Abstract
Diabetic kidney disease (DKD) is the leading cause of end-stage renal disease and a main contributing factor for cardiovascular morbidity and mortality in patients with diabetes mellitus. Strategies employed to delay the progression of this pathology focus on the control of traditional risk factors, such as hyperglycemia, and elevated blood pressure. Although the intimate mechanisms involved in the onset and progression of DKD remain incompletely understood, inflammation is currently recognized as one of the main underlying processes. Untangling the mechanisms involved in the appearing of a harmful inflammatory response in the diabetic patient is crucial for the development of new therapeutic strategies. In this review, we focus on the inflammation-related pathogenic mechanisms involved in DKD and in the therapeutic utility of new anti-inflammatory strategies.
Introduction
Diabetic kidney disease (DKD) is a frequent complication in patients with diabetes mellitus (DM) and constitutes the first cause of kidney disease and an important risk factor for cardiovascular disease in this population (1, 2). DKD is characterized by a plethora of alterations including hemodynamic and metabolic abnormalities, the activation of the renin–angiotensin system (RAS), oxidative stress, and fibrosis, which together trigger the elevation of systemic and intraglomerular pressure, and the appearing of various symptoms related to the development of kidney failure: glomerular hypertrophy, proteinuria, and decreased glomerular filtration (3). Until recently, kidney involvement was only considered as the result of alterations in hemodynamic and metabolic factors; however, recent advances show that DKD is a complex and multifactorial process.
An increasing number of clinical and experimental studies have pointed out that inflammation contributes to the development and progression of DKD (4). Therefore, inflammatory mediators may represent new targets for the development of therapeutic strategies for the treatment of this complication. In this review, we discuss the pathophysiologic and therapeutic implications of the inflammatory cytokines in DKD.
Inflammatory Cytokines in The Setting of DKD
Diverse inflammatory parameters are predictors of the evolution of the DM (5, 6), including the initiation and progression of DKD (4). These include inflammatory cytokines which, in addition to their role as regulators of the immune response, also exert important actions as cardinal effectors of injury. The increase in the levels of these molecules in the diabetic patient leads to microvascular complications such as the development of nephropathy (7–9). Proinflammatory cytokines, both circulating and those synthesized and secreted by inflammatory cells in the renal tissue, are elevated in patients with DKD (10–13), being directly associated with urinary albumin excretion (UAE) levels and with clinical markers of glomerular and tubulointerstitial damage (14, 15) (Figure 1).
Figure 1
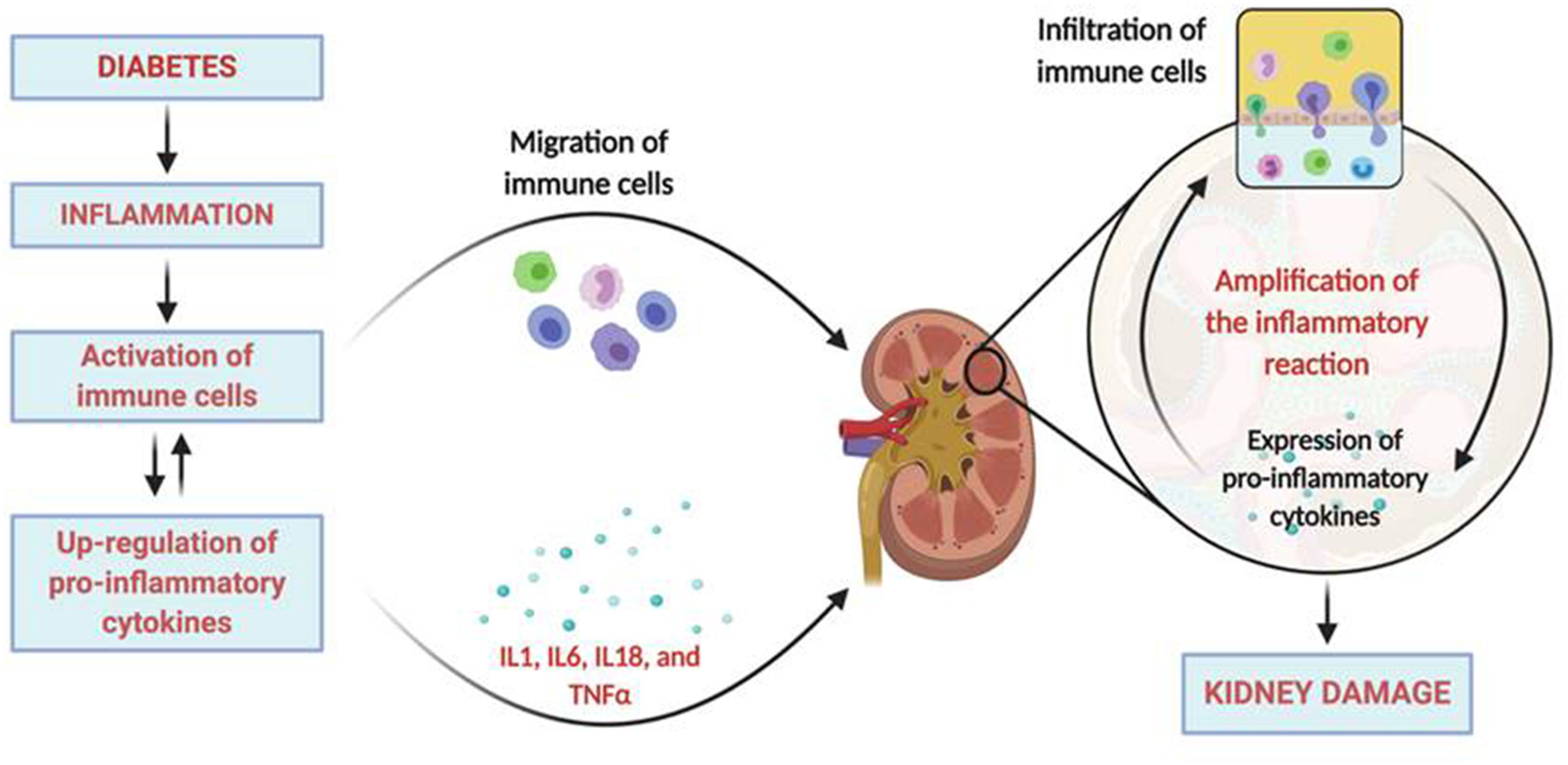
Inflammatory pathophysiology in diabetic kidney disease. The diabetic milieu drives to the development of inflammation that includes the activation of immune cells and the upregulation of pro-inflammatory cytokines. Activated immune cells migrates and infiltrates the renal tissue locally producing more inflammatory mediators and chemokines that recruit more immune cells into the kidney. Moreover, activated resident renal cells can also produce additional proinflammatory mediators, contributing to sustained inflammation, and the induction of kidney damage. Created with BioRender.com.
Urinary levels of interleukin (IL) 6 and IL8 are significantly increased in microalbuminuric diabetic patients with early progressive renal function decline when compared to microalbuminuric or normoalbuminuric subjects with stable renal function (16). IL6 levels are upregulated in patients with DM and nephropathy (17); in the same way, the expression of mRNA encoding IL6 is also increased in renal cells (glomerular, epithelial, mesangial), as well as in kidney infiltrating-cells in patients with DKD compared to diabetic patients without kidney disease (17). Importantly, the expression levels of IL6 mRNA are positively related with the severity of mesangial expansion, a characteristic histological manifestation of DKD.
IL18 is another potent pro-inflammatory cytokine that exhibits elevated serum and urinary levels in patients with DKD, showing significant and direct correlations with UAE levels and with the evolution of albuminuria (7, 18, 19). Additionally, serum levels of IL18 are associated with urinary β-2 microglobulin, a marker of tubular dysfunction (18). IL18 is capable of regulating the synthesis of pro-inflammatory molecules such as IL1 and tumor necrosis factor (TNF) α, and interferon (IFN) ɤ (20), in turn increasing chemokine receptors in mesangial cells (21). Furthermore, IL8 increases the expression of intercellular adhesion molecule 1 (ICAM-1) (22) and promotes endothelial cell apoptosis (23). In addition to infiltrating cells, kidney tubular cells of patients with DKD also express increased levels of IL18 (24), which is related to the activation of the mitogen-activated protein kinase (MAPK) pathways by transforming growth factor (TGF) β (25).
Both serum and urinary levels of the cytokine TNFα are also elevated in patients with DKD (26). Moreover, this increase parallels the progression of renal damage, which may indicate a relationship with the development and progression of renal impairment in the diabetic patient (7, 9, 27). In these subjects, the rise in TNFα has been related with renal damage mediated by cytotoxic effects (26). Similar to IL18, TNFα is also expressed in endothelial, epithelial, mesangial, and tubular renal cells (28, 29). Importantly, animal models of diabetes present increased levels of TNFα in kidney glomeruli and tubules (9, 30–33) being directly and independently related with UAE (32).
Animal models of DKD show upregulation of IL1 expression in many types of kidney cells (31, 34). This increment has been related with the expression of ICAM-1, the vascular cell adhesion molecule-1 (VCAM-1), and the endothelial-leukocyte adhesion molecule-1 (ELAM-1) (35, 36), molecules involved in chemotaxis and adhesion processes. Rat kidney mesangial cells are stimulated to produce prostaglandin E2 after being incubated with recombinant IL1 (37) in response to Ang II (38), which is related to the appearing of abnormalities in intraglomerular hemodynamics. Finally, IL1 has been also related with the production of hyaluronan in the proximal tubule (39), which is related with the development of experimental hypercellularity (40).
The cytokine IL17A is a member of the IL17 family mainly produced by activated T helper (Th) lymphocytes Th17 and, at lesser extent, by macrophages, neutrophils, natural killer, dendritic, and mast cells. Circulating IL17A levels have been related with the severity of kidney disease, showing a progressive decrease from subjects with normal glucose tolerance to subjects with DM with and without DN (41, 42). In opposite with these findings, other authors have found increased IL17A plasma values in patients with DN compared to healthy controls (43). These paradoxical findings might by the fact that the modulatory effects of IL17A on inflammation may be dependent on disease context, tissue, isoform, and receptor-ligand interactions (44).
Pathophysiologic Implications
The pathophysiological implications of the inflammation on DKD occur at various levels. In a first level, inflammatory cytokines, acting in a paracrine or autocrine form, trigger the epithelial-to-mesenchymal transition process in the kidney (45), causing extracellular matrix accumulation. Secondly, the up-regulation of chemoattractant cytokines and adhesion molecules stimulates the attraction of circulating cells and facilitate their infiltration into the renal tissue. Finally, these cells amplify the inflammatory reaction, generating more cytokines and other mediators that contribute to the development and progression of renal injury (Figure 1).
The first clue suggesting the existence of a pathogenic role of these molecules in the DKD was obtained in vitro by incubating macrophages in a glomerular basement membrane derived from diabetic rats; these macrophages presented increased production of IL1 and TNFα when compared to macrophages incubated with membranes from normal animals (8).
The interleukins IL1, IL6, and IL8, have been clearly related to renal disorders in DKD (31, 34). IL1 has been linked with the proliferation of mesangial cells and matrix synthesis, thereby altering the renal architecture, with increased permeability of vascular endothelial cells, and with intraglomerular hemodynamic abnormalities secondary to alterations in prostaglandin production (37, 46). In DKD patients, the expression of mRNA molecules encoding IL6 is positively related with the severity of mesangial expansion (17). The up-regulation of this cytokine have been also related to another functional and structural abnormalities related to DKD including increased vascular endothelium permeability (46), mesangial expansion and fibronectin expression (47), thickening of the glomerular basement membrane (48, 49), and renal hypertrophy (50). High serum and urine levels of the IL18 in type 2 diabetes have been proposed as early predictors of renal dysfunction, being related with the appearing of macroalbuminuria (18) and with the triggering of MAPK pathways by TGF-β (24). High serum TNFα levels have been clearly related with the pathophysiology of DKD including the development of renal hypertrophy and hyperfiltration (33). Levels of sodium retention, which is present in the early stages of DKD, has been related with urinary TNFα (51). Sodium retention in turn induce the expression of TGFβ and the development of renal hypertrophy (52). Moreover, the rise in both urinary and kidney-expressed TNFα levels precede the rise in albuminuria (53). These harmful effects elicited by TNFα in the kidney are mediated by the cytotoxicity on glomerular, mesangial, and epithelial cells (54–57). TNFα also change the permeability of endothelial cells, altering the equilibrium between vasoconstriction and vasodilation and the intraglomerular blood flow, which reduces glomerular filtration rate (GFR) independently from alterations in hemodynamic factors (58, 59). Moreover, TNFα also increases ROS levels in kidney cells independently of hemodynamic mechanisms, altering the glomerular capillary wall and, consequently, increasing UAE (51, 59, 60).
IL17A exerts mainly proinflammatory responses via the activation of the NF-κB pathway and downstream regulation of proinflammatory genes (61). In podocytes in vitro, IL17A increased the expression of IL 6 and TNFα, particularly under conditions of high glucose concentration (62). Similarly, cultured tubular epithelial cells stimulated with this cytokine also present up-regulation of proinflammatory gene expression and monocyte chemoattractant protein 1 (MCP1) production (62, 63). Importantly, IL17A also induced epithelial-to-mesenchymal transition in cultured proximal tubular epithelial cells (64), indicating that this could be another mechanism of renal damage triggered by this cytokine. In experimental models, administration of IL17A in mice significantly upregulated kidney Mcp-1 and Rantes gene expression and the recruitment of inflammatory cells to the kidney (65). In leptin deficient BTBR ob/ob mice, the administration of a neutralizing anti-IL17A antibody resulted in an inhibition of NF-κB activation (66). Similarly, neutralization of IL17A also decreased proinflammatory genes and inflammatory cell infiltration in an experimental angiotensin II-induced renal damage model (67). Taken together, these observations suggest that local IL17A production in diabetic kidneys could activate resident renal cells to produce proinflammatory cytokines and chemokines, such as MCP-1, thereby amplifying the inflammatory response. Although many studies have addressed circulating or urinary IL17A levels in DN patients, the determination of local renal levels of IL17A has not been investigated yet.
Inflammation As A Therapeutic Target in DKD
Up to the present time, the main therapeutic strategy to slow the development and progression of DKD focus on the tight regulation of glucose levels and blood pressure by the utilization of RAS blockers: angiotensin converting enzyme inhibitors (ACEi) and angiotensin II receptor blockers (ARBs). Although this therapeutic approximation reduces the risk of nephropathy progression, they do not completely halt the evolution toward end-stage renal disease (ESRD), with a still significant residual renal risk.
The important role played by inflammation, and specifically the involvement of inflammatory cytokines in the pathophysiology of ERD, constitute a new therapeutic opportunity in DKD to improve kidney function by the pharmacological reduction of the levels of these molecules (Table 1). Interestingly, the reduction in proteinuria and the slowdown in the progression of diabetic and non-diabetic nephropathies derived from RAS blockade has been related not only to hemodynamic/antihypertensive but also to anti-inflammatory/antifibrotic effects. This anti-inflammatory effect is mediated by the inhibition of NF-κB dependent pathways (68), which include the production of the pro-inflammatory cytokines IL6 and TNFα (69).
Table 1
| Drug | Primary target | Main outcomes in DKD | Anti-inflammatory effects | References |
|---|---|---|---|---|
| RAS blockers | Inhibition of ACE or blockade of angiotensin II receptor. | Reduce proteinuria and the progression of nephropathy. | Inhibition of NF-κB, MCP1 gene expression, and macrophage infiltration. | (68, 69) |
| SGLT2 inhibitors | Blockade of glucose reabsorption by SGLT2 at the proximal tubule. | Improved glycemic control. Slower progression of kidney disease and lower rates of clinically relevant renal events. | Reduction of inflammation by targeting the IL1ß and reduction of hsCRP, TNFα, IL6, and IFNγ. | (70–90) |
| DPP4 inhibitors and GLP-1 receptor agonists | Stimulation of glucose-dependent insulin release. | Improved glycemic control and body weight reductions. Renoprotective actions | Reduction in levels of inflammatory markers including CRP, TNFα, IL6, and IL18. | (91–97) |
| Pentoxifylline | Inhibition of phosphodiesterases. | Reduced progression of renal disease and proteinuria. | Downregulation of NF-κB signaling and reduction of inflammatory biomarkers. | (98–114) |
Therapeutic strategies in the DKD with potential anti-inflammatory properties.
RAAS, Renin-Angiotensin Aldosterone System; ACEI, angiotensin converting enzyme; NF-κB, nuclear factor-κB; MCP1, monocyte chemoattractant protein 1; SGLT2, type 2 sodium-glucose cotransporter; IL, interleukin; hsCRP, high sensitivity C reactive protein; TNFα, tumor necrosis factor α; IFN-γ, interferon γ; DPP4, dipeptidyl-peptidase-4; GLP-1, glucagon-like peptide-1.
In recent years, new antidiabetic drugs have emerged (Table 1). These drugs include SGLT2i (sodium-glucose cotransporter type 2 inhibitors), GLP-1RA (glucagon-like peptide-1 receptor agonist), and DPP-4i (dipeptidyl peptidase-4 inhibitors). These compounds improve albuminuria and other traits of DKD in diabetic patients but also have been demonstrated to exert anti-inflammatory effects with potential benefits in the delay of the progression of DKD. The group of SGLT2i (canagliflozin, dapagliflozin, and empagliflozin, among others) are very effective hypoglycemic agents that, beyond glycemic control, have shown potent cardiovascular and renal protective effects in the diabetic patient (70–72). The underlying mechanisms of these effects are not fully understood, but recent findings suggest that are related with the modulation of inflammatory cytokines at renal and systemic levels. A few small clinical pilot studies have shown reductions of inflammatory markers in type 2 diabetes patients treated with SGLT2i. In this sense, mildly reductions in serum concentrations of C-reactive protein (CRP), TNFα, IL6, IL1β, and IFN-γ were observed in diabetic patients treated with canagliflozin, dapagliflozin, and empagliflozin (74–80). While robust data on short and mainly long-term effects of SGLT2i are obtained from clinical studies with a larger number of patients, most of what is known about the anti-inflammatory actions of these drugs comes from experimental approximations, which have made possible to delve into the underlying mechanisms of this beneficial immunomodulatory effect. In HK2 cells, a line of human kidney proximal tubule cells, empagliflozin attenuated the expression of Toll-like receptor-4 (TLR4) induced by high glucose, NF-κB and activator protein1 (AP1) transcription factors binding to nuclear DNA, and secretion of collagen IV and IL6 (81). In vascular endothelial cells, canaglifozin inhibited IL1β-stimulated IL6 and MCP1 secretion by AMP-activated protein kinase dependent and independent mechanisms (82). In obese diabetic or prediabetic animals, SGLT2i reduces albuminuria and tubulointerstitial injury (83–85). Thereby, the slowing in the progression of renal complications in prediabetic rats treated with dapagliflozin was related with the suppression of renal inflammation (84). The expression of TNFα, IL1β, and IL6, as well as the infiltration of cells into atheromatous plaques, were reduced in the atherosclerosis mice model ApoE −/− after treatment with empagliflozin (86). Empagliflozin achieved significant reductions in albuminuria in animal models with type 1 diabetes that were related with lower levels of renal NF-κB, MCP1, and IL6 (87). The inhibition of SGLT2 in experimental type 2 diabetes models, reduced glomerular macrophage infiltration and sclerosis (88) and attenuated the overexpression of NOX4, TGFβ, osteopontin, and MCP1 in the tubular cells induced by high glucose (89).
Several clinical trials suggest that the anti-diabetic drugs DPP-4i and GLP-1RA also possess beneficial renal effects (90, 91), potentially modulating the inflammatory response. The DPP-4i vildagliptin and sitagliptin reduced the levels of markers of inflammation in diabetic patients (92). Administration of GLP-1RA, exenatide or dulaglutide, reduced the levels of CRP in diabetic patients (93). Different clinical trials are presently evaluating the effects of these anti-diabetic therapeutic agents on systemic and renal inflammation: GLP-1RA (exenatide or liraglutide) vs. a DPP-4i (NCT02150707) and liraglutide (LIRALBU; NCT02545738 and NCT01847313). In the meantime, the renoprotective effects derived from the treatment with GLP-1RA and DPP-4i have been demonstrated in the experimental setting employing animal models with kidney disease. The DPP-4i linagliptin reduced albuminuria, glomerulosclerosis, and tubulointerstitial fibrosis, independently of the glycemic control, in a mouse model of DKD (94). Sitagliptin and linagliptin also suppressed the activity of NLRP3/inflammasome in an experimental model of nephropathy (95). The model of type 2 diabetes db/db mice reduced albuminuria levels and mesangial expansion after treatment with the GLP-1RA exenatide (96). This treatment also reduced the levels of inflammatory cytokines and adhesion molecules in type 1 in diabetic rats (97).
The utilization of microRNAs (miRNAs) to regulate Th17 cell differentiation by targeting transcription factors like orphan receptor γt (RORγt) and the activation of STAT-3 or key cytokines involved in this process have been tested in different models of autoimmune disease (115–118). In the context of kidney disease, a recent study described that miRNA-155 deficiency promoted nephrin acetylation and decreased renal damage in a hyperglycemia-induced nephropathy mice model; importantly these effects were associated with a reduction in IL17A production through the up-regulation of the suppressor of cytokine signaling 1 (SOCS1) expression (119). Similarly, miR-155 knockout mice presented a significant reduction of the Th17 immune response, less severe nephritis, and reduced histologic and functional injury in an experimental model of nephritis (120).
Another potential therapeutic opportunity is the administration of neutralizing antibodies against IL17A for the treatment of chronic human inflammatory diseases is being tested in several ongoing clinical trials (121, 122). Anti-IL17A biological agents like secukinumab, ixekizumab, and brodalumab were more effective than diverse anti-TNF alpha agents such as infliximab, adalimumab, and etanercept, but less so than certolizumab. Unfortunately, although it seems clear that IL17A could play an important role in the inflammatory component of kidney disease, there are no clinical trials focused on the effects of anti-IL17A antibodies on renal inflammatory diseases like DKD or even lupus nephritis (123).
Pentoxifylline (PTX) is a methyl-xanthine clinically used for the treatment of intermittent claudication with anti-inflammatory properties. Both experimental and clinical studies in diabetic patients support the renal protective action of PTX, evidenced by a decrease in proteinuria and, in some cases, an improvement in GFR (98–101, 108). It is important to note that this antiproteinuric capacity is related to an anti-inflammatory effect (102–107), being associated with significant reductions in TNFα levels (108, 109). Similarly, CKD patients report a reduction in TNFα, fibrinogen, and CRP levels and a stabilization in kidney function after PTX treatment (110). In these same patients, 1-year treatment of PTX in addition to the ARB background therapy resulted in a reduction in proteinuria and urinary levels of TNFα and MCP1 (111). In the PREDIAN trial, DKD patients under RAS blockade and treated with PTX also presented a stabilization in the progression of kidney disease after 2 years of follow-up (112). Importantly, this outcome was accompanied by reductions in proteinuria and urinary TNFα levels. Two recent meta-analysis in patients with DKD indicate, on the one hand, that the antiproteinuric effect of PTX in these patients is due to a lower production of pro-inflammatory cytokines (113) and, on the other hand, that PTX additively reduces both proteinuria and TNFα levels in patients treated with RAS inhibitors (114).
Discussion
The dramatic increase in the incidence of diabetes has prompted new therapeutic strategies to treat DKD. Inflammation has revealed as a key factor in the development and progression of this complication, allowing the development of therapeutic approaches focused on the modulation of inflammatory processes. However, at the present time, clinical experience on the inhibition of inflammatory molecules and pathways in diabetic patients is scarce and more clinical trials are needed to examine their potential renoprotective efficacy.
Statements
Author contributions
JN-G, JD-C, and CF conceived the idea, prepared the final version, and finalized the manuscript. CF searched the literature, prepared the first and last part of the draft, and reviewed and prepared the revised version. FS-Q prepared the third part of the draft. AP-C, AG-L, EM-N, and CM-F reviewed the literature, critically reviewed the final version, and approved the final manuscript. All authors have substantially contributed to the preparation of the manuscript and agree with the study.
Funding
This work was supported by PI07/0870, PI15/00298, PI16/02057, PI16/00024, ISCIII-RETIC REDinREN RD16/0009, Sociedad Española de Nefrología (S.E.N.) and ACINEF. We acknowledge cofunding by Fondo Europeo de Desarrollo Regional (FEDER), Unión Europea (Una forma de hacer Europa). JD-C was recipient of a contract from Miguel Servet Programme (CP20/00122) of the Instituto de Salud Carlos III (ISCIII), CF was a recipient of a research contract from the ACIISI (Agencia Canaria de Investigación, Innovación y Sociedad de la Información-TESIS2018010110). EM-N was recipient of a research contract RD16/0009/0022 from REDinREN.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
RitzERychlíkILocatelliFHalimiS. End-stage renal failure in type 2 diabetes: a medical catastrophe of worldwide dimensions. Am J Kidney Dis. (1999) 34:795–808. 10.1016/S0272-6386(99)70035-1
2.
AtkinsRC. The epidemiology of chronic kidney disease. Kidney Int Suppl. (2005) S14–8. 10.1111/j.1523-1755.2005.09403.x
3.
WolfG. New insights into the pathophysiology of diabetic nephropathy: from haemodynamics to molecular pathology. Eur J Clin Invest. (2004) 34:785–96. 10.1111/j.1365-2362.2004.01429.x
4.
WadaJMakinoH. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci. (2013) 124:139–52. 10.1042/CS20120198
5.
SchmidtMIDuncanBBSharrettARLindbergGSavagePJOffenbacherSet al. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet. (1999) 353:1649–52. 10.1016/S0140-6736(99)01046-6
6.
SprangerJKrokeAMöhligMHoffmannKBergmannMMRistowMet al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. (2003) 52:812–7. 10.2337/diabetes.52.3.812
7.
NavarroJFMoraCMacaMGarcaJ. Inflammatory parameters are independently associated with urinary albumin in type 2 diabetes mellitus. Am J Kidney Dis. (2003) 42:53–61. 10.1016/S0272-6386(03)00408-6
8.
HasegawaGNakanoKSawadaMUnoKShibayamaYIenagaKet al. Possible role of tumor necrosis factor and interleukin-1 in the development of diabetic nephropathy. Kidney Int. (1991) 40:1007–12. 10.1038/ki.1991.308
9.
MoriwakiYYamamotoTShibutaniYAokiETsutsumiZTakahashiSet al. Elevated levels of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism. (2003) 52:605–8. 10.1053/meta.2003.50096
10.
ChowFOzolsENikolic-PatersonDJAtkinsRCTeschGH. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int. (2004) 65:116–28. 10.1111/j.1523-1755.2004.00367.x
11.
NguyenDPingFMuWHillPAtkinsRCChadbanSJ. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology. (2006) 11:226–31. 10.1111/j.1440-1797.2006.00576.x
12.
LimAKMaFYNikolic-PatersonDJKitchingARThomasMCTeschGH. Lymphocytes promote albuminuria, but not renal dysfunction or histological damage in a mouse model of diabetic renal injury. Diabetologia. (2010) 53:1772–82. 10.1007/s00125-010-1757-1
13.
FerenbachDKluthDCHughesJ. Inflammatory cells in renal injury and repair. Semin Nephrol. (2007) 27:250–9. 10.1016/j.semnephrol.2007.02.001
14.
BrunoGMerlettiFBiggeriABargeroGFerreroSPaganoGet al. Progression to overt nephropathy in type 2 diabetes: the casale monferrato study. Diabetes Care. (2003) 26:2150–5. 10.2337/diacare.26.7.2150
15.
FestaAD'AgostinoRHowardGMykkänenLTracyRPHaffnerSM. Inflammation and microalbuminuria in nondiabetic and type 2 diabetic subjects: the insulin resistance atherosclerosis study. Kidney Int. (2000) 58:1703–10. 10.1046/j.1523-1755.2000.00331.x
16.
WolkowPPNiewczasMAPerkinsBFicocielloLHLipinskiBWarramJHet al. Association of urinary inflammatory markers and renal decline in microalbuminuric type 1 diabetics. J Am Soc Nephrol. (2008) 19:789–97. 10.1681/ASN.2007050556
17.
SuzukiDMiyazakiMNakaRKojiTYagameMJindeKet al. In situ hybridization of interleukin 6 in diabetic nephropathy. Diabetes. (1995) 44:1233–8. 10.2337/diab.44.10.1233
18.
NakamuraAShikataKHiramatsuMNakatouTKitamuraTWadaJet al. Serum interleukin-18 levels are associated with nephropathy and atherosclerosis in Japanese patients with type 2 diabetes. Diabetes Care. (2005) 28:2890–5. 10.2337/diacare.28.12.2890
19.
WongCKHoAWTongPCYeungCYKongAPSLunSWMet al. Aberrant activation profile of cytokines and mitogen-activated protein kinases in type 2 diabetic patients with nephropathy. Clin Exp Immunol. (2007) 149:123–31. 10.1111/j.1365-2249.2007.03389.x
20.
OkamuraHTsutsiHKomatsuTYutsudoMHakuraATanimotoTet al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. (1995) 378:88–91. 10.1038/378088a0
21.
SchwarzMWahlMReschKRadekeHH. IFNgamma induces functional chemokine receptor expression in human mesangial cells. Clin Exp Immunol. (2002) 128:285–94. 10.1046/j.1365-2249.2002.01829.x
22.
StuytRJNeteaMGGeijtenbeekTBKullbergBJDinarelloCAvan der MeerJW. Selective regulation of intercellular adhesion molecule-1 expression by interleukin-18 and interleukin-12 on human monocytes. Immunology. (2003) 110:329–34. 10.1046/j.1365-2567.2003.01747.x
23.
MariñoECardierJE. Differential effect of IL-18 on endothelial cell apoptosis mediated by TNF-alpha and Fas (CD95). Cytokine. (2003) 22:142–8. 10.1016/S1043-4666(03)00150-9
24.
FantuzziGReedDADinarelloCA. IL-12-induced IFN-gamma is dependent on caspase-1 processing of the IL-18 precursor. J Clin Invest. (1999) 104:761–7. 10.1172/JCI7501
25.
MiyauchiKTakiyamaYHonjyoJTatenoMHanedaM. Upregulated IL-18 expression in type 2 diabetic subjects with nephropathy: TGF-beta1 enhanced IL-18 expression in human renal proximal tubular epithelial cells. Diabetes Res Clin Pract. (2009) 83:190–9. 10.1016/j.diabres.2008.11.018
26.
ChenYLQiaoYCXuYLingWPanYHHuangYCet al. Serum TNF-α concentrations in type 2 diabetes mellitus patients and diabetic nephropathy patients: a systematic review and meta-analysis. Immunol Lett. (2017) 186:52–8. 10.1016/j.imlet.2017.04.003
27.
KhoshakhlaghPBahrololoumi-ShapourabadiMMohammadiradAAshtaral-NakhaiLMinaieBAbdollahiM. Beneficial effect of phosphodiesterase-5 inhibitor in experimental inflammatory bowel disease; molecular evidence for involvement of oxidative stress. Toxicol Mech Methods. (2007) 17:281–8. 10.1080/15376510601003769
28.
SugimotoHShikataKWadaJHoriuchiSMakinoH. Advanced glycation end products-cytokine-nitric oxide sequence pathway in the development of diabetic nephropathy: aminoguanidine ameliorates the overexpression of tumour necrosis factor-alpha and inducible nitric oxide synthase in diabetic rat glomeruli. Diabetologia. (1999) 42:878–86. 10.1007/s001250051241
29.
JevnikarAMBrennanDCSingerGGHengJEMaslinskiWWuthrichRPet al. Stimulated kidney tubular epithelial cells express membrane associated and secreted TNF alpha. Kidney Int. (1991) 40:203–11. 10.1038/ki.1991.201
30.
NavarroJFMoraCMurosMGarcíaJ. Urinary tumour necrosis factor-alpha excretion independently correlates with clinical markers of glomerular and tubulointerstitial injury in type 2 diabetic patients. Nephrol Dial Transplant. (2006) 21:3428–34. 10.1093/ndt/gfl469
31.
NavarroJFMilenaFJMoraCLeónCGarcíaJ. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J Nephrol. (2006) 26:562–70. 10.1159/000098004
32.
NavarroJFMilenaFJMoraCLeónCClaverieFFloresCet al. Tumor necrosis factor-alpha gene expression in diabetic nephropathy: relationship with urinary albumin excretion and effect of angiotensin-converting enzyme inhibition. Kidney Int Suppl. (2005) 99:S98–102. 10.1111/j.1523-1755.2005.09918.x
33.
DiPetrilloKGesekFA. Pentoxifylline ameliorates renal tumor necrosis factor expression, sodium retention, and renal hypertrophy in diabetic rats. Am J Nephrol. (2004) 24:352–9. 10.1159/000079121
34.
Sassy-PrigentCHeudesDMandetCBélairMFMichelOPerdereauBet al. Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes. (2000) 49:466–75. 10.2337/diabetes.49.3.466
35.
BradyHR. Leukocyte adhesion molecules and kidney diseases. Kidney Int. (1994) 45:1285–300. 10.1038/ki.1994.169
36.
ParkCWKimJHLeeJHKimYSAhnHJShinYSet al. High glucose-induced intercellular adhesion molecule-1 (ICAM-1) expression through an osmotic effect in rat mesangial cells is PKC-NF-kappa B-dependent. Diabetologia. (2000) 43:1544–53. 10.1007/s001250051567
37.
PfeilschifterJPignatWVosbeckKMärkiF. Interleukin 1 and tumor necrosis factor synergistically stimulate prostaglandin synthesis and phospholipase A2 release from rat renal mesangial cells. Biochem Biophys Res Commun. (1989) 159:385–94. 10.1016/0006-291X(89)90003-X
38.
PfeilschifterJMühlH. Interleukin 1 and tumor necrosis factor potentiate angiotensin II- and calcium ionophore-stimulated prostaglandin E2 synthesis in rat renal mesangial cells. Biochem Biophys Res Commun. (1990) 169:585–95. 10.1016/0006-291X(90)90371-S
39.
JonesSJonesSPhillipsAO. Regulation of renal proximal tubular epithelial cell hyaluronan generation: implications for diabetic nephropathy. Kidney Int. (2001) 59:1739–49. 10.1046/j.1523-1755.2001.0590051739.x
40.
MahadevanPLarkinsRGFraserJRFosangAJDunlopME. Increased hyaluronan production in the glomeruli from diabetic rats: a link between glucose-induced prostaglandin production and reduced sulphated proteoglycan. Diabetologia. (1995) 38:298–305. 10.1007/BF00400634
41.
VasanthakumarRMohanVAnandGDeepaMBabuSAravindhanV. Serum IL-9, IL-17, and TGF-beta levels in subjects with diabetic kidney disease (CURES-134). Cytokine. (2015) 72:109–12. 10.1016/j.cyto.2014.10.009
42.
MohamedRJayakumarCChenFFultonDSteppDGansevoortRTet al. Low-dose IL-17 therapy prevents and reverses diabetic nephropathy, metabolic syndrome, and associated organ fibrosis. J Am Soc Nephrol. (2016) 27:745–65. 10.1681/ASN.2014111136
43.
ZhangNTaiJQuZZhangZZhaoSHeJet al. Increased CD4+CXCR5+T folicular helper cells in diabetic nephropathy. Autoimmunity. (2016) 49:405–13. 10.1080/08916934.2016.1196677
44.
GalvanDLDaneshFR. Paradoxical role of IL-17 in progression of diabetic nephropathy. J Am Soc Nephrol. (2016) 27:657–8. 10.1681/ASN.2015070813
45.
LiuY. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. (2011) 7:684–96. 10.1038/nrneph.2011.149
46.
RoyallJABerkowRLBeckmanJSCunninghamMKMatalonSFreemanBA. Tumor necrosis factor and interleukin 1 alpha increase vascular endothelial permeability. Am J Physiol. (1989) 257(6 Pt 1):L399–410. 10.1152/ajplung.1989.257.6.L399
47.
ColemanDLRuefC. Interleukin-6: an autocrine regulator of mesangial cell growth. Kidney Int. (1992) 41:604–6. 10.1038/ki.1992.91
48.
NosadiniRVelussiMBroccoEBruseghinMAbaterussoCSallerAet al. Course of renal function in type 2 diabetic patients with abnormalities of albumin excretion rate. Diabetes. (2000) 49:476–84. 10.2337/diabetes.49.3.476
49.
Dalla VestraMMussapMGallinaPBruseghinMCernigoiAMSallerAet al. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J Am Soc Nephrol. (2005) 16(Suppl. 1):S78–82. 10.1681/ASN.2004110961
50.
ArakiSHanedaMKoyaDSugimotoTIsshikiKChin-KanasakiMet al. Predictive impact of elevated serum level of IL-18 for early renal dysfunction in type 2 diabetes: an observational follow-up study. Diabetologia. (2007) 50:867–73. 10.1007/s00125-006-0586-8
51.
DiPetrilloKCoutermarshBGesekFA. Urinary tumor necrosis factor contributes to sodium retention and renal hypertrophy during diabetes. Am J Physiol Renal Physiol. (2003) 284:F113–21. 10.1152/ajprenal.00026.2002
52.
YuHCBurrellLMBlackMJWuLLDilleyRJCooperMEet al. Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation. (1998) 98:2621–8. 10.1161/01.CIR.98.23.2621
53.
KalantariniaKAwadASSiragyHM. Urinary and renal interstitial concentrations of TNF-alpha increase prior to the rise in albuminuria in diabetic rats. Kidney Int. (2003) 64:1208–13. 10.1046/j.1523-1755.2003.00237.x
54.
OrtizABustosCAlonsoJAlcázarRLópez-ArmadaMJPlazaJJet al. Involvement of tumor necrosis factor-alpha in the pathogenesis of experimental and human glomerulonephritis. Adv Nephrol Necker Hosp. (1995) 24:53–77.
55.
BertaniTAbbateMZojaCCornaDPericoNGhezziVet al. Tumor necrosis factor induces glomerular damage in the rabbit. Am J Pathol. (1989) 134:419–30.
56.
LasterSMWoodJGGoodingLR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol. (1988) 141:2629–34.
57.
BoyleJJWeissbergPLBennettMR. Tumor necrosis factor-alpha promotes macrophage-induced vascular smooth muscle cell apoptosis by direct and autocrine mechanisms. Arterioscler Thromb Vasc Biol. (2003) 23:1553–8. 10.1161/01.ATV.0000086961.44581.B7
58.
BaudLPerezJFriedlanderGArdaillouR. Tumor necrosis factor stimulates prostaglandin production and cyclic AMP levels in rat cultured mesangial cells. FEBS Lett. (1988) 239:50–4. 10.1016/0014-5793(88)80543-X
59.
McCarthyETSharmaRSharmaMLiJZGeXLDileepanKNet al. TNF-alpha increases albumin permeability of isolated rat glomeruli through the generation of superoxide. J Am Soc Nephrol. (1998) 9:433–8.
60.
KoikeNTakamuraTKanekoS. Induction of reactive oxygen species from isolated rat glomeruli by protein kinase C activation and TNF-alpha stimulation, and effects of a phosphodiesterase inhibitor. Life Sci. (2007) 80:1721–8. 10.1016/j.lfs.2007.02.001
61.
CortvrindtCSpeeckaertRMoermanADelangheJRSpeeckaertMM. The role of interleukin-17A in the pathogenesis of kidney diseases. Pathology. (2017) 49:247–58. 10.1016/j.pathol.2017.01.003
62.
MaJLiYJChenXKwanTChadbanSJWuH. Interleukin 17A promotes diabetic kidney injury. Sci Rep. (2019) 9:2264. 10.1038/s41598-019-38811-4
63.
Van KootenCBoonstraJGPaapeMEFossiezFBanchereauJLebecqueSet al. Interleukin-17 activates human renal epithelial cells in vitro is expressed during renal allograft rejection. J Am Soc Nephrol. (1998) 9:1526–34.
64.
DudasPLSagueSLEllosoMMFarrellFX. Proinflammatory/profibrotic effects of interleukin-17A on human proximal tubule epithelium. Nephron Exp Nephrol. (2011) 117:114–23. 10.1159/000320177
65.
OrejudoMRodrigues-DiezRRRodrigues-DiezRGarcia-RedondoASantos-SanchezLRandez-GarbayoJet al. Interleukin 17A participates in renal inflammation associated to experimental and human hypertension. Front Pharmacol. (2019) 10:1015. 10.3389/fphar.2019.01015
66.
LavozCMatusYSOrejudoMCarpioJDDroguettAEgidoJet al. Interleukin-17A blockade reduces albuminuria and kidney injury in an accelerated model of diabetic nephropathy. Kidney Int. (2019) 95:1418–32. 10.1016/j.kint.2018.12.031
67.
SalehMANorlanderAEMadhurMS. Inhibition of interleukin-17A, but not interleukin-17F, signaling lowers blood pressure, and reduces end-organ inflammation in angiotensin ii–induced hypertension. JACC Basic Transl Sci. (2016) 1:606–16. 10.1016/j.jacbts.2016.07.009
68.
LeeFTCaoZLongDMPanagiotopoulosSJerumsGCooperMEet al. Interactions between angiotensin II and NF-kappaB-dependent pathways in modulating macrophage infiltration in experimental diabetic nephropathy. J Am Soc Nephrol. (2004) 15:2139–51. 10.1097/01.ASN.0000135055.61833.A8
69.
SanzABSanchez-NiñoMDRamosAMMorenoJASantamariaBRuiz-OrtegaMet al. NF-kappaB in renal inflammation. J Am Soc Nephrol. (2010) 21:1254–62. 10.1681/ASN.2010020218
70.
WannerCInzucchiSELachinJMFitchettDvon EynattenMMattheusMet al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med. (2016) 375:323–34. 10.1056/NEJMoa1515920
71.
PerkovicVde ZeeuwDMahaffeyKWFulcherGEronduNShawWet al. Canagliflozin and renal outcomes in type 2 diabetes: results from the CANVAS program randomised clinical trials. Lancet Diabetes Endocrinol. (2018) 6:691–704. 10.1016/S2213-8587(18)30141-4
72.
PerkovicVJardineMJNealBBompointSHeerspinkHJLCharytanDMet al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N Engl J Med. (2019) 380:2295–306. 10.1056/NEJMoa1811744
73.
MatsumuraMNakataniYTankaSAokiCSagaraMYanagiKet al. Efficacy of additional canagliflozin administration to type 2 diabetes patients receiving insulin therapy: examination of diurnal glycemic patterns using continuous glucose monitoring (CGM). Diabetes Ther. (2017) 8:821–7. 10.1007/s13300-017-0274-3
74.
GarveyWTVan GaalLLeiterLAVijapurkarUListJCuddihyRet al. Effects of canagliflozin versus glimepiride on adipokines and inflammatory biomarkers in type 2 diabetes. Metabolism. (2018) 85:32–7. 10.1016/j.metabol.2018.02.002
75.
FerranniniERamosSJSalsaliATangWListJF. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. (2010) 33:2217–24. 10.2337/dc10-0612
76.
OkamotoAYokokawaHSanadaHNaitoT. Changes in levels of biomarkers associated with adipocyte function and insulin and glucagon kinetics during treatment with dapagliflozin among obese type 2 diabetes mellitus patients. Drugs R D. (2016) 16:255–61. 10.1007/s40268-016-0137-9
77.
HattoriS. Empagliflozin decreases remnant-like particle cholesterol in type 2 diabetes patients with insulin resistance. J Diabetes Investig. (2018) 9:870–4. 10.1111/jdi.12781
78.
TobitaHSatoSMiyakeTIshiharaSKinoshitaY. Effects of Dapagliflozin on body composition and liver tests in patients with nonalcoholic steatohepatitis associated with type 2 diabetes mellitus: a prospective, open-label, uncontrolled study. Curr Ther Res Clin Exp. (2017) 87:13–9. 10.1016/j.curtheres.2017.07.002
79.
SogaFTanakaHTatsumiKMochizukiYSanoHTokiHet al. Impact of dapagliflozin on left ventricular diastolic function of patients with type 2 diabetic mellitus with chronic heart failure. Cardiovasc Diabetol. (2018) 17:132. 10.1186/s12933-018-0775-z
80.
KimSRLeeSGKimSHKimJHChoiEChoWet al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun. (2020) 11:2127. 10.1038/s41467-020-15983-6
81.
PanchapakesanUPeggKGrossSKomalaMGMudaliarHForbesJet al. Effects of SGLT2 inhibition in human kidney proximal tubular cells–renoprotection in diabetic nephropathy?PLoS ONE. (2013) 8:e54442. 10.1371/journal.pone.0054442
82.
ManciniSJBoydDKatwanOJStrembitskaAAlmabroukTAKennedySet al. Canagliflozin inhibits interleukin-1β-stimulated cytokine and chemokine secretion in vascular endothelial cells by AMP-activated protein kinase-dependent and -independent mechanisms. Sci Rep. (2018) 8:5276. 10.1038/s41598-018-23420-4
83.
IshibashiYMatsuiTYamagishiS. Tofogliflozin, a highly selective inhibitor of SGLT2 blocks proinflammatory and proapoptotic effects of glucose overload on proximal tubular cells partly by suppressing oxidative stress generation. Horm Metab Res. (2016) 48:191–5. 10.1055/s-0035-1555791
84.
JaikumkaoKPongchaidechaAChueakulaNThongnakLOWanchaiKChatsudthipongVet al. Dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, slows the progression of renal complications through the suppression of renal inflammation, endoplasmic reticulum stress and apoptosis in prediabetic rats. Diabetes Obes Metab. (2018) 20:2617–26. 10.1111/dom.13441
85.
IshibashiYMatsuiTYamagishiSI. Tofogliflozin, a selective inhibitor of sodium-glucose cotransporter 2, suppresses renal damage in KKAy/Ta mice, obese and type 2 diabetic animals. Diab Vasc Dis Res. (2016) 13:438–41. 10.1177/1479164116657304
86.
HanJHOhTJLeeGThongnakLOWanchaiKChatsudthipongVet al. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE −/− mice fed a western diet. Diabetologia. (2017) 60:364–76. 10.1007/s00125-016-4158-2
87.
VallonVGerasimovaMRoseMAMasudaTSatrianoJMayouxEet al. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol. (2014) 306:F194–204. 10.1152/ajprenal.00520.2013
88.
LinBKoibuchiNHasegawaYSuetaDToyamaKUekawaKet al. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc Diabetol. (2014) 13:148. 10.1186/s12933-014-0148-1
89.
TeramiNOgawaDTachibanaHHatanakaTWadaJNakatsukaAet al. Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS ONE. (2014) 9:e100777. 10.1371/journal.pone.0100777
90.
HattoriS. Sitagliptin reduces albuminuria in patients with type 2 diabetes. Endocr J. (2011) 58:69–73. 10.1507/endocrj.K10E-382
91.
SakataKHayakawaMYanoYTamakiNYokotaNEtoTet al. Efficacy of alogliptin, a dipeptidyl peptidase-4 inhibitor, on glucose parameters, the activity of the advanced glycation end product (AGE) - receptor for AGE (RAGE) axis and albuminuria in Japanese type 2 diabetes. Diabetes Metab Res Rev. (2013) 29:624–30. 10.1002/dmrr.2437
92.
BarbieriMRizzoMRMarfellaRBoccardiVEspositoAPansiniAet al. Decreased carotid atherosclerotic process by control of daily acute glucose fluctuations in diabetic patients treated by DPP-IV inhibitors. Atherosclerosis. (2013) 227:349–54. 10.1016/j.atherosclerosis.2012.12.018
93.
FerdinandKCWhiteWBCalhounDALonnEMSagerPTBrunelleRet al. Effects of the once-weekly glucagon-like peptide-1 receptor agonist dulaglutide on ambulatory blood pressure and heart rate in patients with type 2 diabetes mellitus. Hypertension. (2014) 64:731–7. 10.1161/HYPERTENSIONAHA.114.03062
94.
KanasakiK. The role of renal dipeptidyl peptidase-4 in kidney disease: renal effects of dipeptidyl peptidase-4 inhibitors with a focus on linagliptin. Clin Sci. (2018) 132:489–507. 10.1042/CS20180031
95.
JoCHKimSParkJSKimGH. Anti-Inflammatory Action of Sitagliptin and Linagliptin in Doxorubicin Nephropathy. Kidney Blood Press Res. (2018) 43:987–9. 10.1159/000490688
96.
KawanamiDMatobaKSangoKUtsunomiyaK. Incretin-Based Therapies for Diabetic Complications: Basic Mechanisms and Clinical Evidence. Int J Mol Sci. (2016) 17:1223. 10.3390/ijms17081223
97.
KoderaRShikataKKataokaHUTakatsukaTMiyamotoSSasakiMet al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia. (2011) 54:965–78. 10.1007/s00125-010-2028-x
98.
BlagosklonnaiaIAVMamedovRKozlovVVEmanuél'VLKudriashovaMI. Effect of trental on indices kidney function in diabetes mellitus. Probl Endokrinol. (1982) 28:3–8.
99.
AminorroayaAJanghorbaniMRezvanianHAminianTGharaviMAminiM. Comparison of the effect of pentoxifylline and captopril on proteinuria in patients with type 2 diabetes mellitus. Nephron Clin Pract. (2005) 99:c73–7. 10.1159/000083417
100.
Rodríguez-MoránMGuerrero-RomeroF. Pentoxifylline is as effective as captopril in the reduction of microalbuminuria in non-hypertensive type 2 diabetic patients–a randomized, equivalent trial. Clin Nephrol. (2005) 64:91–7. 10.5414/CNP64091
101.
NavarroJFMoraCMurosMGarcíaJ. Additive antiproteinuric effect of pentoxifylline in patients with type 2 diabetes under angiotensin II receptor blockade: a short-term, randomized, controlled trial. J Am Soc Nephrol. (2005) 16:2119–26. 10.1681/ASN.2005010001
102.
DohertyGMJensenJCAlexanderHRBureshCMNortonJA. Pentoxifylline suppression of tumor necrosis factor gene transcription. Surgery. (1991) 110:192–8.
103.
VoisinLBreuilléDRuotBRallièreCRambourdinFDalleMet al. Cytokine modulation by PX differently affects specific acute phase proteins during sepsis in rats. Am J Physiol. (1998) 275:R1412–9. 10.1152/ajpregu.1998.275.5.R1412
104.
StrutzFHeegMKochsiekTSiemersGZeisbergMMüllerGA. Effects of pentoxifylline, pentifylline and gamma-interferon on proliferation, differentiation, and matrix synthesis of human renal fibroblasts. Nephrol Dial Transplant. (2000) 15:1535–46. 10.1093/ndt/15.10.1535
105.
Abdel-SalamOMBaiuomyAREl-ShenawySMArbidMS. The anti-inflammatory effects of the phosphodiesterase inhibitor pentoxifylline in the rat. Pharmacol Res. (2003) 47:331–40. 10.1016/S1043-6618(03)00002-1
106.
Dávila-EsquedaMEMartínez-MoralesF. Pentoxifylline diminishes the oxidative damage to renal tissue induced by streptozotocin in the rat. Exp Diabesity Res. (2004) 5:245–51. 10.1080/154386090897974
107.
Navarro-GonzálezJFMora-FernándezC. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol. (2008) 19:433–42. 10.1681/ASN.2007091048
108.
NavarroJFMoraCRiveroAGallegoEChahinJMacíaMet al. Urinary protein excretion and serum tumor necrosis factor in diabetic patients with advanced renal failure: effects of pentoxifylline administration. Am J Kidney Dis. (1999) 33:458–63. 10.1016/S0272-6386(99)70182-4
109.
Rodriguez-MoránMGonzález-GonzálezGBermúdez-BarbaMVMedina de la GarzaCETamez-PérezHEMartínez-MartínezFJet al. Effects of pentoxifylline on the urinary protein excretion profile of type 2 diabetic patients with microproteinuria: a double-blind, placebo-controlled randomized trial. Clin Nephrol. (2006) 66:3–10. 10.5414/CNP66003
110.
GoicoecheaMGarcía de VinuesaSQuirogaBVerdallesUBarracaDYusteCet al. Effects of pentoxifylline on inflammatory parameters in chronic kidney disease patients: a randomized trial. J Nephrol. (2012) 25:969–75. 10.5301/jn.5000077
111.
LinSLChenYMChiangWCWuKDTsaiTJ. Effect of pentoxifylline in addition to losartan on proteinuria and GFR in CKD: a 12-month randomized trial. Am J Kidney Dis. (2008) 52:464–74. 10.1053/j.ajkd.2008.05.012
112.
Navarro-GonzálezJFMora-FernándezCMuros de FuentesMChahinJMéndezMLGallegoEet al. Effect of pentoxifylline on renal function and urinary albumin excretion in patients with diabetic kidney disease: the PREDIAN trial. J Am Soc Nephrol. (2015) 26:220–9. 10.1681/ASN.2014010012
113.
McCormickBBSydorAAkbariAFergussonDDoucetteSKnollG. The effect of pentoxifylline on proteinuria in diabetic kidney disease: a meta-analysis. Am J Kidney Dis. (2008) 52:454–63. 10.1053/j.ajkd.2008.01.025
114.
TianMLShenYSunZLZhaY. Efficacy and safety of combining pentoxifylline with angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker in diabetic nephropathy: a meta-analysis. Int Urol Nephrol. (2015) 47:815–22. 10.1007/s11255-015-0968-2
115.
ZhuEWangXZhengBWangQHaoJChenSet al. miR-20b suppresses Th17 differentiation and the pathogenesis of experimental autoimmune encephalomyelitis by targeting RORgammat and STAT3. J. Immunol. (2014) 192:5599–609. 10.4049/jimmunol.1303488
116.
QuXZhouJWangTHanJMaLYuHet al. MiR-30a inhibits Th17 differentiation and demyelination of EAE mice by targeting the IL-21R. Brain Behav Immun. (2016) 57:193–9. 10.1016/j.bbi.2016.03.016
117.
LiBWangXChoiIYWangYCLiuSPhamATet al. MIR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J Clin Investig. (2017) 127:3702–16. 10.1172/JCI94012
118.
NaghavianRGhaediKKiani-EsfahaniAGanjalikhani-HakemiMEtemadifarMNasr-EsfahaniMH. miR-141 and miR-200a, revelation of new possible players in modulation of Th17/treg differentiation and pathogenesis of multiple sclerosis. PLoS ONE. (2015) 10:e0124555. 10.1371/journal.pone.0124555
119.
LinXYouYWangJQinYHuangPYangF. MicroRNA-155 deficiency promotes nephrin acetylation and attenuates renal damage in hyperglycemia-induced nephropathy. Inflammation. (2015) 38:546–54. 10.1007/s10753-014-9961-7
120.
KrebsCFKapfferSPaustHJSchmidtTBennsteinSBPetersAet al. MicroRNA-155 drives T H 17 immune response and tissue injury in experimental crescentic GN. J Am Soc Nephrol. (2013) 24:1955–65. 10.1681/ASN.2013020130
121.
MeasePVan der HeijdeDLandeweRMpofuSRahmanPTahirHet al. Secukinumab improves active psoriatic arthritis symptoms and inhibits radiographic progression: primary results from the randomised, double-blind, phase III FUTURE 5 study. Ann Rheum Dis. (2018) 77:890–7. 10.1136/annrheumdis-2017-212687
122.
SbidianEChaimaniAGarcia-DovalIDoGHuaCMazaudCet al. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochrane Database Syst Rev. (2017) 12:CD011535. 10.1002/14651858.CD011535.pub2
123.
LavozCRayego-MateosSOrejudoMOpazo-RíosLMarchantVMarquez-ExpositoLet al. Could IL-17A be a novel therapeutic target in diabetic nephropathy?J Clin Med. (2020) 9:272. 10.3390/jcm9010272
Summary
Keywords
diabetic kidney disease, inflammation, inflammatory cytokines, SGLT2i, GLP-1RA, DPP-4i, pentoxifylline
Citation
Donate-Correa J, Ferri CM, Sánchez-Quintana F, Pérez-Castro A, González-Luis A, Martín-Núñez E, Mora-Fernández C and Navarro-González JF (2021) Inflammatory Cytokines in Diabetic Kidney Disease: Pathophysiologic and Therapeutic Implications. Front. Med. 7:628289. doi: 10.3389/fmed.2020.628289
Received
11 November 2020
Accepted
24 December 2020
Published
22 January 2021
Volume
7 - 2020
Edited by
Michele Provenzano, University of Catanzaro, Italy
Reviewed by
Claudia Torino, Italian National Research Council, Italy; Antonio Bellasi, Papa Giovanni XXIII Hospital, Italy
Updates
Copyright
© 2021 Donate-Correa, Ferri, Sánchez-Quintana, Pérez-Castro, González-Luis, Martín-Núñez, Mora-Fernández and Navarro-González.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan F. Navarro-González jnavgon@gobiernodecanarias.org
This article was submitted to Nephrology, a section of the journal Frontiers in Medicine
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.