 Ceylan Onursal
Ceylan Onursal Elisabeth Dick
Elisabeth Dick Ilias Angelidis
Ilias Angelidis Herbert B. Schiller
Herbert B. Schiller Claudia A. Staab-Weijnitz
Claudia A. Staab-Weijnitz- Comprehensive Pneumology Center, Institute of Lung Biology and Disease, Helmholtz-Zentrum München, Member of the German Center of Lung Research (DZL), Munich, Germany
In addition to providing a macromolecular scaffold, the extracellular matrix (ECM) is a critical regulator of cell function by virtue of specific physical, biochemical, and mechanical properties. Collagen is the main ECM component and hence plays an essential role in the pathogenesis and progression of chronic lung disease. It is well-established that many chronic lung diseases, e.g., chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF) primarily manifest in the elderly, suggesting increased susceptibility of the aged lung or accumulated alterations in lung structure over time that favour disease. Here, we review the main steps of collagen biosynthesis, processing, and turnover and summarise what is currently known about alterations upon lung ageing, including changes in collagen composition, modification, and crosslinking. Recent proteomic data on mouse lung ageing indicates that, while the ER-resident machinery of collagen biosynthesis, modification and triple helix formation appears largely unchanged, there are specific changes in levels of type IV and type VI as well as the two fibril-associated collagens with interrupted triple helices (FACIT), namely type XIV and type XVI collagens. In addition, levels of the extracellular collagen crosslinking enzyme lysyl oxidase are decreased, indicating less enzymatically mediated collagen crosslinking upon ageing. The latter contrasts with the ageing-associated increase in collagen crosslinking by advanced glycation endproducts (AGEs), a result of spontaneous reactions of protein amino groups with reactive carbonyls, e.g., from monosaccharides or reactive dicarbonyls like methylglyoxal. Given the slow turnover of extracellular collagen such modifications accumulate even more in ageing tissues. In summary, the collective evidence points mainly toward age-induced alterations in collagen composition and drastic changes in the molecular nature of collagen crosslinks. Future work addressing the consequences of these changes may provide important clues for prevention of lung disease and for lung bioengineering and ultimately pave the way to novel targeted approaches in lung regenerative medicine.
Introduction
The extracellular matrix (ECM) is a highly dynamic non-cellular component of tissues that provides a complex structural network which serves as a scaffold for adherent and migrating cells. It is mainly composed of collagens, glycoproteins, proteoglycans, glycosaminoglycans, and several other components. By virtue of sequestered growth factors and ECM binding receptors at the surface of adherent cells, the ECM affects a plethora of cellular processes including cell proliferation, differentiation and migration and thus acts as a critical regulator of cell function (1, 2). It is well-established that the ECM plays an important role in the pathogenesis and progression of chronic lung disease (3, 4). Many chronic lung diseases, e.g., chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF) primarily manifest in the elderly, suggesting increased susceptibility of the aged lung or accumulated alterations in lung structure over time that favour disease. How the ECM changes during ageing, however, has not been comprehensively assessed or discussed.
Constituting between 30 and 70% of ECM protein in all tissue types, collagen is the main component of the ECM (5) and even forms the most abundant human protein class in general. Mutations and polymorphisms in genes encoding structural collagen chains as well as collagen biosynthetic proteins are associated with disease affecting the full age range. Some cause fatal congenital disorders leading to death very early in life, others result in premature or accelerated ageing in adolescents and adults, and yet others only become evident in the elderly where they lead to a high burden of multi-morbidity, reduced quality of life, and lower life expectancy (6). Probably the most widely known collagen-related disorders result in drastic bone and cartilage abnormalities, as e.g., brittle-bone (osteogenesis imperfecta) or Caffey disease, characterised by increased bone fragility or episodes of excessive bone formation, respectively (1). Other frequent effects of collagenopathies are skin alterations, visual defects and hearing loss, muscle weakness, vessel abnormalities and kidney disease (1).
Pulmonary manifestations of such collagen mutations and polymorphisms have received less attention, probably because the most severe lung abnormalities in such patients are caused by defects in chest formation and rib fractures, i.e., are of origin secondary to bone and cartilage defects (1, 6, 7). Nevertheless, altered collagen synthesis or turnover by other than genetic causes are frequent hallmarks of chronic lung disease and contribute considerably to disease progression, severity, morbidity, and mortality (3, 4). In lung cancer, for instance, dysregulated collagen expression and crosslinking appear to favour tumour progression by providing a permissive, pro-invasive, and pro-inflammatory environment (8). In pulmonary fibrosis, irrespective of disease aetiology, excessive collagen deposition in the alveolar space is the ultimate pathological feature leading to increasing dyspnoea and progressive lung function decline (9–14). In contrast, COPD/emphysema is characterised by increased degradation of ECM proteins by matrix metalloproteinases (MMPs) and neutrophil elastase, targeting primarily collagens and an unrelated major ECM protein, elastin, respectively (15).
Given that collagen is the most abundant protein type in the body, it is not surprising that collagen has been a subject of research for about 100 years by now. What is striking, however, is how little we know, nonetheless. The latest member of the collagen protein family, type XXVIII collagen encoded by COL28A1, has only been reported in the year 2006 (16). Similarly, new proteins acting in collagen biosynthesis and modification have only been discovered and characterised in the last two decades (17–21). Also, even though collagen is known to undergo excessive post-translational modification (PTM), both intra- and extracellularly, these PTMs have not been comprehensively mapped and the biological function of the majority of the PTMs remains unclear (22). Equally, it is poorly understood how collagen biosynthesis and turnover change during normal ageing and how such changes may affect the function of adherent cells, lung repair, susceptibility to disease, disease progression and comorbidities.
This review aims to draw attention to the complexity of collagen synthesis, processing, and degradation and the importance of these processes in lung ageing and chronic lung disease. To set the stage, we first provide an overview of collagen types and key steps of collagen biosynthesis, processing, and maturation. We then summarise what is known about collagen alterations in the ageing lung and what can be tentatively inferred from studies in other organs. Ultimately, we believe that a better understanding of these mechanisms may provide important clues for prevention of lung disease and for lung bioengineering and pave the way to novel targeted approaches in lung regenerative medicine.
Collagen Types and Structure
The collagen suprafamily comprises to date 28 known highly diverse collagen types in vertebrates, characterised by the presence of triple-helical collagenous domains. Collagens are divided into several subtypes depending on their domain structure and their macromolecular assembly (Figure 1): (A) Fibril-forming collagens (I, II, III, V, XI, XXIV, XXVII), (B) fibril-associated collagens with interrupted triple helices (FACITs, IX, XII, XIV, XVI, XIX, XX, XXI, XXII), (C) network-forming collagens (IV, VIII, X), (D) transmembrane collagens (XIII, XVII, XXIII, XXV), (E) endostatin-producing collagens or multiplexins (XV, XVIII), (F) anchoring fibrils (VII), and (G) beaded-filament-forming collagen (VI). Types XXVI and XXVIII do not fit well in any category (16, 23–25). Depending on the collagen type, collagens can assemble as homotrimers or heterotrimers. For instance, type III collagen is a homotrimer of three identical α1 chains (encoded by COL3A1), type I collagen typically assembles from two α1 chains (COL1A1) and one α2 chain (COL1A2) and type VI collagen from even three distinct chains (COL6A1, COL6A2, and COL6A3 or COL6A5 or COL6A6) (22, 26–28). Interestingly, altered chain stoichiometry has been described for several pathologies including fibrosis and may affect biochemical and biophysical properties of collagen (29–34).
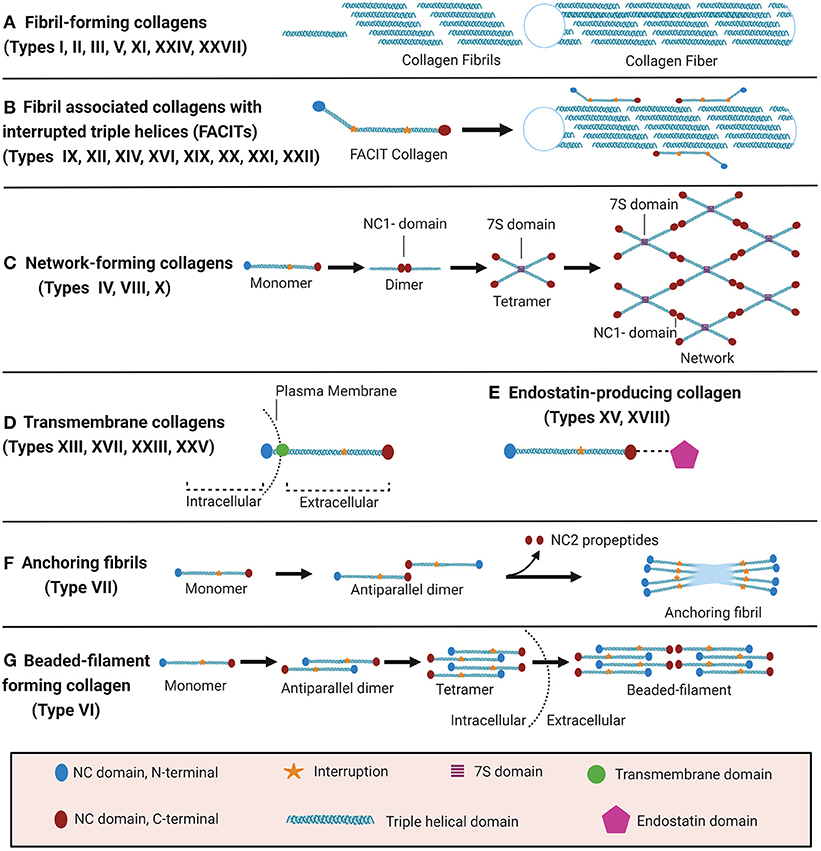
Figure 1. Classification of collagens based on supramolecular assembly. Schematic overview of the major forms of supramolecular assembly of collagen. For some non-collagenous (NC) domains, specific names are established in the literature and therefore specified in the figure: In the collagen IV network (C), the so-called 7S and NC1 domains represent critical nodes. In anchoring fibrils (F), the C-terminal domain which is cleaved off upon fibril formation is termed NC2. Figure was created with Biorender.com.
Collagen Domains and Macromolecular Assembly
The unifying feature of all collagens is the triple-helical collagenous domain, which is composed of three so-called α-chains consisting of amino acid repeats of (Gly-X-Y)n. The smallest amino acid glycine (Gly) can face the interior part of the triple helix while still allowing for a close association of the three chains. X and Y are often proline (Pro) or hydroxylated proline, 3-hydroxyproline (3-Hyp) or 4-hydroxyproline (4-Hyp), respectively (35, 36). While 4-Hyp in position Y of the Gly-X-Y repeat is frequently found in all collagen types and well-established as a major contributor to collagen thermodynamic stability (37–41), 3-Hyp has so far only been unambiguously detected in very few defined X positions of Gly-X-Y in collagen chains of type I, II, IV, and V, and 3-Hyp function is much less understood (42–46).
Frequent non-collagenous (NC) domains, e.g., in FACIT, beaded-filament forming and anchoring fibril collagens, are fibronectin type III, von Willebrand, thrombospondin (TSP) and Kunitz domains. The physiological function of these domains is incompletely understood. Fibronectin type III and von Willebrand domains seem to facilitate protein-protein interactions between collagens and other structural ECM molecules or growth factors in the extracellular space (47, 48). TSP domains mediate heparin and metal ion binding and may provide protection against blood clotting e.g., in subendothelial basement membranes (49, 50). The C-terminal Kunitz domains in type VI and VII collagens are cleaved off in the extracellular space (51–53). For type VI collagen, this cleavage product, endotrophin, stimulates tumour growth and angiogenesis, mediating many of the tumour-promoting effects of type VI collagen (54–56). Endotrophin is thus classified as a matricryptin or matrikine, a class of biologically active peptides derived from proteolytic processing of extracellular matrix proteins (57).
Even though all collagens are multi-domain proteins, the extent to which a collagen consists of collagenous and NC domains can differ drastically and the overall collagen domain architecture determines the macromolecular assembly (Figure 1). Type I collagen, like all fibril-forming collagens, contains a central uninterrupted collagenous domain as a major part of the polypeptide, flanked by relatively short NC domains, the N- and C-terminal telo- and propeptides (25). In contrast, FACITs can contain <10% collagenous domains and associate to the surface of collagen fibrils but do not form such fibrils by themselves (23, 27). Membrane collagens comprise an NC cytoplasmic domain, a transmembrane domain, and extracellular repeats of triple-helical collagenous domains (27). Membrane collagen types XIII, XXIII, and XXV are also collectively referred to as membrane-associated collagens with interrupted triple helices (MACITs) (58, 59). Notably, the C-terminal ectodomains of all membrane collagens can be proteolytically shed off the cell surface (23, 27, 60), and are classified as matrikines or matricryptins (27, 57, 61). Another collagen family that serves as a source for matricryptins are the multiplexins or endostatin-producing collagens. This family comprises collagen types XV and XVIII which generate restin and endostatin, respectively, upon proteolytic cleavage, both known for their potent antiangiogenic properties (24, 25, 27, 57, 58, 61, 62). The prototypical network-forming collagen type IV, a major basement membrane constituent, is encoded by in total 6 genes (COL4A1-COL4A6) the products of which form heterotrimers consisting of the chain combinations (α1)2α2(IV), α3α4α5(IV), or (α5)2α6 (IV) (23, 24, 27). The so-called 7S and NC1 domains are critical nodes in the collagen IV network and stabilised by covalent bonds, namely by LOXL2-mediated crosslinks between lysines in the 7S domain and the unique sulfilimine crosslink (-S=N-) in the NC1 domain (63–65). Also collagens VIII and X can assemble to form networks in tissues, but the molecular determinants have not yet been studied in similar detail (27). Type VII collagen is the major component of anchoring fibrils that are essential for the integrity of the dermo-epidermal junction in skin (24, 25, 27). It consists of two adjacent collagenous triple-helical domains which are flanked by a rather long (140 kDa) NC domain harbouring von Willebrand domains and fibronectin type III repeats at the N-terminus (NC1) and a much shorter (30 kDa) NC domain at the C-terminus (NC2) (27, 66). The initial formation of these anchoring fibrils from homotrimeric type VII collagen molecules involves antiparallel alignment of two collagen VII molecules at the level of their C-termini (NC2 domains), followed by enzymatic cleavage of the NC2 domain and stabilisation of the dimer by disulfide bonds (66). Finally, beaded-filament-forming collagens are strictly only represented by type VI collagen. Suprastructural assembly occurs as follows: Two heterotrimeric type VI collagen molecules dimerize in an antiparallel fashion via interaction of their central triple-helical domains. The protruding N-and C-terminal globular domains on both sides register in parallel with another dimer, forming a tetramer which is stabilised by disulphide bonds Finally, tetramers assemble end-to-end to generate the beaded-filaments (Figure 1) (23, 67). The collagen types XXVI and XXVIII are sometimes mentioned within the context of this class, even if they do not fit well in any category (16, 23–25). Type XXVIII collagen shares some sequence homology with type VI collagen (16). Type XXVI collagen, however, is uncharacteristically small for a collagen, comprises only two very short collagenous domains, and shares few similarities with any of the other described collagens (24).
Collagen Biosynthesis
Collagen biosynthesis is a highly complex process starting with transcription of collagen genes followed by translation and translocation of the nascent polypeptide chain to the rough ER (rER), co-translational modification and folding, trafficking across the Golgi network, secretion, and finally, extracellular processing and maturation (36, 68) (cf. Figure 2). This process is best described for type I collagen which will be used as an example for synthesis of a heterotrimeric fibril-forming collagen in the following.
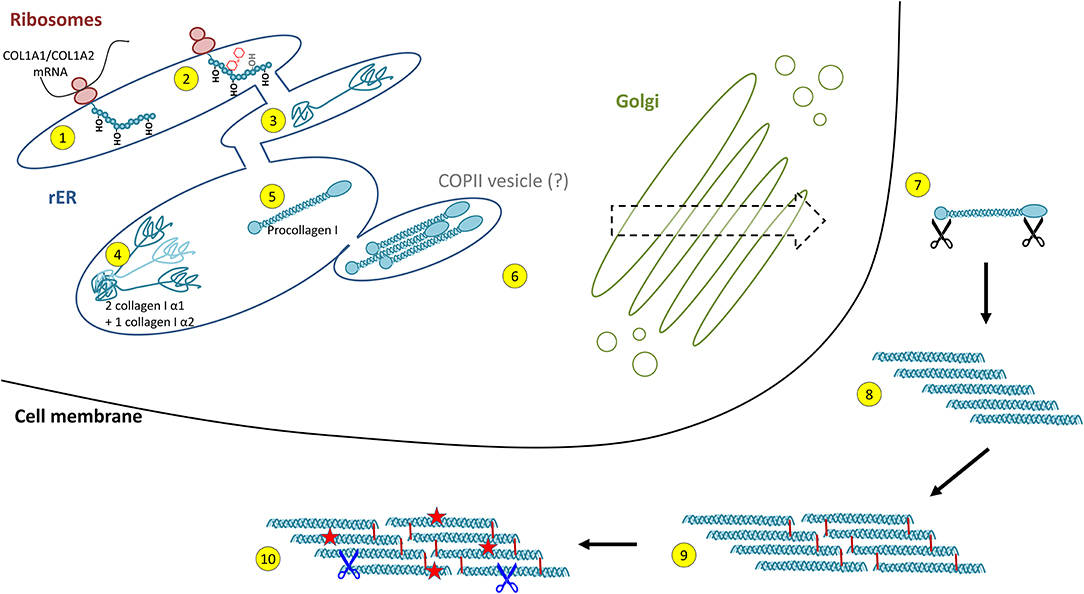
Figure 2. Intracellular collagen biosynthesis and extracellular maturation of collagen I. (1) Cotranslational prolyl-4- and lysyl-hydroxylation of the nascent collagen polypeptide chain in the rough endoplasmic reticulum (rER) is followed by (2) glycosylation and prolyl-3-hydroxylation and (3) folding of the C- and N-terminal propeptides. (4) For collagen I, two properly folded α1 chain C-propeptides assemble with one α2 chain C-propeptide, forming the triple helix nucleus. (5) Triple helix formation occurs in a zipper-like fashion and is dependent on peptidyl-prolyl isomerases and collagen chaperones. (6) Collagen triple helices are transported via the trans-Golgi network and finally secreted into the extracellular space. This was believed to occur via COPII vesicles, a concept that has been challenged recently. (7) In the extracellular space, propeptide cleavage involving at least three proteases (black scissors) triggers (8) auto-assembly of collagen fibrils. (9) Finally, fibrils are stabilised by crosslinking. (10) The mature collagen fibres are subject to insults (red stars) and degradation by extracellular proteases (blue scissors, see Figure 4).
Translation and Co-translational Modification of the Nascent Polypeptides
Following transcription of COL1A1 and COL1A2 genes, the mRNA is translated into the polypeptide at the rER where the nascent polypeptide chain is extended into the lumen of the rER. Proper folding in the rER requires several enzymes and molecular chaperones essential for post-translational modifications (PTMs) and the formation of triple-helical procollagen molecules (36). First, numerous PTMs are introduced into the nascent unfolded polypeptide chain in a co-translational fashion. These include lysyl and prolyl hydroxylations and hydroxylysyl glycosylations, modifications that are mediated by several collagen-specific enzymes which exist in defined multiprotein complexes with other chaperones and protein folding catalysts [for an excellent review, see Ishikawa and Bächinger (36)]. Many of these PTMs are essential for proper stability, assembly and secretion of procollagen, as well as for the final supramolecular structure of these molecules (24, 25, 27, 36). Numerous enzymes catalysing PTMs have been identified. In vertebrates, prolyl-4-hydroxylation is mediated by one of three prolyl-4-hydroxylases (encoded by P4HA1, P4HA2, and P4HA3) which form complexes with protein disulphide isomerase (PDI). In the lung, at least one of these prolyl-4-hydroxylases, P4HA3, is upregulated in IPF and has been put forward as a potential drug target (69). Most members of the collagen prolyl-3-hydroxylase (P3H1-P3H4) family equally form multimeric complexes with other collagen-modifying proteins and chaperones. Of these, P3H1, P3H2, and P3H3 have been shown to exert prolyl-3-hydroxylase activity, while P3H4 (also termed SC65) forms a trimeric complex with P3H3 and lysyl hydroxylase 1 (LH1), but has no prolyl-3-hydroxylase activity of its own (19, 20). Lysyl hydroxylases comprise three enzymes termed LH1-LH3 or procollagen-lysine,2-oxoglutarate 5-dioxygenases 1 to 3 (PLOD1-PLOD3, e.g., Uniprot database) (36). LH1, as already mentioned above, exists in a complex with P3H3 and P3H4, and preferentially hydroxylates lysines in the triple-helical collagenous regions (70). The resulting 5-hydroxylysine residues may be subject to O-glycosylation by glycosyl transferases (36, 70, 71). In contrast, LH2, which associates with FKBP10 (FKBP65) is responsible for hydroxylation of lysines in the non-collagenous telopeptide regions of fibrillar collagens (70, 72). Notably, telopeptide lysines and hydroxylysine are subject to extracellular lysyl oxidase (LOX)-mediated crosslinking and the hydroxylation status of the involved lysines strongly affects nature and stability of the crosslink (for selected examples for crosslinks, see Figure 3) (36, 71–73). LH3 is known to hydroxylate lysines in type IV and V collagens but also exerts glycosyl transferase activity (36, 70, 71). Finally, in addition to LH3, two collagen glycosyl transferases, GLT25D1 and GLT25D2 mediate O-glycosylation of hydroxylysines via the 5-hydroxyl group. Here, hydroxylysines can be either O-glycosylated with a monosaccharide (β-d-galactopyranose, Gal) or a disaccharide (α-d-glucopyranosyl-(1->2)-β-d-galactopyranose, GlcGal), resulting in galactosylhydroxylysine (GHyl) or glucosylgalactosyl-hydroxylysine (GGHyl), respectively (36, 70, 71).
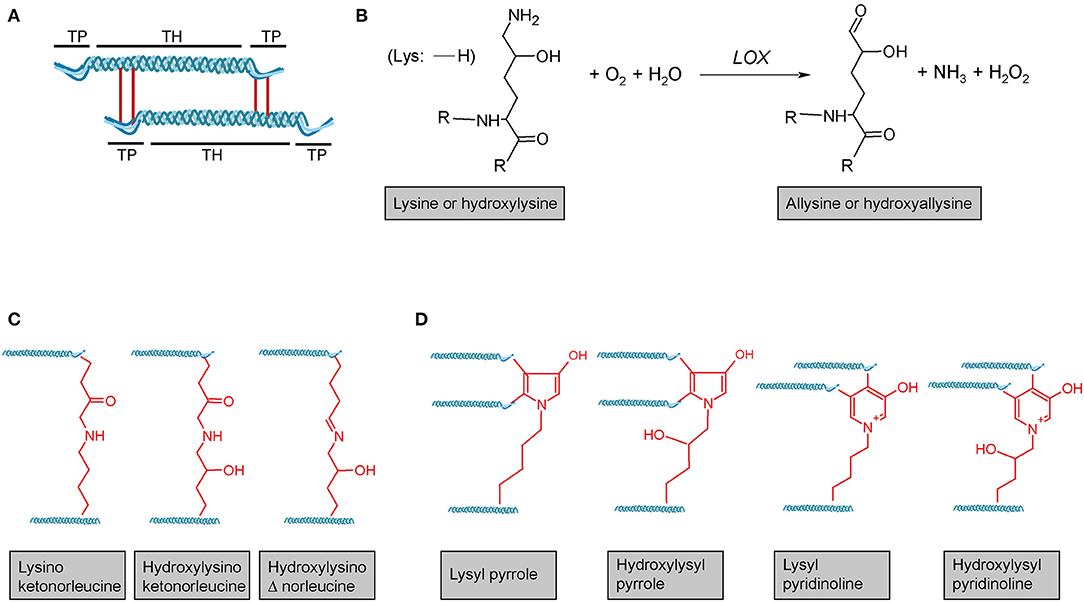
Figure 3. Lysyl oxidase-mediated crosslinking. (A) Schematic representation of telopeptide (TP) and triple-helical (TH) crosslink sites of two adjacent tropocollagen molecules in a collagen fibril. (B) Lysyl oxidase (LOX) enzymes initially catalyse the oxidative deamination of the ε-amino group of a lysine or hydroxylysine, yielding a highly reactive aldehyde group. This entails subsequent reactions with primarily other (hydroxy)lysines and rearrangements, ultimately resulting in different (C) divalent and (D) trivalent collagen crosslinks. R = continued polypeptide chain. Structures were generated using ACD/Chemsketch freeware.
Triple Helix Formation
Triple helix formation is preceded by folding of the N- and C-terminal propeptides and chain selection via the trimerization domains (36, 68, 74, 75), a process supported by a plethora of general ER-folding folding catalysts including Grp78 (BiP), Grp94, PDI, calreticulin, calnexin, and CypB (36). Following chain selection, triple helix formation is initiated and proceeds in a zipper-like fashion. Given the proportionally high number of proline residues in collagen, it is not surprising that one of the rate-limiting steps in triple helix formation is the cis-trans isomerization of proline residues catalysed by rER-resident peptidyl-prolyl isomerases (PPIases) (36, 68). Only the trans-proline conformation allows a linear prolongation of the triple helix (76). The PPIases FKBP10 (FKBP65), CypB as well as FKBP14 (FKBP22) appear to play critical roles in that context (36, 68, 77–79). In addition, heat shock protein 47 (HSP47 or SERPINH1) functions as an important collagen-specific chaperone in collagen modification, triple helix formation, and export from the ER to the Golgi (36, 68, 80–82).
Trafficking From the rER via the Trans-Golgi Network to the Extracellular Space
Procollagen secretion is dependent on coat protein complex II (COPII) vesicle-mediated transport from the rER to the Golgi. Typically, COPII vesicles are not bigger than 60–80 nm in diameter (83), while a completely folded and fairly rigid procollagen molecule can be up to 500 nm in length (84–88). Therefore, a long-held concept involved the transport of procollagen molecules via specialised, enlarged, COPII vesicles (85, 89–93). This concept, however, has been challenged recently: Analysis of endogenous and engineered GFP-tagged procollagen by live-cell imaging did not provide any evidence for dynamic large carrier vesicles between the ER and the Golgi but instead rather supports a model of direct interconnections between organelles or the presence of less well-characterised intermediate carriers (88, 94).
Extracellular Processing and Maturation of Collagen
Extracellular processing and maturation of collagen depends very much on the collagen type and the nature of supramolecular assembly (cf. Figure 1) but is best described for type I collagen and other fibrillar collagens. Here, secretion is followed by the cleavage of the N- and C-terminal propeptides by specific procollagen proteinases, including bone morphogenetic protein 1 (BMP1), members of the ADAMTS protease family, and the more recently discovered meprins (23, 25, 27, 95). Notably, enzymatic cleavage of propeptides occurs for many non-fibrillar collagens, too, including the above-mentioned membrane collagens and multiplexins, also involves MMPs and cathepsins, and is the source of matrikines or matricryptins, collagen-derived fragments with diverse biological activities (57, 61). For fibrillar collagens, propeptide cleavage is followed by spontaneous, entropy-driven, quarter-staggered assembly of the tropocollagen molecules into fibrils (96, 97). Finally, intra- and intermolecular crosslinks, catalysed by enzymes of the lysyl oxidase (LOX) and transglutaminase (TGM) family, stabilise fibrillar collagens in the extracellular space (98–101). In particular the enzymes LOX and LOX-like 2 (LOXL2) have been shown to crosslink fibrillar collagens both intra- and inter-molecularly (101) and LOXL2 has been put forward as a potential therapeutic drug target in IPF (102). Further collagen-associated proteins like the antioxidant proteins extracellular superoxide dismutase or glutathione peroxidase 3 (GPX3) may protect these long-lived molecules from oxidative damage (103–106).
Collagen Turnover
In normal tissue homeostasis, the ECM is subject to a constant and dynamic, albeit typically slow, turnover, in which collagen is degraded and newly synthesised (107). The rate of collagen turnover differs drastically between tissue types. For instance, human cartilage collagen has a half-life of about 117 years, invertebral disc collagen 95 years, and skin collagen 15 years (108, 109). Even if, to our knowledge, the half-life of human lung collagen has not been determined, numerous studies have addressed lung collagen turnover in human and experimental animal lung tissue. Most studies point toward a remarkable level of constant de novo collagen synthesis and the presence of two collagen pools with different degradation rates; a pool of probably newly synthesised collagen which is subject to considerable degradation, and a pool of heavily crosslinked stable collagen which is more resistant to degradation (110–117). Considering the tightly wound triple-helical structure as well as the complex supramolecular structures described above, it is not surprising that only members of two protease families are capable of collagen degradation, namely MMPs and cathepsins. The so far described collagen degradation pathways can be categorised in extracellular and intracellular pathways. These processes are regarded as different from propeptide cleavage which is part of the normal collagen maturation pathway and reflects rates of de novo collagen synthesis rather than collagen turnover. Nevertheless, a strict distinction may represent an oversimplification. Notably, both processes can liberate biologically active peptide fragments, i.e., matricryptins or matrikines (57, 61).
Extracellular Collagen Degradation
Extracellular collagen degradation is mainly attributed to matrix metalloproteinases (MMPs) and cathepsin K. MMPs are extracellular Proteolytic Zinc-dependent endopeptidases which collectively degrade all major ECM molecules. Collagenolytic activity has been observed for MMP1, MMP2, MMP7, MMP8, MMP9, MMP13, MMP14, and MMP19 (118, 119). MMP-mediated degradation of interstitial collagens, in particular of types I-III is best-described, but several MMPs (e.g., MMP2, MMP7, MMP9) also degrade basement membrane type IV collagen (118, 120–122). For a comprehensive overview of MMPs and their collagen substrates, the interested reader is referred to Visse and Nagase (123) and Jobin et al. (124). The C-terminal hemopexin domain of MMPs is crucial for collagen degradation as it not only recognises and binds the substrate but also unwinds the collagen structure in order to access the cleavage site (125, 126). In contrast, the cysteine protease cathepsin K (CatK) is probably the most effective protease for the degradation of extracellular fibrillar collagen (127, 128) because it can also target triple-helical collagen directly, without the need for unwinding of the helix (127).
Uptake of Collagen and Intracellular Collagen Degradation
Uptake of collagen into the cell can occur by phagocytosis of an intact collagen fibril (129) or by macropinocytosis or receptor-mediated endocytosis of already cleaved collagen particles. In order to pass the cell membrane, collagen fibrils are recognised by integrins which triggers the process of phagocytosis (130). The so far observed integrins being involved are α1β1- and α2β1-integrin (131), as well as α10β1- and α11β1-integrins (132, 133). Initial fragmentation of fibrillar collagen is mediated by membrane-bound MMP14 (also termed MT-MMP1) (134).
Smaller collagen fragments, e.g., derived by extracellular MMP- or CatK-mediated degradation, can be taken up by two main pathways, macropinocytosis and receptor-mediated endosomal uptake. In macropinocytosis, solubilized collagen particles are internalised within actin-mediated endocytosis resulting in collagen-containing vacuoles (135, 136). Receptor-mediated endosomal uptake of collagen fragments is dependent on the urokinase plasminogen activator receptor-associated protein (uPARAP/Endo180 or C-type mannose receptor 2, MRC2). This endocytic mannose receptor mediates the internalisation of MMP-cleaved collagen fragments into clathrin-coated vesicles into fibroblasts (137–139). Receptors mediating endosomal collagen uptake in macrophages are macrophage mannose receptor 1 (MRC1) and lactadherin (MFGE8) (140, 141).
For intracellular collagen degradation, both pathways converge at the level of fusion with lysosomes. As to collagen-degrading proteases, lysosomes contain a range of cathepsins, including cathepsins B, D, K, and L, which cleave collagen into low-molecular-weight peptides (142–144). An overview of their known collagen substrates is given in Table 1. Notably, the same pathway can be used for newly synthesised collagen prior to secretion, for instance, when collagen is misfolded or otherwise “defective” (149, 150). In this so-called lysosome-dependent macroautophagy, lysosomes may fuse with ER- or Golgi-derived vesicles (151, 152).
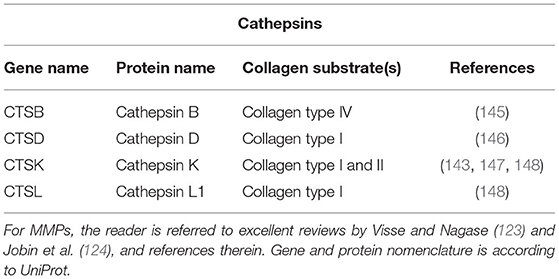
Table 1. Cathepsins and their collagen substrates.
Baseline Changes in Collagen Synthesis and Maturation During Mouse Lung Ageing
In order to examine baseline changes, i.e., changes in the absence of environmental stimuli, in collagen biosynthesis, maturation and degradation (Figures 2, 4) upon ageing, we took advantage of a recently published single-cell RNA-Seq and multi-omics analysis data set on the ageing mouse lung (Figure 5, Table 2) (160). Following extraction of data for all relevant proteins, we observed no significant changes for proteins of the ER-resident machinery of collagen biosynthesis, modification, and triple helix formation (e.g., P4H and PLOD enzymes, SERPINH1, FKBP10, PPIB, Figure 5A). In contrast, we found expression of several collagen subtypes deregulated upon normal ageing: The type VI collagen chains COL6A5 and COL6A6, as well as COL14A1, were significantly downregulated, while levels of COL4A3 and COL16A1 were significantly increased. Furthermore, levels of the collagen crosslinking enzyme lysyl oxidase (LOX) were decreased (Figures 5A,B). Table 2 provides a summary of these findings including a comparison with what is currently known about corresponding changes in human tissues upon ageing.
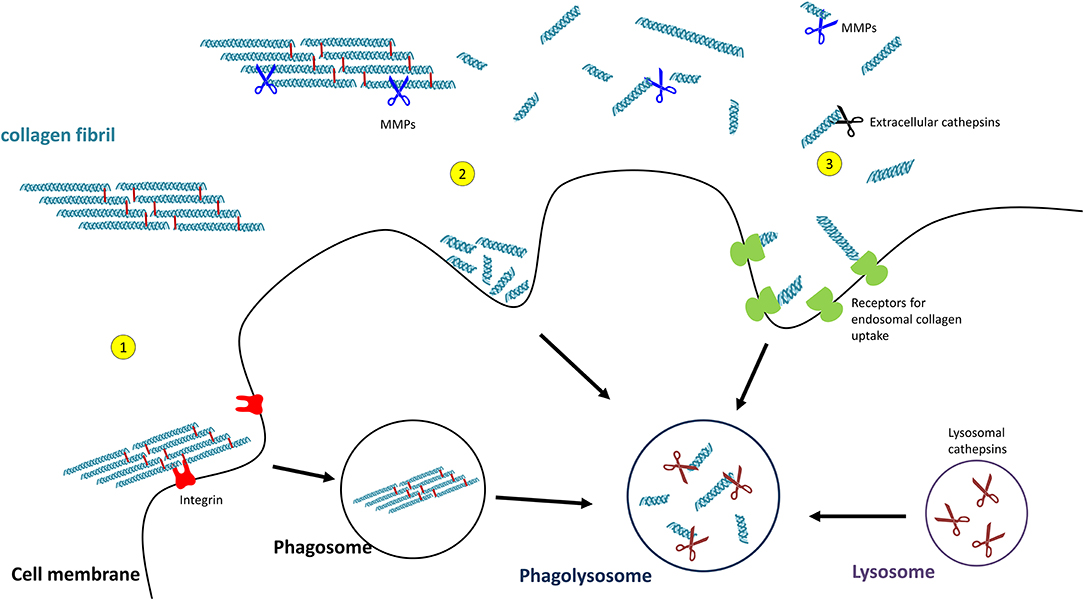
Figure 4. Pathways of collagen degradation. Both extracellular degradation by MMPs and extracellular cathepsins on the one hand and intracellular degradations by (1) collagen phagocytosis, (2) macropinocytosis, or (3) receptor-mediated endocytosis occurs.
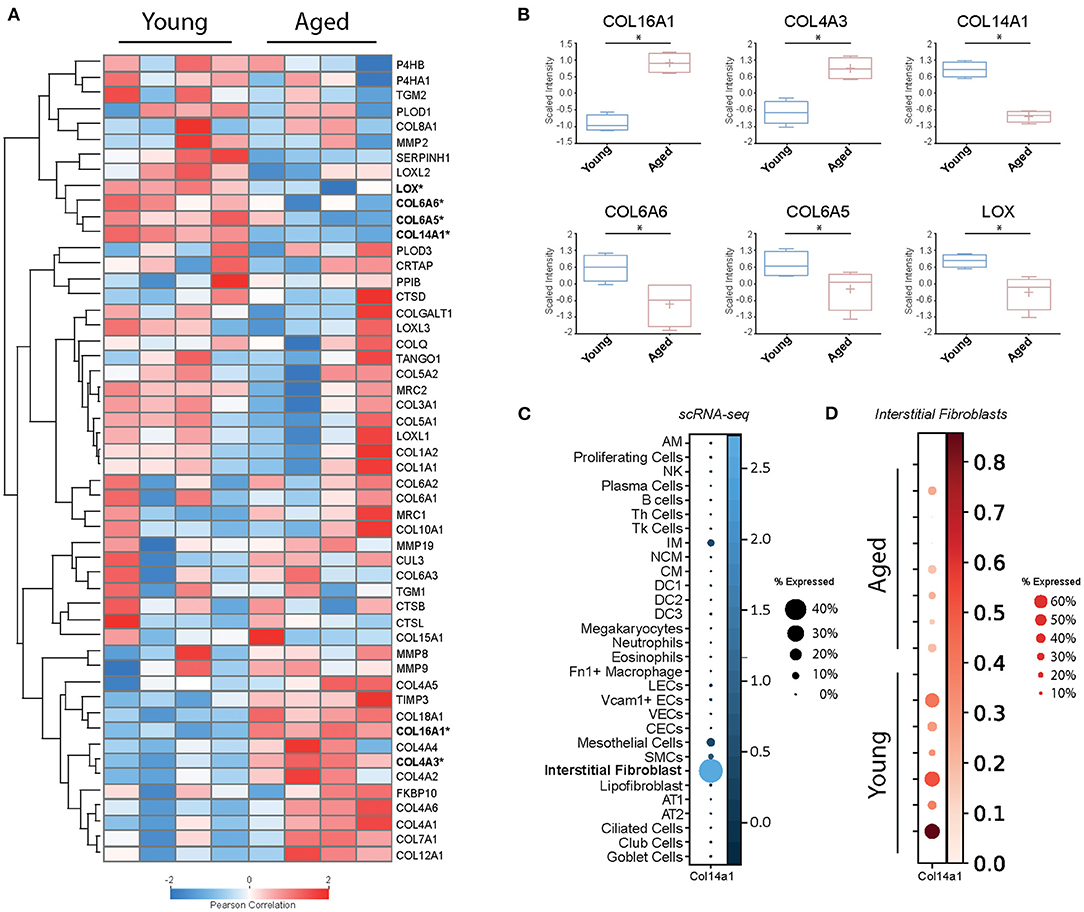
Figure 5. Baseline changes of proteins involved in collagen biosynthesis, trafficking, processing, and degradation. (A) Heatmap of normalised mean intensity values of 52 regulated extracellular matrix proteins (whole lung proteome, mass-spec) grouped by unsupervised hierarchical clustering and Z-scored (Pearson's correlation). (B) Boxplots of 6 significantly regulated proteins with age. (C) The dot plot shows mRNA expression specificity of Col14a1 in scRNA-seq data of whole lung homogenate. (D) Dotplot indicating mRNA expression levels between aged and young mice within the lung interstitial fibroblasts cluster (* = Student's t-test significant, FDR<10%).
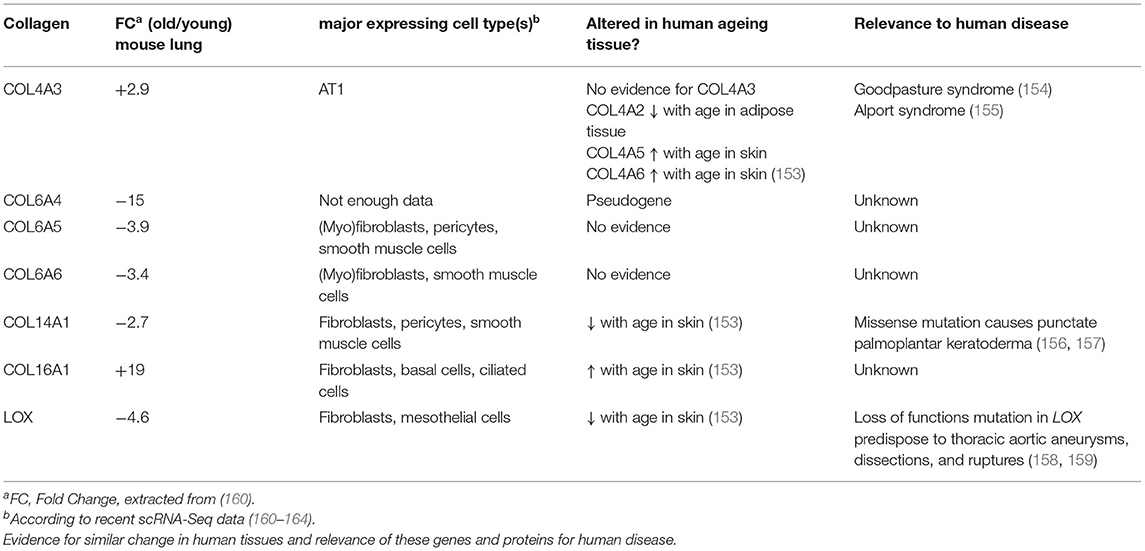
Table 2. Overview on collagens and collagen biosynthetic proteins observed to be regulated with age in the mouse lung.
Downregulation of Type VI Collagen Chains COL6A5, COL6A6
Type VI collagen differs from most other collagens by its unique supramolecular assembly, the formation of characteristic beaded microfilaments in the ECM, a property which it only shares with type XXVI (COL26A1) and type XXVIII collagen (COL28A1) (23) (Figure 1). Type VI collagen is a component of the basement membrane in lung, muscle and skin (165), but can also localise to the pericellular matrix, e.g., in tendon (166, 167). A number of studies, including our own, indicate an important role of type VI collagen for adhesion, migration, proliferation, death, and dysfunction of adherent cells (77, 166–174). Typically, type VI collagen consists of the α1(VI), α2(VI), and α3(VI) chains, which assemble in a 1:1:1 ratio and are encoded by Col6a1, Col6a2, and Col6a3, respectively (166). These three chains are found in all connective tissues, they are by far the most abundant type VI collagen chains, including in the lung, and their levels are not altered during lung ageing in the mouse (160).
Expression of the comparatively recently discovered type VI collagen chains α4(VI), α5(VI), and α6(VI), in contrast, is more restricted, but at least two of these chains are consistently expressed in foetal, new-born, and adult lung (28, 175). An important difference between the murine and human genes is that the human COL6A4 gene is disrupted and not functional. In mice, expression of Col6a4 is mostly observed in new-born tissue—including lung—but lost in all adult tissues except for uterus and ovaries (28, 175, 176). The complete loss of COL6A4 from young to old mouse lungs may therefore correspond to remnant Col6a4 from developmental expression, which is increasingly degraded during normal collagen turnover in a mouse's lifetime, becoming undetectable in the old lung (160). In contrast, COL6A5 and COL6A6 are both expressed in adult human lung, even at higher levels than in foetal lung, arguing for a role in adult lung function (28).
Based on sequence similarities, co-purification and colocalization analysis, both COL6A5 and COL6A6 are predicted to assemble with COL6A1 and COL6A2 as an alternative chain to COL6A3 (28, 175, 177). However, strong biochemical evidence in support of this hypothesis is still lacking. Even though some of the α5(VI) (COL6A5) and α6(VI) (COL6A6) chains may be engaged in type VI collagen triple helix formation, in the lung the α3(VI) chain is more abundant by several orders of magnitude, and expression of Col6a3 is unchanged during ageing. Thus, an α5/α6(VI) -> α3(VI) chain switch from young to old lung tissue is unlikely to represent a major event affecting the general function of type VI collagen. Interestingly, type VI collagen, in particular the α3(VI) chain (COL6A3), has been attributed a role in stemness and promotion of tumour growth. Therefore, local replacement of α5/α6(VI) by α3(VI) chains during ageing may modulate stem cell niches, maybe even contribute to stem cell exhaustion, a major hallmark of ageing (178, 179). But also presence of non-triple-helical (NTH) COL6A5 and COL6A6 polypeptides is conceivable, as evidence for alternative NTH collagen gene products is emerging in the literature, e.g., for COL6A1 (22, 180) and COL4A1 (181, 182).
Downregulation of Type XIV Collagen (COL14A1)
Type XIV collagen belongs to the FACITs (Figure 1) and plays an important role in fibrillogenesis of type I collagen, in particular during development (183–187). Similar to downregulation of COL14A1 in the ageing mouse lung, downregulation of COL14A1 has also been described for human skin ageing (153). Several studies support a role of COL14A1 in connecting the basement membrane to the subepithelial interstitial matrix (185, 188, 189). Like COL6A4 mentioned above, COL14A1 is developmentally regulated in some tissues, e.g., in tendon or the heart (189–192).
Interestingly, several reports in the past have independently argued for an important role of COL14A1 in the maintenance of normal epithelial cell turnover and differentiation in adult tissues. For instance, a missense mutation in COL14A1 at a highly conserved amino acid residue (Pro1502Leu) leads to punctate palmoplantar keratoderma, a skin disease characterised by aberrant squamous cell differentiation leading to hyperkeratosis in the cornified layer (156, 157). In renal cell carcinoma (RCC), which arises from epithelial cells, frequent hypermethylation of COL14A1 has been observed which resulted in transcriptional silencing; knockdown of COL14A1 increased the growth rate of RCC cell lines (193). COL14A1 is also notably downregulated in oesophageal squamous cell carcinoma (194). Collectively, these observations point toward an anti-proliferative or tumour suppressor function of COL14A1.
With this documented role on epithelial cell survival and differentiation, it is at first glance surprising that recent single-cell RNA-Seq data consistently show expression of COL14A1 by interstitial fibroblasts, not epithelial cells (160–164) (Figure 5C). A study on fibroblast heterogeneity even revealed that expression of COL14A1 marks a specific matrix-producing fibroblast subtype which increases in cell number in murine lung fibrosis (195). As lung fibrosis is characterised by loss of normal alveolar type I and type II cells and atypical epithelial differentiation (196), it may be speculated that overexpression and extracellular deposition of fibroblast-generated COL14A1 contributes to these epithelial events and therefore to loss of normal alveolar structure. In addition, loss of Col14a1 in the ageing mouse lung correlates with changes in cell type composition in mouse airway epithelial cells (160). Overall, it seems that deregulation or loss of paracrine COL14A1 has profound consequences on epithelial survival and differentiation and may therefore contribute to the observed changes during ageing. These observations warrant future studies on the exact distribution and molecular function of COL14A1 in lung ageing and disease.
Upregulation of Type IV Collagen (COL4A3)
Type IV collagen is a major structural component of the basement membrane and represents the prototypical network-forming collagen. Six genes (Col4a1-Col4a6) encode for six distinct type IV collagen chains which can assemble into three heterotrimeric molecular isoforms, namely α1(IV)2α2(IV), α3(IV)α4(IV)α5(IV), and α5(IV)2α6(IV) (197). At least the first two heterotrimeric forms have been reported in the lung, the major one being α12α2(IV) (22, 198). Interestingly, Angelidis et al. (160) found that, even if only COL4A3 passed the threshold for significance, protein levels of all six type IV collagen chains were increased in the ageing lung. These changes went in parallel with alterations of other basement membrane-specific proteins, like decrease of FRAS1, FREM1, FREM2, and COL14A1 discussed above. Intriguingly, the authors also found that transcript levels for all type IV collagen genes anti-correlated with protein levels, i.e., were downregulated with ageing. While downregulation of type IV collagen chain transcripts occurs consistently upon ageing of human tissue and cells (153, 199, 200), the post-transcriptional mechanisms that control levels of type IV collagen protein may be tissue-specific: While, similar to the results in ageing mouse lung, Karttunen et al. reported type IV collagen protein to be increased in human kidney upon ageing (201), in skin, type IV collagen protein was found to decrease upon ageing, in parallel to transcript levels (200). Interestingly, a decrease of serum type IV collagen has been described upon ageing (202) which may in part reflect increased type IV collagen retention and deposition in tissue. Overall, type IV collagen turnover, however, as measured by the formation product P4NP7S and the degradation product C4M in serum, seems to be stable during ageing (203).
Upregulation of Type XVI Collagen (COL16A1)
Just like type XIV collagen, also type XVI collagen belongs to the FACIT family of collagens and forms homotrimeric triple helices (23, 204). It is a minor collagen component which shows a tissue-dependent versatility for incorporation into different collagen suprastructures (205, 206). Its distribution in lung ECM is unknown, but judging from recent scRNA-Seq data, where COL16A1 is found expressed by all fibroblast subtypes as well as a broad range of epithelial cells, it is likely to be a constituent of both the basement membrane and the interstitial matrix (160–164). Even though COL16A1 has been observed to be increased upon ageing also in human skin (153), its potential role in the ageing process has not received much attention. Tajima et al. found that increased COL16A1 expression in dermal fibroblasts correlated with cell growth arrest of these cells (207). In contrast, induction of COL16A1 expression promotes proliferation and invasion of cancer cells (205, 208–210). Similar to our speculations on COL14A1, there may be analogous paracrine effects by fibroblast-generated COL16A1, albeit in reverse directions: Loss of fibroblast-generated COL14A1 on the one hand, as well as induction of COL16A1 deposition by fibroblasts on the other, may lead to increased cell proliferation and aberrant differentiation of adjacent epithelial cells and thus contribute to tumorigenesis and tumour invasion. Notably, this is reminiscent of a proposed link between ageing and cancer where senescent fibroblasts secrete factors, including extracellular matrix components that promote epithelial tumorigenesis (211, 212). Clearly, future studies are needed to decipher the role of COL14A1 and COL16A1 in this context.
Downregulation of Lysyl Oxidase
Lysyl oxidase (LOX) is a copper-dependent protein-lysine-6-oxidase, activity of which is critical for stabilisation of extracellular fibrillar collagen (71, 213). During fibril formation, LOX catalyses the oxidative deamination of specific lysine and hydroxylysine residues in the N- and C-telopeptides of fibrillar collagens, resulting in the corresponding aldehyde forms (71) (Figure 3B). These reactive intermediates trigger a series of subsequent condensation reactions between triple-helical subunits of collagen fibres, ultimately leading to divalent or trivalent crosslinks.
Several studies have shown that downregulation of LOX in the context of ageing is not restricted to the lung. For instance, LOX expression is reduced with age in human, rat and monkey skin (153, 214, 215), in urogenital tissues of female mice (216) and in rat aorta (217). Notably, loss of function mutations in LOX predispose to aortic aneurysms and age is the most important risk factor for aortic aneurysms (158, 159), supporting the concept that loss of LOX upon ageing may have direct effects on ECM integrity and tissue stiffness. Downregulation of LOX upon ageing appears to correlate with fewer reducible difunctional LOX-derived crosslinks in skin, aorta and lung (218, 219), even though, to our knowledge, no study has assessed this correlation directly. This contrasts with non-enzymatic collagen crosslinking which increases with age and will be discussed in more detail below (220). Overall, also taking into account recent findings that the same collagen lysines are targeted by LOX-mediated and non-enzymatic collagen crosslinking events (221), there seems to be a shift from LOX-mediated to advanced-glycation end product (AGE) crosslinks upon ageing.
In cancer research, LOX has received considerable attention, both owing to tumour growth and invasion-promoting properties on the one hand and to tumour suppressor functions on the other hand (222, 223). LOX is synthesised as a pro-enzyme and activation requires removal of the pro-peptide, notably by the same enzymes that cleave off the C-terminal pro-peptide of type I collagen (BMP1/Tolloid-like proteinases) (71). The mature protein is overexpressed in various cancer types including lung adenocarcinoma, typically correlates with poor prognosis, and has been described to create a stiffer microenvironment which supports tumour growth and metastasis (222, 224–230). In contrast, the tumour-suppressing activities of LOX are attributed to the released pro-peptide (222, 223, 231, 232).
The Impact of Ageing on Collagen Degradation
Few studies have directly assessed changes in collagen synthesis and degradation upon ageing. Several early studies, measuring the collagen synthesis rate in tissues of young and aged experimental animals, including lungs, as well as ex vivo culture of human skin biopsies, consistently found evidence for a decreased synthesis rate with ageing tissue (233–235). Mays et al., in addition, studied how much of the newly synthesised collagen is degraded and found that, in aged rats, a larger percentage was directly subjected to degradation than in young rats (233). Thus, in older age not only less collagen would be synthesised, but also newly synthesised collagen would be more prone to direct degradation. This concept, however, has been challenged by a recent study on old vs. young mouse lungs where the authors demonstrate that aged mice have higher overall levels of total collagen and lower levels of a collagen endocytic receptor termed mannose receptor, C-type 2 (MRC2), the major receptor for fibroblast-mediated intracellular degradation of collagen (236). Notably, in the proteomics data set presented here, we found lower levels of MRC2 in three out of four aged animals, albeit without reaching statistical significance (Figure 5A), which may reflect the limited statistical power of the proteomics data set. Furthermore, levels of most major lung collagens (types I, III, IV) remained unchanged or were even increased in old vs. young mouse lungs (Figure 5) (160). Overall, this indicates that mature collagen is very stable in the adult lung.
Partially supporting this concept, we observed no consistent changes for collagen-degrading proteases in the data set on mouse lung ageing presented here (Figure 5A) (160). Neither levels of the detected MMPs and TIMPs (MMP2, MMP8, MMP9, MMP19, TIMP3) nor levels of the detected cathepsins (CTSB, CTSD, CTSL) were significantly altered (160). Of course, this is only circumstantial evidence as (a) this study did not provide a comprehensive quantification of all lung collagenases and their inhibitors, (b) levels per se cannot be equated with enzymatic activity, and (c) accessibility of cleavage sites in the collagen substrate itself can be masked or altered upon ageing. As to enzymatic MMP activity, Calabresi et al. have shown that natural ageing of rat lungs is accompanied by decreases in MMP1 and MMP2 activity in parallel with moderately increased levels of the MMP inhibitors TIMP1 and TIMP2 levels (237), suggesting even a loss of collagenase activity upon ageing. Furthermore, and maybe even more importantly, there is evidence suggesting that the degradability of collagen is affected by ageing-related collagen modification and crosslinking (e.g., non-enzymatic crosslinking by advanced glycation end-products, AGEs, discussed below) and addition or removal of glycosaminoglycans (238–243). Thus, protease activity by itself may not be the decisive factor after all, but in fact the accessibility of the corresponding collagen cleavage sites.
It is to date unclear, whether these findings reflect the human situation. Collagen degradation upon ageing in the human lung has not been directly assessed. A number of studies have measured circulating levels of MMPs and TIMPs and deduced changes in ECM turnover upon ageing. However, the reported findings are not consistent (244–248) and even if such changes may be of importance for collagen degradation within the vascular wall, it is unclear how peripheral levels of MMP and TIMPs correlate with the integrity of the ECM in other tissues. Obviously, tissue-resident levels and activities of proteases in correlation with ECM integrity of the studied human tissue are more relevant to examine in that context, but such studies are much scarcer. An immunohistochemical study of human lungs found an age-dependent increase of TIMP2+ cells, but not MMP2+ cells, in the lung (249), providing some evidence for decreased MMP-mediated collagen degradation in the aged lung. Notably, in a recent pioneering study of human skin ageing, the authors correlated matrisome changes with the assessment of ECM integrity by single harmonic generation and two-photon autofluorescence imaging (214). Among other changes, they found decreased MMP2 and increased TIMP3 levels in aged skin. The mechanisms underlying skin ageing, however, involve environmental insults, e.g., exposure to UV light, microorganisms, and mechanical stress that do not apply to the lung.
Undoubtedly, more research is warranted to elucidate mechanisms of collagen and ECM degradation upon ageing of the lung. Nevertheless, taken together, the current evidence supports an age-dependent decline of lung collagen degradation, suggesting that, also in the lung, collagen is a long-lived molecule and thus susceptible to the accumulation of damage with time.
Non-enzymatic Collagen Crosslinking—Advanced Glycation Endproducts
In addition to LOX- and TGM-mediated, i.e., enzymatic crosslinking mentioned above, also non-enzymatic crosslinking of collagen occurs. The probably most important one in the context of human disease and ageing is the reaction of collagen with sugars or sugar metabolites which results in the formation of so-called advanced glycation endproducts (AGEs). These products are principally not restricted to collagen. In the best-described classical Maillard reaction, any free amino group, e.g., the ?-amino group of a lysine or the protein N-terminus, reacts with the carbonyl group of a reducing sugar to form a Schiff base. This unstable Schiff base is spontaneously rearranged into a more stable keto amine intermediate, the so-called Amadori product (Figure 6A). This early glycation product is in equilibrium with glucose and still reversible. However, a small part of these Amadori products undergo subsequent, predominantly oxidative, events resulting in the irreversible formation of different protein adducts and protein crosslinks, collectively termed AGEs. As the latter reactions occur over months and years, such adducts and crosslinks accumulate in long-lived proteins like extracellular collagens, including in the lung, and strongly correlate with age (220, 250–253). Figure 6A shows the classical Maillard reaction and some of the best-characterised and most often analysed AGEs are depicted in Figure 6B [Nε-(1-carboxyethyl)lysine, CEL; hydroimidazolone, MG-H1] and Figure 6C (lysine-arginine cross-links pentosidine and glucosepane; methylglyoxal lysine dimer, MOLD).
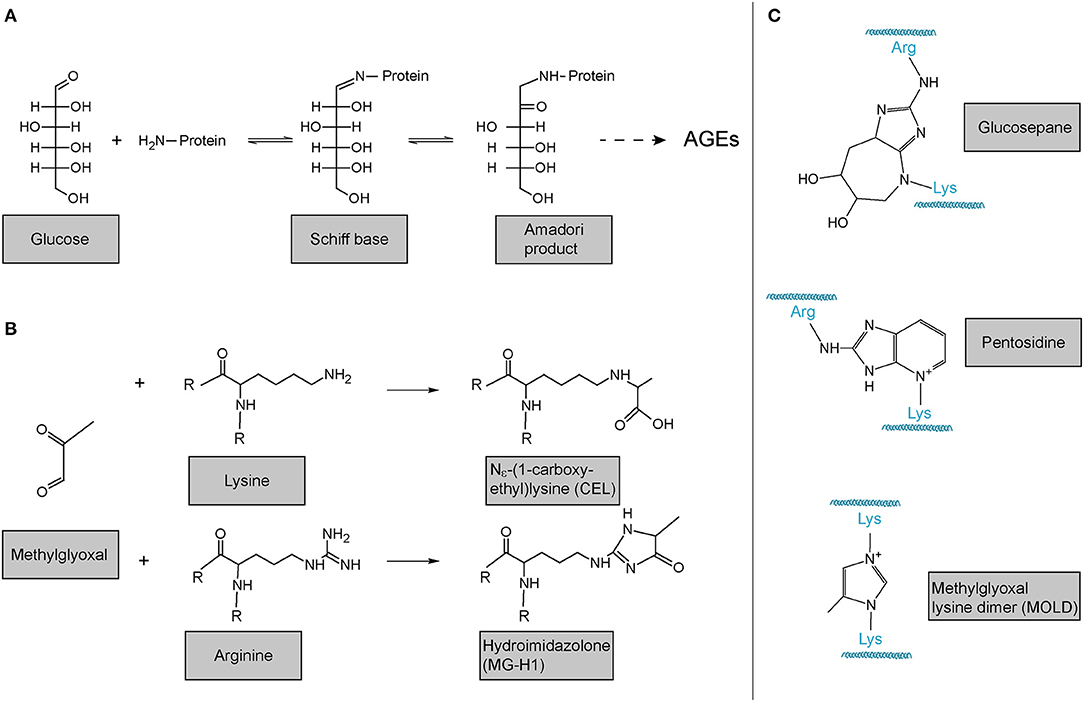
Figure 6. Advanced glycation endproducts (AGE)-mediated collagen modification and crosslinking. (A) In the classical Maillard reaction, a reducing sugar (here: glucose) reacts with an amino group in a protein, e.g., the protein's amino terminus or amino groups of the side chains arginine or lysine. The resulting Schiff base rearranges to a ketoamine, the so-called Amadori product. Subsequent reactions of this initial product result in the irreversible formation of different protein adducts and crosslinks, collectively termed advanced glycation endproducts (AGEs). (B) Reactions of the dicarbonyl methylglyoxal with protein lysines or arginines results in the formation of protein adducts as e.g., Nε-(1-carboxyethyl)lysine (CEL) or the hydroimidazolone MG-H1. (C) Structures of some of the most common AGE-mediated crosslinks. Structures were generated using ACD/Chemsketch freeware.
Notably, while the above-described reactions of proteins with reducing sugars are the best-described, AGEs can also be formed by the reaction of lysines and arginines with other carbonyl compounds, in particular highly reactive dicarbonyl compounds (254, 255). Accumulation of such compounds and its consequences is also referred to as “dicarbonyl stress” in the literature. Methylglyoxal (Figure 6B) is an abundant representative of these dicarbonyl compounds. Methylglyoxal is generated during the formation of AGEs in the context of the above-described glycation of proteins by reducing sugars, but also, in fact more importantly, endogenously as a result of spontaneous degradation of triosephosphates, degradation of glycated proteins, oxidation of aminoacetone in threonine catabolism, and peroxidation of lipids (256–258). In the glycation reaction (Figure 6B), methylglyoxal is about 20.000 times more reactive than glucose. It is thus not surprising that an enzymatic detoxification system has evolved to control intracellular methylglyoxal levels: The glyoxalase system, which consists of the enzymes glyoxalase 1 and 2 (Glo1 and Glo2) and a catalytic amount of reduced glutathione, catalyses the formation of lactate from methylglyoxal (256–259). Interestingly, evidence is emerging that methylglyoxal plays an important role in ageing and age-related disease: In the nematode Caenorhabditis elegans, a model organism frequently used in fundamental research on ageing and lifespan-extending mechanisms, glyoxalase activity is markedly reduced upon ageing and overexpression of the C. elegans orthologue of Glo1 increased lifespan (260, 261). Glyoxalase expression and/or activity has been reported to decrease upon ageing in human arterial tissue (262), in red blood cells (263), in human brain (256, 264), as well as in mouse lung (160). Methylglyoxal-protein adducts are increased in ageing human lens and skin (265, 266). Also, studies in rats have shown that overexpression of Glo1 protects from age-related renal dysfunction (267). Importantly, even if generated intracellularly, methylglyoxal can easily target the extracellular matrix because it is freely membrane-permeable (268) and has a relatively long range of diffusion—its tissue levels are therefore critically dependent on local intracellular glyoxalase activity (257). In summary, it is highly plausible that methylglyoxal and the glyoxalase system also contribute to molecular alterations of the ECM and to lung dysfunction upon ageing, even though direct evidence for this is lacking to this day.
Identification and quantification of AGEs are difficult tasks. First of all, AGEs are derived from many different precursor molecules and represent a highly heterogeneous group of compounds (253, 269). New variants of AGEs are still being discovered (270) and it is likely that not all AGE-protein adducts and crosslinks are known in molecular detail. For the analysis of clinical samples, early detection methods took advantage of the fluorescent properties of many AGEs: Collagen from connective tissue was extracted and solubilized by collagenase digestion and fluorescence measured [≈370 nm excitation/440 nm emission (271)]. More recently, autofluorescence of non-pigmented skin has been shown to correlate with collagen-linked fluorescence, concentrations of the fluorescent AGE pentosidine (Figure 6C), concentrations of the non-fluorescent AGEs Nε-(1-carboxymethyl)lysine (CML) and Nε-(1-carboxyethyl)lysine (CEL, Figure 6B), as well as with age, diabetes duration, and diabetic complications (272–278). Even though clearly a convenient non-invasive technique, any technique based on fluorescence will be limited in specificity and sensitivity, as not all AGEs are fluorescent including major representatives like CML and CEL and interference by other fluorescent molecules can occur, e.g., by NAD(P)H. Numerous attempts have been made to establish immunochemical detection of AGEs in tissue, albeit yielding inconsistent results due to lack of specificity, sample processing artefacts, and contamination by glycated blocking proteins (279–282). Therefore, quantitative results obtained by enzyme-linked immunosorbent assays (ELISAs), for instance, must be interpreted with caution.
The state-of-the-art detection method for AGEs in tissue and clinical samples is high performance or ultra-high performance liquid chromatography combined with mass spectrometry (279). New trending approaches, collectively referred to as “Maillard proteomics” are expected to revolutionise our understanding of glycation site specificity of AGE adducts, a prerequisite to gain an understanding of their function (269).
Even though the association between increases in AGE-mediated crosslinking, increased collagen stiffness, and decreased solubility with ageing is well-established (251), to the best of our knowledge surprisingly few studies have assessed AGEs and collagen AGE adducts in the lung. This may reflect the technical difficulties of specific AGE quantification pointed out above. Nevertheless, measurements of AGE-related fluorescence in the context of two animal studies support the concept that also in lung collagen AGE-load increases with age and anti-correlates with enzymatic solubility (220, 283). Interestingly, a study using the bleomycin-induced mouse model of lung fibrosis suggests that inhibition of AGE formation may protect from lung fibrosis (284). In reverse, accumulation of AGE load in the lung upon ageing may therefore represent one of the reasons why idiopathic lung fibrosis predominantly occurs in the elderly. Resonating with this concept, several reports indicate that diabetes mellitus, where increased blood glucose levels lead to increased tissue levels of AGEs, increases the risk for pulmonary fibrosis (285–288).
The Consequences of “AGEd” Collagen
But what can be the consequences of non-enzymatically glycated—or “AGEd”—collagen? There is evidence that AGEs increase collagen fibril stiffness and attenuate collagen turnover by, on the one hand, inhibiting degradation by MMPs and cathepsin K (239, 243, 289), and, on the other hand, inhibiting phagocytosis pathways (290). Furthermore, experiments with in vitro generated AGE-crosslinked collagen, by e.g., incubating collagen with ribose or methylglyoxal and measuring mechanical properties, indicate that in particular molecular sliding of collagen fibrils and fibres is affected by this type of crosslinking (291, 292). Hence, most likely “AGEd” fibrillar collagen contributes to the generally observed AGE-induced reduction of viscoelasticity of connective tissues accompanied by increased mechanical fragility (254, 291, 293–295).
In addition to changes in collagen turnover and biomechanics, AGE-mediated modification and crosslinking of collagen has the potential to mask important collagen-cell or collagen-protein-interaction sites with direct effects on adherent cell behaviour and function. Indeed, several studies with diverse cell types support this concept. For instance, non-enzymatically glycated collagen exhibits reduced affinity to heparin and keratan sulphate proteoglycans, resulting in diminished adhesion of B cells and reduced migration of endothelial cells (296). Similarly, methylglyoxal-modified type IV collagen displays reduced affinity to integrins, leading to attenuated adhesion of renal glomerular cells, and reduced attachment, viability, and angiogenic activity of endothelial cells (297, 298). Also neurons cultured on glycated type I collagen, show altered morphology and function in comparison to culture on normal type I collagen including reduced neuron interconnection and increased release of pro-inflammatory stimuli like nitrite and TNF-α (299). Finally, it has been reported that AGE-modified collagen fails to interact with discoidin domain receptor 2 and thus loses the capacity to upregulate expression of LOX in osteoblasts (300). Notably, even if direct evidence is lacking, this provides a potential mechanism underlying the above-described loss of LOX upon ageing in the lung and highlights a potential direct relationship between accumulation of AGE-mediated modifications/crosslinks and downregulation of LOX-mediated crosslinks.
Interestingly, molecular modelling studies using a collagen type I microfibril have put forward a number of candidate inter- and intramolecular Lys-Arg pairs that fulfil the requirements in terms of configuration and distance (301) and change in enthalpy (302) to allow for the formation of glucosepane crosslinks. In both studies, the authors identified more potentially AGE-crosslinked arginines and lysines within binding sites for integrins, proteoglycans, MMPs and other proteins. Collectively, these findings emphasise that “AGEd” collagen has a major impact on adherent cell function in many different tissues and clearly contributes to disease, warranting similar studies in the lung.
Effects Mediated Through the Receptor for Advanced Glycation Endproducts
AGEs are ligands for the receptor for advanced glycation endproducts (RAGE), a membrane-bound receptor of the immunoglobulin family (303, 304). Originally discovered in 1992 as an AGE-binding receptor (305), several additional ligands have been identified including S100/calgranulins, amyloid fibrils, amphoterins, and Mac-1. RAGE can thus be viewed as a relatively promiscuous pro-inflammatory pattern recognition receptor (303, 304). RAGE is abundantly expressed in the lung, at baseline predominantly in alveolar type I cells toward the basal membrane, but it can be activated in many other cell types upon exposure to RAGE ligands (304). Numerous studies have established associations of RAGE and its soluble decoy receptor sRAGE with lung injury and disease (303, 304, 306, 307).
RAGE-mediated downstream signalling depends on the identity and oligomerization state of the ligand and the tissue context. Ligand-engaged RAGE can trigger oxidative stress and multiple signalling cascades including p21 ras, erk1/2 (p44/p42) MAP kinases, p38 and SAPK/JNK MAP kinases, rho GTPases, phosphoinositol-3 kinase, and the JAK/STAT pathways. This leads predominantly to sustained activation of NF-κB- and STAT-dependent gene transcription (304, 308–310). As, typically, studies have been performed using soluble and globular glycated proteins such as serum albumin or ovalbumin (305, 311–313), it is not entirely clear whether “AGEd” collagen is capable of inducing these pathways. Studies using glycated collagen have established a RAGE-dependent link between “AGEd” collagen and apoptosis of mesenchymal cells. For instance, Nϵ(1-carboxymethyl) lysine (CML)-collagen (derived from reaction of collagen with glyoxal instead of methylglyoxal, Figure 6B), but not control collagen, induces fibroblast and osteoblast apoptosis via the RAGE/p38/JNK axis, but largely independent of NF-κB signalling (314, 315). Interestingly, some evidence indicates that RAGE promotes adhesion to collagen without the need for AGE-mediated modification, even though the glycation status of the collagen used was not reported in these studies (316, 317).
Conclusions
There can be no doubt that collagen plays an important role in lung physiology and pathology. Even though the collagen family of proteins has been the subject of studies for many decades it is striking how little we still know about collagen composition changes and molecular alterations in lung ageing and disease. Here, we provided a comprehensive review on mechanisms of collagen biosynthesis, processing, modifications and crosslinking and how these change upon ageing. Collectively, a picture emerges where ageing (1) leaves the normal ER-resident collagen biosynthesis machinery largely unaffected, but (2) results in distinct changes in collagen composition and (3) changes in the molecular nature of collagen crosslinks. Clearly, these observations warrant future work to address the mechanistic consequences of these changes in the context of lung ageing and disease. Ultimately, such studies may be critical to the field of lung regenerative medicine.
Author Contributions
CO, ED, and CS-W wrote the manuscript and prepared figures. HS and IA provided data on lung ageing and prepared Figure 5. All authors proofread and edited the manuscript.
Funding
This work was supported in part by the Friedrich-Baur-Stiftung (Reg.-Nr. 51/16, grant to CS-W), the Deutsche Forschungsgemeinschaft (DFG) within the Research Training Group DFG GRK2338 (ED and CS-W), the Helmholtz Association, and the German Center for Lung Research (DZL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Arseni L, Lombardi A, Orioli D. From structure to phenotype: impact of collagen alterations on human health. Int J Mol Sci. (2018) 19:1407. doi: 10.3390/ijms19051407
2. Kular JK, Basu S, Sharma RI. The extracellular matrix: structure, composition, age-related differences, tools for analysis and applications for tissue engineering. J Tissue Eng. (2014) 5:2041731414557112. doi: 10.1177/2041731414557112
3. Burgstaller G, Oehrle B, Gerckens M, White ES, Schiller HB, Eickelberg O. The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J. (2017) 50:1601805. doi: 10.1183/13993003.01805-2016
4. Zhou Y, Horowitz JC, Naba A, Ambalavanan N, Atabai K, Balestrini J, et al. Extracellular matrix in lung development, homeostasis and disease. Matrix Biol. (2018) 73:77–104. doi: 10.1016/j.matbio.2018.03.005
5. Beachley VZ, Wolf MT, Sadtler K, Manda SS, Jacobs H, Blatchley MR, et al. Tissue matrix arrays for high-throughput screening and systems analysis of cell function. Nat Methods. (2015) 12:1197–204. doi: 10.1038/nmeth.3619
6. Romero-Ortuno R, Kenny RA, McManus R. Collagens and elastin genetic variations and their potential role in aging-related diseases and longevity in humans. Exp Gerontol. (2020) 129:110781. doi: 10.1016/j.exger.2019.110781
7. Jobling R, D'Souza R, Baker N, Lara-Corrales I, Mendoza-Londono R, Dupuis L, et al. The collagenopathies: review of clinical phenotypes and molecular correlations. Curr Rheumatol Rep. (2014) 16:394. doi: 10.1007/s11926-013-0394-3
8. Gotte M, Kovalszky I. Extracellular matrix functions in lung cancer. Matrix Biol. (2018) 73:105–21. doi: 10.1016/j.matbio.2018.02.018
9. Blackwell TS, Tager AM, Borok Z, Moore BB, Schwartz DA, Anstrom KJ, et al. Future directions in idiopathic pulmonary fibrosis research. An NHLBI workshop report. Am J Respir Crit Care Med. (2014) 189:214–22. doi: 10.1164/rccm.201306-1141WS
10. Kim DS, Collard HR, King TE Jr. Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thoracic Soc. (2006) 3:285–92. doi: 10.1513/pats.200601-005TK
11. Renzoni E, Srihari V, Sestini P. Pathogenesis of idiopathic pulmonary fibrosis: review of recent findings. F1000 Prime Rep. (2014) 6:69. doi: 10.12703/P6-69
12. Selman M, King TE, Pardo A, American Thoracic S European Respiratory S P. American College of Chest P. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Internal Med. (2001) 134:136–51. doi: 10.7326/0003-4819-134-2-200101160-00015
13. Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med. (2014) 189:1161–72. doi: 10.1164/rccm.201312-2221PP
14. Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. (2014) 9:157–79. doi: 10.1146/annurev-pathol-012513-104706
15. Churg A, Zhou S, Wright JL. Series “matrix metalloproteinases in lung health and disease”: matrix metalloproteinases in COPD. Eur Respir J. (2012) 39:197–209. doi: 10.1183/09031936.00121611
16. Veit G, Kobbe B, Keene DR, Paulsson M, Koch M, Wagener R. Collagen XXVIII, a novel von Willebrand factor A domain-containing protein with many imperfections in the collagenous domain. J Biol Chem. (2006) 281:3494–504. doi: 10.1074/jbc.M509333200
17. Vranka JA, Sakai LY, Bachinger HP. Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J Biol Chem. (2004) 279:23615–21. doi: 10.1074/jbc.M312807200
18. Tiainen P, Pasanen A, Sormunen R, Myllyharju J. Characterization of recombinant human prolyl 3-hydroxylase isoenzyme 2, an enzyme modifying the basement membrane collagen IV. J Biol Chem. (2008) 283:19432–9. doi: 10.1074/jbc.M802973200
19. Gruenwald K, Castagnola P, Besio R, Dimori M, Chen Y, Akel NS, et al. Sc65 is a novel endoplasmic reticulum protein that regulates bone mass homeostasis. J Bone Mineral Res. (2014) 29:666–75. doi: 10.1002/jbmr.2075
20. Heard ME, Besio R, Weis M, Rai J, Hudson DM, Dimori M, et al. Sc65-null mice provide evidence for a novel endoplasmic reticulum complex regulating collagen lysyl hydroxylation. PLoS Genet. (2016) 12:e1006002. doi: 10.1371/journal.pgen.1006002
21. Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. (2006) 127:291–304. doi: 10.1016/j.cell.2006.08.039
22. Merl-Pham J, Basak T, Knüppel L, Ramanujam D, Athanason M, Behr J, et al. Quantitative proteomic profiling of extracellular matrix and site-specific collagen post-translational modifications in an in vitro model of lung fibrosis. Matrix Biol Plus. (2019) 1:100005. doi: 10.1016/j.mbplus.2019.04.002
23. Kadler KE, Baldock C, Bella J, Boot-Handford RP. Collagens at a glance. J Cell Sci. (2007) 120(Pt 12):1955–8. doi: 10.1242/jcs.03453
24. Gordon MK, Hahn RA. Collagens. Cell Tissue Res. (2010) 339:247–57. doi: 10.1007/s00441-009-0844-4
25. Sorushanova A, Delgado LM, Wu Z, Shologu N, Kshirsagar A, Raghunath R, et al. The collagen suprafamily: from biosynthesis to advanced biomaterial development. Adv Mater. (2019) 31:e1801651. doi: 10.1002/adma.201801651
26. Canty EG, Kadler KE. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci. (2005) 118(Pt 7):1341–53. doi: 10.1242/jcs.01731
27. Ricard-Blum S. The collagen family. Cold Spring Harbor Perspect Biol. (2011) 3:a004978. doi: 10.1101/cshperspect.a004978
28. Fitzgerald J, Rich C, Zhou FH, Hansen U. Three novel collagen VI chains, alpha4(VI), alpha5(VI), and alpha6(VI). J Biol Chem. (2008) 283:20170–80. doi: 10.1074/jbc.M710139200
29. Han S, Makareeva E, Kuznetsova NV, DeRidder AM, Sutter MB, Losert W, et al. Molecular mechanism of type I collagen homotrimer resistance to mammalian collagenases. J Biol Chem. (2010) 285:22276–81. doi: 10.1074/jbc.M110.102079
30. Kuznetsova NV, McBride DJ, Leikin S. Changes in thermal stability and microunfolding pattern of collagen helix resulting from the loss of alpha2(I) chain in osteogenesis imperfecta murine. J Mol Biol. (2003) 331:191–200. doi: 10.1016/S0022-2836(03)00715-0
31. Sharma U, Carrique L, Vadon-Le Goff S, Mariano N, Georges RN, Delolme F, et al. Structural basis of homo- and heterotrimerization of collagen I. Nat Commun. (2017) 8:14671. doi: 10.1038/ncomms14671
32. Rojkind M, Giambrone MA, Biempica L. Collagen types in normal and cirrhotic liver. Gastroenterology. (1979) 76:710–9. doi: 10.1016/S0016-5085(79)80170-5
33. Kilian O, Pfeil U, Wenisch S, Heiss C, Kraus R, Schnettler R. Enhanced alpha 1(I) mRNA expression in frozen shoulder and dupuytren tissue. Eur J Med Res. (2007) 12:585–90. Available online at: https://europepmc.org/article/med/18024269
34. Roberts-Pilgrim AM, Makareeva E, Myles MH, Besch-Williford CL, Brodeur AC, Walker AL, et al. Deficient degradation of homotrimeric type I collagen, alpha1(I)3 glomerulopathy in oim mice. Mol Genet Metab. (2011) 104:373–82. doi: 10.1016/j.ymgme.2011.07.025
35. Kirkness MW, Lehmann K, Forde NR. Mechanics and structural stability of the collagen triple helix. Curr Opin Chem Biol. (2019) 53:98–105. doi: 10.1016/j.cbpa.2019.08.001
36. Ishikawa Y, Bachinger HP. A molecular ensemble in the rER for procollagen maturation. Biochim Biophys Acta. (2013) 1833:2479–91. doi: 10.1016/j.bbamcr.2013.04.008
37. Holster T, Pakkanen O, Soininen R, Sormunen R, Nokelainen M, Kivirikko KI, et al. Loss of assembly of the main basement membrane collagen, type IV, but not fibril-forming collagens and embryonic death in collagen prolyl 4-hydroxylase I null mice. J Biol Chem. (2007) 282:2512–9. doi: 10.1074/jbc.M606608200
38. Kobayashi Y, Sakai R, Kakiuchi K, Isemura T. Physicochemical analysis of (Pro-Pro-Gly)n with defined molecular weight–temperature dependence of molecular weight in aqueous solution. Biopolymers. (1970) 9:415–25. doi: 10.1002/bip.1970.360090404
39. Sakakibara S, Inouye K, Shudo K, Kishida Y, Kobayashi Y, Prockop DJ. Synthesis of (Pro-Hyp-Gly) n of defined molecular weights. Evidence for the stabilization of collagen triple helix by hydroxypyroline. Biochim Biophys Acta. (1973) 303:198–202. doi: 10.1016/0005-2795(73)90164-5
40. Burjanadze TV. Thermodynamic substantiation of water-bridged collagen structure. Biopolymers. (1992) 32:941–9. doi: 10.1002/bip.360320805
41. Rappu P, Salo AM, Myllyharju J, Heino J. Role of prolyl hydroxylation in the molecular interactions of collagens. Essays Biochem. (2019) 63:325–35. doi: 10.1042/EBC20180053
42. Weis MA, Hudson DM, Kim L, Scott M, Wu JJ, Eyre DR. Location of 3-hydroxyproline residues in collagen types I, II, III, and V/XI implies a role in fibril supramolecular assembly. J Biol Chem. (2010) 285:2580–90. doi: 10.1074/jbc.M109.068726
43. Pokidysheva E, Tufa S, Bresee C, Brigande JV, Bachinger HP. Prolyl 3-hydroxylase-1 null mice exhibit hearing impairment and abnormal morphology of the middle ear bone joints. Matrix Biol. (2013) 32:39–44. doi: 10.1016/j.matbio.2012.11.006
44. Pokidysheva E, Zientek KD, Ishikawa Y, Mizuno K, Vranka JA, Montgomery NT, et al. Posttranslational modifications in type I collagen from different tissues extracted from wild type and prolyl 3-hydroxylase 1 null mice. J Biol Chem. (2013) 288:24742–52. doi: 10.1074/jbc.M113.464156
45. Vranka JA, Pokidysheva E, Hayashi L, Zientek K, Mizuno K, Ishikawa Y, et al. Prolyl 3-hydroxylase 1 null mice display abnormalities in fibrillar collagen-rich tissues such as tendons, skin, and bones. J Biol Chem. (2010) 285:17253–62. doi: 10.1074/jbc.M110.102228
46. Pokidysheva E, Boudko S, Vranka J, Zientek K, Maddox K, Moser M, et al. Biological role of prolyl 3-hydroxylation in type IV collagen. Proc Natl Acad Sci USA. (2014) 111:161–6. doi: 10.1073/pnas.1307597111
47. Xu ER, Blythe EE, Fischer G, Hyvonen M. Structural analyses of von Willebrand factor C domains of collagen 2A and CCN3 reveal an alternative mode of binding to bone morphogenetic protein-2. J Biol Chem. (2017) 292:12516–27. doi: 10.1074/jbc.M117.788992
48. Campbell ID, Spitzfaden C. Building proteins with fibronectin type III modules. Structure. (1994) 2:333–7. doi: 10.1016/S0969-2126(00)00034-4
49. Bolten SN, Rinas U, Scheper T. Heparin: role in protein purification and substitution with animal-component free material. Appl Microbiol Biotechnol. (2018) 102:8647–60. doi: 10.1007/s00253-018-9263-3
50. Leppanen VM, Tossavainen H, Permi P, Lehtio L, Ronnholm G, Goldman A, et al. Crystal structure of the N-terminal NC4 domain of collagen IX, a zinc binding member of the laminin-neurexin-sex hormone binding globulin (LNS) domain family. J Biol Chem. (2007) 282:23219–30. doi: 10.1074/jbc.M702514200
51. Rattenholl A, Pappano WN, Koch M, Keene DR, Kadler KE, Sasaki T, et al. Proteinases of the bone morphogenetic protein-1 family convert procollagen VII to mature anchoring fibril collagen. J Biol Chem. (2002) 277:26372–8. doi: 10.1074/jbc.M203247200
52. Bruckner-Tuderman L, Nilssen O, Zimmermann DR, Dours-Zimmermann MT, Kalinke DU, Gedde-Dahl T Jr, et al. Immunohistochemical and mutation analyses demonstrate that procollagen VII is processed to collagen VII through removal of the NC-2 domain. J Cell Biol. (1995) 131:551–9. doi: 10.1083/jcb.131.2.551
53. Aigner T, Hambach L, Soder S, Schlotzer-Schrehardt U, Poschl E. The C5 domain of Col6A3 is cleaved off from the Col6 fibrils immediately after secretion. Biochem Biophys Res Commun. (2002) 290:743–8. doi: 10.1006/bbrc.2001.6227
54. Park J, Scherer PE. Adipocyte-derived endotrophin promotes malignant tumor progression. J Clin Investig. (2012) 122:4243–56. doi: 10.1172/JCI63930
55. Nanda A, Carson-Walter EB, Seaman S, Barber TD, Stampfl J, Singh S, et al. TEM8 interacts with the cleaved C5 domain of collagen alpha 3(VI). Cancer Res. (2004) 64:817–20. doi: 10.1158/0008-5472.CAN-03-2408
56. Chen P, Cescon M, Bonaldo P. Collagen VI in cancer and its biological mechanisms. Trends Mol Med. (2013) 19:410–7. doi: 10.1016/j.molmed.2013.04.001
57. Ricard-Blum S, Salza R. Matricryptins and matrikines: biologically active fragments of the extracellular matrix. Exp Dermatol. (2014) 23:457–63. doi: 10.1111/exd.12435
58. Izzi V, Heljasvaara R, Heikkinen A, Karppinen SM, Koivunen J, Pihlajaniemi T. Exploring the roles of MACIT and multiplexin collagens in stem cells and cancer. Semin Cancer Biol. (2020) 62:134–48. doi: 10.1016/j.semcancer.2019.08.033
59. Tu H, Huhtala P, Lee HM, Adams JC, Pihlajaniemi T. Membrane-associated collagens with interrupted triple-helices (MACITs): evolution from a bilaterian common ancestor and functional conservation in C. elegans. BMC Evolut Biol. (2015) 15:281. doi: 10.1186/s12862-015-0554-3
60. Franzke CW, Bruckner-Tuderman L, Blobel CP. Shedding of collagen XVII/BP180 in skin depends on both ADAM10 and ADAM9. J Biol Chem. (2009) 284:23386–96. doi: 10.1074/jbc.M109.034090
61. Ricard-Blum S, Vallet SD. Matricryptins network with matricellular receptors at the surface of endothelial and tumor cells. Front Pharmacol. (2016) 7:11. doi: 10.3389/fphar.2016.00011
62. Ramchandran R, Dhanabal M, Volk R, Waterman MJ, Segal M, Lu H, et al. Antiangiogenic activity of restin, NC10 domain of human collagen XV: comparison to endostatin. Biochem Biophys Res Commun. (1999) 255:735–9. doi: 10.1006/bbrc.1999.0248
63. Vanacore R, Ham AJ, Voehler M, Sanders CR, Conrads TP, Veenstra TD, et al. A sulfilimine bond identified in collagen IV. Science. (2009) 325:1230–4. doi: 10.1126/science.1176811
64. Bhave G, Cummings CF, Vanacore RM, Kumagai-Cresse C, Ero-Tolliver IA, Rafi M, et al. Peroxidasin forms sulfilimine chemical bonds using hypohalous acids in tissue genesis. Nat Chem Biol. (2012) 8:784–90. doi: 10.1038/nchembio.1038
65. Anazco C, Lopez-Jimenez AJ, Rafi M, Vega-Montoto L, Zhang MZ, Hudson BG, et al. Lysyl oxidase-like-2 cross-links collagen IV of glomerular basement membrane. J Biol Chem. (2016) 291:25999–6012. doi: 10.1074/jbc.M116.738856
66. Brittingham R, Colombo M, Ito H, Steplewski A, Birk DE, Uitto J, et al. Single amino acid substitutions in procollagen VII affect early stages of assembly of anchoring fibrils. J Biol Chem. (2005) 280:191–8. doi: 10.1074/jbc.M406210200
67. Ricard-Blum S, Dublet B, van der Rest M. Unconventional Collagens: Types 6, 7, 8, 9, 10, 12, 14, 16, and 19. Oxford, UK: Oxford University Press (2000).
68. Ishikawa Y, Boudko S, Bachinger HP. Ziploc-ing the structure: triple helix formation is coordinated by rough endoplasmic reticulum resident PPIases. Biochim Biophys Acta. (2015) 1850:1983–93. doi: 10.1016/j.bbagen.2014.12.024
69. Luo Y, Xu W, Chen H, Warburton D, Dong R, Qian B, et al. A novel profibrotic mechanism mediated by TGFbeta-stimulated collagen prolyl hydroxylase expression in fibrotic lung mesenchymal cells. J Pathol. (2015) 236:384–94. doi: 10.1002/path.4530
70. Gjaltema RA, Bank RA. Molecular insights into prolyl and lysyl hydroxylation of fibrillar collagens in health and disease. Crit Rev Biochem Mol Biol. (2017) 52:74–95. doi: 10.1080/10409238.2016.1269716
71. Yamauchi M, Sricholpech M. Lysine post-translational modifications of collagen. Essays Biochem. (2012) 52:113–33. doi: 10.1042/bse0520113
72. Gjaltema RA, van der Stoel MM, Boersema M, Bank RA. Disentangling mechanisms involved in collagen pyridinoline cross-linking: the immunophilin FKBP65 is critical for dimerization of lysyl hydroxylase 2. Proc Natl Acad Sci USA. (2016) 113:7142–7. doi: 10.1073/pnas.1600074113
73. Eyre DR, Weis MA, Wu JJ. Advances in collagen cross-link analysis. Methods. (2008) 45:65–74. doi: 10.1016/j.ymeth.2008.01.002
74. Boudko SP, Sasaki T, Engel J, Lerch TF, Nix J, Chapman MS, et al. Crystal structure of human collagen XVIII trimerization domain: a novel collagen trimerization fold. J Mol Biol. (2009) 392:787–802. doi: 10.1016/j.jmb.2009.07.057
75. Khoshnoodi J, Cartailler JP, Alvares K, Veis A, Hudson BG. Molecular recognition in the assembly of collagens: terminal noncollagenous domains are key recognition modules in the formation of triple helical protomers. J Biol Chem. (2006) 281:38117–21. doi: 10.1074/jbc.R600025200
76. Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. (2007) 3:619–29. doi: 10.1038/nchembio.2007.35
77. Knüppel L, Heinzelmann K, Lindner M, Hatz R, Behr J, Eickelberg O, et al. FK506-binding protein 10 (FKBP10) regulates lung fibroblast migration via collagen VI synthesis. Respir Res. (2018) 19:67. doi: 10.1186/s12931-018-0768-1
78. Staab-Weijnitz CA, Fernandez IE, Knuppel L, Maul J, Heinzelmann K, Juan-Guardela BM, et al. FK506-binding protein 10, a potential novel drug target for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2015) 192:455–67. doi: 10.1164/rccm.201412-2233OC
79. Ishikawa Y, Bachinger HP. A substrate preference for the rough endoplasmic reticulum resident protein FKBP22 during collagen biosynthesis. J Biol Chem. (2014) 289:18189–201. doi: 10.1074/jbc.M114.561944
80. Ishida Y, Nagata K. Hsp47 as a collagen-specific molecular chaperone. Methods Enzymol. (2011) 499:167–82. doi: 10.1016/B978-0-12-386471-0.00009-2
81. Drogemuller C, Becker D, Brunner A, Haase B, Kircher P, Seeliger F, et al. A missense mutation in the SERPINH1 gene in Dachshunds with osteogenesis imperfecta. PLoS Genet. (2009) 5:e1000579. doi: 10.1371/journal.pgen.1000579
82. Smith T, Ferreira LR, Hebert C, Norris K, Sauk JJ. Hsp47 and cyclophilin B traverse the endoplasmic reticulum with procollagen into pre-Golgi intermediate vesicles. A role for Hsp47 and cyclophilin B in the export of procollagen from the endoplasmic reticulum. J Biol Chem. (1995) 270:18323–8. doi: 10.1074/jbc.270.31.18323
83. Lee MC, Miller EA, Goldberg J, Orci L, Schekman R. Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol. (2004) 20:87–123. doi: 10.1146/annurev.cellbio.20.010403.105307
84. Stephens DJ. Cell biology: collagen secretion explained. Nature. (2012) 482:474–5. doi: 10.1038/482474a
85. Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgartel C, Schekman R, et al. Ubiquitin-dependent regulation of COPII coat size and function. Nature. (2012) 482:495–500. doi: 10.1038/nature10822
86. Saito K, Katada T. Mechanisms for exporting large-sized cargoes from the endoplasmic reticulum. Cell Mol Life Sci. (2015) 72:3709–20. doi: 10.1007/s00018-015-1952-9
87. Malhotra V, Erlmann P, Nogueira C. Procollagen export from the endoplasmic reticulum. Biochem Soc Trans. (2015) 43:104–7. doi: 10.1042/BST20140286
88. McCaughey J, Stephens DJ. ER-to-Golgi transport: a sizeable problem. Trends Cell Biol. (2019) 29:940–53. doi: 10.1016/j.tcb.2019.08.007
89. Saito K, Chen M, Bard F, Chen S, Zhou H, Woodley D, et al. TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell. (2009) 136:891–902. doi: 10.1016/j.cell.2008.12.025
90. Ma W, Goldberg J. TANGO1/cTAGE5 receptor as a polyvalent template for assembly of large COPII coats. Proc Natl Acad Sci USA. (2016) 113:10061–6. doi: 10.1073/pnas.1605916113
91. Raote I, Ortega Bellido M, Pirozzi M, Zhang C, Melville D, Parashuraman S, Zimmermann T, et al. TANGO1 assembles into rings around COPII coats at ER exit sites. J Cell Biol. (2017) 216:901–9. doi: 10.1083/jcb.201608080
92. Raote I, Ortega-Bellido M, Santos AJ, Foresti O, Zhang C, Garcia-Parajo MF, et al. TANGO1 builds a machine for collagen export by recruiting and spatially organizing COPII, tethers and membranes. Elife. (2018) 7:e32723. doi: 10.7554/eLife.32723
93. Venditti R, Scanu T, Santoro M, Di Tullio G, Spaar A, Gaibisso R, et al. Sedlin controls the ER export of procollagen by regulating the Sar1 cycle. Science. (2012) 337:1668–72. doi: 10.1126/science.1224947
94. McCaughey J, Stevenson NL, Cross S, Stephens DJ. ER-to-Golgi trafficking of procollagen in the absence of large carriers. J Cell Biol. (2019) 218:929–48. doi: 10.1083/jcb.201806035
95. Prox J, Arnold P, Becker-Pauly C. Meprin alpha and meprin beta: procollagen proteinases in health and disease. Matrix Biol. (2015) 44–46:7–13. doi: 10.1016/j.matbio.2015.01.010
96. Kadler KE, Holmes DF, Trotter JA, Chapman JA. Collagen fibril formation. Biochem J. (1996) 316(Pt 1):1–11. doi: 10.1042/bj3160001
97. Williams BR, Gelman RA, Poppke DC, Piez KA. Collagen fibril formation. Optimal in vitro conditions and preliminary kinetic results. J Biol Chem. (1978) 253:6578–85. doi: 10.1016/S0021-9258(19)46970-6
98. Bowness JM, Folk JE, Timpl R. Identification of a substrate site for liver transglutaminase on the aminopropeptide of type III collagen. J Biol Chem. (1987) 262:1022–4. doi: 10.1016/S0021-9258(19)75743-3
99. Bowness JM, Tarr AH, Wiebe RI. Transglutaminase-catalysed cross-linking: a potential mechanism for the interaction of fibrinogen, low density lipoprotein and arterial type III procollagen. Thrombosis Res. (1989) 54:357–67. doi: 10.1016/0049-3848(89)90094-7
100. Shweke N, Boulos N, Jouanneau C, Vandermeersch S, Melino G, Dussaule JC, et al. Tissue transglutaminase contributes to interstitial renal fibrosis by favoring accumulation of fibrillar collagen through TGF-beta activation and cell infiltration. Am J Pathol. (2008) 173:631–42. doi: 10.2353/ajpath.2008.080025
101. Vallet SD, Ricard-Blum S. Lysyl oxidases: from enzyme activity to extracellular matrix cross-links. Essays Biochem. (2019) 63:349–64. doi: 10.1042/EBC20180050
102. Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. (2010) 16:1009–17. doi: 10.1038/nm.2208
103. Kliment CR, Englert JM, Gochuico BR, Yu G, Kaminski N, Rosas I, et al. Oxidative stress alters syndecan-1 distribution in lungs with pulmonary fibrosis. J Biol Chem. (2009) 284:3537–45. doi: 10.1074/jbc.M807001200
104. Kliment CR, Tobolewski JM, Manni ML, Tan RJ, Enghild J, Oury TD. Extracellular superoxide dismutase protects against matrix degradation of heparan sulfate in the lung. Antioxidants Redox Signal. (2008) 10:261–8. doi: 10.1089/ars.2007.1906
105. Schamberger AC, Schiller HB, Fernandez IE, Sterclova M, Heinzelmann K, Hennen E, et al. Glutathione peroxidase 3 localizes to the epithelial lining fluid and the extracellular matrix in interstitial lung disease. Sci Rep. (2016) 6:29952. doi: 10.1038/srep29952
106. Zelko IN, Folz RJ. Extracellular superoxide dismutase attenuates release of pulmonary hyaluronan from the extracellular matrix following bleomycin exposure. FEBS Lett. (2010) 584:2947–52. doi: 10.1016/j.febslet.2010.05.025
107. Bradley K, McConnell-Breul S, Crystal RG. Collagen in the human lung. Quantitation of rates of synthesis and partial characterization of composition. J Clin Investig. (1975) 55:543–50. doi: 10.1172/JCI107961
108. Sivan SS, Wachtel E, Tsitron E, Sakkee N, van der Ham F, Degroot J, et al. Collagen turnover in normal and degenerate human intervertebral discs as determined by the racemization of aspartic acid. J Biol Chem. (2008) 283:8796–801. doi: 10.1074/jbc.M709885200
109. Verzijl N, DeGroot J, Thorpe SR, Bank RA, Shaw JN, Lyons TJ, et al. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. (2000) 275:39027–31. doi: 10.1074/jbc.M006700200
111. Decaris ML, Gatmaitan M, FlorCruz S, Luo F, Li K, Holmes WE, et al. Proteomic analysis of altered extracellular matrix turnover in bleomycin-induced pulmonary fibrosis. Mol Cell Proteomics. (2014) 13:1741–52. doi: 10.1074/mcp.M113.037267
112. Last JA, Reiser KM. Biosynthesis of collagen crosslinks. III. In vivo labeling and stability of lung collagen in rats with bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. (1989) 1:111–7. doi: 10.1165/ajrcmb/1.2.111
113. Last JA, Summers P, Reiser KM. Biosynthesis of collagen crosslinks. II. In vivo labelling and stability of lung collagen in rats. Biochim Biophys Acta. (1989) 990:182–9. doi: 10.1016/S0304-4165(89)80032-7
114. Laurent GJ. Rates of collagen synthesis in lung, skin and muscle obtained in vivo by a simplified method using [3H]proline. Biochem J. (1982) 206:535–44. doi: 10.1042/bj2060535
115. Laurent GJ. Lung collagen: more than scaffolding. Thorax. (1986) 41:418–28. doi: 10.1136/thx.41.6.418
116. McAnulty RJ, Laurent GJ. Collagen synthesis and degradation in vivo. Evidence for rapid rates of collagen turnover with extensive degradation of newly synthesized collagen in tissues of the adult rat. Collagen Relat Res. (1987) 7:93–104. doi: 10.1016/S0174-173X(87)80001-8
117. McKleroy W, Lee TH, Atabai K. Always cleave up your mess: targeting collagen degradation to treat tissue fibrosis. Am J Physiol Lung Cell Mol Physiol. (2013) 304:L709–21. doi: 10.1152/ajplung.00418.2012
118. Craig VJ, Zhang L, Hagood JS, Owen CA. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. (2015) 53:585–600. doi: 10.1165/rcmb.2015-0020TR
119. Afratis NA, Selman M, Pardo A, Sagi I. Emerging insights into the role of matrix metalloproteases as therapeutic targets in fibrosis. Matrix Biol. (2018) 68–69:167–79. doi: 10.1016/j.matbio.2018.02.007
120. Lauer-Fields JL, Juska D, Fields GB. Matrix metalloproteinases and collagen catabolism. Biopolymers. (2002) 66:19–32. doi: 10.1002/bip.10201
121. Henriet P, Emonard H. Matrix metalloproteinase-2: Not (just) a “hero” of the past. Biochimie. (2019) 166:223–32. doi: 10.1016/j.biochi.2019.07.019
122. Wilson CL, Matrisian LM. Matrilysin: an epithelial matrix metalloproteinase with potentially novel functions. Int J Biochem Cell Biol. (1996) 28:123–36. doi: 10.1016/1357-2725(95)00121-2
123. Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circul Res. (2003) 92:827–39. doi: 10.1161/01.RES.0000070112.80711.3D
124. Jobin PG, Butler GS, Overall CM. New intracellular activities of matrix metalloproteinases shine in the moonlight. Biochim Biophys Acta. (2017) 1864(11 Pt A):2043–55. doi: 10.1016/j.bbamcr.2017.05.013
125. Chung L, Dinakarpandian D, Yoshida N, Lauer-Fields JL, Fields GB, Visse R, et al. Collagenase unwinds triple-helical collagen prior to peptide bond hydrolysis. EMBO J. (2004) 23:3020–30. doi: 10.1038/sj.emboj.7600318
126. Manka SW, Carafoli F, Visse R, Bihan D, Raynal N, Farndale RW, et al. Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1. Proc Natl Acad Sci USA. (2012) 109:12461–6. doi: 10.1073/pnas.1204991109
127. Aguda AH, Panwar P, Du X, Nguyen NT, Brayer GD, Brömme D. Structural basis of collagen fiber degradation by cathepsin K. Proc Natl Acad Sci USA. (2014) 111:17474–9. doi: 10.1073/pnas.1414126111
128. Panwar P, Du X, Sharma V, Lamour G, Castro M, Li H, et al. Effects of cysteine proteases on the structural and mechanical properties of collagen fibers. J Biol Chem. (2013) 288:5940–50. doi: 10.1074/jbc.M112.419689
129. Everts V, van der Zee E, Creemers L, Beertsen W. Phagocytosis and intracellular digestion of collagen, its role in turnover and remodelling. Histochem J. (1996) 28:229–45. doi: 10.1007/BF02409011
130. Lee W, Sodek J, McCulloch CA. Role of integrins in regulation of collagen phagocytosis by human fibroblasts. J Cell Physiol. (1996) 168:695–704. doi: 10.1002/(SICI)1097-4652(199609)168:3<695::AID-JCP22>3.0.CO;2-X
131. Jokinen J, Dadu E, Nykvist P, Kapyla J, White DJ, Ivaska J, et al. Integrin-mediated cell adhesion to type I collagen fibrils. J Biol Chem. (2004) 279:31956–63. doi: 10.1074/jbc.M401409200
132. Heino J. Cellular signaling by collagen-binding integrins. Adv Exp Med Biol. (2014) 819:143–55. doi: 10.1007/978-94-017-9153-3_10
133. Zeltz C, Gullberg D. The integrin-collagen connection–a glue for tissue repair? J Cell Sci. (2016) 129:653–64. doi: 10.1242/jcs.180992
134. Lee H, Overall CM, McCulloch CA, Sodek J. A critical role for the membrane-type 1 matrix metalloproteinase in collagen phagocytosis. Mol Biol Cell. (2006) 17:4812–26. doi: 10.1091/mbc.e06-06-0486
135. von Delwig A, Hilkens CM, Altmann DM, Holmdahl R, Isaacs JD, Harding CV, et al. Inhibition of macropinocytosis blocks antigen presentation of type II collagen in vitro and in vivo in HLA-DR1 transgenic mice. Arthritis Res Ther. (2006) 8:R93. doi: 10.1186/ar1964
136. Yamazaki S, Su Y, Maruyama A, Makinoshima H, Suzuki J, Tsuboi M, et al. Uptake of collagen type I via macropinocytosis cause mTOR activation and anti-cancer drug resistance. Biochem Biophys Res Commun. (2020) 526:191–8. doi: 10.1016/j.bbrc.2020.03.067
137. Kjoller L, Engelholm LH, Hoyer-Hansen M, Dano K, Bugge TH, Behrendt N. uPARAP/endo180 directs lysosomal delivery and degradation of collagen IV. Exp Cell Res. (2004) 293:106–16. doi: 10.1016/j.yexcr.2003.10.008
138. Madsen DH, Engelholm LH, Ingvarsen S, Hillig T, Wagenaar-Miller RA, Kjoller L, et al. Extracellular collagenases and the endocytic receptor, urokinase plasminogen activator receptor-associated protein/Endo180, cooperate in fibroblast-mediated collagen degradation. J Biol Chem. (2007) 282:27037–45. doi: 10.1074/jbc.M701088200
139. Madsen DH, Ingvarsen S, Jurgensen HJ, Melander MC, Kjoller L, Moyer A, et al. The non-phagocytic route of collagen uptake: a distinct degradation pathway. J Biol Chem. (2011) 286:26996–7010. doi: 10.1074/jbc.M110.208033
140. Atabai K, Jame S, Azhar N, Kuo A, Lam M, McKleroy W, et al. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J Clin Investig. (2009) 119:3713–22. doi: 10.1172/JCI40053
141. Martinez-Pomares L, Wienke D, Stillion R, McKenzie EJ, Arnold JN, Harris J, et al. Carbohydrate-independent recognition of collagens by the macrophage mannose receptor. Eur J Immunol. (2006) 36:1074–82. doi: 10.1002/eji.200535685
142. Fonovic M, Turk B. Cysteine cathepsins and extracellular matrix degradation. Biochim Biophys Acta. (2014) 1840:2560–70. doi: 10.1016/j.bbagen.2014.03.017
143. Kafienah W, Bromme D, Buttle DJ, Croucher LJ, Hollander AP. Human cathepsin K cleaves native type I and II collagens at the N-terminal end of the triple helix. Biochem J. (1998) 331(Pt 3):727–32. doi: 10.1042/bj3310727
144. Song F, Wisithphrom K, Zhou J, Windsor LJ. Matrix metalloproteinase dependent and independent collagen degradation. Front Biosci. (2006) 11:3100–20. doi: 10.2741/2036
145. Buck MR, Karustis DG, Day NA, Honn KV, Sloane BF. Degradation of extracellular-matrix proteins by human cathepsin B from normal and tumour tissues. Biochem J. (1992) 282(Pt 1):273–8. doi: 10.1042/bj2820273
146. Scott PG, Pearson H. Cathepsin D: specificity of peptide-bond cleavage in type-I collagen and effects on type-III collagen and procollagen. Eur J Biochem. (1981) 114:59–62. doi: 10.1111/j.1432-1033.1981.tb06172.x
147. Garnero P, Borel O, Byrjalsen I, Ferreras M, Drake FH, McQueney MS, et al. The collagenolytic activity of cathepsin K is unique among mammalian proteinases. J Biol Chem. (1998) 273:32347–52. doi: 10.1074/jbc.273.48.32347
148. Nosaka AY, Kanaori K, Teno N, Togame H, Inaoka T, Takai M, et al. Conformational studies on the specific cleavage site of Type I collagen (alpha-1) fragment (157-192) by cathepsins K and L by proton NMR spectroscopy. Bioorg Med Chem. (1999) 7:375–9. doi: 10.1016/S0968-0896(98)00227-2
149. Berg RA, Schwartz ML, Crystal RG. Regulation of the production of secretory proteins: intracellular degradation of newly synthesized “defective” collagen. Proc Natl Acad Sci USA. (1980) 77:4746–50. doi: 10.1073/pnas.77.8.4746
150. Bienkowski RS. Intracellular degradation of newly synthesized collagen. Rev Sobre Biol Cell. (1989) 21:423–43.
151. Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-beta1. J Biol Chem. (2012) 287:11677–88. doi: 10.1074/jbc.M111.308460
152. Klionsky DJ, Eskelinen EL, Deretic V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes. wait, I'm confused. Autophagy. (2014) 10:549–51. doi: 10.4161/auto.28448
153. Glass D, Vinuela A, Davies MN, Ramasamy A, Parts L, Knowles D, et al. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. (2013) 14:R75. doi: 10.1186/gb-2013-14-7-r75
154. Turner N, Mason PJ, Brown R, Fox M, Povey S, Rees A, et al. Molecular cloning of the human Goodpasture antigen demonstrates it to be the alpha 3 chain of type IV collagen. J Clin Investig. (1992) 89:592–601. doi: 10.1172/JCI115625
155. Hudson BG, Kalluri R, Gunwar S, Weber M, Ballester F, Hudson JK, et al. The pathogenesis of Alport syndrome involves type IV collagen molecules containing the alpha 3(IV) chain: evidence from anti-GBM nephritis after renal transplantation. Kidney Int. (1992) 42:179–87. doi: 10.1038/ki.1992.276
156. Guo BR, Zhang X, Chen G, Zhang JG, Sun LD, Du WD, et al. Exome sequencing identifies a COL14A1 mutation in a large Chinese pedigree with punctate palmoplantar keratoderma. J Med Genet. (2012) 49:563–8. doi: 10.1136/jmedgenet-2012-100868
157. Zhang XJ, Li M, Gao TW, He PP, Wei SC, Liu JB, et al. Identification of a locus for punctate palmoplantar keratodermas at chromosome 8q24.13-8q24.21. J Investig Dermatol. (2004) 122:1121–5. doi: 10.1111/j.0022-202X.2004.22507.x
158. Guo DC, Regalado ES, Gong L, Duan X, Santos-Cortez RL, Arnaud P, et al. LOX mutations predispose to thoracic aortic aneurysms and dissections. Circul Res. (2016) 118:928–34. doi: 10.1161/CIRCRESAHA.115.307130
159. Lee VS, Halabi CM, Hoffman EP, Carmichael N, Leshchiner I, Lian CG, et al. Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc Natl Acad Sci USA. (2016) 113:8759–64. doi: 10.1073/pnas.1601442113
160. Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun. (2019) 10:963. doi: 10.1038/s41467-019-08831-9
161. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. (2020) 6:eaba1983. doi: 10.1126/sciadv.aba1983
162. Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv. (2020) 6:eaba1972. doi: 10.1126/sciadv.aba1972
163. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med. (2019) 199:1517–36. doi: 10.1101/296608
164. Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J. (2019) 54:1802441. doi: 10.1183/13993003.02441-2018
165. Bober M, Enochsson C, Collin M, Morgelin M. Collagen VI is a subepithelial adhesive target for human respiratory tract pathogens. J Innate Immun. (2010) 2:160–6. doi: 10.1159/000232587
166. Cescon M, Gattazzo F, Chen P, Bonaldo P. Collagen VI at a glance. J Cell Sci. (2015) 128:3525–31. doi: 10.1242/jcs.169748
167. Bonnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol. (2011) 7:379–90. doi: 10.1038/nrneurol.2011.81
168. Aumailley M, Specks U, Timpl R. Cell adhesion to type-VI collagen. Biochem Soc Trans. (1991) 19:843–7. doi: 10.1042/bst0190843
169. Klein G, Muller CA, Tillet E, Chu ML, Timpl R. Collagen type VI in the human bone marrow microenvironment: a strong cytoadhesive component. Blood. (1995) 86:1740–8. doi: 10.1182/blood.V86.5.1740.bloodjournal8651740
170. Atkinson JC, Ruhl M, Becker J, Ackermann R, Schuppan D. Collagen VI regulates normal and transformed mesenchymal cell proliferation in vitro. Exp Cell Res. (1996) 228:283–91. doi: 10.1006/excr.1996.0328
171. Perris R, Kuo HJ, Glanville RW, Bronner-Fraser M. Collagen type VI in neural crest development: distribution in situ and interaction with cells in vitro. Dev Dyn. (1993) 198:135–49. doi: 10.1002/aja.1001980207
172. Iyengar P, Espina V, Williams TW, Lin Y, Berry D, Jelicks LA, et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J Clin Investig. (2005) 115:1163–76. doi: 10.1172/JCI23424
173. Ruhl M, Sahin E, Johannsen M, Somasundaram R, Manski D, Riecken EO, et al. Soluble collagen VI drives serum-starved fibroblasts through S phase and prevents apoptosis via down-regulation of Bax. J Biol Chem. (1999) 274:34361–8. doi: 10.1074/jbc.274.48.34361
174. Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet. (2003) 35:367–71. doi: 10.1038/ng1270
175. Gara SK, Grumati P, Urciuolo A, Bonaldo P, Kobbe B, Koch M, et al. Three novel collagen VI chains with high homology to the alpha3 chain. J Biol Chem. (2008) 283:10658–70. doi: 10.1074/jbc.M709540200
176. Gara SK, Grumati P, Squarzoni S, Sabatelli P, Urciuolo A, Bonaldo P, et al. Differential and restricted expression of novel collagen VI chains in mouse. Matrix Biol. (2011) 30:248–57. doi: 10.1016/j.matbio.2011.03.006
177. Fitzgerald J, Holden P, Hansen U. The expanded collagen VI family: new chains and new questions. Connect Tissue Res. (2013) 54:345–50. doi: 10.3109/03008207.2013.822865
178. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
179. Meiners S, Eickelberg O, Konigshoff M. Hallmarks of the ageing lung. Eur Respir J. (2015) 45:807–27. doi: 10.1183/09031936.00186914
180. Sato T, Takano R, Tokunaka K, Saiga K, Tomura A, Sugihara H, et al. Type VI collagen alpha1 chain polypeptide in non-triple helical form is an alternative gene product of COL6A1. J Biochem. (2018) 164:173–81. doi: 10.1093/jb/mvy040
181. Sugiyama H, Tokunaka K, Hayashi T, Imamura Y, Morita M, Yamato M. Non-triple helical form of Type IV collagen alpha1 chain. Heliyon. (2015) 1:e00051. doi: 10.1016/j.heliyon.2015.e00051
182. Shin Y, Moriya A, Tohnishi Y, Watanabe T, Imamura Y. Basement membrane-like structures containing NTH alpha1(IV) are formed around the endothelial cell network in a novel in vitro angiogenesis model. Am J Physiol Cell Physiol. (2019) 317:C314–25. doi: 10.1152/ajpcell.00353.2018
183. Schuppan D, Cantaluppi MC, Becker J, Veit A, Bunte T, Troyer D, et al. Undulin, an extracellular matrix glycoprotein associated with collagen fibrils. J Biol Chem. (1990) 265:8823–32. doi: 10.1016/S0021-9258(19)38962-8
184. Brown JC, Golbik R, Mann K, Timpl R. Structure and stability of the triple-helical domains of human collagen XIV. Matrix Biol. (1994) 14:287–95. doi: 10.1016/0945-053X(94)90194-5
185. Agarwal P, Zwolanek D, Keene DR, Schulz JN, Blumbach K, Heinegard D, et al. Collagen XII and XIV, new partners of cartilage oligomeric matrix protein in the skin extracellular matrix suprastructure. J Biol Chem. (2012) 287:22549–59. doi: 10.1074/jbc.M111.335935
186. Gerecke DR, Meng X, Liu B, Birk DE. Complete primary structure and genomic organization of the mouse Col14a1 gene. Matrix Biol. (2004) 22:595–601. doi: 10.1016/j.matbio.2003.11.005
187. Walchli C, Koch M, Chiquet M, Odermatt BF, Trueb B. Tissue-specific expression of the fibril-associated collagens XII and XIV. J Cell Sci. (1994) 107(Pt 2):669–81.
188. Bader HL, Lambert E, Guiraud A, Malbouyres M, Driever W, Koch M, et al. Zebrafish collagen XIV is transiently expressed in epithelia and is required for proper function of certain basement membranes. J Biol Chem. (2013) 288:6777–87. doi: 10.1074/jbc.M112.430637
189. Thierry L, Geiser AS, Hansen A, Tesche F, Herken R, Miosge N. Collagen types XII and XIV are present in basement membrane zones during human embryonic development. J Mol Histol. (2004) 35:803–10. doi: 10.1007/s10735-004-1132-y
190. Ansorge HL, Meng X, Zhang G, Veit G, Sun M, Klement JF, et al. Type XIV collagen regulates fibrillogenesis: premature collagen fibril growth and tissue dysfunction in null mice. J Biol Chem. (2009) 284:8427–38. doi: 10.1074/jbc.M805582200
191. Tao G, Levay AK, Peacock JD, Huk DJ, Both SN, Purcell NH, et al. Collagen XIV is important for growth and structural integrity of the myocardium. J Mol Cell Cardiol. (2012) 53:626–38. doi: 10.1016/j.yjmcc.2012.08.002
192. Young BB, Gordon MK, Birk DE. Expression of type XIV collagen in developing chicken tendons: association with assembly and growth of collagen fibrils. Dev Dyn. (2000) 217:430–9.doi: 10.1002/(SICI)1097-0177(200004)217:4<430::AID-DVDY10>3.0.CO;2-5
193. Morris MR, Ricketts C, Gentle D, Abdulrahman M, Clarke N, Brown M, et al. Identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma. Oncogene. (2010) 29:2104–17. doi: 10.1038/onc.2009.493
194. Li J, Wang X, Zheng K, Liu Y, Li J, Wang S, et al. The clinical significance of collagen family gene expression in esophageal squamous cell carcinoma. PeerJ. (2019) 7:e7705. doi: 10.7717/peerj.7705
195. Xie T, Wang Y, Deng N, Huang G, Taghavifar F, Geng Y, et al. Single-cell deconvolution of fibroblast heterogeneity in mouse pulmonary fibrosis. Cell Reports. (2018) 22:3625–40. doi: 10.1016/j.celrep.2018.03.010
196. Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. (2016) 1:e90558. doi: 10.1172/jci.insight.90558
197. Mak KM, Mei R. Basement membrane type IV collagen and laminin: an overview of their biology and value as fibrosis biomarkers of liver disease. Anatomical Rec. (2017) 300:1371–90. doi: 10.1002/ar.23567
198. Miner JH. Chapter 4: Basement membranes. In: Mecham R, editor. The Extracellular Matrix: An Overview. Berlin: Springer-Verlag (2011). p. 117–45.
199. Lago JC, Puzzi MB. The effect of aging in primary human dermal fibroblasts. PLoS ONE. (2019) 14:e0219165. doi: 10.1371/journal.pone.0219165
200. Vazquez F, Palacios S, Aleman N, Guerrero F. Changes of the basement membrane and type IV collagen in human skin during aging. Maturitas. (1996) 25:209–15. doi: 10.1016/S0378-5122(96)01066-3
201. Karttunen T, Risteli J, Autio-Harmainen H, Risteli L. Effect of age and diabetes on type IV collagen and laminin in human kidney cortex. Kidney Int. (1986) 30:586–91. doi: 10.1038/ki.1986.225
202. Tamaro G, Moretti M, Marchi P, Mangiarotti M, Pozzato G. Serum type IV collagen and prolyl hydroxylase levels: effect of gender and age. Clin Chim Acta. (1994) 228:205–10. doi: 10.1016/0009-8981(94)90290-9
203. Kehlet SN, Willumsen N, Armbrecht G, Dietzel R, Brix S, Henriksen K, et al. Age-related collagen turnover of the interstitial matrix and basement membrane: implications of age- and sex-dependent remodeling of the extracellular matrix. PLoS ONE. (2018) 13:e0194458. doi: 10.1371/journal.pone.0194458
204. Grässel S, Timpl R, Tan EM, Chu ML. Biosynthesis and processing of type XVI collagen in human fibroblasts and smooth muscle cells. Eur J Biochem. (1996) 242:576–84. doi: 10.1111/j.1432-1033.1996.0576r.x
205. Grässel S, Bauer RJ. Collagen XVI in health and disease. Matrix Biol. (2013) 32:64–73. doi: 10.1016/j.matbio.2012.11.001
206. Kassner A, Hansen U, Miosge N, Reinhardt DP, Aigner T, Bruckner-Tuderman L, et al. Discrete integration of collagen XVI into tissue-specific collagen fibrils or beaded microfibrils. Matrix Biol. (2003) 22:131–43. doi: 10.1016/S0945-053X(03)00008-8
207. Tajima S, Akagi A, Tanaka N, Ishibashi A, Kawada A, Yamaguchi N. Expression of type XVI collagen in cultured skin fibroblasts is related to cell growth arrest. FEBS Lett. (2000) 469:1–4. doi: 10.1016/S0014-5793(00)01241-2
208. Bedal KB, Grassel S, Oefner PJ, Reinders J, Reichert TE, Bauer R. Collagen XVI induces expression of MMP9 via modulation of AP-1 transcription factors and facilitates invasion of oral squamous cell carcinoma. PLoS ONE. (2014) 9:e86777. doi: 10.1371/journal.pone.0086777
209. Ratzinger S, Grässel S, Dowejko A, Reichert TE, Bauer RJ. Induction of type XVI collagen expression facilitates proliferation of oral cancer cells. Matrix Biol. (2011) 30:118–25. doi: 10.1016/j.matbio.2011.01.001
210. Senner V, Ratzinger S, Mertsch S, Grassel S, Paulus W. Collagen XVI expression is upregulated in glioblastomas and promotes tumor cell adhesion. FEBS Lett. (2008) 582:3293–300. doi: 10.1016/j.febslet.2008.09.017
211. Kang J, Chen W, Xia J, Li Y, Yang B, Chen B, et al. Extracellular matrix secreted by senescent fibroblasts induced by UVB promotes cell proliferation in HaCaT cells through PI3K/AKT and ERK signaling pathways. Int J Mol Med. (2008) 21:777–84. doi: 10.3892/ijmm.21.6.777
212. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. (2001) 98:12072–7. doi: 10.1073/pnas.211053698
213. Herchenhan A, Uhlenbrock F, Eliasson P, Weis M, Eyre D, Kadler KE, et al. Lysyl oxidase activity is required for ordered collagen fibrillogenesis by tendon cells. J Biol Chem. (2015) 290:16440–50. doi: 10.1074/jbc.M115.641670
214. McCabe MC, Hill RC, Calderone K, Cui Y, Yan Y, Quan T, et al. Alterations in extracellular matrix composition during aging and photoaging of the skin. Matrix Biol Plus. (2020) 8:100041. doi: 10.1016/j.mbplus.2020.100041
215. Szauter KM, Cao T, Boyd CD, Csiszar K. Lysyl oxidase in development, aging and pathologies of the skin. Pathol Biol. (2005) 53:448–56. doi: 10.1016/j.patbio.2004.12.033
216. Jiang Y, Zong W, Luan H, Liu JH, Zhang AZ, Li XL, et al. Decreased expression of elastin and lysyl oxidase family genes in urogenital tissues of aging mice. J Obstetr Gynaecol Res. (2014) 40:1998–2004. doi: 10.1111/jog.12425
217. Behmoaras J, Slove S, Seve S, Vranckx R, Sommer P, Jacob MP. Differential expression of lysyl oxidases LOXL1 and LOX during growth and aging suggests specific roles in elastin and collagen fiber remodeling in rat aorta. Rejuvenation Res. (2008) 11:883–9. doi: 10.1089/rej.2008.0760
218. Reiser KM. Influence of age and long-term dietary restriction on enzymatically mediated crosslinks and nonenzymatic glycation of collagen in mice. J Gerontol. (1994) 49:B71–9. doi: 10.1093/geronj/49.2.B71
219. Reiser KM, Hennessy SM, Last JA. Analysis of age-associated changes in collagen crosslinking in the skin and lung in monkeys and rats. Biochim Biophys Acta. (1987) 926:339–48. doi: 10.1016/0304-4165(87)90220-0
220. Rolewska P, Al-Robaiy S, Navarrete Santos A, Simm A, Silber RE, Bartling B. Age-related expression, enzymatic solubility and modification with advanced glycation end-products of fibrillar collagens in mouse lung. Exp Gerontol. (2013) 48:29–37. doi: 10.1016/j.exger.2012.04.012
221. Hudson DM, Archer M, King KB, Eyre DR. Glycation of type I collagen selectively targets the same helical domain lysine sites as lysyl oxidase-mediated cross-linking. J Biol Chem. (2018) 293:15620–7. doi: 10.1074/jbc.RA118.004829
222. Johnston KA, Lopez KM. Lysyl oxidase in cancer inhibition and metastasis. Cancer Lett. (2018) 417:174–81. doi: 10.1016/j.canlet.2018.01.006
223. Wang TH, Hsia SM, Shih YH, Shieh TM. Association of smoking, alcohol use, and betel quid chewing with epigenetic aberrations in cancers. Int J Mol Sci. (2017) 18:1210. doi: 10.3390/ijms18061210
224. Cox TR, Gartland A, Erler JT. Lysyl oxidase, a targetable secreted molecule involved in cancer metastasis. Cancer Res. (2016) 76:188–92. doi: 10.1158/0008-5472.CAN-15-2306
225. Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. (2006) 440:1222–6. doi: 10.1038/nature04695
226. Gong R, Lin W, Gao A, Liu Y, Li J, Sun M, et al. Forkhead box C1 promotes metastasis and invasion of non-small cell lung cancer by binding directly to the lysyl oxidase promoter. Cancer Sci. (2019) 110:3663–76. doi: 10.1111/cas.14213
227. Hou X, Du H, Quan X, Shi L, Zhang Q, Wu Y, et al. Silibinin inhibits NSCLC metastasis by targeting the EGFR/LOX pathway. Front Pharmacol. (2018) 9:21. doi: 10.3389/fphar.2018.00021
228. Osawa T, Ohga N, Akiyama K, Hida Y, Kitayama K, Kawamoto T, et al. Lysyl oxidase secreted by tumour endothelial cells promotes angiogenesis and metastasis. Br J Cancer. (2013) 109:2237–47. doi: 10.1038/bjc.2013.535
229. Tang H, Leung L, Saturno G, Viros A, Smith D, Di Leva G, et al. Lysyl oxidase drives tumour progression by trapping EGF receptors at the cell surface. Nat Commun. (2017) 8:14909. doi: 10.1038/ncomms14909
230. Wilgus ML, Borczuk AC, Stoopler M, Ginsburg M, Gorenstein L, Sonett JR, et al. Lysyl oxidase: a lung adenocarcinoma biomarker of invasion and survival. Cancer. (2011) 117:2186–91. doi: 10.1002/cncr.25768
231. Min C, Kirsch KH, Zhao Y, Jeay S, Palamakumbura AH, Trackman PC, et al. The tumor suppressor activity of the lysyl oxidase propeptide reverses the invasive phenotype of Her-2/neu-driven breast cancer. Cancer Res. (2007) 67:1105–12. doi: 10.1158/0008-5472.CAN-06-3867
232. Wu M, Min C, Wang X, Yu Z, Kirsch KH, Trackman PC, et al. Repression of BCL2 by the tumor suppressor activity of the lysyl oxidase propeptide inhibits transformed phenotype of lung and pancreatic cancer cells. Cancer Res. (2007) 67:6278–85. doi: 10.1158/0008-5472.CAN-07-0776
233. Mays PK, McAnulty RJ, Campa JS, Laurent GJ. Age-related changes in collagen synthesis and degradation in rat tissues. Importance of degradation of newly synthesized collagen in regulating collagen production. Biochem J. (1991) 276(Pt 2):307–13. doi: 10.1042/bj2760307
234. Neuberger A, Perrone JC, Slack HG. The relative metabolic inertia of tendon collagen in the rat. Biochem J. (1951) 49:199–204. doi: 10.1042/bj0490199
235. Uitto J. A method for studying collagen biosynthesis in human skin biopsies in vitro. Biochim Biophys Acta. (1970) 201:438–45. doi: 10.1016/0304-4165(70)90163-7
236. Podolsky MJ, Yang CD, Valenzuela CL, Datta R, Huang SK, Nishimura SL, et al. Age-dependent regulation of cell-mediated collagen turnover. JCI Insight. (2020) 5:e137519. doi: 10.1172/jci.insight.137519
237. Calabresi C, Arosio B, Galimberti L, Scanziani E, Bergottini R, Annoni G, et al. Natural aging, expression of fibrosis-related genes and collagen deposition in rat lung. Exp Gerontol. (2007) 42:1003–11. doi: 10.1016/j.exger.2007.06.016
238. Bishop PN, Holmes DF, Kadler KE, McLeod D, Bos KJ. Age-related changes on the surface of vitreous collagen fibrils. Investig Ophthalmol Visual Sci. (2004) 45:1041–6. doi: 10.1167/iovs.03-1017
239. DeGroot J, Verzijl N, Wenting-Van Wijk MJ, Bank RA, Lafeber FP, Bijlsma JW, et al. Age-related decrease in susceptibility of human articular cartilage to matrix metalloproteinase-mediated degradation: the role of advanced glycation end products. Arthritis Rheumatism. (2001) 44:2562–71. doi: 10.1002/1529-0131(200111)44:11<2562::AID-ART437>3.0.CO;2-1
240. Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. (2006) 114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854
241. Lee DH, Oh JH, Chung JH. Glycosaminoglycan and proteoglycan in skin aging. J Dermatol Sci. (2016) 83:174–81. doi: 10.1016/j.jdermsci.2016.05.016
242. Li Y, Liu Y, Xia W, Lei D, Voorhees JJ, Fisher GJ. Age-dependent alterations of decorin glycosaminoglycans in human skin. Sci Rep. (2013) 3:2422. doi: 10.1038/srep02422
243. Panwar P, Butler GS, Jamroz A, Azizi P, Overall CM, Bromme D. Aging-associated modifications of collagen affect its degradation by matrix metalloproteinases. Matrix Biol. (2018) 65:30–44. doi: 10.1016/j.matbio.2017.06.004
244. Bonnema DD, Webb CS, Pennington WR, Stroud RE, Leonardi AE, Clark LL, et al. Effects of age on plasma matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs). J Cardiac Failure. (2007) 13:530–40. doi: 10.1016/j.cardfail.2007.04.010
245. Komosinska-Vassev K, Olczyk P, Winsz-Szczotka K, Kuznik-Trocha K, Klimek K, Olczyk K. Age- and gender-dependent changes in connective tissue remodeling: physiological differences in circulating MMP-3, MMP-10, TIMP-1 and TIMP-2 level. Gerontology. (2011) 57:44–52. doi: 10.1159/000295775
246. Manicourt DH, Fujimoto N, Obata K, Thonar EJ. Serum levels of collagenase, stromelysin-1, and TIMP-1. Age- and sex-related differences in normal subjects and relationship to the extent of joint involvement and serum levels of antigenic keratan sulfate in patients with osteoarthritis. Arthritis Rheumatism. (1994) 37:1774–83. doi: 10.1002/art.1780371211
247. Sundstrom J, Evans JC, Benjamin EJ, Levy D, Larson MG, Sawyer DB, et al. Relations of plasma total TIMP-1 levels to cardiovascular risk factors and echocardiographic measures: the Framingham heart study. Euro Heart J. (2004) 25:1509–16. doi: 10.1016/j.ehj.2004.05.029
248. Tayebjee MH, Lip GY, Blann AD, Macfadyen RJ. Effects of age, gender, ethnicity, diurnal variation and exercise on circulating levels of matrix metalloproteinases (MMP)-2 and−9, their inhibitors, tissue inhibitors of matrix metalloproteinases (TIMP)-1 and−2. Thrombosis Res. (2005) 115:205–10. doi: 10.1016/j.thromres.2004.08.023
249. Vitenberga Z, Pilmane M. Age-related lung tissue remodeling due to the local distribution of MMP-2, TIMP-2, TGF-beta and Hsp70. Biotech Histochem. (2018) 93:239–48. doi: 10.1080/10520295.2017.1421322
250. Fishman SL, Sonmez H, Basman C, Singh V, Poretsky L. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: a review. Mol Med. (2018) 24:59. doi: 10.1186/s10020-018-0060-3
251. Monnier VM, Mustata GT, Biemel KL, Reihl O, Lederer MO, Zhenyu D, et al. Cross-linking of the extracellular matrix by the maillard reaction in aging and diabetes: an update on “a puzzle nearing resolution”. Ann NY Acad Sci. (2005) 1043:533–44. doi: 10.1196/annals.1333.061
252. Singh R, Barden A, Mori T, Beilin L. Advanced glycation end-products: a review. Diabetologia. (2001) 44:129–46. doi: 10.1007/s001250051591
253. Vistoli G, De Maddis D, Cipak A, Zarkovic N, Carini M, Aldini G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): an overview of their mechanisms of formation. Free Radical Res. (2013) 47(Suppl. 1):3–27. doi: 10.3109/10715762.2013.815348
254. Jost T, Zipprich A, Glomb MA. Analysis of advanced glycation endproducts in rat tail collagen and correlation to tendon stiffening. J Agric Food Chem. (2018) 66:3957–65. doi: 10.1021/acs.jafc.8b00937
255. Nowotny K, Castro JP, Hugo M, Braune S, Weber D, Pignitter M, et al. Oxidants produced by methylglyoxal-modified collagen trigger ER stress and apoptosis in skin fibroblasts. Free Radical Biol Med. (2018) 120:102–13. doi: 10.1016/j.freeradbiomed.2018.03.022
256. Maessen DE, Stehouwer CD, Schalkwijk CG. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin Sci. (2015) 128:839–61. doi: 10.1042/CS20140683
257. Rabbani N, Xue M, Thornalley PJ. Methylglyoxal-induced dicarbonyl stress in aging and disease: first steps towards glyoxalase 1-based treatments. Clin Sci. (2016) 130:1677–96. doi: 10.1042/CS20160025
258. Thornalley PJ. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems–role in ageing and disease. Drug Metab Drug Interactions. (2008) 23:125–50. doi: 10.1515/DMDI.2008.23.1-2.125
259. Thornalley PJ. The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem J. (1990) 269:1–11. doi: 10.1042/bj2690001
260. Morcos M, Du X, Pfisterer F, Hutter H, Sayed AA, Thornalley P, et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell. (2008) 7:260–9. doi: 10.1111/j.1474-9726.2008.00371.x
261. Schlotterer A, Kukudov G, Bozorgmehr F, Hutter H, Du X, Oikonomou D, et al. C. elegans as model for the study of high glucose- mediated life span reduction. Diabetes. (2009) 58:2450–6. doi: 10.2337/db09-0567
262. Kirk JE. The glyoxalase I activity of arterial tissue in individuals of various ages. J Gerontol. (1960) 15:139–41. doi: 10.1093/geronj/15.2.139
263. McLellan AC, Thornalley PJ. Glyoxalase activity in human red blood cells fractioned by age. Mech Ageing Dev. (1989) 48:63–71. doi: 10.1016/0047-6374(89)90026-2
264. Kuhla B, Boeck K, Luth HJ, Schmidt A, Weigle B, Schmitz M, et al. Age-dependent changes of glyoxalase I expression in human brain. Neurobiol Aging. (2006) 27:815–22. doi: 10.1016/j.neurobiolaging.2005.04.006
265. Ahmed N, Thornalley PJ, Dawczynski J, Franke S, Strobel J, Stein G, et al. Methylglyoxal-derived hydroimidazolone advanced glycation end-products of human lens proteins. Investig Ophthalmol Visual Sci. (2003) 44:5287–92. doi: 10.1167/iovs.03-0573
266. Fan X, Sell DR, Zhang J, Nemet I, Theves M, Lu J, et al. Anaerobic vs aerobic pathways of carbonyl and oxidant stress in human lens and skin during aging and in diabetes: a comparative analysis. Free Radical Biol Med. (2010) 49:847–56. doi: 10.1016/j.freeradbiomed.2010.06.003
267. Ikeda Y, Inagi R, Miyata T, Nagai R, Arai M, Miyashita M, et al. Glyoxalase I retards renal senescence. Am J Pathol. (2011) 179:2810–21. doi: 10.1016/j.ajpath.2011.08.023
268. Thornalley PJ. Modification of the glyoxalase system in human red blood cells by glucose in vitro. Biochem J. (1988) 254:751–5. doi: 10.1042/bj2540751
269. Soboleva A, Schmidt R, Vikhnina M, Grishina T, Frolov A. Maillard proteomics: opening new pages. Int J Mol Sci. (2017) 18:2677. doi: 10.3390/ijms18122677
270. Sakasai-Sakai A, Takata T, Suzuki H, Maruyama I, Motomiya Y, Takeuchi M. Immunological evidence for in vivo production of novel advanced glycation end-products from 1,5-anhydro-D-fructose, a glycogen metabolite. Sci Rep. (2019) 9:10194. doi: 10.1038/s41598-019-46333-2
271. Monnier VM, Kohn RR, Cerami A. Accelerated age-related browning of human collagen in diabetes mellitus. Proc Natl Acad Sci USA. (1984) 81:583–7. doi: 10.1073/pnas.81.2.583
272. Gerrits EG, Lutgers HL, Kleefstra N, Graaff R, Groenier KH, Smit AJ, et al. Skin autofluorescence: a tool to identify type 2 diabetic patients at risk for developing microvascular complications. Diabetes Care. (2008) 31:517–21. doi: 10.2337/dc07-1755
273. Koetsier M, Lutgers HL, de Jonge C, Links TP, Smit AJ, Graaff R. Reference values of skin autofluorescence. Diabetes Technol Therapeutics. (2010) 12:399–403. doi: 10.1089/dia.2009.0113
274. Lutgers HL, Graaff R, Links TP, Ubink-Veltmaat LJ, Bilo HJ, Gans RO, et al. Skin autofluorescence as a noninvasive marker of vascular damage in patients with type 2 diabetes. Diabetes Care. (2006) 29:2654–9. doi: 10.2337/dc05-2173
275. Meerwaldt R, Graaff R, Oomen PHN, Links TP, Jager JJ, Alderson NL, et al. Simple non-invasive assessment of advanced glycation endproduct accumulation. Diabetologia. (2004) 47:1324–30. doi: 10.1007/s00125-004-1451-2
276. Meerwaldt R, Links T, Graaff R, Thorpe SR, Baynes JW, Hartog J, et al. Simple noninvasive measurement of skin autofluorescence. Ann NY Acad Sci. (2005) 1043:290–8. doi: 10.1196/annals.1333.036
277. Noordzij MJ, Mulder DJ, Oomen PH, Brouwer T, Jager J, Castro Cabezas M, et al. Skin autofluorescence and risk of micro- and macrovascular complications in patients with type 2 diabetes mellitus-a multi-centre study. Diabetic Med. (2012) 29:1556–61. doi: 10.1111/dme.12005
278. van Waateringe RP, Slagter SN, van Beek AP, van der Klauw MM, van Vliet-Ostaptchouk JV, Graaff R, et al. Skin autofluorescence, a non-invasive biomarker for advanced glycation end products, is associated with the metabolic syndrome and its individual components. Diabetol Metab Syndrome. (2017) 9:42. doi: 10.1186/s13098-017-0241-1
279. de Vos LC, Lefrandt JD, Dullaart RP, Zeebregts CJ, Smit AJ. Advanced glycation end products: an emerging biomarker for adverse outcome in patients with peripheral artery disease. Atherosclerosis. (2016) 254:291–9. doi: 10.1016/j.atherosclerosis.2016.10.012
280. Koito W, Araki T, Horiuchi S, Nagai R. Conventional antibody against Nepsilon-(carboxymethyl)lysine (CML) shows cross-reaction to Nepsilon-(carboxyethyl)lysine (CEL): immunochemical quantification of CML with a specific antibody. J Biochem. (2004) 136:831–7. doi: 10.1093/jb/mvh193
281. Nagai R, Fujiwara Y, Mera K, Motomura K, Iwao Y, Tsurushima K, et al. Usefulness of antibodies for evaluating the biological significance of AGEs. Ann NY Acad Sci. (2008) 1126:38–41. doi: 10.1196/annals.1433.001
282. Xiao Y, Isaacs SN. Enzyme-linked immunosorbent assay (ELISA) and blocking with bovine serum albumin (BSA)–not all BSAs are alike. J Immunol Methods. (2012) 384:148–51. doi: 10.1016/j.jim.2012.06.009
283. Bellmunt MJ, Portero M, Pamplona R, Muntaner M, Prat J. Age-related fluorescence in rat lung collagen. Lung. (1995) 173:177–85. doi: 10.1007/BF00175658
284. Chen L, Wang T, Wang X, Sun BB, Li JQ, Liu DS, et al. Blockade of advanced glycation end product formation attenuates bleomycin-induced pulmonary fibrosis in rats. Respir Res. (2009) 10:55. doi: 10.1186/1465-9921-10-55
285. Ehrlich SF, Quesenberry CP Jr, Van Den Eeden SK, Shan J, Ferrara A. Patients diagnosed with diabetes are at increased risk for asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, and pneumonia but not lung cancer. Diabetes Care. (2010) 33:55–60. doi: 10.2337/dc09-0880
286. Enomoto T, Usuki J, Azuma A, Nakagawa T, Kudoh S. Diabetes mellitus may increase risk for idiopathic pulmonary fibrosis. Chest. (2003) 123:2007–11. doi: 10.1378/chest.123.6.2007
287. Gribbin J, Hubbard R, Smith C. Role of diabetes mellitus and gastro-oesophageal reflux in the aetiology of idiopathic pulmonary fibrosis. Respir Med. (2009) 103:927–31. doi: 10.1016/j.rmed.2008.11.001
288. Hu Y, Ma Z, Guo Z, Zhao F, Wang Y, Cai L, et al. Type 1 diabetes mellitus is an independent risk factor for pulmonary fibrosis. Cell Biochem Biophys. (2014) 70:1385–91. doi: 10.1007/s12013-014-0068-4
289. Panwar P, Lamour G, Mackenzie NC, Yang H, Ko F, Li H, et al. Changes in structural-mechanical properties and degradability of collagen during aging-associated modifications. J Biol Chem. (2015) 290:23291–306. doi: 10.1074/jbc.M115.644310
290. Chong SA, Lee W, Arora PD, Laschinger C, Young EW, Simmons CA, et al. Methylglyoxal inhibits the binding step of collagen phagocytosis. J Biol Chem. (2007) 282:8510–20. doi: 10.1074/jbc.M609859200
291. Gautieri A, Passini FS, Silvan U, Guizar-Sicairos M, Carimati G, Volpi P, et al. Advanced glycation end-products: mechanics of aged collagen from molecule to tissue. Matrix Biol. (2017) 59:95–108. doi: 10.1016/j.matbio.2016.09.001
292. Li Y, Fessel G, Georgiadis M, Snedeker JG. Advanced glycation end-products diminish tendon collagen fiber sliding. Matrix Biol. (2013) 32:169–77. doi: 10.1016/j.matbio.2013.01.003
293. Haus JM, Carrithers JA, Trappe SW, Trappe TA. Collagen, cross-linking, and advanced glycation end products in aging human skeletal muscle. J Appl Physiol. (2007) 103:2068–76. doi: 10.1152/japplphysiol.00670.2007
294. Reddy GK. AGE-related cross-linking of collagen is associated with aortic wall matrix stiffness in the pathogenesis of drug-induced diabetes in rats. Microvasc Res. (2004) 68:132–42. doi: 10.1016/j.mvr.2004.04.002
295. Snedeker JG, Gautieri A. The role of collagen crosslinks in ageing and diabetes - the good, the bad, the ugly. Muscles Ligaments Tendons J. (2014) 4:303–8. doi: 10.11138/mltj/2014.4.3.303
296. Reigle KL, Di Lullo G, Turner KR, Last JA, Chervoneva I, Birk DE, et al. Non-enzymatic glycation of type I collagen diminishes collagen-proteoglycan binding and weakens cell adhesion. J Cell Biochem. (2008) 104:1684–98. doi: 10.1002/jcb.21735
297. Dobler D, Ahmed N, Song L, Eboigbodin KE, Thornalley PJ. Increased dicarbonyl metabolism in endothelial cells in hyperglycemia induces anoikis and impairs angiogenesis by RGD and GFOGER motif modification. Diabetes. (2006) 55:1961–9. doi: 10.2337/db05-1634
298. Pedchenko VK, Chetyrkin SV, Chuang P, Ham AJ, Saleem MA, Mathieson PW, et al. Mechanism of perturbation of integrin-mediated cell-matrix interactions by reactive carbonyl compounds and its implication for pathogenesis of diabetic nephropathy. Diabetes. (2005) 54:2952–60. doi: 10.2337/diabetes.54.10.2952
299. Bufalo MC, Almeida ME, Franca IA, Zambelli VO, Martins Sant'anna MB, Kimura LF, et al. Advanced glycation endproducts produced by in vitro glycation of type I collagen modulate the functional and secretory behavior of dorsal root ganglion cells cultivated in two-dimensional system. Exp Cell Res. (2019) 382:111475. doi: 10.1016/j.yexcr.2019.06.020
300. Khosravi R, Sodek KL, Faibish M, Trackman PC. Collagen advanced glycation inhibits its discoidin domain receptor 2 (DDR2)-mediated induction of lysyl oxidase in osteoblasts. Bone. (2014) 58:33–41. doi: 10.1016/j.bone.2013.10.001
301. Gautieri A, Redaelli A, Buehler MJ, Vesentini S. Age- and diabetes-related nonenzymatic crosslinks in collagen fibrils: candidate amino acids involved in advanced glycation end-products. Matrix Biol. (2014) 34:89–95. doi: 10.1016/j.matbio.2013.09.004
302. Collier TA, Nash A, Birch HL, de Leeuw NH. Preferential sites for intramolecular glucosepane cross-link formation in type I collagen: a thermodynamic study. Matrix Biol. (2015) 48:78–88. doi: 10.1016/j.matbio.2015.06.001
303. Buckley ST, Ehrhardt C. The receptor for advanced glycation end products (RAGE) and the lung. J Biomed Biotechnol. (2010) 2010:917108. doi: 10.1155/2010/917108
304. Oczypok EA, Perkins TN, Oury TD. All the “RAGE” in lung disease: the receptor for advanced glycation endproducts (RAGE) is a major mediator of pulmonary inflammatory responses. Paediatric Respir Rev. (2017) 23:40–9. doi: 10.1016/j.prrv.2017.03.012
305. Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem. (1992) 267:14987–97. doi: 10.1016/S0021-9258(18)42137-0
306. Haider SH, Oskuei A, Crowley G, Kwon S, Lam R, Riggs J, et al. Receptor for advanced glycation end-products and environmental exposure related obstructive airways disease: a systematic review. Eur Respir Rev. (2019) 28:2677. doi: 10.1183/16000617.0096-2018
307. Su X, Looney MR, Gupta N, Matthay MA. Receptor for advanced glycation end-products (RAGE) is an indicator of direct lung injury in models of experimental lung injury. Am J Physiol Lung Cell Mol Physiol. (2009) 297:L1–5. doi: 10.1152/ajplung.90546.2008
308. Ramasamy R, Yan SF, Herold K, Clynes R, Schmidt AM. Receptor for advanced glycation end products: fundamental roles in the inflammatory response: winding the way to the pathogenesis of endothelial dysfunction and atherosclerosis. Ann NY Acad Sci. (2008) 1126:7–13. doi: 10.1196/annals.1433.056
309. Rojas A, Anazco C, Gonzalez I, Araya P. Extracellular matrix glycation and receptor for advanced glycation end-products activation: a missing piece in the puzzle of the association between diabetes and cancer. Carcinogenesis. (2018) 39:515–21. doi: 10.1093/carcin/bgy012
310. Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, et al. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. (1994) 269:9889–97. doi: 10.1016/S0021-9258(17)36966-1
311. Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, et al. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem. (1999) 274:31740–9. doi: 10.1074/jbc.274.44.31740
312. Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, et al. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J Clin Investig. (2001) 108:1853–63. doi: 10.1172/JCI11951
313. Schmidt AM, Yan SD, Yan SF, Stern DM. The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta. (2000) 1498:99–111. doi: 10.1016/S0167-4889(00)00087-2
314. Alikhani M, Alikhani Z, Boyd C, MacLellan CM, Raptis M, Liu R, et al. Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone. (2007) 40:345–53. doi: 10.1016/j.bone.2006.09.011
315. Alikhani Z, Alikhani M, Boyd CM, Nagao K, Trackman PC, Graves DT. Advanced glycation end products enhance expression of pro-apoptotic genes and stimulate fibroblast apoptosis through cytoplasmic and mitochondrial pathways. J Biol Chem. (2005) 280:12087–95. doi: 10.1074/jbc.M406313200
316. Demling N, Ehrhardt C, Kasper M, Laue M, Knels L, Rieber EP. Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. (2006) 323:475–88. doi: 10.1007/s00441-005-0069-0
Keywords: collagen, extracellular matrix, ageing, crosslinking, chronic lung disease, advanced-glycation end products, lysyl oxidase
Citation: Onursal C, Dick E, Angelidis I, Schiller HB and Staab-Weijnitz CA (2021) Collagen Biosynthesis, Processing, and Maturation in Lung Ageing. Front. Med. 8:593874. doi: 10.3389/fmed.2021.593874
Received: 11 August 2020; Accepted: 24 March 2021;
Published: 20 May 2021.
Edited by:
Gary Peter Anderson, The University of Melbourne, AustraliaReviewed by:
Eleni Papakonstantinou, Aristotle University of Thessaloniki, GreeceBeatrice Ludovica Ritondo, University of Rome Tor Vergata, Italy
Copyright © 2021 Onursal, Dick, Angelidis, Schiller and Staab-Weijnitz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia A. Staab-Weijnitz, c3RhYWItd2Vpam5pdHpAaGVsbWhvbHR6LW11ZW5jaGVuLmRlb3JjaWQub3JnLzAwMDAtMDAwMi0xMjExLTc4MzQ=
†These authors have contributed equally to this work