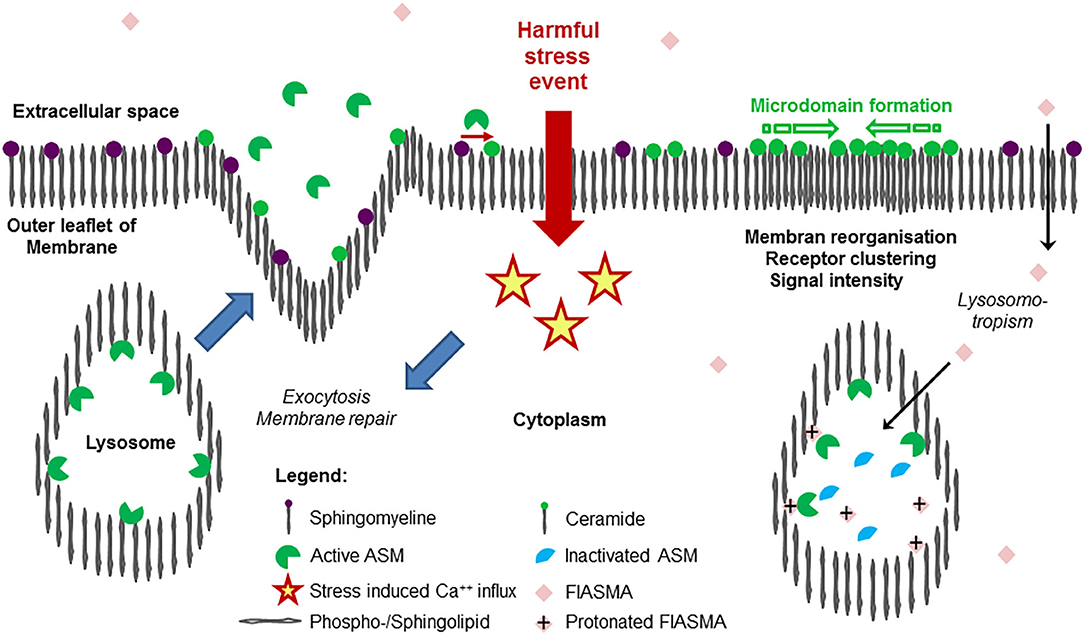
Figure 1. Release of acid sphingomyelinase (ASM) in the course of stress response and mode of action of its inhibition. Exposition of a cell membrane to external harmful stress events such as endogenous or exogenous stimuli (bacterial endotoxin, pro-inflammatory cytokines, pore-forming toxins, Table 2) is followed by intracellular Ca++ influx, triggering the exocytosis of lysosomes, where ASM was bound to the inner lysosomal membrane by SAP domain. In the extracellular space, ASM mediates hydrolysis of SM (abundantly embedded into the outer leaflet of the membrane), generates ceramide, facilitating remodeling, and repair of the membrane (restoring integrity), as well as pathogen entry. As a hallmark of ceramide-induced signal transduction, due to the trend for self-aggregation and formation of ceramide-enriched microdomains, subunits of receptor proteins are reorganized to functionally active receptor complexes such as TLR4, TNFR, etc. As a result, an increase in signaling quality and intensity is observed, controlling an adequate cellular response to external harmful stimuli. On the right panel, the mode of action of cationic amphiphilic substances is illustrated: in an uncharged form at physiological pH value, the compounds diffuse across the cellular membrane through the cytoplasma into the lysosome, where the weak basic nitrogen atom of the compound is protonated because of the acid pH value of the lysosome. The protonated compound interacts with the sapsonin domain of ASM, detaching them from the membrane, then undergoing proteolytic inactivation. Following stimulation and release, the inactivated ASM is unable to contribute to ceramide generation at the outer leaflet of the membrane. Due to similar physicochemical properties, a long list of cationic amphiphilic substances with a broad range of clinical indications, but most of them used in daily care as antidepressive drugs, are found to function as functional inhibitors of acid sphingomyelinase (FIASMA) and to effectively control stress-induced ceramide generation. Most of the available FIASMAs are licensed for medical use in humans, are minimally toxic, and may therefore be applied for disease states associated with increased activity of ASM.
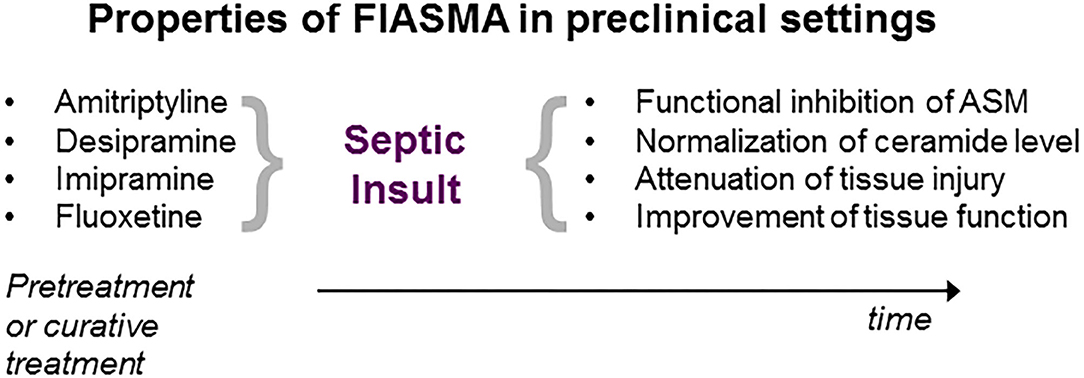
Figure 2. Properties of ASM inhibition in preclinical sepsis research. Administration of the inhibitor either prior (pretreatment) or following septic insult (curative). For details, see text.
Exposition of immune cells to ROS resulted also in a rapid and transient, Ca++-dependent translocation of ASM to the outer leaflet of cellular membranes—mediated by exocytosis of lysosomes—and formation of ceramide-enriched platforms (59). On the other hand, these microdomains are required for Pseudomonas aeruginosa-induced activation of NADPH oxidase and production of ROS, demonstrating a positive feedback mechanism for amplification of ASM-mediated redox signaling (43).
On a molecular level, the generation of ceramide is an integral part of intrinsic repair mechanisms following perforation of the plasmalemma by pathogenic pore-forming toxins, whereby lysosomes fuse with the plasma membrane. Hereby, lysosomes externalize their contents including acid sphingomyelinase and facilitate exposure to the abundant SM at the outer membrane bilayer (70, 72). Subsequent formation of ceramide-enriched, topically restricted domains in the outer leaflet of the lipid bilayer is an essential step contributing to tighter packing of the membrane, forwarding a negative curvature and inward vesiculation of the damaged area. At the end, the resulting internal degradation contributes to cellular integrity and survival (72, 73), e.g., after exposition of cells toward listeriolysin O or pneumolysin (74).
Microdomain Formation, Protein Organization, and Ceramide Signaling
In cultured immune cells, exposure to endotoxin led to activation of ASM, generation of ceramide, phosphorylation of PKC-ζ, assembly of Toll-like receptor 4 (TLR4) within lipid rafts, activation of the stress-responsive kinases, and release of tumor necrosis factor-α (TNF-α). These pathogenetic mechanisms could be abrogated by CD14 blockade or inhibition of ASM but reversed by treatment with the central effector molecule ceramide (51). In addition to other proteins associated with the lipid raft, ASM is contributing to TLR4 signaling triggered by endotoxin and non-microbial endogenous ligands (75) (Figure 1).
Moreover, ASM is involved in most effective host membrane remodeling during enteropathogenic infection with Shigella spp. resulting in decreased binding of pathogens to epithelial cells and therefore, impeding abovementioned or other pathogens from reinfection, which was proposed as a novel stress-responsive cell-autonomous defense mechanism (76).
In concert with the endogenous danger signal adenosine triphosphate (ATP), ASM is involved in secretion of membrane TNF-α within microvesicles bypassing conventional pathways. These cytokine-carrying microvesicles are biologically more potent than soluble TNF-α in vivo, evolving significant lung inflammation in mice, which might have crucial implications for the biological activity of this prototypically proinflammatory cytokine (77).
Ceramide as Second Messenger
Beyond the membrane reorganizing capacities (78), ceramides also act as second messengers transducing cellular signals (79). These functions widely differ among the diverse cell types, ranging from induction of senescence to apoptosis. As an example, the role of ceramide generation and the function of generated ceramides in mitochondria were extensively studied (80): organelle-specific accumulation of ceramide is critically involved in progression of mitochondrial apoptosis and mitophagy, defining ceramides as a bona fide transducer of mitochondrial (dys-)function [excellently reviewed in (80) and references cited therein].
In the next paragraphs, consequences of ASM depletion in the course of local or systemic infections with the presentation of interesting phenotypes will be discussed. In 1995, the generation of ASM-deficient mice was of great value for studying the pathogenesis and treatment not only of type A/B-Niemann-Pick disease (see below) but also for investigations into the role of ASM in ceramide signaling, induction of apoptosis, and ceramide-induced tissue damage (33).
Hyperresponsiveness
Mice, genetically lacking ASM (33), showed an exaggerated response to polymicrobial sepsis with an increased bacterial burden, an enhanced phagocytotic activity, a more pronounced cytokine storm and decreased survival rate (81). Moreover, on a functional level, leukocyte-endothelial interaction was found diminished in ASMKO animals corresponding to a distinct leukocytes' phenotype with respect to rolling and sticking as well as expression of cellular surface proteins (81). A similar phenotype was found in cultivated lung epithelial cells with controlled ASM activity, where increased neutrophil recruitment, elevated levels of cytokine mRNA, and a pro-oxidative shift could be observed (82).
Moreover, the availability of ASMKO mice provides the opportunity of a better understanding of ceramide generation for immune response on a molecular level. In a fundamental experimental setting, injection of endotoxin and its putative effector TNF-α, into mice induced disseminated apoptosis in endothelium of intestine, lung, fat tissue, and thymus accompanied with cytokine release and ceramide increase. This endothelial cell death was ASM dependent, since ASMKO mice were protected against endothelial apoptosis and animal death (83).
In mice and men, complete loss of function of ASM resulted in a diminished CD1d-restricted antigen presentation of invariant natural killer cells (iNKT), decreased levels of this cell population and resistance to iNKT cell-mediated inflammatory conditions, supporting the concept of a tight link between cellular sphingolipid metabolism and immunity (84).
Pneumonia in ASM-Deficient Patients
Niemann-Pick disease (types A and B) is an autosomal recessive lysosomal storage disorder caused by biallelic mutations in the SMPD1 gene resulting in a severe human disease state characterized by deficient ASM activity (85). A recent study reported around 185 mutations with ASM-deficient NPD worldwide (86), while the detection of new mutations is not yet completed (87). The majority of mutations are predicted to affect proper folding, stability, as well as membrane binding of the enzyme (29). Type B patients are characterized by hepatosplenomegaly and progressive alterations of the respiratory system, but the central nervous system is usually less affected, which is more profound in type A, resulting in early death (~2 years of age) (85). Lung involvement is the most important prognostic factor in NPD-B, with recurrent respiratory infections starting in infancy being the major cause of morbidity and mortality (82, 88, 89). In lung epithelial cells, decreasing ASM activity by 50% leads to an increased neutrophil recruitment via elevated levels of cytokine expression, both at baseline and in response to bacterial stimulation. Instead of preventing the host defense responses, decreased ASM activity results in an inflammatory response even in the absence of infection supporting the hypothesis of a chronic inflammatory state impairing host defense mechanisms (82). Despite the low incidence of progressive pulmonary disease in a cohort of more than 100 patients diagnosed with NPD-B, pneumonia was the leading cause of death of juvenile patients (<21 years) (90, 91). On the one hand, pathophysiology of the pulmonary disease is presumably related to the accumulation of SM in alveolar macrophages, on the other hand, inflammation, abnormal surfactant catabolism, as well as composition contributing to lung abnormalities was shown in ASMKO mice (92, 93). In parallel, endogenous lipid pneumonia, interstitial fibrosis, and accumulation of foamy macrophages were found in human lung biopsies (94) supporting the hypothesis that the lung is a primarily affected organ of NPD-B contributing to morbidity and mortality (90, 95).
In a brief summary of this section, ASM is a highly conserved stress-responsive enzyme, activity of which is triggered by a variety of harmful events including infection and inflammation. As a key consequence of activation, there is a disorganization of the previously strict segregation of the enzyme (located in the lysosome) and the corresponding substrate (SM in the outer leaflet of cellular membranes). Besides switching the current pH status by two orders of magnitudes, the enzyme is capable of rapid and transient ceramide formation upon activation. The resulting self-aggregation of ceramide molecules is reorganizing cellular membranes. Now it is time to put this critical event of signal amplification as a general mechanism into the scene of stress response, membrane repair, oxidative stress, proper maturation of phagolysosomes, and regulation of host defense against invading pathogens. As our buddy—keeping us well and fine by its integration in imperative actions of life—the enzyme and its regulation of translocation and activity received also value for further elucidation with respect to pathophysiology and pathogenesis of a series of diseases.
ASM is A Dubious Friend
Invasion of Pathogens
Non-human sphingomyelinases are essential factors for virulence of extracellular, facultative, or obligate intracellular pathogens. These enzymes contribute to phagosomal escape or phagosomal maturation avoidance and even immune response evasion (96). Whether the activity profile of these prokaryotic proteins contribute to the analyzed hydrolysis rate of SM to ceramide in conventional assays in samples obtained from patients with blood stream infection (Box 1) is still discussed controversially. Some prominent and representative mechanistic approaches are shown here in an exemplary manner.
Infections by Staphylococcus aureus are a major clinical problem ranging from mild infections (skin and soft-tissue) to severe and even lethal infections (e.g., pneumonia, endocarditis, sepsis, osteomyelitis, etc.). S. aureus stimulates ASM via CD44-triggered release of ROS, resulting in ceramide release, clustering of CD44 in ceramide-enriched membrane platforms, co-activation of GTPases, and translocation of linker proteins with subsequent rapid rearrangement of the cytoskeleton. In the absence of either CD44 or ASM, reduced internalization of macrophages is counteracted by a reduced killing capacity (44).
A major toxin of S. aureus (α-toxin) caused in bone marrow-derived macrophages ASM-dependent ceramide formation, release of cathepsin B and D from lysosomes, inflammasome activation, and induction of pro-inflammatory cytokines, which could be abrogated by pharmacological ASM-inhibition (45). In in vitro experiments with endothelial cells, ASM activation by α-toxin was linked to degradation of tight junctions, which could be blocked by pharmacological inhibition. However, most importantly, in in vivo experiments, genetic deficiency prevented severe degradation of tight junctions in the lung and edema formation (46). Combination of antibiotic treatment and ASM inhibition are proposed to exhibit synergistic and super-additive effects (47).
Inflammasomes are important for host defense against invading pathogens; ASM activation is critically involved in the activation of endothelial inflammasomes including recruitment of adapter proteins and caspases (97–99) and subsequent oxidative signaling by lipid raft-associated redox platforms, release of cytokines, activation of stress kinases, and altering tight junctions in epithelial cells (45).
For exposition of proteins, non-enveloped viruses undergo partial uncoating in order to get access to the cytoplasm by membrane lysis. Cellular ASM is a critical player in this process, since ASM is induced and hijacked by mimicking wound removal processes facilitating the adenovirus further membrane disruption and infection. In addition, stimulation of Ca++ influx and lysosomal exocytosis are key steps for efficient membrane penetration of the virus (71). Most recently, influenza A virus infection was found to be associated with suppression of ASM activity in cultured alveolar epithelial cells, whereby depletion of SM content either in the epithelial membrane or in the virus envelope impaired virus infection and reduced virus entry as well as reduced virus infectivity and impaired its attachment and internalization, respectively. Inhibition of ASM by desipramine did not affect influenza virus infection (100), which is in line with the observation that ceramide generated by de novo synthesis might play an antiviral role (101).
For meningococcal internalization into brain endothelial cells, transient ASM activation and ceramide release are also determinative factors for invasiveness among a defined set of pathogenic isolates of Neisseria meningitidis (102).
In the pathogenesis of cystic fibrosis, abrogation of ceramide accumulation by inhaled functional inhibitor of ASM (FIASMA, see below) restored normal ceramide concentrations in murine bronchial epithelial cells, reduced age-dependent pulmonary inflammation and deposits of DNA in bronchi, as well as prevented infection with P. aeruginosa (103, 104). ASM activation (by endotoxin) and subsequent ceramide formation play a pivotal role in Escherichia coli-induced apoptosis of immature dendritic cells, a phenomenon contributing to sepsis-induced immunosuppression (105). In liver specimen, inhibition of activity in endotoxin challenged mice correlated with a reduced rate of hepatocellular apoptosis (34).
It is also known that ceramide-containing microparticles in packed red blood cells contribute to adverse effects following transfusion, which was abrogated by treatment with a FIASMA during storage time (106).
ASM activation is a critical factor for redirection of TNF-α trafficking, thus the cytokine is embedded in microvesicles bypassing conventional pathways in a highly potent manner. Rerouting has crucial implications for the activity profile of the pacemaker cytokine of inflammation, inducing a significant TNF-dependent inflammation status and allowing long-range TNF signaling to target cells more effectively than soluble TNF, which is of particular interest therapeutically targeting TNF in acute inflammatory diseases (77).
Marker and Mediator in Sepsis and Pneumonia
Endotoxin challenge in mice resulted in a mild, 2-fold increase of ASM plasma activity, which was accompanied by release of cytokines (107). These data were confirmed by a similar observation of an increase of ceramide content in lipoproteins paralleled by an increased activity of circulating ASM (108).
Intratracheal administration of an ASM inhibitor in a model of acute lung injury improved lung function and decreased pulmonary inflammation (109). In critically ill preterm infants with acute lung injury, beyond other markers and mediators, ASM activity was found to be increased in tracheal aspirates (110). Inhibition of NF-κB function attenuated pulmonary inflammation of acute respiratory distress syndrome in a neonatal piglet model with decreased leukocyte concentrations in bronchoalveolar lavage, reduced ASM activity, and subsequently decreased ceramide levels (111). This emphasizes the pivotal role of sphingolipid signaling controlling pulmonary edema formation and lung function.
In patients with community-acquired pneumonia (CAP), a plasma decrease of SM was found to be closely associated with an increase in ceramides (16:0, 18:0, 24:1), but normalized toward clinical remission (112). Furthermore, the sphingolytic activity of plasma-secreted ASM was nearly 3-fold increased with same tendency for normalization. A similar course was also observed with respect to gene expression rate of SMPD1 in circulating leukocytes, supporting the concept of a disease-relevant regulation of ASM expression in CAP at both, protein and mRNA level. These observations might qualify ASM as a potential target for host-directed treatments to reduce end-organ damage in pneumonia (112).
Endothelial Integrity
Cultivated endothelial cells are affected by serum obtained from patients with sepsis, resulting in clustering of receptors relevant for signal transduction and suppression of a stress-sensitive transcript marker of these cells (i.e., ADAMTS13), which is abrogated by direct and indirect inhibition of ASM activity (113). Both, plasma activity and amount of ASM were found to be increased in septic patients dependent on clinical severity (34, 113). Thus, ASM is involved in the dysregulation of ceramide metabolism in endothelial cells leading to macrodomain formation, cytotoxicity, and downregulation of ADAMTS13 expression (113), which is held to function as an adverse effect to endothelial dysfunction and microthrombus formation in sepsis (114). These results obtained in in vitro experimentation were recently confirmed in a monocentric clinical study, where a plasma decrease of the endothelium-stabilizing mediator sphingosine-1-phosphate (S1P) was strongly negatively associated with an increase in ceramide levels (115). Moreover, the association with severity of clinical course could be outlined by a highly powerful value of an integrative analysis of the S1P/Cer ratio for prediction of unfavorable outcome, superior to established severity scores (SOFA) and HLA-DR expression on circulating monocytes (115).
Role of Alternative Splicing and SNP in Sepsis and Depression
Beside predominant localization in lysosomes especially of monocytic, hepatic, and endothelial cells, and delocalization of the protein to the outer leaflet of cellular membranes in response to multiple stimuli including pathogens (116), encoding mRNASMPD1 is undergoing alternative splicing as an independent matter of stress response. A series of SMPD1 mRNA splice isoforms are described to date, but only ASM-1 as the full-length transcript, Ref_mRNA NM_000543 has been shown to be translated into a catalytically active protein (14, 117, 118). It might be speculated, that the extensive variability is a marker of negative regulation with highest variability in healthy controls but decreasing in a stress condition such as severe infection by skipping of exon 3 and/or elongation of exon 2 (119). Thus, an association of specific pattern of alternatively spliced SMPD1 transcripts with disease severity (healthy and non-infectious patients undergoing intensive care as controls vs. sepsis) was observed (119). At the end, alternative splicing of SMPD1 might act in a dominant negative manner upon overexpression making alternative splicing a promising target in regulation of ASM activity (118, 119). Interestingly, a similar phenomenon was observed in patients with major depression, a disease, where despite clear associations of ASM activity with the severity of the disease (120) the regulatory mechanisms and cause-effect links are not well-understood (121). Strikingly, alternative splicing of SMPD1 was also found to be reduced in depressed patients, and most interestingly, normalized upon subchronical treatment with FIASMA in both, patients and healthy volunteers (118, 122).
There are a small number of reports on polymorphisms of the SMPD1 gene locus: V36A, A487V, and G508R as well as hexanucleotide repeat in the signaling peptide (123–127). In contrast to the variety of missense mutations, there is only a minor effect of these sequence variations on overall activity and function of the enzyme, but they might increase the susceptibility for common diseases such as allergy (125). The G508A transition—exchange of an uncharged with a charged amino acid—is discussed to be significant for recognition of a potential phosphorylation site, which is relevant for the control of protein activation and secretion (35, 52). In line with this hypothesis, G508A is associated with plasma-secreted activity in a gene-dosage-dependent manner (125), since subjects homozygous for the minor A allele displayed half of the plasma activity compared with the major G allele. Whether this observation on a molecular level might contribute to control the susceptibility to infections is still a matter of debate, but as discussed below, a decreased ASM activity is associated with lower respiratory tract infections (90, 91).
In brief, ASM is also held responsible for internalization of pathogens (bacteria and viruses). In acute and chronic infection, inhibition of ASM improved outcome in (pre-)clinical studies in patients with cystic fibrosis and CAP. ASM inhibition also diminished inflammation-responsive fibrogenesis following sepsis in a preclinical study. A multilevel regulation with association to favorable outcome was observed in patients undergoing CAP. Due to the facts that (i) the impact of single nucleotide polymorphisms and alternative splicing of ASM with respect to susceptibility and progression of infection is not fully understood and (ii) pharmacological inhibition of the enzyme resulted in improved outcome from chronic infection, the role of ASM should not be evaluated as exclusively favorable without any restriction. Our friend is becoming dubious and needs more critical consideration.
But ASM is Also Worrying
In a series of clinical and preclinical studies, ASM activity was positively associated with the severity of the disease. In a small, mixed population from an intensive care unit (ICU), ASM activity was found to be increased and to remain elevated in the presence of a low level of procalcitonin-discriminating non-surviving patients after systemic inflammation. Also, a posttrauma effect with a significant increase in ASM after surgery parallel to a postoperative increase of procalcitonin and C-reactive protein was observed (128). Strikingly, patients with severe sepsis exhibit an enhanced, 2-fold sphingolytic activity in comparison with controls. A further increase was associated with greater risk by the severity of illness and with fatal outcome (34). Supporting the hypothesis that ASM activity is highly correlated to inflammation, we also found ASM activity mildly increased in patients with rheumatoid arthritis, which was associated with oxidant activity, markers of inflammation, and endothelial activation (129). Interestingly, treatment of these patients by biologicals with TNF-α-binding capacity suppressed the increase completely to levels as found in healthy controls along with improvement of the clinical condition (130). These data support the concept that activation of circulating ASM may play a critical role in the development of apoptosis and organ failure in inflammation-associated disease, especially sepsis. An inhibition of ASM should be explored further as a potential target in the complicated puzzle of sepsis. At sepsis diagnosis, leukocyte-associated ceramide content as a result of ASM activity was significantly elevated and correlated to TNF-α concentration as both a marker of cytokine release as well as mediator of ASM activation (131). Ceramide concentration was highly predictive for risk of development of organ failure (131).
In hemophagocytic lymphohistiocytosis (HLH), a rare systemic inflammatory syndrome resulting from unrestrained immune cell activation, levels of circulating ASM were found elevated. Also, there was a shift in ceramide and sphingosine ratio (increase), while levels of S1P were decreased (132). Interestingly, an elevated ratio between ceramide and sphingosine was predictive for unfavorable outcome (132). Data were confirmed in a small cohort with a 10–20-fold increase of ASM activity with a trend to normalization during recovery (133).
As an indirect measure for ASM-induced unfavorable outcome, fluoxetine and desipramine reduced in an lipopolysaccharide (LPS)-induced model of septic shock the levels of circulating TNF-α similar to prednisolone, accompanied with an improved outcome to untreated controls (134). In murine models of peritonitis and endotoxemia following LPS challenge, amitriptyline-treated mice were protected from overwhelming cytokine release (KC, MCP-1) and from pulmonary edema as well as exhibiting improved survival. Anti-inflammatory effect of amitriptyline treatment is reflected by increased IL-10 levels and decreased accumulation of immune cells at the site of infection (135).
Eryptosis
In patients with hyperbilirubinemia, as often observed in the acute phase of severe infection due to sepsis-induced cholestasis and also a long-term consequence of survivors (136, 137), high levels of conjugated bilirubin may also lead to progressive sclerosing cholangitis (138) and are able to stimulate suicidal death of human erythrocytes. As an underlying mechanism, bilirubin triggered rapid Ca++ influx resulting in the release of ASM, formation of ceramide, and subsequent translocation of phosphatidylserine to the erythrocyte surface (139), a phenomenon, which is also compatible with the anemia status of these patients exerting an increased mortality rate (140).
Platelets
Ceramide accumulation over time, generated by ASM in stored and aged platelets, caused lung injury in endotoxin-challenged mice, examined by neutrophil accumulation, endothelial barrier dysfunction, and histological evidence of tissue injury (141). Interestingly, this adverse effect of pulmonary complications following transfusion could be overcome by ASM inhibition (141).
Mechanisms and Effects of ASM Inhibition
A broad panel of cationic amphiphilic compounds is known to inhibit the activity of ASM by lysosomotropism due to their unique physicochemical properties, which was firstly observed by Albouz et al. (142). Interaction with the membrane-embedding N-terminal saposin domain of ASM caused detachment from the inner lysosomal membrane and a consecutive proteolysis of the enzyme (143, 144). As a result, there is a significant decrease in sphingolytic activity, therefore these compounds are termed as FIASMA (36). The compounds differ markedly in molecular structure. A prediction of inhibitory capacity is available by a structure-property-activity relation (SPAR) model in order to specify the structural and physicochemical characteristics including variables referring to pKa, logP, as well as a factor depicting the steric hindrance of the most basic nitrogen atom of the compound modulating the free presentation of a protonated nitrogen atom at the inner lysosomal surface (145). It is noteworthy that these compounds are licensed for medical use in humans, are minimally toxic, and in use for a broad range of clinical indications, including the treatment of intensive care patients. There is a long list of FIASMA of ASM including amitriptyline, imipramine, desipramine, doxepine, fluoxetine, maprotiline, nortriptyline, paroxetine, sertraline, suloctidil, terfenadine, and famotidine, to mention a few (145) (Figure 1).
Furthermore, evaluating lysosomotropic and ASM-inhibiting activities in appropriate cell culture models are an applicable approach of newly designed substances to identify novel compounds with anti-apoptotic and anti-inflammatory capacity, which results ultimately in a decreased response of prototypic inflammatory mediators. At the end, in new candidate drugs, based on established SPAR models, physicochemical and biological properties will be selected and identified to enrich the pool of compounds, which might be beneficial for the treatment of the adverse effects of ASM upon activation (146).
In the next paragraph, there is a short overview on the proposed beneficial effects of ASM inhibition in a series of conditions associated with development of organ failure and long-term effects of sepsis sequelae, classified by the affected tissue.
Muscles, Diaphragm—Heart, and Cardiovascular System
Cardiac dysfunction, in particular of the left ventricle, is a common and early event in sepsis and is strongly associated with an increase in patients' mortality. Surrogates of cardiomyopathy cardiac function, ceramide formation, markers of oxidative stress, as well as troponin I levels were found to be improved in FIASMA-treated animals in a semi-lethal peritonitis model (147). Interestingly, in this study, an activation of de novo synthesis of ceramides could be identified to be responsible for cardiac ceramide increase (147). In an in vitro and ex vivo experimental setting, Ferreira et al. showed that mouse myotubes and diaphragm muscle fiber bundles are sensitive to ASM treatment mediated by release of mitochondrial ROS, resulting in significant depression of diaphragm force and accelerated fatigue in a time and concentration manner (148, 149). Also, the p47(phox) subunit of NADPH oxidase is held responsible to play an important role on oxidant-mediated diaphragm weakness triggered by ASM (150). Similar results could be obtained in a chronic malfunction of skeletal muscles and dysregulation of sphingolipid turnover in insulin-responsive tissues at old age. In parallel with a progressive increase of ceramide content and Cer/SM ratio during aging of rats, there is a decrease of insulin responsiveness, which can be overcome by ASM inhibition (151). Interestingly, in muscle tissue, the counteracting activity of sphingosine-1-phosphate is once more evident, promoting cell survival, Ca++ mobilization, fiber growth and repair, as well as fatigue resistance [excellently reviewed in (152)].
ASMKO mice are protected from TNF-α-induced hypotension and tachycardia (153), which might be regarded as clinical symptoms of systemic inflammation (154). One hypothesis for this surprising observation might be a ceramide-triggered activation of NOS, resulting in vasodilation and hypovolemic shock (153), which was in a similar manner also observed for ceramide generation by the neutral isoform of sphingomyelinase (155). Therefore, pharmacological inhibition of ASM-triggered ceramide generation and presumed prevention of shock should be considered to contribute to the overall improved survival (153) in the acute phase as observed in a series of peritonitis models in mice (147, 156).
Lung
Pulmonary outcome following endotoxin challenge in mice was improved by ASM inhibition, resulting in attenuating alveolar collapse (157). Inhibition of ASM was identified as a possible target in acute lung injury and pulmonary edema, induced by administration of platelet-activating factor and endotoxin (158). Fluoxetine and desipramine reduced in ovalbumin-sensitized rats the number of migrated immune cells into bronchalveolar lavage fluid but did not exert anti-inflammatory activity by attenuation of bronchial hyperreactivity (134).
Liver Tissue—Long-Term Effects on Tissue Function and Fibrosis Following Sepsis
Long-term sepsis survivors might develop hepatocellular/hepatobiliary injury and fibrosis (159–162). ASM, also an important regulator of hepatocyte apoptosis and hepatic stellate cell (HSC) activation (163), is linked to the promotion of liver dysfunction in the acute phase as well as to fibrogenesis in the long term. In both, the acute and the postacute phase, pharmacological inhibition of ASM displayed a beneficial effect on oxidative stress levels, hepatobiliary function, macrophage infiltration, hepatic stellate activation, and overall survival (156). ASM inhibition exhibited a protective effect on liver function in the acute phase, and the reduction of HSC activation diminished development of sepsis-associated liver fibrosis in the postacute phase of sepsis (156). In this context, dysregulation of hepatic biotransformation capacity, especially of the cytochrome P450 (CYP) system, represents an important distress factor during host response (164–166). Thus, these enzymes are estimated to be responsible for metabolizing >75% of drugs which are in daily clinical use. Pharmacological inhibition of ASM has an important impact on expression and activity of different hepatic CYP enzymes using an animal model of polymicrobial sepsis in the acute as well as in the postsepsis phase (167).
In murine models of peritonitis and endotoxemia following LPS challenge, amitriptyline-treated mice were protected from overwhelming cytokine release (KC, MCP-1) and from pulmonary edema as well as exhibited improved survival. Anti-inflammatory effect of amitriptyline treatment is reflected by increased IL-10 levels and decreased accumulation of immune cells at the site of infection (135).
In all studies enrolling patients with severe infection, sepsis and multiple organ failure as well as in all reports modeling the disease continuum ex vivo or in vivo, we found a clear association of ASM activity with morbidity and unfavorable outcome. These observations are independent from the underlying disease such as peritonitis, pneumonia, and HHL. On the other hand, inhibition of ASM activity is capable to prevent destructive events of overwhelming immune activation in all affected tissues and circulating cells, be they nucleated or not. Here, we come to the point for further consideration of the presumed beneficial effects of ASM inhibition in daily clinical care, because a broad panel of drugs is exhibiting ASM inhibition.
Conclusions and Further Perspectives
The expenditure for locally restricted or disseminated ceramide generation by ASM for effective host response is remarkable. The multifaceted roles of ceramide in this context, specific function in individual tissues and organs, the flux and interconnections of lipid mediators with agonistic functions requires further investigation and redefining. Nonetheless, there is conclusive preclinical evidence that the conserved stress enzyme ASM plays an essential role in the pathogenesis of sepsis/host response and that its inhibition might improve the outcome. However, the gap between preclinical and clinical trials has not been convincingly closed so far. Therefore, we suggest as a first step that a retrospective analysis of sepsis patients in the intensive care unit, who are coincidentally treated with FIASMAs, should be initiated to overcome the missing link from bench to bedside. Our knowledge gained from preclinical experiments indicates that FIASMAs might be promising candidates for future pharmacological studies targeting ASM in sepsis and host response keeping in mind that these drugs are already FDA approved and just need to be repurposed.
FIASMA—From Unattended ASM Inhibition at ICU to an Intended Use
FIASMA are a class of drugs, which are widely used for (often chronic) treatment of a number of symptoms such as major depression, neuropathic pain, fibromyalgia, etc., to mention a few (168). From that list, major depression was recently ranked as the third leading burden of non-fatal diseases worldwide with a prevalence of >10% in all analyzed regions and with an increasing incidence to be first during the next decade (169, 170). In the Global Burden of Disease survey for 2017, more than 264 million cases were reported worldwide (169).
In Germany, the 12-month prevalence of major depression is 12% in adults, of whom the majority is treated with antidepressant drugs (171).
In cultured PBMC from healthy volunteers treated either with imipramine or amitriptyline in a therapeutical dosage, activity of ASM is rapidly reduced to levels around 30% of original activity over a period of 3 weeks. Interestingly, removal of the drugs resulted in a gradual and prolonged, but complete normalization of activity over a period of 5–6 days (172). Considering this scenario combined with the facts that (i) daily use medication is often or regularly spared on ICU admission and (ii) that drugs initially used at the newly admitted ICU patient might also exert an inhibitory capacity on ASM, it seems quite possible that the septic patients at least in the early days following admission might be subdivided in separate groups: one with decreased ASM activity due to antecedent treatment with FIASMA due to other indication and gradual normalization of ASM activity, one with inhibition of ASM activity only just at ICU submission, and one without any interference with ASM activity. In addition, it might be further speculated that the presented ASM activity in circulation is also superimposed by the conserved mechanisms of stress response as discussed in detail previously.
This scenario in mind, there are some principal opportunities, risks, as well as consequences examining host response and its unintended, but presumptive treatment by FIASMA.
1. For a more precise interpretation of results from clinical sepsis studies, the definition of the as-yet unknown biasing factor of ASM inhibition by consideration of both ASM activity and inhibition by FIASMA due to other indication prior to hospitalization is needed (anamnesis, documentation of prehospital medication, identification of FIASMA therein). In sepsis trials, depression at least (and treatment thereof) should be defined as a significant comorbidity.
2. The incidence of sepsis is determined including clinical parameters of organ dysfunction in a cohort undergoing prehospital treatment with FIASMA compared with those without. Answering this question will also result in the assessment, whether FIASMA-treated patients are at higher risk for a severe course due to putative impairment of phagolysosomal elimination of invading pathogens or—on the contrary—at lower risk for development of tissue damage and organ failure due to potential beneficial effects of FIASMA. From an epidemiological point of view, two studies came up with interesting results: in a general population (~60,000 individuals) with self-reported anxiety and depression symptoms, severe depression was found to be associated with an increased risk for blood stream infection, moderate were not. However, an increased mortality risk was found for the later subcohort only; unfortunately, no data on antidepressant treatment were given in this study (173). Similar results were obtained from a Danish cohort study, where the underlying depressive disorder was assessed either by psychiatric diagnosis or by at least two antidepressant prescription redemptions within a 6-month period prior to hospitalization due to sepsis (174).
3. On ICU, ASM activity is also determined in septic patients treated with FIASMA due to other indications, comparison with an untreated group, and association with clinical parameters.
4. Ultimately, there is also a need for placebo-controlled trial with a carefully selected FIASMA (on the basis of data from observational trials) for a prospective investigation of the proposed beneficial effect of resulting ASM inhibition with respect to severity (development, duration, resolution of organ failure) and overall outcome.
Drug Repurposing
Repurposing of established drugs to treat both, common, or rare diseases is becoming an increasingly attractive and fast-track approach because it involves the use of compounds with known pharmacokinetic and safety profiles, with potentially lower overall development costs and shorter development timelines due to existing approval by the regulatory authorities (175). In the case of ASM, it is of remarkable importance since there is a long list of potential candidates available (36), and both, the structure-activity relationship (145) as well as the underlying mechanism of effective inhibition are known (144). Also, there are promising results from preclinical and clinical studies as discussed in this review, and there are some more outcomes using an explorative design for identification of beneficial effect of ASM inhibition.
Out of around 800 unique three-drug combinations, two sets were identified to effectively inhibit Ebola virus entry into human cells and were further validated for inhibition of live Ebola virus infection—at least two drugs of the triple exhibiting effective ASM inhibitory capacity (176).
There is also an excellent review [Beckmann et al., (177) and references cited therein], critically outlining the effectiveness of amitriptyline in a series of serious conditions such as cancer, infection, and metabolic and neurological diseases, all of them assessed as ASM-related diseases. For this drug, potential new applications for therapeutical treatment are demonstrated which might also be considered a general opportunity, since amitriptyline is proposed to function PARS PRO TOTO for this group of drugs exhibiting inhibitory capacity for ASM. Beside other drugs, these studies revealed that amitriptyline is a promising candidate for further consideration for the treatment of infectious diseases and overwhelming host response. However, adverse effects of a missing residual activity as shown by incompetence for phagocytosis and increased bacterial burden were also observed (65, 81). On the other hand, there is a clear association of ASM activity with the severity of sepsis and unfavorable outcome (34, 113, 128). It has to be mentioned that in heterogeneous mice by FIASMA treatment, a temporary status similar to complete loss of function was shown, underlining the effectiveness of functional ASM inhibition (167). A residual activity of 15% is insufficient to prevent clinical features of Niemann-Pick diseases over time (17).
It is of major interest that fluoxetine, a widely used antidepressant drug, efficiently inhibited the entry and propagation of SARS-CoV-2 in the cell culture model without cytotoxic effects and also exerted potent antiviral activity against two currently circulating influenza A virus subtypes, an effect which was also observed upon treatment with the FIASMAs amiodarone and imipramine (178).
The recently proposed concept of a stress/injury-induced lysosomal exocytosis (59) as the major source of extracellularly circulating ASM (38) is in line (i) with a restoration of dependency for Zn++ ions, since due to rapid displacement of these ions after reaching the extracellular space, an additional regulating factor participates in the resolution of ASM activity beyond the outfall of the lysosomal content, and (ii) the effectiveness of activity decreases in circulation upon treatment with FIASMA, resulting in intralysosomal proteolysis of the mature ASM protein and subsequent release of inactive protein fragments without sphingolytic activity, and (iii) missing evidence of phosphorylated ASM in circulation of a patient undergoing stress response. However, the validation of this concept in septic patients still needs further examination, e.g., by comparison of the fragmentation and phosphorylation pattern of ASM isolated from both compartments, lysosomes and plasma, obtained from patients undergoing treatment with FIASMA. Another important point with respect to the dosage of FIASMA for treatment of host response is the fact of similarity to that of the original indication (often major depression) due to a very similar mode of action regarding ceramide generation.
Résumé and Take Home Messages
During an episode of sepsis, a broad panel of cell, tissue, and organ response is controlled by stress-induced ceramide generation; there is a broad understanding of potentially harmful effects of ceramide generation during sepsis and there is a broad panel of well-established and approved drugs with effectiveness for ASM inhibition (Box 2), at the end encouraging systematic studies for detailed examination of an unattended or an intended inhibition of ASM during sepsis to improve patients outcome.
Box 2. Take Home messages.
• ASM activity in circulation is a marker and mediator of harmful events, increased in patients with sepsis and associated with the clinical course regarding development, duration, and resolution of organ failure and outcome;
• In septic patients, different phenotypes with respect to ASM activity might be expected due to antecedent treatment with drugs for other indications (i.e., major depression etc.), which are also inhibiting ASM by lysosomotropic mechanisms;
• Further investigation in upcoming clinical studies is necessary to examine the potential consequences of an unattended ASM inhibition;
• Intended inhibition of ASM, also by repurposed drugs, is a promising approach to control the adverse effects of an overwhelming ASM activity associated with unfavorable outcome in these patients.
Methods
The objective of this narrative review is to evaluate the changes of sphingomyelinase activity during infection and host response and the discussion of translation into the field of translational sepsis research including development of new strategies for diagnosis and treatment. A search in the main biomedical databases (PubMed, Medline, Scopus, and Web of science) was conducted for a 20-year period ending in July 2020, focused on primary research articles in the field of interest. Keyword search for abstracts and titles included (“sphingomyelinase” OR “SMPD1”) AND (“sepsis” OR “inflammation” OR “infection”). The search identified 723 references, which were selected by particular importance for this review. The prescreened references were completed by other publications related to the issue. We apologize if a valuable work of any appreciated colleagues could not be included due to space limitations and the narrow scope of this review.
Author Contributions
RC and H-YC drafted the manuscript. Both authors revised the final version of the manuscript.
Funding
This work was supported by internal institutional funds from the Department of Anesthesiology and Intensive Care Medicine and Interdisciplinary Center for Clinical Research (IZKF), Jena University Hospital, Germany.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Quintern LE, Weitz G, Nehrkorn H, Tager JM, Schram AW, Sandhoff K. Acid sphingomyelinase from human urine: purification and characterization. Biochim Biophys Acta. (1987) 922:323–36. doi: 10.1016/0005-2760(87)90055-5
3. Quintern LE, Zenk TS, Sandhoff K. The urine from patients with peritonitis as a rich source for purifying human acid sphingomyelinase and other lysosomal enzymes. Biochim Biophys Acta. (1989) 1003:121–4. doi: 10.1016/0005-2760(89)90244-0
4. Yamanaka T, Hanada E, Suzuki K. Acid sphingomyelinase of human brain. improved purification procedures and characterization. J Biol Chem. (1981) 256:3884–9.
5. Yamaji-Hasegawa A, Tsujimoto M. Asymmetric distribution of phospholipids in biomembranes. Biol Pharm Bull. (2006) 29:1547–53. doi: 10.1248/bpb.29.1547
6. Pike LJ. The challenge of lipid rafts. J Lipid Res. (2009) 50:S323–8. doi: 10.1194/jlr.R800040-JLR200
7. Ikeda M, Kihara A, Igarashi Y. Lipid asymmetry of the eukaryotic plasma membrane: functions and related enzymes. Biol Pharm Bull. (2006) 29:1542–6. doi: 10.1248/bpb.29.1542
8. Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. (2018) 19:175–91. doi: 10.1038/nrm.2017.107
9. Airola MV, Hannun YA. Sphingolipid metabolism and neutral sphingomyelinases. Handb Exp Pharmacol. (2013) 215:57–76. doi: 10.1007/978-3-7091-1368-4_3
10. Goni FM, Alonso A. Sphingomyelinases: enzymology and membrane activity. FEBS Lett. (2002) 531:38–46. doi: 10.1016/S0014-5793(02)03482-8
11. Marchesini N, Hannun YA. Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem Cell Biol. (2004) 82:27–44. doi: 10.1139/o03-091
12. Henry B, Ziobro R, Becker KA, Kolesnick R, Gulbins E. Acid sphingomyelinase. Handb Exp Pharmacol. (2013) 215:77–88. doi: 10.1007/978-3-7091-1368-4_4
13. Zeidan YH, Hannun YA. The acid sphingomyelinase/ceramide pathway: biomedical significance and mechanisms of regulation. Curr Mol Med. (2010) 10:454–66. doi: 10.2174/156652410791608225
14. Schuchman EH, Suchi M, Takahashi T, Sandhoff K, Desnick RJ. Human acid sphingomyelinase. isolation, nucleotide sequence and expression of the full-length and alternatively spliced cDNAs. J Biol Chem. (1991) 266:8531–9
15. Schuchman EH, Levran O, Pereira LV, Desnick RJ. Structural organization and complete nucleotide sequence of the gene encoding human acid sphingomyelinase (SMPD1). Genomics. (1992) 12:197–205. doi: 10.1016/0888-7543(92)90366-Z
16. Newrzella D, Stoffel W. Molecular cloning of the acid sphingomyelinase of the mouse and the organization and complete nucleotide sequence of the gene. Biol Chem Hoppe Seyler. (1992) 373:1233–8. doi: 10.1515/bchm3.1992.373.2.1233
17. Simonaro CM, Park JH, Eliyahu E, Shtraizent N, McGovern MM, Schuchman EH. Imprinting at the SMPD1 locus: implications for acid sphingomyelinase-deficient Niemann-Pick disease. Am J Hum Genet. (2006) 78:865–70. doi: 10.1086/503750
18. Newrzella D, Stoffel W. Functional analysis of the glycosylation of murine acid sphingomyelinase. J Biol Chem. (1996) 271:32089–95. doi: 10.1074/jbc.271.50.32089
19. Ferlinz K, Hurwitz R, Moczall H, Lansmann S, Schuchman EH, Sandhoff K. Functional characterization of the N-glycosylation sites of human acid sphingomyelinase by site-directed mutagenesis. Eur J Biochem. (1997) 243:511–7. doi: 10.1111/j.1432-1033.1997.511_1a.x
20. Ni X, Morales CR. The lysosomal trafficking of acid sphingomyelinase is mediated by sortilin and mannose 6-phosphate receptor. Traffic. (2006) 7:889–902. doi: 10.1111/j.1600-0854.2006.00429.x
21. Freeman SJ, Davidson DJ, Shankaran P, Callahan JW. Monoclonal antibodies against human placental sphingomyelinase. Biosci Rep. (1983) 3:545–50. doi: 10.1007/BF01120698
22. Callahan JW, Jones CS, Davidson DJ, Shankaran P. The active site of lysosomal sphingomyelinase: evidence for the involvement of hydrophobic and ionic groups. J Neurosci Res. (1983) 10:151–63. doi: 10.1002/jnr.490100205
23. Lansmann S, Ferlinz K, Hurwitz R, Bartelsen O, Glombitza G, Sandhoff K. Purification of acid sphingomyelinase from human placenta: characterization and N-terminal sequence. FEBS Lett. (1996) 399:227–31. doi: 10.1016/S0014-5793(96)01331-2
24. Merrill AH Jr, Jones DD. An update of the enzymology and regulation of sphingomyelin metabolism. Biochim Biophys Acta. (1990) 1044:1–12. doi: 10.1016/0005-2760(90)90211-F
25. Cremesti AE, Goni FM, Kolesnick R. Role of sphingomyelinase and ceramide in modulating rafts: do biophysical properties determine biologic outcome? FEBS Lett. (2002) 531:47–53. doi: 10.1016/S0014-5793(02)03489-0
26. Linke T, Wilkening G, Lansmann S, Moczall H, Bartelsen O, Weisgerber J, et al. Stimulation of acid sphingomyelinase activity by lysosomal lipids and sphingolipid activator proteins. Biol Chem. (2001) 382:283–90. doi: 10.1515/BC.2001.035
27. Lansmann S, Schuette CG, Bartelsen O, Hoernschemeyer J, Linke T, Weisgerber J, et al. Human acid sphingomyelinase. Eur J Biochem. (2003) 270:1076–88. doi: 10.1046/j.1432-1033.2003.03435.x
28. Lee CY, Tamura T, Rabah N, Lee DY, Ruel I, Hafiane A, et al. Carboxyl-terminal disulfide bond of acid sphingomyelinase is critical for its secretion and enzymatic function. Biochemistry. (2007) 46:14969–78. doi: 10.1021/bi700817g
29. Gorelik A, Illes K, Heinz LX, Superti-Furga G, Nagar B. Crystal structure of mammalian acid sphingomyelinase. Nat Commun. (2016) 7:12196. doi: 10.1038/ncomms12196
30. Brady RO, Kanfer JN, Mock MB, Fredrickson DS. The metabolism of sphingomyelin. II Evidence of an enzymatic deficiency in niemann-pick diseae. Proc Natl Acad Sci USA. (1966) 55:366–9. doi: 10.1073/pnas.55.2.366
31. Schissel SL, Schuchman EH, Williams KJ, Tabas I. Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J Biol Chem. (1996) 271:18431–6. doi: 10.1074/jbc.271.31.18431
32. Spence MW, Byers DM, Palmer FB, Cook HW. A new Zn2+-stimulated sphingomyelinase in fetal bovine serum. J Biol Chem. (1989) 264:5358–63
33. Horinouchi K, Erlich S, Perl DP, Ferlinz K, Bisgaier CL, Sandhoff K, et al. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat Genet. (1995) 10:288–93. doi: 10.1038/ng0795-288
34. Claus RA, Bunck AC, Bockmeyer CL, Brunkhorst FM, Losche W, Kinscherf R, et al. Role of increased sphingomyelinase activity in apoptosis and organ failure of patients with severe sepsis. FASEB J. (2005) 19:1719–21. doi: 10.1096/fj.04-2842fje
35. Zeidan YH, Hannun YA. Activation of acid sphingomyelinase by protein kinase Cdelta-mediated phosphorylation. J Biol Chem. (2007) 282:11549–61. doi: 10.1074/jbc.M609424200
36. Kornhuber J, Tripal P, Reichel M, Muhle C, Rhein C, Muehlbacher M, et al. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cell Physiol Biochem. (2010) 26:9–20. doi: 10.1159/000315101
37. Bose RN, Moghaddas S, Belkacemi L, Tripathi S, Adams NR, Majmudar P, et al. Absence of activation of DNA repair genes and excellent efficacy of phosphaplatins against human ovarian cancers: implications to treat resistant cancers. J Med Chem. (2015) 58:8387–401. doi: 10.1021/acs.jmedchem.5b00732
38. Andrews NW. Solving the secretory acid sphingomyelinase puzzle: insights from lysosome-mediated parasite invasion and plasma membrane repair. Cell Microbiol. (2019) 21:e13065. doi: 10.1111/cmi.13065
39. Dreschers S, Franz P, Dumitru C, Wilker B, Jahnke K, Gulbins E. Infections with human rhinovirus induce the formation of distinct functional membrane domains. Cell Physiol Biochem. (2007) 20:241–54. doi: 10.1159/000104170
40. Grassme H, Riehle A, Wilker B, Gulbins E. Rhinoviruses infect human epithelial cells via ceramide-enriched membrane platforms. J Biol Chem. (2005) 280:26256–62. doi: 10.1074/jbc.M500835200
41. Miller ME, Adhikary S, Kolokoltsov AA, Davey RA. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J Virol. (2012) 86:7473–83. doi: 10.1128/JVI.00136-12
42. Ng CG, Griffin DE. Acid sphingomyelinase deficiency increases susceptibility to fatal alphavirus encephalomyelitis. J Virol. (2006) 80:10989–99. doi: 10.1128/JVI.01154-06
43. Zhang Y, Li X, Carpinteiro A, Gulbins E. Acid sphingomyelinase amplifies redox signaling in Pseudomonas aeruginosa-induced macrophage apoptosis. J Immunol. (2008) 181:4247–54. doi: 10.4049/jimmunol.181.6.4247
44. Li C, Wu Y, Riehle A, Orian-Rousseau V, Zhang Y, Gulbins E, et al. Regulation of Staphylococcus aureus infection of macrophages by CD44, reactive oxygen species, and acid sphingomyelinase. Antioxid Redox Signal. (2018) 28:916–34. doi: 10.1089/ars.2017.6994
45. Ma J, Gulbins E, Edwards MJ, Caldwell CC, Fraunholz M, Becker KA. Staphylococcus aureus alpha-toxin induces inflammatory cytokines via lysosomal acid sphingomyelinase and ceramides. Cell Physiol Biochem. (2017) 43:2170–84. doi: 10.1159/000484296
46. Becker KA, Fahsel B, Kemper H, Mayeres J, Li C, Wilker B, et al. Staphylococcus aureus alpha-toxin disrupts endothelial-cell tight junctions via acid sphingomyelinase and ceramide. Infect Immun. (2018) 86:e00606–17. doi: 10.1093/med/9780198738381.003.0024
47. Peng H, Li C, Kadow S, Henry BD, Steinmann J, Becker KA, et al. Acid sphingomyelinase inhibition protects mice from lung edema and lethal Staphylococcus aureus sepsis. J Mol Med. (2015) 93:675–89. doi: 10.1007/s00109-014-1246-y
48. Grassme H, Gulbins E, Brenner B, Ferlinz K, Sandhoff K, Harzer K, et al. Acidic sphingomyelinase mediates entry of N. gonorrhoeae into nonphagocytic cells Cell. (1997) 91:605–15. doi: 10.1016/S0092-8674(00)80448-1
49. Hauck CR, Grassme H, Bock J, Jendrossek V, Ferlinz K, Meyer TF, et al. Acid sphingomyelinase is involved in CEACAM receptor-mediated phagocytosis of neisseria gonorrhoeae. FEBS Lett. (2000) 478:260–6. doi: 10.1016/S0014-5793(00)01851-2
50. Nelson JB, O'Hara SP, Small AJ, Tietz PS, Choudhury AK, Pagano RE, et al. Cryptosporidium parvum infects human cholangiocytes via sphingolipid-enriched membrane microdomains. Cell Microbiol. (2006) 8:1932–45. doi: 10.1111/j.1462-5822.2006.00759.x
51. Cuschieri J, Bulger E, Billgrin J, Garcia I, Maier RV. Acid sphingomyelinase is required for lipid Raft TLR4 complex formation. Surg Infect. (2007) 8:91–106. doi: 10.1089/sur.2006.050
52. Jenkins RW, Canals D, Idkowiak-Baldys J, Simbari F, Roddy P, Perry DM, et al. Regulated secretion of acid sphingomyelinase: implications for selectivity of ceramide formation. J Biol Chem. (2010) 285:35706–18. doi: 10.1074/jbc.M110.125609
53. Dumitru CA, Carpinteiro A, Trarbach T, Hengge UR, Gulbins E. Doxorubicin enhances TRAIL-induced cell death via ceramide-enriched membrane platforms. Apoptosis. (2007) 12:1533–41. doi: 10.1007/s10495-007-0081-9
54. Erdreich-Epstein A, Tran LB, Bowman NN, Wang H, Cabot MC, Durden DL, et al. Ceramide signaling in fenretinide-induced endothelial cell apoptosis. J Biol Chem. (2002) 277:49531–7. doi: 10.1074/jbc.M209962200
55. Jacobi J, Garcia-Barros M, Rao S, Rotolo JA, Thompson C, Mizrachi A, et al. Targeting acid sphingomyelinase with anti-angiogenic chemotherapy. Cell Signal. (2017) 29:52–61. doi: 10.1016/j.cellsig.2016.09.010
56. Bezombes C, Grazide S, Garret C, Fabre C, Quillet-Mary A, Muller S, et al. Rituximab antiproliferative effect in B-lymphoma cells is associated with acid-sphingomyelinase activation in raft microdomains. Blood. (2004) 104:1166–73. doi: 10.1182/blood-2004-01-0277
57. Haimovitz-Friedman A, Kan CC, Ehleiter D, Persaud RS, McLoughlin M, Fuks Z, et al. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med. (1994) 180:525–35. doi: 10.1084/jem.180.2.525
58. Llacuna L, Mari M, Garcia-Ruiz C, Fernandez-Checa JC, Morales A. Critical role of acidic sphingomyelinase in murine hepatic ischemia-reperfusion injury. Hepatology. (2006) 44:561–72. doi: 10.1002/hep.21285
59. Li X, Gulbins E, Zhang Y. Oxidative stress triggers Ca-dependent lysosome trafficking and activation of acid sphingomyelinase. Cell Physiol Biochem. (2012) 30:815–26. doi: 10.1159/000341460
60. Cremesti A, Paris F, Grassme H, Holler N, Tschopp J, Fuks Z, et al. Ceramide enables fas to cap and kill. J Biol Chem. (2001) 276:23954–61. doi: 10.1074/jbc.M101866200
61. Rotolo JA, Zhang J, Donepudi M, Lee H, Fuks Z, Kolesnick R. Caspase-dependent and -independent activation of acid sphingomyelinase signaling. J Biol Chem. (2005) 280:26425–34. doi: 10.1074/jbc.M414569200
62. Rotolo J, Stancevic B, Zhang J, Hua G, Fuller J, Yin X, et al. Anti-ceramide antibody prevents the radiation gastrointestinal syndrome in mice. J Clin Invest. (2012) 122:1786–90. doi: 10.1172/JCI59920
63. Schramm M, Herz J, Haas A, Kronke M, Utermohlen O. Acid sphingomyelinase is required for efficient phago-lysosomal fusion. Cell Microbiol. (2008) 10:1839–53. doi: 10.1111/j.1462-5822.2008.01169.x
64. Utermohlen O, Herz J, Schramm M, Kronke M. Fusogenicity of membranes: the impact of acid sphingomyelinase on innate immune responses. Immunobiology. (2008) 213:307–14. doi: 10.1016/j.imbio.2007.10.016
65. Utermohlen O, Karow U, Lohler J, Kronke M. Severe impairment in early host defense against Listeria monocytogenes in mice deficient in acid sphingomyelinase. J Immunol. (2003) 170:2621–8. doi: 10.4049/jimmunol.170.5.2621
66. Ghosh S, Bhattacharyya S, Das S, Raha S, Maulik N, Das DK, et al. Generation of ceramide in murine macrophages infected with Leishmania donovani alters macrophage signaling events and aids intracellular parasitic survival. Mol Cell Biochem. (2001) 223:47–60. doi: 10.1023/A:1017996609928
67. Scheel-Toellner D, Wang K, Craddock R, Webb PR, McGettrick HM, Assi LK, et al. Reactive oxygen species limit neutrophil life span by activating death receptor signaling. Blood. (2004) 104:2557–64. doi: 10.1182/blood-2004-01-0191
68. Schissel SL, Keesler GA, Schuchman EH, Williams KJ, Tabas I. The cellular trafficking and zinc dependence of secretory and lysosomal sphingomyelinase, two products of the acid sphingomyelinase gene. J Biol Chem. (1998) 273:18250–9. doi: 10.1074/jbc.273.29.18250
69. Church LD, Hessler G, Goodall JE, Rider DA, Workman CJ, Vignali DA, et al. TNFR1-induced sphingomyelinase activation modulates TCR signaling by impairing store-operated Ca2+ influx. J Leukoc Biol. (2005) 78:266–78. doi: 10.1189/jlb.1003456
70. Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, et al. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol. (2010) 189:1027–38. doi: 10.1083/jcb.201003053
71. Luisoni S, Suomalainen M, Boucke K, Tanner LB, Wenk MR, Guan XL, et al. Co-option of membrane wounding enables virus penetration into cells. Cell Host Microbe. (2015) 18:75–85. doi: 10.1016/j.chom.2015.06.006
72. Andrews NW, Almeida PE, Corrotte M. Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol. (2014) 24:734–42. doi: 10.1016/j.tcb.2014.07.008
73. Draeger A, Babiychuk EB. Ceramide in plasma membrane repair. Handb Exp Pharmacol. (2013) 216:341–53. doi: 10.1007/978-3-7091-1511-4_17
74. Schoenauer R, Larpin Y, Babiychuk EB, Drucker P, Babiychuk VS, Avota E, et al. Down-regulation of acid sphingomyelinase and neutral sphingomyelinase-2 inversely determines the cellular resistance to plasmalemmal injury by pore-forming toxins. FASEB J. (2019) 33:275–85. doi: 10.1096/fj.201800033R
75. Plociennikowska A, Hromada-Judycka A, Borzecka K, Kwiatkowska K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. (2015) 72:557–81. doi: 10.1007/s00018-014-1762-5
76. Tawk C, Nigro G, Rodrigues Lopes I, Aguilar C, Lisowski C, Mano M, et al. Stress-induced host membrane remodeling protects from infection by non-motile bacterial pathogens. EMBO J. (2018) 37:e98529. doi: 10.15252/embj.201798529
77. Soni S, O'Dea KP, Tan YY, Cho K, Abe E, Romano R, et al. ATP redirects cytokine trafficking and promotes novel membrane TNF signaling via microvesicles. FASEB J. (2019) 33:6442–55. doi: 10.1096/fj.201802386R
78. Ventura AE, Mestre B, Silva LC. Ceramide domains in health and disease: a biophysical perspective. Adv Exp Med Biol. (2019) 1159:79–108. doi: 10.1007/978-3-030-21162-2_6
79. Albeituni S, Stiban J. Roles of ceramides and other sphingolipids in immune cell function and inflammation. Adv Exp Med Biol. (2019) 1161:169–91. doi: 10.1007/978-3-030-21735-8_15
80. Hernandez-Corbacho MJ, Salama MF, Canals D, Senkal CE, Obeid LM. Sphingolipids in mitochondria. Biochim Biophys Acta Mol Cell Biol Lipids. (2017) 1862:56–68. doi: 10.1016/j.bbalip.2016.09.019
81. Jbeily N, Suckert I, Gonnert FA, Acht B, Bockmeyer CL, Grossmann SD, et al. Hyperresponsiveness of mice deficient in plasma-secreted sphingomyelinase reveals its pivotal role in early phase of host response. J Lipid Res. (2013) 54:410–24. doi: 10.1194/jlr.M031625
82. MacFadden-Murphy E, Roussel L, Martel G, Berube J, Rousseau S. Decreasing SMPD1 activity in BEAS-2B bronchial airway epithelial cells results in increased NRF2 activity, cytokine synthesis and neutrophil recruitment. Biochem Biophys Res Commun. (2017) 482:645–50. doi: 10.1016/j.bbrc.2016.11.087
83. Haimovitz-Friedman A, Cordon-Cardo C, Bayoumy S, Garzotto M, McLoughlin M, Gallily R, et al. Lipopolysaccharide induces disseminated endothelial apoptosis requiring ceramide generation. J Exp Med. (1997) 186:1831–41. doi: 10.1084/jem.186.11.1831
84. Melum E, Jiang X, Baker KD, Macedo MF, Fritsch J, Dowds CM, et al. Control of CD1d-restricted antigen presentation and inflammation by sphingomyelin. Nat Immunol. (2019) 20:1644–55. doi: 10.1038/s41590-019-0504-0
85. Schuchman EH, Wasserstein MP. Types A and B niemann-pick disease. Pediatr Endocrinol Rev. (2016) 13(Suppl. 1):674–81. doi: 10.1016/j.ymgme.2016.12.008
86. Ranganath P, Matta D, Bhavani GS, Wangnekar S, Jain JM, Verma IC, et al. Spectrum of SMPD1 mutations in Asian-Indian patients with acid sphingomyelinase (ASM)-deficient Niemann-Pick disease. Am J Med Genet A. (2016) 170:2719–30. doi: 10.1002/ajmg.a.37817
87. Manshadi MD, Kamalidehghan B, Keshavarzi F, Aryani O, Dadgar S, Arastehkani A, et al. Four novel p.N385K, p.V36A, c.1033-1034insT and c.1417-1418delCT mutations in the sphingomyelin phosphodiesterase 1 (SMPD1) gene in patients with types A and B niemann-pick disease (NPD). Int J Mol Sci. (2015) 16:6668–76. doi: 10.3390/ijms16046668
88. Arda IS, Gencoglu A, Coskun M, Ozbek N, Demirhan B, Hicsonmez A. A very unusual presentation of Niemann-Pick disease type B in an infant: similar findings to congenital lobar emphysema. Eur J Pediatr Surg. (2005) 15:283–6. doi: 10.1055/s-2004-830362
89. von Ranke FM, Pereira Freitas HM, Mancano AD, Rodrigues RS, Hochhegger B, Escuissato D, et al. Pulmonary involvement in niemann-pick disease: a state-of-the-art review. Lung. (2016) 194:511–8. doi: 10.1007/s00408-016-9893-0
90. McGovern MM, Lippa N, Bagiella E, Schuchman EH, Desnick RJ, Wasserstein MP. Morbidity and mortality in type B Niemann-Pick disease. Genet Med. (2013) 15:618–23. doi: 10.1038/gim.2013.4
91. McGovern MM, Avetisyan R, Sanson BJ, Lidove O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J Rare Dis. (2017) 12:41. doi: 10.1186/s13023-017-0572-x
92. Dhami R, He X, Gordon RE, Schuchman EH. Analysis of the lung pathology and alveolar macrophage function in the acid sphingomyelinase–deficient mouse model of Niemann-Pick disease. Lab Invest. (2001) 81:987–99. doi: 10.1038/labinvest.3780311
93. Ikegami M, Dhami R, Schuchman EH. Alveolar lipoproteinosis in an acid sphingomyelinase-deficient mouse model of Niemann-Pick disease. Am J Physiol Lung Cell Mol Physiol. (2003) 284:L518–25. doi: 10.1152/ajplung.00258.2002
94. Nicholson AG, Florio R, Hansell DM, Bois RM, Wells AU, Hughes P, et al. Pulmonary involvement by Niemann-Pick disease. a report of six cases histopathology. Lung. (2006) 48:596–603. doi: 10.1111/j.1365-2559.2006.02355.x
95. Cassiman D, Packman S, Bembi B, Turkia HB, Al-Sayed M, Schiff M, et al. Cause of death in patients with chronic visceral and chronic neurovisceral acid sphingomyelinase deficiency (Niemann-Pick disease type B and B variant): literature review and report of new cases. Mol Genet Metab. (2016) 118:206–13. doi: 10.1016/j.ymgme.2016.05.001
96. Flores-Diaz M, Monturiol-Gross L, Naylor C, Alape-Giron A, Flieger A. Bacterial sphingomyelinases and phospholipases as virulence factors. Microbiol Mol Biol Rev. (2016) 80:597–628. doi: 10.1128/MMBR.00082-15
97. Grassme H, Carpinteiro A, Edwards MJ, Gulbins E, Becker KA. Regulation of the inflammasome by ceramide in cystic fibrosis lungs. Cell Physiol Biochem. (2014) 34:45–55. doi: 10.1159/000362983
98. Boini KM, Xia M, Koka S, Gehr TW, Li PL. Instigation of NLRP3 inflammasome activation and glomerular injury in mice on the high fat diet: role of acid sphingomyelinase gene. Oncotarget. (2016) 7:19031–44. doi: 10.18632/oncotarget.8023
99. Koka S, Xia M, Chen Y, Bhat OM, Yuan X, Boini KM, et al. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. (2017) 13:336–44. doi: 10.1016/j.redox.2017.06.004
100. Audi A, Soudani N, Dbaibo G, Zaraket H. Depletion of host and viral sphingomyelin impairs influenza virus infection. Front Microbiol. (2020) 11:612. doi: 10.3389/fmicb.2020.00612
101. Soudani N, Hage-Sleiman R, Karam W, Dbaibo G, Zaraket H. Ceramide suppresses influenza a virus replication in vitro. J Virol. (2019) 93:e00053–19. doi: 10.1128/JVI.00053-19
102. Simonis A, Hebling S, Gulbins E, Schneider-Schaulies S, Schubert-Unkmeir A. Differential activation of acid sphingomyelinase and ceramide release determines invasiveness of Neisseria meningitidis into brain endothelial cells. PLoS Pathog. (2014) 10:e1004160. doi: 10.1371/journal.ppat.1004160
103. Teichgraber V, Ulrich M, Endlich N, Riethmuller J, Wilker B, De Oliveira-Munding CC, et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med. (2008) 14:382–91. doi: 10.1038/nm1748
104. Becker KA, Riethmuller J, Luth A, Doring G, Kleuser B, Gulbins E. Acid sphingomyelinase inhibitors normalize pulmonary ceramide and inflammation in cystic fibrosis. Am J Respir Cell Mol Biol. (2010) 42:716–24. doi: 10.1165/rcmb.2009-0174OC
105. Falcone S, Perrotta C, De Palma C, Pisconti A, Sciorati C, Capobianco A, et al. Activation of acid sphingomyelinase and its inhibition by the nitric oxide/cyclic guanosine 3',5'-monophosphate pathway: key events in Escherichia coli-elicited apoptosis of dendritic cells. J Immunol. (2004) 173:4452–63. doi: 10.4049/jimmunol.173.7.4452
106. Hoehn RS, Jernigan PL, Japtok L, Chang AL, Midura EF, Caldwell CC, et al. Acid sphingomyelinase inhibition in stored erythrocytes reduces transfusion-associated lung inflammation. Ann Surg. (2017) 265:218–26. doi: 10.1097/SLA.0000000000001648
107. Wong ML, Xie B, Beatini N, Phu P, Marathe S, Johns A, et al. Acute systemic inflammation up-regulates secretory sphingomyelinase in vivo: a possible link between inflammatory cytokines and atherogenesis. Proc Natl Acad Sci USA. (2000) 97:8681–6. doi: 10.1073/pnas.150098097
108. Lightle S, Tosheva R, Lee A, Queen-Baker J, Boyanovsky B, Shedlofsky S, et al. Elevation of ceramide in serum lipoproteins during acute phase response in humans and mice: role of serine-palmitoyl transferase. Arch Biochem Biophys. (2003) 419:120–8. doi: 10.1016/j.abb.2003.08.031
109. von Bismarck P, Wistadt CF, Klemm K, Winoto-Morbach S, Uhlig U, Schutze S, et al. Improved pulmonary function by acid sphingomyelinase inhibition in a newborn piglet lavage model. Am J Respir Crit Care Med. (2008) 177:1233–41. doi: 10.1164/rccm.200705-752OC
110. Laube M, Amann E, Uhlig U, Yang Y, Fuchs HW, Zemlin M, et al. Inflammatory mediators in tracheal aspirates of preterm infants participating in a randomized trial of inhaled nitric oxide. PLoS ONE. (2017) 12:e0169352. doi: 10.1371/journal.pone.0169352
111. von Bismarck P, Klemm K, Wistadt CF, Winoto-Morbach S, Uhlig U, Schutze S, et al. Surfactant “fortification” by topical inhibition of nuclear factor-kappaB activity in a newborn piglet lavage model. Crit Care Med. (2007) 35:2309–18. doi: 10.1097/01.CCM.0000281472.47067.45
112. Arshad H, Alfonso JCL, Franke R, Michaelis K, Araujo L, Habib A, et al. Decreased plasma phospholipid concentrations and increased acid sphingomyelinase activity are accurate biomarkers for community-acquired pneumonia. J Transl Med. (2019) 17:365. doi: 10.1186/s12967-019-2112-z
113. Chung HY, Hupe DC, Otto GP, Sprenger M, Bunck AC, Dorer MJ, et al. Acid sphingomyelinase promotes endothelial stress response in systemic inflammation and sepsis. Mol Med. (2016) 22:412–23. doi: 10.2119/molmed.2016.00140
114. Bockmeyer CL, Reuken PA, Simon TP, Budde U, Losche W, Bauer M, et al. ADAMTS13 activity is decreased in a septic porcine model. significance for glomerular thrombus deposition Thromb Haemost. (2011) 105:145–53. doi: 10.1160/TH10-03-0153
115. Wu X, Hou J, Li H, Xie G, Zhang X, Zheng J, et al. Inverse correlation between plasma sphingosine-1-phosphate and ceramide concentrations in septic patients and their utility in predicting mortality. Shock. (2019) 51:718–24. doi: 10.1097/SHK.0000000000001229
116. Jenkins RW, Canals D, Hannun YA. Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal. (2009) 21:836–46. doi: 10.1016/j.cellsig.2009.01.026
117. Quintern LE, Schuchman EH, Levran O, Suchi M, Ferlinz K, Reinke H, et al. Isolation of cDNA clones encoding human acid sphingomyelinase: occurrence of alternatively processed transcripts. EMBO J. (1989) 8:2469–73. doi: 10.1002/j.1460-2075.1989.tb08382.x
118. Rhein C, Tripal P, Seebahn A, Konrad A, Kramer M, Nagel C, et al. Functional implications of novel human acid sphingomyelinase splice variants. PLoS ONE. (2012) 7:e35467. doi: 10.1371/journal.pone.0035467
119. Kramer M, Quickert S, Sponholz C, Menzel U, Huse K, Platzer M, et al. Alternative splicing of SMPD1 in sepsis. PLoS ONE. (2015) 10:e0124503. doi: 10.1371/journal.pone.0124503
120. Gulbins E, Palmada M, Reichel M, Luth A, Bohmer C, Amato D, et al. Acid sphingomyelinase-ceramide system mediates effects of antidepressant drugs. Nat Med. (2013) 19:934–8. doi: 10.1038/nm.3214
121. Muhle C, Wagner CJ, Farber K, Richter-Schmidinger T, Gulbins E, Lenz B, et al. Secretory acid sphingomyelinase in the serum of medicated patients predicts the prospective course of depression. J Clin Med. (2019) 8:846. doi: 10.3390/jcm8060846
122. Rhein C, Reichel M, Kramer M, Rotter A, Lenz B, Muhle C, et al. Alternative splicing of SMPD1 coding for acid sphingomyelinase in major depression. J Affect Disord. (2017) 209:10–5. doi: 10.1016/j.jad.2016.09.019
123. Wan Q, Schuchman EH. A novel polymorphism in the human acid sphingomyelinase gene due to size variation of the signal peptide region. Biochim Biophys Acta. (1995) 1270:207–10. doi: 10.1016/0925-4439(95)00050-E
124. Rhein C, Naumann J, Muhle C, Zill P, Adli M, Hegerl U, et al. The acid sphingomyelinase sequence variant p.A487V is not associated with decreased levels of enzymatic activity. JIMD Rep. (2013) 8:1–6. doi: 10.1007/8904_2012_147
125. Reichel M, Richter-Schmidinger T, Muhle C, Rhein C, Alexopoulos P, Schwab SG, et al. The common acid sphingomyelinase polymorphism p.G508R is associated with self-reported allergy. Cell Physiol Biochem. (2014) 34:82–91. doi: 10.1159/000362986
126. Rhein C, Reichel M, Muhle C, Rotter A, Schwab SG, Kornhuber J. Secretion of acid sphingomyelinase is affected by its polymorphic signal peptide. Cell Physiol Biochem. (2014) 34:1385–401. doi: 10.1159/000366345
127. Rhein C, Muhle C, Kornhuber J, Reichel M. Alleged detrimental mutations in the smpd1 gene in patients with niemann-pick disease. Int J Mol Sci. (2015) 16:13649–52. doi: 10.3390/ijms160613649
128. Kott M, Elke G, Reinicke M, Winoto-Morbach S, Schadler D, Zick G, et al. Acid sphingomyelinase serum activity predicts mortality in intensive care unit patients after systemic inflammation: a prospective cohort study. PLoS ONE. (2014) 9:e112323. doi: 10.1371/journal.pone.0112323
129. Hanaoka BY, Ormseth MJ, Michael Stein C, Banerjee D, Nikolova-Karakashian M, Crofford LJ. Secretory sphingomyelinase (S-SMase) activity is elevated in patients with rheumatoid arthritis. Clin Rheumatol. (2018) 37:1395–9. doi: 10.1007/s10067-017-3824-1
130. Chung HY, Wickel J, Stuhlmuller B, Kinne RW, Claus RA. Acid sphingomyelinase activity is elevated in the serum of rheumatoid arthritis patients, suppressed by anti-TNF-alpha treatment. Clin Exp Rheumatol. (2019) 37(Suppl) 122:18–9.
131. Delogu G, Famularo G, Amati F, Signore L, Antonucci A, Trinchieri V, et al. Ceramide concentrations in septic patients: a possible marker of multiple organ dysfunction syndrome. Crit Care Med. (1999) 27:2413–7. doi: 10.1097/00003246-199911000-00015
132. Jenkins RW, Clarke CJ, Lucas JTJr, Shabbir M, Wu BX, Simbari F, et al. Evaluation of the role of secretory sphingomyelinase and bioactive sphingolipids as biomarkers in hemophagocytic lymphohistiocytosis. Am J Hematol. (2013) 88:E265–72. doi: 10.1002/ajh.23535
133. Takahashi T, Abe T, Sato T, Miura K, Takahashi I, Yano M, et al. Elevated sphingomyelinase and hypercytokinemia in hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. (2002) 24:401–4. doi: 10.1097/00043426-200206000-00016
134. Roumestan C, Michel A, Bichon F, Portet K, Detoc M, Henriquet C, et al. Anti-inflammatory properties of desipramine and fluoxetine. Respir Res. (2007) 8:35. doi: 10.1186/1465-9921-8-35
135. Xia BT, Beckmann N, Winer LK, Pugh AM, Pritts TA, Nomellini V, et al. Amitriptyline reduces inflammation and mortality in a murine model of sepsis. Cell Physiol Biochem. (2019) 52:565–79. doi: 10.33594/000000040
136. Zhai R, Sheu CC, Su L, Gong MN, Tejera P, Chen F, et al. Serum bilirubin levels on ICU admission are associated with ARDS development and mortality in sepsis. Thorax. (2009) 64:784–90. doi: 10.1136/thx.2009.113464
137. Recknagel P, Gonnert FA, Westermann M, Lambeck S, Lupp A, Rudiger A, et al. Liver dysfunction and phosphatidylinositol-3-kinase signalling in early sepsis: experimental studies in rodent models of peritonitis. PLoS Med. (2012) 9:e1001338. doi: 10.1371/journal.pmed.1001338
138. Kulaksiz H, Heuberger D, Engler S, Stiehl A. Poor outcome in progressive sclerosing cholangitis after septic shock. Endoscopy. (2008) 40:214–8. doi: 10.1055/s-2007-967024
139. Lang E, Gatidis S, Freise NF, Bock H, Kubitz R, Lauermann C, et al. Conjugated bilirubin triggers anemia by inducing erythrocyte death. Hepatology. (2015) 61:275–84. doi: 10.1002/hep.27338
140. Reade MC, Weissfeld L, Angus DC, Kellum JA, Milbrandt EB. The prevalence of anemia and its association with 90-day mortality in hospitalized community-acquired pneumonia. BMC Pulm Med. (2010) 10:15. doi: 10.1186/1471-2466-10-15
141. McVey MJ, Kim M, Tabuchi A, Srbely V, Japtok L, Arenz C, et al. Acid sphingomyelinase mediates murine acute lung injury following transfusion of aged platelets. (2017). 312:L625–37. doi: 10.1152/ajplung.00317.2016
142. Albouz S, Hauw JJ, Berwald-Netter Y, Boutry JM, Bourdon R, Baumann N. Tricyclic antidepressants induce sphingomyelinase deficiency in fibroblast and neuroblastoma cell cultures. Biomedicine. (1981) 35:218–20
143. Hurwitz R, Ferlinz K, Sandhoff K. The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol Chem Hoppe Seyler. (1994) 375:447–50. doi: 10.1515/bchm3.1994.375.7.447
144. Kolzer M, Werth N, Sandhoff K. Interactions of acid sphingomyelinase and lipid bilayers in the presence of the tricyclic antidepressant desipramine. FEBS Lett. (2004) 559:96–8. doi: 10.1016/S0014-5793(04)00033-X
145. Kornhuber J, Tripal P, Reichel M, Terfloth L, Bleich S, Wiltfang J, et al. Identification of new functional inhibitors of acid sphingomyelinase using a structure-property-activity relation model. J Med Chem. (2008) 51:219–37. doi: 10.1021/jm070524a
146. Blaess M, Bibak N, Claus RA, Kohl M, Bonaterra GA, Kinscherf R, et al. NB 06: from a simple lysosomotropic aSMase inhibitor to tools for elucidating the role of lysosomes in signaling apoptosis and LPS-induced inflammation. Eur J Med Chem. (2018) 153:73–104. doi: 10.1016/j.ejmech.2017.09.021
147. Chung HY, Kollmey AS, Schrepper A, Kohl M, Blass MF, Stehr SN, et al. Adjustment of dysregulated ceramide metabolism in a murine model of sepsis-induced cardiac dysfunction. Int J Mol Sci. (2017) 18:839. doi: 10.3390/ijms18040839
148. Ferreira LF, Moylan JS, Gilliam LA, Smith JD, Nikolova-Karakashian M, Reid MB. Sphingomyelinase stimulates oxidant signaling to weaken skeletal muscle and promote fatigue. Am J Physiol Cell Physiol. (2010) 299:C552–60. doi: 10.1152/ajpcell.00065.2010
149. Ferreira LF, Moylan JS, Stasko S, Smith JD, Campbell KS, et al. Sphingomyelinase depresses force and calcium sensitivity of the contractile apparatus in mouse diaphragm muscle fibers. J Appl Physiol 1985. (2012). 112:1538–45. doi: 10.1152/japplphysiol.01269.2011
150. Bost ER, Frye GS, Ahn B, Ferreira LF. Diaphragm dysfunction caused by sphingomyelinase requires the p47(phox) subunit of NADPH oxidase. Respir Physiol Neurobiol. (2015) 205:47–52. doi: 10.1016/j.resp.2014.10.011
151. Hassouneh LK, Timofiychuk OA, Babenko NA. Acid sphingomyelinase inhibitors, imipramine and zoledronic acid, increase skeletal muscle tissue sensitivity to insulin action at old age. Gen Physiol Biophys. (2018) 37:163–74. doi: 10.4149/gpb_2017020
152. Cordeiro AV, Silva VRR, Pauli JR, da Silva ASR, Cintra DE, Moura LP, et al. The role of sphingosine-1-phosphate in skeletal muscle: physiology, mechanisms, and clinical perspectives. J Cell Physiol. (2019) 234:10047–59. doi: 10.1002/jcp.27870
153. Reiss LK, Raffetseder U, Gibbert L, Drescher HK, Streetz KL, Schwarz A, et al. Reevaluation of lung injury in TNF-induced shock: the role of the acid sphingomyelinase. Mediators Inflamm. (2020) 2020:3650508. doi: 10.1155/2020/3650508
154. Fink MP, Heard SO. Laboratory models of sepsis and septic shock. J Surg Res. (1990) 49:186–96. doi: 10.1016/0022-4804(90)90260-9
155. Barsacchi R, Perrotta C, Bulotta S, Moncada S, Borgese N, Clementi E. Activation of endothelial nitric-oxide synthase by tumor necrosis factor-alpha: a novel pathway involving sequential activation of neutral sphingomyelinase, phosphatidylinositol-3' kinase, and Akt. Mol Pharmacol. (2003) 63:886–95. doi: 10.1124/mol.63.4.886
156. Chung HY, Witt CJ, Jbeily N, Hurtado-Oliveros J, Giszas B, Lupp A, et al. Acid sphingomyelinase inhibition prevents development of sepsis sequelae in the murine liver. Sci Rep. (2017) 7:12348. doi: 10.1038/s41598-017-11837-2
157. Okuro RT, Machado MN, Casquilho NV, Jardim-Neto A, Roncally-Carvalho A, Atella GC, et al. The role of sphingolipid metabolism disruption on lipopolysaccharide-induced lung injury in mice. Pulm Pharmacol Ther. (2018) 50:100–10. doi: 10.1016/j.pupt.2018.04.008
158. Goggel R, Winoto-Morbach S, Vielhaber G, Imai Y, Lindner K, Brade L, et al. PAF-mediated pulmonary edema: a new role for acid sphingomyelinase and ceramide. Nat Med. (2004) 10:155–60. doi: 10.1038/nm977
159. Koch A, Horn A, Duckers H, Yagmur E, Sanson E, Bruensing J, et al. Increased liver stiffness denotes hepatic dysfunction and mortality risk in critically ill non-cirrhotic patients at a medical ICU. Crit Care. (2011) 15:R266. doi: 10.1186/cc10543
160. Nesseler N, Launey Y, Aninat C, Morel F, Malledant Y, Seguin P. Clinical review: the liver in sepsis. Crit Care. (2012) 16:235. doi: 10.1186/cc11381
161. Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol. (2017) 14:55–66. doi: 10.1038/nrgastro.2016.168
162. Horvatits T, Drolz A, Trauner M, Fuhrmann V. Liver injury and failure in critical illness. Hepatology. (2019) 70:2204–15. doi: 10.1002/hep.30824
163. Quillin RC III, Wilson GC, Nojima H, Freeman CM, Wang J, Schuster RM, et al. Inhibition of acidic sphingomyelinase reduces established hepatic fibrosis in mice. Hepatol Res. (2015) 45:305–14. doi: 10.1111/hepr.12352
164. Harbrecht BG, Frye RF, Zenati MS, Branch RA, Peitzman AB. Cytochrome P-450 activity is differentially altered in severely injured patients. Crit Care Med. (2005) 33:541–6. doi: 10.1097/01.CCM.0000155989.54344.E0
165. Kruger PS, Freir NM, Venkatesh B, Robertson TA, Roberts MS, Jones M. A preliminary study of atorvastatin plasma concentrations in critically ill patients with sepsis. Intensive Care Med. (2009) 35:717–21. doi: 10.1007/s00134-008-1358-3
166. Shah RR, Smith RL. Inflammation-induced phenoconversion of polymorphic drug metabolizing enzymes: hypothesis with implications for personalized medicine. Drug Metab Dispos. (2015) 43:400–10. doi: 10.1124/dmd.114.061093
167. Chung HY, Witt CJ, Hurtado-Oliveros J, Wickel J, Graler MH, Lupp A, et al. Acid sphingomyelinase inhibition stabilizes hepatic ceramide content and improves hepatic biotransformation capacity in a murine model of polymicrobial sepsis. Int J Mol Sci. (2018) 19:3163. doi: 10.3390/ijms19103163
168. Hauser W, Bernardy K, Uceyler N, Sommer C. Treatment of fibromyalgia syndrome with antidepressants: a meta-analysis. JAMA. (2009) 301:198–209. doi: 10.1001/jama.2008.944
169. Disease GBD, Injury I, Prevalence C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet. (2018) 392:1789–858. doi: 10.1016/S0140-6736(18)32279-7
171. Cipriani A, Salanti G, Furukawa TA, Egger M, Leucht S, Ruhe HG, et al. Antidepressants might work for people with major depression: where do we go from here? Lancet Psychiatry. (2018) 5:461–3. doi: 10.1016/S2215-0366(18)30133-0
172. Kornhuber J, Medlin A, Bleich S, Jendrossek V, Henkel AW, Wiltfang J, et al. High activity of acid sphingomyelinase in major depression. J Neural Transm. (2005) 112:1583–90. doi: 10.1007/s00702-005-0374-5
173. Askim A, Gustad LT, Paulsen J, Reitan SK, Mehl A, Mohus RM, et al. Anxiety and depression symptoms in a general population and future risk of bloodstream infection: the HUNT study. Psychosom Med. (2018) 80:673–9. doi: 10.1097/PSY.0000000000000619
174. Davydow DS, Ribe AR, Pedersen HS, Vestergaard M, Fenger-Gron M. The association of unipolar depression with thirty-day mortality after hospitalization for infection: a population-based cohort study in Denmark. J Psychosom Res. (2016) 89:32–8. doi: 10.1016/j.jpsychores.2016.08.006
175. Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. (2019) 18:41–58. doi: 10.1038/nrd.2018.168
176. Sun W, He S, Martinez-Romero C, Kouznetsova J, Tawa G, Xu M, et al. Synergistic drug combination effectively blocks ebola virus infection. Antiviral Res. (2017) 137:165–72. doi: 10.1016/j.antiviral.2016.11.017
177. Beckmann N, Sharma D, Gulbins E, Becker KA, Edelmann B. Inhibition of acid sphingomyelinase by tricyclic antidepressants and analogons. Front Physiol. (2014) 5:331. doi: 10.3389/fphys.2014.00331
178. Schloer S, Brunotte L, Goretzko J, Mecate-Zambrano A, Korthals N, Gerke V, et al. Targeting the endolysosomal host-SARS-CoV-2 interface by clinically licensed functional inhibitors of acid sphingomyelinase (FIASMA) including the antidepressant fluoxetine. Emerg Microbes Infect. 9:2245–2255. doi: 10.1101/2020.07.27.222836
Keywords: sphingomyelinase (SMase), ceramide (CER), sepsis, organ failure (OF), inhibitor, FIASMA
Citation: Chung H-Y and Claus RA (2021) Keep Your Friends Close, but Your Enemies Closer: Role of Acid Sphingomyelinase During Infection and Host Response. Front. Med. 7:616500. doi: 10.3389/fmed.2020.616500
Received: 12 October 2020; Accepted: 03 December 2020;
Published: 21 January 2021.
Edited by:
Alessandro Russo, University of Pisa, ItalyReviewed by:
Liana C. Silva, University of Lisbon, PortugalKatrin Anne Becker, University of Duisburg-Essen, Germany
Copyright © 2021 Chung and Claus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf A. Claus, cmFsZi5jbGF1c0BtZWQudW5pLWplbmEuZGU=