 Yuri Battaglia1*
Yuri Battaglia1* Fulvio Fiorini2Cristiano Azzini3
Fulvio Fiorini2Cristiano Azzini3 Pasquale Esposito4,5Alessandro De vito3Antonio Granata6Alda Storari1Renzo Mignani7
Pasquale Esposito4,5Alessandro De vito3Antonio Granata6Alda Storari1Renzo Mignani7- 1Division of Nephrology and Dialysis, St. Anna University Hospital, Ferrara, Italy
- 2Division of Nephrology and Dialysis, “Santa Maria della Misericordia” Hospital, Rovigo, Italy
- 3Department of Neuroscience and Rehabilitation, Ferrara University Hospital, Ferrara, Italy
- 4Division of Nephrology, Dialysis and Transplantation, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Ospedale Policlinico San Martino, Genoa, Italy
- 5Department of Internal Medicine, University of Genoa, Genoa, Italy
- 6Division of Nephrology and Dialysis, “Cannizzaro” Hospital, Catania, Italy
- 7Division of Nephrology and Dialysis Department, Infermi Hospital, Rimini, Italy
Fabry Disease (FD), a rare and progressive, X-linked lysosomal storage disorder, is caused by mutations in the α-galactosidase A (GLA) gene which leads to enzymatic deficiency of GLA. Misdiagnosed and undiagnosed FD cases are common for the variable FD phenotype, ranging from asymptomatic and/or impairment of single organs, which is typically seen in females and in patients with late-onset mutation, to multiple organ disease, which is frequently found in males with classic GLA mutation. Consequently, for an early diagnosis and an efficient treatment of FD, three different strategies of screening, new-born screening, high-risk screening and familiar screening, have been conducted. However, most of FD screening in the CKD population has been carried out in hemodialysis patients and kidney transplant recipients, for whom the renal damage is already irreversible, so the effectiveness of enzymatic replacement therapy is limited and delayed therapeutic intervention results in worse long-term outcomes. This review investigates the actual strategies of screening initiatives for the identification of FD, examining in detail those performed in CKD patients not on dialysis.
Introduction
Screening is considered one of the main processes to detect healthy patients affected by latent or early symptomatic stages of the disease (1). It allows an early diagnosis and an effective treatment of the disease, reducing the cost of medical care and improving the quality of life.
According to the classic Wilson and Jungner criteria1, this procedure takes into account whether the disease is adequately understood and is characterized by specific findings, including a delay between the detection of risk factors and the onset of the disease.
Due to the fact that chronic kidney disease (CKD) occurs asymptomatically and is clinically silent in early stages, large-scale national screening programs (2) have been only conducted in some countries, showing more than 10% presence of markers for renal damage (3).
However, a global awareness campaign, promoted by international and national societies of nephrology to reduce the impact of CKD, confirmed that risk factors, such as high blood pressure, were found in roughly 10% of both adult and adolescent populations (4–6).
Although the etiology of CKD is primarily due to diabetes and hypertension (7–9), a considerable proportion of unknown CKD cases are caused by undetected genetic diseases, including Fabry Disease (FD).
This review analyzes the recent developments on different approaches of screening FD programs with a specific focus on those which were performed on not dialysis-dependent chronic kidney disease (NDD-CKD) patients.
Pathogenesis and Clinical Manifestation of Fabry Disease
Over the last 20 years, an increasing amount of the Nephrology community's attention has been captured by FD, a hereditary, X-linked, lysosomal storage disease (10). It is caused by deficiency of α-galactosidase A (GLA) and the gene encoding GLA is located on long arm of X-chromosome (Xq21-22) (11). The enzyme deficiency leads to the accumulation of glycosphingolipids, mainly globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-GB3), in lysosomes of skin, eye, kidney, heart, brain, and the peripheral nervous system (12–14). FD is a progressive disease that worsens with the age. Clinical manifestations ensue during childhood or early adolescence with angiokeratomas, lenticular and corneal opacity, microalbuminuria or proteinuria, gastrointestinal pain, recurrent fever, alterations of thermoregulation (i.e. hypohidrosis or anhidrosis) and in the peripheral nervous system (15).
In adulthood, patients show manifestations of FD in the cardiac, cerebrovascular, and renal systems (16–18). In particular, renal impairment is one of most pervasive signs of FD (19). Firstly, a glomerular hyperfiltration, likely due to the activation of the sympathetic nervous system, increased oxidative stress and inflammatory cytokines, associated with a mesangial cell proliferation occurs (20). Later, early morphologic findings of impaired renal function, such as the development of a focal and segmental glomerulosclerosis and vascular changes, are observed (21). With the progression of the disease, proteinuria and progressive renal failure leading to dialysis (ESRD) appears in ~50% and 20% of male and female FD patients, respectively (22).
This clinical picture, which is considered the classic type of FD, leads to death of male patients during the fourth or fifth decade of life (23). By contrast, female patients commonly show milder involvement due to random inactivation of the X-chromosome, determining the development of organs which are chimeras of normal and affected cells (24).
Besides the classic type, milder and late-onset forms of FD have been described as atypical variants. They are characterized by non-specific symptoms and/or involvement of single organ, also defined cardiac and renal variants (25). Therefore, diagnosis of FD in patients with atypical variants is often difficult and it is generally made in the more advanced stages of disease (26).
However, early diagnosis is compulsory in patients with FD since early therapeutic interventions, such as enzymatic replacement therapy and chaperone therapy, significantly stabilize the disease and prevent the progression of renal, cerebrovascular and cardiovascular complications (27–29).
Screening in CKD Patients Not on Dialysis
Unfortunately, the true prevalence of FD is not well defined. Initially, underestimated prevalence (1:117,000–1:833,00) of FD was found in general population (30–32), mainly because of the lack of clear genotype-phenotype correlation. Subsequently, large new-born and high-risk screening studies showed higher FD prevalence than previously expected. A recent re-analysis of studies for pathogenic GLA mutation, conducted on high-risk populations, showed that the prevalence of FD in 5,491 (4,054 males and 1,437 female) patients with no definitive cause of left ventricular hypertrophy was 0.94% in males and 0.90% in females and among the 5,978 (3,904 males and 2,074 females) young patients with unexplained stroke, 0.13% were males and 0.14% were females (33) (more details are described in the next paragraphs). Nevertheless, few researchers have addressed the problem of screening in NDD-CKD population to detect patients affected by FD, determining its prevalence.
Although the ERBP guidelines (34), in 2013, recommended screening for FD in NDD-CKD patients, no study had been published to support this ungraded statement at the time. However, a singular study performed by Andrade et al. (35), screened 499 renal male patients (of which 141 not on dialysis, 159 on hemodialysis, 59 on peritoneal dialysis, and 138 with a kidney transplant) by plasma GLA assay in a large Canadian province (population ~4.25 million). No new FD cases were found despite the fact that multiple racial populations, including 71 white and 26 Asian, were exanimated.
For the first time in 2016, Turkmen et al. (36) showed in their TURKFAB Study the FD prevalence was 0.95% (3/313) in stage 1–5 NDD-CKD patients with unknown etiology of 10 Turk centers. Furthermore, 8 other patients with both FD and CKD were identified from two index patients, for a total of 11 cases of 386 screened people.
To date, only four FD screening studies reported data about CKD patients, the TURKFAB Study (T1) described above (36), one conducted in the Aegean region of Turkey (T2) (37), one in Taiwan (T3) (38) and, recently, one in Canada (C1) (39) (Table 1).
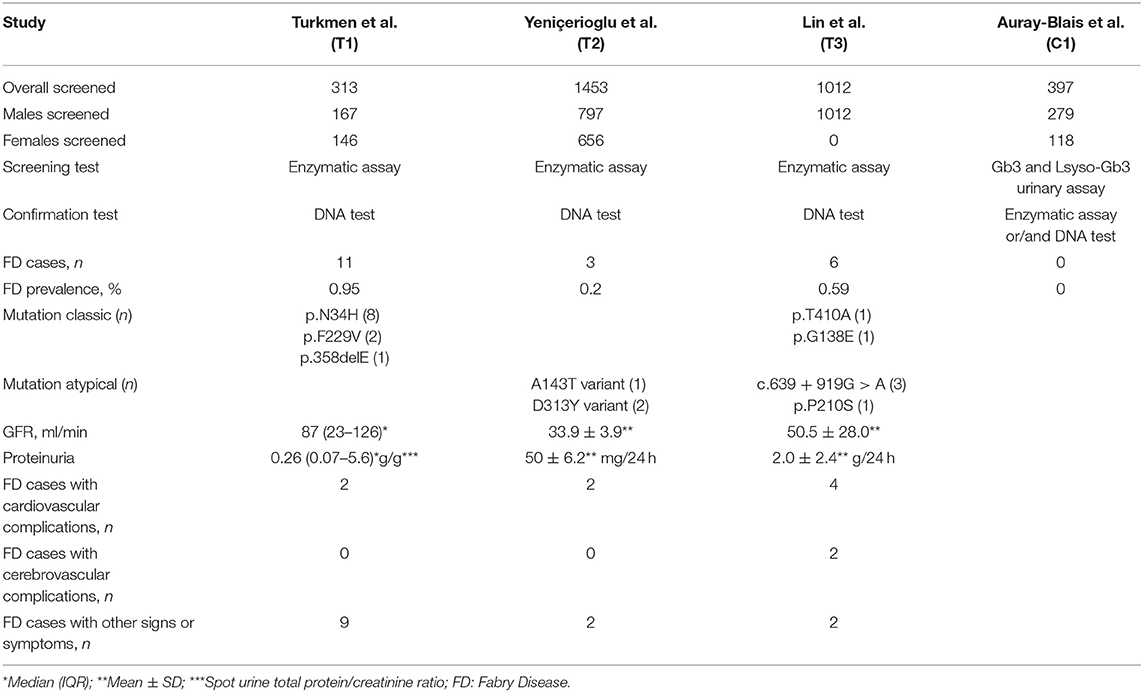
Table 1. Characteristics of Fabry disease screening studies in chronic kidney disease patients not on dialysis.
In a total of 3,175 (313 in T1; 1,453 in T2; 1,012 in T3; 397 in C1) follow-ups of stable CKD (3–5 stages in T2 and C1; 1–5 stages in T1 and T3) patients in 23 centers (10 in T1; 7 in T2; 2 in T3; 4 in C1), the prevalence ranged between 0% and 0.95%, of which 0.4–1.8% and 0.0–1.0% in 2,255 males and 920 females, respectively.
Surprisingly, a slightly lower average age of 16 male cases (47 years) compared with 4 female cases (49 years) was identified. This finding is in contradiction with the suggestion of performing FD screening in men under 50 years and in all ages for women, according to ERBP working groups (34). Furthermore, stratifying the cases by type of GLA mutation, 9 males with classic mutations were younger than 7 males with atypical mutations (35 and 62 years, respectively). This result seems to indicate that FD screening should be conducted in men of all ages to identify not only the classical GLA mutations, but also the late-onset GLA mutations.
All CKD patients were screened by analyzing GLA activity in dried venous blood spots (DBS) and, when enzyme activity was lower than 1.2 micromole/L/h, the GLA gene was sequenced, except for 397 patients of C1 study in which an innovative method, the dosage of Gb3 and lyso-GB3 in dried urine spots, was used. Although the enzymatic activity is normal or slightly lower in roughly 30% of FD females, only one study (T3) excluded female to avoid false-negative results. Accordingly, the prevalence of FD could be underestimated in approximately one-third (females: 146 in T1; 656 in T2) of screened CKD patients. Furthermore, in the Canadian study (C1) 43% of suspected FD patients, screened with the urine test, died in the course of study before an enzymatic or genetic test was performed.
Ancillary, Nakagawa et al. (40) screened 2,325 Japanese patients affected by typical cardiac, renal, or neurological FD complications, including 374 (16.1%) subjects with proteinuria of which 129 (34.5%) were female and 245 (65.5%) were male. Of ten patients with GLA mutations (6/10 were pathogenic), two had a nephrotic range proteinuria, namely a 37-year-old female with juvenile minor ischemic stroke and a 56-year-old male with CKD. Although data of renal function among the screened are missing, this study highlights the importance of investigations not only in CKD patients with unknown causes, but also in the presence of proteinuria, an early finding of renal damage. Furthermore, the results of the ongoing study, HISTOFAB2 (ClinicalTrials.gov identifier: NCT03869554), might improve the understanding of the FD screening benefits also in CKD patients with undetermined etiology after a renal biopsy.
Finally, in order to help identify suspected FD patients, efficient and convenient screening protocols have been proposed (34, 41) and on the basis of available data, a comprehensive flow-chart is shown in Figure 1.
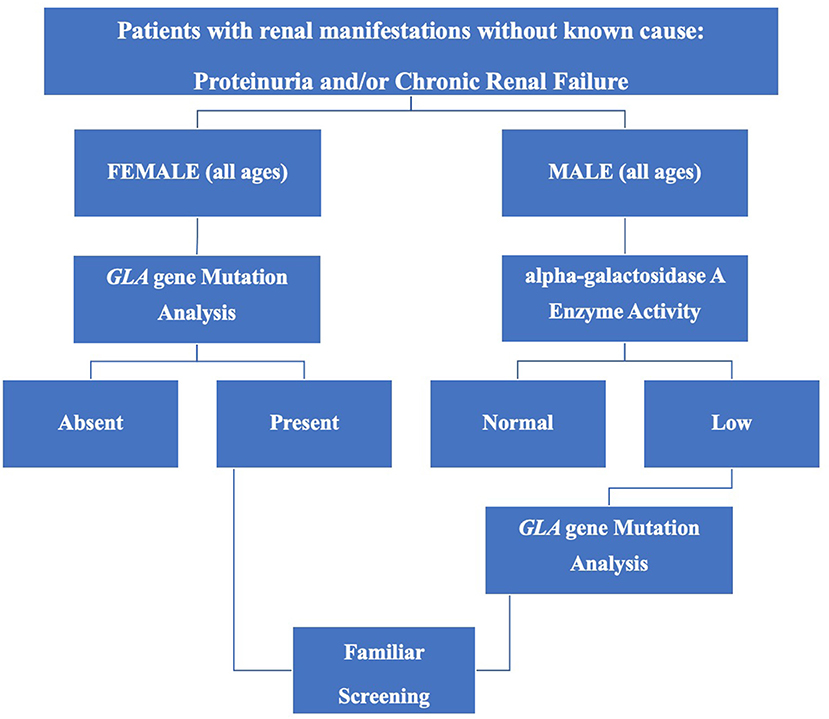
Figure 1. Screening for Fabry disease in chronic kidney disease patients not on dialysis.
High-Risk Screening in Dialysis Patients and Kidney Transplant Recipients
In CKD population, most screening studies have been carried out in patients receiving hemodialysis treatment or after renal transplantation (42). Doheny et al. (33) showed an FD prevalence of 0.21% in 23,954 male dialysis patients (of these 66% had the classic phenotype and 34% had late-onset phenotype) and 0.15% in 12,866 female dialysis patients (of these 68.4 and 31.6% had classic and late-onset phenotype, respectively) among the 27 reviewed studies. Also, 5 (0.24%) pathogenic mutations among the 2,031 KTR males were reported; and, none among the 1,043 KTR females was found. By contrast, other recent studies demonstrated a higher percentage of GLA mutations in females compared to males (43). Notably, of the 265 KTR screened, Veroux et al. (44) identified six patients (one male and five female) with a pathogenic GLA mutation (2.3%) and one female patient with a benign variant of GLA mutation (0.3%). This higher incidence (2.3%) of KTRs with undetected FD could be explained by the presence of many clusters of FD in the population and by the homogeneity of cohort.
Finally, high-risk screening in KTRs and dialysis patients is able to facilitate the detection of unrecognized FD patients even though the CKD and FD stages in these patients are remarkably advanced with a high risk of adverse outcomes and a poor efficacy of delayed treatment.
Family Screening
In order to increase the diagnostic rate of FD, currently, another different approach, the family screening, is suggested (45). It is crucial for X-linked transmission of FD. Indeed, for an index case with FD, a pedigree on average three generations is usually performed and approximately five family members are identified (46). Secondary analysis of some studies reported that the prevalence of FD among relatives of FD patients was about 2%, with higher percentage in males (47). By contrast, in a recent family screening study (48), of 1,214 relatives of 71 CKD patients with FD, it was found that the majority of the 115 (9.74%) family members with a GLA mutation were women (66.1%) and under 44 of age (74%). This conflicting data is not surprising since most of the screenings were performed with GLA activity dosage and/or with female exclusion. It may be related to lower costs of enzymatic analysis, which is adequate for screening diagnosis in males, compared to genetic sequencing, which is mandatory in females that often have normal GLA activity because of lyonization (22). However, the GLA genotyping should be performed in the patients screened with null or low GLA activity, to distinguish among the classic, the late-onset, and the benign mutations (49).
On the other hand, other studies reported only the number of cases, ranging between 8 and 24 cases (50–53). Recently, in a kidney transplant (KTR) screening study (44), seven FD probands' relatives were evaluated, finding ten females (five sisters, four daughters, and one nephew) and two males (one brother and one son) with pathogenic GLA mutation. 5/12 FD relatives received an early treatment before irreversible organ damage. This data confirms that the family screenings are able to delay the progression of FD and lead to better long-term outcomes.
New-Born Screening
In Northern Italy, Spada et al. conducted the first screening study to detect FD in the newborns, finding an incidence of GLA deficiency at 1/3,100. Most of identified GLA mutations were likely to result in atypical phenotype (54). Subsequently, several new-born screening studies confirmed higher prevalence of both FD classic males (ranging from 1:22,000 to 1:40,000) and FD atypical phenotype (1:1,000–1:3,000 in males and 1:6,000–1:40,000 in females) compared to other strategy of screening (55, 56).
However, according to the European Renal Best Practice (ERBP) (34) and the KDIGO controversies conference (45), the new-born screening to diagnose FD patients is not justified in general population for various clinical and ethical issues, including the high prevalence of atypical forms observed in the infants, as reported above. Although Hsu et al. highlighted that patients with late-onset phenotype are likely to have a silent progression of irreversible organ damage (57), the optimal time for screening late-onset variants remains debated. The best strategy to avoid irreversible damage in FD adults is an early diagnosis and effective treatment, but only a better characterization of the natural history of atypical FD forms could make the use of newborn screening advantageous.
Conclusion
Although expert working groups strongly recommend improvement in the screening process, few FD screening studies have been published for NDD-CKD patients and the data of FD prevalence is inconclusive. However, the importance of early detection of both CKD and FD is fundamental to start efficient treatment and to reduce the rate of morbidity and mortality. Clinicians should become conscious of the fact that FD needs to be taken into account in the differential diagnosis of CKD patients with unknown etiology even without other FD manifestations. Further multicenter systematic large-scale screening studies must be encouraged to assess the value of this screening strategy and to determine the true prevalence of FD in NDD-CKD patients.
Author Contributions
YB: conceptualization. CA and AD: investigation. YB and PE: writing and editing. FF and AG: supervision. AS and RM: validation. All authors have read and agreed to the published version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
1. ^Available online at: https://apps.who.int/iris/bitstream/10665/37650/17/WHO_PHP_34.pdf (accessed December 1, 2020).
2. ^Available online at: https://www.clinicaltrials.gov/ct2/show/study/NCT03869554?term=screening&cond=Fabry$+$Disease&draw=2&rank=5 (accessed December 1, 2020).
References
1. Morrison AS. Screening. In: Rothman KJ, Greenland S, editors. Modern Epidemiology. 2nd ed. Philadelphia: Lippincott Williams & Wilkins (1998). p. 499–518.
2. Couser WG, Remuzzi G, Mendis S, Tonelli M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int. (2011) 80:1258–70. doi: 10.1038/ki.2011.368
3. GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. (2020) 395:709–33. doi: 10.1016/S0140-6736(20)30045-3
4. Battaglia Y, Esposito P, Corrao S, Russo L, Balducci A, Storari A, et al. On behalf of the Italian Kidney Foundation “FIR-Onlus”. Evaluation of hypertension, proteinuria, and abnormalities of body weight in italian adolescents participating in the world kidney days. Kidney Blood Press Res. (2020) 45:286–96. doi: 10.1159/000502547
5. Battaglia Y, Russo L, Spadola R, Russo D, Local Coordinators. Awareness of kidney diseases in general population and in high school students. Italian report for World Kidney Days 2010-2011. J Nephrol. (2012) 25:843–6. doi: 10.5301/JN.2012.9486
6. Russo D, Del Prete M, Battaglia Y, Russo L. Risk for chronic kidney disease in high school students: Italian report for world kidney day 2008-2009. J Nephrol. (2011) 24:250–3. doi: 10.5301/JN.2011.6370
7. Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al. Chronic kidney disease: global dimension and perspectives. Lancet. (2013) 382:260–72 doi: 10.1016/S0140-6736(13)60687-X
8. Garofalo C, Borrelli S, Pacilio M, Minutolo R, Chiodini P, De Nicola L, et al. Hypertension and prehypertension and prediction of development of decreased estimated GFR in the general population: a meta-analysis of cohort studies. Am J Kidney Dis. (2016) 67:89–97. doi: 10.1053/j.ajkd.2015.08.027
9. Garofalo C, Borrelli S, Minutolo R, Chiodini P, De Nicola L, Conte G. A systematic review and meta-analysis suggests obesity predicts onset of chronic kidney disease in the general population. Kidney Int. (2017) 91:1224–35. doi: 10.1016/j.kint.2016.12.013
11. Kornreich R, Desnick RJ, Bishop DF. Nucleotide sequence of the human α-actosidase A. Nucleic Acids Res. (1989) 17:3301. doi: 10.1093/nar/17.8.3301
12. Duro G, Zizzo C, Cammarata G, Burlina A, Burlina A, Polo G, et al. Mutations in the GLA gene and LysoGb3: is it really Anderson-Fabry disease? Int J Mol Sci. (2018) 19:3726. doi: 10.3390/ijms19123726
13. Desnick RJ, Ioannou YA, Eng CM. Alpha-galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. 8th ed. New York, NY: McGraw-Hill (2001). p. 3733–74.
14. Bernardini A, Camporeale A, Pieroni M, Pieruzzi F, Figliozzi S, Lusardi P, et al. Atrial dysfunction assessed by cardiac magnetic resonance as an early marker of Fabry cardiomyopathy. JACC Cardiovasc Imaging. (2020) 13:2262–4. doi: 10.1016/j.jcmg.2020.05.011
15. Colomba P, Scalia S, Cammarata G, Zizzo C, Francofonte D, Savica V, et al. Fabry disease, a complex pathology not easy to diagnose. Cardiogenetics. (2015) 5:5612. doi: 10.4081/cardiogenetics.2015.5612
16. Cocozza S, Olivo G, Riccio E, Russo C, Pontillo G, Ugga L, et al. Corpus callosum involvement: a useful clue for differentiating Fabry disease from multiple sclerosis. Neuroradiology. (2017) 59:563–70. doi: 10.1007/s00234-017-1829-8
17. Pisani A, Petruzzelli Annicchiarico L, Pellegrino A, Bruzzese D, Feriozzi S, Imbriaco M, et al. Parapelvic cysts, a distinguishing feature of renal Fabry disease. Nephrol Dial Transplant. (2018) 33:318–23. doi: 10.1093/ndt/gfx009
18. Figliozzi S, Camporeale A, Pieroni M, Pieruzzi F, Namdar M, Lusardi P, et al. Progressive electrocardiographic changes in parallel with cardiac magnetic resonance findings in fabry disease. Eur Heart J. (2020) 41 (Suppl. 2):ehaa946.2125. doi: 10.1093/ehjci/ehaa946.2125
19. Battaglia Y, Scalia S, Rinaldi R, Storari A, Mignani R, Russo D, et al. Identification of new α-galactosidase A mutation responsible for Fabry disease: a case report. Clin Nephrol. (2019) 91:126–8. doi: 10.5414/CN109501
20. Riccio E, Sabbatini M, Bruzzese D, Annicchiarico Petruzzelli L, Pellegrino A, Spinelli L, et al. Glomerular hyperfiltration: an early marker of nephropathy in Fabry disease. Nephron. (2019) 141:10–7. doi: 10.1159/000493469
21. Svarstad E, Marti HP. The changing landscape of Fabry disease. Clin J Am Soc Nephrol. (2020) 15:569–76. doi: 10.2215/CJN.09480819
22. Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry registry. Fabry Registry Mol Genet Metab. (2008) 93:112. doi: 10.1016/j.ymgme.2007.09.013
23. Branton M, Schiffmann R, Kopp JB. Natural history and treatment of renal involvement in Fabry disease. J Am Soc Nephrol. (2002) 13 (Suppl. 2):S139–43 doi: 10.1097/01.ASN.0000016683.73778.78
24. Echevarria L, Benistan K, Toussaint A, Dubourg O, Hagege AA, Eladari D, et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. (2015) 89:44–54. doi: 10.1111/cge.12613
25. Sessa A, Meroni M, Battini G, Amato M, Righetti M, Bertella M, et al. ‘Atypical’ clinical variants of Anderson-Fabry disease. Nephron. (2001) 89:469–70 doi: 10.1159/000046124
26. Hoffmann B, Mayetepek E. Fabry disease-often seen, rarely diagnosed. Dtsch Arztebl Int. (2009) 106:440–7. doi: 10.3238/arztebl.2009.0440
27. Tøndel C, Bostad L, Larsen KK, Hirth A, Vikse BE, Houge G, et al. Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol. (2013) 24:137–48. doi: 10.1681/ASN.2012030316
28. Chimenti C, Nencini P, Pieruzzi F, Feriozzi S, Mignani R, Pieroni M, et al. The GALA project: practical recommendations for the use of migalastat in clinical practice on the basis of a structured survey among Italian experts. Orphanet J Rare Dis. (2020) 15:86. doi: 10.1186/s13023-020-1318-8
29. Mignani R, Pieroni M, Pisani A, Spada M, Battaglia Y, Verrecchia E, et al. New insights from the application of the FAbry STabilization indEX in a large population of Fabry cases. Clin Kidney J. (2018) 12:65–70. doi: 10.1093/ckj/sfy108
30. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. (1999) 281:249–54. doi: 10.1001/jama.281.3.249
31. Fuller M, Meikle PJ, Hopwood JJ. Epidemiology of lysosomal storage diseases: an overview. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis Ltd. (2006).
32. Pinto R, Caseiro C, Lemos M, Lopes L, Fontes A, Ribeiro H, et al. Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet. (2004) 12:87–92. doi: 10.1038/sj.ejhg.5201044
33. Doheny D, Srinivasan R, Pagant S, Chen B, Yasuda M, Desnick RJ. Fabry disease: prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995–2017. J Med Genet. (2018) 55:261–8. doi: 10.1136/jmedgenet-2017-105080
34. Terryn W, Cochat P, Froissart R, Ortiz A, Pirson Y, Poppe B, et al. Fabry nephropathy: indications for screening and guidance for diagnosis and treatment by the European renal best practice. Nephrol Dial Transplant. (2013) 28:505–17. doi: 10.1093/ndt/gfs526
35. Andrade J, Waters PJ, Singh RS, Levin A, Toh BC, Vallance HD, et al. Screening for Fabry disease in patients with chronic kidney disease: limitations of plasma alpha-galactosidase assay as a screening test. Clin J Am Soc Nephrol. (2008) 3:139–45. doi: 10.2215/CJN.02490607
36. Turkmen K, Guclu A, Sahin G, Kocyigit I, Demirtas L, Erdur FM, et al. The prevalence of Fabry disease in patients with chronic kidney disease in Turkey: the TURKFAB study. Kidney Blood Press Res. (2016) 41:1016–24. doi: 10.1159/000452605
37. Yeniçerioglu Y, Akdam H, Dursun B, Alp A, Saglam Eyiler F, Akin D, et al. Screening Fabry's disease in chronic kidney disease patients not on dialysis: a multicenter study. Ren Fail. (2017) 39:104–11. doi: 10.1080/0886022X.2016.1254656
38. Lin CJ, Chien YH, Lai TS, Shih HM, Chen YC, Pan CF, et al. Results of Fabry disease screening in male pre-end stage renal disease patients with unknown etiology found through the platform of a chronic kidney disease education program in a Northern Taiwan Medical Center. Kidney Blood Press Res. (2018) 43:1636–45. doi: 10.1159/000494678
39. Auray-Blais C, Lavoie P, Abaoui M, Côté AM, Boutin M, Akbari A, et al. High-risk screening for Fabry disease in a Canadian cohort of chronic kidney disease patients. Clin Chim Acta. (2020) 501:234–40. doi: 10.1016/j.cca.2019.10.045
40. Nakagawa N, Sawada J, Sakamoto N, Takeuchi T, Takahashi F, Maruyama JI, et al. High-risk screening for Anderson-Fabry disease in patients with cardiac, renal, or neurological manifestations. J Hum Genet. (2019) 64:891–8. doi: 10.1038/s10038-019-0633-1
41. Auray-Blais C, Millington DS, Young SP, Clarke JT, Schiffmann R. Proposed high-risk screening protocol for Fabry disease in patients with renal and vascular disease. J Inherit Metab Dis. (2009) 32:303–8. doi: 10.1007/s10545-009-1055-6
42. Linthorst GE, Bouwman MG, Wijburg FA, Aerts JM, Poorthuis BJ, Hollak CE. Screening for Fabry disease in high-risk populations: a systematic review. J Med Genet. (2010) 47:217–22. doi: 10.1136/jmg.2009.072116
43. Erdogmus S, Kutlay S, Kumru G, Ors Sendogan D, Erturk S, Keven K, et al. Fabry disease screening in patients with kidney transplant: a single-center study in Turkey. Exp Clin Transplant. (2020) 18:444–9. doi: 10.6002/ect.2019.0279
44. Veroux M, Monte IP, Rodolico MS, Corona D, Bella R, Basile A, et al. Screening for Fabry disease in kidney transplant recipients: experience of a multidisciplinary team. Biomedicines. (2020) 8:396. doi: 10.3390/biomedicines8100396
45. Schiffmann R, Hughes DA, Linthorst GE, Ortiz A, Svarstad E, Warnock DG, et al. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a “kidney disease: improving global outcomes” (KDIGO) controversies conference. Kidney Int. (2017) 91:284–93. doi: 10.1016/j.kint.2016.10.004
46. Laney DA, Fernhoff PM. Diagnosis of Fabry disease via analysis of family history. J Genet Couns. (2008) 17:79–83. doi: 10.1007/s10897-007-9128-x
47. Nampoothiri S, Yesodharan D, Bhattacherjee A, Ahamed H, Puri RD, Gupta N, et al. Fabry disease in India: a multicenter study of the clinical and mutation spectrum in 54 patients. JIMD Rep. (2020) 56:82–94. doi: 10.1002/jmd2.12156
48. Sodré LSS, Huaira RMNH, Colugnati FAB, Carminatti M, Braga LSS, Coutinho MP, et al. Screening of family members of chronic kidney disease patients with Fabry disease mutations: a very important and underrated task. J Bras Nefrol. (2020). doi: 10.1590/2175-8239-jbn-2020-0080. [Epub ahead of print].
49. Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, et al. Natural history of Fabry renal disease. Influence of a-galactosidase A activity and genetic mutations on clinical course. Medicine. (2001) 81:122–38. doi: 10.1097/00005792-200203000-00003
50. Bolasco P, Sitzia I, Murtas S. Fabry disease in Southern Sardinia: epidemiological results from screening in an extensive area. Giornale Italiano di Nefrologia. (2017) 34:83–102.
51. Herrera J, Miranda CS. Prevalence of Fabry's disease within hemodialysis patients in Spain. Clin Nephrol. (2014) 81:112–20. doi: 10.5414/CN108053
52. Gutiérrez-Amavizca BE, Orozco-Castellanos R, Padilla-Gutiérrez JR, Valle Y, Figuera LE. Pedigree analysis of Mexican families with Fabry disease as a powerful tool for identification of heterozygous females. Genet Mol Res. (2014) 13:6752–8. doi: 10.4238/2014.August.28.19
53. Silva CAB, Barreto FC, Reis MA, Moura Junior JA, Cruz CMS. Targeted screening of Fabry disease in male hemodialysis patients in Brazil highlights importance of family screening. Nephron. (2016) 134:221–30. doi: 10.1159/000448740
54. Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, et al. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. (2006) 79:31–40. doi: 10.1086/504601
55. Wasserstein MP, Caggana M, Bailey SM, Desnick RJ, Edelmann L, Estrella L, et al. The New York pilot newborn screening program for lysosomal storage diseases: report of the first 65,000 infants. Genet Med. (2019) 21:631–40. doi: 10.1038/s41436-018-0129-y
56. Colon C, Ortolano S, Melcon-Crespo C, Alvarez JV, Lopez-Suarez OE, Couce ML, et al. Newborn screening for Fabry disease in the north-west of Spain. Eur J Pediatr. (2017) 176:1075–81. doi: 10.1007/s00431-017-2950-8
Keywords: Fabry disease, chronic kidney disease, screening, prevention, lysosomal disorders
Citation: Battaglia Y, Fiorini F, Azzini C, Esposito P, De vito A, Granata A, Storari A and Mignani R (2021) Deficiency in the Screening Process of Fabry Disease: Analysis of Chronic Kidney Patients Not on Dialysis. Front. Med. 8:640876. doi: 10.3389/fmed.2021.640876
Received: 12 December 2020; Accepted: 05 January 2021;
Published: 09 February 2021.
Edited by:
Michele Provenzano, University of Catanzaro, ItalyReviewed by:
Adamasco Cupisti, University of Pisa, ItalySilvio Borrelli, University of Campania Luigi Vanvitelli, Italy
Copyright © 2021 Battaglia, Fiorini, Azzini, Esposito, De vito, Granata, Storari and Mignani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuri Battaglia, eXVyaS5iYXR0YWdsaWFAb3NwZmUuaXQ=