 Dominic Stanculescu
Dominic Stanculescu Lars Larsson
Lars Larsson Jonas Bergquist
Jonas Bergquist- 1Independent Researcher, Sint Martens Latem, Belgium
- 2Basic and Clinical Muscle Biology, Department of Physiology and Pharmacology, Karolinska Institute, Solna, Sweden
- 3Analytical Chemistry and Neurochemistry, Department of Chemistry – Biomedical Center, Uppsala University, Uppsala, Sweden
- 4The Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) Collaborative Research Centre at Uppsala University, Uppsala, Sweden
Here the hypothesis is advanced that maladaptive mechanisms that prevent recovery in some intensive care unit (ICU) patients may also underlie Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Specifically, these mechanisms are: (a) suppression of the pituitary gland's pulsatile secretion of tropic hormones, and (b) a “vicious circle” between inflammation, oxidative and nitrosative stress (O&NS), and low thyroid hormone function. This hypothesis should be investigated through collaborative research projects.
Introduction
Critical illness refers to the physiological response to virtually any severe injury or infection, such as sepsis, liver disease, HIV infection, head injury, pancreatitis, burns, cardiac surgery, etc. (1). Researchers make a distinction between the acute phase of critical illness—in the first hours or days following severe trauma or infection; and the chronic or prolonged phase—in the case of patients that survive the acute phase but for unknown reasons do not start recovering and continue to require intensive care (i.e., “chronic ICU patients”). Independent of the nature of the critical illness, the acute phase is associated with an excessive response of pro-inflammatory cytokines (2) and is characterized by a uniform dysregulation of the endocrine axes (3). In prolonged critical illness, this dysregulation is maintained even once the initial inflammatory surge has settled (4). Regardless of the initial injury or infection, patients that suffer from prolonged critical illness experience profound muscular weakness, cognitive impairment, loss of lean body mass, pain, increased vulnerability to infection, skin breakdown, etc. (1, 5, 6). Whereas, the acute phase is considered to be an adaptive response to the severe stress of injury or infection (shifting energy and resources to essential organs and repair), the physiological mechanisms in the prolonged phase are now increasingly considered to be maladaptive responses to the stress of severe injury or infection, hindering recovery (7–10). Some have also suggested that the non-recovery from endocrine disturbances could explain the development of “post-intensive care syndrome” (PICS) (11); i.e., “the cognitive, psychiatric and/or physical disability after treatment in ICUs” (12, 13).
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a debilitating, multi-system disease of unclear etiology (14, 15). The most common peri-onset events reported by patients are infection-related episodes (64%), stressful incidents (39%), and exposure to environmental toxins (20%) (16). “Impaired function, post-exertional malaise (an exacerbation of some or all of an individual's ME/CFS symptoms after physical or cognitive exertion, or orthostatic stress that leads to a reduction in functional ability), and unrefreshing sleep” are considered to be core symptoms (14). The severity of the symptoms varies: “very severely affected patients experience profound weakness, almost constant pain, severe limitations to physical and mental activity, sensory hypersensitivity (light, touch, sound, smell, and certain foods), and hypersensitivity to medications” (17). We have listed a few hall mark symptoms that are often found in critically ill patients in chronic intensive care (ICU) patients and ME/CFS patients (Table 1).
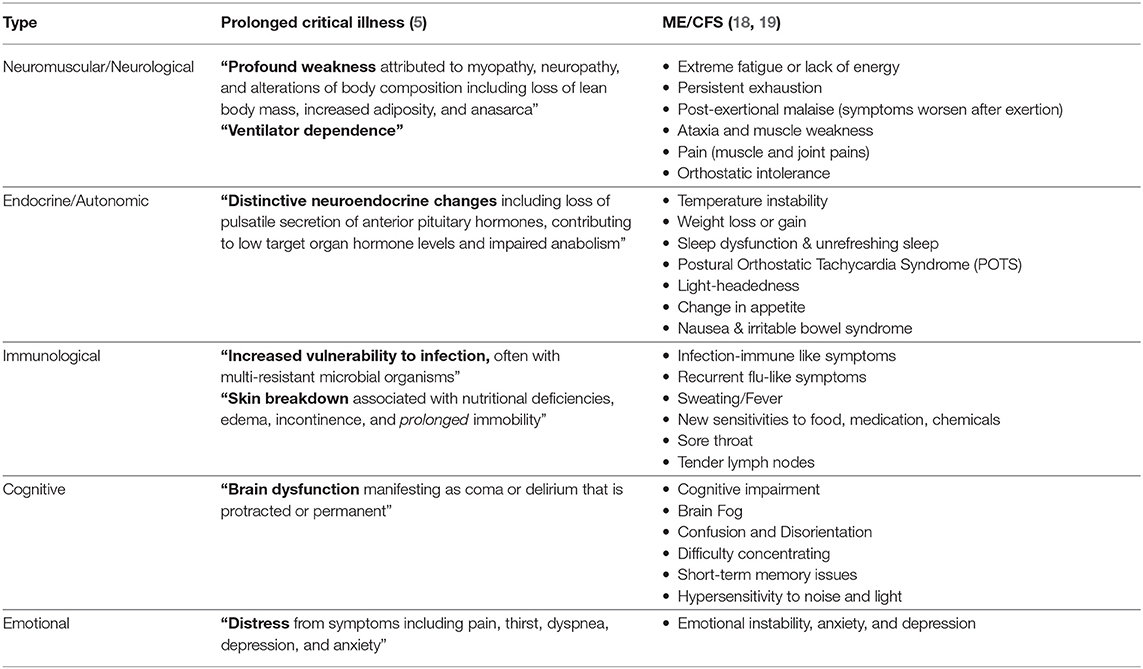
Table 1. Comparison of the typical clinical picture of ICU patients and patients with ME/CFS.
Here the hypothesis is advanced that maladaptive mechanisms that prevent recovery in some ICU patients also underlie ME/CFS. Specifically, these mechanisms are: (a) suppression of the pituitary gland's pulsatile secretion of tropic hormones, and (b) a “vicious circle” between inflammation, oxidative and nitrosative stress (O&NS), and low thyroid hormone function. These mechanisms characterize prolonged critical illness regardless of the nature of the initial severe injury or infection (3, 8–10); similarly, we propose that these mechanisms could underlie the perpetuation of illness in ME/CFS regardless of the nature of the peri-onset event (i.e., infection, stressful incident, exposure to environmental toxins, or other). We provide an overview of these mechanisms in ICU patients and discuss their relevance for understanding ME/CFS. We also bring findings from fibromyalgia into the discussion here because ME/CFS and fibromyalgia are often jointly considered in the literature (20, 21); fibromyalgia is similarly a syndrome that is medically unexplained, often comorbid with ME/CFS, and “shares the core symptoms of fatigue, sleep problems and cognitive difficulties” (22). Additional research projects are required to investigate the validity of this hypothesis building on the findings from critical illness and ME/CFS summarized here.
This hypothesis may be particularly relevant in light of the current COVID-19 pandemic. Many COVID-19 patients continue to experience a variety of debilitating symptoms despite successfully defeating the virus—termed “post COVID-19 syndrome” or “long COVID-19”—that resemble ME/CFS (23–26).
Suppression of Pulsatile Pituitary Secretions
Endocrine patterns observed during the initial acute phase of critical illness (in the first few hours or days) differ markedly from those observed during prolonged critical illness (after a few days) (27, 28). Indeed, the acute phase is characterized by increased release of pituitary hormones; the prolonged phase is characterized by suppression of the release of pituitary hormones. Simultaneously, hormone half-life and hormone up-take by the peripheral tissues differ markedly between these two phases (4, 29). This biphasic pattern of the endocrine system during critical illness, however, is not readily observable in single or average measurements of circulating tropic and non-tropic hormone concentrations—which are a function of both hormone release and elimination from the blood stream. This pattern was thus only discovered in the early 1990s with measurements of the frequency and amplitude of pituitary secretions (i.e., pulsatility) performed as often as every 10 min over 24 h on ICU patients (29). The pulsatility of tropic hormone secretion is part of the signaling to the peripheral glands and thus considered a determining factor of hormone function (i.e., impact on target glands or tissues), in addition to overall volume of hormone release (30, 31). The finding that pulsatile pituitary secretions are suppressed during prolonged critical illness was critical in understanding the physiology of the syndrome and the curious failure of patients to recover (32). We describe the biphasic endocrine patterns during acute and prolonged critical illness for each of the main endocrine axes in further detail below, as well as the implications for the autonomic nervous system, metabolism and the immune system. We also provide evidence suggesting that the endocrine patterns observed in prolonged critical illness also underlie ME/CFS.
The Adreno-Cortical Axis (HPA Axis)
The adreno-cortical axis—also called hypothalamic-pituitary-adrenal (HPA) axis—is the body's primary stress management system. The HPA axis responds to physical and mental challenges in part by controlling the body's glucocorticoids levels, notably cortisol (33). Cortisol in turn modulates inflammation response, cardiovascular function and glucose metabolism (34). An inability to deal with stress, proneness to exaggerated immune responses and weight loss are associated with hypocortolism or poor HPA axis function (35–38). The HPA axis also regulates mineralocorticoids that, in turn, regulate water and electrolyte balance (i.e., blood pressure). Low blood pressure and dizziness upon standing up are associated with a compromised HPA axis (35). Finally, the HPA axis (in addition to the gonadotropic axis not covered here) also contributes to the production of androgens, notably DHEA and testosterone, which are steroids that impact muscle mass, fat storage, pain, brain function and many other physiological traits. Low androgens are associated with muscle fatigue, joint pain, and noise intolerance (39–42).
In normal conditions, the adrenal gland secretes cortisol during the day in pulses, with the highest amounts in the early morning hours and lower amounts at night. The hypothalamus signals to the pituitary with corticotrophin-releasing hormone (CRH), and to a lesser extent, arginine vasopressin (AVP), to produce adrenocorticotropic hormone (ACTH). This is in turn signals the adrenals to release cortisol and other hormones. Most cortisol circulating in the blood is bound to carrier molecules (29, 43). Production of cortisol is regulated by an inhibitory feedback loop. When free circulating cortisol attaches to glucocorticoid receptors on the hypothalamus and pituitary, these glands reduce production of CRH and AVP, and ACTH, respectively. The number and affinity of glucocorticoid receptors is thus considered one of the most important determining factors in the regulation of the HPA axis (43)
In Critical Illness
During the acute phase of critical illness, plasma cortisol concentrations rise rapidly. Increased cortisol availability is considered a vital response that allows for fluid retention, increased cardiac output and blood pressure, and induces an appropriate immune response while protecting against excessive inflammation (29, 44, 45). Until recently believed to be the result of increased cortisol production by the adrenals, it is now known that high cortisol availability during this phase of critical illness is in fact largely driven by two peripheral mechanisms: a decrease in the abundance and affinity of the cortisol carrier molecules in circulation, and a slowing of cortisol breakdown in the liver and kidney (29, 34, 44, 46, 47). Via inhibitory feedback loops, these higher cortisol concentrations suppress the HPA axis at the central level: the secretions of CRH and AVP by the hypothalamus and of ACTH by the pituitary fall, leading to an eventual drop in plasma cortisol levels (48).
Whereas, in critically ill patients that begin to recover, the HPA axis essentially normalizes within 28 days of illness, in cases of prolonged critical illness ACTH levels (surprisingly) continue to be depressed despite dropping cortisol levels (49, 50). Why and how this central suppression of ACTH is maintained is not clear and continues to be debated. Pro-inflammatory cytokines and O&NS likely play a leading role. Cytokines can mediate tissue-specific changes in the abundance and affinity of glucocorticoid receptors—which are major factors determining the activity of the HPA axis (2, 44). Specifically, the cytokine IL-1β is known to modulate CRH release by the hypothalamus; TNF-α is known to impair ACTH release by the pituitary; and TNF-α is also known to impair cortisol production by the adrenal glands (2).
Without sufficient pulsatile stimulation by the tropic hormone ACTH, adrenal glands begin to atrophy and lose zonational structure. This is evidenced in the post-mortem dissection of patients that had been critically ill for a few weeks, but not in the patients that quickly died from their illness or trauma (34, 51). The weakening of adrenal glands not only compromises patients' ability to cope with external stressors but also permits excessive inflammatory responses. In sum, the initial beneficial increase in cortisol availability induced by peripheral mechanisms during the acute phase of critical illness leads to a suppression of the HPA axis at the central-level from which a subset of patients appears unable to escape (Figure 1).
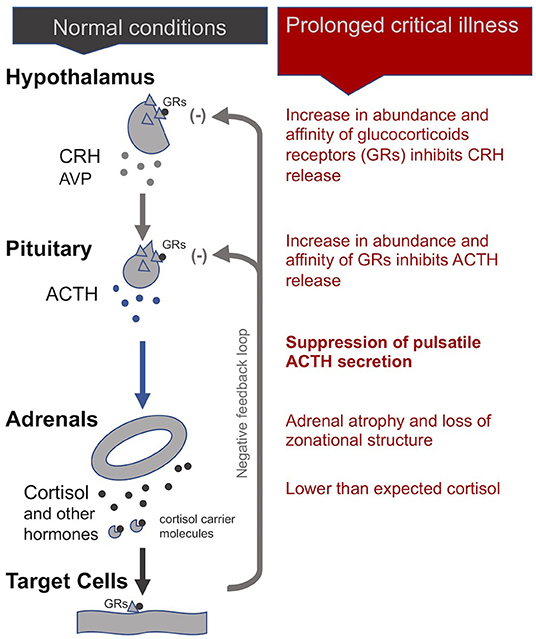
Figure 1. The adreno-cortical axis (HPA axis) during normal conditions and prolonged critical illness.
In ME/CFS
Dysfunction of the HPA axis has been documented extensively in ME/CFS patients since the early 1980s (52–63). Researchers have observed decreased baseline cortisol levels, blunted HPA axis responses to physical and psychological stressors, reduced HPA axis responsivity to provocation tests (such as CRH and ACTH administration), and a heightened inhibitory feedback loop (consistent with a higher abundance and affinity of glucocorticoid receptors at the level of the pituitary and hypothalamus). Strikingly, the magnitude of HPA axis dysfunction becomes more pronounced with illness duration and is associated with symptom severity (43, 64). Very few have studied pulsatility of ACTH release: one study of 36 study-pairs found no statistically significant differences in ACTH pulsatility between ME/CFS and matched controls (65), while another found a differential pattern of ACTH release over 24-h periods (66). Variations in the study-participants' severity of illness—and methods used to control for these—may explain these apparently contradictory findings. Several studies have found the morning peak of ACTH is missing or weak in ME/CFS patients (43). A recent study assessing secretory events of cortisol found that CFS/ME patients have the same number of secretory events but secrete lower quantities in early morning hours (67). Significantly, a group of ME/CFS patients were found to have 50% smaller adrenals than controls (68), resembling adrenal atrophy in prolonged critical illness.
ME/CFS researchers have also proposed models to explain the persistence of a suppressed HPA axis (33, 69, 70). Essentially, a short stress (i.e., a burst of cortisol) will produce a small perturbation in the glucocorticoid receptor concentration on the central glands that quickly returns to normal levels. However, long, repeated stress—from which the system doesn't have time to recover—leads to a persistent high glucocorticoid receptor concentration, forcing the HPA axis to an alternate steady state. More recent models of the HPA axis have also included non-genomic feedback-controls (71), the endogenous effects of circadian rhythm (72), and interactions with the gonadotropic axis and the immune system (73, 74) to explain how HPA axis suppression is maintained even after the initial stress is gone.
HPA axis dysfunction is also present in the majority of fibromyalgia patients (75–77). Various mechanisms have been suggested, including depressed secretion of CRH by the hypothalamus, a deficiency of CRH receptors on the pituitary, and adrenal atrophy due to chronic under-stimulation by reduced ACTH levels (78).
Moreover, the dysfunction of the HPA axis in ME/CFS and fibromyalgia has also been associated with pro-inflammatory cytokines and O&NS (43, 55, 79, 80). A recent paper considering the bidirectional relationship between the function of the HPA axis and inflammation finds that immune-inflammatory and O&NS pathways induce HPA axis dysfunction in ME/CFS (81); the direction of causality is analogous to inflammatory pathways inducing endocrine dysfunctions in critical illness. Others have similarly theorized that local inflammation in the hypothalamus leads to a disturbed HPA axis in ME/CFS (82).
In sum, the HPA axis dysfunctions in ME/CFS are not unlike the dysfunctions in prolonged critical illness. However, to our knowledge a comprehensive study of the pituitary pulsatile secretions of ACTH in ME/CFS patients—which proved revelatory in understanding prolonged critical illness—does not yet exist. The relationship between the pituitary's pulsatile ACTH secretions, severity of illness, the integrity and function of adrenal glands and resulting physiological alterations in ME/CFS thus remains largely unexplored.
The Somatotropic Axis (HPS Axis)
The somatotropic axis—also called hypothalamic-pituitary-somatotropic (HPS) axis—plays important roles in growth and development of children, but also contributes to a variety of physiological pathways in adults, including balancing catabolic (i.e., the break-down of molecules and tissues) and anabolic activities (i.e., the building of molecules and tissue) (4). An HPS axis dysfunction is known to cause loss of muscle and bone mass, induces weakness (29), and impacts gut mucosa integrity as well as glucose and fat metabolism (83). Low energy, exhaustion, mental fatigue, weak muscle strength as well as poor recovery after physical activity are associated with an inhibited HPS axis function (42, 84, 85).
Uniquely, in the case of the HPS axis, the hypothalamus sends both stimulating (+) and inhibiting (-) signals to the pituitary for the production of growth hormone (GH): these are, respectively, the GH-releasing hormone (GHRH) and the GH-inhibiting hormone (GHIH, also called somatostatin) (4). In addition, ghrelin, mostly produced by the gut, also stimulates GH production by the pituitary. In normal conditions, GH is released by the pituitary in a pulsatile fashion under the control of these three signals, with peaks of GH levels alternating with virtually undetectable valleys in 3- to 5-h intervals over the course of the day (29). GH in turn has direct effects on some tissues and also stimulates the production of insulin-like growth hormone-1 (IGF-1), mostly by the liver. Nearly all of the IGF-1 hormones in the plasma are bound to IGF-binding proteins (IGFBP). IGF-1 and GH exert inhibitory feedback on the hypothalamus and the pituitary to maintain homeostasis. The half-life of GH is only 10 to 20 min, whereas the half-life of IGF-1 is more than 12 h. Thus, IGF-1 plasma concentrations are regularly used as proxies for GH secretion in clinical settings. This, however, overlooks the function of the pulsatile secretion of GH on the balance of anabolic and catabolic activities in the body (4).
In Critical Illness
In the acute phase of critical illness, the pituitary produces more GH: higher peaks, lower valleys and increased pulse frequencies (86). The rapid onset of two main peripheral mechanisms explain this finding: First, under the influence of cytokines, the liver expresses fewer GH receptors (i.e., becomes resistant to GH) and thus produces less IGF-1. Second, alterations in IGF binding proteins results in IGF-1 being cleared out faster from the system (i.e., IGF-1 has a shorter half-life) (87). The lower IGF-1 concentrations resulting from these two peripheral mechanisms will—via the feedback loop inherent to the axis—spur more GH production (29). The resulting increase in catabolic activity during the acute phase of critical illness serves to mobilize amino acids derived from the breakdown of peripheral tissues, such as skeletal muscle and bone, for use by the central organs (4).
However, if a critically ill patient fails to recover within a few days, GH secretion becomes erratic and almost non-pulsatile. Experiments have demonstrated that this is largely due to a lack of stimulation of the hypothalamus and pituitary by the hormone ghrelin. There is also evidence for changes in the relative amounts of GHIH and GHRH signals from the hypothalamus (4). As for the peripheral hormone, IGF-1, its levels are low or normal in prolonged critical illness. The liver's resistance to GH (which previously suppressed IGF-1 production during the acute phase of critical illness) does not persist during prolonged critical illness (29, 87). However, without a concomitant pulsatile release of GH, the anabolic function of IGF-1 becomes inhibited (4).
In sum, although the increase in catabolic activity during the acute phase of critical illness may initially be beneficial because it serves to mobilize amino acids, the perpetuation of the imbalance in catabolic vs. anabolic activity (due in part to the loss of the pulsatile function of GH) during prolonged critical illness may be considered maladaptive (Figure 2). The imbalance in catabolic relative to anabolic activity in prolonged critical illness leads to protein break-down in skeletal muscle, liver, kidney and heart, reducing their cell mass and leading to impaired function (7). These processes are ultimately reflected in muscle and bone wasting typically present in prolonged critical illness (88, 89).
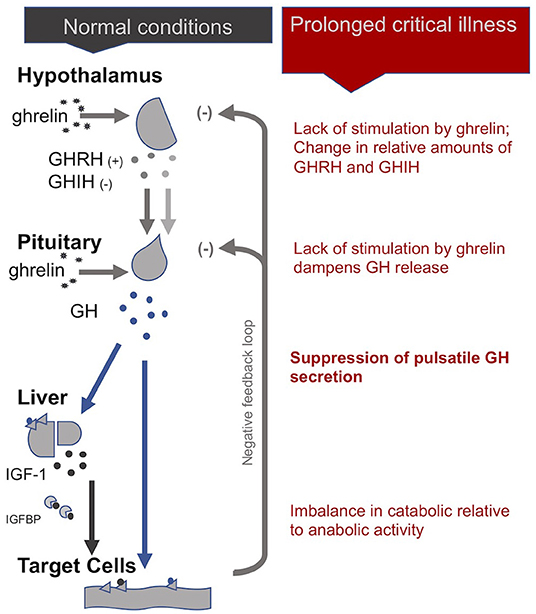
Figure 2. The somatotropic axis (HPS axis) during normal conditions and prolonged critical illness.
In ME/CFS
GH regulation in ME/CFS has been studied since the 1990s. The findings are mixed, but almost none addresses the question of the pulsatility of GH release. Some described low nocturnal GH secretion (90, 91), while others have found normal levels of 24-h urinary GH excretion (92). Some have found reduced response to induced hypoglycemia (90, 91), while others describe normal GH responses to stimulation (93). One study describes unaffected diurnal patterns of GH release in ME/CFS, but it focused on assessing basal levels rather than the nature of secretory patterns (i.e., pulsatile vs. erratic) and may not have accounted for variations in the severity of illness of patients (66). In terms of IGF-1, there are no consistent differences between ME/CFS patients and controls (93, 94), which is consistent with findings from prolonged critical illness.
Studies in fibromyalgia show relative GH deficiency (76, 78, 95–99) and low or low-normal IGF-1 levels (95, 96, 100). Interestingly, some studies showed that fibromyalgia patients “failed to exhibit a GH response to exercise” (97, 101), consistent with a loss in pulsatility of GH release.
In sum, endocrine observations in ME/CFS are not unlike HPS axis dysfunctions found in prolonged critical illness. To our knowledge the pituitary pulsatile secretions of GH in ME/CFS patients has not been comprehensively studied. The relationship between the pituitary's pulsatile GH secretions, severity of illness and the balance between catabolic and anabolic activities in ME/CFS thus remains largely undiscovered.
The Thyrotropic Axis (HPT Axis)
The thyrotropic axis—also called hypothalamic-pituitary-thyroid (HPT) axis—regulates the basal rate of our metabolism. Dysfunctions of the HPT axis are associated with tiredness, stiffness, constipation, dry skin and weight gain, among a myriad of other hypothyroid-like symptoms (35, 42).
In normal conditions, an inhibitory feedback loop works to maintain stable circulating thyroid hormone concentrations according to a daily rhythm (102). When unbound circulating thyroid hormone concentrations dip below a certain threshold, the hypothalamus produces thyrotropin-releasing hormones (TRH) in order to signal the pituitary to produce thyroid stimulating hormone (TSH), which in turn signals the thyroid gland to produce more thyroid hormones.
In Critical Illness
Dysfunctions of the HPT axis during critical illness have been studied extensively. Starting in the early 1970s, clinicians working in ICUs observed that patients with a wide range of critical conditions had low plasma concentrations of the active form of thyroid hormones (T3) relative to plasma concentrations of inactivated thyroid hormones reverse T3 (rT3) (103–105). They gave this condition the name “non-thyroidal illness syndrome” (NTIS), also called “euthyroid sick syndrome” or “low T3 syndrome.” While NTIS was initially considered to be beneficial in critical illness—i.e., a state of “protective” down-regulation of metabolism during times of duress (106) —it is increasingly seen as maladaptive and hampering the recovery of patients in the case of prolonged critical illness (9, 10, 29, 103, 104, 107, 108).
During acute and early stages of critical illness, peripheral mechanisms involving cytokines (notably IL-1β, IL-6, TNF-α) lead to the quick depression of thyroid hormone activity (104, 105, 109–111) to help conserve energy resources (48, 104). The mechanisms include the alterations in the amount and affinity of thyroid hormone binding globulines in the blood (112–114); modifications in the expression of the transporters that carry the thyroid hormone into the cells (115, 116); the down- and up-regulation of deiodinase enzymes that convert the thyroid hormone into active and inactive forms, respectively (113, 117); and the variation in the quantity and isoforms of cellular thyroid hormone receptors present (notably in the liver, adipose tissue and muscle) (118–120). An alteration in any of these steps—which determine thyroid hormone function—can lead to large time- and tissue-specific adjustments in cellular metabolism (121, 122)—even without, or with only minor, changes in the blood concentrations of thyroid hormones (121, 123, 124).
During prolonged critical illness these peripheral mechanisms are supplemented by central mechanisms that also depress thyroid hormone function (125, 126). Cytokines (notably IL-12 and IL-18), in association with other signaling factors (including leptin, glucocorticoids, etc.), are believed to up-regulate the deiodinase enzymes D1 and D2 in the hypothalamus resulting in higher local levels of T3 that inhibit TRH release irrespective of circulating thyroid hormone concentrations (10, 127, 128). Moreover, cytokines (notably IL-1b and TNF-α) also suppress the release of TSH by the pituitary (129, 130). Finally, by reducing iodine uptake and thyroid hormone excretion, cytokines (notably IL-1) also impact the activity of the thyroid gland itself (103, 113). Together, these mechanisms can alter the inhibitory feedback mechanisms of the HPT axis (i.e., its “set-point”) during prolonged critical illness. Single measurements of circulating TSH, however, are ineffective in revealing such alterations in the set-point of the HPT axis.
In sum, an initial beneficial alteration of thyroid hormone activity in the periphery during acute critical illness is followed by a cytokine-mediated central suppression of the HPT axis resulting in a virtual complete loss of pulsatile TSH secretion (29). Peripheral mechanisms (notably variations in the conversion and transport of thyroid hormones) may further modulate thyroid hormone function in time- and tissue-specific ways resulting in complex physiological alterations in these patients (Figure 3) —not readily observable in blood concentrations of thyroid hormones. How these alterations of the HPT axis persist as well as their broader implications on metabolism and the immune system are further described below (see section A “Vicious Circle” Perpetuating Illness).
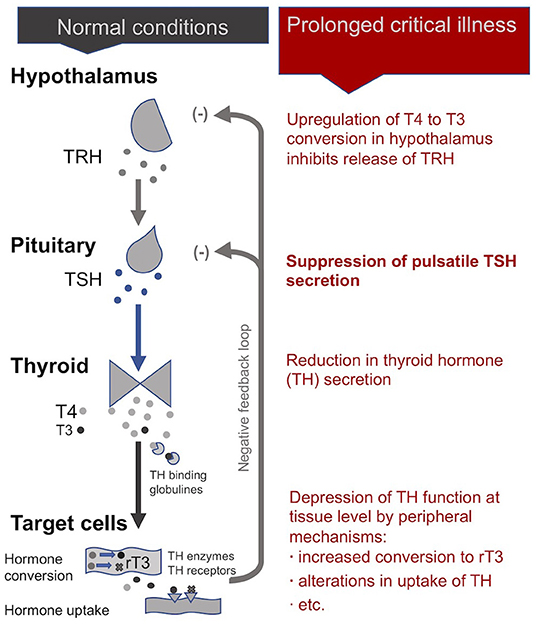
Figure 3. The thyrotropic axis (HPT axis) during normal conditions and prolonged critical illness.
In ME/CFS
Dysfunctions of the HPT axis have long been suspected to play a role in ME/CFS (77, 131–134) and fibromyalgia (135–140). A recent study showed that ME/CFS patients had similar TSH levels as controls, but lower Free T3, Total T4, and Total T3, which the authors suggest resembles NTIS (141)—the typical feature of critically ill patients in ICUs described above.
In sum, alterations of the HPT axis in ME/CFS resemble dysfunctions found in prolonged critical illness. However, there does not to our knowledge exist a thorough study of the pulsatility of pituitary TSH secretion events in ME/CFS patients, nor a study of the tissue-specific alterations in thyroid hormone function—which proved revelatory in understanding prolonged critical illness. The relationship between the TSH axis dysfunctions, severity of illness, hypometabolic state and organ/tissue specific symptoms in ME/CFS thus remains largely unexplored.
Intermediate Conclusions
The endocrine axes control many of the most fundamental physiological processes; their suppression is associated with a myriad of symptoms (see Table 2). Essentially, the suppression of pulsatile pituitary secretions of ACTH, GH, and TSH are central to prolonged critical illness. Inflammatory pathways play a role in inducing and maintaining this suppression irrespective of the nature of the original illness or trauma (see Table 3). The resulting endocrine patterns may be considered maladaptive and have wide ranging implications, including dysfunction of the balance between anabolic and catabolic processes, metabolism, and the regulation of the immune system. The physiological parallels between ME/CFS and prolonged critical illness would suggest that the suppression of pulsatile pituitary secretions of these tropic hormones might also underlie ME/CFS, and that the severity of ME/CFS might be a function of the strength of the mechanism; this however remains largely unstudied. In the next section we provide an overview of a model from critical illness that explains the perpetuation of these endocrine dysfunctions and we describe the relevance of the model for understanding ME/CFS.
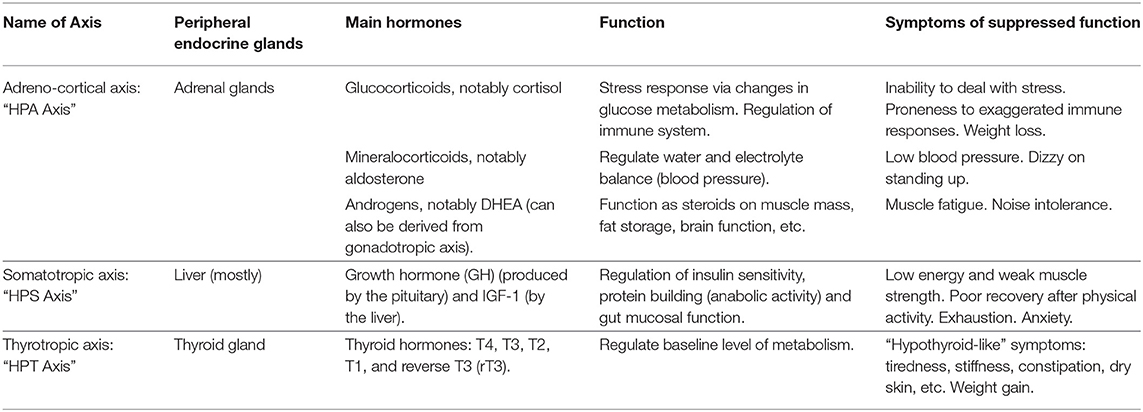
Table 2. Summary of endocrine axes and function of the main hormones in adults.
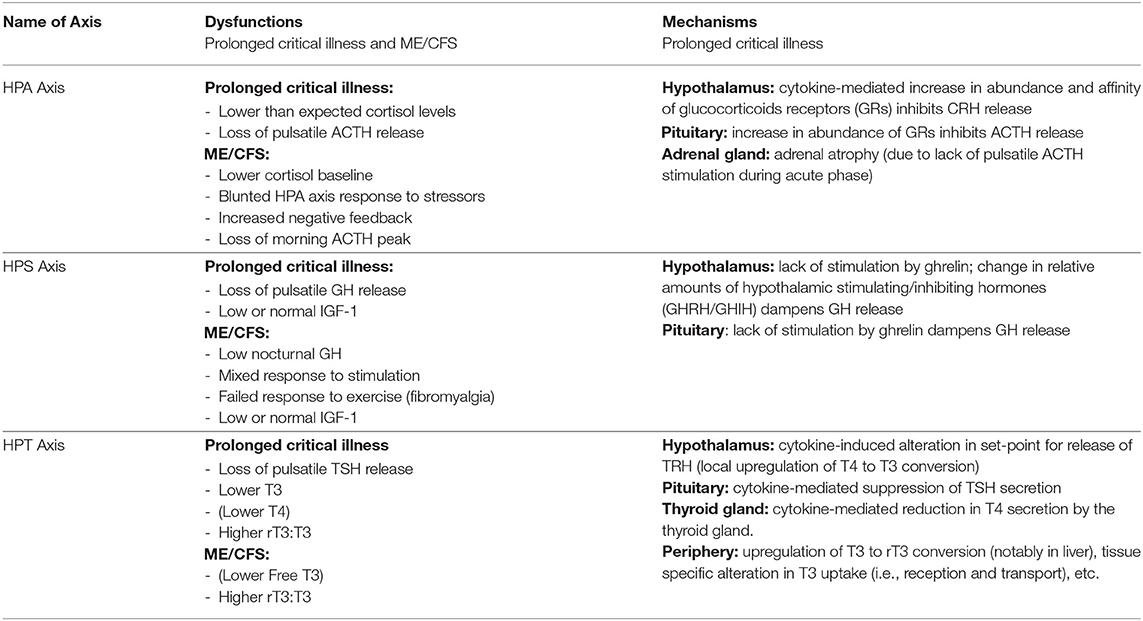
Table 3. Summary of endocrine dysfunctions and mechanisms in critical illness and ME/CFS.
A “Vicious Circle” Perpetuating Illness
Based on nearly five decades of research, critical illness researchers have proposed a model that describes how NTIS is maintained by reciprocal relationships between inflammation (notably pro-inflammatory cytokines), O&NS and reduced thyroid hormone function, forming a “vicious circle” (9, 10) (Figure 4). This model can help to explain the perplexing failure to recover of some critically ill patients in ICUs that survive their initial severe illness or injury. We describe the main elements of this model in a simplified manner below, as well as the implications for metabolism and the immune system. We also provide evidence suggesting that the “vicious circle” observed in prolonged critical illness also underlies ME/CFS.
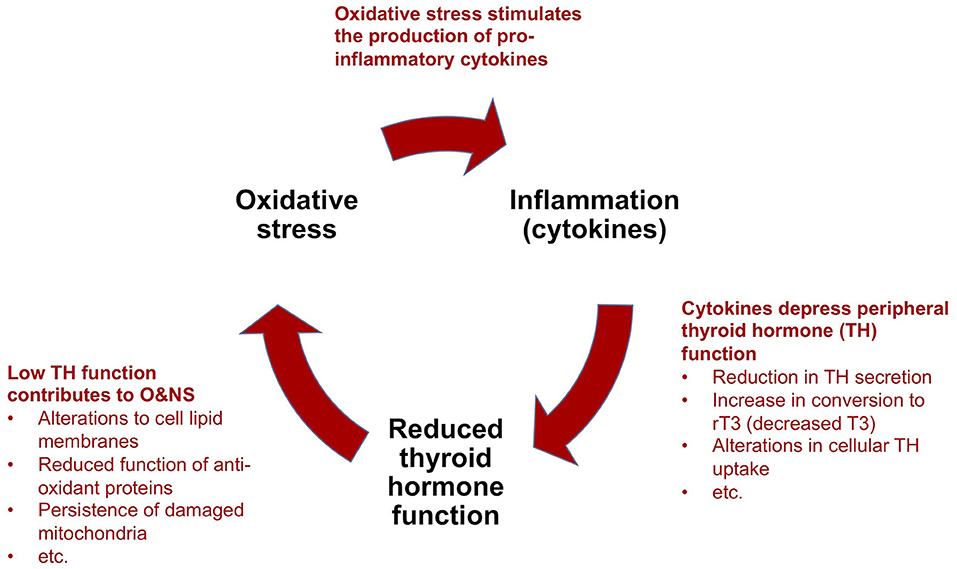
Figure 4. Simplified model to explain the perpetuation of prolonged critical illness: a “vicious circle”.
In Prolonged Critical Illness
The key elements of the suggested “vicious circle” in prolonged critical illness include the following mechanisms:
(a) Cytokines depress thyroid hormone function: As described above [see section The thyrotropic axis (HPT Axis) In Critical Illness], in acute and early stages of critical illness, various peripheral mechanisms involving cytokines lead to the quick depression of thyroid hormone activity in tissue-specific ways. In prolonged critical illness, cytokines in association with other signaling factors targeting the hypothalamus, as well as the pituitary and the thyroid glands, also inhibit thyroid hormone production. The relative sequence and importance of these various mechanisms in depressing the HPT axis and thyroid hormone function in different tissues and phases of critical illness are the subject of most NTIS publications (10, 104, 105). Notwithstanding the effect of other mechanisms, alterations in the activity of the deiodinase enzymes lead to a decrease in T3 and an increase in rT3 and thus a reduction in thyroid hormone function in peripheral tissues during prolonged critical illness [based on biopsies on ICU patients who died (142) and studies on mice (143, 144)]. Circulating thyroid hormone concentrations, however, only reveal the “tip of the iceberg” of the alterations occurring at the tissue level (141, 145), which thus are often missed altogether in clinical settings (146).
(b) Low thyroid hormone function contributes to oxidative and nitrosative stress: The relationship between thyroid hormone function and O&NS is complex, and both hyperthyroidism and hypothyroidism have been associated with oxidative stress (147). Nonetheless, it seems clear that depressed thyroid hormone function hinders tissue cells from maintaining a healthy O&NS balance. Mechanisms include alterations to the lipid concentration of the cell membranes that maintain the cell's O&NS balance (148), and reduced function of two proteins (Uncoupling Proteins-2 and -3) with anti-oxidant properties (149). Moreover, in low thyroid hormone function conditions, mitochondria damaged by O&NS are not cleared out of cells (9). In turn, it appears that oxidative stress depletes the glutathione required by the abovementioned deiodinase enzymes for the conversion of T4 into T3 (104). Similarly, competition for, and the resulting depletion of the trace mineral selenium—a component of both the deiodinase and the anti-oxidant enzymes (150) —may amplify the self-perpetuating link between increased oxidative stress and low thyroid hormone function.
(c) Oxidative and nitrosative stress stimulate the production of pro-inflammatory cytokines: The final mechanism which completes the “vicious circle” in prolonged critical illness is the link between O&NS and inflammation. O&NS stimulates the production of pro-inflammatory cytokines, notably leptin, resistin, TNF-α and IL-6 (151). In turn, pro-inflammatory cytokines (notably IL-6) further increase O&NS by triggering the production of superoxide radicals (104, 152). There is thus a tendency for O&NS and pro-inflammatory cytokines to perpetuate each other as well.
In sum, according to a model proposed by critical illness researchers, a “vicious circle” involving O&NS, pro-inflammatory cytokines, and low thyroid hormone function—as well as reciprocal relationships across these elements—can perpetuate a hypometabolic and inflammatory state, and thus help to explain why some critically ill patients fail to recover.
In ME/CFS
Similar patterns of O&NS, cytokines, and low thyroid hormone function have recently been documented in ME/CFS patients providing the elements for a similar “vicious circle.” We briefly summarize the findings from ME/CFS research relevant to each of these elements.
Reduced thyroid hormone function: An immune-mediated loss of thyroid hormone function in ME/CFS has long been suspected (132). As mentioned above [see section: The thyrotropic axis (HPT Axis) In ME/CFS], a recent study confirmed that CFS patients have lower circulating levels of Free T3, Total T4, and Total T3 than controls (141). Moreover, this study found a significantly higher ratio of rT3 to T3 hormones. These findings imply a depressed thyroid hormone function resembling NTIS. Given the possible tissue-specific alterations in thyroid hormone activity resulting from peripheral mechanisms, the authors suggest these circulating levels only reflect the “tip of the iceberg” of genuine T3 deficits in target tissues.
Oxidative & nitrosative stress: Numerous studies have found increased O&NS in ME/CFS and identified this as a factor in the observed metabolic dysfunction (153, 154). Indeed, Pall proposed a model that describes a “vicious circle” involving oxidative stress and cytokines in ME/CFS a decade ago (cf. the “NO/ONOO-Cycle”) (155). Researchers also suggest that high lactate and low glutathione levels found in the brains of ME/CFS patients likely derive from similar mechanisms involving oxidative stress (156). A recent study described the relationship between O&NS and immune-inflammatory pathways in ME/CFS (80).
Pro-inflammatory cytokines: Neuro-inflammation is central to ME/CFS, and many researchers have tried to develop diagnostic biomarkers for ME/CFS based on cytokine profiles of patients (157, 158). Montoya et al. found that some 17 cytokines were positively correlated with the severity of ME/CFS, of which 13 are pro-inflammatory. Similarly, circulatory levels of pro-inflammatory cytokines are altered in fibromyalgia patients (159). However, others have argued that given the innumerable sources of potential variance in the measurement of cytokines, it is “unlikely that a consistent and replicable diagnostic cytokine profile will ever be discovered” for ME/CFS (160). It may therefore be ineffectual to compare the cytokine profiles of ME/CFS and prolonged critical illness patients.
In sum, given the presence of reduced thyroid hormone function, O&NS and pro-inflammatory cytokines in ME/CFS, the “vicious circle” model proposed by critical illness researchers to explain prolonged critical illness may also help to understand why ME/CFS patients fail to recover.
Implications of the “Vicious Circle” and Its Elements
Reduced thyroid hormone function, increased O&NS and pro-inflammatory cytokines discovered in prolonged critical illness as well as in ME/CFS have important implications notably on metabolism, organ function, immune responses and the endocrine system. These are further described below:
Reduced thyroid hormone function: The prolonged down-regulation of thyroid hormone activity certainly has implications for the immune system. Authors describe the profound effects of circulating thyroid hormone levels on the activity of monocytes, lymphocytes macrophages, neutrophils, dendritic cells and natural killer cells; as well as cytokines (161–170). Notably, depressed thyroid levels appear to depress the activity of natural killer cells (171)—a signature finding in ME/CFS (172). Such immune dysfunctions might explain other pathologies, such as viral reactivation observed in ICU patients (173–175) and suspected in ME/CFS patients (176, 177). Experimenting on rats, researchers have shown that depressed thyroid hormone levels occur in a specific sequence, manifesting (from first to last) in the liver, kidney, brain, heart and adipose tissues (145). An implication of a tissue-specific down-regulation of thyroid hormone activity is differential impact on organ function. Some ME/CFS practitioners have argued that tissue-specific modulation of T3 can help explain the disparate and evolving symptoms in ME/CFS and fibromyalgia (133, 134, 138, 140). In aggregate, depressed thyroid hormone function would engender a general hypometabolic state. Finally, thyroid hormone function also impacts other endocrine axes as well (178, 179)—notably the HPA axis—setting the stage for further complex interactions between the various endocrine axes and the immune system.
Oxidative & nitrosative stress: The implications of chronic oxidative stress in the body are widely documented. In addition to inducing inflammation, oxidative stress causes cell damage and disrupts normal cellular transcription and signaling mechanisms (9). O&NS has been shown to cause mitochondrial damage during critical illness (180) and ME/CFS (153).
Pro-inflammatory cytokines: Researchers are finding that the more than 100 different cytokines play a part in determining the function of hormones through both central and peripheral mechanisms (32). As described in the previous section, cytokines are likely culprits in the central (i.e., hypothalamic and pituitary) suppression of the HPA, HPS and HPT axes in prolonged critical illness (29). Pro-inflammatory cytokines and inflammation also hinder normal mitochondrial function during critical illness (181). The alterations in cytokines found in critical illness likely have many further implications that have yet to be fully understood (182) which is also the case for ME/CFS (183).
Intermediate Conclusions
In sum, critical illness researchers have proposed that the self-perpetuating relationships between inflammation (notably pro-inflammatory cytokines), O&NS and low thyroid hormone function explains the maintenance of illness in some ICU patients following severe injury or infection. Given that the same elements of such a “vicious circle” have also been documented in ME/CFS, we suggest that the model can also explain the failure of ME/CFS patients to recover. Moreover, these elements have been shown to have profound implications on metabolism, as well as on the function of the immune and endocrine systems—which in in turn could explain the myriad of symptoms in prolonged critical illness and ME/CFS.
Relationship to Other Hypotheses of ME/CFS Pathogenesis
Our hypothesis that maladaptive mechanisms which prevent recovery in prolonged critical illness also underlie ME/CFS complements several other hypotheses of ME/CFS pathogenesis. In this section we provide an initial and non-exhaustive discussion of some of these complementarities.
Allostatic overload: Some researchers consider ME/CFS to be a maladaptive response to physical, infectious, and/or emotional stressors. They describe an “allostatic overload” (i.e., the cumulative effect of stressful situations exceeding a person's ability to cope) or a “‘crash’ in the stress system” (184, 185). Our hypothesis fits into this theoretical framework and offers an explanation for the possible underlying physiological mechanisms by drawing on the research from critical care medicine.
Hypothalamic endocrine suppression: Researchers have suggested that hypothalamic endocrine suppression could explain ME/CFS (132, 186) and fibromyalgia (187–189). Our thesis upholds this hypothesis and seeks to strengthen it by suggesting that the controversy around the existence of central endocrine suppression in ME/CFS may be resolved by studying the pulsatile secretions of the pituitary—rather than single or average measurements of circulating tropic and non-tropic hormone concentrations, which can fail to discern the dysfunctions of the endocrine axes.
Anomalies in thyroid hormone function: Numerous clinical practitioners and researchers believe that anomalies in thyroid hormone function—including changes in the conversion of thyroid hormones, a resistance of thyroid hormone receptors at cellular level, etc. —contribute to ME/CFS and fibromyalgia (133–141). Indeed, practitioners have written about their successes in treating ME/CFS patients with thyroid hormone supplements (42, 77, 188, 190–194); and patients have published books on their experiences (195–197). Our hypothesis complements this reasoning: we propose that both the central and peripheral mechanisms altering thyroid hormone function during critical illness (c.f. NTIS, euthyroid sick syndrome or “low T3 syndrome”) also occur in ME/CFS. Moreover, by applying a model from critical illness, we suggest that low thyroid hormone function is one element of a “vicious circle” perpetuating illness in ME/CFS.
Viral Reactivation: It has long been suggested that viral reactivation plays a role in ME/CFS, particularly reactivation of Epstein-Barr virus (EBV) and cytomegalovirus (CMV) (176, 177). Similarly, high incidences of viral reactivation have also been observed in ICU patients, notably in patients with sepsis and prolonged critical illness. ICU researchers propose that this viral reactivation is a result of immune suppression occurring during critical illness (173–175). Thus, critical illness research would suggest that viral reactivation is a secondary pathology in ME/CFS—except in cases in which the viral infection was the onset event.
Viral infection: Viral infection is recognized to be a leading onset event of ME/CFS (16, 198–201). This is particularly concerning in the context of the COVID-19 pandemic. Many COVID-19 patients continue to experience a variety of debilitating symptoms after defeating the virus that resemble ME/CFS. Building on our hypothesis, we would suggest that post COVID-19 syndrome is evidence of a maladaptive response to the stress of infection akin to that experienced in prolonged critical illness and ME/CFS.
Chronic inflammation: Researchers have found that chronic inflammation—auto-immune, allergic or bacterial/viral—underlies ME/CFS (194, 202, 203). Others also ascribe the perpetuation of ME/CFS to the relationship between inflammation and O&NS (80, 155). Our hypothesis is largely complementary to these findings and associated theories. Indeed, following a cytokine surge during the acute phase of critical illness, inflammation is believed to persist in the case of prolonged critical illness (4). Moreover, pro-inflammatory cytokines and O&NS are elements in the “vicious circle” model of prolonged critical illness, which we propose also serves to understand the perpetuation of illness in ME/CFS patients.
Neuroinflammation of the brain: ME/CFS is associated with inflammation of the brain (hence the name myalgic encephalomyelitis) (204, 205). Some have specifically proposed that inflammation of the hypothalamus underlies ME/CFS (81, 82). Similarly, alterations of the endocrine axes through mechanisms mediated by pro-inflammatory cytokines which impact the hypothalamus and pituitary are central to prolonged critical illness (see section Suppression of Pulsatile Pituitary Secretions).
Energy metabolic defect: Researchers have found impairment in energy production (205, 206), reduced mitochondrial activity (207–209) and irregularities in the metabolites of ME/CFS patients (210, 211) —suggesting that they experience a hypometabolic or “dauer” state (212). Our hypothesis is compatible with analyses that emphasize metabolic defects in ME/CFS. Indeed, the suppression of pituitary secretions, depressed thyroid hormone function, O&NS and immune system dysfunction—hallmarks of prolonged critical illness—have severe impacts on metabolism, including on glucose utilization and mitochondrial activity (see section A “Vicious Circle” Perpetuating Illness). Certainly, prolonged critical illness resembles a hypometabolic “dauer” state as well.
Genetic predisposition: Research also suggests there may a genetic element in the pathogenesis of ME/CFS (213–216). Our hypothesis is compatible with a possible genetic predisposition for ME/CFS. Indeed, it is not known why some critically ill patients succumb to prolonged critical illness while others begin recovery (217, 218); genetics may play a role. The findings from the field of ME/CFS in the area of genetics might inform the field of critical illness in this regard.
In sum, our hypothesis is largely complementary to hypotheses that emphasize metabolic, hormonal and/or immune dysfunctions in the pathogenesis of ME/CFS. Our hypothesis—drawing from research on critical illness—integrates these dysfunctions into a single framework and provides arguments for the direction of causality between them.
Conclusion
Decades of research in the field of critical medicine have demonstrated that in response to the stress of severe infection or injury, endocrine axes experience profound alterations. An assessment of the pituitary's pulsatile secretions reveals that in the subset of patients which survive their severe infection or injury but do not begin recovery (i.e., prolonged critically ill patients), the suppression of endocrine axes is maintained irrespective of the initial severe infection or injury. Recent pathological models propose that mechanisms involving pro-inflammatory cytokines, O&NS and low thyroid hormone function explain the perpetuation of these endocrine dysfunctions (i.e., a “vicious circle”).
The symptoms, physiological abnormalities and endocrine patterns observed in severe ME/CFS are not unlike those of prolonged critical illness. Moreover, the same elements of a “vicious circle” also exist in ME/CFS. However, unlike in critical illness, the pituitary's pulsatile secretion and its relationships to metabolic and immune functions remain largely unstudied in ME/CFS.
Without excluding possible predisposing genetic or environmental factors, we propose the hypothesis that the maladaptive mechanisms that prevent recovery of prolonged critically ill patients also underlie ME/CFS. The severity of ME/CFS illness may be a function of the strength of these mechanisms; very severe ME/CFS most resembles prolonged critical illness. We further argue that this hypothesis should be investigated through collaborative research projects building on the findings from critical illness and ME/CFS. If this hypothesis is validated, past trials to break the “vicious circle” that perpetuates critical illness, and the early successes to reactivate the pulsatile secretion of the pituitary in ICU patients, may provide avenues for a cure for ME/CFS—including cases onset by infections. Certainly, given the similarities described above, active collaboration between critical illness and ME/CFS researchers could lead to improved outcomes for both conditions.
Finally, we suggest that immediate collaborative efforts should be sought among the researcher community in order to conduct longitudinal studies with the aim of identifying similarities and differences across prolonged critical illness, post-ICU syndrome, ME/CFS, fibromyalgia and long-COVID in relation to the hormonal axes, O&NS and pro-inflammatory response with the objective of discovering diagnostic and therapeutic targets mitigating the functional disability that these conditions induce.
Data Availability Statement
The original contributions generated for the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
DS wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
Open Medicine Foundation and Swedish Research Council (2015-4870 (JB)) are acknowledged for support.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Text throughout this hypothesis is reproduced from DS's blogposts on Health Rising (219–221) licensed Creative Common–Attribution CC BY.
Abbreviations
ACTH, Adrenocorticotropic hormone; ARV, Arginine vasopressin; CRH, Corticotrophin-releasing hormone; DHEA, Dehydroepiandrosterone; GH, Growth hormone; GHIH, Growth hormone inhibiting hormone; GHRH, Growth hormone releasing hormone; HPA, hypothalamus-pituitary-adrenal axis: “Adreno-cortical axis”; HPS, Hypothalamic-pituitary-somatotropic axis: “Somatropic axis”; HPT, Hypothalamic-pituitary-thyroid: “Thyrotropic axis”; ICU, Intensive Care Unit; IGF-1, Insulin like growth hormone-1; IGFBP, Insulin like growth hormone binding proteins; ME/CFS, Myalgic Encephalomyelitis/Chronic Fatigue Syndrome; NTIS, Non-thyroidal illness syndrome; O&NS, oxidative and nitrosative stress; PICS, Post-intensive care syndrome; POTS, Postural Orthostatic Tachycardia Syndrome; TRH, Thyrotropin-releasing hormone; TSH, Thyroid stimulating hormone.
References
1. Loss SH, Nunes DSL, Franzosi OS, Salazar GS, Teixeira C, Vieira SRR. Chronic critical illness: are we saving patients or creating victims? Rev Bras Ter Intensiva. (2017) 29:87–95. doi: 10.5935/0103-507X.20170013
2. Marik PE. Mechanisms and clinical consequences of critical illness associated adrenal insufficiency. Curr Opin Crit Care. (2007) 13:363–9. doi: 10.1097/MCC.0b013e32818a6d74
3. Langouche L, Van den Berghe G. Hypothalamic-pituitary hormones during critical illness: a dynamic neuroendocrine response. Handb Clin Neurol. (2014) 124:115–26. doi: 10.1016/B978-0-444-59602-4.00008-3
4. Mesotten D, Van den Berghe G. Changes within the growth hormone/insulin-like growth factor I/IGF binding protein axis during critical illness. Endocrinol Metab Clin North Am. (2006) 35:793–805. doi: 10.1016/j.ecl.2006.09.010
5. Nelson JE, Cox CE, Hope AA, Carson SS. Chronic critical illness. Am J Respir Crit Care Med. (2010) 182:446–54. doi: 10.1164/rccm.201002-0210CI
6. Vanhorebeek I, Latronico N, Van den Berghe G. ICU-acquired weakness. Intensive Care Med. (2020) 46:637–53. doi: 10.1007/s00134-020-05944-4
7. Weekers F, Van den Berghe G. Endocrine modifications and interventions during critical illness. Proc Nutr Soc. (2004) 63:443–50. doi: 10.1079/PNS2004373
8. Boonen E, Van den Berghe G. Endocrine responses to critical illness: novel insights and therapeutic implications. J Clin Endocrinol Metab. (2014) 99:1569–82. doi: 10.1210/jc.2013-4115
9. Mancini A, Di Segni C, Raimondo S, Olivieri G, Silvestrini A, Meucci E, et al. Thyroid hormones, Oxidative stress, and inflammation. Mediators Inflamm. (2016) 2016:6757154. doi: 10.1155/2016/6757154
10. Chatzitomaris A, Hoermann R, Midgley JE, Hering S, Urban A, Dietrich B, et al. Thyroid allostasis–adaptive responses of thyrotropic feedback control to conditions of strain, stress, and developmental programming. Front Endocrinol. (2017) 8:163. doi: 10.3389/fendo.2017.00163
11. Van Aerde N, Van Dyck L, Vanhorebeek I, Van den Berghe G. Endocrinopathy of the critically Ill. In: Preiser J-C, Herridge M, Azoulay E, editors. Post-Intensive Care Syndrome. Cham: Springer International Publishing. (2020) p. 125-43. doi: 10.1007/978-3-030-24250-3_9
12. Rawal G, Yadav S, Kumar R. Post-intensive care syndrome: an overview. J Transl Int Med. (2017) 5:90–2. doi: 10.1515/jtim-2016-0016
13. Smith S, Rahman O. Post intensive care syndrome. In: StatPearls Treasure Island, FL: StatPearls Publishing. (2020).
14. Institute of Medicine. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness. Washington, DC: The National Academies Press, (2015).
15. Komaroff AL. Advances in understanding the pathophysiology of chronic fatigue syndrome. JAMA. (2019) 322:499–500. doi: 10.1001/jama.2019.8312
16. Chu L, Valencia IJ, Garvert DW, Montoya JG. Onset patterns and course of myalgic encephalomyelitis/chronic fatigue syndrome. Front Pediatr. (2019) 7:12. doi: 10.3389/fped.2019.00012
17. Centers for Disease Control and Prevention. (2019). Clinical Care of Patients with ME/CFS - Severely Affected Patients. Available online at: https://www.cdc.gov/me-cfs/healthcare-providers/clinical-care-patients-mecfs/severely-affected-patients.html. (accessed September 6, 2020).
18. Nacul L, Authier J, Scheibenbogen C, Lorusso L, Helland I, Alegre Martin J, et al. EUROPEAN ME NETWORK (EUROMENE) expert consensus on the diagnosis, service provision and care of people with ME/CFS in Europe. Preprints. (2020).
19. Open Medicine Foundation. (2020). Symptoms of ME/CFS. Available online at: https://www.omf.ngo/symptoms-mecfs (accessed October 9, 2020).
20. Meeus M, Ickmans K, Struyf F, Kos D, Lambrecht L, Willekens B, et al. What is in a name? Comparing diagnostic criteria for chronic fatigue syndrome with or without fibromyalgia. Clin Rheumatol. (2016) 35:191–203. doi: 10.1007/s10067-014-2793-x
21. Teodoro T, Edwards MJ, Isaacs JD. A unifying theory for cognitive abnormalities in functional neurological disorders, fibromyalgia and chronic fatigue syndrome: systematic review. J Neurol Neurosurg Psychiatry. (2018) 89:1308–19. doi: 10.1136/jnnp-2017-317823
22. Natelson BH. Myalgic encephalomyelitis/chronic fatigue syndrome and fibromyalgia: definitions, similarities, and differences. Clin Ther. (2019) 41:612–8. doi: 10.1016/j.clinthera.2018.12.016
23. Greenhalgh T, Knight M, A'Court C, Buxton M, Husain L. Management of post-acute covid-19 in primary care. BMJ. (2020) 370:m3026. doi: 10.1136/bmj.m3026
24. Wildwing T, Holt N. The neurological symptoms of long COVID-19: a comparison with other neurological conditions and implications for healthcare services. medRxiv [Preprint]. (2020). doi: 10.1101/2020.07.21.20158816
25. Dani M, Dirksen A, Taraborrelli P, Torocastro M, Panagopoulos D, Sutton R, et al. Autonomic dysfunction in ‘long COVID’: rationale, physiology and management strategies. Clin Med. (2020). doi: 10.7861/clinmed.2020-0896. [Epub ahead of print].
26. Davis HE, Assaf GS, McCorkell L, Wei H, Low RJ, Re'em Y, et al. Characterizing long COVID in an International Cohort: 7 months of symptoms and their impact. medRxiv [Preprint]. (2020). doi: 10.1101/2020.12.24.20248802
27. Van den Berghe GH. Acute and prolonged critical illness are two distinct neuroendocrine paradigms. Verh K Acad Geneeskd Belg. (1998) 60:487–518; discussion −20.
28. Vanhorebeek I, Van den Berghe G. The neuroendocrine response to critical illness is a dynamic process. Crit Care Clin. (2006) 22:1–15. doi: 10.1016/j.ccc.2005.09.004
29. Van den Berghe G. On the neuroendocrinopathy of critical illness. Perspectives for feeding and novel treatments. Am J Respir Crit Care Med. (2016) 194:1337–48. doi: 10.1164/rccm.201607-1516CI
30. Veldhuis JD. Mechanisms and Biological Significance of Pulsatile Hormone Secretion. Symposium Proceedings. London. Novartis Found Symp. (2000) 227:1–4.
31. Keenan DM, Veldhuis JD. Pulsatility of Hypothalamo-Pituitary Hormones: A Challenge in Quantification. Physiology. (2016) 31:34–50. doi: 10.1152/physiol.00027.2015
32. Van den Berghe G. Novel insights into the neuroendocrinology of critical illness. Eur J Endocrinol. (2000) 143:1–13. doi: 10.1530/eje.0.1430001
33. Gupta S, Aslakson E, Gurbaxani BM, Vernon SD. Inclusion of the glucocorticoid receptor in a hypothalamic pituitary adrenal axis model reveals bistability. Theor Biol Med Model. (2007) 4:8. doi: 10.1186/1742-4682-4-8
34. Téblick A, Peeters B, Langouche L, Van den Berghe G. Adrenal function and dysfunction in critically ill patients. Nat Rev Endocrinol. (2019) 15:417–27. doi: 10.1038/s41574-019-0185-7
35. Litin SC. Mayo Clinic Family Health Book 5th Edition: Completely Revised and Updated. Rochester, MN: Mayo Clinic Press (2018). 1392 p.
36. McEwen BS, Biron CA, Brunson KW, Bulloch K, Chambers WH, Dhabhar FS, et al. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Brain Res Rev. (1997) 23:79–133. doi: 10.1016/S0165-0173(96)00012-4
37. Stoppelbein L, Greening L, Fite PJ. Brief report: role of cortisol in posttraumatic stress symptoms among mothers of children diagnosed with cancer. J Pediatr Psychol. (2010) 35:960–5. doi: 10.1093/jpepsy/jsp139
38. Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Annals N Y Acad Sci. (2012) 1261:55–63. doi: 10.1111/j.1749-6632.2012.06633.x
39. Simoens VL, Hébert S. Cortisol suppression and hearing thresholds in tinnitus after low-dose dexamethasone challenge. BMC Ear Nose Throat Disorders. (2012) 12:4. doi: 10.1186/1472-6815-12-4
40. Hasson D, Theorell T, Bergquist J, Canlon B. Acute stress induces hyperacusis in women with high levels of emotional exhaustion. PLoS ONE. (2013) 8:e52945–e. doi: 10.1371/journal.pone.0052945
41. Rendina DN, Ryff CD, Coe CL. Precipitous dehydroepiandrosterone declines reflect decreased physical vitality and function. J Gerontol A Biol Sci Med Sci. (2017) 72:747–53. doi: 10.1093/gerona/glw135
42. Hertoghe T. Atlas of Endocrinology for Hormone Therapy. 2nd ed. Luxembourg: International Medical Books (2019).
43. Tomas C, Newton J, Watson S. A review of hypothalamic-pituitary-adrenal axis function in chronic fatigue syndrome. ISRN Neurosci. (2013) 2013:784520. doi: 10.1155/2013/784520
44. Boonen E, Bornstein SR, Van den Berghe G. New insights into the controversy of adrenal function during critical illness. Lancet Diabetes Endocrinol. (2015) 3:805–15. doi: 10.1016/S2213-8587(15)00224-7
45. Bergquist M, Huss F, Fredén F, Hedenstierna G, Hästbacka J, Rockwood AL, et al. Altered adrenal and gonadal steroids biosynthesis in patients with burn injury. Clin Mass Spectrometry. (2016) 1:19–26. doi: 10.1016/j.clinms.2016.10.002
46. Peeters B, Boonen E, Langouche L, Van den Berghe G. The HPA axis response to critical illness: new study results with diagnostic and therapeutic implications. Mol Cell Endocrinol. (2015) 408:235–40. doi: 10.1016/j.mce.2014.11.012
47. Peeters B, Meersseman P, Vander Perre S, Wouters PJ, Debaveye Y, Langouche L, et al. ACTH and cortisol responses to CRH in acute, subacute, and prolonged critical illness: a randomized, double-blind, placebo-controlled, crossover cohort study. Intensive Care Med. (2018) 44:2048–58. doi: 10.1007/s00134-018-5427-y
48. Boonen E, Van den Berghe G. Novel insights in the HPA-axis during critical illness. Acta Clinica Belgica. (2014) 69:397–406. doi: 10.1179/2295333714Y.0000000093
49. Peeters B, Langouche L, Van den Berghe G. Adrenocortical stress response during the course of critical illness. Compr Physiol. (2017) 8:283–98. doi: 10.1002/cphy.c170022
50. Peeters B, Meersseman P, Vander Perre S, Wouters PJ, Vanmarcke D, Debaveye Y, et al. Adrenocortical function during prolonged critical illness and beyond: a prospective observational study. Intensive Care Med. (2018) 44:1720–9. doi: 10.1007/s00134-018-5366-7
51. Boonen E, Langouche L, Janssens T, Meersseman P, Vervenne H, De Samblanx E, et al. Impact of duration of critical illness on the adrenal glands of human intensive care patients. J Clin Endocrinol Metabol. (2014) 99:4214–22. doi: 10.1210/jc.2014-2429
52. Poteliakhoff A. Adrenocortical activity and some clinical findings in acute and chronic fatigue. J Psychosomatic Res. (1981) 25:91–5. doi: 10.1016/0022-3999(81)90095-7
53. Demitrack MA, Dale JK, Straus SE, Laue L, Listwak SJ, Kruesi MJ, et al. Evidence for impaired activation of the hypothalamic-pituitary-adrenal axis in patients with chronic fatigue syndrome. J Clin Endocrinol Metab. (1991) 73:1224–34. doi: 10.1210/jcem-73-6-1224
54. Scott LV, Medbak S, Dinan TG. Blunted adrenocorticotropin and cortisol responses to corticotropin-releasing hormone stimulation in chronic fatigue syndrome. Acta Psychiatr Scand. (1998) 97:450–7. doi: 10.1111/j.1600-0447.1998.tb10030.x
55. De Becker P, De Meirleir K, Joos E, Campine I, Van Steenberge E, Smitz J, et al. Dehydroepiandrosterone (DHEA) response to i.v. ACTH in patients with chronic fatigue syndrome. Horm Metab Res. (1999) 31:18–21. doi: 10.1055/s-2007-978690
56. Cleare AJ, Miell J, Heap E, Sookdeo S, Young L, Malhi GS, et al. Hypothalamo-pituitary-adrenal axis dysfunction in chronic fatigue syndrome, and the effects of low-dose hydrocortisone therapy. J Clin Endocrinol Metab. (2001) 86:3545–54. doi: 10.1210/jcem.86.8.7735
57. Gaab J, Huster D, Peisen R, Engert V, Heitz V, Schad T, et al. Hypothalamic-pituitary-adrenal axis reactivity in chronic fatigue syndrome and health under psychological, physiological, and pharmacological stimulation. Psychosom Med. (2002) 64:951–62. doi: 10.1097/00006842-200211000-00012
58. Jerjes WK, Cleare AJ, Wessely S, Wood PJ, Taylor NF. Diurnal patterns of salivary cortisol and cortisone output in chronic fatigue syndrome. J Affect Disord. (2005) 87:299–304. doi: 10.1016/j.jad.2005.03.013
59. Segal TY, Hindmarsh PC, Viner RM. Disturbed adrenal function in adolescents with chronic fatigue syndrome. J Pediatr Endocrinol Metab. (2005) 18:295–301. doi: 10.1515/JPEM.2005.18.3.295
60. Van Den Eede F, Moorkens G, Hulstijn W, Van Houdenhove B, Cosyns P, Sabbe BG, et al. Combined dexamethasone/corticotropin-releasing factor test in chronic fatigue syndrome. Psychol Med. (2008) 38:963–73. doi: 10.1017/S0033291707001444
61. Van Den Eede F, Moorkens G, Van Houdenhove B, Cosyns P, Claes SJ. Hypothalamic-pituitary-adrenal axis function in chronic fatigue syndrome. Neuropsychobiology. (2007) 55:112–20. doi: 10.1159/000104468
62. Papadopoulos AS, Cleare AJ. Hypothalamic-pituitary-adrenal axis dysfunction in chronic fatigue syndrome. Nat Rev Endocrinol. (2011) 8:22–32. doi: 10.1038/nrendo.2011.153
63. Craddock TJ, Fritsch P, Rice MA Jr, del Rosario RM, Miller DB, et al. A role for homeostatic drive in the perpetuation of complex chronic illness: Gulf War Illness and chronic fatigue syndrome. PLoS ONE. (2014) 9:e84839. doi: 10.1371/journal.pone.0084839
64. Gaab J, Engert V, Heitz V, Schad T, Schurmeyer TH, Ehlert U. Associations between neuroendocrine responses to the Insulin Tolerance Test and patient characteristics in chronic fatigue syndrome. J Psychosom Res. (2004) 56:419–24. doi: 10.1016/S0022-3999(03)00625-1
65. Crofford LJ, Young EA, Engleberg NC, Korszun A, Brucksch CB, McClure LA, et al. Basal circadian and pulsatile ACTH and cortisol secretion in patients with fibromyalgia and/or chronic fatigue syndrome. Brain Behav Immun. (2004) 18:314–25. doi: 10.1016/j.bbi.2003.12.011
66. Di Giorgio A, Hudson M, Jerjes W, Cleare AJ. 24-hour pituitary and adrenal hormone profiles in chronic fatigue syndrome. Psychosom Med. (2005) 67:433-40. doi: 10.1097/01.psy.0000161206.55324.8a
67. Pednekar DD, Amin MR, Fekri Azgomi H, Aschbacher K, Crofford LJ, Faghih RT. Characterization of cortisol dysregulation in fibromyalgia and chronic fatigue syndromes: a state-space approach. IEEE Trans Biomed Eng. (2020) 67:3163–72. doi: 10.1109/TBME.2020.2978801
68. Scott LV, Teh J, Reznek R, Martin A, Sohaib A, Dinan TG. Small adrenal glands in chronic fatigue syndrome: a preliminary computer tomography study. Psychoneuroendocrinology. (1999) 24:759–68. doi: 10.1016/S0306-4530(99)00028-1
69. Ben-Zvi A, Vernon SD, Broderick G. Model-based therapeutic correction of hypothalamic-pituitary-adrenal axis dysfunction. PLoS Comput Biol. (2009) 5:e1000273. doi: 10.1371/journal.pcbi.1000273
70. Sedghamiz H, Morris M, Craddock TJA, Whitley D, Broderick G. High-fidelity discrete modeling of the HPA axis: a study of regulatory plasticity in biology. BMC Syst Biol. (2018) 12:76. doi: 10.1186/s12918-018-0599-1
71. Zarzer CA, Puchinger MG, Kohler G, Kugler P. Differentiation between genomic and non-genomic feedback controls yields an HPA axis model featuring hypercortisolism as an irreversible bistable switch. Theor Biol Med Model. (2013) 10:65. doi: 10.1186/1742-4682-10-65
72. Hosseinichimeh N, Rahmandad H, Wittenborn AK. Modeling the hypothalamus-pituitary-adrenal axis: a review and extension. Math Biosci. (2015) 268:52–65. doi: 10.1016/j.mbs.2015.08.004
73. Craddock TJ, Del Rosario RR, Rice M, Zysman JP, Fletcher MA, Klimas NG, et al. Achieving remission in gulf war illness: a simulation-based approach to treatment design. PLoS ONE. (2015) 10:e0132774. doi: 10.1371/journal.pone.0132774
74. Morris MC, Cooney KE, Sedghamiz H, Abreu M, Collado F, Balbin EG, et al. Leveraging prior knowledge of endocrine immune regulation in the therapeutically relevant phenotyping of women with chronic fatigue syndrome. Clin Ther. (2019) 41:656–74 e4. doi: 10.1016/j.clinthera.2019.03.002
75. Kirnap M, Colak R, Eser C, Ozsoy O, Tutuş A, Kelestimur F. A comparison between low-dose (1 μg), standard-dose (250 μg) ACTH stimulation tests and insulin tolerance test in the evaluation of hypothalamo–pituitary–adrenal axis in primary fibromyalgia syndrome. Clin Endocrinol. (2001) 55:455–9. doi: 10.1046/j.1365-2265.2001.01373.x
76. Riedel W, Schlapp U, Leck S, Netter P, Neeck G. Blunted ACTH and cortisol responses to systemic injection of corticotropin-releasing hormone (CRH) in fibromyalgia: role of somatostatin and CRH-binding protein. Ann N Y Acad Sci. (2002) 966:483–90. doi: 10.1111/j.1749-6632.2002.tb04251.x
77. Holtorf K. Diagnosis and Treatment of Hypothalamic-Pituitary-Adrenal (HPA) Axis Dysfunction in Patients with Chronic Fatigue Syndrome (CFS) and Fibromyalgia (FM). J Chronic Fatigue Syndrome. (2008) 14:59–88. doi: 10.1300/J092v14n03_06
78. Gupta A, Silman AJ. Psychological stress and fibromyalgia: a review of the evidence suggesting a neuroendocrine link. Arthritis Res Ther. (2004) 6:98–106. doi: 10.1186/ar1176
79. Jason LA, Porter N, Herrington J, Sorenson M, Kubow S. Kindling and oxidative stress as contributors to myalgic encephalomyelitis/chronic fatigue syndrome. J Behav Neurosci Res. (2009) 7:1–17.
80. Morris G, Maes M. Oxidative and nitrosative stress and immune-inflammatory pathways in patients with Myalgic Encephalomyelitis (ME)/Chronic Fatigue Syndrome (CFS). Curr Neuropharmacol. (2014) 12:168–85. doi: 10.2174/1570159X11666131120224653
81. Morris G, Anderson G, Maes M. Hypothalamic-pituitary-adrenal hypofunction in Myalgic Encephalomyelitis (ME)/Chronic Fatigue Syndrome (CFS) as a consequence of activated immune-inflammatory and oxidative and nitrosative pathways. Mol Neurobiol. (2017) 54:6806–19. doi: 10.1007/s12035-016-0170-2
82. Hatziagelaki E, Adamaki M, Tsilioni I, Dimitriadis G, Theoharides TC. Myalgic encephalomyelitis/chronic fatigue syndrome-metabolic disease or disturbed homeostasis due to focal inflammation in the hypothalamus? J Pharmacol Exp Ther. (2018) 367:155–67. doi: 10.1124/jpet.118.250845
83. Elijah IE, Branski LK, Finnerty CC, Herndon DN. The GH/IGF-1 system in critical illness. Best Pract Res Clin Endocrinol Metab. (2011) 25:759–67. doi: 10.1016/j.beem.2011.06.002
84. Wallymahmed ME, Foy P, Shaw D, Hutcheon R, Edwards RH, MacFarlane IA. Quality of life, body composition and muscle strength in adult growth hormone deficiency: the influence of growth hormone replacement therapy for up to 3 years. Clin Endocrinol (Oxf). (1997) 47:439–46. doi: 10.1046/j.1365-2265.1997.2801076.x
85. van Dam PS. Neurocognitive function in adults with growth hormone deficiency. Horm Res. (2005) 64(Suppl. 3):109–14. doi: 10.1159/000089326
86. Van den Berghe G. Impact of critical illness on the growth hormone/insulin growth factor system in relation to other endocrine responses. In: Houston MS, Holly JMP, Feldman EL, editors. IGF and Nutrition in Health and Disease. Totowa, NJ: Humana Press. (2004) p. 291-309. doi: 10.1007/978-1-59259-795-6_16
87. Van den Berghe G. Endocrine evaluation of patients with critical illness. Endocrinol Metab Clin North Am. (2003) 32:385–410. doi: 10.1016/S0889-8529(03)00005-7
88. Baxter RC. Changes in the IGF–IGFBP axis in critical illness. Best Pract Res Clin Endocrinol Metabol. (2001) 15:421–34. doi: 10.1053/beem.2001.0161
89. Van den Berghe G, Wouters P, Weekers F, Mohan S, Baxter RC, Veldhuis JD, et al. Reactivation of pituitary hormone release and metabolic improvement by infusion of growth hormone-releasing peptide and thyrotropin-releasing hormone in patients with protracted critical illness. J Clin Endocrinol Metab. (1999) 84:1311–23. doi: 10.1210/jc.84.4.1311
90. Berwaerts J, Moorkens G, Abs R. Secretion of growth hormone in patients with chronic fatigue syndrome. Growth Horm IGF Res. (1998) 8(Suppl. B):127–9. doi: 10.1016/S1096-6374(98)80036-1
91. Moorkens G, Berwaerts J, Wynants H, Abs R. Characterization of pituitary function with emphasis on GH secretion in the chronic fatigue syndrome. Clin Endocrinol. (2000) 53:99–106. doi: 10.1046/j.1365-2265.2000.01049.x
92. Allain TJ, Bearn JA, Coskeran P, Jones J, Checkley A, Butler J, et al. Changes in growth hormone, insulin, insulinlike growth factors (IGFs), and IGF-binding protein-1 in chronic fatigue syndrome. Biol Psychiatry. (1997) 41:567–73. doi: 10.1016/S0006-3223(96)00074-1
93. Cleare AJ, Sookdeo SS, Jones J, O'Keane V, Miell JP. Integrity of the growth hormone/insulin-like growth factor system is maintained in patients with chronic fatigue syndrome. J Clin Endocrinol Metab. (2000) 85:1433–9. doi: 10.1210/jc.85.4.1433
94. The GK, Bleijenberg G, van der Meer JW. The effect of acclydine in chronic fatigue syndrome: a randomized controlled trial. PLoS Clin Trials. (2007) 2:e19. doi: 10.1371/journal.pctr.0020019
95. Bennett RM, Clark SR, Campbell SM, Burckhardt CS. Low levels of somatomedin C in patients with the fibromyalgia syndrome. A possible link between sleep and muscle pain. Arthritis Rheum. (1992) 35:1113–6. doi: 10.1002/art.1780351002
96. Bennett RM, Cook DM, Clark SR, Burckhardt CS, Campbell SM. Hypothalamic-pituitary-insulin-like growth factor-I axis dysfunction in patients with fibromyalgia. J Rheumatol. (1997) 24:1384–9.
97. Paiva ES, Deodhar A, Jones KD, Bennett R. Impaired growth hormone secretion in fibromyalgia patients: evidence for augmented hypothalamic somatostatin tone. Arthritis Rheum. (2002) 46:1344–50. doi: 10.1002/art.10209
98. Cuatrecasas G, Gonzalez MJ, Alegre C, Sesmilo G, Fernandez-Sola J, Casanueva FF, et al. High prevalence of growth hormone deficiency in severe fibromyalgia syndromes. J Clin Endocrinol Metab. (2010) 95:4331–7. doi: 10.1210/jc.2010-0061
99. Rigamonti AE, Grugni G, Arreghini M, Capodaglio P, De Col A, Agosti F, et al. GH responsiveness to combined gh-releasing hormone and arginine administration in obese patients with fibromyalgia syndrome. Int J Endocrinol. (2017) 2017:3106041. doi: 10.1155/2017/3106041
100. Bennett RM, Clark SC, Walczyk J. A randomized, double-blind, placebo-controlled study of growth hormone in the treatment of fibromyalgia. Am J Med. (1998) 104:227–31. doi: 10.1016/S0002-9343(97)00351-3
101. Ross RL, Jones KD, Bennett RM, Ward RL, Druker BJ, Wood LJ. Preliminary evidence of increased pain and elevated cytokines in fibromyalgia patients with defective growth hormone response to exercise. Open Immunol J. (2010) 3:9–18. doi: 10.2174/1874226201003010009
102. Fisher DA. Physiological variations in thyroid hormones: physiological and pathophysiological considerations. Clin Chem. (1996) 42:135–9. doi: 10.1093/clinchem/42.1.135
103. De Groot LJ. Dangerous dogmas in medicine: the nonthyroidal illness syndrome. J Clin Endocrinol Metab. (1999) 84:151–64. doi: 10.1210/jcem.84.1.5364
104. Wajner SM, Maia AL. new insights toward the acute non-thyroidal illness syndrome. Front Endocrinol. (2012) 3:8. doi: 10.3389/fendo.2012.00008
105. Warner MH, Beckett GJ. Mechanisms behind the non-thyroidal illness syndrome: an update. J Endocrinol. (2010) 205:1–13. doi: 10.1677/JOE-09-0412
106. Carter JN, Eastman CJ, Corcoran JM, Lazarus L. Effect of severe, chronic illness on thyroid function. Lancet. (1974) 2:971–4. doi: 10.1016/S0140-6736(74)92070-4
107. Plikat K, Langgartner J, Buettner R, Bollheimer LC, Woenckhaus U, Scholmerich J, et al. Frequency and outcome of patients with nonthyroidal illness syndrome in a medical intensive care unit. Metabolism. (2007) 56:239–44. doi: 10.1016/j.metabol.2006.09.020
108. Boelen A, Kwakkel J, Fliers E. Beyond low plasma T3: local thyroid hormone metabolism during inflammation and infection. Endocr Rev. (2011) 32:670–93. doi: 10.1210/er.2011-0007
109. Boelen A, Platvoet-Ter Schiphorst MC, Wiersinga WM. Association between serum interleukin-6 and serum 3,5,3′-triiodothyronine in nonthyroidal illness. J Clin Endocrinol Metabol. (1993) 77:1695–9. doi: 10.1210/jcem.77.6.8263160
110. Davies PH, Black EG, Sheppard MC, Franklyn JA. Relation between serum interleukin-6 and thyroid hormone concentrations in 270 hospital in-patients with non-thyroidal illness. Clin Endocrinol (Oxf). (1996) 44:199–205. doi: 10.1046/j.1365-2265.1996.668489.x
111. Wajner SM, Goemann IM, Bueno AL, Larsen PR, Maia AL. IL-6 promotes nonthyroidal illness syndrome by blocking thyroxine activation while promoting thyroid hormone inactivation in human cells. J Clin Invest. (2011) 121:1834–45. doi: 10.1172/JCI44678
112. Bartalena L, Farsetti A, Flink IL, Robbins J. Effects of interleukin-6 on the expression of thyroid hormone-binding protein genes in cultured human hepatoblastoma-derived (Hep G2) cells. Mol Endocrinol. (1992) 6:935–42. doi: 10.1210/mend.6.6.1323058
113. Bartalena L, Bogazzi F, Brogioni S, Grasso L, Martino E. Role of cytokines in the pathogenesis of the euthyroid sick syndrome. Eur J Endocrinol. (1998) 138 6:603–14. doi: 10.1530/eje.0.1380603
114. Afandi B, Vera R, Schussler GC, Yap MG. Concordant decreases of thyroxine and thyroxine binding protein concentrations during sepsis. Metabolism. (2000) 49:753–4. doi: 10.1053/meta.2000.6239
115. Bartalena L, Robbins J. Variations in thyroid hormone transport proteins and their clinical implications. Thyroid. (1992) 2:237–45. doi: 10.1089/thy.1992.2.237
116. Mebis L, Paletta D, Debaveye Y, Ellger B, Langouche L, D'Hoore A, et al. Expression of thyroid hormone transporters during critical illness. Eur J Endocrinol. (2009) 161:243. doi: 10.1530/EJE-09-0290
117. Huang SA, Mulcahey MA, Crescenzi A, Chung M, Kim BW, Barnes C, et al. Transforming growth factor-beta promotes inactivation of extracellular thyroid hormones via transcriptional stimulation of type 3 iodothyronine deiodinase. Mol Endocrinol. (2005) 19:3126–36. doi: 10.1210/me.2005-0173
118. Kwakkel J, Wiersinga WM, Boelen A. Interleukin-1beta modulates endogenous thyroid hormone receptor alpha gene transcription in liver cells. J Endocrinol. (2007) 194:257–65. doi: 10.1677/JOE-06-0177
119. Rodriguez-Perez A, Palos-Paz F, Kaptein E, Visser TJ, Dominguez-Gerpe L, Alvarez-Escudero J, et al. Identification of molecular mechanisms related to nonthyroidal illness syndrome in skeletal muscle and adipose tissue from patients with septic shock. Clin Endocrinol. (2008) 68:821–7. doi: 10.1111/j.1365-2265.2007.03102.x
120. Lado-Abeal J, Romero A, Castro-Piedras I, Rodriguez-Perez A, Alvarez-Escudero J. Thyroid hormone receptors are down-regulated in skeletal muscle of patients with non-thyroidal illness syndrome secondary to non-septic shock. Eur J Endocrinol. (2010) 163:765–73. doi: 10.1530/EJE-10-0376
121. Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, et al. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. (2008) 29:898–938. doi: 10.1210/er.2008-0019
122. De Groot LJ. The non-thyroidal illness syndrome [Updated 2015 Feb 1]. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, et al., editors. Endotext. South Dartmouth, MA: MDText.com, Inc. (2000).
123. Mendoza A, Hollenberg AN. New insights into thyroid hormone action. Pharmacol Ther. (2017) 173:135–45. doi: 10.1016/j.pharmthera.2017.02.012
124. Cicatiello AG, Di Girolamo D, Dentice M. Metabolic effects of the intracellular regulation of thyroid hormone: old players, new concepts. Front Endocrinol (Lausanne). (2018) 9:474. doi: 10.3389/fendo.2018.00474
125. Fliers E, Bianco AC, Langouche L, Boelen A. Thyroid function in critically ill patients. Lancet Diabetes Endocrinol. (2015) 3:816–25. doi: 10.1016/S2213-8587(15)00225-9
126. Moura Neto A, Zantut-Wittmann DE. Abnormalities of thyroid hormone metabolism during systemic illness: the low T3 syndrome in different clinical settings. Int J Endocrinol. (2016) 2016:2157583. doi: 10.1155/2016/2157583
127. Boelen A, Kwakkel J, Thijssen-Timmer DC, Alkemade A, Fliers E, Wiersinga WM. Simultaneous changes in central and peripheral components of the hypothalamus-pituitary-thyroid axis in lipopolysaccharide-induced acute illness in mice. J Endocrinol. (2004) 182:315–23. doi: 10.1677/joe.0.1820315
128. Joseph-Bravo P, Jaimes-Hoy L, Charli JL. Regulation of TRH neurons and energy homeostasis-related signals under stress. J Endocrinol. (2015) 224:R139–59. doi: 10.1530/JOE-14-0593
129. Harel G, Shamoun DS, Kane JP, Magner JA, Szabo M. Prolonged effects of tumor necrosis factor-alpha on anterior pituitary hormone release. Peptides. (1995) 16:641–5. doi: 10.1016/0196-9781(95)00019-G
130. Wassen FW, Moerings EP, Van Toor H, De Vrey EA, Hennemann G, Everts ME. Effects of interleukin-1 beta on thyrotropin secretion and thyroid hormone uptake in cultured rat anterior pituitary cells. Endocrinology. (1996) 137:1591–8. doi: 10.1210/endo.137.5.8612490
131. Teitelbaum J, Bird B. Effective treatment of severe chronic fatigue. J Musculoskeletal Pain. (1995) 3:91–110. doi: 10.1300/J094v03n04_11
132. Fuite J, Vernon SD, Broderick G. Neuroendocrine and immune network re-modeling in chronic fatigue syndrome: an exploratory analysis. Genomics. (2008) 92:393–9. doi: 10.1016/j.ygeno.2008.08.008
133. Holtorf K. Thyroid hormone transport into cellular tissue. J Restorative Med. (2014) 3:53–68. doi: 10.14200/jrm.2014.3.0104
134. Holtorf K. Peripheral thyroid hormone conversion and its impact on TSH and metabolic activity. J Restorative Med. (2014) 3:30–52. doi: 10.14200/jrm.2014.3.0103
135. Neeck G, Riedel W. Thyroid function in patients with fibromyalgia syndrome. J Rheumatol. (1992) 19:1120–2.
136. Lowe JC. Thyroid status of 38 fibromyalgia patients. Clin Bull Myofascial Therapy. (1996) 2:47–64. doi: 10.1300/J425v02n01_07
137. Lowe JC, Reichman AJ, Honeyman GS, Yellin J. Thyroid status of fibromyalgia patients. Clin Bull Myofascial Therapy. (1998) 3:69–70. doi: 10.1300/J425v03n01_08
138. Lowe JC, Yellin JG. The Metabolic Treatment of Fibromyalgia. Boulder, CO: McDowell Publishing Company (2000). 1260 p.
139. Garrison RL, Breeding PC. A metabolic basis for fibromyalgia and its related disorders: the possible role of resistance to thyroid hormone. Medical Hypotheses. (2003) 61:182–9. doi: 10.1016/S0306-9877(02)00294-3
140. Lowe JC, Yellin J. Inadequate thyroid hormone regulation as the main mechanism of fibromyalgia: a review of the evidence. Thyroid Sci. (2008) 3:R1–14.
141. Ruiz-Núñez B, Tarasse R, Vogelaar EF, Janneke Dijck-Brouwer DA, Muskiet FAJ. Higher prevalence of “low t3 syndrome” in patients with chronic fatigue syndrome: a case–control study. Front Endocrinol. (2018) 9:97. doi: 10.3389/fendo.2018.00097
142. Mebis L, Langouche L, Visser T, van den Berghe G. The type II iodothyronine deiodinase is up-regulated in skeletal muscle during prolonged critical illness. J Clin Endocrinol Metabolism. (2007) 92:3330–3. doi: 10.1210/jc.2007-0510
143. Boelen A, van der Spek AH, Bloise F, de Vries EM, Surovtseva OV, van Beeren M, et al. Tissue thyroid hormone metabolism is differentially regulated during illness in mice. J Endocrinol. (2017) 233:25–36. doi: 10.1530/JOE-16-0483
144. Fontes KN, Cabanelas A, Bloise FF, de Andrade CBV, Souza LL, Wilieman M, et al. Differential regulation of thyroid hormone metabolism target genes during non-thyroidal illness syndrome triggered by fasting or sepsis in adult mice. Front Physiol. (2017) 8:828. doi: 10.3389/fphys.2017.00995
145. Donzelli R, Colligiani D, Kusmic C, Sabatini M, Lorenzini L, Accorroni A, et al. Effect of hypothyroidism and hyperthyroidism on tissue thyroid hormone concentrations in rat. Eur Thyroid J. (2016) 5:27–34. doi: 10.1159/000443523
146. Dietrich JW, Landgrafe-Mende G, Wiora E, Chatzitomaris A, Klein HH, Midgley JE, et al. Calculated parameters of thyroid homeostasis: emerging tools for differential diagnosis and clinical research. Front Endocrinol. (2016) 7:57. doi: 10.3389/fendo.2016.00057
147. Resch U, Helsel G, Tatzber F, Sinzinger H. Antioxidant status in thyroid dysfunction. Clin Chem Lab Med. (2002) 40:1132–4. doi: 10.1515/cclm.2002.198
148. Cano-Europa E, Blas-Valdivia V, Franco-Colin M, Ortiz-Butron R. The relationship between thyroid states, oxidative stress and cellular damage. In: Lushchak V, editor. Oxidative Stress and Diseases. London: IntechOpen. (2012) p. 414-36.
149. Bianco AC, Sheng XY, Silva JE. Triiodothyronine amplifies norepinephrine stimulation of uncoupling protein gene transcription by a mechanism not requiring protein synthesis. J Biol Chem. (1988) 263:18168–75. doi: 10.1016/S0021-9258(19)81340-6
150. Wajner SM, Rohenkohl HC, Serrano T, Maia AL. Sodium selenite supplementation does not fully restore oxidative stress-induced deiodinase dysfunction: implications for the nonthyroidal illness syndrome. Redox Biol. (2015) 6:436–45. doi: 10.1016/j.redox.2015.09.002
151. Chatterjee S. Chapter two - oxidative stress, inflammation, and disease. In: Dziubla T. Butterfield DA, editors. Oxidative Stress and Biomaterials. Academic Press. (2016) p. 35-58. doi: 10.1016/B978-0-12-803269-5.00002-4
152. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. (2007) 39:44–84. doi: 10.1016/j.biocel.2006.07.001
153. Morris G, Maes M. Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Metab Brain Dis. (2014) 29:19–36. doi: 10.1007/s11011-013-9435-x
154. Armstrong CW, McGregor NR, Lewis DP, Butt HL, Gooley PR. Metabolic profiling reveals anomalous energy metabolism and oxidative stress pathways in chronic fatigue syndrome patients. Metabolomics. (2015) 11:1626–39. doi: 10.1007/s11306-015-0816-5
155. Pall ML. Chapter 2: The NO/ONOO? cycle mechanism as the cause of chronic fatigue syndrome/myalgia encephalomyelitis. In: Svoboda E, Zelenjcik K, editors. Chronic Fatigue Syndrome: Symptoms, Causes and Prevention. New York, NY: Nova Science (2009).
156. Shungu DC, Weiduschat N, Murrough JW, Mao X, Pillemer S, Dyke JP, et al. Increased ventricular lactate in chronic fatigue syndrome. III. Relationships to cortical glutathione and clinical symptoms implicate oxidative stress in disorder pathophysiology. NMR Biomed. (2012) 25:1073–87. doi: 10.1002/nbm.2772
157. Montoya JG, Holmes TH, Anderson JN, Maecker HT, Rosenberg-Hasson Y, Valencia IJ, et al. Cytokine signature associated with disease severity in chronic fatigue syndrome patients. Proc Natl Acad Sci USA. (2017) 114:E7150–E8. doi: 10.1073/pnas.1710519114
158. Hornig M, Montoya JG, Klimas NG, Levine S, Felsenstein D, Bateman L, et al. Distinct plasma immune signatures in ME/CFS are present early in the course of illness. Sci Adv. (2015) 1:e1400121. doi: 10.1126/sciadv.1400121
159. Hernandez ME, Becerril E, Perez M, Leff P, Anton B, Estrada S, et al. Proinflammatory cytokine levels in fibromyalgia patients are independent of body mass index. BMC Res Notes. (2010) 3:156. doi: 10.1186/1756-0500-3-156
160. VanElzakker MB, Brumfield SA, Lara Mejia PS. Neuroinflammation and cytokines in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): a critical review of research methods. Front Neurol. (2018) 9:1033. doi: 10.3389/fneur.2018.01033
161. Balazs C, Leovey A, Szabo M, Bako G. Stimulating effect of triiodothyronine on cell-mediated immunity. Eur J Clin Pharmacol. (1980) 17:19–23. doi: 10.1007/BF00561672
162. Pillay K. Congenital hypothyroidism and immunodeficiency: evidence for an endocrine-immune interaction. J Pediatr Endocrinol Metab. (1998) 11:757–61. doi: 10.1515/JPEM.1998.11.6.757
163. Klecha AJ, Genaro AM, Gorelik G, Barreiro Arcos ML, Silberman DM, Schuman M, et al. Integrative study of hypothalamus-pituitary-thyroid-immune system interaction: thyroid hormone-mediated modulation of lymphocyte activity through the protein kinase C signaling pathway. J Endocrinol. (2006) 189:45–55. doi: 10.1677/joe.1.06137
164. Klein JR. The immune system as a regulator of thyroid hormone activity. Exp Biol Med (Maywood). (2006) 231:229–36. doi: 10.1177/153537020623100301
165. Hans VH, Lenzlinger PM, Joller-Jemelka HI, Morganti-Kossmann MC, Kossmann T. Low T3 syndrome in head-injured patients is associated with prolonged suppression of markers of cell-mediated immune response. Eur J Trauma. (2005) 31:359–68. doi: 10.1007/s00068-005-2068-y
166. Hodkinson CF, Simpson EE, Beattie JH, O'Connor JM, Campbell DJ, Strain JJ, et al. Preliminary evidence of immune function modulation by thyroid hormones in healthy men and women aged 55-70 years. J Endocrinol. (2009) 202:55–63. doi: 10.1677/JOE-08-0488
167. Straub RH, Cutolo M, Buttgereit F, Pongratz G. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med. (2010) 267:543–60. doi: 10.1111/j.1365-2796.2010.02218.x
168. Jara EL, Munoz-Durango N, Llanos C, Fardella C, Gonzalez PA, Bueno SM, et al. Modulating the function of the immune system by thyroid hormones and thyrotropin. Immunol Lett. (2017) 184:76–83. doi: 10.1016/j.imlet.2017.02.010
169. Bilal MY, Dambaeva S, Kwak-Kim J, Gilman-Sachs A, Beaman KD. A role for iodide and thyroglobulin in modulating the function of human immune cells. Front Immunol. (2017) 8:1573. doi: 10.3389/fimmu.2017.01573
170. van der Spek AH, Surovtseva OV, Jim KK, van Oudenaren A, Brouwer MC, Vandenbroucke-Grauls C, et al. Regulation of intracellular triiodothyronine is essential for optimal macrophage function. Endocrinology. (2018) 159:2241–52. doi: 10.1210/en.2018-00053
171. De Vito P, Incerpi S, Pedersen JZ, Luly P, Davis FB, Davis PJ. Thyroid hormones as modulators of immune activities at the cellular level. Thyroid. (2011) 21:879–90. doi: 10.1089/thy.2010.0429
172. Klimas NG, Salvato FR, Morgan R, Fletcher MA. Immunologic abnormalities in chronic fatigue syndrome. J Clin Microbiol. (1990) 28:1403–10. doi: 10.1128/JCM.28.6.1403-1410.1990
173. Textoris J, Mallet F. Immunosuppression and herpes viral reactivation in intensive care unit patients: one size does not fit all. Crit Care. (2017) 21:230–. doi: 10.1186/s13054-017-1803-1
174. Coşkun O, Yazici E, Sahiner F, Karakaş A, Kiliç S, Tekin M, et al. Cytomegalovirus and Epstein–Barr virus reactivation in the intensive care unit. Medizinische Klinik - Intensivmedizin und Notfallmedizin. (2017) 112:239–45. doi: 10.1007/s00063-016-0198-0
175. Walton AH, Muenzer JT, Rasche D, Boomer JS, Sato B, Brownstein BH, et al. Reactivation of multiple viruses in patients with sepsis. PLoS ONE. (2014) 9:e98819. doi: 10.1371/journal.pone.0098819
176. Rasa S, Nora-Krukle Z, Henning N, Eliassen E, Shikova E, Harrer T, et al. Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J Transl Med. (2018) 16:268. doi: 10.1186/s12967-018-1644-y
177. Kerr JR. Epstein-barr virus induced gene-2 upregulation identifies a particular subtype of chronic fatigue syndrome/myalgic encephalomyelitis. Front Pediatr. (2019) 7:59. doi: 10.3389/fped.2019.00059
178. Lizcano F, Rodríguez JS. Thyroid hormone therapy modulates hypothalamo-pituitary-adrenal axis. Endocr J. (2011) 58:137–42. doi: 10.1507/endocrj.K10E-369
179. Sánchez-Franco F, Fernández L, Fernández G, Cacicedo L. Thyroid hormone action on ACTH secretion. Horm Metab Res. (1989) 21:550–2. doi: 10.1055/s-2007-1009285
180. Preiser JC, Ichai C, Orban JC, Groeneveld AB. Metabolic response to the stress of critical illness. Br J Anaesth. (2014) 113:945–54. doi: 10.1093/bja/aeu187
181. McBride MA, Owen AM, Stothers CL, Hernandez A, Luan L, Burelbach KR, et al. The metabolic basis of immune dysfunction following sepsis and trauma. Front Immunol. (2020) 11:1043. doi: 10.3389/fimmu.2020.01043
182. van der Spek AH, Fliers E, Boelen A. Thyroid hormone metabolism in innate immune cells. J Endocrinol. (2017) 232:R67–R81. doi: 10.1530/JOE-16-0462
183. Bynke A, Julin P, Gottfries C-G, Heidecke H, Scheibenbogen C, Bergquist J. Autoantibodies to beta-adrenergic and muscarinic cholinergic receptors in Myalgic Encephalomyelitis (ME) patients—A validation study in plasma and cerebrospinal fluid from two Swedish cohorts. Brain Behav Immunity Health. (2020) 7:100107. doi: 10.1016/j.bbih.2020.100107
184. Van Houdenhove B, Van Den Eede F, Luyten P. Does hypothalamic-pituitary-adrenal axis hypofunction in chronic fatigue syndrome reflect a ‘crash’ in the stress system? Med Hypotheses. (2009) 72:701–5. doi: 10.1016/j.mehy.2008.11.044
185. Arroll MA. Allostatic overload in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Med Hypotheses. (2013) 81:506–8. doi: 10.1016/j.mehy.2013.06.023
186. Neeck G, Crofford LJ. Neuroendocrine peturbations in fibromyalgia and chronic fatigue syndrome. Rheumatic Disease Clinics North America. (2000) 26:989–1002. doi: 10.1016/S0889-857X(05)70180-0
187. Adler GK, Manfredsdottir VF, Creskoff KW. Neuroendocrine abnormalities in fibromyalgia. Current Pain Headache Reports. (2002) 6:289–98. doi: 10.1007/s11916-002-0050-5
188. Teitelbaum J. From Fatigued to Fantastic!: A Clinically Proven Program to Regain Vibrant Health and Overcome Chronic Fatigue and Fibromyalgia: Avery. (2007). 424 p.
189. Riedel W, Layka H, Neeck G. Secretory pattern of GH, TSH, thyroid hormones, ACTH, cortisol, FSH, and LH in patients with fibromyalgia syndrome following systemic injection of the relevant hypothalamic-releasing hormones. Z Rheumatol. (1998) 57(Suppl. 2):81–7. doi: 10.1007/s003930050242
190. Wilson ED. Wilson's Syndrome: The Miracle of Feeling Well. Houston, TX: Cornerstone Publishing Company (1991). 346 p.
191. Honeyman-Lowe G, Lowe JC. Your Guide to Metabolic Health. McDowell Health-Science Books. Boulder, CO: LLC (2003). 384 p.
192. Skinner G. Diagnosis and Management of Hypothyroidism. West Midlands: Louise Lorne (2003). 201 p.
193. Durrant-Peatfield B. Your Thyroid and How to Keep it Healthy. London: Hammersmith Press Limited (2006). 264 p.
194. Myhill S. Diagnosis and Treatment of Chronic Fatigue Syndrome and Myalgic Encephalitis, 2nd ed.: It's Mitochondria, Not Hypochondria. White River Junction, VT: Chelsea Green Publishing (2018). 432 p.
195. Holmes D. Tears Behind Closed Doors: Failure to Diagnose a Thyroid Condition. Wolverhampton: Normandi Publishing Limited (2002). 270 p.
196. Bowthorpe JA. Stop the Thyroid Madness: A Patient Revolution Against Decades of Inferior Thyroid Treatment. Fredericksburg, TX: Laughing Grape Pub. (2008). 293 p.
197. Robinson P. Recovering with T3: My Journey from Hypothyroidism to Good Health Using the T3 Thyroid Hormone. Elephant in the Room Books (2018). 288 p.
198. Fremont M, Metzger K, Rady H, Hulstaert J, De Meirleir K. Detection of herpesviruses and parvovirus B19 in gastric and intestinal mucosa of chronic fatigue syndrome patients. In Vivo. (2009) 23:209–13.
199. Katz BZ, Shiraishi Y, Mears CJ, Binns HJ, Taylor R. Chronic fatigue syndrome after infectious mononucleosis in adolescents. Pediatrics. (2009) 124:189–93. doi: 10.1542/peds.2008-1879
200. Katz BZ, Jason LA. Chronic fatigue syndrome following infections in adolescents. Curr Opin Pediatr. (2013) 25:95–102. doi: 10.1097/MOP.0b013e32835c1108
201. Hickie I, Davenport T, Wakefield D, Vollmer-Conna U, Cameron B, Vernon SD, et al. Post-infective and chronic fatigue syndromes precipitated by viral and non-viral pathogens: prospective cohort study. BMJ. (2006) 333:575. doi: 10.1136/bmj.38933.585764.AE
202. Blomberg J, Gottfries CG, Elfaitouri A, Rizwan M, Rosén A. Infection elicited autoimmunity and myalgic encephalomyelitis/chronic fatigue syndrome: an explanatory model. Front Immunol. (2018) 9:229. doi: 10.3389/fimmu.2018.00229
203. Morris G, Berk M, Galecki P, Maes M. The emerging role of autoimmunity in myalgic encephalomyelitis/chronic fatigue syndrome (ME/cfs). Mol Neurobiol. (2014) 49:741–56. doi: 10.1007/s12035-013-8553-0
204. Glassford JA. The neuroinflammatory etiopathology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Front Physiol. (2017) 8:88. doi: 10.3389/fphys.2017.00088
205. Tomas C, Newton J. Metabolic abnormalities in chronic fatigue syndrome/myalgic encephalomyelitis: a mini-review. Biochem Soc Trans. (2018) 46:547–53. doi: 10.1042/BST20170503
206. Keller BA, Pryor JL, Giloteaux L. Inability of myalgic encephalomyelitis/chronic fatigue syndrome patients to reproduce VO(2)peak indicates functional impairment. J Transl Med. (2014) 12:104. doi: 10.1186/1479-5876-12-104
207. Myhill S, Booth NE, McLaren-Howard J. Chronic fatigue syndrome and mitochondrial dysfunction. Int J Clin Exp Med. (2009) 2:1–16.
208. Esfandyarpour R, Kashi A, Nemat-Gorgani M, Wilhelmy J, Davis RW. A nanoelectronics-blood-based diagnostic biomarker for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Proc Natl Acad Sci U S A. (2019) 116:10250–7. doi: 10.1073/pnas.1901274116
209. Vernon SD, Whistler T, Cameron B, Hickie IB, Reeves WC, Lloyd A. Preliminary evidence of mitochondrial dysfunction associated with post-infective fatigue after acute infection with Epstein Barr virus. BMC Infect Dis. (2006) 6:15. doi: 10.1186/1471-2334-6-15
210. Fluge O, Mella O, Bruland O, Risa K, Dyrstad SE, Alme K, et al. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight. (2016) 1:e89376. doi: 10.1172/jci.insight.89376
211. Germain A, Ruppert D, Levine SM, Hanson MR. Metabolic profiling of a myalgic encephalomyelitis/chronic fatigue syndrome discovery cohort reveals disturbances in fatty acid and lipid metabolism. Mol Biosyst. (2017) 13:371–9. doi: 10.1039/C6MB00600K
212. Naviaux RK, Naviaux JC, Li K, Bright AT, Alaynick WA, Wang L, et al. Metabolic features of chronic fatigue syndrome. Proc Natl Acad Sci USA. (2016) 113:E5472–80. doi: 10.1073/pnas.1607571113
213. Kashi AA, Davis RW, Phair RD. The IDO metabolic trap hypothesis for the etiology of ME/CFS. Diagnostics (Basel). (2019) 9:82. doi: 10.3390/diagnostics9030082
214. Perez M, Jaundoo R, Hilton K, Del Alamo A, Gemayel K, Klimas NG, et al. Genetic predisposition for immune system, hormone, and metabolic dysfunction in Myalgic encephalomyelitis/chronic fatigue syndrome: a pilot study. Front Pediatrics. (2019) 7:206. doi: 10.3389/fped.2019.00206
215. Carlo-Stella N, Badulli C, De Silvestri A, Bazzichi L, Martinetti M, Lorusso L, et al. A first study of cytokine genomic polymorphisms in CFS: positive association of TNF-857 and IFNgamma 874 rare alleles. Clin Exp Rheumatol. (2006) 24:179–82.
216. Vangeel E, Van Den Eede F, Hompes T, Izzi B, Del Favero J, Moorkens G, et al. Chronic fatigue syndrome and DNA hypomethylation of the glucocorticoid receptor gene promoter 1F region: associations with HPA axis hypofunction and childhood Trauma. Psychosom Med. (2015) 77:853–62. doi: 10.1097/PSY.0000000000000224
217. Azoulay E, Vincent JL, Angus DC, Arabi YM, Brochard L, Brett SJ, et al. Recovery after critical illness: putting the puzzle together-a consensus of 29. Crit Care. (2017) 21:296. doi: 10.1186/s13054-017-1887-7
218. Szubski CR, Tellez A, Klika AK, Xu M, Kattan MW, Guzman JA, et al. Predicting discharge to a long-term acute care hospital after admission to an intensive care unit. Am J Crit Care. (2014) 23:e46–53. doi: 10.4037/ajcc2014985
219. Stanculescu D. “Neither Dying, nor Recovering”: Learning from ICUs to Solve ME/CFS and Fibromyalgia – A Synopsis [Internet]. Health Rising, (2019 November 21). Available online at: https://www.healthrising.org/blog/2019/11/21/neither-dying-nor-recovering_icus-fibromyalgia-chronic-fatigue-syndrome/ (accessed September 6, 2020).
220. Stanculescu D. The relevance of research on critical illnesses for chronic fatigue syndrome ME/CFS: a vicious cycle between cytokines, oxidative stress and thyroid hormones [Internet]. Health Rising. (2019). Available online at: https://www.healthrising.org/blog/2019/07/24/chronic-fatigue-syndrome-ntis-low-t3-vicious-circle/ (accessed September 9, 2020).
221. Stanculescu D. Neuroendocrine dysfunctions in prolonged critical illness: relevance for chronic fatigue syndrome ME/CFS and Fibromyalgia Pt I [Internet]. Health Rising. (2019). Available oline at: https://www.healthrising.org/blog/2019/09/05/neuroendocrine-dysfunctions-and-treatments-in-prolonged-critical-illness-relevance-for-chronic-fatigue-syndrome-me-cfs-and-fibromyalgia/ (accessed September 6, 2020).
Keywords: myalgic encephalomyelitis, critical illness, non-thyroidal illness syndrome, low t-3 syndrome, pituitary, cytokines, oxidative and nitrosative stress, post-intensive care syndrome
Citation: Stanculescu D, Larsson L and Bergquist J (2021) Hypothesis: Mechanisms That Prevent Recovery in Prolonged ICU Patients Also Underlie Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Front. Med. 8:628029. doi: 10.3389/fmed.2021.628029
Received: 10 November 2020; Accepted: 08 January 2021;
Published: 28 January 2021.
Edited by:
Nuno Sepulveda, Charité–Universitätsmedizin Berlin, GermanyReviewed by:
Jose Alegre-Martin, Vall d'Hebron University Hospital, SpainKlaus Wirth, Sanofi, Germany
Jonathan Kerr, Norfolk and Norwich University Hospital, United Kingdom
Copyright © 2021 Stanculescu, Larsson and Bergquist. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonas Bergquist, Sm9uYXMuQmVyZ3F1aXN0QGtlbWkudXUuc2U=