 Leen J. M. Heyens
Leen J. M. Heyens Dana Busschots1,2
Dana Busschots1,2 Sven Francque
Sven Francque- 1Faculty of Health and Life Sciences, Hasselt University, Hasselt, Belgium
- 2School of Nutrition and Translational Research in Metabolism, NUTRIM, Maastricht University, Maastricht, Netherlands
- 3Department of Gastro-Enterology and Hepatology, Ziekenhuis Oost-Limburg, Genk, Belgium
- 4Division of Gastroenterology and Hepatology, Department of Internal Medicine, Maastricht University Medical Centre, Maastricht, Netherlands
- 5Department of Gastroenterology and Hepatology, University Hospital Katholieke Universiteit (KU) Leuven, Leuven, Belgium
- 6Department of Gastroenterology and Hepatology, Antwerp University Hospital, Antwerp, Belgium
- 7Laboratory of Experimental Medicine and Paediatrics, Faculty of Medicine and Health Sciences, University of Antwerp, Antwerp, Belgium
An increasing percentage of people have or are at risk to develop non-alcoholic fatty liver disease (NAFLD) worldwide. NAFLD comprises different stadia going from isolated steatosis to non-alcoholic steatohepatitis (NASH). NASH is a chronic state of liver inflammation that leads to the transformation of hepatic stellate cells to myofibroblasts. These cells produce extra-cellular matrix that results in liver fibrosis. In a normal situation, fibrogenesis is a wound healing process that preserves tissue integrity. However, sustained and progressive fibrosis can become pathogenic. This process takes many years and is often asymptomatic. Therefore, patients usually present themselves with end-stage liver disease e.g., liver cirrhosis, decompensated liver disease or even hepatocellular carcinoma. Fibrosis has also been identified as the most important predictor of prognosis in patients with NAFLD. Currently, only a minority of patients with liver fibrosis are identified to be at risk and hence referred for treatment. This is not only because the disease is largely asymptomatic, but also due to the fact that currently liver biopsy is still the golden standard for accurate detection of liver fibrosis. However, performing a liver biopsy harbors some risks and requires resources and expertise, hence is not applicable in every clinical setting and is unsuitable for screening. Consequently, different non-invasive diagnostic tools, mainly based on analysis of blood or other specimens or based on imaging have been developed or are in development. In this review, we will first give an overview of the pathogenic mechanisms of the evolution from isolated steatosis to fibrosis. This serves as the basis for the subsequent discussion of the current and future diagnostic biomarkers and anti-fibrotic drugs.
Introduction
Non-alcoholic fatty liver disease (NAFLD) refers to the presence of liver steatosis in the absence of factors that are known to induce lipid accumulation in hepatocytes, such as alcohol consumption or the use of steatogenic drugs. The diagnosis requires > 5% of the hepatocytes containing lipid droplets on histology or the amount of fat in the liver to exceed 5% of the total liver weight (1). Currently, NAFLD is the most common chronic liver disease with an estimated prevalence of 25% worldwide (2). The prevalence of NAFLD parallels the global increase in overweight and obesity which is the result of an increase of caloric intake over expenditure that leads to an increase in body mass index (BMI) (3). As a result, NAFLD will become the most common cause of liver cirrhosis, decompensated liver disease or hepatocellular carcinoma (HCC) in the short term, and thus the most important indication for liver transplantation (4).
NAFLD comprises a broad spectrum of liver lesions but also has extra-hepatic consequences. These extra-hepatic complications, including cardiovascular disease, diabetes, and non-liver malignancies, are responsible for a significant part of NAFLD-attributable morbidity and mortality (5, 6). Furthermore, there is a considerable impact on the quality of life (7, 8). With respect to the liver, the NAFLD spectrum consists of the following entities: isolated fatty liver (non-alcoholic fatty liver, NAFL); and non-alcoholic steatohepatitis (NASH), i.e., steatosis accompanied by chronic inflammation and cell damage, histologically characterized by lobular inflammation and ballooning of hepatocytes, the latter being the driving force of fibrosis that can evolve to cirrhosis and decompensated cirrhosis (Figure 1). HCC can also develop, even in non-cirrhotic NAFLD (9). The rate of disease progression is usually slow. About 20% of patients with NAFLD will develop NASH in three to seven years (10), which is considered the potentially progressive form of the disease (11). About 9 to 25% of individuals with NASH develop cirrhosis over a 10 to 20 year period (12).
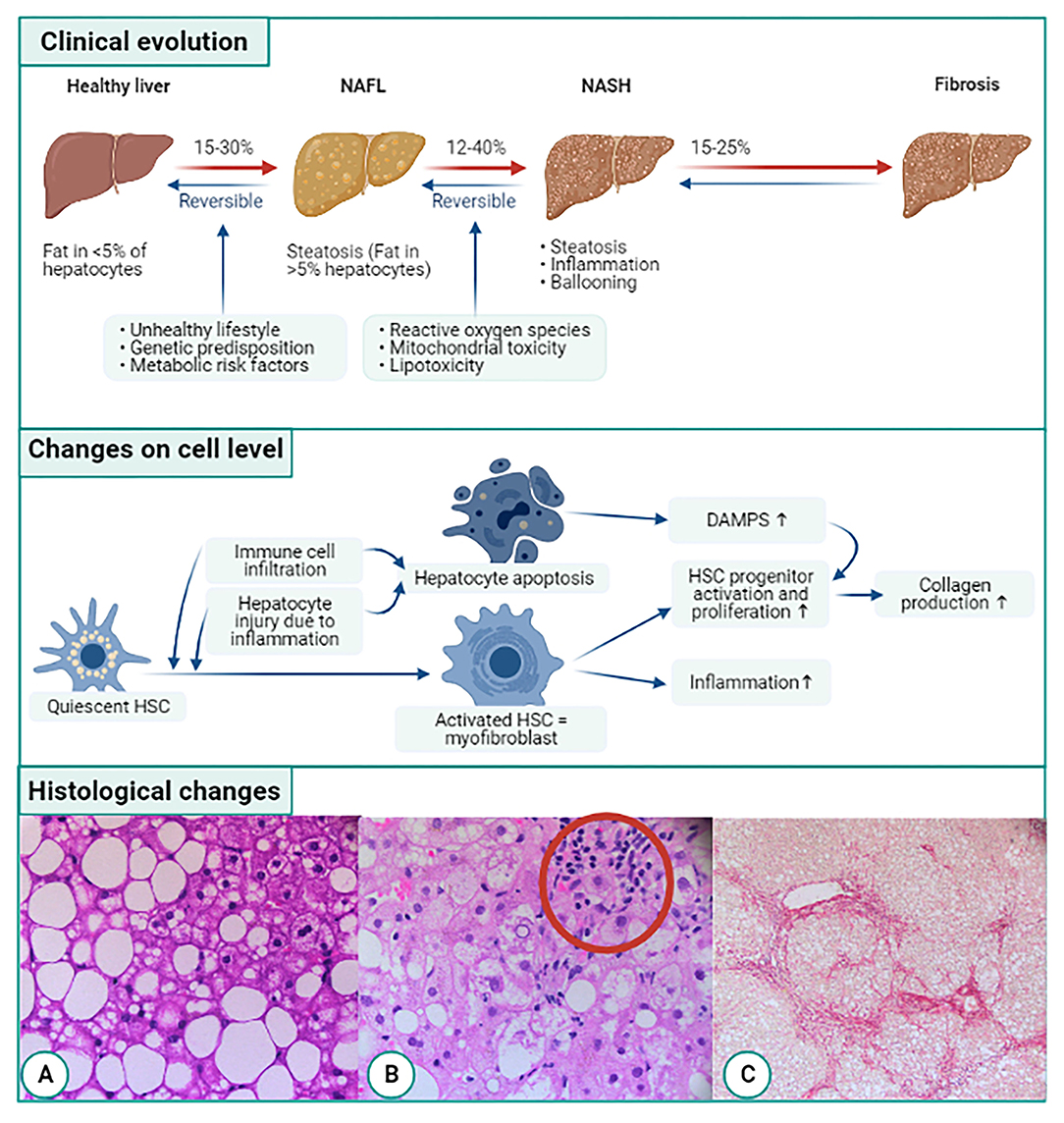
Figure 1. Overview of evolution of NAFLD related fibrogenesis on clinical, cellular, and histological level. On the clinical level NAFLD starts of as simple steatosis (NAFL). The abnormal amount of liver fat triggers inflammation by infiltrating immune cells and secretion of cytokines. This is called non-alcoholic steatohepatitis or NASH which can cause liver fibrosis. On cellular level, quiescent hepatic stellate cells (HSCs) are activated by immune cell infiltration and hepatocyte injury due to inflammation. The activated HSC transdifferentiates into collagen producing myofibroblasts furthermore the myofibroblasts trigger HSC progenitor proliferation and activation. Another consequence of the immune cell infiltration and hepatocyte injury is apoptosis of hepatocytes, leading to the release of damage-associated patterns (DAMPs). DAMPs also activate hepatic progenitor cells. Both the myofibroblasts and HSCs will start producing collagen. On a histological level, first fat accumulates in the liver (A). This leads to the infiltration of immune cells (B) and ballooning and eventually liver fibrosis occurs (C). Histological pictures courtesy of Dr. P. Van Eyken, pathologist, Ziekenhuis Oost-Limburg, Genk, Belgium.
Unfortunately, it is very hard to know which NASH patient will progress to cirrhosis due to the complex multifactorial etiology of NASH determined by genetic, epigenetic, lifestyle, and nutritional factors (13). However, the stage of liver fibrosis is the strongest predictor for liver-related mortality and development of other comorbidities (14–18), with an increase if fibrosis (F) is ≥ 2 on a scale of 0–4 as proposed by the NASH Clinical Research Network Scoring System (NASH CRN) (19). Accordingly, NASH patients with ≥F2 are considered the target population for pharmacological treatment and are most likely to benefit from antifibrotic drugs. Regression of stage F1 fibrosis is more likely with simple lifestyle changes and treatment of the metabolic comorbidities (20). Therefore, it is important to correctly diagnose the stage of liver fibrosis, preferably with non-invasive methods. Additionally, regression of advanced fibrosis should be the primary hepatic endpoint in clinical studies for antifibrotic drugs (21). Therefore, this review will tackle diagnostic methods to determine the stage of liver fibrosis and antifibrotic drugs that can reduce advanced fibrosis.
Pathogenesis
As understanding the pathogenesis of NAFLD and NAFLD-related fibrosis is vital in the development of biomarkers for its diagnosis and in finding targets for its treatment, we first review the most important aspects of NAFLD-related fibrogenesis.
From Liver Steatosis to NASH
Weight gain, often caused by an unhealthy lifestyle with a high-calorie diet and decreased physical activity, is one of the most important factors in the development of NAFLD. The liver plays a critical role in maintaining the metabolic balance that comes under pressure with a high caloric intake and low energy expenditure. Lipid overload, as seen in NAFLD, is a major contributor to the development of lipotoxicity (Figure 1A). Lipotoxicity accelerates the development of progressive inflammation, oxidative stress, and fibrosis (22). The excess energy consumed is usually stored in the form of fat in both subcutaneous and visceral depots. This capacity of the adipose tissue to store fat is genetically determined. When this capacity is exceeded, the adipose tissue experiences an overload and becomes damaged. This results in adipose tissue insulin resistance (IR) and inflammation of the tissue with imbalances in the secretion of adipokines and other inflammatory mediators (23), which causes a low-grade systemic inflammation (24). Together with ectopic fat accumulation, this leads to IR in the muscles and liver (25). The IR causes a disbalance in the homeostasis of glucose and lipid metabolism. As a result, more free fatty acids (FFA) that have to be processed by the liver, enter the circulation (26). Another consequence is that dietary carbohydrates (especially fructose) are absorbed by the liver and converted to FFA by de novo lipogenesis. About 40% of the fat that builds up in the liver comes from dietary carbohydrates and fat. The other 60% is derived from the dysfunctional adipose tissue (26). Thus, IR leads to an increase in FFA flux, leading to a toxic effect on the liver (27). The FFA are normally broken down in the mitochondria by beta-oxidation. Due to the FFA overload, the mitochondria are overwhelmed, and this leads to mitochondrial uncoupling. As a result, they produce reactive oxygen species (ROS) (22, 26). Combined with the dysfunctional adipose tissue and endotoxins from the gut, this leads to a pro-inflammatory and apoptotic climate in the liver, causing NASH (26–28). The Kupffer cells, the resident macrophages of the liver, as well as infiltrating immune cells, contribute to the inflammatory state of NASH. Kupffer cells absorb large amounts of FFA, which drives them toward an inflammatory phenotype. This leads to the secretion of inflammatory cytokines such as interleukin (IL)-6, tumor necrosis factor (TNF)-α and IL-10. Both IL-6 and TNF-α are associated with NASH progression (29, 30).
Taken together, NASH is the result of a complex interplay between different factors like genetic variation and obesity, which leads to a profibrotic climate in the liver (31).
From NASH to Liver Fibrosis
Immune responses in chronic liver diseases like NAFLD, not only lead to the restoration of tissue function but also to tissue injury. An overactive or exaggerated immune response can result in organ dysfunction and leads to the deposition of fibrotic tissue in parallel to the cell loss (31). These immune responses comprise both innate and adaptive responses (32). For example, neutrophil infiltration is often seen in histologic samples of NASH patients (Figure 1B). Additionally, patients with NASH and advanced fibrosis related to NASH have a higher neutrophil/lymphocyte ratio than patients without NASH (33, 34). Likewise, CD8+ lymphocytes have also been seen in the inflammatory infiltrate in NASH (35).
The inflammation caused by NASH causes hepatocyte death and apoptosis. The dying hepatocytes release damage-associated molecular patterns (DAMPs). The DAMPs, including nucleic acids, intracellular proteins, and adenosine triphosphate (ATP), send a danger signal to the surrounding cells (36). The danger signal activates the hepatic progenitor cells (HPCs). Apoptosis, on the other hand, produces low levels of DAMPs since most of the cell content will be retained in an apoptotic body. These apoptotic bodies will be phagocytosed by hepatic stellate cells (HSCs) and Kupffer cells. This induces a pro-fibrogenic response. Additionally, the DNA from the apoptotic hepatocytes triggers the activation of Toll-Like-Receptor (TLR)-9 on HSCs and collagen production (37).
Infiltration of the immune cells activates the trans-differentiation of HSCs into collagen-producing myofibroblasts (28). Usually, this process is involved in tissue repair upon short-term injury. When liver injury occurs, the HSCs are activated and differentiate from the quiescent phenotype to proliferative and contractile myofibroblasts (38). In their quiescent stage, HSCs store retinoids and synthesize glial fibrillary acidic protein (GFAP). When activated, a gradual loss of retinoids and GFAP coincides with their development into myofibroblasts with the synthesis of extracellular matrix (ECM) products like type I, type III, and type IV collagen but also hyaluronic acid (HA) (39). Levels of the glycosaminoglycan polymer HA increase with the amount of liver fibrosis (40). The collagen accumulation is accompanied by a rise in metalloproteinases (MMPs) such as MMP-9, which break down ECM products (41, 42). The combination of active and overexpressed MMP-9 and build-up of type III collagen leads to an abundance of cleaved type III collagen products like plasma N-terminal propeptide of type III procollagen (PIIIPN) or neo-epitope PRO-C3 (43, 44). Normally the MMPs are kept in check by tissue inhibitors of metalloproteinase (TIMPs). There are four TIMPs of which TIMP-1 is secreted by macrophages and fibroblasts (45). In murine fibrotic livers, likewise to the increase in MMPs, high concentrations of TIMP-1 were found (46). This creates a disturbance in the MMP/TIMP balance and, therefore a shift toward ECM synthesis and thus fibrogenesis (47). Alpha-2 macroglobulin (A2M) causes the balance to tip even further toward fibrogenesis by also inhibiting the MMPs (48). In addition to the ECM products, myofibroblasts also synthesize α-smooth muscle actin (α-SMA) (49). Ramzy et al. indicated that an increase in α-SMA marks the activation of HSCs (50).
During differentiation, the characteristic star-like shape of the HSCs changes to a more droplet form. The process is then balanced by the counteracting anti-fibrotic mechanisms resulting in the inactivation or apoptosis of the myofibroblasts and scar resolution. In chronic diseases, like NAFLD, there is an imbalance in these processes. The imbalance will cause persistent activation of proliferating, contractile, and migrating fibroblasts. This leads to the excessive production of ECM. The abundance of ECM will destroy the physiological architecture of the liver (51). The regulators of this balance are non-parenchymal cells (NPCs) like Kupffer cells and other immune cells, which are, as mentioned above, recruited to the site by the death and apoptosis of hepatocytes (52). NPCs will start producing pro-fibrogenic cytokines. On a molecular basis, a complex network of cytokine-induced pathways arises to coordinate the pro-fibrogenic cell interactions. The proposed signaling pathways associated with HSC activation and fibrosis progression are the transforming growth factor beta (TGF-β), platelet-derived growth factor (PDGF), inflammasome (NLRP3)-caspase 1, and the WNT/β-catenin (28).
From Liver Fibrosis to Liver Cirrhosis
Progression of liver fibrosis to liver cirrhosis varies between people depending on environmental and host factors (53). Cirrhosis is a consequence of long-standing fibrogenesis that results in the encapsulation of injured liver parenchyma by a collagenous scar. Histologically, cirrhosis is characterized by fibrotic septa that connect the portal tracts with each other and with the central veins (Figure 1C). This leads to a disconnection of the hepatocytes from the central vein, creating islands of hepatocytes. Vascular changes also occur, including loss of sinusoidal fenestrae and appearance of a basal membrane, or so-called capillarisation of the liver sinusoids, another hallmark of cirrhosis (54). The changes in liver structure ultimately lead to an increase of intravascular resistance within the portal system and decreased hepatic perfusion (55). The consequence is a loss of liver function (56).
Molecular Signaling Pathways Involved in Liver Fibrogenesis
TGF-β Signaling
TGF-β, together with PDGF, is the most potent inducer of hepatic fibrosis (57). The TGF-β superfamily consists of 33 members, of which TGF-β1 plays an essential role in liver fibrogenesis (58). The consequences of TGF-β1 signaling are inhibition of HSC apoptosis and induction of HSCs to produce excessive amounts of ECM proteins like fibronectin and collagen types I, II, and IV (59). Additionally, the production of matrix-degrading proteins is inhibited by TGF-β1 (60). In patients with hepatic fibrosis, increased concentrations of TGF-β1 correlate with the severity of fibrosis (61, 62).
TGF-β1 mainly exerts its effects via small Mothers Against Decapentaplegic (SMAD)-dependent pathways. The SMAD family is divided into three groups based on their functions. First, there are the receptor-regulated SMADs (R-SMADs), which include SMAD1, SMAD2, SMAD3, SMAD5, and SMAD8. Secondly, SMAD4 is the only member of the common SMAD (co-SMAD). The third group consists of the inhibitory SMADs (i-SMADs) and includes SMAD6 and SMAD7. The R-SMADs bind to membrane bound serine/threonine receptors and are activated by their kinase activity. Co-SMADs act as co-factors and attach to the R-SMADs to form a complex that will translocate to the nucleus of the cell. i-SMADs, on the other hand, counteract the effect of the R-SMADs (63). SMAD3 and SMAD4 have been found to be pro-fibrotic, whereas SMAD2 and SMAD7 are protective (64). SMAD3 induces hepatocyte death and lipid accumulation, especially in NASH (65). SMAD4 even enhances fibrogenesis by promoting SMAD3 activity. SMAD7, on the other hand, downregulates SMAD3 (66, 67). On their turn, SMADs also act as signal integrators and interact with the mitogen-activated protein kinases (MAPK) and nuclear factor kappa beta (NFκB) pathway (68).
PDGF Signaling
PDGF is a growth factor that promotes HSCs division and proliferation (28). The PDGF family consists of four members: PDGF-A, -B, -C, and -D (69). In healthy circumstances, PDGF is produced by platelets. When liver injury occurs, Kupffer cells recruited to the site of inflammation secrete PDGF (70). All PDGF members and its receptors (PDGF-R) are overexpressed in the case of liver fibrosis, and the activity increases with the degree of liver fibrosis (71–74). For example, PDGF-C activates the TGF-β/SMAD3 pathway in mice, leading to HSC proliferation, collagen production, and eventually fibrosis (72). However, of the four members, PDGF-B and -D are the most potent in activating the downstream pathways extracellular signal-regulated protein kinase/mitogen-activated protein kinase (Erk/MAPK) and protein kinase B (Akt/PKB). The activation leads to HSC proliferation (71, 75). PDGF-A expression was increased in hepatocytes from fibrotic livers compared to normal livers (76). In HSCs, on the other hand, although they express both receptors, only PDGF-BR expression was upregulated during HSC activation both in vitro as in vivo (77, 78).
The NLRP3 Inflammasome Caspase-1 Pathway
Inflammasomes are multiprotein complexes that sense danger signals like DAMPs and pathogen-associated molecular patterns (PAMPs) from damaged cells and pathogens (79). There are multiple inflammasomes implicated in liver disorders, but the nucleotide-binding oligomerization domain (NOD)-like receptor protein 3 (NLRP3) inflammasome has been studied most extensively (80–82). The NLRP3 inflammasome is activated in a two-step process. First, a bacterial signal, for example lipopolysaccharide (LPS), upregulates Nlrp3 expression via the NFκB-pathway (83). This, in turn, will enable a second signal, e.g., a DAMP, to activate the NLRP3 inflammasome. Once activated, the inflammasome binds with the adaptor molecule ASC to mediate caspase-1 cleavage, thereby activating the enzyme (79, 84). Caspase-1 activates pro-inflammatory cytokines IL-1β and IL-18 by proteolysis, though also activates the cytosolic protein gasdermin D (GSDMD) (85). GSDMD in a cleaved form will create pores in the plasma membrane of cells (86). This induces pyroptotic cell death and, consequently, the release of IL-1β and IL-18 (86–88).
In NAFLD, the NLRP3 inflammasome has been found to negatively regulate disease progression (89). In early NAFLD models, mRNA upregulation of the NLRP3 inflammasome components, like Nlrp3, Asc, and Casp1, was found. However, no active inflammasomes were found, indicating that not enough signals were present in a fatty liver to properly activate the inflammasome (90, 91). In NASH, on the other hand, IL-1 β will stimulate the production of inflammatory cytokines, thus aggravating the already existing inflammation (92). In a mouse knock-in model of the NLRP3 inflammasome, inflammation was increased, and simultaneously a high neutrophil infiltration was found. In addition, NLRP3 also induced HSC activation and collagen deposition, thereby causing liver fibrosis (80). Blockage of NLRP3 resulted in a reduction of liver inflammation and fibrosis in an experimental mouse model of NASH (93). It is clear that NLRP3 is involved in the pathogenesis of liver fibrosis with NAFLD. Nevertheless, additional studies are necessary to provide a better insight into these mechanisms.
Wnt/β-Catenin Signaling
The Wnt signaling pathway consists of canonical and non-canonical arms and regulates a large number of cellular functions (94). The canonical pathway exerts anti-lipid formation and anti-inflammatory effects, while the non-canonical pathway promotes fat formation, lipid accumulation, and inflammation (95). An imbalance between these two pathways has been associated with NAFLD by triggering lipotoxicity and fibrogenesis (96, 97). More specifically, the Wnt signaling pathway promotes hepatic fibrosis by enhancing HSC activation and survival, and upregulation of TGF-β/SMAD pathways (49).
Other Mechanisms That Contribute to Liver Fibrogenesis
Gut Liver Axis
About 70% of the liver's blood supply comes from the intestines. This blood circulation enables the liver to interact with products derived from the intestines, like bacterial DNA, LPS, or intact bacteria due to an increased intestinal barrier permeability (98). Normally, the Kupffer cells will clear the endotoxins, maintaining the immune tolerance and homeostasis. Alteration of the gut microbiome, gut permeability, and Kupffer cell responsivity can alter this balance (31). Moreover, fructose, a compound frequently found in sugar beverages, has been shown to promote a leaky gut and liver fibrosis. Fructose induces the ethanol-inducible cytochrome P450-2E1-mediated oxidative and nitrative stress (99). In addition, the bacterial products can bind to the TLRs in the liver, thereby inducing liver inflammation. This causes the progression of liver disease due to the fact that the TLRs will activate the NFκβ and the c-Jun N-terminal kinase (JNK) pathways (100). Most of the studies are, however, performed in mice, and more research in humans is necessary (101). A study conducted by Kapil et al. in humans indicated that small intestinal bacterial overgrowth and TLR signaling are involved with liver histology in NAFLD (102). In a study by Boursier et al. it was shown that NAFLD severity was associated with gut microbiome alterations and shifts in the metabolic function of the microbiome (103). More specifically, they found that the Ruminococcus bacteria were independently associated with fibrosis (103). These first results in human trials concerning the microbial environment are leading to further investigations of the influence of the gut microbiome in people with metabolic disorders like NAFLD (104).
Genetic Mechanisms
In addition to environmental factors, genes play a role in NAFLD (1). Several genes have been identified through genome-wide association studies (105–107). Amongst those, Patatin-like phospholipase domain containing 3 (PNPLA3) and transmembrane 6 superfamily member 2 (TM6SF2) seem to have the biggest impact (108). The PNPLA3 gene has been most extensively studied. It is located on chromosome 22 and encodes a 481 amino acid protein that mediates triacylglycerol hydrolysis. The I148M variant of PNPLA3 (rs738409) is strongly associated with NAFLD in adults but also in obese children and adolescents (109, 110). In a mouse model of NAFLD, overexpression of the I148M variant of the PNPLA3 gene caused hepatic steatosis (111). However, the exact mechanism is not yet known (105). The TM6SF2 gene, located on chromosome 19, plays a role in the progression of NAFLD. A single nucleotide polymorphism (rs58542926) replacing a cytosine by a thymine in position 167 has been linked to an increased hepatic triglyceride content (112). This specific gene variant has also been associated with fibrosis progression (113). Both PNLPA3 and TM6SF2 thus exert an additive effect on NASH and significant fibrosis (114).
Diagnosis of Liver Fibrosis
As previously stated, the stage of fibrosis is the most essential determinant of liver-related progression and mortality, and a key indicator for the development of other comorbidities like type 2 diabetes (T2DM) and cardiovascular disease, indicating the need to correctly diagnose fibrosis (115).
Liver Biopsy
Liver biopsy is currently considered as the gold standard for the diagnosis and histological assessment of NAFLD (1). Unfortunately, due to its invasive nature, a biopsy is not suited for screening purposes and cannot be implemented early in the diagnostic path of potential patients (116). It is mostly reserved for patients with a high risk of advanced liver disease during long-term follow-up, to distinguish NASH from NAFL and to determine the extent of liver fibrosis (117, 118). Additionally, a biopsy is still required in more advanced stages of drug development for NASH to assess treatment efficacy (119, 120).
Histological Scoring Systems for Liver Biopsy Samples
There are different histological scoring systems for classifying liver biopsy samples (121). However, the most widely used scores are the NASH CRN including the NAFLD Activity Score (NAS) and the Steatosis-Activity-Fibrosis (SAF) score (Table 1) (19, 122). The NAS scoring system is initially developed for use in clinical studies, and a definition of NASH has been based on this score. The score ranges from 0 to 8 (Table 1) and is composed of the unweighted sum of steatosis, ballooning, and lobular inflammation. A score between 0 and 2 corresponds to no NASH, 3–4 is borderline NASH, and definite NASH has a score between 5 and 8. Yet, there are several remarks concerning this scoring system. Firstly, this definition of activity does not distinguish steatosis separately from necroinflammation. Secondly, lobular inflammation outweighs ballooning, while ballooning is an essential feature of the NASH definition. Thirdly, the grading of the ballooning is based on the number of ballooned cells, without a clear definition of how to assess ballooning. This causes a greater opportunity for interobserver variability in NASH diagnosis. The SAF score, on the other hand, assesses steatosis (S) separately from activity (A), and of course, also fibrosis (F). This scoring system was developed by the Fatty Liver: Inhibition of Progression (FLIP) consortium. The activity score is, in this case, a combination of lobular inflammation and ballooning both scored from 0 to 2, overcoming the problem of one criterium outweighing the other. Additionally, a clear definition of ballooning is given. If the size of the hepatocyte is twice as big as usual, it is considered as severe ballooning (123). Although both the NAS and SAF score have a comparable fibrosis grading system, the SAF score may potentially be more appropriate for routine diagnosis and clinical trials as it comes with an easy to use diagnostic algorithm and better-defined criteria leading to less interobserver variability (117). However, future comparative studies are needed to determine which scoring system is the most potent in scoring NAFLD related fibrosis. In contrast to differences in the concepts of activity and the scoring of the features of ballooning and lobular inflammation, the scoring of fibrosis is the same in both NASH CRN and FLIP SAF (except for the subclassification of F1 in NASH CRN). Stage one (F1) of NASH CRN system is composed of three subclasses, namely: F1a stands for mild perisinusoidal/pericellular fibrosis, F2a is moderate perisinusoidal/pericellular, and F1c is portal/periportal fibrosis. For the FLIP SAF system, the subclasses of F1 were pooled into one stage of mild perisinusoidal/pericellular fibrosis. Stage two (F2) correlates with perisinusoidal/pericellular and portal/periportal fibrosis. Next, stage three (F3) corresponds to bridging fibrosis. Lastly, stage four (F4) stands for liver cirrhosis. In a clinical situation, people speak of significant and advanced fibrosis; in this case significant fibrosis stands for ≥F2 and advanced for ≥F3. It is thus different from the MetaVir score designed for the staging of liver fibrosis caused by viral hepatitis where F1 does not have subclassifications, and for the other stage's fibrosis expansion should be located in the portal zones (124, 125). Consequently, when reviewing literature, one should pay attention to the scoring system used as a reference golden standard when studying non-invasive biomarkers.
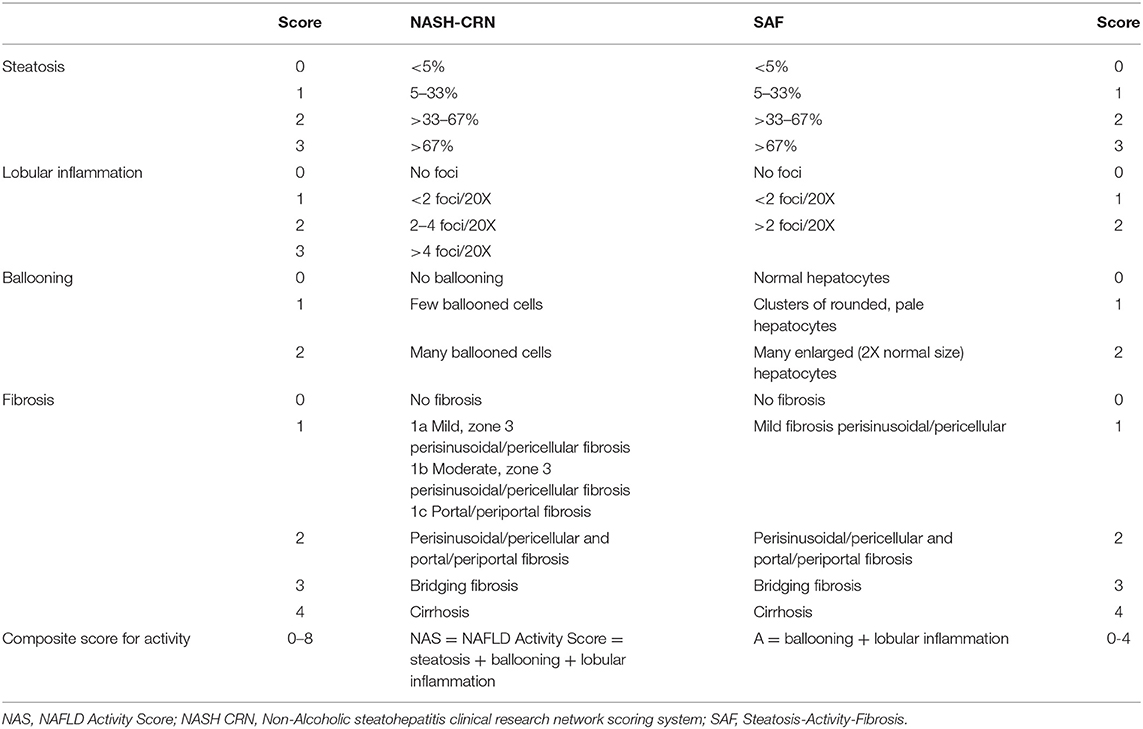
Table 1. Comparison between the histologic scoring of NAFLD according to NASH CRN system and SAF system (18, 120).
Outside these commonly used scoring systems, there are also other more granular scoring systems though they are not used as the golden reference standard in studies with non-invasive biomarkers. For instance, the Ishak staging system ranging from 0 to 6 with 6 being cirrhosis, was previously one of the most frequently used fibrosis scoring systems in clinical trials for different etiologies of liver disease. The Ishak fibrosis stages reflect more scarring than each preceding stage. Succession from one stage to the next represents progressively more advanced liver disease (126). Another scoring system is the EPoS staging system developed by the Elucidating (E) Pathways (P) of (o) Steatohepatitis (S) consortium. This system is based on e-slides, histological glass slide images that have been turned into electronic files. It includes, similarly to the Ishak system, seven stages ranging from 0 to 6. In a first study presented at the International Liver Congress of 2018, the EPoS scoring system showed promising results in terms of interobserver reproducibility (127).
Limitations of a Liver Biopsy
Although being the golden standard, a liver biopsy also has several limitations. The procedure comes with some discomfort and risks. As for the incidence of pain, this was reported to be 20%, though when a mildly unpleasant feeling was included in the assessment, the incidence increased to 84% (128). The incidence of severe complications and mortality was found to be between 0.3 and 0.57% and 0.01, respectively (129–131). Furthermore, the interpretation of the biopsy requires a high level of expertise and training; hence experienced physicians need to perform it. Liver biopsies are also prone to sampling error with discordance of one stage or more of 41% in a study with paired biopsies (132). This is due to the fact that a biopsy sample is only 1:50.000 of the liver mass. Fibrosis is not spread uniformly throughout the liver, which leads to this sampling error (132). Another problem in the assessment of histological liver biopsy samples is inter- and intra-observer variability (128). Evaluation of fibrosis is mostly consistent among observers. The evaluation of inflammatory activity, on the other hand, was inconsistent at a high rate in a study performed by Younossi et al. (133). Moreover, NASH can mimic other liver diseases, therefore the possibility of another etiology needs to be kept in mind (117).
Non-invasive Tests for the Detection of Liver Fibrosis
Non-invasive assessment of liver fibrosis can overcome some of the limitations of the biopsy and can be implemented and used for screening of NAFLD. There is currently an intensive search for biomarkers in NAFLD. Although stand-alone biomarkers are unlikely to provide the complex set of information that a liver biopsy offers, they can, if accurate and validated, provide an alternative to the biopsy to assess specific aspects of the disease. As outlined before, liver fibrosis is one of these crucial features, and non-invasive assessment of liver fibrosis has made significant advances in the last two decades. Currently, non-invasive assessment of liver fibrosis is composed of two different approaches: a biological approach based on the quantification of biomarkers (mostly in serum) and a physical approach based on the measurement of liver stiffness (117). A combination of the biological and physical approach results in a greater accuracy compared to the individual strategies to identify liver fibrosis, without the necessity of doing a liver biopsy (134, 135).
Methodological Aspects of Non-invasive Tests
As outlined previously, for a correct interpretation of the data, one should first of all look at the fibrosis scoring system that has been used in the design and validation of the non-invasive test. Hence, the non-invasive tests for NAFLD should be tested against the NASH CRN or SAF grading systems and not the MetaVir. Though, one should keep in mind the differences between the NASH CRN or SAF score. Moreover, as the non-invasive tests are validated against a liver biopsy, they cannot outperform the golden standard. Second, the values of non-invasive scores mostly show substantial overlap between histological fibrosis stages. Therefore, although often proposed for that purpose, non-invasive scores are not very accurate in predicting a precise corresponding histological fibrosis stage and hence cannot be used to diagnose the histological fibrosis stage of a given patient. According to the cut-off chosen, based on a given combination of specificity and sensitivity, non-invasive scores are useful to rule-in or rule-out significant or advanced fibrosis or cirrhosis, or conversely, the absence thereof, with NPV and PPV depending on the prevalence of the condition in the population that is studied. So, the result of a non-invasive test informs you about the likelihood of finding e.g., significant fibrosis, or the absence thereof, in a given patient, but does not tell you the patient has F2.
Liver Stiffness Measurement
Vibration Controlled Transient Elastography
The physical approach to assess fibrosis consists of measuring liver stiffness, which is a physical characteristic of the liver tissue, influenced by (but not equalling) the stage of liver fibrosis. Liver stiffness can be assessed by VCTETM, as measured by the FibroScan® device, was shown to correlate with liver fibrosis in a cross-sectional analysis of patients with viral hepatitis and is now widely used as a technique to non-invasively assess liver fibrosis in various liver diseases and different circumstances, including not only screening and baseline assessment but also follow-up and assessment of treatment response (136, 137). VCTETM measures liver fibrosis via the velocity of a low-frequency (50 Hz) elastic shear wave (induced by a mechanical pulse) propagating through the liver (117). The probe uses pulse-echo ultrasound (US) to follow the propagation of the shear wave and measures its velocity. The velocity of the wave depends, amongst others, on the amount of liver fibrosis. It is a straightforward, non-invasive, and easy to use technique. The area covered by the VCTETM measurement has a volume that is 100 times bigger than an average liver biopsy sample (138). Choosing the cut-off value for the VCTETM has to be done with care and depends on the clinical situation. Low cut-off values for the VCTETM, for example 7.9 kPa, have higher negative predictive values (NPV) than positive predictive values (PPV), meaning it can more precisely rule out more severe stages of fibrosis and rule in the absence of fibrosis. In contrast, higher cut-offs have an increase in the PPV and can, therefore, be more reliably used to rule-in more severe stages of fibrosis (139). A recent meta-analysis by Hsu et al. using the following thresholds 6.2, 7.6, 8.8, and 11.8 kPa, showed a pooled area under the receiving operating curve (AUROC) of 0.82, 0.87, 0.84, and 0.83 (with 95% CI) for diagnosing ≥F1, ≥F2, ≥F3, and F4, respectively (Table 2) (140).
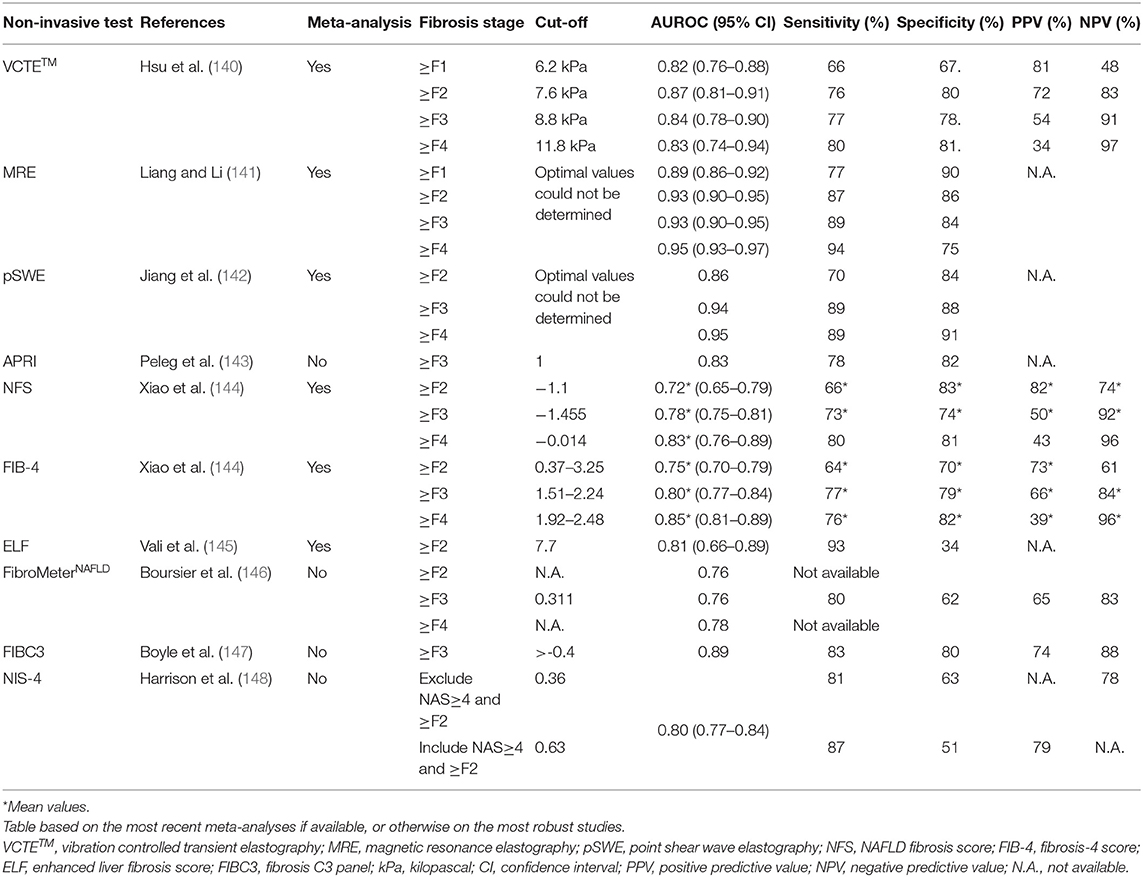
Table 2. Overview of the accuracy indices of the different non-invasive diagnostic tools for NAFLD-related liver fibrosis.
VCTETM has been found to be a cost-effective surveillance strategy to evaluate the presence of fibrosis (138). However, there are some limitations when using VCTETM measurements. Factors influencing the results of the FibroScan® measurements are ascites, elevated central venous pressure, and obesity. Gross ascites prevents an accurate measurement of liver stiffness by VCTETM. Fluid and adipose tissue attenuate the elastic wave (149–151). To overcome the latter problem, the extra-large (XL) probe was developed. It is able to assess the degree of fibrosis more accurately, though it may not be superior to the standard medium probe in obese patients (152, 153). The XL probe has a more sensitive US transducer, larger vibration amplitude, deeper focal length, and deeper signal penetration (tissue depth >35–75 mm) (154).
Liver inflammation may also reduce the accuracy of the test, as it can increase the VCTETM value by 1.3 to 3 times. This is illustrated by the rapid decline of liver stiffness after successful eradication of viral hepatitis C in a time frame that is too short to allow for substantial fibrosis regression (155). The pattern of fibrosis also differs between diseases, and the staging systems differ accordingly, as outlined previously. Accuracy and cut-offs need hence to be defined in a disease-specific way. Operator experience, sex, and metabolic syndrome can influence the FibroScan® measurements too (156, 157). A study performed by Vuppalanchi et al. demonstrated a failure rate of 5.5% because of excess skin to liver capsule distance, machine error, and invalid readings. Another study performed by the same research group indicated that fasting of at least 3 h in necessary (158). Without fasting, a significant increase (26 ± 25%, p = 0.02) in VCTETM was seen (159). Nonetheless, with sufficient operator experience, the failure rate and unreliability can be minimized (117, 160).
An extra feature recently added to the FibroScan® device is the possibility to measure the amount of liver fat. Since this an important characteristic of NAFLD, assessment of steatosis is therefore crucial. The fat content can be measured via the Controlled Attenuation Parameter (CAPTM). The CAPTM can be determined by the ultrasonic attenuation on the FibroScan® device at a frequency of 3.5 MHz on a go-and-return path (161, 162).
Other Ultrasound-Based Elastography Methods
There are several other ultrasound (US)-based methods available to determine liver elasticity (163). US elastography makes use of two different techniques, namely strain imaging or shear wave imaging. Strain imaging is used with strain elastography (SE) and acoustic radiation force impulse (ARFI) (164). Shear-wave imaging is the same technique as in the FibroScan® device (165). They have been less extensively studied in the context of NAFLD, but data on their accuracy are increasingly reported along with their use in clinical practice (166).
Point shear wave elastography (pSWE) is an ARFI-based technique that uses a short-duration, high-intensity acoustic pulse to displace tissue perpendicular to the longitudinal waves of the tissue surface (167). Next, the transducer detects the tissue displacement within a focal point along the radiation force resulting in the measurement of tissue stiffness. The big advantage of pSWE is that additional equipment is not necessary. pSWE can be incorporated in an US machine with brightness-mode. Next, direct anatomical visualization is possible, avoiding the areas with large blood vessels or parts of the biliary system (168). This implies, however, that a radiologist or sonographer is usually needed to perform the pSWE as a specific anatomical and technical expertise is necessary to interpret the visual images (169). Furthermore, in contrast to VCTETM, pSWE has a lower failure rate of 1–2% due to the fact that it is not limited by the presence of ascites (142, 170). A meta-analysis that compared VCTETM and pSWE showed that both provide excellent diagnostic accuracies for the diagnosis of advanced fibrosis and cirrhosis (Table 2) (142). However, a recent study conducted by Leong et al. comparing VCTETM with pSWE for diagnosis of fibrosis stage in a biopsy-proven cohort found that VCTETM outperformed pSWE. (171). Especially for the diagnosis of ≥F2 and ≥F3, the AUROC for VCTETM, respectively, 0.83 and 0.83, was higher than that of pSWE (0.72 and 0.69) (171).
Magnetic Resonance Elastography
Magnetic resonance elastography (MRE) is a magnetic resonance imaging-based method for quantitatively imaging tissue stiffness. These measurements can be taken rapidly during breath-hold acquisition mode. Even in the early stages, MRE can be used to detect NAFLD. The diagnostic accuracy of MRE for liver fibrosis and steatosis is higher than VCTETM and CAPTM (172). The pooled summary receiver operating characteristics (SROC) curve of MRE in 12 studies, including 910 patients with biopsy-proven NAFLD, was 0.89 for ≥F1, 0.93 for ≥F2, 0.93 for ≥F3, and 0.95 for F4, respectively (Table 2) (141). Nonetheless, due to high-performance costs, MRE is usually not performed routinely to screen patients for NAFLD (172).
Non-invasive Score Calculations to Detect Liver Fibrosis
The biological approach via non-invasive score calculation is composed of routinely measured clinical and laboratory variables that can aid in predicting liver fibrosis. Different scores have been proposed to calculate the risk of fibrosis. The aspartate aminotransferase (AST)-to-platelet ratio index (APRI), developed initially for hepatitis C infection, has been suggested for predicting significant fibrosis in NASH (117, 143). The NAFLD Fibrosis Score (NFS) has been demonstrated to be useful as a prognostic marker for fibrosis. Advanced fibrosis can be excluded with an NPV of 93% when using a low cut-off value and having a high PPV 90% (117). A meta-analysis conducted by Xiao et al. found an AUROC value of 0.78 for the exclusion of advanced fibrosis (Table 2) (144). The Fibrosis-4 (FIB-4) score was also designed as a parameter of fibrosis detection in patients with hepatitis C infection. This index is not influenced by BMI and is composed of routinely available laboratory data (AST, alanine aminotransferase (ALT) and platelets) (117). The FIB-4 had an AUROC of 0.80 for diagnosing advanced fibrosis, a sensitivity of 77%, a specificity of 79%, a PPV of 66%, and an NPV of 84% (Table 2) (144). The Enhanced Liver Fibrosis (ELF) score developed for the detection of liver fibrosis has good accuracy for the non-invasive diagnosis of advanced fibrosis in NAFLD (145). The ELF test is a panel consisting of the following markers: PIIINP, HA, and TIMP1 (173, 174). One side note, although it still has the same name, the test components and formula have been altered throughout the years. Nevertheless, not all studies reflect the accuracy of the current ELF test (145). A recent meta-analysis by Vali et al. in biopsy-proven NAFLD patients examined the accuracy of the ELF test for the diagnosis of advanced and significant liver fibrosis and NASH (145). At the recommended cut-off of 7.7, a high sensitivity (93%) was found, though specificity was limited. When the high cut-off was used (9.8), a higher specificity of 86% was reached. The cut-off value should thus be decided based on the purpose of the test in a specific clinical situation (the so-called context of use) (145).
FibroMeters, a family of blood tests specifically designed for each cause of chronic liver disease, were commercialized by Echosens (175). Although a FibroMeterNAFLD is available, the FibroMeterV2G, developed for hepatitis C is more accurate in NAFLD with an AUROC of respectively 0.76 and 0.8 for the detection of F≥3 (Table 2) (146). This is probably due to the fact that the FibroMeterNAFLD only contains AST, ALT, platelets, glucose, and ferritin, whereas the FibroMeterV2G uses AST, urea, platelets, prothrombin time, HA, and A2M. These last two are direct markers of liver fibrosis, while the others are indirect markers (146). In a biopsy-proven NAFLD cohort, the FibroMeterVCTE was tested for accuracy. FibroMeterVCTE combines the results of the VCTETM and FibroMeterV2G markers in one test. In this cohort, the FibroMeterVCTE was significantly more accurate than the FibroMeter or VCTETM alone (AUROC: 0.87 ± 0.012, p ≤ 0.005) (135).
Other parameters or scores that have been proposed for the detection of NAFLD related liver fibrosis are PRO-C3 and NIS4. Measurement of type III collagen neo-epitopes (PRO-C3) as a single diagnostic marker or as part of a panel has shown to have reasonable accuracy in assessing NAFLD disease stage and activity (147). When used as a single marker, PRO-C3 performed equally to simple panels like the FIB-4 (176). This might be due to the fact that PRO-C3 is more a product of active fibrogenesis instead of static collagen accumulation. PRO-C3 might therefore be helpful to detect patients with active liver fibrogenesis (177, 178). When on the other hand, used in the FIBC3 panel in combination with age, BMI, T2DM, platelets, it was able to distinguish advanced fibrosis (≥F3) with an AUROC of 0.89, a specificity of 80%, sensitivity of 83%, PPV of 74% and an NPV of 88%, respectively, for a cut-off value of >-0.4 (147). In three independent cohorts with suspected NASH, the non-invasive blood-based diagnostic test NIS-4 was developed and validated to detect patients with NAS≥4 and ≥F2 (148). The NIS4 panel comprised of the following NASH-associated biomarkers: miR-34a-5p, A2M, YKL-40, and glycated hemoglobin (HbA1c) (179, 180). The exact functions of miR-34a-5p and YKL-40 in the development of fibrosis are not yet fully understood, though their levels are elevated in patients with liver fibrosis (181–183). In the pooled validation cohort, NAS≥4 ≥F2 patients were excluded with a cut-off of 0.36 and this with a sensitivity and specificity of 81.5 and 63%, respectively. To include NAS≥4 and ≥F2 patients, a NIS4 value of 0.63 was used. This resulted in a sensitivity of 87.1% and a specificity of 50.7%. Furthermore, the NIS4 algorithm experiences no influence of age, sex, BMI, or liver enzyme concentrations (148).
Nonetheless, scoring systems have their limits. There is no single threshold for non-invasive tests that has the perfect balance between sensitivity and specificity. Up to now, the scores are more used as a first-line risk determination, without the necessity of doing a liver biopsy. For example, the NFS works best in distinguishing advanced vs. non-advanced or any with no fibrosis (172). In 25 to 30% of the patients, however, the NFS score is intermediate (117). A recent study indicated that the NFS and FIB-4 scores were better compared to the other scores (BARD, APRI, and AST/ALT ratio) to determine fibrosis, and as good as MRE in predicting the presence of advanced fibrosis in patients with biopsy-proven NAFLD (172).
Future Biomarkers for Liver Fibrosis
Up to now, no accurate serum biomarkers are available to detect a precise stage of fibrosis. Fortunately, a lot of research is carried out on this topic. For example, Mac 2-binding protein glycan isomer (M2BPGi) is secreted by HSCs to act as a messenger for Kupffer cells during fibrosis progression (184). M2BPGi can therefore act as biomarker for detection of liver fibrosis (185). A study by Nah and colleagues indicated that M2BPGi can exclude advanced fibrosis with a sensitivity of 80% and specificity of 77.9% and a NPV of 98.9% with an AUROC of 0.85 when compared to MRE (186). Serum autotaxin (ATX) may also be a potential serum biomarker for liver fibrosis with NAFLD ADDIN EN.CITE (187, 188). ATX is responsible for the transformation of lysophosphatidylcholine to lysophosphatidate (189). The latter is involved in the process of cell migration, neurogenesis, angiogenesis, smooth muscle contraction, platelet aggregation, and wound healing (190, 191). Sinusoidal endothelial liver cells process ATX, therefore, it is thought that in the case of chronic liver injury, ATX metabolism is impaired. First results within a cohort of NAFLD patients with fibrosis show that ATX can select patients who require further evaluation. The diagnostic accuracy was, however, lower than that of MRE (187). More recently a study was published by Kimura et al. on the possible biomarker thrombospondin 2 (TSP2) for the detection of liver fibrosis with NAFLD. TSP2 is involved in multiple processes such as collagen/fibrin formation. TSP2 had an AUROC of 0.82 for prediction of ≥F3 (192). Lastly, the serum marker type IV collage 7s can be used to diagnose significant fibrosis with an AUROC of 0.832, a sensitivity of 91.4%, and a NPV of 87.9% with a cut-off value of ≥5.2 ng/mL (193).
A recent study by Caussy et al. demonstrated that a combination of 10 metabolites consisting of eight lipids (5α-androstan-3β monosulfate, pregnanediol-3-glucuronide, androsterone sulfate, epiandrosterone sulfate, palmitoleate, dehydroisoandrosterone sulfate, 5α-androstan-3β disulfate, and glycocholate), one amino acid (taurine), and one carbohydrate (fructose) could detect advanced fibrosis. With an AUROC of the metabolite combination of 0.94 and a sensitivity and specificity of, respectively, 90 and 79%, the metabolites performed better than the FIB-4 (0.78) and NFS (0.84) for the detection of advanced liver fibrosis (194). However, they used mass spectrometry to analyse the metabolites, which is not easily accessible and expensive and therefore not (yet) applicable in a routine clinical situation (195). However, this type of biomarkers, requiring more sophisticated techniques and all kinds of omics approaches, might show the way ahead to increase the accuracy over the currently available tools. To support biomarker development for the detection of liver fibrosis, two large projects, one in the United States of Amerika called Non-Invasive Biomarkers of Metabolic Liver Disease (NIMBLE), and one in Europe called the Liver Investigation: Testing Marker Utility in Steatohepatitis (LITMUS), have been set-up (196).
The Use of Non-invasive Biomarkers in a Clinical Situation and in Trials
A non-invasive test alone has a certain accuracy depending on the context of use (147). Notwithstanding, when used in a sequential way or at the same time, the accuracy of the non-invasive tests increases significantly. The most appropriate combination is probably one with a biological test in combination with liver stiffness measurements. In a recent study by Boursier et al. different stepwise combinations were tested. The sequential combinations of the FIB-4 followed by FibroMeterVCTE and the VCTETM followed by the FibroMeterVCTE provided a diagnostic accuracy of 90%. A liver biopsy to confirm the results was only needed in 20% of the cases (135). Another study conducted by Srivastava et al. in a primary care cohort tested a 2-step algorithm that combined the FIB-4, followed by, if needed, the ELF test (197). Use the 2-step algorithm improved detection of advanced fibrosis and cirrhosis by 4.9-fold (197). Davyduke et al. piloted a FIB-4 first strategy, followed by a VCTETM when classified as high risk (198). When using this strategy, only 15% of the patients needed to be referred for further assessment (198). With a probabilistic decision model of a cohort of 1000 NAFLD patients, different sequential combinations of the non-invasive tests were simulated, and costs were compared. The price per case of advanced fibrosis was significantly lower when using a sequential combination (£8,932 for FIB-4/ELF, £9,083 for FIB-4/VCTETM) compared to the standard of care (£25,543) (199). Proving that the sequential combinations of non-invasive tests are cost-effective, reduce unnecessary referral and detect advanced fibrosis without the necessity of doing a liver biopsy, which can be useful for inclusion in clinical trials (199–201).
Currently, efficacy assessment in clinical trials requires histology hence biopsy in phase II trials that need to provide data to go into phase III, and for the interim analysis for conditional approval in phase III, with regression of fibrosis of 1 stage without worsening of fibrosis as one of the endpoints for regulatory approval in non-cirrhotic NASH (119, 120). Efficacy assessment in earlier phase II trials, on the other hand, can be based on non-invasive biomarkers, and trial sponsors are encouraged to collect data on biomarker response in late phase II and in phase III trials to inform future trial design. Several non-invasive markers of fibrosis have been used in several trials and mostly serve to support the data on histology. For example, in the pirfenidone (PFD) study, they evaluated the antifibrotic effects with, similarly, for the cenicriviroc (CVC) study where they used the NFS, FIB-4, APRI, and ELF scores (196, 202).
Much more data and analyses are needed to couple responses in histology to responses in biomarkers and to define criteria of response in terms of biomarkers, i.e., what magnitude of change, absolute and or relative, is clinically meaningful and correlates with a histological response or with another endpoint concerned clinically relevant and resulting in clinically significant benefit.
Anti-Fibrotic Drugs
NAFLD management is centered on lifestyle modifications, weight loss, and habitual physical activity. Weight loss promotes fat reduction and NAFLD remission. A bodyweight reduction of 3 to 5% improves steatosis, and a decrease of 10% improves necroinflammation and fibrosis (1). However, dietary and lifestyle changes are hard to maintain. As a result, there is a need for appropriate drugs to treat NAFLD. The target of treatment is still a matter of debate. Fibrosis regression (mostly defined by at least one stage improvement according to NASH CRN), as a highly potential surrogate marker for clinical benefit, is one of the endpoints approved by the regulatory authorities for phase III trials in NASH (119, 120). Unfortunately, there are currently no drugs approved specifically for the treatment of liver fibrosis by the United States of America Food and Drug Administration (FDA) or European Medicines Agency (EMA) (203), despite the increased insight into the molecular and cellular mechanisms of liver fibrosis.
Current Options for NAFLD Treatment
Besides vitamin E, the only drugs that can be recommended for the treatment of NAFLD, are drugs already used for the treatment of T2DM treatment and/or obesity. Most of these drugs have a direct effect on NASH, and via this, an indirect effect on fibrosis regression (although direct antifibrotic effects might even so be present) since NASH and fibrosis are strongly intertwined.
Drugs used to treat T2DM with effects on NAFLD histology are thiazolidinediones (TZDs), glucagon-like peptide-1 receptor agonist (GLP-1 RA) and sodium-glucose cotransporter- (SGLT)-2 inhibitors (117, 203–205). TZDs like pioglitazone act on peroxisome proliferator-activated receptors (PPARs), mainly on the PPARγ isoform. If PPARγ binds to the retinoid X receptor, it has powerful insulin-sensitizing properties in adipose tissue. Although PPARγ is poorly expressed in the hepatocytes, it still exerts anti-steatogenic effects (206). In rat livers, depletion of PPARγ led to a decrease in fibrogenesis (207). However, the effect was not so strong in human trials with TZDs. TZDs cause a reduction in liver fat, despite some overall weight gain, which is reflective of an improvement in adipose tissue function and goes along with a redistribution of fat from visceral to subcutaneous fat storage (208). PPARγ is also implicated in the activation state of HSC, so TZDs can also have direct effects on fibrogenesis. Based on the histological improvements seen with pioglitazone, several guidelines recommend the use of TZDs in patients with liver biopsy-proven NASH and T2DM (209). In the PIVENS trial, after 96 weeks of pioglitazone treatment, no improvement in fibrosis stage was seen when compared to placebo (210).
GLP-1 RAs improve glycaemic control via a decreased glucagon secretion, slowed gastric emptying, glucose-dependent insulin secretion, enhanced satiety, reduction in body weight, and BMI (204, 211–213). They also lead to the improvement of the hepatic markers ALT, AST, and gamma-glutamyl transferase (GGT). Presently, GLP-1 RA use is only recommended in case of a high BMI (>27 kg/m2) and comorbidities like diabetes and arterial hypertension (AHT). However, NASH should be added to the list of comorbidities associated with obesity (117). In the LEAN trial, the efficacy of 48 weeks of liraglutide, a GLP-1 RA, was investigated with NASH regression without worsening of fibrosis as the primary endpoint. Despite the significant difference in the resolution of NASH between liraglutide and placebo, no significant difference in fibrosis score was detected (214). Another GLP-1 RA, semaglutide, was tested in patients at risk for NAFLD development for 104 weeks. Semaglutide significantly reduced ALT after 28 to 20 weeks of treatment (215). In a recent study by Legry et al. the effect of semaglutide in mice with induced NASH was researched. This showed a reduction in NAS though it did not reduce fibrosis (216). These results were confirmed in human trials, semaglutide was significant on the resolution of NASH, though not on the fibrosis endpoint after 72 weeks of treatment (217). Despite having an efficacy on NASH resolution, the TZD pioglitazone and GLP-1 RA semaglutide were not able to reach significance on the ≥1 point fibrosis reduction endpoint. Notwithstanding, a decrease in the mean fibrosis score has been reported (218, 219).
SGLT-2 is a class of oral antidiabetics that reduces hyperglycaemia by promoting urinary excretion of glucose without affecting insulin secretion (220). In rodent models of T2DM, the SGLT-2 inhibitor ipragliflozin prevented the development of NASH (221). Not only in rodent models SGLT-2 inhibitors proved their effect of NAFLD development, in patients with T2DM who received dapagliflozin or empagliflozin a decrease in hepatic steatosis was seen. However, in these studies, no effects on liver fibrosis were detected (205, 222). A recent meta-analysis by Mantovani et al. also confirmed the significant effect on hepatic steatosis, though up to now, no results on the histological response of SGLT-2 inhibitors are available, at least not from randomized placebo-controlled trials (223, 224).
Therapy in Development
As mentioned above, there are currently no approved drugs for NAFDL treatment, yet potent drugs are coming. The first drugs are already in phase III trials and are expected to be on the market by the end of 2020 (117). As mentioned, fibrosis regression of at least one point without worsening of NASH, as a likely reasonable surrogate for a clinically meaningful benefit, is one of the two regulatory endpoints for conditional approval in non-cirrhotic NASH. Obeticholic acid (OCA) is so far the only drug that demonstrated efficacy on this endpoint in phase III. OCA is a first-in-class selective farnesoid X receptor (FXR) agonist with anti-cholestatic and hepato-protective properties (225). The FXR is a bile acid nuclear receptor that plays a role in lipoprotein and glucose metabolism, hepatic regeneration, and regulation of hepatic inflammation. Activation of the FXR receptor in mice has been shown to inhibit NLRP3 inflammasome activation in hepatocytes, thereby preventing disease progression (226). OCA was FDA approved in 2016 for the treatment of primary biliary cholangitis, though it is currently in a phase III trial to test the effects and safety in NAFLD (Regenerate Study) (209). In a first interim analysis of the Regenerate study, OCA thus demonstrated statistically significant fibrosis regression of at least one point with an effect size of 11% in phase III after 72 weeks on 25 mg. OCA did, however, not meet the endpoint of NASH resolution (227).
Other drugs that are currently being tested for the treatment of fibrosis are lanifibranor, PFD, and CVC. Lanifibranor (IVA337) is a drug that activates each of the three PPAR isoforms (228). These isoforms play an essential role in the regulation of cellular differentiation, development, and tumorigenesis throughout the body. The drug has both anti-fibrotic and anti-inflammatory effects and is also beneficial for metabolic changes. Currently, lanifibranor is going into phase III, based on the significant results the drug demonstrated on both resolution of NASH and improvement of fibrosis and the combination of both (229–231). PFD is an oral antifibrotic drug approved for the treatment of idiopathic pulmonary fibrosis. In a recent study by Poo et al. the effect of prolonged-release formulation (PR-PFD) plus standard of care was tested in patients with advanced liver fibrosis (PROMETEO Study). In 35% of the patients, a significant reduction of fibrosis was seen, leading to the conclusion that PR-PFD is efficacious and safe in patients with advanced liver disease. Moreover, it showed promising antifibrotic effects (196). CVC, a drug that targets macrophages in the liver by inhibiting the C-C chemokine receptors CCR2 and CCR5 (232), met this endpoint as a key secondary endpoint in phase II after 1 year of treatment with an effect size of 9.6% (p = 0.023) but was not significant at 2 years and showed no efficacy on NASH resolution (202, 233). A phase III trial (AURORA) is still ongoing evaluating the effect of CVC in NASH patients with F2 or F3 (234).
Different molecules failed, despite pre-clinical studies being positive. For instance, selonsertib is an apoptosis signal-regulating kinase 1 inhibitor that has been demonstrated in patients with moderate-to-severe NASH to reduce fibrosis, steatosis, and progression to cirrhosis (235). However, in several phase III clinical trials, selonsertib did not achieve the endpoints for the reduction of fibrosis (236). This also applies to simtuzumab, a monoclonal antibody against lysyl oxidase-like 2 that is involved in fibrogenesis. In the phase IIb trials, simtuzumab was unable to reduce fibrosis (237). Likewise, for the galactin-3 inhibitor GR-MD-02, phase II trials failed to show efficacy in NASH and reduction of liver fibrosis (238, 239). Correspondingly also elafibranor, a dual PPAR agonist that showed promising results in the reduction of NASH and liver fibrosis in a phase II trial, failed to replicate these results in a phase III trial. Therefore, the trial has been ended (240). Recent suggestions made by Ratziu et al. stated that these failed trials were caused by the rush to move compounds into clinical development without thoroughly being investigated in the pre-clinical trials (241). More attention should be paid to optimize treatment dose and regimen correctly, but most importantly, the results of small studies should be interpreted with care (241).
Future Therapeutic Targets
Although recent clinical trials have been promising in treating NAFLD-related liver fibrosis, the overall efficacy of these drugs has been modest. Only a minority of patients achieved treatment response (242). Possible new targets can be found in the gut microbiome. As stated, it plays an important role in fibrogenesis. Therefore, targeting the GLA by modulating the gut-microbiome can be a promising therapeutic approach in NAFLD (243). In mice, whole-body deletion of nucleotide-binding oligomerization domain-containing (NOD)2 caused an increase in liver steatosis and fibrosis. NOD2 can therefore potentially engage the GLA to protect against steatosis, fibrosis, and gut dysbiosis (244). Another possible treatment option is the blocking of PDGF. Blocking of PDGF signaling ameliorates experimental liver fibrogenesis. PDGF signaling can be blocked by regulation of the isoforms, regulation of the receptor binding, and finally, by inhibiting the signaling pathways (245). PDGFR kinase activity blocking is one of the most efficient ways to block the signaling pathway of PDGF. Several kinase inhibitors have been developed, though they are not entirely specific. Imatinib mesylate (Gleevec®) effectively inhibits PDGFR signaling in CCl4-treated mice leading to improved liver regeneration in vivo and induced apoptosis of HSCs both in vivo and in vitro (246). In a pig serum-induced rat model of liver fibrosis, characterized by a slow progression of fibrosis, like in a human situation, imatinib had an effect in the early stages of liver fibrosis (247). Finally, Wnt3a, a canonical Wnt ligand, could be used as a future therapeutic target. In a study conducted by Wang et al. where LRP6 mutant mice were treated with Wnt3a, liver inflammation was reduced, indicating the ant-inflammatory role of Wnt3a (97).
Conclusion
Progressive liver fibrosis in NAFLD can lead to cirrhosis and liver-related morbidity and mortality and is also the strongest predictor of overall mortality. Halting fibrosis progression and regression of existing fibrosis are hence essential goals for treatment. Liver biopsy is still the gold standard for the staging of liver fibrosis. Accurate non-invasive diagnosis of liver fibrosis that can replace liver biopsy in most of the circumstances is obviously needed, both for initial diagnosis and monitoring of evolution over time and response to treatment. Non-invasive biological markers and liver stiffness measurement, alone or in combination, are extensively studied and further developed and validated. Currently, anti-fibrotic drugs are in development, and some have promising results that can lead to the prevention of liver fibrosis and delaying or even halting the development of cirrhosis. Moreover, based on the available data and international guidelines, a multi-disciplinary approach to treat and guide NAFLD patients is recommended due to the association with other metabolic features. These developments will potentially have an extensive impact on global health and on the healthcare costs that accompanies the rising incidence of NAFLD-related fibrosis. Furthermore, it has the possibility to lower the increasing incidence of HCC. Nonetheless, a better understanding of the complex pathology of NAFLD-related fibrogenesis is necessary to identify new targets for treatment and to find markers that will lead to new diagnostic methods that can accurately detect disease severity and fibrosis stages and its evolution over time.
Author Contributions
LH collected the data and drafted the first version of the paper. All authors listed have made a substantial, direct and intellectual contribution to the review, and approved it for publication.
Conflict of Interest
DB has received travel grants from AbbVie and Gilead Sciences and research grants from Gilead Sciences. GR has received research grants from AbbVie, Janssen Pharmaceuticals, MSD, and has acted as a consultant/advisor for AbbVie, BMS, Gilead Sciences and MSD.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Ph.D. authors of this paper are part of the Limburg Clinical Research Center (LCRC), supported by the foundation Limburg Sterk Merk, province of Limburg, Flemish government, Hasselt University, Ziekenhuis Oost-Limburg, and Jessa Hospital.
References
1. Perumpail BJ, Khan MA, Yoo ER, Cholankeril G, Kim D, Ahmed A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J Gastroenterol. (2017) 23:8263–76. doi: 10.3748/wjg.v23.i47.8263
2. Younossi ZM, Blissett D, Blissett R, Henry L, Stepanova M, Younossi Y, et al. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology. (2016) 64:1577–86. doi: 10.1002/hep.28785
3. Polyzos SA, Kountouras J, Mantzoros CS. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metab Clin Exp. (2019) 92:82–97. doi: 10.1016/j.metabol.2018.11.014
4. Hussain A, Patel PJ, Rhodes F, Srivastava A, Patch D, Rosenberg W. Decompensated cirrhosis is the commonest presentation for NAFLD patients undergoing liver transplant assessment. Clin Med. (2020) 20:313–8. doi: 10.7861/clinmed.2019-0250
5. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. (2015) 62 (1 Suppl):S47–64. doi: 10.1016/j.jhep.2014.12.012
6. Francque SM, van der Graaff D, Kwanten WJ. Non-alcoholic fatty liver disease and cardiovascular risk: pathophysiological mechanisms and implications. J Hepatol. (2016) 65:425–43. doi: 10.1016/j.jhep.2016.04.005
7. Balp MM, Krieger N, Przybysz R, Way N, Cai J, Zappe D, et al. The burden of non-alcoholic steatohepatitis (NASH) among patients from Europe: a real-world patient-reported outcomes study. JHEP Rep. (2019) 1:154–61. doi: 10.1016/j.jhepr.2019.05.009
8. McSweeney L, Breckons M, Fattakhova G, Oluboyede Y, Vale L, Ternent L, et al. Health-related quality of life and patient-reported outcome measures in NASH-related cirrhosis. JHEP Rep. (2020) 2:100099. doi: 10.1016/j.jhepr.2020.100099
9. Calzadilla Bertot L, Adams LA. The natural course of non-alcoholic fatty liver disease. Int J Mol Sci. (2016) 17:774. doi: 10.3390/ijms17050774
10. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol. (2018) 69:896–904. doi: 10.1016/j.jhep.2018.05.036
11. Bian Z, Ma X. Liver fibrogenesis in non-alcoholic steatohepatitis. Front Physiol. (2012) 3:248. doi: 10.3389/fphys.2012.00248
12. Kumar R, Priyadarshi RN, Anand U. Non-alcoholic fatty liver disease: growing burden, adverse outcomes and associations. J Clin Transl Hepatol. (2020) 8:76–86. doi: 10.14218/JCTH.2019.00051
13. Schuppan D, Surabattula R, Wang XY. Determinants of fibrosis progression and regression in NASH. J Hepatol. (2018) 68:238–50. doi: 10.1016/j.jhep.2017.11.012
14. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta-analysis. Hepatology. (2017) 65:1557–65. doi: 10.1002/hep.29085
15. Younossi ZM, Stepanova M, Rafiq N, Makhlouf H, Younoszai Z, Agrawal R, et al. Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver-related mortality. Hepatology. (2011) 53:1874–82. doi: 10.1002/hep.24268
16. Ekstedt M, Hagström H, Nasr P, Fredrikson M, Stål P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. (2015) 61:1547–54. doi: 10.1002/hep.27368
17. Sebastiani G, Alshaalan R, Wong P, Rubino M, Salman A, Metrakos P, et al. Prognostic value of non-invasive fibrosis and steatosis tools, hepatic venous pressure gradient (HVPG) and histology in nonalcoholic steatohepatitis. PLoS ONE. (2015) 10:e0128774. doi: 10.1371/journal.pone.0128774
18. Leung JC, Loong TC, Wei JL, Wong GL, Chan AW, Choi PC, et al. Histological severity and clinical outcomes of nonalcoholic fatty liver disease in nonobese patients. Hepatology. (2017) 65:54–64. doi: 10.1002/hep.28697
19. Kleiner DE, Brunt EM, Wilson LA, Behling C, Guy C, Contos M, et al. Association of histologic disease activity with progression of nonalcoholic fatty liver disease. JAMA Netw Open. (2019) 2:e1912565. doi: 10.1001/jamanetworkopen.2019.12565
20. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol. (2015) 13:643–54.e1-9; quiz e39-40. doi: 10.1016/j.cgh.2014.04.014
21. Torok NJ, Dranoff JA, Schuppan D, Friedman SL. Strategies and endpoints of antifibrotic drug trials: Summary and recommendations from the AASLD emerging trends conference, Chicago, June 2014. Hepatology. (2015) 62:627–34. doi: 10.1002/hep.27720
22. Musso G, Cassader M, Paschetta E, Gambino R. Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis. Gastroenterology. (2018) 155:282–302.e8. doi: 10.1053/j.gastro.2018.06.031
23. Gastaldelli A, Cusi K. From NASH to diabetes and from diabetes to NASH: mechanisms and treatment options. JHEP Rep. (2019) 1:312–28. doi: 10.1016/j.jhepr.2019.07.002
24. Lefere S, Tacke F. Macrophages in obesity and non-alcoholic fatty liver disease: crosstalk with metabolism. JHEP Rep. (2019) 1:30–43. doi: 10.1016/j.jhepr.2019.02.004
25. Gastaldelli A, Cusi K, Pettiti M, Hardies J, Miyazaki Y, Berria R, et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology. (2007) 133:496–506. doi: 10.1053/j.gastro.2007.04.068
26. Brunt EM, Wong VW, Nobili V, Day CP, Sookoian S, Maher JJ, et al. Nonalcoholic fatty liver disease. Nat Rev Dis Primers. (2015) 1:15080. doi: 10.1038/nrdp.2015.80
27. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metab Clin Exp. (2016) 65:1038–48. doi: 10.1016/j.metabol.2015.12.012
28. Roehlen N, Crouchet E, Baumert TF. Liver fibrosis: mechanistic concepts and therapeutic perspectives. Cells. (2020) 9:875. doi: 10.3390/cells9040875
29. Tomita K, Tamiya G, Ando S, Ohsumi K, Chiyo T, Mizutani A, et al. Tumour necrosis factor alpha signalling through activation of kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut. (2006) 55:415–24. doi: 10.1136/gut.2005.071118
30. Van Herck MA, Weyler J, Kwanten WJ, Dirinck EL, De Winter BY, Francque SM, et al. The differential roles of T cells in non-alcoholic fatty liver disease and obesity. Front Immunol. (2019) 10:82. doi: 10.3389/fimmu.2019.00082
31. Tanwar S, Rhodes F, Srivastava A, Trembling PM, Rosenberg WM. Inflammation and fibrosis in chronic liver diseases including non-alcoholic fatty liver disease and hepatitis C. World J Gastroenterol. (2020) 26:109–33. doi: 10.3748/wjg.v26.i2.109
32. Paquissi FC. Immune imbalances in non-alcoholic fatty liver disease: from general biomarkers and neutrophils to interleukin-17 axis activation and new therapeutic targets. Front Immunol. (2016) 7:490. doi: 10.3389/fimmu.2016.00490
33. Yilmaz H, Yalcin KS, Namuslu M, Celik HT, Sozen M, Inan O, et al. Neutrophil-Lymphocyte ratio (NLR) could be better predictor than C-reactive protein (CRP) for liver fibrosis in non-alcoholic steatohepatitis(NASH). Ann Clin Lab Sci. (2015) 45:278–86. doi: 10.1097/MEG.0000000000001393
34. Wong L, Bozhilov K, Hernandez B, Kwee S, Chan O, Ellis L, et al. Underlying liver disease and advanced stage liver cancer are associated with elevated neutrophil-lymphocyte ratio. Clin Mol Hepatol. (2019) 25:305–16. doi: 10.3350/cmh.2019.0004
35. Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology. (2014) 59:1393–405. doi: 10.1002/hep.26937
36. Mihm S. Danger-Associated molecular patterns (DAMPs): molecular triggers for sterile inflammation in the liver. Int J Mol Sci. (2018) 19:3104. doi: 10.3390/ijms19103104
37. Watanabe A, Hashmi A, Gomes DA, Town T, Badou A, Flavell RA, et al. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology. (2007) 46:1509–18. doi: 10.1002/hep.21867
38. Affo S, Yu LX, Schwabe RF. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu Rev Pathol. (2017) 12:153–86. doi: 10.1146/annurev-pathol-052016-100322
39. Rojkind M, Giambrone MA, Biempica L. Collagen types in normal and cirrhotic liver. Gastroenterology. (1979) 76:710–9. doi: 10.1016/S0016-5085(79)80170-5
40. Neuman MG, Cohen LB, Nanau RM. Hyaluronic acid as a non-invasive biomarker of liver fibrosis. Clin Biochem. (2016) 49:302–15. doi: 10.1016/j.clinbiochem.2015.07.019
41. Arthur MJ. Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. (2000) 279:G245–9. doi: 10.1152/ajpgi.2000.279.2.G245
42. Roderfeld M. Matrix metalloproteinase functions in hepatic injury and fibrosis. Matrix Biol. (2018) 68–9:452–62. doi: 10.1016/j.matbio.2017.11.011
43. Veidal SS, Vassiliadis E, Barascuk N, Zhang C, Segovia-Silvestre T, Klickstein L, et al. Matrix metalloproteinase-9-mediated type III collagen degradation as a novel serological biochemical marker for liver fibrogenesis. Liver Int. (2010) 30:1293–304. doi: 10.1111/j.1478-3231.2010.02309.x
44. Byrne CD, Targher G. What's new in NAFLD pathogenesis, biomarkers and treatment? Nat Rev Gastroenterol Hepatol. (2020) 17:70–1. doi: 10.1038/s41575-019-0239-2
45. Robert S, Gicquel T, Victoni T, Valença S, Barreto E, Bailly-Maître B, et al. Involvement of matrix metalloproteinases (MMPs) and inflammasome pathway in molecular mechanisms of fibrosis. Biosci Rep. (2016) 36:e00360. doi: 10.1042/BSR20160107
46. Roeb E, Purucker E, Breuer B, Nguyen H, Heinrich PC, Rose-John S, et al. TIMP expression in toxic and cholestatic liver injury in rat. J Hepatol. (1997) 27:535–44. doi: 10.1016/S0168-8278(97)80359-5
47. Yoshiji H, Kuriyama S, Miyamoto Y, Thorgeirsson UP, Gomez DE, Kawata M, et al. Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology. (2000) 32:1248–54. doi: 10.1053/jhep.2000.20521
48. Kawser CA, Iredale JP, Winwood PJ, Arthur MJ. Rat hepatic stellate cell expression of alpha2-macroglobulin is a feature of cellular activation: implications for matrix remodelling in hepatic fibrosis. Clin Sci. (1998) 95:179–86. doi: 10.1042/cs0950179
49. Miao CG, Yang YY, He X, Huang C, Huang Y, Zhang L, et al. Wnt signaling in liver fibrosis: progress, challenges and potential directions. Biochimie. (2013) 95:2326–35. doi: 10.1016/j.biochi.2013.09.003
50. Ramzy M, Abdelghany H, Zenhom N, El-Tahawy N. Effect of histone deacetylase inhibitor on epithelial-mesenchymal transition of liver fibrosis. IUBMB Life. (2018) 70:511. doi: 10.1002/iub.1742
51. Elpek G. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J Gastroenterol. (2014) 20:7260–76. doi: 10.3748/wjg.v20.i23.7260
52. Vonghia L, Michielsen P, Francque S. Immunological mechanisms in the pathophysiology of non-alcoholic steatohepatitis. Int J Mol Sci. (2013) 14:19867–90. doi: 10.3390/ijms141019867
53. Zhou WC, Zhang QB, Qiao L. Pathogenesis of liver cirrhosis. World J Gastroenterol. (2014) 20:7312–24. doi: 10.3748/wjg.v20.i23.7312
54. Iwakiri Y, Shah V, Rockey DC. Vascular pathobiology in chronic liver disease and cirrhosis - current status and future directions. J Hepatol. (2014) 61:912–24. doi: 10.1016/j.jhep.2014.05.047
55. Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. (2008) 371:838–51. doi: 10.1016/S0140-6736(08)60383-9
56. Saffioti F, Pinzani M. Development and regression of cirrhosis. Dig Dis. (2016) 34:374–81. doi: 10.1159/000444550
57. Dewidar B, Meyer C, Dooley S, Meindl-Beinker AN. TGF-β in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells. (2019) 8:1419. doi: 10.3390/cells8111419
58. Dropmann A, Dediulia T, Breitkopf-Heinlein K, Korhonen H, Janicot M, Weber SN, et al. TGF-β1 and TGF-β2 abundance in liver diseases of mice and men. Oncotarget. (2016) 7:19499–518. doi: 10.18632/oncotarget.6967
59. Kanzler S, Lohse AW, Keil A, Henninger J, Dienes HP, Schirmacher P, et al. TGF-beta1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am J Physiol. (1999) 276:G1059–68. doi: 10.1152/ajpgi.1999.276.4.G1059
60. Kim KK, Sheppard D, Chapman HA. TGF-β1 signaling and tissue fibrosis. Cold Spring Harb Perspect Biol. (2018) 10:ea022293. doi: 10.1101/cshperspect.a022293
61. Mahmoud AA, Bakir AS, Shabana SS. Serum TGF-β, serum MMP-1, and HOMA-IR as non-invasive predictors of fibrosis in Egyptian patients with NAFLD. Saudi J Gastroenterol. (2012) 18:327–33. doi: 10.4103/1319-3767.101132
62. Vonghia L, Magrone T, Verrijken A, Michielsen P, Van Gaal L, Jirillo E, et al. Peripheral and hepatic vein cytokine levels in correlation with non-alcoholic fatty liver disease (NAFLD)-related metabolic, histological, and haemodynamic features. PLoS ONE. (2015) 10:e0143380. doi: 10.1371/journal.pone.0143380
63. Itoh S, Itoh F, Goumans MJ, Ten Dijke P. Signaling of transforming growth factor-beta family members through smad proteins. Eur J Biochem. (2000) 267:6954–67. doi: 10.1046/j.1432-1327.2000.01828.x
64. Xu F, Liu C, Zhou D, Zhang L. TGF-β/SMAD pathway and its regulation in hepatic fibrosis. J Histochem Cytochem. (2016) 64:157–67. doi: 10.1369/0022155415627681
65. Yang L, Roh YS, Song J, Zhang B, Liu C, Loomba R, et al. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology. (2014) 59:483–95. doi: 10.1002/hep.26698
66. Lan HY, Chung ACK. Transforming growth factor-β and smads. Contrib Nephrol. (2011) 170:75–82. doi: 10.1159/000324949
67. Inagaki Y, Okazaki I. Emerging insights into transforming growth factor β smad signal in hepatic fibrogenesis. Gut. (2007) 56:284–92. doi: 10.1136/gut.2005.088690
68. Derynck R, Zhang YE. Smad-dependent and smad-independent pathways in TGF-beta family signalling. Nature. (2003) 425:577–84. doi: 10.1038/nature02006
70. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. (2010) 30:245–57. doi: 10.1055/s-0030-1255354
71. Borkham-Kamphorst E, van Roeyen CR, Ostendorf T, Floege J, Gressner AM, Weiskirchen R. Pro-fibrogenic potential of PDGF-D in liver fibrosis. J Hepatol. (2007) 46:1064–74. doi: 10.1016/j.jhep.2007.01.029
72. Lee JI, Wright JH, Johnson MM, Bauer RL, Sorg K, Yuen S, et al. Role of Smad3 in platelet-derived growth factor-C-induced liver fibrosis. Am J Physiol Cell Physiol. (2016) 310:C436–45. doi: 10.1152/ajpcell.00423.2014
73. Kostallari E, Hirsova P, Prasnicka A, Verma VK, Yaqoob U, Wongjarupong N, et al. Hepatic stellate cell-derived platelet-derived growth factor receptor-alpha-enriched extracellular vesicles promote liver fibrosis in mice through SHP2. Hepatology. (2018) 68:333–48. doi: 10.1002/hep.29803
74. Pinzani M, Milani S, Herbst H, DeFranco R, Grappone C, Gentilini A, et al. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am J Pathol. (1996) 148:785–800.
75. Czochra P, Klopcic B, Meyer E, Herkel J, Garcia-Lazaro JF, Thieringer F, et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J Hepatol. (2006) 45:419–28. doi: 10.1016/j.jhep.2006.04.010
76. Lim BJ, Lee WK, Lee HW, Lee KS, Kim JK, Chang HY, et al. Selective deletion of hepatocyte platelet-derived growth factor receptor α and development of liver fibrosis in mice. Cell Commun Signal. (2018) 16:93. doi: 10.1186/s12964-018-0306-2
77. Breitkopf K, Roeyen C, Sawitza I, Wickert L, Floege J, Gressner AM. Expression patterns of PDGF-A, -B, -C and -D and the PDGF-receptors alpha and beta in activated rat hepatic stellate cells (HSC). Cytokine. (2005) 31:349–57. doi: 10.1016/j.cyto.2005.06.005
78. Wong L, Yamasaki G, Johnson RJ, Friedman SL. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J Clin Invest. (1994) 94:1563–9. doi: 10.1172/JCI117497
79. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
80. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. (2014) 59:898–910. doi: 10.1002/hep.26592
81. Wang Q, Wei S, Zhou S, Qiu J, Shi C, Liu R, et al. Hyperglycemia aggravates acute liver injury by promoting liver-resident macrophage NLRP3 inflammasome activation via the inhibition of AMPK/mTOR-mediated autophagy induction. Immunol Cell Biol. (2020) 98:54–66. doi: 10.1111/imcb.12297
82. Pierantonelli I, Rychlicki C, Agostinelli L, Giordano DM, Gaggini M, Fraumene C, et al. Lack of NLRP3-inflammasome leads to gut-liver axis derangement, gut dysbiosis and a worsened phenotype in a mouse model of NAFLD. Sci Rep. (2017) 7:12200. doi: 10.1038/s41598-017-17187-3
83. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
84. Luan J, Ju D. Inflammasome: a double-edged sword in liver diseases. Front Immunol. (2018) 9:2201. doi: 10.3389/fimmu.2018.02201
85. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. (2017) 277:61–75. doi: 10.1111/imr.12534
86. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
87. Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P. The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur J Immunol. (2018) 48:584–92. doi: 10.1002/eji.201747404
88. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
89. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. (2012) 482:179–85. doi: 10.1038/nature10809
90. Sui YH, Luo WJ, Xu QY, Hua J. Dietary saturated fatty acid and polyunsaturated fatty acid oppositely affect hepatic NOD-like receptor protein 3 inflammasome through regulating nuclear factor-kappa B activation. World J Gastroenterol. (2016) 22:2533–44. doi: 10.3748/wjg.v22.i8.2533
91. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. (2011) 54:133–44. doi: 10.1002/hep.24341
92. Wan X, Xu C, Yu C, Li Y. Role of NLRP3 inflammasome in the progression of NAFLD to NASH. Can J Gastroenterol Hepatol. (2016) 2016:6489012. doi: 10.1155/2016/6489012
93. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
94. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. (2009) 17:9–26. doi: 10.1016/j.devcel.2009.06.016
95. Wang XM, Wang XY, Huang YM, Chen X, Lü MH, Shi L, et al. Role and mechanisms of action of microRNA-21 as regards the regulation of the WNT/β-catenin signaling pathway in the pathogenesis of non-alcoholic fatty liver disease. Int J Mol Med. (2019) 44:2201–12. doi: 10.3892/ijmm.2019.4375
96. Wang JN, Li L, Li LY, Yan Q, Li J, Xu T. Emerging role and therapeutic implication of Wnt signaling pathways in liver fibrosis. Gene. (2018) 674:57–69. doi: 10.1016/j.gene.2018.06.053
97. Wang S, Song K, Srivastava R, Dong C, Go GW, Li N, et al. Nonalcoholic fatty liver disease induced by noncanonical Wnt and its rescue by Wnt3a. Faseb J. (2015) 29:3436–45. doi: 10.1096/fj.15-271171
98. Hu H, Lin A, Kong M, Yao X, Yin M, Xia H, et al. Intestinal microbiome and NAFLD: molecular insights and therapeutic perspectives. J Gastroenterol. (2020) 55:142–58. doi: 10.1007/s00535-019-01649-8
99. Cho YE, Kim DK, Seo W, Gao B, Yoo SH, Song BJ. Fructose promotes leaky gut, endotoxemia, and liver fibrosis through ethanol-inducible cytochrome P450-2E1-mediated oxidative and nitrative stress. Hepatology. (2019). doi: 10.1002/hep.30652
100. Szabo G, Velayudham A, Romics L Jr, Mandrekar P. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: the role of toll-like receptors 2 and 4. Alcohol Clin Exp Res. (2005) 29 (11 Suppl):140s−5s. doi: 10.1097/01.alc.0000189287.83544.33
101. Schwenger KJ, Clermont-Dejean N, Allard JP. The role of the gut microbiome in chronic liver disease: the clinical evidence revised. JHEP Rep. (2019) 1:214–26. doi: 10.1016/j.jhepr.2019.04.004
102. Kapil S, Duseja A, Sharma BK, Singla B, Chakraborti A, Das A, et al. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J Gastroenterol Hepatol. (2016) 31:213–21. doi: 10.1111/jgh.13058
103. Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. (2016) 63:764–75. doi: 10.1002/hep.28356
104. Scorletti E, Afolabi PR, Miles EA, Smith DE, Almehmadi A, Alshathry A, et al. Design and rationale of the INSYTE study: a randomised, placebo controlled study to test the efficacy of a synbiotic on liver fat, disease biomarkers and intestinal microbiota in non-alcoholic fatty liver disease. Contemp Clin Trials. (2018) 71:113–23. doi: 10.1016/j.cct.2018.05.010
105. Namjou B, Lingren T, Huang Y, Parameswaran S, Cobb BL, Stanaway IB, et al. GWAS and enrichment analyses of non-alcoholic fatty liver disease identify new trait-associated genes and pathways across eMERGE Network. BMC Med. (2019) 17:135. doi: 10.1186/s12916-019-1364-z
106. Chalasani N, Guo X, Loomba R, Goodarzi MO, Haritunians T, Kwon S, et al. Genome-wide association study identifies variants associated with histologic features of nonalcoholic fatty liver disease. Gastroenterology. (2010) 139:1567–76:76.e1–6. doi: 10.1053/j.gastro.2010.07.057
107. Kitamoto T, Kitamoto A, Yoneda M, Hyogo H, Ochi H, Nakamura T, et al. Genome-wide scan revealed that polymorphisms in the PNPLA3, SAMM50, and PARVB genes are associated with development and progression of nonalcoholic fatty liver disease in Japan. Hum Genet. (2013) 132:783–92. doi: 10.1007/s00439-013-1294-3
108. Xia MF, Ling Y, Bian H, Lin HD, Yan HM, Chang XX, et al. I148M variant of PNPLA3 increases the susceptibility to non-alcoholic fatty liver disease caused by obesity and metabolic disorders. Aliment Pharmacol Ther. (2016) 43:631–42. doi: 10.1111/apt.13521
109. Santoro N, Kursawe R, D'Adamo E, Dykas DJ, Zhang CK, Bale AE, et al. A common variant in the patatin-like phospholipase 3 gene (PNPLA3) is associated with fatty liver disease in obese children and adolescents. Hepatology. (2010) 52:1281–90. doi: 10.1002/hep.23832
110. Anstee QM, Darlay R, Cockell S, Meroni M, Govaere O, Tiniakos D, et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort*. J Hepatol. (2020) 73:505–15. doi: 10.1016/j.jhep.2020.04.003
111. Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. (2012) 122:4130–44. doi: 10.1172/JCI65179
112. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg-Hansen A, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. (2014) 46:352–6. doi: 10.1038/ng.2901
113. Liu YL, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JB, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. (2014) 5:4309. doi: 10.1038/ncomms5309
114. Koo BK, Joo SK, Kim D, Bae JM, Park JH, Kim JH, et al. Additive effects of PNPLA3 and TM6SF2 on the histological severity of non-alcoholic fatty liver disease. J Gastroenterol Hepatol. (2018) 33:1277–85. doi: 10.1111/jgh.14056
115. Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. (2015) 149:389–97.e10. doi: 10.1053/j.gastro.2015.04.043
116. Vilar-Gomez E, Chalasani N. Non-invasive assessment of non-alcoholic fatty liver disease: clinical prediction rules and blood-based biomarkers. J Hepatol. (2018) 68:305–15. doi: 10.1016/j.jhep.2017.11.013
117. Francque S, Lanthier N, Verbeke L, Reynaert H, Van Steenkiste C, Vonghia L, et al. The Belgian association for study of the liver guidance document on the management of adult and paediatric non-alcoholic fatty liver disease. Acta Gastro Enterol Belgica. (2018) 81:55–81.
118. Lv S, Jiang S, Liu S, Dong Q, Xin Y, Xuan S. Noninvasive quantitative detection methods of liver fat content in nonalcoholic fatty liver disease. J Clin Transl Hepatol. (2018) 6:217–21. doi: 10.14218/JCTH.2018.00021
119. FDA. Noncirrhotic Nonalcoholic Steatohepatitis With Liver Fibrosis: Developing Drugs for Treatment: Guidance for Industry. FDA (2018).
120. European Medicine Agency. Draft Reflection Paper on Regulatory Requirements for the Development of Medicinal Products for Chronic Non-infectious Liver Diseases (PBC, PSC, NASH). European Medicine Agency (2018).
121. Goodman ZD. Grading and staging systems for inflammation and fibrosis in chronic liver diseases. J Hepatol. (2007) 47:598–607. doi: 10.1016/j.jhep.2007.07.006
122. Hjelkrem M, Stauch C, Shaw J, Harrison SA. Validation of the non-alcoholic fatty liver disease activity score. Aliment Pharmacol Ther. (2011) 34:214–8. doi: 10.1111/j.1365-2036.2011.04695.x
123. Nascimbeni F, Bedossa P, Fedchuk L, Pais R, Charlotte F, Lebray P, et al. Clinical validation of the FLIP algorithm and the SAF score in patients with non-alcoholic fatty liver disease. J Hepatol. (2020) 72:828–38. doi: 10.1016/j.jhep.2019.12.008
124. Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR cooperative study group. Hepatology. (1996) 24:289–93. doi: 10.1002/hep.510240201
125. Chengxi L, Rentao L, Wei Z. Progress in non-invasive detection of liver fibrosis. Cancer Biol Med. (2018) 15:124–36. doi: 10.20892/j.issn.2095-3941.2018.0018
126. Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, et al. Histological grading and staging of chronic hepatitis. J Hepatol. (1995) 22:696–9. doi: 10.1016/0168-8278(95)80226-6
127. Bedossa P, Arola J, Davies S, Gouw ASHm, Guido M, Lackner C editors. The EPoS Staging System is a Reproducible 7-tier Fibrosis Score for NAFLD Adapted Both to Glass Slides and Digitized Images (e-slides). Paris: European Assocation for the Study of the Liver; International Liver Congress (2018).
128. Sumida Y, Nakajima A, Itoh Y. Limitations of liver biopsy and non-invasive diagnostic tests for the diagnosis of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. (2014) 20:475–85. doi: 10.3748/wjg.v20.i2.475
129. Cadranel JF, Rufat P, Degos F. Practices of liver biopsy in France: results of a prospective nationwide survey. For the group of epidemiology of the French association for the study of the liver (AFEF). Hepatology. (2000) 32:477–81. doi: 10.1053/jhep.2000.16602
130. Bravo AA, Sheth SG, Chopra S. Liver biopsy. N Engl J Med. (2001) 344:495–500. doi: 10.1056/NEJM200102153440706
131. van der Poorten D, Kwok A, Lam T, Ridley L, Jones DB, Ngu MC, et al. Twenty-year audit of percutaneous liver biopsy in a major Australian teaching hospital. Intern Med J. (2006) 36:692–9. doi: 10.1111/j.1445-5994.2006.01216.x
132. Ratziu V, Charlotte F, Heurtier A, Gombert S, Giral P, Bruckert E, et al. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology. (2005) 128:1898–906. doi: 10.1053/j.gastro.2005.03.084
133. Younossi ZM, Gramlich T, Liu YC, Matteoni C, Petrelli M, Goldblum J, et al. Nonalcoholic fatty liver disease: assessment of variability in pathologic interpretations. Mod Pathol. (1998) 11:560–5. doi: 10.1016/S0016-5085(99)70506-8
134. Boursier J, Vergniol J, Sawadogo A, Dakka T, Michalak S, Gallois Y, et al. The combination of a blood test and fibroscan improves the non-invasive diagnosis of liver fibrosis. Liver Int. (2009) 29:1507–15. doi: 10.1111/j.1478-3231.2009.02101.x
135. Boursier J, Guillaume M, Leroy V, Irlès M, Roux M, Lannes A, et al. New sequential combinations of non-invasive fibrosis tests provide an accurate diagnosis of advanced fibrosis in NAFLD. J Hepatol. (2019) 71:389–96. doi: 10.1016/j.jhep.2019.04.020
136. Colletta C, Smirne C, Fabris C, Toniutto P, Rapetti R, Minisini R, et al. Value of two noninvasive methods to detect progression of fibrosis among HCV carriers with normal aminotransferases. Hepatology. (2005) 42:838–45. doi: 10.1002/hep.20814
137. Moreno-Otero R, Trapero-Marugan M, Mendoza J. Liver fibrosis assessment by transient elastography in hepatitis C patients with normal alanine aminotransferase. Gut. (2006) 55:1055–6.
138. Wilder J, Patel K. The clinical utility of FibroScan((R)) as a noninvasive diagnostic test for liver disease. Med Dev. (2014) 7:107–14. doi: 10.2147/MDER.S46943
139. Wong VW, Vergniol J, Wong GL, Foucher J, Chan HL, Le Bail B, et al. Diagnosis of fibrosis and cirrhosis using liver stiffness measurement in nonalcoholic fatty liver disease. Hepatology. (2010) 51:454–62. doi: 10.1002/hep.23312
140. Hsu C, Caussy C, Imajo K, Chen J, Singh S, Kaulback K, et al. Magnetic resonance vs transient elastography analysis of patients with nonalcoholic fatty liver disease: a systematic review and pooled analysis of individual participants. Clin Gastroenterol Hepatol. (2019) 17:630–7.e8. doi: 10.1016/j.cgh.2018.05.059
141. Liang Y, Li D. Magnetic resonance elastography in staging liver fibrosis in non-alcoholic fatty liver disease: a pooled analysis of the diagnostic accuracy. BMC Gastroenterol. (2020) 20:89. doi: 10.1186/s12876-020-01234-x
142. Jiang W, Huang S, Teng H, Wang P, Wu M, Zhou X, et al. Diagnostic accuracy of point shear wave elastography and transient elastography for staging hepatic fibrosis in patients with non-alcoholic fatty liver disease: a meta-analysis. BMJ Open. (2018) 8:e021787. doi: 10.1136/bmjopen-2018-021787
143. Peleg N, Issachar A, Sneh-Arbib O, Shlomai A. AST to Platelet Ratio Index and fibrosis 4 calculator scores for non-invasive assessment of hepatic fibrosis in patients with non-alcoholic fatty liver disease. Digest Liver Dis. (2017) 49:1133–8. doi: 10.1016/j.dld.2017.05.002
144. Xiao G, Zhu S, Xiao X, Yan L, Yang J, Wu G. Comparison of laboratory tests, ultrasound, or magnetic resonance elastography to detect fibrosis in patients with nonalcoholic fatty liver disease: a meta-analysis. Hepatology. (2017) 66:1486–501. doi: 10.1002/hep.29302
145. Vali Y, Lee J, Boursier J, Spijker R, Löffler J, Verheij J, et al. Enhanced liver fibrosis test for the non-invasive diagnosis of fibrosis in patients with NAFLD: a systematic review and meta-analysis. J Hepatol. (2020) 73:252–62. doi: 10.1016/j.jhep.2020.03.036
146. Boursier J, Vergniol J, Guillet A, Hiriart JB, Lannes A, Le Bail B, et al. Diagnostic accuracy and prognostic significance of blood fibrosis tests and liver stiffness measurement by FibroScan in non-alcoholic fatty liver disease. J Hepatol. (2016) 65:570–8. doi: 10.1016/j.jhep.2016.04.023
147. Boyle M, Tiniakos D, Schattenberg JM, Ratziu V, Bugianessi E, Petta S, et al. Performance of the PRO-C3 collagen neo-epitope biomarker in non-alcoholic fatty liver disease. JHEP Rep. (2019) 1:188–98. doi: 10.1016/j.jhepr.2019.06.004
148. Harrison SA, Ratziu V, Boursier J, Francque S, Bedossa P, Majd Z, et al. A blood-based biomarker panel (NIS4) for non-invasive diagnosis of non-alcoholic steatohepatitis and liver fibrosis: a prospective derivation and global validation study. Lancet Gastroenterol Hepatol. (2020) 5:970–85. doi: 10.1016/S2468-1253(20)30252-1
149. Millonig G, Friedrich S, Adolf S, Fonouni H, Golriz M, Mehrabi A, et al. Liver stiffness is directly influenced by central venous pressure. J Hepatol. (2010) 52:206–10. doi: 10.1016/j.jhep.2009.11.018
150. Myers RP, Pomier-Layrargues G, Kirsch R, Pollett A, Beaton M, Levstik M, et al. Discordance in fibrosis staging between liver biopsy and transient elastography using the FibroScan XL probe. J Hepatol. (2012) 56:564–70. doi: 10.1016/j.jhep.2011.10.007
151. Verlinden W. Real-Time 2D Shear Wave Elastography: Applications and Confounding Factors. Chapter 2: The Effect of Patient Position and Fluid State on Liver and Spleen Stiffness and the Reproducibility of Liver Stiffness Measurements by Real-Time 2D Shear Wave Elastography. [PhD], Antwerp University, Antwerp (2016)
152. de Ledinghen V, Wong VW, Vergniol J, Wong GL, Foucher J, Chu SH, et al. Diagnosis of liver fibrosis and cirrhosis using liver stiffness measurement: comparison between M and XL probe of FibroScan(R). J Hepatol. (2012) 56:833–9. doi: 10.1016/j.jhep.2011.10.017
153. Wong VW, Vergniol J, Wong GL, Foucher J, Chan AW, Chermak F, et al. Liver stiffness measurement using XL probe in patients with nonalcoholic fatty liver disease. Am J Gastroenterol. (2012) 107:1862–71. doi: 10.1038/ajg.2012.331
154. Tsai E, Lee TP. Diagnosis and evaluation of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis, including noninvasive biomarkers and transient elastography. Clin Liver Dis. (2018) 22:73–92. doi: 10.1016/j.cld.2017.08.004
155. Verlinden W, Francque S, Michielsen P, Vanwolleghem T. Successful antiviral treatment of chronic hepatitis C leads to a rapid decline of liver stiffness without an early effect on spleen stiffness. Hepatology. (2016) 64:1809–10. doi: 10.1002/hep.28610
156. Castera L, Foucher J, Bernard PH, Carvalho F, Allaix D, Merrouche W, et al. Pitfalls of liver stiffness measurement: a 5-year prospective study of 13,369 examinations. Hepatology. (2010) 51:828–35. doi: 10.1002/hep.23425
157. Roulot D, Czernichow S, Le Clesiau H, Costes JL, Vergnaud AC, Beaugrand M. Liver stiffness values in apparently healthy subjects: influence of gender and metabolic syndrome. J Hepatol. (2008) 48:606–13. doi: 10.1016/j.jhep.2007.11.020
158. Vuppalanchi R, Siddiqui MS, Van Natta ML, Hallinan E, Brandman D, Kowdley K, et al. Performance characteristics of vibration-controlled transient elastography for evaluation of nonalcoholic fatty liver disease. Hepatology. (2018) 67:134–44. doi: 10.1002/hep.29489
159. Vuppalanchi R, Weber R, Russell S, Gawrieh S, Samala N, Slaven JE, et al. Is fasting necessary for individuals with nonalcoholic fatty liver disease to undergo vibration-controlled transient elastography? Am J Gastroenterol. (2019) 114:995–7. doi: 10.14309/ajg.0000000000000116
160. Gaia S, Carenzi S, Barilli AL, Bugianesi E, Smedile A, Brunello F, et al. Reliability of transient elastography for the detection of fibrosis in non-alcoholic fatty liver disease and chronic viral hepatitis. J Hepatol. (2011) 54:64–71. doi: 10.1016/j.jhep.2010.06.022
161. Sasso M, Miette V, Sandrin L, Beaugrand M. The controlled attenuation parameter (CAP): a novel tool for the non-invasive evaluation of steatosis using fibroscan. Clin Res Hepatol Gastroenterol. (2012) 36:13–20. doi: 10.1016/j.clinre.2011.08.001
162. Baumeler S, Jochum W, Neuweiler J, Bergamin I, Semela D. Controlled attenuation parameter for the assessment of liver steatosis in comparison with liver histology: a single-centre real life experience. Swiss Med Wkly. (2019) 149:w20077. doi: 10.4414/smw.2019.20077
163. Sigrist RMS, Liau J, Kaffas AE, Chammas MC, Willmann JK. Ultrasound elastography: review of techniques and clinical applications. Theranostics. (2017) 7:1303–29. doi: 10.7150/thno.18650
164. Ophir J, Céspedes I, Ponnekanti H, Yazdi Y, Li X. Elastography: a quantitative method for imaging the elasticity of biological tissues. Ultrason Imaging. (1991) 13:111–34. doi: 10.1177/016173469101300201
165. Garra BS. Elastography: history, principles, and technique comparison. Abdom Imaging. (2015) 40:680–97. doi: 10.1007/s00261-014-0305-8
166. Tapper EB, Lok AS. Use of liver imaging and biopsy in clinical practice. N Engl J Med. (2017) 377:756–68. doi: 10.1056/NEJMra1610570
167. Nightingale K. Acoustic radiation force impulse (ARFI) imaging: a review. Curr Med Imaging Rev. (2011) 7:328–39. doi: 10.2174/157340511798038657
168. Honda Y, Yoneda M, Imajo K, Nakajima A. Elastography techniques for the assessment of liver fibrosis in non-alcoholic fatty liver disease. Int J Mol Sci. (2020) 21:4039. doi: 10.3390/ijms21114039
169. Han A, Labyed Y, Sy EZ, Boehringer AS, Andre MP, Erdman JW, et al. Inter-sonographer reproducibility of quantitative ultrasound outcomes and shear wave speed measured in the right lobe of the liver in adults with known or suspected non-alcoholic fatty liver disease. Eur Radiol. (2018) 28:4992–5000. doi: 10.1007/s00330-018-5541-9
170. Ferraioli G, Tinelli C, Lissandrin R, Zicchetti M, Bernuzzi S, Salvaneschi L, et al. Ultrasound point shear wave elastography assessment of liver and spleen stiffness: effect of training on repeatability of measurements. Eur Radiol. (2014) 24:1283–9. doi: 10.1007/s00330-014-3140-y
171. Leong WL, Lai LL, Nik Mustapha NR, Vijayananthan A, Rahmat K, Mahadeva S, et al. Comparing point shear wave elastography (ElastPQ) and transient elastography for diagnosis of fibrosis stage in non-alcoholic fatty liver disease. J Gastroenterol Hepatol. (2020) 35:135–41. doi: 10.1111/jgh.14782
172. Imajo K, Kessoku T, Honda Y, Tomeno W, Ogawa Y, Mawatari H, et al. Magnetic resonance imaging more accurately classifies steatosis and fibrosis in patients with nonalcoholic fatty liver disease than transient elastography. Gastroenterology. (2016) 150:626–37.e7. doi: 10.1053/j.gastro.2015.11.048
173. Guha IN, Parkes J, Roderick P, Chattopadhyay D, Cross R, Harris S, et al. Noninvasive markers of fibrosis in nonalcoholic fatty liver disease: validating the European liver fibrosis panel and exploring simple markers. Hepatology. (2008) 47:455–60. doi: 10.1002/hep.21984
174. Rosenberg WM, Voelker M, Thiel R, Becka M, Burt A, Schuppan D, et al. Serum markers detect the presence of liver fibrosis: a cohort study. Gastroenterology. (2004) 127:1704–13. doi: 10.1053/j.gastro.2004.08.052
175. Guillaume M, Moal V, Delabaudiere C, Zuberbuhler F, Robic MA, Lannes A, et al. Direct comparison of the specialised blood fibrosis tests FibroMeter(V2G) and enhanced liver fibrosis score in patients with non-alcoholic fatty liver disease from tertiary care centres. Aliment Pharmacol Ther. (2019) 50:1214–22. doi: 10.1111/apt.15529
176. Leeming D, Grove J, Kaye P, Hoad C, Francis S, Nielsen M, et al. Estimation of serum “true collagen type III formation”(Pro-C3) levels as a marker of non-alcoholic steatohepatitis in a prospective cohort. J Hepatol. (2017) 66:S154. doi: 10.1016/S0168-8278(17)30582-2
177. Luo Y, Oseini A, Gagnon R, Charles ED, Sidik K, Vincent R, et al. An evaluation of the collagen fragments related to fibrogenesis and fibrolysis in nonalcoholic steatohepatitis. Sci Rep. (2018) 8:12414. doi: 10.1038/s41598-018-30457-y
178. Karsdal MA, Henriksen K, Nielsen MJ, Byrjalsen I, Leeming DJ, Gardner S, et al. Fibrogenesis assessed by serological type III collagen formation identifies patients with progressive liver fibrosis and responders to a potential antifibrotic therapy. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G1009–17. doi: 10.1152/ajpgi.00283.2016
179. Cermelli S, Ruggieri A, Marrero JA, Ioannou GN, Beretta L. Circulating MicroRNAs in patients with chronic hepatitis c and non-alcoholic fatty liver disease. PLoS ONE. (2011) 6:e23937. doi: 10.1371/journal.pone.0023937
180. Lin B, Ma Y, Wu S, Liu Y, Liu L, Wu L. Novel serum biomarkers for noninvasive diagnosis and screening of nonalcoholic fatty liver disease-related hepatic fibrosis. Omics. (2019) 23:181–9. doi: 10.1089/omi.2019.0035
181. Johansen JS, Christoffersen P, Møller S, Price PA, Henriksen JH, Garbarsch C, et al. Serum YKL-40 is increased in patients with hepatic fibrosis. J Hepatol. (2000) 32:911–20. doi: 10.1016/S0168-8278(00)80095-1
182. Kumagai E, Mano Y, Yoshio S, Shoji H, Sugiyama M, Korenaga M, et al. Serum YKL-40 as a marker of liver fibrosis in patients with non-alcoholic fatty liver disease. Sci Rep. (2016) 6:35282. doi: 10.1038/srep35282
183. Yamada H, Suzuki K, Ichino N, Ando Y, Sawada A, Osakabe K, et al. Associations between circulating microRNAs (miR-21, miR-34a, miR-122 and miR-451) and non-alcoholic fatty liver. Clin Chim Acta. (2013) 424:99–103. doi: 10.1016/j.cca.2013.05.021
184. Shirabe K, Bekki Y, Gantumur D, Araki K, Ishii N, Kuno A, et al. Mac-2 binding protein glycan isomer (M2BPGi) is a new serum biomarker for assessing liver fibrosis: more than a biomarker of liver fibrosis. J Gastroenterol. (2018) 53:819–26. doi: 10.1007/s00535-017-1425-z
185. Jang SY, Tak WY, Park SY, Kweon YO, Lee YR, Kim G, et al. Diagnostic efficacy of serum Mac-2 binding protein glycosylation isomer and other markers for liver fibrosis in non-alcoholic fatty liver diseases. Ann Lab Med. (2021) 41:302–9. doi: 10.3343/alm.2021.41.3.302
186. Nah EH, Cho S, Kim S, Kim HS, Cho HI. Diagnostic performance of Mac-2 binding protein glycosylation isomer (M2BPGi) in screening liver fibrosis in health checkups. J Clin Lab Anal. (2020) 34:e23316. doi: 10.1002/jcla.23316
187. Honda Y, Imajo K, Kobayashi T, Kessoku T, Ogawa Y, Tomeno W, et al. Autotaxin is a valuable biomarker for the prediction of liver fibrosis in patients with non-alcoholic fatty liver disease. Hepatol Res. (2019) 49:1136–46. doi: 10.1111/hepr.13382
188. Fujimori N, Umemura T, Kimura T, Tanaka N, Sugiura A, Yamazaki T, et al. Serum autotaxin levels are correlated with hepatic fibrosis and ballooning in patients with non-alcoholic fatty liver disease. World J Gastroenterol. (2018) 24:1239–49. doi: 10.3748/wjg.v24.i11.1239
189. Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, et al. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J Biol Chem. (2002) 277:39436–42. doi: 10.1074/jbc.M205623200
190. Moolenaar WH. Lysophospholipids in the limelight: autotaxin takes center stage. J Cell Biol. (2002) 158:197–9. doi: 10.1083/jcb.200206094
191. Moolenaar WH, van Meeteren LA, Giepmans BN. The ins and outs of lysophosphatidic acid signaling. Bioessays. (2004) 26:870–81. doi: 10.1002/bies.20081
192. Kimura T, Tanaka N, Fujimori N, Yamazaki T, Katsuyama T, Iwashita Y, et al. Serum thrombospondin 2 is a novel predictor for the severity in the patients with NAFLD. Liver Int. (2021) 41:505–14. doi: 10.1111/liv.14776
193. Ogawa Y, Honda Y, Kessoku T, Tomeno W, Imajo K, Yoneda M, et al. Wisteria floribunda agglutinin-positive Mac-2-binding protein and type 4 collagen 7S: useful markers for the diagnosis of significant fibrosis in patients with non-alcoholic fatty liver disease. J Gastroenterol Hepatol. (2018) 33:1795–803. doi: 10.1111/jgh.14156
194. Caussy C, Ajmera VH, Puri P, Hsu CL, Bassirian S, Mgdsyan M, et al. Serum metabolites detect the presence of advanced fibrosis in derivation and validation cohorts of patients with non-alcoholic fatty liver disease. Gut. (2019) 68:1884–92. doi: 10.1136/gutjnl-2018-317584
195. Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. (2009) 81:6656–67. doi: 10.1021/ac901536h
196. Poo JL, Torre A, Aguilar-Ramírez JR, Cruz M, Mejía-Cuán L, Cerda E, et al. Benefits of prolonged-release pirfenidone plus standard of care treatment in patients with advanced liver fibrosis: PROMETEO study. Hepatol Int. (2020) 14:817–7. doi: 10.1007/s12072-020-10069-3
197. Srivastava A, Gailer R, Tanwar S, Trembling P, Parkes J, Rodger A, et al. Prospective evaluation of a primary care referral pathway for patients with non-alcoholic fatty liver disease. J Hepatol. (2019) 71:371–8. doi: 10.1016/j.jhep.2019.03.033
198. Davyduke T, Tandon P, Al-Karaghouli M, Abraldes JG, Ma MM. Impact of implementing a “FIB-4 first” strategy on a pathway for patients with NAFLD referred from primary care. Hepatol Commun. (2019) 3:1322–33. doi: 10.1002/hep4.1411
199. Srivastava A, Jong S, Gola A, Gailer R, Morgan S, Sennett K, et al. Cost-comparison analysis of FIB-4, ELF and fibroscan in community pathways for non-alcoholic fatty liver disease. BMC Gastroenterol. (2019) 19:122. doi: 10.1186/s12876-019-1039-4
200. Broussier T, Lannes A, Zuberbuhler F, Oberti F, Fouchard I, Hunault G, et al. Simple blood fibrosis tests reduce unnecessary referrals for specialized evaluations of liver fibrosis in NAFLD and ALD patients. Clin Res Hepatol Gastroenterol. (2020) 44:349–55. doi: 10.1016/j.clinre.2019.07.010
201. Anstee QM, Lawitz EJ, Alkhouri N, Wong VW, Romero-Gomez M, Okanoue T, et al. Noninvasive tests accurately identify advanced fibrosis due to NASH: baseline data from the STELLAR trials. Hepatology. (2019) 70:1521–30. doi: 10.1002/hep.30842
202. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. (2018) 67:1754–67. doi: 10.1002/hep.29477
203. Leoni S, Tovoli F, Napoli L, Serio I, Ferri S, Bolondi L. Current guidelines for the management of non-alcoholic fatty liver disease: a systematic review with comparative analysis. World J Gastroenterol. (2018) 24:3361–73. doi: 10.3748/wjg.v24.i30.3361
204. Snyder HS, Sakaan SA, March KL, Siddique O, Cholankeril R, Cummings CD, et al. Non-alcoholic fatty liver disease: a review of anti-diabetic pharmacologic therapies. J Clin Transl Hepatol. (2018) 6:168–74. doi: 10.14218/JCTH.2017.00050
205. Shimizu M, Suzuki K, Kato K, Jojima T, Iijima T, Murohisa T, et al. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes Obes Metab. (2019) 21:285–92. doi: 10.1111/dom.13520
206. Panebianco C, Oben JA, Vinciguerra M, Pazienza V. Senescence in hepatic stellate cells as a mechanism of liver fibrosis reversal: a putative synergy between retinoic acid and PPAR-gamma signalings. Clin Exp Med. (2017) 17:269–80. doi: 10.1007/s10238-016-0438-x
207. Wang Z, Xu J-P, Zheng Y-C, Chen W, Sun Y-W, Wu Z-Y, et al. Peroxisome proliferator-activated receptor gamma inhibits hepatic fibrosis in rats. Hepatobil Pancreat Dis Int. (2011) 10:64–71. doi: 10.1016/S1499-3872(11)60009-X
208. Cholankeril R, Patel V, Perumpail BJ, Yoo ER, Iqbal U, Sallam S, et al. Anti-Diabetic medications for the pharmacologic management of NAFLD. Diseases. (2018) 6:93. doi: 10.3390/diseases6040093
209. Sumida Y, Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterol. (2018) 53:362–76. doi: 10.1007/s00535-017-1415-1
210. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. (2010) 362:1675–85. doi: 10.1056/NEJMoa0907929
211. Mills EP, Brown KPD, Smith JD, Vang PW, Trotta K. Treating nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus: a review of efficacy and safety. Ther Adv Endocrinol Metab. (2018) 9:15–28. doi: 10.1177/2042018817741852
212. Fan H, Pan Q, Xu Y, Yang X. Exenatide improves type 2 diabetes concomitant with non-alcoholic fatty liver disease. Arq Bras Endocrinol Metab. (2013) 57:702–8. doi: 10.1590/S0004-27302013000900005
213. Naftalin RJ. A computer model simulating human glucose absorption and metabolism in health and metabolic disease states. F1000Research. (2016) 5:647. doi: 10.12688/f1000research.8299.1
214. Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. (2016) 387:679–90. doi: 10.1016/S0140-6736(15)00803-X
215. Newsome P, Francque S, Harrison S, Ratziu V, Van Gaal L, Calanna S, et al. Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment Pharmacol Ther. (2019) 50:193–203. doi: 10.1111/apt.15316
216. Legry VPP, Stankovic-Valentin N, Parroche P, Descamps E, Belanger C, Hum D editor. The combination of elafibranor and semaglutide drastically improves fibrosing steatohepatitis and distinctly modulates liver inflammatory signature. in International Liver Conference. London (2020). doi: 10.1016/S0168-8278(20)30584-5
217. Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. (2021) 384:1113–24. doi: 10.1056/NEJMoa2028395
218. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, et al. Long-Term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med. (2016) 165:305–15. doi: 10.7326/M15-1774
219. Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. (2008) 135:1176–84. doi: 10.1053/j.gastro.2008.06.047
220. Tahrani AA, Barnett AH, Bailey CJ. SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol. (2013) 1:140–51. doi: 10.1016/S2213-8587(13)70050-0
221. Tahara A, Takasu T. SGLT2 inhibitor ipragliflozin alone and combined with pioglitazone prevents progression of nonalcoholic steatohepatitis in a type 2 diabetes rodent model. Physiol Rep. (2019) 7:e14286. doi: 10.14814/phy2.14286
222. Kuchay MS, Krishan S, Mishra SK, Farooqui KJ, Singh MK, Wasir JS, et al. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: a randomized controlled trial (E-LIFT Trial). Diabetes Care. (2018) 41:1801–8. doi: 10.2337/dc18-0165
223. Mantovani A, Petracca G, Csermely A, Beatrice G, Targher G. Sodium-Glucose cotransporter-2 inhibitors for treatment of nonalcoholic fatty liver disease: a meta-analysis of randomized controlled trials. Metabolites. (2020) 11:22. doi: 10.3390/metabo11010022
224. Akuta N, Kawamura Y, Fujiyama S, Sezaki H, Hosaka T, Kobayashi M, et al. SGLT2 inhibitor treatment outcome in nonalcoholic fatty liver disease complicated with diabetes mellitus: the long-term effects on clinical features and liver histopathology. Intern Med. (2020) 59:1931–7. doi: 10.2169/internalmedicine.4398-19
225. Abenavoli L, Falalyeyeva T, Boccuto L, Tsyryuk O, Kobyliak N. Obeticholic acid: a new era in the treatment of nonalcoholic fatty liver disease. Pharmaceuticals. (2018) 11:104. doi: 10.3390/ph11040104
226. Han CY, Rho HS, Kim A, Kim TH, Jang K, Jun DW, et al. FXR Inhibits endoplasmic reticulum stress-induced NLRP3 inflammasome in hepatocytes and ameliorates liver injury. Cell Rep. (2018) 24:2985–99. doi: 10.1016/j.celrep.2018.07.068
227. Younossi Z, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Positive results from REGENERATE: a phase 3 international, randomized, placebo-controlled study evaluating obeticholic acid treatment for NASH: 938. Am J Gastroenterol. (2019) 114:S546. doi: 10.1136/gutjnl-2019-BSGAbstracts.205
228. Wettstein G, Luccarini JM, Poekes L, Faye P, Kupkowski F, Adarbes V, et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol Commun. (2017) 1:524–37. doi: 10.1002/hep4.1057
229. Boubia B, Poupardin O, Barth M, Binet J, Peralba P, Mounier L, et al. Design, synthesis, and evaluation of a novel series of indole sulfonamide peroxisome proliferator activated receptor (PPAR) alpha/gamma/delta triple activators: discovery of lanifibranor, a new antifibrotic clinical candidate. J Med Chem. (2018) 61:2246–65. doi: 10.1021/acs.jmedchem.7b01285
230. Francque SBP, Abdelmalek MF, Anstee QM, Bugianesi E, Ratziu V, Huot-Marchand P, et al. A randomised, double-blind, placebo-controlled, multi-centre, dose-range, proof-of-concept, 24-week treatment study of lanifibranor in adult subjects with non-alcoholic steatohepatitis: design of the NATIVE study. Contemp Clin Trials. (2020) 98:106170. doi: 10.1016/j.cct.2020.106170
231. Francque S, Bedossa P, Ratziu V, Anstee Q, Bugianesi E, Lanthier N. The panPPAR agonist lanifibranor results in highly significant improvements in both resolution of NASH and regression of fibrosis at 24 weeks in non-cirrhotic NASH: results of the NATIVE Phase 2b trial. Hepatology. (2020).
232. Tacke F. Cenicriviroc for the treatment of non-alcoholic steatohepatitis and liver fibrosis. Exp Opin Invest Drugs. (2018) 27:301–11. doi: 10.1080/13543784.2018.1442436
233. Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z, et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b CENTAUR study. Hepatology. (2020) 72:892–905. doi: 10.1002/hep.31108
234. Anstee QM, Neuschwander-Tetri BA, Wong VW, Abdelmalek MF, Younossi ZM, Yuan J, et al. Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: AURORA Phase 3 study design. Contemp Clin Trials. (2020) 89:105922. doi: 10.1016/j.cct.2019.105922
235. Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: a randomized, phase 2 trial. Hepatology. (2017) 67:549–59. doi: 10.1002/hep.29514
236. Harrison SA, Wong VW, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J Hepatol. (2020) 73:26–39. doi: 10.1016/j.jhep.2020.02.027
237. Harrison SA, Abdelmalek MF, Caldwell S, Shiffman ML, Diehl AM, Ghalib R, et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology. (2018) 155:1140–53. doi: 10.1053/j.gastro.2018.07.006
238. Harrison SA, Dennis A, Fiore MM, Kelly MD, Kelly CJ, Paredes AH, et al. Utility and variability of three non-invasive liver fibrosis imaging modalities to evaluate efficacy of GR-MD-02 in subjects with NASH and bridging fibrosis during a phase-2 randomized clinical trial. PLoS ONE. (2018) 13:e0203054. doi: 10.1371/journal.pone.0203054
239. Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, et al. Effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology. (2020) 158:1334–45.e5. doi: 10.1053/j.gastro.2019.11.296
240. AASLD. RESOLVE-IT phase 3 trial of elafibranor in NASH: Final results of the week 72 interim surrogate efficacy analysis. Hepatology. (2020).
241. Ratziu V, Friedman SL. Why do so many NASH trials fail? Gastroenterology. (2020). doi: 10.1053/j.gastro.2020.05.046
242. Lemoinne S, Friedman SL. New and emerging anti-fibrotic therapeutics entering or already in clinical trials in chronic liver diseases. Curr Opin Pharmacol. (2019) 49:60–70. doi: 10.1016/j.coph.2019.09.006
243. Lee NY, Yoon SJ, Han DH, Gupta H, Youn GS, Shin MJ, et al. Lactobacillus and Pediococcus ameliorate progression of non-alcoholic fatty liver disease through modulation of the gut microbiome. Gut Microbes. (2020) 11:882–99. doi: 10.1080/19490976.2020.1712984
244. Cavallari JF, Pokrajac NT, Zlitni S, Foley KP, Henriksbo BD, Schertzer JD. NOD2 in hepatocytes engages a liver-gut axis to protect against steatosis, fibrosis, and gut dysbiosis during fatty liver disease in mice. Am J Physiol Endocrinol Metab. (2020) 319:E305–14. doi: 10.1152/ajpendo.00181.2020
245. Borkham-Kamphorst E, Weiskirchen R. The PDGF system and its antagonists in liver fibrosis. Cytok Growth Factor Rev. (2016) 28:53–61. doi: 10.1016/j.cytogfr.2015.10.002
246. Kuo WL, Yu MC, Lee JF, Tsai CN, Chen TC, Chen MF. Imatinib mesylate improves liver regeneration and attenuates liver fibrogenesis in CCL4-treated mice. J Gastrointest Surg. (2012) 16:361–9. doi: 10.1007/s11605-011-1764-7
Keywords: NAFLD, liver fibrosis, liver biopsy, non-invasive assessment, liver stiffness
Citation: Heyens LJM, Busschots D, Koek GH, Robaeys G and Francque S (2021) Liver Fibrosis in Non-alcoholic Fatty Liver Disease: From Liver Biopsy to Non-invasive Biomarkers in Diagnosis and Treatment. Front. Med. 8:615978. doi: 10.3389/fmed.2021.615978
Received: 10 October 2020; Accepted: 22 March 2021;
Published: 14 April 2021.
Edited by:
Peter Olinga, University of Groningen, NetherlandsReviewed by:
Ludovico Abenavoli, University of Catanzaro, ItalyTakefumi Kimura, National Institutes of Health (NIH), United States
Copyright © 2021 Heyens, Busschots, Koek, Robaeys and Francque. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sven Francque, c3Zlbi5mcmFuY3F1ZUB1emEuYmU=